
Circulating Molecular Chaperones in Subjects with Amnestic Mild Cognitive Impairment and Alzheimer’s Disease: Data from the Zabùt Aging Project
Abstract
Molecular chaperones play essential roles in many processes such as cell differentiation, tissue homeostasis, and organ remodeling. Recent data indicate that chaperones can act as cytoprotectants for brain cells during the progression of neurodegenerative diseases, including Alzheimer’s disease (AD). However, very few data on the levels of chaperones in dementia, including its prodromal phases, have been reported. In this study, we used biological samples and epidemiological data collected during the Zabùt Aging Project (a prospective, community-based, cohort study of normal/pathological aging conducted in Sicily, Italy, with a follow-up of ten years) to determine if there is an association between plasma levels of the chaperones Hsp60, Hsp70, and Hsp90 with amnestic mild cognitive impairment (aMCI) and AD. Twenty-six aMCI individuals, 26 AD and 26 controls, matched for age and sex, were enrolled. After adjustment for education, subjects with AD showed significantly higher levels of Hsp60 than aMCI (OR = 1.16, 95% CI 1.04–1.30) and controls (OR = 1.12, 95% CI 1.03–1.22), while Hsp70 was significantly higher only in AD (OR = 1.84, 95% CI 1.09–3.10) than controls. In contrast, circulating levels of Hsp90 were significantly diminished in aMCI (OR = 0.69, 95% CI 0.52–0.91) and AD (OR = 0.51, 95% CI 0.35–0.75) compared to controls. However, these results were no longer significant after adjustment for multiple comparisons. Although the results lost significance after adjustment for multiple comparisons, they are encouraging despite the smallness of the sample and new studies should be carried out with larger populations to determine to what extent sequential measurement of serum chaperones in aMCI and AD can be trusted as indicators of disease status and progression.
INTRODUCTION
Alzheimer’s disease (AD) is the most common type of dementia accounting for nearly 60% of all cases [1]. According to the NIA-AA criteria of 2011 [2], the diagnosis of AD dementia is confirmed when the symptoms have a gradual onset over months to years and a clear-cut history of worsening of cognition which interfere with daily living, social and professional activities.
AD is a proteinopathy whose neuropathology consists of cortical extracellular deposits of amyloid protein called senile or neuritic plaques, and intraneuronal inclusions, called neurofibrillary tangles formed by accumulation of abnormal filaments of the tau protein, both found predominately in the brain regions involved in memory and learning [3, 4].
Given that AD is a proteinopathy, it follows that molecular chaperones must be involved in some way, since they are key players in the maintenance of protein homeostasis. Molecular chaperones, many of which are heat shock proteins (Hsps), are the main components of the chaperoning system and play a variety of roles, in addition to maintaining protein homeostasis, and thus they ensure correct cell differentiation, tissue homeostasis, and organ remodeling during the entire lifespan [5]. The chaperones, also referred to as Hsps, 60– KDa (Hsp60, or HSPD1), 70– KDa Hsp (Hsp70, or HSPA1), and 90– KDa Hsp (Hsp90, or HSPC1) are among the most important in brain tissue as indicated by the fact that they are highly responsive in stressed neurons and activated glia [6].
Molecular chaperones overexpression is considered often a mechanism that protects cells from stressful events such as heat shock, oxidative stress, and hypoxia. An increase in the intracellular levels of chaperones is at times followed by their active secretion, via various pathways, into intercellular fluids [7]. Secreted chaperones may reach the circulation, in which they can be detected and measured. During acute and chronic stress in nervous tissue, molecular chaperones mediate the refolding or degradation of stress-damaged proteins, protecting the nervous cells from potential deleterious effects of unfolded-damaged proteins that can form aggregates, and in turn promoting cell resistance and recovery [8]. As a consequence, the cytoprotective effects of the heat shock response are seen as an attractive tool for chaperone-driven therapies, i.e., chaperonotherapy [9], as supported also by the fact that Hsp60 has already been shown to efficiently inhibit amyloid-β (Aβ) aggregation [10]. Indeed, AD, as other neurodegenerative diseases, is characterized by the accumulation of aberrantly folded protein species, including Aβ [11– 14] and it is highly probable that a failure of the chaperoning system could contribute to the pathogenic mechanism of AD.
Very few data are available regarding the circulating levels of Hsp60, Hsp70, and Hsp90 in subjects with dementia, and even less information exists pertaining to subjects with mild cognitive impairment (MCI), a clinical syndrome that is today considered a prodromal phase of AD [15, 16]. Therefore, it is justified to consider chaperones as disease biomarkers because by monitoring their levels with time, information may be obtained on the progression and severity of regional brain pathology in subjects with dementia. Furthermore, the results should provide tenets for research aimed at elucidating the possible role, cytoprotective and/or pathogenic, of chaperones in AD.
The aims of this study were to: 1) measure levels of Hsp60, Hsp70, and Hsp90 in plasma of patients with cognitive impairment of the AD-type, compared to controls; 2) establish if there is association between Hsps levels and clinical conditions (i.e., amnestic MCI [aMCI] and AD); and 3) determine if there is a relationship between Hsps levels and other biochemical parameters.
MATERIALS AND METHODS
Subject recruitment and diagnostic criteria
Twenty-six AD individuals, 26 aMCI and 26 CONS (cognitively normal individuals without functional impairment) matched for age and sex, were enrolled during the 10-year follow-up of the Zabùt Aging Project (ZAP). The latter is a population-based cohort study conducted in low-educated, rural subjects living in Sambuca di Sicilia, a small Sicilian town in the province of Agrigento, Italy.
All subjects were evaluated with a comprehensive, standard protocol, including a detailed anamnesis with questions concerning housing, lifestyles, life habits, and physical functioning. Furthermore, participants underwent a clinical examination by physicians who assessed past and present health status including psychiatric and neurological examinations. Finally, a comprehensive cognitive battery covering memory, attention/visuo-constructional abilities, language, and executive functioning was carried out by psychologists trained in neuropsychology.
The diagnosis of dementia was made according to the DSM-IV-TR criteria [17] and probable AD was diagnosed according to the criteria established by the National Institute on Aging and the Alzheimer’s Association [2]. Exclusion criteria were: dementia due to other general medical conditions (DSM-IV-TR code 294.1), such as head trauma, severe metabolic or endocrine diseases, brain tumor, stroke, normal pressure hydrocephalus, and other neurological disorders as well as subjects with a history of alcohol or substance abuse or dependence, with uncertain dementia, with delirium and with psychiatric disorders preceding the onset of dementia.
aMCI was classified according to modified Petersen’s criteria [18] as follows: 1) self and/or informant report of memory problems; 2) impaired performance in memory test according to age- and education-adjusted Italian normative data as well as normality cut-off scores (≥95% of the lower tolerance limit of the normal population distribution) [19]; 3) preserved general cognitive functions (Mini-Mental State Examination age- and education-adjusted scores) [20]; 4) cognitive deterioration, representing a decline from a previously higher ability level (Clinical Dementia Rating = 0.5) [21]; 5) generally preserved basic activities of daily living (ADL) [22] with minimal impairment regarding instrumental ADL (IADL) [23]; 6) no dementia (DSM-IV TR criteria) [17] or clinical history of stroke. Subjects with aMCI and AD underwent also routine 1.5T MRI scan to exclude relevant vascular lesions, which would affect cognition, as previously detailed [24].
Behavioral symptoms were assessed using the total composite score (range 0– 144) of the Neuropsychiatric Inventory (NPI) [25], a fully structured caregiver interview with 12 sub-scales measuring different behavioral symptoms of dementia, with higher scores indicating greater behavioral burden. To assess the risk of malnutrition we used the Mini Nutritional Assessment (MNA), a validated nutrition screening and assessment tool for geriatric patients age 65 and above [26].
All subjects underwent blood screening for dementia, genomic DNA extraction and APOE genotyping as previously described [27]. All analyses were conducted on E4-carrying (E2/E4, E3/E4, E4/E4) versus non-E4 carrying genotypes. The study was in agreement with the principles outlined in the Helsinki declaration and all participants or their caregiver provided written informed consent. The ZAP study design was approved by the local ethical committee ASP– 1 (“Azienda Sanitaria Provinciale”, i.e., provincial health authority) of Agrigento, Italy, and general authorization for the genetic data treatment was provided by the Italian data Protection Authority.
Biochemical parameters and comorbidity
Venous blood samples were obtained in the morning after an overnight fast. Plasma total cholesterol (TC), triglycerides (TG), high-density lipoprotein cholesterol (HDL), and glucose levels were measured using standard enzymatic methods. Low density lipoprotein cholesterol (LDL) was calculated using Friedewald’s formula: LDL (mg/dL) = TC (mg/dL) – TG (mg/dL)/5 – HDL (mg/dL). High sensitivity C-reactive protein was measured by a chemiluminescent immunoassay (Immulite, Medical Systems, Italy).
The following vascular diseases were assessed:
– hypertension (blood pressure≥140/90 mmHg or current use of antihypertensive medications) [28];
– dyslipidemia (fasting TC≥240 mg/dL and/or HDL <40 mg/dL in men and <50 mg/dL in women, LDL≥160 mg/dL, and/or TG≥150 mg/dL) [29];
– obesity body mass index (BMI)≥30 kg/m2 [30],
– diabetes mellitus (fasting blood glucose levels≥110 mg/dL and/or current use of hypoglycemic drugs) [29];
– atrial fibrillation and ischemic heart disease (myocardial infarction, angina, coronary artery bypass grafting or angioplasty or stenting) had to be supported by clinical and/or instrumental records.
Finally, the Cumulative Illness Rating Scale (CIRS) was used to quantify somatic comorbidity. It assigns a score to the total burden of illness of 13 body systems ranging from no disease (score = 1) to life-threatening disease (score = 5) [31].
Measurement of Hsps
ELISA test was performed as previously described [32]. A blood sample from each subject was collected into EDTA-treated tubes. After a centrifugation for 10 min at 2,000xg using a refrigerated centrifuge, plasma was collected, aliquoted, and stored at – 20°C until use. Hsp90α, Hsp70, and Hsp60 were measured using commercial quantitative, sandwich, enzyme-linked immunosorbent assay kit (EIA, Enzo Life Sciences, Vinci, Italy), according to the manufacturer’s instructions. The standards were diluted in standard diluent to generate standard curves for each protein, ranging from 0.625 ng/ml to 4 ng/ml for Hsp90α, from 0.20 to 125 ng/ml for Hsp70, and from 3.125 to 100 ng/ml for Hsp60. Then, 100μl of standards and samples were added to the immunoassay plates and incubated at 23°C for 1 h. After diluting the primary and secondary antibodies according to the manufacturer’s instructions, 100μl of specific primary antibody for each protein was added to each well and incubated at 23°C for 1 h. Subsequently, 100μl of specific horseradish peroxidase conjugate secondary antibody was added to the plate and incubated at 23°C for 30 min followed by 100μl of 3,3′,5,5′– tetramethylbenzidine substrate for 15 min in the dark.
Finally, 100μl of Stop Solution was added, and absorbance was measured at 450 nm with a microplate photometric reader (DV990BV4, GDV, Milan, Italy). Sample concentration was calculated by interpolating the sample measurement in the standard curve. The sensitivity of the human Hsp90α EIA kit has been determined to be 0.05 ng/ml, for human Hsp70 EIA kit has been reported to be 0.09 ng/ml, while for human Hsp60 EIA kit was determined to be 3.125 ng/ml.
Statistical analysis
Normal distribution and homogeneity of variables were tested with Kolmogorov-Smirnov and Levene’s test, respectively. Mean or median values, SDs or interquartile ranges were calculated for quantitative variables with normal or not normal distribution. Differences among normal variables were compared by one-way analysis of variance (ANOVA) with Scheffe’s post-hoc test, Mann-Whitney test for not normal variables, and Chi2 test for categorical variables. The variables which were significantly associated with study outcomes (aMCI and AD) by univariate analysis (p≤0.05) were fitted with backward multiple logistic regression models, calculating odds ratios (OR) with 95% confidence intervals (95% CI). Model 1 was adjusted only for education, while model 2 was further adjusted for continuous and dichotomic variables, which were found to be significant at univariate analysis. Due to collinearity (two variables were collinear if p≤0.05 and V-Cramer ≥0.5) the following variables were excluded in Model 2: disease duration, MMSE, ADL lost and IADL lost. All statistical analyses were carried out using STATA v14.2 software. For all analyses, significance level was set at p≤0.05.
RESULTS
Clinical features in subjects with AD, aMCI, and CONS
Demographic and clinical features of participants are shown in Table 1. ANOVA revealed a significant education-effect between groups. However, after Scheffe’s post-hoc analysis only AD showed a significantly higher median education than aMCI at pair comparison (p = 0.012). A significant difference in disease length between groups was found, with Scheffe’s post-hoc analysis revealing a longer disease length in AD respect to aMCI (p = 0.012). Although AD showed a higher prevalence of APOE4 allele than aMCI and CONS, no differences were found in APOE4 allele distribution between groups. Regarding functional disability, there was a significantly higher number of ADL and IADL lost within groups. In particular, after Scheffe’s post-hoc analysis AD showed a significant ADL impairment compared with aMCI and CONS (p≤0.001 for both comparisons), while no significant difference was found between aMCI and CONS (p = 0.76). Furthermore, Scheffe’s post-hoc analysis revealed a significant IADL impairment only in AD with respect to aMCI and CONS (p≤0.001 for both comparisons).
Table 1
Demographic and clinical characteristics of CONS, aMCI and AD*
| Total n = 78 | CONS n = 26 | aMCI n = 26 | AD n = 26 | p | Post-hoc | |
| Age | 80.4 (5.5) | 80.5 (5.2) | 80.0 (5.3) | 80.8 (6.1) | 0.868 | n.s. |
| Education, median y (range) | 4 (3– 5) | 5 (3– 6) | 3 (2– 5) | 5 (4– 6) | 0.040 | AD > aMCI |
| Male, n (%) | 53 (68) | 18 (69) | 17 (65) | 18 (69) | 0.943 | n.s. |
| Disease length; median y (range) | 2 (1– 6) | n.a. | 2 (1– 3) | 6 (4– 8) | <0.001 | AD > aMCI |
| APOE4 allele, n (%) | 4 (5) | 0 (0) | 1 (4) | 3 (11) | 0.230 | n.s. |
| ADL lost | 1.1 (1.8) | 0.2 (0.4) | 0.4 (0.6) | 2.9 (2.1) | <0.001 | AD > aMCI, CONS |
| IADL lost | 2.5 (3.1) | 0.5 (0.6) | 1.1 (1.4) | 6.3 (2.1) | <0.001 | AD > aMCI, CONS |
| MMSE | 21.9 (7.2) | 27.2 (3.3) | 24.1 (2.9) | 14.5 (7.0) | <0.001 | AD < aMCI, CONS |
| NPI total score | 2.9 (2.4) | 1.2 (1.3) | 2.5 (1.6) | 5.1 (2.3) | <0.001 | AD > aMCI > CONS |
| MNA score, median (range) | 24 (20– 25) | 25 (24– 26) | 24 (23– 26) | 18 (17– 21) | 0.031 | AD < CONS |
*Except were specified data are expressed as mean (SD). CONS, controls; aMCI, amnestic mild cognitive impairment; AD, Alzheimer’s disease; ADL, Activities of Daily Living; IADL, Instrumental Activities of Daily Living; MMSE, Mini-Mental State Examination; NPI, Neuropsychiatric Inventory; MNA, Mini Nutritional Assessment; n.s., not significant.
Concerning global cognition, ANOVA revealed significant differences in MMSE score within groups, with AD showing the lowest performance compared with aMCI and CONS (p≤0.001 for both comparisons). Regarding behavioral symptoms, there were significant differences between groups; in particular, after Scheffe’s post-hoc analysis AD showed a higher NPI score than aMCI and CONS (p≤0.001 for both comparisons), and aMCI had a significant higher NPI score than CONS (p = 0.038). Finally, there were significant differences in MNA scores between groups; in particular, after Scheffe’s post-hoc analysis AD showed lower MNA score than CONS (p = 0.022).
Biochemical parameters and comorbidity in subjects with AD, aMCI and CONS
Results of biochemical parameters and comorbidity are presented in Table 2. There were not significant differences after ANOVAs concerning glycemia, CT, HDL, LDL, TG, and hs-CRP between groups.
Table 2
Biochemical parameters and comorbidity in CONS, aMCI and AD
| Biochemical variables, mean (SD) | Total n = 78 | CONS n = 26 | aMCI n = 26 | AD n = 26 | p | Post-hoc |
| Glycemia, mg/dL | 97.2 (44.9) | 89.5 (25.3) | 96.4 (19.5) | 105.8 (71.0) | 0.426 | n.s. |
| CT mg/dL | 193.6 (37.1) | 200.6 (33.0) | 188.1 (34.8) | 192.1 (43.2) | 0.361 | n.s. |
| HDL mg/dL | 56.9 (15.6) | 57.2 (13.4) | 56.2 (14.5) | 57.3 (19.0) | 0.960 | n.s. |
| LDL mg/dL | 116.1 (32.4) | 122.1 (28.4) | 110.8 (27.5) | 115.2 (40.5) | 0.452 | n.s. |
| TG mg/dL | 101.9 (38.2) | 106.0 (33.5) | 101.4 (47.5) | 98.1 (32.8) | 0.764 | n.s. |
| hs-CRP mg/L | 0.4 (0.4) | 0.3 (0.3) | 0.4 (0.3) | 0.5 (0.5) | 0.565 | n.s. |
| HSP 60 (ng/ml) | 15.4 (9.7) | 12.3 (6.5) | 11.9 (4.3) | 21.9 (12.9) | <0.001 | AD > aMCI, CONS |
| HSP 70 (ng/ml) | 4.3 (1.5) | 3.8 (1.2) | 4.2 (1.1) | 4.9 (1.9) | 0.018 | AD > CONS |
| HSP 90 (ng/ml) | 15.1 (2.9) | 17.0 (2.4) | 15.0 (2.1) | 13.2 (2.7) | <0.001 | AD < aMCI < CONS |
| Comorbidity, n (%) | ||||||
| Hypertension | 57 (73) | 20 (77) | 23 (88) | 14 (54) | 0.016 | AD < aMCI |
| Dyslipidemia | 44 (56) | 13 (50) | 14 (54) | 17 (65) | 0.508 | AD > CONS |
| Diabetes | 13 (17) | 3 (11) | 6 (23) | 4 (15) | 0.524 | n.s. |
| Obesity | 32 (41) | 12 (46) | 12 (46) | 8 (31) | 0.454 | n.s. |
| AF | 1 (1) | 0 (0) | 0 (0) | 1 (4) | 0.363 | n.s. |
| CHD | 5 (9) | 0 (0) | 5 (19) | 0 (0) | 0.005 | AD, CONS < aMCI |
| CVD | 1 (1) | 0 (0) | 0 (0) | 1 (4) | 0.363 | n.s. |
| CIRS total score, mean (SD) | 20.8 (3.0) | 19.2 (2.8) | 21.1 (2.5) | 22.1 (3.0) | 0.002 | AD > CONS |
CONS, controls; aMCI, amnestic mild cognitive impairment; AD, Alzheimer’s disease; CT, total cholesterol, HDL, high density lipoprotein; LDL, low density lipoprotein; TG, triglycerides; hs-CRP, high sensitivity C-reactive protein; HSP, heat shock protein; AF, atrial fibrillation; CHD, coronary heart disease; CVD, cardiovascular disease; CIRS, Cumulative Illness Rating Scale; n.s., not significant.
The mean plasma levels were 15.4 ng/ml (SD = 9.7) for Hsp60, 4.3 ng/ml (SD = 1.5) for Hsp70, and 15.1 ng/ml (SD = 2.9) for Hsp90. The most common comorbidity was hypertension (73%, n = 57), followed by dyslipidemia (56%, n = 44), and obesity (41%, n = 32). Furthermore, a mean CIRS total score of 20.8 (SD = 3.0) was revealed. AD patients showed significantly higher Hsp60 levels compared to aMCI and CONS (21.9 versus 11.9 and 12.3 ng/ml, respectively; p≤0.001); higher Hsp70 levels than CONS (4.9 versus 3.8 ng/ml; p = 0.017); and lower Hsp90 levels than aMCI (13.2 versus 15.0 ng/ml; p = 0.032) and aMCI lower Hsp90 values than CONS (15.0 versus 17.0 ng/ml; p≤0.001). Comorbidity: AD showed a lower frequency of hypertension than aMCI (54% versus 88%; p = 0.006). Moreover, compared to CONS, AD showed a higher prevalence of dyslipidemia (65% versus 50%; p = 0.037) and a higher CIRS total score (22.1 versus 19.2; p = 0.001).
Table 3
Multivariate logistic regression analysis: association of Hsp levels (continuous variables) in subjects with aMCI and AD
| Model | aMCI versus CONS OR (IC95%) | AD versus aMCI OR (IC95%) | AD versus CONS OR (IC95%) | |
| Hsp60 (per unit increase) | 1 | 0.99 (0.89–1.10) | 1.16** (1.04–1.30) | 1.12** (1.03–1.22) |
| 2 | 0.93 (0.81–1.06) | 1.20 (0.99–1.44) | 1.11 (0.95–1.30) | |
| Hsp70 (per unit increase) | 1 | 1.30 (0.76–2.21) | 1.52 (0.94–2.45) | 1.84* (1.09–3.10) |
| 2 | 1.23 (0.68–2.25) | 1.85 (0.81–4.21) | 1.24 (0.45–3.41) | |
| Hsp90 (per unit increase) | 1 | 0.69* (0.52–0.91) | 0.73* (0.55–0.96) | 0.51** (0.35–0.75) |
| 2 | 0.76 (0.57–1.03) | 0.76 (0.47–1.24) | 0.57 (0.30–1.05) |
*p < 0.05; **p < 0.01; ***p < 0.001. Model 1 corrected for education. Model 2 corrected for education, NPI, MNA, CIRS.
Logistic regression models adjusted for education (Model 1) revealed significantly higher Hsp60 values in AD versus aMCI (OR = 1.16, 95% CI 1.04–1.30) and AD versus CONS (OR = 1.12, 95% CI 1.03–1.22); and higher Hsp70 values in AD versus CONS (OR = 1.84, 95% CI 1.09–3.10) (see Table 3). Contrarily, lower levels of Hsp90 were found in aMCI versus CONS (OR = 0.69, 95% CI 0.52–0.91), AD versus aMCI (OR = 0.73, 95% CI 0.55–0.96) and AD versus CONS (OR = 0.51, 95% CI 0.35–0.75). However, results between aMCI and AD versus CONS were no longer significant after adjustment for multiple comparisons (Model 2: adjusted for education, NPI, MNA, and CIRS) (see Table 3).
DISCUSSION
In this study, we investigated the relationship between plasma levels of Hsp60, Hsp70, and Hsp90 and either clinical features or other biochemical parameters in three samples, aMCI, AD, and controls. The results regarding clinical features showed a significantly higher median education and longer disease length in AD than in aMCI. AD subjects showed a significant ADL and IADL impairment as compared with aMCI and CONS; and aMCI showed a significant ADL impairment compared to controls. We also evaluated global cognitive performance and AD patients showed lower performance than aMCI and controls.
Concerning behavioral symptoms, AD showed lower MNA score than CONS and higher NPI score than aMCI and CONS; and aMCI had a significant higher NPI score than controls. These data coincide with the well-known, typical indicators of AD.
The biochemical analyses revealed that all the groups examined were similar, namely no significant difference among them were detected for glycemia, CT, HDL, LDL, TG, and hs-CRP levels. However, a significant prevalence of dyslipidemia was registered in AD subjects compared to controls. In addition, AD patients showed a higher CIRS total score than controls. Several studies that explored the association between lipids and/or lipid-lowering treatment and AD indicate a harmful effect of dyslipidemia on AD risk and progression, although this association still remains controversial [33]. Functional cell biology studies support a critical involvement of lipid raft cholesterol in the modulation of Aβ precursor protein processing by β-secretase and γ-secretase resulting in altered Aβ production [34]. On the contrary, it has been demonstrated that the presence of high-cholesterol results in increasing generation of plasmin, an Aβ-degrading enzyme [35]. Inflammation and oxidative stress may provoke dyslipidemia [36] with consequences on brain cholesterol homeostasis, neuroinflammation, production of neurofibrillary tangles and, thereby, aggravating excitotoxicity [37]. The influence of ROS may be detrimental to lipids, proteins, and nucleic acids, which results in structural and functional changes such as protein misfolding and, eventually, in necrotic or apoptotic cell death [38, 39]. An accumulation of misfolded proteins in the aging brain results in oxidative and inflammatory damage, which in turn leads to energy failure and synaptic dysfunction, pathological features found in AD [38, 40].
Our study showed that circulating Hsp60 levels were significantly higher in AD compared to aMCI and CONS, and the chaperonin plasma levels were associated with AD even after adjusting for education. However, the statistical significance of this association disappeared after adjustment for confounding factors. Hsp60 is classically described as a mitochondrial protein that assists the correct folding of other mitochondrial proteins [41]. The regulation of mitochondrial turnover and function becomes impaired as a consequence of age and this has a preponderant role in AD, in which it has been shown that mitochondrial dysfunction (e.g., reduced membrane potential, increased permeability, increased ROS production as a consequence of both amyloid-β protein precursor and Aβ overexpression) does occur [42, 43]. We found Hsp60 to be increased in AD and this could be part of a mechanism to restore mitochondrial homeostasis in which the chaperonin would be neuroprotective. There is evidence supporting this assumption. For example, it has been demonstrated that in a human neuroblastoma cell line, induced expression of the chaperonin prevented intracellular-amyloid-induced inhibition of complex IV, reducing apoptosis [44]. Recently, in vivo experiments demonstrated that Hsp60 has the capacity to drive mitochondrial biogenesis through activation of the peroxisome proliferator-activated receptor γ coactivator–1α [45], while decreased Hsp60 levels were found in hippocampus of mice fed with high fat diet and the decrease was associated with AD pathogenesis [46]. In another study, it has been shown that treatment of rats with Oxotremorine-M, a non-selective muscarinic acetylcholine receptor agonist, significantly increased the levels of Hsp60, Hsp70, and Hsp90 in hippocampus [47]. Moreover, a loss of function of the chaperonin via oxidation by Aβ1–42 caused an increase in protein misfolding whereas higher Hsp60 expression in the cerebellum of AD rats reversed the deficiency of Purkinje cells [48, 49]. This multiplicity of roles in various anatomical-histological locations is in agreement with the demonstration that Hsp60 not only occurs in the mitochondria, but also in the cytosol, the plasma-cell membrane, the cell surface, the extracellular space, and the peripheral blood with roles in various physiological and pathological processes pertaining to protein homeostasis and other unrelated phenomena [50].
To our knowledge, this is the first report on the association between plasma Hsp60 and AD. Hsp60 upregulation in brain of AD patients was paralleled by a significant increase of the protein in AD lymphocytes [51]. The increased levels of the chaperonin were associated with a cytoprotective role against nitrosative stress [51].
Hsp60 overexpression may be followed by its secretion from stressed cells into the extracellular space and circulation [7], which makes the chaperonin a strong candidate as biomarker of cell stress. Accordingly, blood levels of Hsp60 might reflect the pathological status of nervous system cells, thus representing putative biomarker of neurodegeneration.
Of all Hsps, Hsp70 is the most strongly induced after oxidative stress, anticancer drugs, or protein damage-misfolding caused by stress [52]. Hsp70 molecules are found in various cellular compartments (nucleus, mitochondria, cytosol), extracellularly, and in circulation [6]. Hsp70 has the ability to inhibit apoptosis and to trigger the activation of the immune response by stimulating both innate and adaptive immunities [53]. Increased Hsp70 expression levels have been found in the brain of patients affected by AD, suggesting an association between this chaperone, AD pathophysiology, and neuroprotective response [54]. For instance, Hsp70 was identified as a protector against Aβ accumulation and its overexpression rescued neurons from Aβ-mediated neurotoxicity [54, 55]. In the present study, we investigated Hsp70 plasma levels in AD, aMCI, and controls. Our results showed higher Hsp70 levels in AD patients compared to controls. Logistic regression analysis demonstrated that this increase was associated with AD after correction for education, but this result was not confirmed after multiple comparisons. To our knowledge, our paper is the first to report the association between Hsp70 plasma levels and AD. Other studies reported an association between HSP70–2 (HSPA1B) gene expression with non-cognitive symptoms in late-onset AD or a correlation between Hsp70 mRNA in mononuclear blood cells with dementia of the AD type [56, 57]. Increased plasma Hsp70 levels were associated with cognitive decline in language and executive functions in MCI due to AD and vascular aetiology and, probably, this increase was a way to protect neurons and glial cells [58, 59]. Moreover, similarly to Hsp60, Hsp70 has been found to be elevated in AD lymphocytes compared to controls, probably to counteract nitrosative stress [51].
We observed a significant decrease of Hsp90 levels in AD and aMCI compared to CONS, and in AD compared to aMCI. The Hsp90 reduction was associated with aMCI and AD after controlling for education, but also in this case, results were no longer significant after multiple comparisons. Hsp90 is the most abundant protein in eukaryotic cells under unstressed conditions, and shares the same characteristics with other molecular chaperones as it plays an important role in the folding of many proteins pertaining for instance to essential signaling pathways, and in the refolding of denatured proteins after stress [60, 61]. In AD brains, levels of Hsp90 were increased in both the cytosolic and membranous fractions, and Hsp90 was co-localized with amyloid plaques [55]. These observations suggest that Hsp-induced microglial activation might play a neuroprotective role by facilitating Aβ clearance and cytokine production [62]. Hsp90 can tightly bind and down-regulate the activity of HSF1 but in the presence of cellular stress HSF1 dissociates from Hsp90, inducing heat shock response genes, such as those encoding the proteins Hsp27, Hsp40, Hsp70, and Hsp90. The expression of these chaperone genes expands the buffering capacity of the cell and restores protein homeostasis under stressful conditions [63]. In agreement with our results, a significant decrease of Hsp90 serum levels has been demonstrated in AD and aMCI patients [64]. Our data suggest that Hsp90 decrease in blood is a sign of increasing protein aggregation in AD in agreement with existent literature data [64] and, probably also unveils weak defense mechanisms against oxidative stress. Further studies should investigate if Hsp90 diminution is due to post-translational modifications, and if miRNA is involved in translation regulation.
The strength of our work is the case-control study design, the adjustment for multiple potential confounders, and the inclusion of subjects drawn from a population-based study, with inherent reduction of selection bias. However, some limitations of the present study should be pointed out. First, the relatively small sample size of the groups might increase the likelihood of spurious associations and lack of significance (e.g., the negative associations between Hsp levels and aMCI/AD obtained after multiple comparisons). Second, aMCI and dementia were diagnosed according to clinical criteria, with inevitably some uncertainty about the rate of misclassification.
Without postmortem confirmation and without the use of cerebrospinal fluid biomarkers of the AD-type neuropathological process, a comprehensive clinical protocol was used including complete neuropsychological assessment and the clinical classification of AD and aMCI was supported by brain imaging. Third, although analyses were adjusted for major potential confounders, it should be noted that the observed interrelationships might not be a direct cause-effect relationship due to complex interactive factors (the presence of age-related inflammation for example), and Hsps levels might reflect a nonspecific disease state, with a subsequent peripheral increase. However, aMCI and AD did not differ from CONS in hs-CRP levels, a marker of acute inflammation, as well as in lipids fractions and the majority of comorbid disorders evaluated. Lastly, the cross-sectional study design precludes making causal inferences about the relationship between Hsps levels and the study outcome.
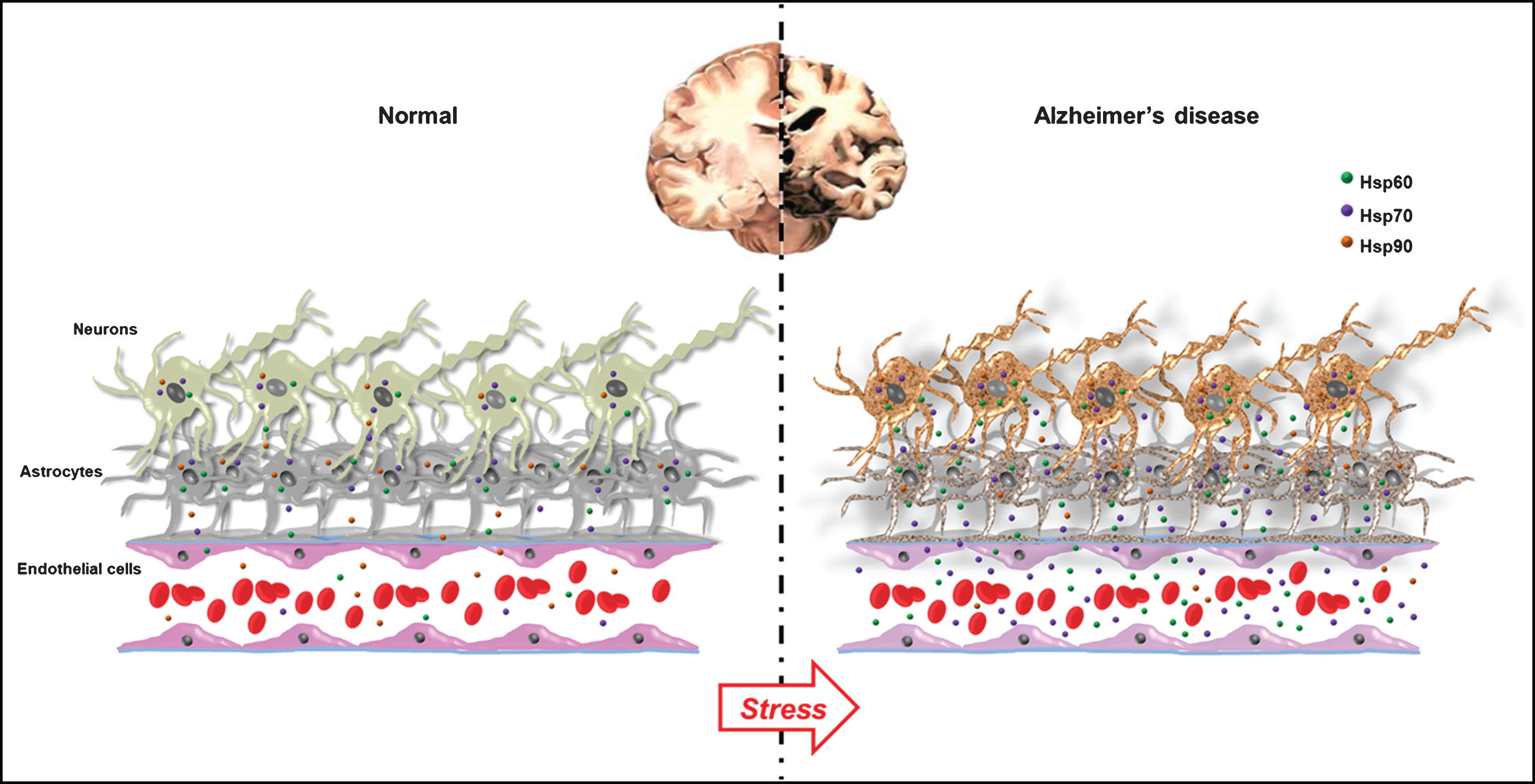
In conclusion, from the data discussed above we can hypothesize that Hsp60 and Hsp70 are up-regulated in response to cellular stress likely to protect brain cells against stress, in particular against misfolded proteins aggregation and deposition. On the other hand, Hsp90 downregulation could be related to the failure of the instauration of defense mechanisms against proteotoxic stress, which could facilitate the progression of neurodegeneration. Therefore, we can build a working hypothesis assuming that the measurement of these Hsps as cell stress biomarkers in blood will reflect the degree and extent of brain damage, thus providing a useful tool for the diagnosis, prognosis, and early treatment of AD (see Fig. 1).
Fig.1
Stress, heat shock proteins (molecular chaperones), and neurons: normal versus Alzheimer’s disease (AD) brains. The figure summarizes our working hypothesis derived from our results and based on literature data. Under normal conditions, Hsp60, Hsp70, and Hsp90 are ubiquitously expressed at their physiological levels and they can be released into the extracellular environment to serve, for example, as cell to cell communicators. As reported in the text, in AD, Hsp60 and Hsp70 are overexpressed in stressed neurons and/or activated glia. The degree of Hsp60, Hsp70 and Hsp90 expression may reflect the neuropathological abnormalities present in AD brain. We hypothesize that Hsp60 and Hsp70 are upregulated in response to cellular stress to protect brain cells against stress, and counteract misfolded proteins aggregation and deposition. On the contrary, Hsp90 downregulation could be related to the failure of the instauration of defense mechanisms against proteotoxic stress, which would facilitate the progression of neurodegeneration. The three chaperones, following extracellular accumulation, can cross the blood-brain barrier by a mechanism yet to be elucidated and, thus, be present in the circulation, making them easily accessible as biomarkers providing information on what is going on in the cells of origin, e.g., stressed AD neurons.
Future perspectives
The results obtained suggest that we are at the dawn of a new research era that will produce data crucial to understand the molecular mechanisms responsible for the involvement of molecular chaperones, such as Hsp60, Hsp70, and Hsp90, in the pathogenic deviations observed in cell differentiation, tissue homeostasis, and organ remodeling in some anatomical areas of the nervous system, preceding the onset of dementia symptoms. Problems still to be solved are the following: Is the increase/decrease of Hsps in plasma always a reliable indication of an analogous change in intracellular concentration? If Hsp secretion and its deviations from the physiological patterns turn out to be crucial for pathogenesis, which are the cells and the secretory pathways involved? Which is the mechanism underpinning the crossing of the blood-brain barrier by chaperones? Could chaperonotherapy targeting the Hsps studied reduce histopathological signs and clinical symptoms of early dementia, at least in pre-clinical models?
The last interesting point will be the measurement of chaperone levels in the cerebrospinal fluid and the determination of the distribution of the chaperones in brain cells and tissues, which will give the possibility to check the relationships that may occur between the AD-related neurodegenerative process and Hsp quantities and distribution.
These questions illustrate the complexity of the pathogenic picture to investigate in the near future and emphasize the pressing need to continue this research topic. Similarly, Hsps should be investigated in other dementia types and neurodegenerative conditions in which molecular chaperones surely play key roles, either defensive or pathogenic. To make advances in this area of medical research, large population-based prospective studies are required to evaluate the diagnostic and prognostic value of Hsp60, Hsp70, and Hsp90, as well as to gather the basic information required to develop specific chaperonotherapy for each condition. These possibilities are currently under intense scrutiny in our laboratories and in those of many others, ensuring a bright future for research on this serious and widespread neurodegenerative conditions.
ACKNOWLEDGMENTS
This work was completely supported by a Grant project for young researcher 2007 (GR–2007–686973) from the Italian Ministry of Health to R.M., who is the PI of the ZAP, and to A.B.C., who is the responsible of lipid analysis and genotyping. Part of this work was carried out using instruments provided by the IEMEST through the Italian Programme for Research and Competitiveness 2007–2013 grant awarded to the project titled “Cyber Brain –Polo di innovazione” (PONa3_00210). A.J.L.M, and E.C. de M. were partially supported by IMET. This work was done under the agreement between IEMEST (Italy) and IMET (USA) (this is IMET contribution number 18–011).
The authors thank all participants and all persons working in the Zabùt Aging Project for data collection and management.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0825r1)
REFERENCES
[1] | Fratiglioni L , Winblad B , von Strauss E ((2007) ) Prevention of Alzheimer’s disease and dementia. Major findings from the Kungsholmen Project. Physiol Behav 92: , 98–104. |
[2] | McKhann GM , Knopman DS , Chertkow H , Hyman BT , Jack CR Jr , Kawas CH , Klunk WE , Koroshetz WJ , Manly JJ , Mayeux R , Mohs RC , Morris JC , Rossor MN , Scheltens P , Carrillo MC , Thies B , Weintraub S , Phelps CH ((2011) ) The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 263–269. |
[3] | Maiti P , Manna J , Veleri S , Frautschy S ((2014) ) Molecular chaperone dysfunction in neurodegenerative diseases and effects of curcumin. Biomed Res Int 2014: , 495091. |
[4] | Nelson PT , Alafuzoff I , Bigio EH , Bouras C , Braak H , Cairns NJ , Castellani RJ , Crain BJ , Davies P , Del Tredici K , Duyckaerts C , Frosch MP , Haroutunian V , Hof PR , Hulette CM , Hyman BT , Iwatsubo T , Jellinger KA , Jicha GA , Kovari E , Kukull WA , Leverenz JB , Love S , Mackenzie IR , Mann DM , Masliah E , McKee AC , Montine TJ , Morris JC , Schneider JA , Sonnen JA , Thal DR , Trojanowski JQ , Troncoso JC , Wisniewski T , Woltjer RL , Beach TG ((2012) ) Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J Neuropathol Exp Neurol 71: , 362–381. |
[5] | Macario AJL , Conway de Macario E ((2005) ) Sick chaperones, cellular stress, and disease. N Engl J Med 353: , 1489–1501. |
[6] | Marino Gammazza A , Bavisotto CC , Barone R , Conway de Macario E , Macario AJL ((2016) ) Alzheimer’s disease and molecular chaperones: Current knowledge and the future of chaperonotherapy. Curr Pharm Des 22: , 4040–4049. |
[7] | Caruso Bavisotto C , Cappello F , Macario AJL , Conway de Macario E , Logozzi M , Fais S , Campanella C ((2017) ) Exosomal HSP60: A potentially useful biomarker for diagnosis, assessing prognosis, and monitoring response to treatment. Expert Rev Mol Diagn 17: , 815–822. |
[8] | Koren J , 3rd , Jinwal UK , Lee DC , Jones JR , Shults CL , Johnson AG , Anderson LJ , Dickey CA ((2009) ) Chaperone signalling complexes in Alzheimer’s disease. J Cell Mol Med 13: , 619–630. |
[9] | Cappello F , Marino Gammazza A , Palumbo Piccionello A , Campanella C , Pace A , Conway de Macario E , Macario AJL ((2014) ) Hsp60 chaperonopathies and chaperonotherapy: Targets and agents. Expert Opin Ther Targets 18: , 185–208. |
[10] | Mangione MR , Vilasi S , Marino C , Librizzi F , Canale C , Spigolon D , Bucchieri F , Fucarino A , Passantino R , Cappello F , Bulone D , San Biagio PL ((2016) ) Hsp60, amateur chaperone in amyloid-beta fibrillogenesis. Biochim Biophys Acta 1860: , 2474–2483. |
[11] | Bayer TA ((2015) ) Proteinopathies, a core concept for understanding and ultimately treating degenerative disorders? Eur Neuropsychopharmacol 25: , 713–724. |
[12] | Kampinga HH , Bergink S ((2016) ) Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol 15: , 748–759. |
[13] | Luheshi LM , Crowther DC , Dobson CM ((2008) ) Protein misfolding and disease: From the test tube to the organism. Curr Opin Chem Biol 12: , 25–31. |
[14] | Selkoe DJ ((2004) ) Cell biology of protein misfolding: The examples of Alzheimer’s and Parkinson’s diseases. Nat Cell Biol 6: , 1054–1061. |
[15] | Albert MS , DeKosky ST , Dickson D , Dubois B , Feldman HH , Fox NC , Gamst A , Holtzman DM , Jagust WJ , Petersen RC , Snyder PJ , Carrillo MC , Thies B , Phelps CH ((2011) ) The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 270–279. |
[16] | Mariani E , Monastero R , Mecocci P ((2007) ) Mild cognitive impairment: A systematic review. J Alzheimers Dis 12: , 23–35. |
[17] | ((2017) ) Diagnostic and Statistical Manual of Mental Disorders (DSM). Knowledge Organization 44: , 668–676. |
[18] | Winblad B , Palmer K , Kivipelto M , Jelic V , Fratiglioni L , Wahlund LO , Nordberg A , Backman L , Albert M , Almkvist O , Arai H , Basun H , Blennow K , de Leon M , DeCarli C , Erkinjuntti T , Giacobini E , Graff C , Hardy J , Jack C , Jorm A , Ritchie K , van Duijn C , Visser P , Petersen RC ((2004) ) Mild cognitive impairment–beyond controversies, towards a consensus: Report of the International Working Group on Mild Cognitive Impairment. J Intern Med 256: , 240–246. |
[19] | Carlesimo GA , Caltagirone C , Gainotti G ((1996) ) The Mental Deterioration Battery: Normative data, diagnostic reliability and qualitative analyses of cognitive impairment. The Group for the Standardization of the Mental Deterioration Battery. Eur Neurol 36: , 378–384. |
[20] | Folstein MF , Folstein SE , McHugh PR ((1975) ) “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12: , 189–198. |
[21] | Morris JC ((1993) ) The Clinical Dementia Rating (CDR): Current version and scoring rules. Neurology 43: , 2412–2414. |
[22] | Katz S , Ford AB , Moskowitz RW , Jackson BA , Jaffe MW ((1963) ) Studies of illness in the aged. The Index of ADL: A standardized measure of biological and psychosocial function. JAMA 185: , 914–919. |
[23] | Lawton MP , Brody EM ((1969) ) Assessment of older people: Self-maintaining and instrumental activities of daily living. Gerontologist 9: , 179–186. |
[24] | Camarda C , Torelli P , Camarda R , Battaglini I , Gagliardo C , Monastero R ((2015) ) Isolated, subtle, neurological abnormalities in neurologically and cognitively healthy aging subjects. J Neurol 262: , 1328–1339. |
[25] | Cummings JL , Mega M , Gray K , Rosenberg-Thompson S , Carusi DA , Gornbein J ((1994) ) The Neuropsychiatric Inventory: Comprehensive assessment of psychopathology in dementia. Neurology 44: , 2308–2314. |
[26] | Vellas B , Villars H , Abellan G , Soto ME , Rolland Y , Guigoz Y , Morley JE , Chumlea W , Salva A , Rubenstein LZ , Garry P ((2006) ) Overview of the MNA–Its history and challenges. J Nutr Health Aging 10: , 453–463; discussion 463-455. |
[27] | Monastero R , Mariani E , Camarda C , Ingegni T , Averna MR , Senin U , Camarda R , Mecocci P ((2006) ) Association between apolipoprotein E epsilon4 allele and apathy in probable Alzheimer’s disease. Acta Psychiatr Scand 113: , 59–63. |
[28] | Chobanian AV1 , Bakris GL , Black HR , Cushman WC , Green LA , Izzo JL Jr , Jones DW , Materson BJ , Oparil S , Wright JT Jr , Roccella EJ ; National Heart Lung, and Blood Institute Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National High Blood Pressure Education Program Coordinating Committee ((2003) ) The Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure: The JNC 7 report. JAMA 289: , 2560–2572. |
[29] | National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) ((2002) ) Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 106: , 3143–3421. |
[30] | ((2000) ) Obesity: Preventing and managing the global epidemic. Report of a WHO consultation. World Health Organ Tech Rep Ser 894: , i-xii, 1–253. |
[31] | Parmelee PA , Thuras PD , Katz IR , Lawton MP ((1995) ) Validation of the Cumulative Illness Rating Scale in a geriatric residential population. J Am Geriatr Soc 43: , 130–137. |
[32] | Marino Gammazza A , Rizzo M , Citarrella R , Rappa F , Campanella C , Bucchieri F , Patti A , Nikolic D , Cabibi D , Amico G , Conaldi PG , San Biagio PL , Montalto G , Farina F , Zummo G , Conway de Macario E , Macario AJL , Cappello F ((2014) ) Elevated blood Hsp60, its structural similarities and cross-reactivity with thyroid molecules, and its presence on the plasma membrane of oncocytes point to the chaperonin as an immunopathogenic factor in Hashimoto’s thyroiditis. Cell Stress Chaperones 19: , 343–353. |
[33] | Reitz C ((2012) ) Dyslipidemia and dementia: Current epidemiology, genetic evidence, and mechanisms behind the associations. J Alzheimers Dis 30: (Suppl 2), S127–145. |
[34] | Grziwa B , Grimm MO , Masters CL , Beyreuther K , Hartmann T , Lichtenthaler SF ((2003) ) The transmembrane domain of the amyloid precursor protein in microsomal membranes is on both sides shorter than predicted. J Biol Chem 278: , 6803–6808. |
[35] | Crameri A , Biondi E , Kuehnle K , Lutjohann D , Thelen KM , Perga S , Dotti CG , Nitsch RM , Ledesma MD , Mohajeri MH ((2006) ) The role of seladin-1/DHCR24 in cholesterol biosynthesis, APP processing and Abeta generation in vivo. EMBO J 25: , 432–443. |
[36] | Pashkow FJ ((2011) ) Oxidative stress and inflammation in heart disease: Do antioxidants have a role in treatment and/or prevention? Int J Inflam 2011: , 514623. |
[37] | Songco A , Rattner W ((1987) ) Reflex anuria. Urology 29: , 432–433. |
[38] | Chen X , Guo C , Kong J ((2012) ) Oxidative stress in neurodegenerative diseases. Neural Regen Res 7: , 376–385. |
[39] | Rizzo M , Kotur-Stevuljevic J , Berneis K , Spinas G , Rini GB , Jelic-Ivanovic Z , Spasojevic-Kalimanovska V , Vekic J ((2009) ) Atherogenic dyslipidemia and oxidative stress: A new look. Transl Res 153: , 217–223. |
[40] | Querfurth HW , LaFerla FM ((2010) ) Alzheimer’s disease. N Engl J Med 362: , 329–344. |
[41] | Gorska M , Marino Gammazza A , Zmijewski MA , Campanella C , Cappello F , Wasiewicz T , Kuban-Jankowska A , Daca A , Sielicka A , Popowska U , Knap N , Antoniewicz J , Wakabayashi T , Wozniak M ((2013) ) Geldanamycin-induced osteosarcoma cell death is associated with hyperacetylation and loss of mitochondrial pool of heat shock protein 60 (hsp60). PLoS One 8: , e71135. |
[42] | Scarpulla RC ((2008) ) Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol Rev 88: , 611–638. |
[43] | Picone P , Nuzzo D , Caruana L , Scafidi V , Di Carlo M ((2014) ) Mitochondrial dysfunction: Different routes to Alzheimer’s disease therapy. Oxid Med Cell Longev 2014: , 780179. |
[44] | Veereshwarayya V , Kumar P , Rosen KM , Mestril R , Querfurth HW ((2006) ) Differential effects of mitochondrial heat shock protein 60 and related molecular chaperones to prevent intracellular beta-amyloid-induced inhibition of complex IV and limit apoptosis. J Biol Chem 281: , 29468–29478. |
[45] | Barone R , Macaluso F , Sangiorgi C , Campanella C , Marino Gammazza A , Moresi V , Coletti D , Conway de Macario E , Macario AJL , Cappello F , Adamo S , Farina F , Zummo G , Di Felice V ((2016) ) Skeletal muscle Heat shock protein 60 increases after endurance training and induces peroxisome proliferator-activated receptor gamma coactivator 1 alpha1 expression. Sci Rep 6: , 19781. |
[46] | Nuzzo D , Picone P , Baldassano S , Caruana L , Messina E , Marino Gammazza A , Cappello F , Mule F , Di Carlo M ((2015) ) Insulin resistance as common molecular denominator linking obesity to Alzheimer’s disease. Curr Alzheimer Res 12: , 723–735. |
[47] | Frinchi M , Scaduto P , Cappello F , Belluardo N , Mudo G ((2018) ) Heat shock protein (Hsp) regulation by muscarinic acetylcholine receptor (mAChR) activation in the rat hippocampus. J Cell Physiol 233: , 6107–6116. |
[48] | Boyd-Kimball D , Sultana R , Poon HF , Lynn BC , Casamenti F , Pepeu G , Klein JB , Butterfield DA ((2005) ) Proteomic identification of proteins specifically oxidized by intracerebral injection of amyloid beta-peptide (1-42) into rat brain: Imlications for Alzheimer’s disease. Neuroscience 132: , 313–324. |
[49] | Jiang YQ , Wang XL , Cao XH , Ye ZY , Li L , Cai WQ ((2013) ) Increased heat shock transcription factor 1 in the cerebellum reverses the deficiency of Purkinje cells in Alzheimer’s disease. Brain Res 1519: , 105–111. |
[50] | Cappello F , Conway de Macario E , Marasa L , Zummo G , Macario AJL ((2008) ) Hsp60 expression, new locations, functions and perspectives for cancer diagnosis and therapy. Cancer Biol Ther 7: , 801–809. |
[51] | Calabrese V , Sultana R , Scapagnini G , Guagliano E , Sapienza M , Bella R , Kanski J , Pennisi G , Mancuso C , Stella AM , Butterfield DA ((2006) ) Nitrosative stress, cellular stress response, and thiol homeostasis in patients with Alzheimer’s disease. Antioxid Redox Signal 8: , 1975–1986. |
[52] | Di Domenico F , Sultana R , Tiu GF , Scheff NN , Perluigi M , Cini C , Butterfield DA ((2010) ) Protein levels of heat shock proteins 27, 32, 60, 70, 90 and thioredoxin-1 in amnestic mild cognitive impairment: An investigation on the role of cellular stress response in the progression of Alzheimer disease. Brain Res 1333: , 72–81. |
[53] | Asea A , Rehli M , Kabingu E , Boch JA , Bare O , Auron PE , Stevenson MA , Calderwood SK ((2002) ) Novel signal transduction pathway utilized by extracellular HSP70: Role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem 277: , 15028–15034. |
[54] | Magrane J , Smith RC , Walsh K , Querfurth HW ((2004) ) Heat shock protein 70 participates in the neuroprotective response to intracellularly expressed beta-amyloid in neurons. J Neurosci 24: , 1700–1706. |
[55] | Evans CG , Wisen S , Gestwicki JE ((2006) ) Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1-42) aggregation in vitro. J Biol Chem 281: , 33182–33191. |
[56] | Clarimon J , Bertranpetit J , Boada M , Tarraga L , Comas D ((2003) ) HSP70-2 (HSPA1B) is associated with noncognitive symptoms in late-onset Alzheimer’s disease. J Geriatr Psychiatry Neurol 16: , 146–150. |
[57] | Wakutani Y , Urakami K , Shimomura T , Takahashi K ((1995) ) Heat shock protein 70 mRNA levels in mononuclear blood cells from patients with dementia of the Alzheimer type. Dementia 6: , 301–305. |
[58] | Lee KS , Chung JH , Oh BH , Hong CH ((2008) ) Increased plasma levels of heat shock protein 70 in patients with vascular mild cognitive impairment. Neurosci Lett 436: , 223–226. |
[59] | Son SJ , Lee KS , Chung JH , Chang KJ , Roh HW , Kim SH , Jin T , Back JH , Kim HJ , Lee Y , Choi SH , Noh JS , Lim KY , Chung YK , Hong CH , Oh BH ((2015) ) Increased plasma levels of heat shock protein 70 associated with subsequent clinical conversion to mild cognitive impairment in cognitively healthy elderly. PLoS One 10: , e0119180. |
[60] | Rappa F , Sciume C , Lo Bello M , Bavisotto CC , Marino Gammazza A , Barone R , Campanella C , David S , Carini F , Zarcone F , Rizzuto S , Lena A , Tomasello G , Uzzo ML , Spatola GF , Bonaventura G , Leone A , Gerbino A , Cappello F , Bucchieri F , Zummo G , Farina F ((2014) ) Comparative analysis of Hsp10 and Hsp90 expression in healthy mucosa and adenocarcinoma of the large bowel. Anticancer Res 34: , 4153–4159. |
[61] | Wandinger SK , Richter K , Buchner J ((2008) ) The Hsp90 chaperone machinery. J Biol Chem 283: , 18473–18477. |
[62] | Kakimura J , Kitamura Y , Takata K , Umeki M , Suzuki S , Shibagaki K , Taniguchi T , Nomura Y , Gebicke-Haerter PJ , Smith MA , Perry G , Shimohama S ((2002) ) Microglial activation and amyloid-beta clearance induced by exogenous heat-shock proteins. FASEB J 16: , 601–603. |
[63] | Zhao H , Michaelis ML , Blagg BS ((2012) ) Hsp90 modulation for the treatment of Alzheimer’s disease. Adv Pharmacol 64: , 1–25. |
[64] | Gezen-Ak D , Dursun E , Hanagasi H , Bilgic B , Lohman E , Araz OS , Atasoy IL , Alaylioglu M , Onal B , Gurvit H , Yilmazer S ((2013) ) BDNF, TNFalpha, HSP90, CFH, and IL-10 serum levels in patients with early or late onset Alzheimer’s disease or mild cognitive impairment. J Alzheimers Dis 37: , 185–195. |


