
Treatment Combinations for Alzheimer’s Disease: Current and Future Pharmacotherapy Options
Abstract
Although Alzheimer’s disease (AD) is the world’s leading cause of dementia and the population of patients with AD continues to grow, no new therapies have been approved in more than a decade. Many clinical trials of single-agent therapies have failed to affect disease progression or symptoms compared with placebo. The complex pathophysiology of AD may necessitate combination treatments rather than monotherapy. The goal of this narrative literature review is to describe types of combination therapy, review the current clinical evidence for combination therapy regimens (both symptomatic and disease-modifying) in the treatment of AD, describe innovative clinical trial study designs that may be effective in testing combination therapy, and discuss the regulatory and drug development landscape for combination therapy. Successful combination therapies in other complex disorders, such as human immunodeficiency virus, may provide useful examples of a potential path forward for AD treatment.
INTRODUCTION
Alzheimer’s disease (AD) is a complex progressive neurodegenerative disorder and the leading cause of dementia [1, 2]. It is estimated that close to 50 million people worldwide are affected by dementia, including AD, and the prevalence of the disease is projected to more than double by 2050 [3].
Risk factors for AD include age, family history, apolipoprotein E ɛ4 genotype, diabetes, hypertension, obesity, hypercholesterolemia, traumatic brain injury, and low education level [1, 2, 4]. Mutations in genes presenilin 1 (PSEN1), presenilin 2 (PSEN2), and amyloid precursor protein (APP) are associated with early-onset autosomal-dominant AD [5].
Alzheimer disease is complex neuropathologically and is characterized by extracellular amyloid plaques, intracellular neurofibrillary tangles, and nerve cell death [6-8]. Amyloid plaques are composed of amyloid-β (Aβ), a cleavage product of the amyloid-β protein precursor (AβPP). AβPP is progressively cleaved by β-secretase (BACE 1) and then γ-secretase to form Aβ [9]. Aβ monomers progressively aggregate into oligomers, fibrils, and insoluble amyloid plaques [6]. Neurofibrillary tangles, the other hallmark protein aggregate in AD, are made up of hyperphosphorylated tau protein. Under normal conditions, tau promotes stabilization of microtubules; when hyperphosphorylated, tau accumulates into “tangles” composed of paired helical filaments [7]. The amyloid cascade hypothesis of AD posits that accumulation of Aβ dysregulates synaptic and neuronal function, creating the intracellular conditions for formation of neurofibrillary tangles, leading to neuronal loss and further compromise of neurotransmitter function [6]. Loss of cholinergic neurons in the basal forebrain (and consequent loss of signaling) is hypothesized to create a cholinergic deficit contributing to short-term memory loss in AD [8]. These complex pathologies may occur sequentially but many are present simultaneously in the brain of the person with AD [6].
MANAGEMENT OF Alzheimer’S disease
Despite the growing population of patients with AD, only five treatment options are currently approved to treat the cognitive symptoms of AD in the United States, the most recent of which (memantine) was approved more than a decade ago [10]. Four of the five standard-of-care treatments are also licensed in the European Union; these include three cholinesterase inhibitors (donepezil, galantamine, and rivastigmine) and one N-methyl-D-aspartate receptor antagonist (memantine; Fig. 1) [11-15]. In 2014, a fifth treatment option consisting of a fixed-dose combination with donepezil and memantine was approved for the treatment of patients with moderate to severe AD dementia who are on stable donepezil therapy [16]. Most therapeutic agents under development over the past 15 years have failed; AD is among the least well-served therapeutic areas for drug treatments. Nearly all trials conducted to date have been monotherapy trials comparing an active agent with placebo with or without a background standard-of-care agent, such as cholinesterase inhibitors or memantine.
Fig.1
Combination therapies in the AD drug development pipeline. 5-HT, 5-hydroxytryptamine (serotonin); Aβ, amyloid-β; AChE1, acetylcholinesterase inhibitor; AD, Alzheimer’s disease; BACE, aspartyl protease β-site amyloid precursor protein cleaving enzyme; GM-CSF, granulocyte-macrophage colony-stimulating factor; MAO, monoamine oxidase; NMDA, N-methyl-D-aspartate; PPAR, peroxisome-proliferator activated receptor; RAGE, receptor for advanced glycation end-products; rTMS, repetitive transcranial magnetic stimulation.
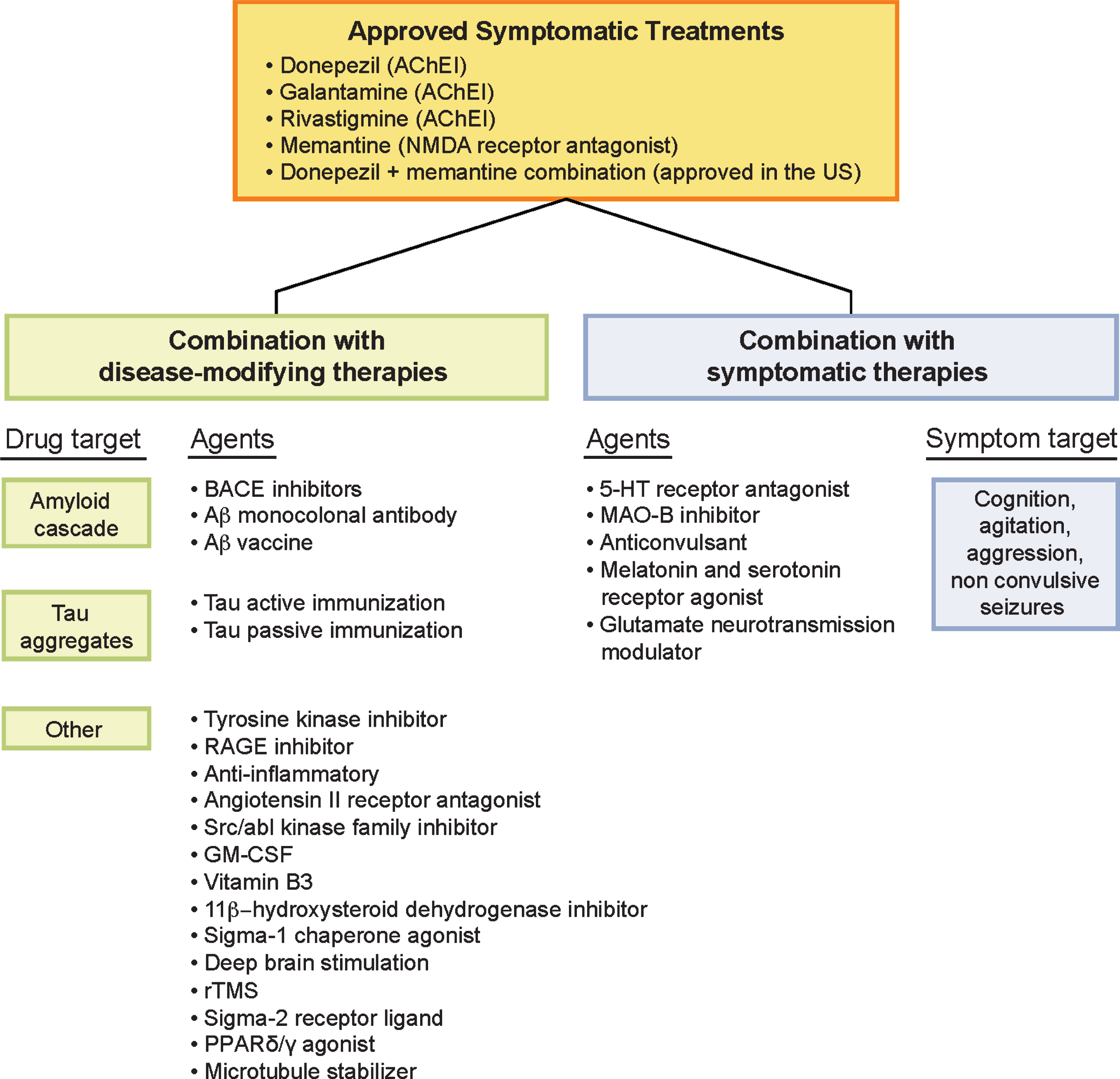
There is an increasing appreciation of the complexity of AD, the diversity of pathology, and the dynamic interactive network of components that make up the disease [17]. Recognition of this complexity suggests that addressing more than one target in the form of combination therapy may be needed for successful AD treatment [18, 19]. Combination therapies have succeeded in other diseases, such as cancer and human immunodeficiency virus-1 (HIV) and may prove to be more effective than targeting one mechanism at a time [20]. Here, we explore the current state of and future possibilities for pharmacodynamic combination therapy for the treatment of AD.
TYPES OF COMBINATION THERAPIES
Combination trials are distinct from add-on trials; the latter are the type typically used for new therapies in AD whereas, in combination therapy trials, two drugs are tested alone, in combination, and in comparison with placebo, usually in a 2×2 trial design. With add-on therapy, a new agent is compared with placebo in patients who are already receiving treatment with a background therapy. Most trials for new therapeutic agents in AD are conducted in patients already receiving cholinesterase inhibitors, memantine, or both and are thus new types of add-on treatment. The benefit of combination trials versus add-on trials is that the combination design allows investigators to analyze the effects of each therapy alone and in concert with others, distinguishing the individual and synergistic effects of the trial therapies.
Treatment combinations can be characterized as pharmacodynamic or pharmacokinetic. Pharmacodynamic combinations are designed to exert multiple effects on disease biology; pharmacokinetic combinations affect a drug’s absorption, distribution, metabolism, or elimination [21]. Examples of pharmacokinetic combinations include levodopa plus a dopamine decarboxylase inhibitor for treatment of Parkinson’s disease [22] and dextromethorphan plus quinidine for treatment of pseudobulbar affect [23]. Pharmacokinetic combinations are not considered further in this review.
Pharmacodynamic combinations for treatment of AD can include symptomatic agents that address the behavioral and cognitive symptoms of AD without changing the underlying disease biology or disease-modifying therapies (DMTs) that change disease course by addressing the underlying biology that leads to nerve cell death [24]. Pharmacodynamic combinations allow the combination of ≥2 symptomatic agents, ≥2 DMTs, or complex combinations of symptomatic therapies and DMTs to treat AD. Current clinical add-on trials generally use standard-of-care agents (cholinesterase inhibitors or memantine) and a new DMT or symptomatic agent; additive effects of the multiple simultaneous therapies may be important for patient outcomes.
Combination therapy can be used flexibly in addressing the drug target, delivery method, or delivery timing. For example, within AD DMT development programs, therapeutic agents could target amyloid, tau, or other disease processes, such as inflammation. The benefit of this type of combination therapy is that it can address ≥2 of these targets (e.g., amyloid- and tau-targeting therapies) or address a single target in two ways (e.g., two amyloid-targeting therapies). Combination therapies can also be designed to use >1 delivery method (e.g., oral and intravenous). Sequential combinations address targets consecutively (e.g., remove amyloid plaque with monoclonal antibody and then follow with a BACE 1 inhibitor to prevent plaque reaccumulation) [25]. Sequential combinations will likely evolve in concert with increased understanding of AD etiology and reflect the complexity of the biology of AD [26].
Another form of combination therapy is the use of multifunctional molecules—single agents that combine >1 activity or >1 target. An example of this is rasagiline, a monoamine oxidase (MAO) B inhibitor used to treat Parkinson’s disease, which, in addition to MAO B inhibition, has neuroprotective effects and effects on amyloid processing [27, 28].
Combination Therapies for Alzheimer’S disease
Several DMTs are currently in clinical studies as add-on therapies to the standard of care, cholinesterase inhibitors or memantine (Table 1). Because these studies allow the DMTs to be added to a background standard-of-care therapy, the analyses will assist in understanding the differences between combination therapy with background therapy compared to background therapy without the experimental agent.
Table 1
Add-On Clinical Trials of Combination Treatments for AD
| Agent | Dose | Type | AD Stage | Phase | Baseline Therapy* | Clinical Trials |
| Disease modifying | ||||||
| Aducanumab | NR | Amyloid passive immunization | Early | III | SOC† | NCT02484547 |
| NCT02477800 | ||||||
| Gantenerumab | NR | Early | II/III | NR‡ | NCT01760005 | |
| III | SOC† | NCT02051608 | ||||
| Crenezumab | NR | Amyloid passive immunization | Early | III | SOC† | NCT02670083 NCT03114657 |
| Solanezumab | NR | Amyloid passive immunization | Early | II/III | NR‡ | NCT01760005 |
| JNJ-54861911 | NR | BACE inhibitor | Early | II/III | NR‡ | NCT01760005 |
| Masitinib | 3.0 or 4.5 mg/kg/d | Selective tyrosine kinase inhibitor | Mild to moderate | III | Rivastigmine and/or memantine | NCT01872598 |
| ALZT-OP1 (cromolyn and ibuprofen) | NR | Anti-amyloid/anti-inflammatory | Early | III | AChEI’s† and/or memantine† | NCT02547818 |
| Flebogama DIF 5% and Albutein 20% | NR | Intravenous human immunoglobulin, human albumin | Mild, moderate | II/III | AChEI’s and/or memantine | NCT01561053 |
| BAN2401 | 2.5, 5.0, or 10.0 mg/kg once biweekly; or 5.0 or 10 mg/kg once a month | Amyloid passive immunization | Early | II | AChEI’s† and/or memantine† | NCT01767311 |
| AADvac-1 | Axon peptide 108 40 μg; 6 doses in 4-week intervals, then 5 doses in 3-month intervals | Tau active immunization | Mild | II | AChEI; memantine (permitted but not required | NCT02579252 |
| ABBV-8E12 | NR | Tau passive immunization | Early | II | SOC† | NCT02880956 |
| Nasal insulin | Insulin 20 IU BID | Peptide hormone | Early | II/III | NR‡ | NCT01767909 |
| Glulisine 20 IU BID | II | AChEI or memantine | NCT02503501 | |||
| Liraglutide | 1.8 mg/d | GLP-1 receptor agonist | Mild | II | AChEI† | NCT01843075 |
| Sargramostim | 250 μg/m2/d for 5 days/week for 3 weeks | GM-CSF | Mild, moderate | II | AChEI† and/or memantine† or Axona† | NCT01409915 |
| Telmisartan | 40 or 80 mg/d | Angiotensin II receptor antagonist | Mild, moderate | II | AChEI and/or memantine | NCT02085265 |
| Nicotinamide | 1500 mg BID | Vitamin B3 | Early | II | SOC† | NCT03061474 |
| Saracatinib | 100 to 125 mg/d | Src/abl kinase family inhibitor | Mild | II | AChEI’s† and/or memantine† and/or antidepressants† | NCT02167256 |
| UE2343 | 10 mg/d | β-hydroxysteroid dehydrogenase inhibitor | Mild | II | AChEI’s† and/or memantine† | NCT02727699 |
| ANAVEX2-73 | 30 or 50 mg/d | Sigma-1 chaperone agonist | Mild, moderate | II | SOC | NCT02244541 NCT02756858 |
| BIIB092 | NR | Tau passive immunization | Early | II | SOC† | NCT03352557 |
| Curcumin and yoga | Curcumin: 800 mg/d; yoga: aerobic or nonaerobic | Dietary supplement/ exercise regimen | MCI | II | NR† | NCT01811381 |
| Deep brain stimulation of the fornix | NR | Procedural intervention | NR | NA | AChEI’s and/or memantine | NCT03290274 |
| Neflamapimod | 40 mg BID | p38 MAPK alpha inhibitor | Early, mild | II | AChEI† or memantine† | NCT03402659 |
| Repetitive transcranial magnetic stimulation | 2 or 4 weeks of once-daily treatment 5 days/week; at 20Hz | Procedural intervention | Mild, moderate | NA | AChEI† | NCT02908815 |
| CT1812 | 280 or 560 mg/d | Sigma-2 receptor ligand | Mild, moderate | I/II | AChEI† or memantine† | NCT02907567 |
| T3D-959 | 3, 10, 30, or 90 mg/d for 2 weeks | PPARδ/γ agonist | Mild, moderate | I/II | NR‡ | NCT02560753 |
| ACI-24 | 340 to 460 μg/mL | Amyloid passive immunization | Mild, moderate | I/II | AChEI | 2008-006257-40 (EudraCT) |
| ACI-35 | NR | Tau active immunization | Mild, moderate | I | AChEI | ISRCTN13033912 (ISRCTN registry) |
| ABvac40 | NR | Amyloid active immunization | Mild, moderate | II | NR† | Unregistered trial ongoing |
| TPI 287 | 2.0, 6.3, or 20 mg/m2 once every 3 weeks | Microtubule stabilizer | Mild, moderate | I | SOC† | NCT01966666 |
| LY3303560 | NR | Tau passive immunization | Early, mild, moderate | AChEI†, memantine,† and/or other AD therapy† | NCT03019536 | |
| NR‡ | NCT02754830 | |||||
| Idalopirdine | 30 or 60 mg/d | 5-HT6 antagonist | Mild, moderate | III | Donepezil 10 mg/d | NCT01955161 |
| 10 or 30 mg/d | Donepezil 10 mg/d | NCT02006641 | ||||
| 30 or 60 mg/d | AChEI | NCT02006654 | ||||
| 60 mg/d | Donepezil 10 mg/d or donepezil 10 mg/d and memantine (IR 20 mg/d or XR 28 mg/d) | NCT02079246 | ||||
| Intepirdine | 35 mg/d | 5-HT6 antagonist | Mild, moderate | III | Donepezil 5 or 10 mg/d | NCT02585934 |
| II | AChEI | NCT02910102 | ||||
| LY3002813 | NR; alone or in combination with LY3202626 | Amyloid passive immunization | Early | II | AChEI and/or memantine | NCT03367403 |
| Symptomatic | ||||||
| Levetiracetam | NR | Anticonvulsant | Mild, moderate | II | Donepezil,† galantamine,† rivastigmine,† or memantine† | NCT02002819 |
| SUVN-502 | NR | 5-HT6 antagonist | Moderate | II | Donepezil and memantine | NCT02580305 |
| Citalopram | 30 mg/d | Selective serotonin reuptake inhibitor | Mild, moderate, severe | III | SOC | NCT00898807 |
| Sertraline | 25 to 125 mg/d (target dose, 100 mg/d) | Selective serotonin reuptake inhibitor | NR | II/III | SOC | NCT00086138 |
| Risperidone | Up to 1.5 mg/d followed by divalproex if agitation persists | Serotonin-dopamine antagonist antipsychotic | NR | IV | NR‡ | NCT00208819 |
| Olanzapine | Up to 7.5 mg/d followed by divalproex if agitation persists | Multi-acting receptor-targeted antipsychotic | NR | IV | NR‡ | NCT00208819 |
| Quetiapine | NR | Multi-acting receptor-targeted antipsychotic | NR | NA | AChEI† | NCT00232570 |
| Brexpiprazole | 1 or 2 mg/d | Partial dopamine receptor agonist | Mild, moderate, severe | II/III | NR | NCT03620981 |
| Aripiprazole | 2, 3, or 6 mg/d | Partial dopamine receptor agonist | Mild, moderate, severe | III | NR‡ | NCT02168920 |
| Rasagiline | 0.5 mg/d, uptitrated to 1 mg/d | Monoamine oxidase B inhibitor | Mild, moderate | II | AChEI† or memantine† | NCT02359552 |
| Piromelatine | 5, 20, or 50 mg/d | Melatonin and serotonin receptor agonist | Mild | II | Prescribed drugs for AD including AChEI’s† | NCT02615002 |
| Riluzole | NR | Glutamate neurotransmission modulator | Mild | II | Donepezil or rivastigmine† or galantamine† | NCT01703117 |
5-HT, 5-hydroxytrytamine (serotonin); AChEI, acetylcholinesterase inhibitor; AD, Alzheimer’s disease; BACE, aspartyl protease β-site amyloid precursor protein cleaving enzyme 1; BID, twice-daily; EudraCT, European Clinical Trials Database; GLP-1, glucagon-like peptide-1; GM-CSF, granulocyte-macrophage colony-stimulating factor; IR, immediate release; MAPK, mitogen-activated protein kinase; MCI, mild cognitive impairment; NA, not available; NR, not reported; PPAR, peroxisome proliferator-activated receptor; SOC, standard-of-care medication(s) for AD (agent/dose not specified); XR, extended release. *Doses of baseline therapy were not reported except where indicated. †Patients who were receiving stable standard-of-care therapy and those not currently receiving therapy were eligible. ‡Available inclusion/exclusion criteria did not note baseline use of AD therapy.
Phase III add-on treatments involving disease-modifying therapies
As of April 2018, nine DMTs are the subject of ongoing or recently completed phase III trials as an add-on to standard-of-care agents (Table 1). One approach taken by several of these putative therapies is to inhibit BACE 1 [9].
A placebo-controlled phase III trial of one BACE 1 inhibitor, verubecestat (MK-8931), in patients with prodromal AD was recently terminated after an initial safety analysis failed to establish a positive risk/benefit ratio [29]. Verubecestat had demonstrated promising findings in a phase I trial by reducing Aβ40 and Aβ42 in the cerebrospinal fluid of healthy subjects and patients with mild to moderate AD [30]. Verubecestat was also investigated in patients with mild to moderate AD, but the development program was terminated because of a lack of positive effect in an interim analysis of the trial (NCT01739348) [31, 32]. This lack of efficacy supports the theory that use of a BACE 1 inhibitor in patients who have accumulated enough Aβ deposition to have dementia is unlikely to have clinical benefit. BACE 1 inhibitors might work in monotherapy in primary prevention or early secondary prevention when Aβ accumulation is incomplete provided that they prove to be safe.
Another method for targeting the amyloid cascade is the use of humanized or fully human monoclonal antibodies (mAbs) that bind and mount an immunologic response against the Aβ peptide, leading to increased amyloid clearance [33]. Based on promising results in phase I/II trials [34-36], three Aβ mAbs (aducanumab, gantenerumab, and crenezumab) are being investigated in placebo-controlled phase III trials as add-on therapy in patients with early (i.e., prodromal) or mild AD. These trials are estimated to be completed between 2019 and 2022.
Several completed Aβ mAb passive immunization studies have not been successful; placebo-controlled phase III trials with solanezumab and bapineuzumab, both of which demonstrated promise in early studies, failed to show clinical benefit as add-on therapy to standard-of-care agents and resulted in termination of their development programs [37-39]. Solanezumab is currently being investigated in a phase III trial in parallel with gantenerumab in the Dominantly Inherited Alzheimer Network Trials Unit (DIAN-TU) trial [40] (Table 1). The DIAN-TU had also been investigating the BACE 1 inhibitor JNJ-54861911; however, this agent was recently removed from the study due to incidences of high elevations of liver enzymes [41].
The second major pathologic hallmark of AD is the formation of intracellular neurofibrillary tangles composed of hyperphosphorylated tau. Tau pathology is characterized primarily by abnormal phosphorylation and other modifications that alter tau structure and lead to formation of tau protein aggregates, associated with neurofibrillary degeneration and dementia [7]. Tau aggregation inhibitors (TAIs) have the potential to prevent or reverse tau aggregation and consequently reduce tau pathology and associated behavioral deficits in patients with AD [42, 43].
A second-generation TAI, TRx0237 (a methylthioninium chloride commonly known as methylene blue), was evaluated in two phase III trials and an ongoing phase III open-label extension study in patients with AD. Patients were either untreated or treated with stable doses of standard-of-care agents. The first study compared TRx0237 (150 or 250 mg/d) with control (placebo plus TRx0237 4 mg twice daily) in 891 patients with mild to moderate AD (Mini-Mental State Examination [MMSE] score of 14–26). This trial failed to show treatment benefit in the co-primary endpoints (Alzheimer’s Disease Assessment Scale–Cognitive subscale [ADAS-Cog] and Alzheimer’s Disease Cooperative Study–Activities of Daily Living) compared with control. Post-hoc analyses suggested that patients not on background therapy tended to have better outcomes [44]. This analysis in favor of monotherapy outcomes requires confirmation in prospective studies.
Although these trials examining the potential efficacy of BACE 1 inhibition, Aβ immunization, and TAI as add-on treatment to standard-of-care agents may provide important clinical data on how to best treat AD, the recent failure of many anti-AD agents to meet primary study endpoints suggests the need for a change in strategy. The complexity and extended timeline of AD pathology may require the combination of different treatment targets to demonstrate clinical efficacy [20].
The high rate of inflammatory changes in AD suggests that co-administration of anti-inflammatory and amyloid- or tau-directed treatments warrants assessment. Masitinib, a selective tyrosine kinase inhibitor, antagonizes mast cell function and indirectly controls proinflammatory mediators, thus potentially targeting the neuroinflammatory pathways believed to play a role in the pathogenesis of AD [45, 46]. Masitinib was investigated as an add-on therapy in a placebo-controlled phase II trial in patients with AD (n=34) receiving stable doses of cholinesterase inhibitors or memantine. Compared with the placebo group, patients receiving masitinib showed significant improvements in the ADAS-Cog and MMSE at weeks 12 and 24. Masitinib was subsequently investigated in a placebo-controlled phase III trial in patients with mild to moderate AD, in which an interim analysis of the trial was anticipated in 2017 [44]. The status of this trial is currently unknown.
Phase III combination treatments involving disease-modifying therapies
There are two DMTs currently in phase III trials that address two targets and represent valid combination therapies: ALZT-OPT1 and Gamunex (immune globulin intravenous (human), 10%; Grifols Therapeutics, Clayton, NC, USA). The ALZT-OPT1 trial, a combination regimen with cromolyn (anti-amyloid agent) and ibuprofen (anti-inflammatory agent), is enrolling patients with early AD who are either receiving or not receiving standard-of-care agents (Table 1). Cromolyn is a treatment for asthma approved by the US Food and Drug Administration (FDA) that bears structural similarity to other anti-amyloid agents and is likely to cross the blood-brain barrier. Cromolyn reduced Aβ fibrilization and oligomerization in vitro and reduced Aβ40 and Aβ42 monomer concentrations in mouse brain; oligomerization and fibrilization were unchanged in vivo. ALZT-OPT1 is a true combination trial in that the combination targets multiple disease pathways (amyloid and inflammation) and includes multiple methods of administration (intranasal inhaler for cromolyn and oral tablet for ibuprofen). ALZT-OPT1 is also an add-on study because it allows patients to continue standard-of-care treatments on stable doses. The study is anticipated to be completed in November 2019.
A phase III combination trial under way for Gamunex uses plasma exchange to deliver human albumin in combination with intravenous immunoglobulin. This combination trial targets amyloid in two ways. Aβ can cross the blood-brain barrier and bind to albumin, effectively sequestering Aβ in the periphery. The hypothesis behind plasma exchange with albumin replacement is that removing and replacing the albumin bound to pathogenic elements will allow further transfer of Aβ out of the central nervous system. Albumin also has anti-oxidant effects that may contribute therapeutic benefit [48]. Combining plasma exchange with infusion of intravenous immunoglobulin (which also binds amyloid) may further increase amyloid clearance from the brain [49]. This study, which enrolled patients receiving stable doses of standard-of-care agents and was scheduled to be completed in December 2017, has not yet reported results.
The receptor for advanced glycation end-products (RAGE), which is expressed by various brain cells and binds and transports Aβ from blood to brain, is thought to play multiple roles in the pathogenesis of AD and has emerged as a potential target for the treatment of the disease [50]. Agents that block Aβ-RAGE interaction may have numerous therapeutic actions including reducing neuroinflammation, improving cognitive decline, and reducing brain Aβ levels. Because RAGE inhibitors act to reduce both inflammation and the concentration of Aβ in the brain, they are considered multifunctional molecules, a combination not of >1 drug but of >1 mechanism of action. However, a phase III clinical trial for the RAGE inhibitor, azeliragon (also known as PF-04494700 and TTP488), did not meet either co-primary efficacy endpoint (improvement in cognitive or functional outcomes) and was terminated [51].
Phase III add-on treatments involving symptomatic agents
Several symptomatic treatments in current or recently completed phase III trials as add-ons to standard-of-care therapies were designed to ameliorate neuropsychiatric symptoms (agitation, aggression, depression, and anxiety) or to enhance the effects of cholinesterase inhibitors by modulating serotonin (Table 1). Most patients with AD develop neuropsychiatric symptoms, but no drug is currently approved by the FDA to treat these symptoms in patients with AD. Symptomatic therapies, such as antidepressants (e.g., citalopram, fluoxetine, sertraline), and second-generation antipsychotics (e.g., risperidone, olanzapine, quetiapine, aripiprazole) are often used off-label to treat behavioral symptoms, although there is mixed evidence on their efficacy in AD [52, 53]. A meta-analysis of 16 placebo-controlled trials indicated that risperidone and olanzapine reduced aggression; however, both drugs were associated with serious adverse events and extrapyramidal symptoms [54] as well as cerebrovascular adverse events, confusion, and sedation [55-58]. In addition, increased mortality has been reported in patients receiving versus not receiving second-generation antipsychotics, with mortality rates approximately two-fold higher in those treated with antipsychotic agents [59].
One second-generation antipsychotic, brexpiprazole, a dopamine D2 receptor partial agonist approved for the treatment of schizophrenia and as add-on therapy to major depressive disorder, is currently being investigated in phase III trials for the treatment of agitation in patients with AD. Two of the phase III trials, in patients with mild to severe AD [60], recently completed. Topline results from these studies showed mixed improvement of agitation symptoms, with one study meeting the primary endpoint (change from baseline in Cohen-Mansfield Agitation Inventory [CMAI]) but not the secondary endpoint (change from baseline Clinical Global Impression–Severity of Illness [CGI-S]) and the second study meeting the secondary (CGI-S) but not the primary endpoint (CMAI) [61]. A third study is expected to be initiated in 2018 [62]. Cholinesterase inhibitors and memantine have both been shown to have psychotropic effects; the add-on design of these trials may be relevant to efficacy [63, 64].
Serotonin (5-hydroxytryptamine [5-HT]) receptor antagonists amplify the cholinergic signal associated with cholinesterase inhibitors and may potentially optimize the treatment of cognitive symptoms in patients with AD [65]. They represent pharmacodynamic combinations with two agents acting on the same mechanism. The selective 5-HT6 antagonist idalopirdine (Lu AE58054) was investigated as an add-on therapy to donepezil in a placebo-controlled phase II trial of patients with AD [66]. Patients receiving combination therapy with idalopirdine and donepezil had significantly improved ADAS-Cog scores compared with patients receiving placebo at week 24. However, idalopirdine as an add-on to donezepil or cholinesterase inhibitors did not meet primary endpoints in any of the three large, long-term, phase III trials in patients with mild to moderate AD [67]. The 5-HT6 antagonist, intepirdine (SB-742457, RVT-101), showed improvements in ADAS-Cog and the Alzheimer’s Disease Cooperative Study Group–Activities of Daily Living scale after 24 weeks of treatment in a donepezil- and placebo-controlled phase II trial in patients with AD not previously receiving treatment with cholinesterase inhibitors or memantine, although this study did not meet co-primary endpoints [68]. Recently, both a phase II gait-and-balance trial and a 24-week phase III trial (add-on to donepezil therapy) in patients with mild to moderate AD failed to meet primary endpoints [69, 70].
Phase II add-on and combination treatments
As of January 2018, 21 DMTs and 7 symptomatic therapies are currently in phase II clinical trials, many of which are trials of add-on therapies with standard-of-care treatment. The mechanisms of action for the agents in these add-on studies include amyloid immunization, tau immunization, glucagon-like peptide-1 receptor agonists, and serotonin receptor agonists. Several of the studies use novel approaches, such as combining a device (i.e., deep brain stimulation, repetitive transcranial magnetic stimulation) with standard-of-care treatment and combining lifestyle changes (i.e., yoga) with dietary supplements (i.e., curcumin; Table 1).
A phase II trial from Eli Lilly will examine the safety and efficacy of amyloid passive immunization (LY3002813) alone and in combination with a BACE 1 inhibitor (LY3202626). Both drugs have undergone clinical trials as monotherapy, several of which are still under way. However, this combination trial will test for a synergistic effect between the two drugs with three treatment arms: intravenous amyloid immunization (LY3002813) plus oral placebo, intravenous amyloid immunization plus BACE 1 inhibitor (LY3202626), and intravenous placebo plus oral placebo. The rationale for this approach is that mounting an immune response to Aβ already in the system and slowing further production of Aβ by inhibiting BACE 1 will have a greater effect on the amyloid cascade than targeting either mechanism alone. Patients enrolled in this trial may also continue standard-of-care treatments. The trial is expected to be completed in late 2020.
Rasagiline, a selective MAO inhibitor [71], is also in a phase II trial in patients with mild to moderate AD. Rasagiline has been shown to have neuroprotective, antiapoptotic action dependent on Bcl-2, protein kinase C, and the proteasome-ubiquitin complex [72]. Importantly, the S-isomer of rasagiline (TVP1002) is 1000 times less potent than an MAO inhibitor and exhibits comparable, neuroprotective activity. In preclinical experiments, rasagiline’s propargylamine moiety protects mitochondrial viability by activating Bcl-2 and protein kinase C and downregulates proapoptotic proteins [72]. Importantly, from an AD therapeutic perspective, compounds containing propargylamine, such as rasagiline, modulate proteolytic cleavage of the AβPP, decreasing Aβ production by activation of the nonamyloidogenic alpha-secretase pathway. This effect involves activation of mitogen-activated protein kinase and protein kinase C [28]. This phase II proof-of-concept (PoC) trial will have results in late 2018. Rasagiline is an example of combination treatment in a single multifunctional agent.
Phase I combination treatments
Four DMTs are currently in phase I trials as add-on therapy to standard-of-care treatment (Table 1). An Aβ-targeting active immunization agent, ABvac40, a passive Aβ immunization agent, LY3002813, and 1 tau active immunization agent, ACI-35, are being investigated in patients with early to moderate AD. Similarly, a tau-targeting agent, TPI-287, is currently being investigated in patients with mild to moderate AD who are either receiving or not receiving standard-of-care therapy. The study completed in November 2017, and initial exploratory analyses suggest a potential MMSE benefit compared with placebo; this may have been because of a larger than expected decline in MMSE score in the placebo group [73].
Clinical Trial Designs
The challenges in AD therapy development and numerous clinical trial failures suggest that new innovative study designs may be needed [10, 74]. In placebo-controlled factorial 2×2 trials, patients are randomized to A plus placebo, B plus placebo, A plus B, or placebo plus placebo [75]. This type of study design enables simultaneous investigation of a combination regimen with two drugs and an analysis of the separate effects of each drug in a well-controlled manner. A modification of this study design is used in the current add-on studies that randomize patients receiving or not receiving standard-of-care agents to placebo or active treatment.
Another trial type is a 3-arm study in which patients are randomized to treatment A, B, or a combination of A and B. Superior efficacy of AB over A suggests that B is active in the combination, and superior efficacy of AB over B indicates that A is contributing to the treatment effect. One advantage of this trial design is that no patients are left untreated. Another advantage is that this type of trial is generally smaller with an easier recruitment process, making it a good choice for an early-stage PoC study. However, because this design makes it more difficult to differentiate the effects of A from those of B and the effects of A or B from a placebo, it is less appropriate for later-stage drug development.
The Investigation of Serial Studies to Predict Your Therapeutic Response With Imaging and Molecular Analysis (I-SPY) model used in breast cancer clinical trials offers an adaptive trial design that could also be used in AD [76]. I-SPY combines two types of trial designs: an “umbrella” trial in which many therapeutic agents are tested in parallel (with treatment groups sharing a protocol and a placebo group) and a “basket” trial in which patients are grouped by genotype or other disease features. The I-SPY trials are designed to use Bayesian modeling to predict whether each agent is likely to meet primary endpoints. The advantages of this type of trial design for AD include the ability to compare effects on biomarkers between therapeutic agents, adaptive dose adjustment for patients, a shared placebo group and trial infrastructure (reducing clinical trial costs), and the opportunity to continuously compare and potentially combine treatment groups. The European Prevention of Alzheimer’s Disease (EPAD) and the DIAN-TU study have trial protocols similar to that of the I-SPY approach [40, 77].
However, AD biology and pathogenesis remain less well known than those of breast cancer, and tissue samples are not readily available to assess treatment effects. Additionally, subgroupings or genotypes of patients with AD may not correlate with similarities in how patients respond to treatment, making the creation and analysis of treatment “baskets” more complex. Finally, the sensitivity of efficacy endpoints is not established and there is no firm consensus on intermediate endpoints that could predict AD treatment efficacy and support accelerated approval.
Several adaptive trials are under way for AD. DIAN-TU is an international trial designed to evaluate DMTs for autosomal dominant AD [78]. The trial began enrolling patients in 2012 to test the two anti-Aβ antibodies solanezumab and gantenerumab. The BACE inhibitor JNJ-54861911 was also being investigated but was removed from the trial as previously mentioned [41] (Table 1). The goal of this trial is not only to test specific agents but also to provide a framework for testing the latest therapies, potentially combining them, and to provide a repository of data and biological samples for future research. Adaptive endpoints include effects on cognitive and clinical measures, potentially implementing innovative disease progression models, a dose escalation algorithm, novel imaging techniques, and an adaptive efficacy assessment that considers early disease biomarkers and later disease cognitive analyses [40].
The EPAD project focuses on prodromal AD, aiming to develop treatments to prevent disease progression and dementia [77]. EPAD comprises the EPAD Longitudinal Cohort Study (EPAD-LCS) and the EPAD-PoC. EPAD-LCS enrolls patients older than 50 years who have participated in other research studies or who have been referred by a clinician and includes annual brain scans and biomarker draws over 44 months [79]. This patient pool will provide information about probability of AD-related decline and serve as a large recruitment pool for the second arm of the study, EPAD-PoC. EPAD-PoC will examine new AD interventions and will draw from the EPAD-LCS patient pool, using inclusion and exclusion criteria based on risk of decline, cognitive test scores, biomarker values, and genetic information collected as part of the EPAD-LCS protocol. These EPAD-PoC trials will identify the patient populations and subpopulations most likely to respond to a particular agent and therefore the ideal patients to recruit for later-stage clinical trials. EPAD-PoC will also use an adaptive Bayesian design to predict success or futility of individual drugs and adaptively randomize patients to the most successful drugs [80].
The adaptive trial designs of both EPAD and DIAN-TU will aid in developing multiple drugs by allowing more direct comparison of drug effects and a more efficient, cost-effective trial framework than traditional, stand-alone clinical trials. The longitudinal and disease progression model aspects of these studies provide the possibility of testing the efficacy of particular drugs or drug combinations at specific stages of disease progression [80].
Regulatory Guidance
Both the FDA and European Medicines Agency have released guidances for industry, highlighting the nonclinical and clinical data needed for combination therapy development [28, 40, 74]. In general, combination trials need to justify the pharmacologic and medical rationale for the combination therapy, as well as demonstrate efficacy and a positive risk-benefit profile. In patients inadequately controlled with a monotherapy (i.e., an add-on scenario), a randomized controlled superiority trial should be done to demonstrate greater efficacy with the combination therapy compared with the respective monotherapies using a 3-arm A versus B versus AB trial design [81]. When co-developing two novel drugs, the FDA recommends a 4-arm A versus B versus AB versus placebo (or approved standard-of-care agent) design in phase II trials and a 2-arm AB versus placebo (or standard-of-care agent) design in phase III trials [82].
There is increasing recognition of the need for timely development of combination therapies and the corresponding need for combination-specific regulation. In particular, the 21st Century Cures Act supports efficient combination therapy development by promoting clear communication regarding market expectations, coordination between research centers, and compliance with good manufacturing processes [83].
The FDA promotes combination drug development only for “serious diseases,” defined as “a disease or condition associated with morbidity that has substantial impact on day-to-day functioning” [82] and has issued specific guidance on combination drug development for cancer, tuberculosis, and HIV infection [84-86]. From a regulatory standpoint, the demonstrated success and continued potential benefit of combination treatment in all of these disorders have paved the way for efficient development of effective combination therapy for AD [83].
DRUG DEVELOPMENT
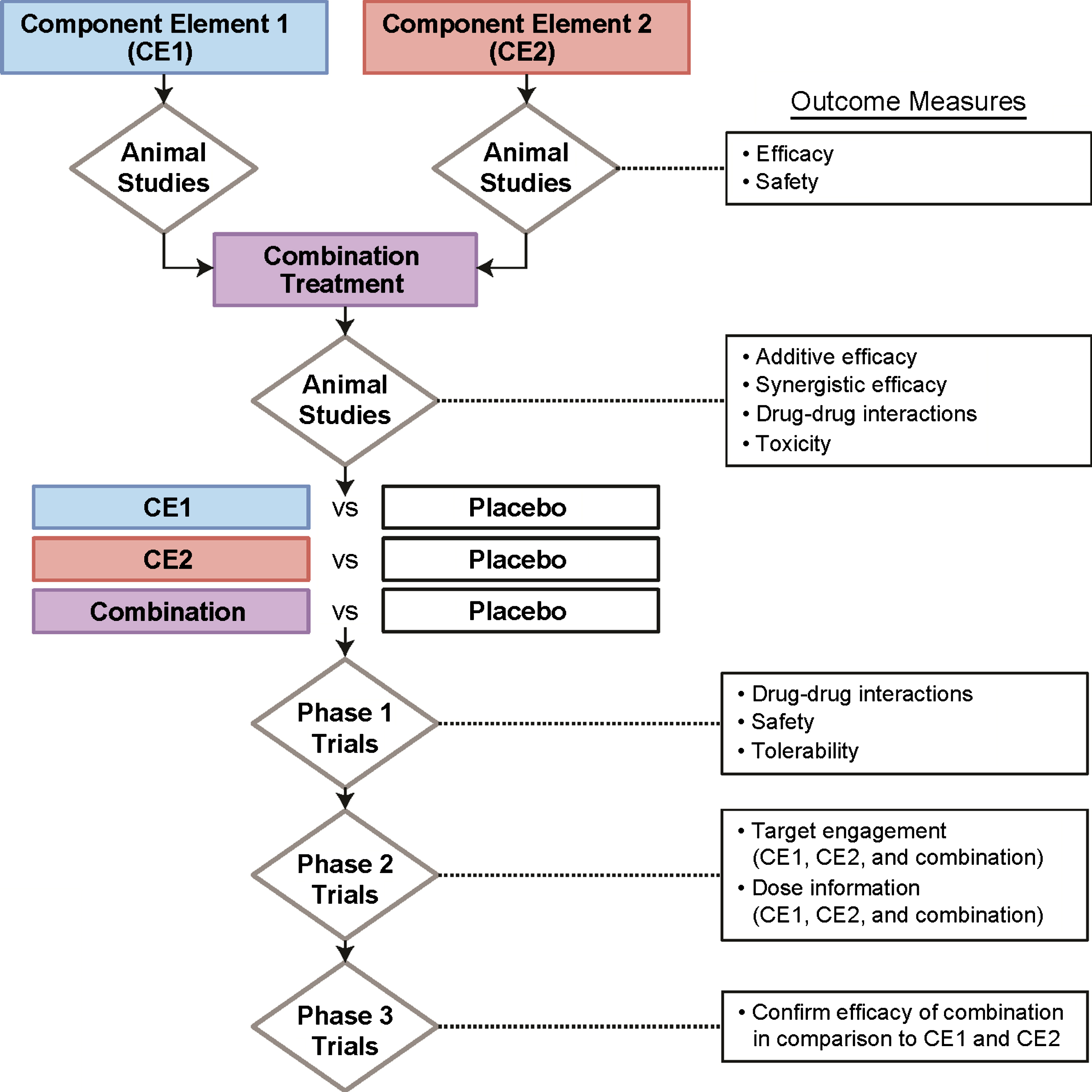
There is no established framework for drug development of combination therapies. Agents might usefully target the same pathway and amplify the effect or target complementary aspects of the disease capitalizing on synergies of effects. Component elements of combination therapies would reasonably be tested in animal model systems to ensure their effects and the combination would be tested to explore additive and synergistic effects (Fig. 2). Failure at the animal model level would represent one part of a decision not to advance a compound further. Assessment of combinations in animals would also allow discovery of potentially important drug-drug interactions or toxicity resulting from the combination. Data will also be needed from phase I first-in-human trials on drug-drug interactions, safety, and tolerability in humans. Phase II learning trials would generate target engagement and dose information on each element and the combination. Phase II allows exploration of dose and dose-response; it is possible that lower doses of individual agents may be used in combination regimens where synergies apply. This outcome might help limit the potential toxicity associated with combinations. If successful in phase II, confirmatory phase III studies would be conducted. Planning combination strategies from the beginning has the advantage of having the candidate agents at the same level of investigation when combinations become appropriate. Combinations of novel agents and better-understood repurposed agents are another means of populating a pipeline of combination treatments. Targets that might be addressed in combination therapy regimens include Aβ, tau, inflammation, and neuroprotection.
Fig.2
Combination drug development.
Conclusions
Given the complexity of AD, treatment of patients remains challenging. The currently approved treatments for AD are limited to cholinesterase inhibitors and memantine or the combination of these agents. Furthermore, despite the early promise of several new drugs, many have failed larger phase III trials, not meeting efficacy endpoints. The high failure rate of the therapies in development for AD stems in large part from the complex pathologic causes of the disease, as well as our incomplete understanding of the relationships among the numerous pathways involved in development of AD and subsequent neurodegeneration, and the potential lack of efficacy of available agents. Although combining cholinesterase inhibitors and memantine has had limited success in the treatment of AD, it is nonetheless likely that targeting multiple pathways may be required for successful treatment. Consequently, additional trials investigating logical combinations of agents should continue to be performed.
Several ongoing clinical trials are investigating DMTs or symptomatic agents as add-ons to background standard-of-care therapy. However, a combination of ≥2 agents that target separate pathways presents an opportunity for treating the disease that may offer synergistic effects, and there are very few of these in trials. Combining therapeutic agents may allow for lower doses of the individual agents, reducing costs and side effects. Innovative and adaptive clinical trial designs may also capture the potential evolution of therapeutic combinations over the long and complex course of disease progression, with one set of agents appropriate for preclinical AD, another for early-stage AD, and yet another for AD dementia. Taken together, the challenges of treating AD have steered the current treatment landscape toward investigating new drugs as an add-on to standard-of-care and repurposing existing drugs indicated for other therapeutic conditions, and also toward combining two agents that target different pathways in parallel, following the example of successful treatment combinations for other serious diseases and conditions, such as cancer and HIV [20].
Acknowledgments
The authors received medical writing assistance for development of this manuscript from Maria Hovenden, PhD, and Lauren Stutzbach, PhD, at Complete Publication Solutions, LLC (North Wales, PA; a CHC Group company), which was funded by Lundbeck. JLC acknowledges support from a COBRE NIH/MIGMS grant (P20GM109025) and Keep Memory Alive.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0766r1).
REFERENCES
[1] | Blennow K , de Leon MJ , Zetterberg H ((2006) ) Alzheimer’s disease. Lancet 368: , 387–403. |
[2] | Alzheimer’s Association ((2016) ) 2016 Alzheimer’s disease facts and figures. Alzheimers Dement 12: , 459–509. |
[3] | Alzheimer’s Disease International, World Alzheimer Report 2016: The global impact of dementia, Alzheimer’s Disease International, https://www.alz.co.uk/research/WorldAlzheimerReport2016.pdf, Accessed on March 17, (2017) . |
[4] | Reitz C , Brayne C , Mayeux R ((2011) ) Epidemiology of Alzheimer disease. Nat Rev Neurol 7: , 137–152. |
[5] | Ringman JM , Goate A , Masters CL , Cairns NJ , Danek A , Graff-Radford N , Ghetti B , Morris JC , Dominantly Inherited Alzheimer Network ((2014) ) Genetic heterogeneity in Alzheimer disease and implications for treatment strategies. Curr Neurol Neurosci Rep 14: , 499. |
[6] | Selkoe DJ , Hardy J ((2016) ) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8: , 595–608. |
[7] | Iqbal K , Alonso Adel C , Chen S , Chohan MO , El-Akkad E , Gong CX , Khatoon S , Li B , Liu F , Rahman A , Tanimukai H , Grundke-Iqbal I ((2005) ) Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta 1739: , 198–210. |
[8] | Mufson EJ , Counts SE , Perez SE , Ginsberg SD ((2008) ) Cholinergic system during the progression of Alzheimer’s disease: therapeutic implications. Expert Rev Neurother 8: , 1703–1718. |
[9] | Yan R , Vassar R ((2014) ) Targeting the beta secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol 13: , 319–329. |
[10] | Cummings JL , Morstorf T , Zhong K ((2014) ) Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther 6: , 37. |
[11] | Aricept ((2015) ) (donepezil hydrochloride). Full Prescribing Information, Eisai Inc., Woodcliff Lake, NJ. |
[12] | Exelon ((2015) ) (rivastigmine tartrate). Full Prescribing Information, Novartis Pharmaceuticals Corporation, East Hanover, NJ. |
[13] | Exelon Patch ((2016) ) (rivastigmine transdermal system). Full Prescribing Information, Novartis Pharmaceuticals Corporation, East Hanover, NJ. |
[14] | Razadyne ((2016) ) (galantamine hydrobromide). Full Prescribing Information, Janssen Pharmaceuticals Inc., Titusville, NJ. |
[15] | Namenda XR ((2014) ) (memantine hydrochloride). Full Prescribing Information, Forest Pharmaceuticals Inc., St. Louis, MO. |
[16] | Namzaric ((2016) ) (memantine and donepezil hydrochlorides). Full Prescribing Information, Allergan USA, Inc., Irvine, CA. |
[17] | Mizuno S , Iijima R , Ogishima S , Kikuchi M , Matsuoka Y , Ghosh S , Miyamoto T , Miyashita A , Kuwano R , Tanaka H ((2012) ) AlzPathway: a comprehensive map of signaling pathways of Alzheimer’s disease. BMC Syst Bio 6: , 52. |
[18] | Schmitt B , Bernhardt T , Moeller HJ , Heuser I , Frolich L ((2004) ) Combination therapy in Alzheimer’s disease: a review of current evidence. CNS Drugs 18: , 827–844. |
[19] | Fessel WJ ((2017) ) Concordance of several subcellular interactions initiates Alzheimer’s dementia: their reversal requires combination treatment. Am J Alzheimers Dis Other Demen 32: , 166–181. |
[20] | Tomaszewski S , Gauthier S , Wimo A , Rosa-Neto P ((2016) ) Combination therapy of anti-tau and anti-amyloid drugs for disease modification in early-stage Alzheimer’s disease: socio-economic considerations modeled on treatments for tuberculosis, HIV/AIDS and breast cancer. J Prev Alzheimers Dis 3: , 164–172. |
[21] | Toews ML , Bylund DB ((2005) ) Pharmacologic principles for combination therapy. Proc Am Thorac Soc 2: , 282–289. |
[22] | Barbeau A , Mars H , Botez MI , Joubert M ((1972) ) Levodopa combined with peripheral decarboxylase inhibition in Parkinson’s disease. Can Med Assoc J 106: , 1169–1174. |
[23] | Pioro EP , Brooks BR , Cummings J , Schiffer R , Thisted RA , Wynn D , Hepner A , Kaye R , Safety Tolerability and Efficacy Results Trial of AVP-923 in PBA Investigators ((2010) ) Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbar affect. Ann Neurol 68: , 693–702. |
[24] | Cummings. J , Fox N ((2017) ) Defining disease modifying therapy for Alzheimer’s disease. J Prev Alzheimers Dis 4: , 109–115. |
[25] | Wang A , Das P , Switzer RC 3rd , Golde TE , Jankowsky JL ((2011) ) Robust amyloid clearance in a mouse model of Alzheimer’s disease provides novel insights into the mechanism of amyloid-beta immunotherapy. J Neurosci 31: , 4124–4136. |
[26] | Jack CR Jr. , Vemuri P , Wiste HJ , Weigand SD , Aisen PS , Trojanowski JQ , Shaw LM , Bernstein MA , Petersen RC , Weiner MW , Knopman DS , Alzheimer’s Disease Neuroimaging Initiative ((2011) ) Evidence for ordering of Alzheimer disease biomarkers. Arch Neurol 68: , 1526–1535. |
[27] | Parkinson Study Group ((2004) ) A controlled, randomized, delayed-start study of rasagiline in early Parkinson disease. Arch Neurol 61: , 561–566. |
[28] | Youdim MB , Weinstock M ((2001) ) Molecular basis of neuroprotective activities of rasagiline and the anti-Alzheimer drug TV3326 [(N-propargyl-(3R)aminoindan-5-YL)-ethyl methyl carbamate]. Cell Mol Neurobiol 21: , 555–573. |
[29] | Merck, Press Release Merck announces discontinuation of APECS study evaluating verubecestat (MK-8931) for the treatment of people with prodromal Alzheimer’s disease, http://www.mrknewsroom.com/news-release/research-and-development-news/merck-announces-discontinuation-apecs-study-evaluating-ve, Accessed on June 7,(2018) . |
[30] | Kennedy ME , Stamford AW , Chen X , Cox K , Cumming JN , Dockendorf MF , Egan M , Ereshefsky L , Hodgson RA , Hyde LA , Jhee S , Kleijn HJ , Kuvelkar R , Li W , Mattson BA , Mei H , Palcza. J , Scott JD , Tanen M , Troyer MD , Tseng JL , Stone JA , Parker EM , Forman MS ((2016) ) The BACE1 inhibitor verubecestat (MK-8931) reduces CNS beta-amyloid in animal models and in Alzheimer’s disease patients. Sci Transl Med 8: , 363ra150. |
[31] | Egan MF , Kost. J , Tariot PN , Aisen PS , Cummings JL , Vellas B , Sur C , Mukai Y , Voss T , Furtek C , Mahoney E , Harper Mozley L , Vandenberghe R , Mo Y , Michelson D ((2018) ) Randomized trial of verubecestat for mild-to-moderate Alzheimer’s disease. N Engl J Med 378: , 1691–1703. |
[32] | Merck, Press Release: Merck announces EPOCH study of verubecestat for the treatment of people with mild to moderate Alzheimer’s disease to stop for lack of efficacy, https://www.businesswire.com/news/home/20170214006278/en/Merck-Announces-EPOCH-Study-Verubecestat-Treatment-People, Accessed on June 7, (2018) . |
[33] | Schenk D , Basi GS , Pangalos MN ((2012) ) Treatment strategies targeting amyloid beta-protein. Cold Spring Harb Perspect Med 2: , a006387. |
[34] | Sevigny. J , Chiao P , Bussiere T , Weinreb PH , Williams L , Maier M , Dunstan R , Salloway S , Chen T , Ling Y , O’Gorman J , Qian F , Arastu M , Li M , Chollate S , Brennan MS , Quintero-Monzon O , Scannevin RH , Arnold HM , Engber T , Rhodes K , Ferrero J , Hang Y , Mikulskis A , Grimm J , Hock C , Nitsch RM , Sandrock A ((2016) ) The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 537: , 50–56. |
[35] | Novakovic D , Feligioni M , Scaccianoce S , Caruso A , Piccinin S , Schepisi C , Errico F , Mercuri NB , Nicoletti F , Nistico R ((2013) ) Profile of gantenerumab and its potential in the treatment of Alzheimer’s disease. Drug Des Devel Ther 7: , 1359–1364. |
[36] | Mackey H , Cho W , Ward M , Fang Y , Suliman S , Ho C , Paul R ((2016) ) Exploratory analyses of cognitive effects of crenezumab in a mild Alzheimer’s disease subpopulation of a randomized, double-blind, placebo controlled, parallel-group phase 2 study (ABBY). Alzheimers Dement 12: , P610. |
[37] | Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Ferris S, Reichert M, Ketter N, Nejadnik B, Guenzler V, Miloslavsky M, Wang D, Lu Y, Lull J, Tudor IC, Liu E, Grundman M, Yuen E, Black R, Brashear HR Bapineuzumab Clinical Trial Investigators ((2014) ) Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 370: , 322–333. |
[38] | H. Lundbeck A/S Press release: Headline conclusions from the first out of three phase III studies on idalopirdine in Alzheimer’s disease, http://investor.lundbeck.com/releasedetail.cfm?releaseid=990541, Accessed on June 12, (2018) . |
[39] | Doody RS , Thomas RG , Farlow M , Iwatsubo T , Vellas B , Joffe S , Kieburtz K , Raman R , Sun X , Aisen PS , Siemers E , Liu-Seifert H , Mohs R , Alzheimer’s Disease Cooperative Study Steering Committee, Solanezumab Study Group ((2014) ) Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med 370: , 311–321. |
[40] | Bateman RJ , Benzinger TL , Berry S , Clifford DB , Duggan C , Fagan AM , Fanning K , Farlow MR , Hassenstab J , McDade EM , Mills S , Paumier K , Quintana M , Salloway SP , Santacruz A , Schneider LS , Wang G , Xiong C , Dian-Tu Pharma Consortium for the Dominantly Inherited Alzheimer Network ((2017) ) The DIAN-TU Next Generation Alzheimer’s prevention trial: adaptive design and disease progression model. Alzheimers Dement 13: , 8–19. |
[41] | Janssen Press Release: Update on Janssen’s BACE Inhibitor Program Regarding the Dominantly Inherited Alzheimer’s Network Trial (DIAN-TU). Janssen, Titusville, NJ. Available at: https://www.janssen.com/update-janssens-bace-inhibitor-program-regarding-DIAN-TU-july-2018. Accessed December 04, 2018. |
[42] | Wischik CM , Harrington CR , Storey JM ((2014) ) Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem Pharmacol 88: , 529–539. |
[43] | Panza F , Solfrizzi V , Seripa D , Imbimbo BP , Lozupone M , Santamato A , Zecca C , Barulli MR , Bellomo A , Pilotto A , Daniele A , Greco A , Logroscino G ((2016) ) Tau-centric targets and drugs in clinical development for the treatment of Alzheimer’s disease. Biomed Res In 2016: , 3245935. |
[44] | Gauthier S , Feldman HH , Schneider LS , Wilcock GK , Frisoni GB , Hardlund JH , Moebius HJ , Bentham P , Kook KA , Wischik DJ , Schelter BO , Davis CS , Staff RT , Bracoud L , Shamsi K , Storey JM , Harrington CR , Wischik CM ((2016) ) Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 388: , 2873–2884. |
[45] | Piette F , Belmin J , Vincent H , Schmidt N , Pariel S , Verny M , Marquis C , Mely J , Hugonot-Diener L , Kinet JP , Dubreuil P , Moussy A , Hermine O ((2011) ) Masitinib as an adjunct therapy for mild-to-moderate Alzheimer’s disease: a randomised, placebo-controlled phase 2 trial. Alzheimers Res Ther 3: , 16. |
[46] | Shaik-Dasthagirisaheb YB , Conti P ((2016) ) The role of mast cells in Alzheimer’s disease. Adv Clin Exp Med 25: , 781–787. |
[47] | Hori Y , Takeda S , Cho H , Wegmann S , Shoup TM , Takahashi K , Irimia D , Elmaleh DR , Hyman BT , Hudry E ((2015) ) A Food and Drug Administration-approved asthma therapeutic agent impacts amyloid beta in the brain in a transgenic model of Alzheimer disease. J Biol Chem 290: , 1966–1978. |
[48] | Roche M , Rondeau P , Singh NR , Tarnus E , Bourdon E ((2008) ) The antioxidant properties of serum albumin. FEBS Lett 582: , 1783–1787. |
[49] | Boada M , Ramos-Fernandez E , Guivernau B , Munoz FJ , Costa M , Ortiz AM , Jorquera JI , Nunez L , Torres M , Paez A ((2016) ) Treatment of Alzheimer disease using combination therapy with plasma exchange and haemapheresis with albumin and intravenous immunoglobulin: rationale and treatment aroach of the AMBAR (Alzheimer Management By Albumin Replacement) study. Neurologia 31: , 473–481. |
[50] | Deane RJ ((2012) ) Is RAGE still a therapeutic target for Alzheimer’s disease? Future Med Chem 4: , 915–925. |
[51] | vTv Therapeutics, Press Release: vTv Therapeutics announces topline results from the first STEADFAST phase 3 study evaluating azeliragon in people with mild Alzheimer’s disease, http://ir.vtvtherapeutics.com/phoenix.zhtml?c=254081&p=irol-newsArticle_print&ID=2341681, Accessed on June 12, (2018) . |
[52] | Marvanova M ((2014) ) Antipsychotic use in elderly patients with dementia: efficacy and safety concerns. Mental Health Clin 4: , 170–176. |
[53] | Henry G , Williamson D , Tampi RR ((2011) ) Efficacy and tolerability of antidepressants in the treatment of behavioral and psychological symptoms of dementia, a literature review of evidence. Am J Alzheimers Dis Other Demen 26: , 169–183. |
[54] | Ballard C , Waite J. ((2006) ) The effectiveness of atypical antipsychotics for the treatment of aggression and psychosis in Alzheimer’s disease. Cochrane Database Syst Rev, CD003476. |
[55] | Schneider LS , Tariot PN , Dagerman KS , Davis SM , Hsiao JK , Ismail MS , Lebowitz BD , Lyketsos CG , Ryan JM , Stroup TS , Sultzer DL , Weintraub D , Lieberman JA , CATIE-AD Study Group ((2006) ) Effectiveness of atypical antipsychotic drugs in patients with Alzheimer’s disease. N Engl J Med 355: , 1525–1538. |
[56] | Layton D , Harris S , Wilton LV , Shakir SA ((2005) ) Comparison of incidence rates of cerebrovascular accidents and transient ischaemic attacks in observational cohort studies of patients prescribed risperidone, quetiapine or olanzapine in general practice in England including patients with dementia. J Psychopharmacol 19: , 473–482. |
[57] | Risperidal® ((2009) ) (risperidone). Full Prescribing Information, Ortho-McNeil-Janssen Pharmaceuticals Inc, Titusville, NJ. |
[58] | ZYPREXA (2018) (olanzapine). Full Prescribing Information, Eli Lilly and Company, Indianapolis, IN. |
[59] | Gill SS , Bronskill SE , Normand SL , Anderson GM , Sykora K , Lam K , Bell CM , Lee PE , Fischer HD , Herrmann N , Gurwitz JH , Rochon PA ((2007) ) Antipsychotic drug use and mortality in older adults with dementia. Ann Intern Med 146: , 775–786. |
[60] | Wang SM , Han C , Lee SJ , Jun TY , Patkar AA , Masand PS , Pae CU ((2016) ) Second generation antipsychotics in the treatment of major depressive disorder: an update. Chonnam Med J 52: , 159–172. |
[61] | Otsuka and Lundbeck, Press Release: Otsuka and Lundbeck announce improvement of agitation symptoms related to Alzheimer’s-type dementia following treatment with brexpiprazole relative to placebo, http://investor.lundbeck.com/releasedetail.cfm?releaseid=1023859, Accessed on June 5, (2018) . |
[62] | Lundbeck and Otsuka, Press Release: Lundbeck and Otsuka will initiate a third phase III trial to evaluate the use of brexpiprazole in the treatment of agitation in patients with dementia of the Alzheimer’s type, http://investor.lundbeck. com/releasedetail.cfm?ReleaseID=1046327, Accessed on June 7, (2018) . |
[63] | Cummings JL ((2008) ) Effect of cholinesterase inhibitors on behavior. Neurobiol Aging 29: , S6. |
[64] | Fox C , Crugel M , Maidment I , Auestad BH , Coulton S , Treloar A , Ballard C , Boustani M , Katona C , Livingston G ((2012) ) Efficacy of memantine for agitation in Alzheimer’s dementia: a randomised double-blind placebo controlled trial. PLoS One 7: , e35185. |
[65] | Upton N , Chuang TT , Hunter AJ , Virley DJ ((2008) ) 5-HT6 receptor antagonists as novel cognitive enhancing agents for Alzheimer’s disease. Neurotherapeutics 5: , 458–469. |
[66] | Wilkinson D , Windfeld K , Colding-Jorgensen E ((2014) ) Safety and efficacy of idalopirdine, a 5-HT6 receptor antagonist, in patients with moderate Alzheimer’s disease (LADDER): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol 13: , 1092–1099. |
[67] | Atri A , Frolich L , Ballard C , Tariot PN , Molinuevo JL , Boneva N , Windfeld K , Raket LL , Cummings JL ((2018) ) Effect of idalopirdine as adjunct to cholinesterase inhibitors on change in cognition in patients with Alzheimer disease: three randomized clinical trials. JAMA 319: , 130–142. |
[68] | Maher-Edwards G , Watson C , Ascher J , Barnett C , Boswell D , Davies J , Fernandez M , Kurz A , Zanetti O , Safirstein B , Schronen JP , Zvartau-Hind M , Gold M ((2015) ) Two randomized controlled trials of SB742457 in mild-to-moderate Alzheimer’s disease. Alzheimers Dement (N Y) 1: , 23–36. |
[69] | Axovant, Press Release: Axovant announces negative topline results of intepirdine phase 3 mindset trial in Alzheimer’s disease, http://investors.axovant.com/news-releases/news-release-details/axovant-announces-negative-topline-results-intepirdine-phase-3, Accessed on June 5, (2018) . |
[70] | Axovant, Press Release: Axovant announces negative results for intepirdine in phase 2b headway and pilot phase 2 gait and balance studies, http://investors.axovant.com/news-releases/news-release-details/axovant-announces-negative-results-intepirdine-phase-2b-headway, Accessed on February 20, (2018) . |
[71] | Youdim MB , Gross A , Finberg JP ((2001) ) Rasagiline [N-propargyl-1R(+)-aminoindan], a selective and potent inhibitor of mitochondrial monoamine oxidase B. Br J Pharmacol 132: , 500–506. |
[72] | Youdim MB , Bar Am O , Yogev-Falach M , Weinreb O , Maruyama W , Naoi M , Amit T ((2005) ) Rasagiline: neurodegeneration, neuroprotection, and mitochondrial permeability transition. J Neurosci Res 79: , 172–179. |
[73] | Cortice Biosciences, Press Release: Cortice Biosciences announces presentation of results from placebo-controlled phase 1 clinical trials evaluating TPI 287 for treatment of Alzheimer’s disease and related tauopathies, https://globenewswire.com/news-release/2017/11/03/1174533/0/en/Cortice-Biosciences-Announces-Presentation-of-Results-from-Placebo-Controlled-Phase-1-Clinical-Trials-Evaluating-TPI-287-for-Treatment-of-Alzheimer-s-Disease-and-Related-Tauopathie.html, Accessed on April 30, (2018) . |
[74] | Hendrix JA , Bateman RJ , Brashear HR , Duggan C , Carrillo MC , Bain LJ , DeMattos R , Katz RG , Ostrowitzki S , Siemers E , Sperling R , Vitolo OV ((2016) ) Challenges, solutions, and recommendations for Alzheimer’s disease combination therapy. Alzheimers Dement 12: , 623–630. |
[75] | Evans SR ((2010) ) Clinical trial structures. J Exp Stroke Transl Med 3: , 8–18. |
[76] | Messmer MF , Wilhelm EE , Shoulson I ((2017) ) I-SPY 2 breast cancer trial as a model for innovation in Alzheimer disease therapies. JAMA Neurol 74: , 1027–1028. |
[77] | European Prevention of Alzheimer’s Dementia Consortium: project objectives, http://ep-ad.org/project-objectives/, Accessed on February 16, (2018) . |
[78] | ClinicalTrials.gov, Dominantly Inherited Alzheimer Network Trial: an opportunity to prevent dementia. A study of potential disease modifying treatments in individuals at risk for or with a type of early onset Alzheimer’s disease caused by a genetic mutation (DIAN-TU), https://www.clinicaltrials.gov/ct2/show/study/NCT01760005, Accessed on February 16, (2018) . |
[79] | University of Edinburgh, European prevention of Alzheimer’s Dementia (EPAD) Longitudinal Cohort Study (LCS) (EPAD LCS), https://clinicaltrials.gov/ct2/show/NCT02804789, Accessed on April 23, (2018) . |
[80] | Ritchie CW , Molinuevo JL , Truyen L , Satlin A , Van der Geyten S , Lovestone S , European Prevention of Alzheimer’s Dementia Consortium ((2016) ) Development of interventions for the secondary prevention of Alzheimer’s dementia: the European Prevention of Alzheimer’s Dementia (EPAD) project. Lancet Psychiatry 3: , 179–186. |
[81] | European Medicines Agency, Guideline on clinical development of fixed combination medicinal products, http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2017/03/WC500224836.pdf, Accessed on June 7, (2018) . |
[82] | US Food and Drug Administration, Guidance for Industry: codevelopment of two or more unmarketed investigational drugs for use in combination, https://www.fda.gov/downloads/drugs/guidances/ucm236669.pdf, Accessed on June 7, (2018) . |
[83] | Hunter NL , Sherman RE ((2017) ) Combination products: modernizing the regulatory paradigm. Nat Rev Drug Discov 16: , 513–514. |
[84] | US Food and Drug Administration, Guidance for Industry: S9 nonclinical evaluation for anticancer pharmaceuticals, https://www.fda.gov/downloads/Drugs/Guidances/ucm085389.pdf, Accessed on June 7, (2018) . |
[85] | US Food and Drug Administration, Human immunodeficiency virus-1 infection: developing antiretroviral drugs for treatment. Guidance for Industry, https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm355128.pdf, Accessed on January 29, (2018) . |
[86] | US Food and Drug Administration, Guidance for Industry: pulmonary tuberculosis: developing drugs for treatment, https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM373580.pdf, Accessed on June 7, (2018) . |


