
Neuro-Immuno-Gene- and Genome-Editing-Therapy for Alzheimer’s Disease: Are We There Yet?
Abstract
Alzheimer’s disease (AD) is a highly complex neurodegenerative disorder and the current treatment strategies are largely ineffective thereby leading to irreversible and progressive cognitive decline in AD patients. AD continues to defy successful treatment despite significant advancements in the field of molecular medicine. Repeatedly, early promising preclinical and clinical results have catapulted into devastating setbacks leading to multi-billion dollar losses not only to the top pharmaceutical companies but also to the AD patients and their families. Thus, it is very timely to review the progress in the emerging fields of gene therapy and stem cell-based precision medicine. Here, we have made sincere efforts to feature the ongoing progress especially in the field of AD gene therapy and stem cell-based regenerative medicine. Further, we also provide highlights in elucidating the molecular mechanisms underlying AD pathogenesis and describe novel AD therapeutic targets and strategies for the new drug discovery. We hope that the quantum leap in the scientific advancements and improved funding will bolster novel concepts that will propel the momentum toward a trajectory leading to a robust AD patient-specific next generation precision medicine with improved cognitive function and excellent life quality.
INTRODUCTION
Human brain is a highly complex vital organ comprising of various cell types including neurons, astrocytes, oligodendrocyets, tanycytes, and microglia. These various cell types are specifically arranged in a highly complex architectural network, work together in a highly ordered and orchestrated fashion, and are regulated spatiotemporally. During the course of the normal development, the molecular mechanisms underlying normal homeostasis are very robust at young age. However, with age decline these molecular mechanisms undergo gradual senescence thereby causing a wide variety of neurodegenerative disorders and progressive cognitive decline and dementia. One of the most prevalent neurodegenerative disorder with huge socioeconomic burden is Alzheimer’s disease (AD). Although AD was discovered by Alois Alzheimer in 1906, the precise molecular mechanisms underlying AD pathogenesis are still not very clear. As a result, despite significant efforts, an effective treatment for AD remains elusive. Currently, there are an estimated 5.5 million Americans suffering from this devastating disease with an annual healthcare burden of $259 billion. The worldwide incidence of AD is rising at an alarming pace and it is expected that by 2050 the number of Americans with AD will surpass 16 million with ∼$1.1 trillion in healthcare costs. These alarming statistics are an eye opener, and with the race against time, it is absolutely essential to develop novel therapeutic strategies to delay the onset, halt the progression, and improve the cognitive decline in AD patients.
Here we describe the recent developments and efforts in the emerging field of neuro-immuno-gene therapy and gene editing and their potential applications to develop precision patient-specific regenerative therapy for the treatment of AD. Since there are excellent review articles on gene therapy vectors as well as gene editing, the major emphasis of our review will be on the AD preclinical or clinical gene therapy studies. We describe AD therapeutic targets that are excellent candidates for developing novel genome editing as well as immune-gene therapies. Further, we address the most desirable features in AAV that will enhance the gene transfer efficiency to maximize the therapeutic efficacy of novel AD gene therapies.
TARGETING AMYLOID-β (Aβ)
Extensive research has shown that AD is characterized by amyloid plaques which are produced as a result of sequential cleavage of the amyloid-β protein precursor (AβPP) by the β- and γ-secretases [1]. There are multiple forms of Aβ including Aβ34, Aβ40, Aβ42, etc. Although Aβ40 is the most abundant physiological form, Aβ42 that is produced at lower levels is the main culprit and is critically important for Aβ accumulation in the brain. Aberrant accumulation of Aβ aggregates acts as a trigger for AD pathogenesis. To disrupt this crucial pathological pathway, multiple attempts have been made with varying degrees of success. Autophagy plays a crucial role in neurodegeneration and AD pathogenesis. Pickford et al. have shown that an autophagy related protein Beclin1 is reduced in AD patients during the early phase of the disease and regulates Aβ accumulation in mice [2]. Their studies using APP +Becn1 +/– mice indicate that Beclin 1 deficiency caused altered microglial response to Aβ, reduced autophagy, increased extracellular and intraneuronal Aβ deposition, synaptodendritic degeneration, and neuronal loss. Lentivirus-mediated overexpression of Beclin 1 in the frontal cortex and hippocampus led to decreased amyloid pathology. These interesting findings suggest that Beclin 1 is an attractive AD therapeutic target and that improving autophagy by restoring Beclin 1 may lead to a novel AD therapy.
Targeting Aβ is an attractive approach to ameliorate AD pathogenesis. One of the earliest attempts was made by Zhang et al. wherein they developed a recombinant adeno-associated virus (AAV) expressing CB-ABeta42 (cholera toxin B subunit and Aβ42 fusion protein) [3]. A single administration of AAV-CB-Aβ42 vaccine in PDAPPV7171 transgenic mice induced high levels of anti-Aβ42 antibodies either by the intranasal, intramuscular, or oral routes. High levels of anti-Aβ42 antibodies led to a significant reduction in cerebral Aβ burden, decreased plaque-associated astrocytosis and improved memory as well as cognitive function. Subsequently, Fukuchi et al. developed an AAV encoding anti-Aβ single-chain antibody and injected it into the corticohippocampal regions of Tg2576 mice [4]. Their results indicate sustained expression of scFv for one year without neurotoxicity. Although, there was a reduction in the Aβ deposits in the brain of Tg2576 mice, there were no functional cognitive studies performed thereby making the data interpretation little challenging.
Hara et al. have developed an oral vaccine for AD using recombinant AAV vector expressing Aβ1 - 43 [5]. Oral administration of AAVAβ43 or AAV/Aβ in the Tg2576 mice led to marked reduction in Aβ deposition. However, the mechanism by which the antibodies clear Aβ deposits from the brain tissue remain unresolved and no cognitive function studies were performed. Later on Mouri et al. have reported on the histological studies and cognitive function in the Tg2576 mice that received oral vaccination of AAV/Aβ [6]. Their results suggest that cognitive impairments were attenuated without lymphocytic infiltration or microhemorrhage in the brain as compared to non-treated group. Levites et al. have evaluated the therapeutic potential of three recombinant anti-Aβ single-chain variable fragments (scFvs) in APP CRND8-transgenic mice [7]. Recombinant AAV1 expressing either Aβ1 - 16, Aβx - 40, or Aβx - 42 were injected into the ventricles of postnatal day 0 (P0) APP CRND8-transgenic mice. Their data suggest widespread neuronal delivery as well as expression of the anti-Aβ scFv as well as 25–50% decrease in Aβ deposition. Building upon the success of these exciting findings, Levites et al. have generated a monoclonal antibody 3H3 which is capable of binding to a pan-amyloid epitope and when delivered using an AAV in TgCRND8 and ADan mice decreased parenchymal Aβ deposition.
Taking a different approach, Carty et al. investigated the effects of an intracranial administration of AAV5 expressing endothelin-converting enzyme (ECE) on amyloid deposition in 6 months old APP+presenilin-1 (PS1) mice [8]. Their results suggest that upregulation of ECE causes reduction in total amyloid deposition. Similarly, Liu et al. have reported that intramuscular injection of a recombinant AAV8 expressing neprilysin in 3X-Tg-AD mice led to decline in brain Aβ burden [9]. However, both the studies did not report on the cognitive function tests in the treated mice. In a subsequent study, Jiang et al. reported that upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in APPswe/PS1dE9 transgenic AD mouse model [10]. They showed that Aβ1 - 42 stimulation leads to upregulation of TREM2 in microglia in vitro as well as in vivo in wild type mice. Further, they showed that TREM2 directly inhibited microglia-mediated proinflammatory responses after Aβ1 - 42 stimulation. Lentivirus mediated-overexpression of TREM2 in the brain of APPswe/PS1dE9 led to amelioration of Aβ deposition and this was attributed to enhanced microglial phagocytosis rather than attenuation of Aβ production. Overexpression of TREM2 in the brain significantly attenuated neuroinflammation as measured by reduced proinflammatory cytokine expression. These findings suggest that TREM2 could be a potential therapeutic target for developing novel AD therapies.
Most recently, short Aβ peptides have been shown to attenuate Aβ42 toxicity in vivo [11]. In these comprehensive studies, Moore et al. initially expressed multiple copies of Aβ36 - 40 and Aβ42 - 43 in Drosophila melanogaster using PhiC31-mediated transgenesis to evaluate inherent toxicity and potential immunomodulatory effects on Aβ42 toxicity. Their data revealed that as compared to Aβ42 the short peptides are not toxic, do not induce locomotor dysfunction and do not accumulate as amyloid. They further demonstrated that AAV2/1-mediated expression of Aβ38 and Aβ40 in nontransgenic mice did not cause abnormal deposition or behavioral alterations. However, the effects of Aβ38 from the rAAV-expressed BRI2 fusion proteins in APP TgCRND8 mice were rather complex since BRI2 expression can modulate Aβ deposition in vivo. In a recent study, intramuscular delivery of AAV-p75NTR (a receptor for Aβ) ectodomain has been shown to attenuate cognitive deficits and AD-like pathology in APP/PS1 transgenic mice [12]. However, further research is warranted to gain a better understanding of the role of p75NTR-mediated improvement in AD pathogenesis.
Sevigny et al. have generated Aducanumab, a human monoclonal antibody that selectively targets aggregated Aβ [13]. Aducanumab has been shown to enter the brain, bind parenchymal Aβ, and reduce soluble and insoluble Aβ in a dose dependent manner in Tg2576 mice. It significantly increased the recruitment of Iba-1-positive microglia to Aβ plaques thereby suggesting FcγR-mediated phagocytosis of antibody-Aβ complexes as a possible clearance mechanism. In the PRIME study involving AD patients, Aducanumab was shown to penetrate brain, decrease Aβ levels in a time and dose-dependent manner, and confer clinical benefit. Although, a human monoclonal antibody Aducanumab that selectively targets aggregated Aβ has shown promising results in the ongoing phase III clinical trials, many other clinical trials have witnessed major setbacks including the recent trials of the humanized monoclonal antibody Bapineuzumab and Solanezumab designed to increase the clearance of soluble Aβ peptides from the brain of the AD patients [14–18]. These major setbacks necessitate the development of the next generation of precision-targeted and AD patient-specific therapies.
TARGETING TAU
Tau plays a critical role not only in AD pathophysiology but also in various other neurodegenerative disorders [19–23]. In order to successfully treat, AD it is critical to simultaneously target both Aβ as well as tau. There have been numerous attempts to eliminate the toxic tau forms either using anti-tau antibodies or by employing gene silencing.
Most recently, Tan et al. have reported the generation of a new tau knockout strain (tauΔex1) using CRISPR/Cas9-mediated genome editing of intron1/exon1 of Mapt gene in C57Bl/6J mice [24]. To introduce a small deletion at the intron-1/exon1junction of MAPT by CRISPR/Cas9 genome editing, a pair of guide RNAs 115 bp up and 18 bp downstream of the transcriptional start codon were used. The CRISPR/Cas9 protein as well as the two guide RNAs were directly injected into the cytoplasm of the fertilized oocytes. Traditionally generation of tau–/– has been achieved by ES cell gene targeting which requires introduction of large selectable marker cassettes in the MAPT coding sequence. The drawback of such an approach is that these selectable markers can create undesirable and inconsistent results. These mice did not display the overt phenotype but showed a significantly reduced susceptibility to excitotic seizures with normal memory formation in young mice.
Utilizing CRISPR-dCas9-activator transgenic SPH mice, Zhou et al. have achieved simultaneous transcriptional activation of multiple genes to directly and efficiently convert astrocytes into functional neurons in vivo [25]. In a series of ambitious experiments, they were able to simultaneously control the expression of ten different genes or ten genetic elements including eight genes and two lncRNAs into the brain by multiplexing different ten sgRNAs into a single AAV vector. The different genes were simultaneously activated to different extents due to their differential basal expression levels, epigenetic status or sgRNA target sites. Their unique approach is potentially applicable to multiple fields including disease modeling, cellular reprogramming as well as gene therapy.
Due to safety concerns and the development of neurovascular side effects and other adverse events, many immunotherapies targeting Aβ were discontinued. Holtzman et al. have previously shown that passive immunization with an anti-Tau antibody HJ8.5 decreases the accumulation of pathological tau in a human P301S tau-expressing transgenic (P301s-tg) mouse model of FTD/tauopathy [26]. To further improve upon their results, they developed an AAV2/8 vector expressing anti-Tau-scFv and demonstrated the expression of anti-scFvs by the neurons and astrocytes [27]. These anti-Tau-scFvs retained their antigen binding specificity and effectively reduced pathological tau accumulation in the hippocampus of P301S-tg mice. These studies highlight the potential of AAV-mediated gene transfer for the therapeutic delivery of not only scFvs but also neurotrophic growth factors, microRNAs, CRISPR/Cas9, or any potential combinations.
TARGETING BACE1
BACE1 overexpression contributes to pathogenesis. BACE1 which acts upstream of γ-secretase in amyloid processing is an attractive AD therapeutic target for reducing the cerebral Aβ levels. There are several BACE1 inhibitors currently being explored for AD therapy. However, recently Merck and Co has halted a pivotal phase II/III trial of its BACE1 inhibitor Verubecestat in more than 2,000 patients with mild to moderate AD [28]. Besides there are multiple BACE1 inhibitor trials including phase III trials for early AD using Elenbecestat (Biogen/Eisai) early and mid AD using AZD3293 (AstraZeneca/Eli Lilly), phase II/III trial of JNJ-54861911 (Johnson & Johnson) for asymptomatic at-risk patients with family history, APOE4 or biomarker positivity and phase II trial of CNP520 (Novartis) for asymptomatic at-risk patients (APOE4) [28]. Atwal et al. have developed a high affinity, phage-derived human antibody that targets BACE1 [29]. Anti-BACE1 antibody blocks the activity of BACE1 in vitro as well as in vivo. Five-month-old Tg2576 mice treated with anti-BACE1 antibody led to a 70% reduction in Aβ1 - 40 and Aβ1 - 42 at doses of 30 and 100 mg/kg. However, when tested in the PS2APP mouse model, it showed reduction in Aβ in the periphery but no reduction in brain Aβ. Further testing in cynomolgus monkeys revealed ∼50% Aβ reduction. Recently Hu et al. have demonstrated that deletion of BACE1 in the adult mouse reverses preformed amyloid deposition and improves cognitive functions [30]. They generated BACE1 conditional knockout BACE1fl/fl and bred these mice with ubiquitin-CreER to induce deletion of BACE1 at adult stage. Interestingly, BACE1 deletion in adult AD mouse model (5xFAD) led to complete reversal of amyloid deposition and led to significant improvement in gliosis and neuritic dystrophy and improvement in cognitive function. These exciting results will hopefully lead to an improved understanding of BACE1 inhibitors in AD patients.
NGF THERAPY
AD is a highly complex neurodegenerative disorder and the current treatment strategies are largely ineffective leading to progressive cognitive decline in AD patients. There are multiple therapeutic targets that could potentially be utilized for either halting or delaying the progression of AD. Tuszynski et al. performed a phase I trial of ex vivo NGF gene delivery in eight individuals with mild AD by implanting autologous fibroblasts genetically modified to express human NGF into the forebrain [31]. These studies led to improvement in the rate of cognitive decline without long term adverse effects. In an interesting preclinical study, Bishop et al. have evaluated the therapeutic potential of AAV2-NGF (CERE-110) for targeted, stable, and sustained NGF delivery and trophic activity in rodent basal forebrain cholinergic neurons. Their studies indicated that NGF transgene delivery to the targeted brain region is reliable, accurate and leads to sustained long-term NGF expression with no loss or buildup of protein. Further, the results indicated that CERE-110 was neuroprotective and neurorestorative to basal forebrain cholinergic neurons in the rat fimbria-fornix lesion and aged rat models as well as had bioactive effects on young rat basal forebrain cholinergic neurons. These and additional studies in the primates led to the initiation of a phase I gene therapy study in which Rafii et al. stereotactically injected CERE-110 in the nucleus basalis of Meynert in AD patients. The AAV2-NGF vector utilized in this study has an expression cassette comprising CAG promoter, human NGF cDNA and a human growth hormone polyadenylation signal. The study demonstrated the safety and tolerability of the approach and led to least cognitive decline, which was dose dependent over a two-year period. Positron emission tomographic imaging and neuropsychological testing revealed no evidence of accelerated cognitive decline. The brain autopsy revealed long term, targeted, gene-mediated NGF expression and bioactivity. These phase 1 studies have paved the way for the future AAV based gene therapy trials for AD as well as other neurodegenerative diseases.
More recently, Eyjolfsdottir et al. have utilized a second-generation encapsulated cell biodelivery device to achieve delivery of nerve growth factor to the cholinergic basal forebrain of AD patients in a first in human study [32]. In their study they engineered a clonal NGF-secreting ARPE-19 cell line using a sleeping beauty transposon expression system. These implants released NGF at the levels ranging from 24.4 ng to 148.8 ng/24 hours than prior to implantation. Although the levels of neurofilament light chain protein increased and GFAP remained stable, the levels of Aβ t-tau and p-tau did not change significantly. A few patients showed improved cognitive performance and EEG. The ECB technology offers a number of advantages including the following: 1) the grafted cells are encapsulated and isolated from the host cells thereby making it easier to remove or replace the graft, 2) unlike viral vectors, the host cells do not undergo any direct genetic manipulation thereby avoiding the risk of mutation and carcinogenesis, and 3) the transgene expression is very stable thereby allowing sustained long term controlled release of NGF. However, these advantages need to be weighed with caution because a follow up study has revealed that MRI results in responders indicated more brain atrophy and neurodegeneration, increase in cerebrospinal fluid (CSF) levels of T-tau and neurofilaments at baseline despite better clinical status, and reduced pathological levels of CSF Aβ1 - 42 [33]. A larger clinical trial will be able to address any further potential concerns.
BDNF THERAPY
BDNF levels have been shown to decline in AD and therefore restoring BDNF levels may offer potential benefits in the AD therapy [34–36]. Chen et al. have investigated the effects of AAV-ProBDNF in APPswe/PS1dE9 mice [37]. Their results indicate that proBDNF enhances memory and learning impairment and also exacerbates brain Aβ burden. However, it is not clear from these studies as to how proBDNF aggravates learning and memory impairment. Osborne et al. have developed a novel gene therapy vector to achieve sustained BDNF signaling in neurons [38]. BDNF signaling occurs through TrkB and p75. However, overexpression of BDNF has been shown to cause downregulation of TrkB receptors. To overcome this limitation, they have designed a novel gene sequence simultaneously coding for BDNF and TrkB receptor that are separated by a short viral-2A sequence. AAV-mediated expression of this novel gene sequence in HEK293 cells and SH-SY5Y cells led to increased BDNF/TrkB-mediated intracellular signaling. It will be interesting to evaluate the therapeutic efficacy of this vector in vivo in multiple AD models. Caccamo et al. found that Aβ deposition interferes with the activity of the cAMP-response element binding protein (CREB) and have reported that lentiviral-mediated CREB binding protein gene transfer increased BDNF levels and ameliorated learning and memory deficits in the 3xTg-AD mouse model [39]. Most recently, Nagahara et al. have evaluated BDNF gene delivery to the entorhinal cortex to treat AD and reduce neurodegeneration and associated memory loss [40]. Using MRI-guided and convection enhanced delivery they were able to achieve accurate AAV2-BDNF delivery to entorhinal cortex of non-human primates.
TARGETING APOE4
ApoE, a lipid binding protein, is produced in the liver and abundantly expressed in the brain especially in astrocytes and microglia where it facilitates neuronal plasticity and synaptogenesis, Aβ fibrillogenesis, and deposition and is also involved in the clearance as well as degradation of Aβ [41–45]. ApoE has three different isoforms, ApoE2, ApoE3, and ApoE4, which differ by only two residues. ApoE2 is neuroprotective, ApoE3 is neutral, while ApoE4 is the most important genetic risk factor for AD. The ApoE mediated influence on AD pathogenesis is a subject of intense investigation. Huang et al. have used embryonic stem cell-derived neurons to show that ApoE secreted by glia stimulates neuronal Aβ production with an ApoE4>ApoE3>ApoE2 potency rank order [46]. They further demonstrated that ApoE binding to ApoE receptors activates dual leucine-zipper kinase (DLK), a MAP-kinase kinase kinase that then activates MKK7 and ERK1/2 MAP kinases. Activated ERK1/2 induces cFos phosphorylation thereby stimulating AP-1, which in turn enhances the transcription of APP and thereby increases Aβ levels. To prove that this mechanism of action operates in vivo, they stereotactically injected AAVs that express a control protein or DN-cFos (Dominant-negative form of cFos) or AAVs that express dCas9 with a control guide RNA or the guide RNA directed to the AP-1 binding sequence of the APP promoter to achieve CRISPRi in vivo. Their results indicate that AP-1 dependent regulation of APP expression operates physiologically in vivo.
To investigate whether ApoE independently influences tau pathology in AD, Shi et al. have developed new P301S/E4 tau transgenic mice, which have significantly higher tau levels [47]. These mice have significantly higher tau levels in the brain and a greater extent of somatodendritic tau redistribution by 3 months of age as compared to P301S/E2, P301S/E3, and P301S/KO mice. By 9 months, P301S mice with different ApoE genotypes display distinct phosphorylated tau protein (p-tau) staining patterns. Interestingly, P301S/E4 mice develop markedly more brain atrophy and neuroinflammation than P301S/E2 and P301S/E3 mice while P301S/KO mice are largely protected from these changes. Overall, their data suggest that ApoE affects tau pathogenesis, neuroinflammation, and tau mediated neurodegeneration independently of Aβ pathology. Thus in order to develop the next generation of precision-targeted AD therapy, it will be necessary to co-target Aβ as well as tau and ApoE.
Dodart et al. have investigated the potential of direct intracerebral administration of lentiviral vectors expressing three common human ApoE isoforms to alter hippocampal Aβ and amyloid burden in the PDAPP mice lacking ApoE model of AD [48]. They demonstrated that intracerebral gene delivery of the lentiviral vectors expressing human ApoE4 constructs results in an increased Aβ burden in PDAPP-apoE–/– mice. They further confirmed that gene delivery of a lentiviral vector expressing human ApoE2 for 5 weeks reduced brain Aβ burden and insoluble Aβ1 - 42 by 30–50% in PDAPP mice expressing endogenous mouse ApoE. In contrast they did not observe a significant reduction in brain Aβ burden 5 weeks post treatment with lenti-ApoE3 or lenti-ApoE4.
Rosenberg et al. have developed AAVrh.10hAPOE2-HA and tested its potential therapeutic efficacy following direct intraparenchymal, intracisternal, and intraventricular delivery in the African Green non-human primates [49]. Their data demonstrated that while all the three routes tested were capable of mediating high level ApoE2 expression in AD relevant regions, intracisternal delivery of AAVrh.10hAPOE2-HA led to wide distribution of ApoE2 with the least invasive surgical intervention. ApoE2 expression was detected in the ependymal cells of the choroid plexus in the ventricles, as well as around the frontal and mid-brain, including the hippocampal region, as well as around the posterior of the brain and the spinal cord. Besides the targeted areas, ApoE2 staining was also observed in the anterior olfactory nucleus, thalamic reticular nucleus, lateral hypothalamus, pineal body in the epithalamus, stria terminalis, pontine gray fibers in the brain stem, Purkinje cells and deep cerebellar nuclei in the cerebellum and gray nuclei of the spinal cord. ApoE2-HA levels in the CSF were quantified by ELISA and were found to be consistently >1μg/ml at day 28 and day 56 post vector delivery.
CYTOKINE GENE THERAPY
Neuroinflammation plays a critical role in the pathogenesis of multiple neurodegenerative disease including AD [50, 51]. Therefore, it would be beneficial to develop strategies to counteract neuroinflammation by utilizing anti-inflammatory cytokines. Targeting neuroinflammation presents an attractive paradigm for developing novel gene therapy strategies for AD. In this regard, earlier attempts were made by Kiyota et al. who reported that AAV serotype 2/1 hybrid-mediated neuronal expression of 7ND gene; a dominant-negative CCL2 mutant in APP/PS1 mice reduced astro/microgliosis and improved spatial learning [52]. In subsequent studies, they evaluated AAV2/1 mediated delivery of the mouse IL4 and IL10 genes to ameliorate cognitive dysfunction in APP+PS1 mice [53, 54]. The hippocampal neurons transduced with AAV2/1-IL4 led to a sustained expression of IL4 reduced astro/microgliosis, enhanced neurogenesis, improved cognitive function, and restored NR2B phosphorylation and cell surface expression in the neurons. IL4 promotes Aβ degradation in mononuclear phagocytes, monocyte-derived macrophages as well as reduces the levels of monomeric and oligomeric Aβ. IL4-mediated enhanced neurogenesis including enhanced neuronal progenitor cell proliferation as well as neuronal differentiation are potentially mediated by the activation of extracellular signaling-regulated kinases, p70 ribosomal protein S6 kinase-survival signaling pathways, and insulin-response substrate 1 and 2 pathways and through protection of neurons from apoptosis through the activation of PI3 kinase and Akt. Further hippocampal injection of AAV-IL10 in APP+PS1 mice led to sustained expression of IL10 and led to significantly reduced astrogliosis and microglial accumulation around thioflavin-S (TS)+ compact plaques and enhanced clearance of Aβ from the brain to the vascular circulation. Surprisingly IL10 expression did not reduce Aβ burden in the mouse brain. It remains to be seen whether AAV-mediated simultaneous co-expression of IL4 and IL10 in the hippocampus of APP+PS1 mice might have a potentially synergistic effect by significantly reducing Aβ burden and robustly improving cognitive function.
In a separate study, Chakrabarty et al. have investigated the potential therapeutic efficacy of AAV2/1-IL10-mediated intracranial expression of murine IL10 in TgCRND8 and Tg2576 AD mouse models [55]. Their studies revealed that IL10 expression leads to increased amyloid loads, decreased levels of immediate early genes and synaptic markers, worsened cognitive behavior, reduced microglial Aβ phagocytosis, and increased ApoE expression and its sequestration within insoluble amyloid plaques. Except for the fact that IL10 did not reduce Aβ burden, these results are contradictory to those reported by Kiyota et al. but consistent with the findings by Guillot-Sestier et al. who have reported that IL10 signaling is elevated in AD patient brains and IL10 deficiency in APP/PS1 +IL10–/– mice mitigates cerebral amyloidosis and partially rescued synaptic toxicity and behavior impairment.
Prior studies have shown that the IL2 knockout mice exhibit impaired learning and memory formation and altered hippocampal development [56]. Alves et al. observed decreased IL2 expression in the hippocampus of AD patients and have therefore developed and tested the potential therapeutic efficacy of an AAV8-IL2 vector in APP/PS1ΔE9 mice [57]. Administration of AAV8-IL2 intraperitoneally led to IL2 secretion, rescued impaired synaptic plasticity and spatial memory impairment, caused an expansion and activation of systemic and brain regulatory T cells and activated the JAK/STAT3 pathway in the hippocampus. Further, IL2 induced astrocytic activation and recruitment of astrocytes around amyloid plaques, decreased amyloid-β42/40 ratio amyloid plaque load and significantly reduced spine density.
Chakrabarty et al. have developed an AAV2/1 vector expressing murine IL4 (AAV2-mIL4) [58]. Surprisingly, stereotactic injection of AAV2-mIL4 into the CA layer of hippocampus at 4 months of age in TgCRND8 mice led to 33.5% increase in amyloid plaques and 41% increase in Aβ42 levels and 55% increase in Aβ40 levels in the SDS extractable Aβ levels, respectively, and 76% increase in Aβ42 levels and 62% increase in Aβ40 levels in the formic acid extractable Aβ levels, respectively. However, APP, APP CTF, BACE, ApoE, and Aβ degrading enzyme levels did not get altered. These results are contradictory to those reported by Kiyota et al. [54] even though both these studies have used AAV2/1, with similar expression cassettes. The only differences include a dose of 2×109 viral particles and the use of APP+PS1 Tg mice by Kiyota et al. [54] whereas Chakrabarty et al. [58] have used a slightly higher titer 2×1010 viral genomes and TgCRND8 mice. There is a need to evaluate these AAV2/1-mIL4 vectors in multiple AD models to resolve the controversial nature of the results. Most recently, Vogelaar et al. have shown that intrathecal IL-4 treatment during the chronic phase of several experimental autoimmune encephalomyelitis models reversed disease progression without affecting inflammation [59].
Chakrabarty et al. have explored the effect of murine tumor necrosis factor alpha (mTNFα) in regulating Aβ accumulation [60]. Recombinant AAV2/1-mTNFα-mediated expression of mTNFα in the hippocampus of 4-month-old APP transgenic TgCRND8 mice resulted in significant reduction in hippocampal Aβ burden. However, no changes in AβPP levels or AβPP processing were observed either in mTNFα expressing APP transgenic mice or in the nontransgenic littermates. Furthermore, analysis of Aβ plaque burden in mTNFα expressing mice showed that even after substantial reduction compared to EGFP expressing age-matched controls, the Aβ plaque burden levels of the treated mice do not decrease to the levels of 4-month-old unmanipulated mice.
In a separate study, Chakrabarty et al. have evaluated the role of IFNγ-mediated neuroinflammation in pathology in transgenic mice. They expressed murine IFNγ in the brains of APP transgenic mice using a recombinant of AAV2/1-mIFN-γ virus [61]. Their results demonstrate that the expression of IFNγ in the brains of 4-month-old APP TgCRND8 mice leads to robust noncell autonomous activation of microglia and astrocytes, upregulation of MHCII, CD11b, CD11c and complement system components, infiltration of Ly-6c positive monocytes and a significant suppression of Aβ deposition. When AAV2/1-mIFN-γ virus was injected into the cerebral ventricles of 36–48 hours old (neonatal day P2) TgCRND8 mice much prior to the onset of plaque deposition, it led to expression largely localized in the choroid plexus and ependymal cells lining the ventricles, along with a few neurons in the hippocampus, cortex, and cerebellum. Quantification of Aβ plaques in the forebrain and hippocampus of AAV2/1-mIFN-γ injected TgCRND8 mice revealed 73% and 70% reduction in plaque burden respectively. When the AAV2/1-mIFN-γ virus was injected in the 4-month-old TgCRND8 mice, Aβ plaque burden was higher than the pretreatment levels. Further, cognitive studies did not reveal a beneficial effect in mIFNγ expressing mice. Therefore, newer strategies are required to simultaneously reduce Aβ plaque burden and improve cognitive function.
To assess the effects of IL-6 on Aβ deposition and AβPP processing, Chakrabarty et al. have developed AAV-mIL-6 [62]. Overexpression of mIL-6 in the brains of APP transgenic TgCRND8 and Tg2576 mice led to extensive gliosis and concurrently attenuated Aβ deposition in TgCRND8 mouse brains. They also found that there was an upregulation of glial phagocytic markers and enhanced microglia-mediated phagocytosis. Surprisingly, mIL-6-induced neuroinflammation had no effect on AβPP processing in TgCRND8 or Tg2576 mice. These results suggest that mIL-6 mediated reactive gliosis may be beneficial during the early AD phase by potentially enhancing Aβ plaque clearance rather than creating a neurotoxic feedback loop, which might exacerbate amyloid pathology.
Intraneuronal accumulation of Aβ peptides is responsible for driving the neurodegenerative disease pathology in AD. Sudol et al. have developed Aβ42-specific intracellular antibodies with and without an intracellular trafficking signal using AAV2 [63]. Testing of these intrabodies in doxycycline inducible Aβ42 expressing cells revealed that only endoplasmic reticulum targeted anti-Aβ42 intrabody was effective in significantly reducing the secreted form of Aβ42. Subsequent testing of rAAV-scFvAβIB and rAAV-scFvAβKDELIB by intrahippocampal injections into the CA1 layer in 3xTg-AD mice revealed that the rAAV-scFvAβKDELIB treatment significantly reduced both extracellular Aβ42 and phospho-Tau. These interesting results support the feasibility of using tropism modified and transcriptionally targeted AAV-mediated expression of Aβ-specific intrabodies to significantly reduce and possibly halt the progression of neurodegeneration in AD patients.
The most popular AD transgenic models utilize overexpression of mutant human AβPP and PS1 to increase Aβ production and mimic AD cognitive impairment and AD pathogenesis [64–67]. However, a major caveat is that overexpression of AβPP causes increased production of not only Aβ40 and Aβ42 but also other AβPP moieties that may have either neurotoxic, neuroprotective, or other signaling functions, which may positively or negatively influence the cognitive function. Therefore, to gain an improved understanding of the role of Aβ40 and Aβ42 in vivo, Lawlor et al. have developed AAV1 vectors encoding BRI-Aβ cDNAs to achieve high level and specific Aβ expression in the absence of AβPP overexpression [68]. Their results indicate that AAV-mediated gene transfer in adult rats leads to elevated Aβ levels and AD like phenotype with learning and memory deficits when both AAV-BRI-Aβ40 and AAV-BRI-Aβ42 are co-injected. Subsequent gene transfer studies in APPCRND8 transgenic mice using AAV1 expressing truncated BRI2 protein lacking the BRI2 - 23 peptide have revealed that BRI2 - 23 sequence is required to suppress Aβ deposition in vivo [69]. In another study, they have compared CNS transduction profiles of AAV serotypes bb2, cy5, rh20, rh39, and rh34 with AAV8 [70]. Their results suggest that as compared to AAV8, more widespread neuronal transduction was observed by employing cy5, rh20, and rh39 while preferential transduction of astrocytes was observed with rh43. Prior results by Klein et al. had suggested that AAV8 has superior transgene expression in the adult rat hippocampus as compared to AAV2 or AAV5 [71]. They also tested an AAV8 vector expressing the P301L form of tau, which led to profound loss of dopamine neurons and caused significant amphetamine-stimulated rotational behavior. These research findings are significant because it will become possible to engineer novel AAV capsids to achieve high-level tissue specific expression.
Independent studies have shown that IL-33 transcript and protein levels are reduced in the brains of late onset AD patients as compared to healthy individuals [72, 73]. To investigate the role of IL-33 in AD pathophysiology Fu et al. injected IL-33 in APP/PS1 transgenic mice and examined the cognitive function as well as measured soluble Aβ and amyloid plaque deposition. Their results indicate that stimulation of IL-33/ST2 signaling reverses synaptic plasticity, rescues contextual memory deficits and reduces Aβ accumulation. Further, IL-33 treatment causes an increase in the anti-inflammatory genes including Arg1 and Fizz1 in the microglia and the macrophages in the brain causing alternative activation state with enhanced Aβ phagocytosis. Their finding that IL-33 mobilizes innate immunity to prevent and clear established Aβ accumulation even at late stage AD potentially represents an attractive approach for the treatment of AD. These exciting results suggest that targeting neuroinflammation has significant benefits and could potentially lead to development of a multi-target AD gene therapy.
AAV9 TOXICITY
Prior studies by Gray et al. have demonstrated that AAV9 is able to transduce approximately twice as many neurons as astrocytes in the adult rodent CNS at doses of 1.25×1012, 1×1013, and 8×1013 vg/kg body weight. Further, they reported that self-complementary vectors were >10 fold more efficient when compared with the single stranded vectors. However, when tested in the non-human primates, they observed reduction in peripheral organ and brain transduction with a preferential glial transduction. Their results indicate that high peripheral tropism, limited neuronal transduction in non-human primates, and the pre-existing neutralizing antibodies are the major caveats to human translation of intravascular AAV9 based gene delivery. Recently, the results from a first-in-human trial of high-dose i.v. AAV9 in infants with SMA were reported [74]. In these trials the patients demonstrated significant improvement in survival and motor function in comparison to the controls. However, in a subset of the patients (4/15) there was an asymptomatic transaminase elevation [35 times the upper limit of the normal range for alanine aminotransferase (ALT) and 37 times for aspartate aminotransferase (AST)] potentially due to T-cell response against capsid-derived peptides presented by the transduced hepatocytes. To better understand the efficiency and safety of this approach, Hinderer et al. have reported severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an AAV9 variant AAV9hu68 expressing human SMN [75]. In yet another study, Hordeaux et al. have reported that the neurotropic properties of AAV-PHP.B are specifically limited to C57BL/6J mice and not to other commonly used mouse strain BALB/cJ or non-human primates [76]. They further reported the potential for serious acute toxicity in the non-human primate after systemic administration of high dose of AAV. Therefore, significant caution needs to be exercised during preclinical testing to thoroughly evaluate the safety and toxicity profiles of all the gene therapy vectors in male as well as female subjects.
AAV IMMUNOGENICITY
AAV serotype comparison for CNS transduction [77, 78]. The most commonly used AAV serotypes for the CNS gene therapy clinical trials include AAV5, AAV9, and AAVrh.10 [79–82]. However, one of the major caveats of the current gene therapy strategies is AAV-specific adaptive immune response. More specifically AAV-neutralizing antibodies will interfere with AAV transduction causing suboptimal transduction rates. Further, cytotoxic CD8 + T cells directed against the AAV capsid would lead to untimely rejection of AAV transduced cells. Since some of the epitopes are common between different serotypes, it may not be possible to use different serotypes to bypass the immune response. Therefore, it is important to develop suitable strategies to overcome vector directed immune responses. In this regard, CpG-depleted AAV vectors have been shown to evade immune detection [83]. In their pioneering studies, Faust et al. initially tested the immunogenicity of AAVrh32.33 in wild type as well as Tlr9 knock out mice. They found that in Tlr9-deficient mice, IFNγ T cell responses toward capsid and transgene antigen were suppressed resulting in minimal cellular infiltrate and stable transgene expression. Recently Hudry et al. have shown that AAV can associate with exosomes (exo-AAV) when the vector is isolated from conditioned media of producer cells and it is more resistant to neutralizing antibodies [84]. Systemic injection of exo-AAV was capable of efficient transduction of the CNS at low vector doses compared to conventional AAV. The ability of exo-AAV to evade neutralizing antibodies while retaining excellent CNS gene transfer is clinically relevant.
AAV NOVEL SEROTYPES AND AAV CAPSID ENGINEERING
One of the major caveats of the current gene therapy vectors is limited CNS gene transfer. CNS gene transfer efficiency and the gene therapy efficacy can be significantly improved by rationally engineering AAV capsid. There have been several studies to maximize the in vivo gene transfer efficiency. Zhong et al. have previously shown that protein tyrosine kinases phosphorylate specific residues on the AAV2 capsid surface resulting in the ubiquitination and degradation of virion prior to reaching the nucleus [85]. Specific mutation of surface exposed AAV2 capsid tyrosine residues to phenylalanine resulted in improved intracellular transport thereby significantly improving AAV-mediated transduction in vitro as well as in vivo [86]. Yang et al. have compared CNS transduction efficiencies of 12 different rAAVs (rAAV 1, 2, 5, 6, 6.2, 7, 8, rh.8, rh.10, rh.39, and rh.43) in 10-week-old mice following systemic delivery. Their results suggest that rAAVrh.8 robustly achieves global CNS transduction with minimal peripheral tissue dissemination as compared to other vectors and may be very useful for the treatment of AD.
Deverman et al. have developed novel AAVs by using CREATE (Cre recombinase-based AAV targeted evolution) for widespread gene transfer to the brain [87]. The CREATE system utilizes a rAAV capsid genome (rAAV-Cap-in-cis-lox) along with a full-length AAV cap gene, controlled by regulatory elements from the AAV rep gene with a Cre-invertible switch. They developed AAV capsid libraries by inserting 7 amino acids of randomized sequence between amino acids 588 and 589 of the AAV9 capsid within the rAAV-Cap-in-cis-lox backbone and delivered the virus libraries to animals with Cre expression in selected cell population. Selective amplification and recovery of sequences that transduced the target population yielded multiple AAV variants. They developed an AAV variant AAV-PHP.B with a 40-fold higher gene transfer efficiency (80% transduction of astrocytes and 50–100% transduction of neurons) in various brain regions as compared to the standard AAV9 [88, 89]. Chan et al. have subsequently developed two more variants AAV-PHP.eB and AAV-PHP.S that efficiently transduced the central and peripheral nervous system, respectively [90]. Morabito et al. have demonstrated AAV-PHP.B-mediated global scale physiological expression of GBA1 gene as an approach for wide protection from synucleinopathy in A53T-SCNA transgenic mice [91]. However, whether the selective neurotropism is also preserved in nonhuman primates or human CNS remains to be seen and needs to be investigated in future studies. In this regard, very recently, Matsuzaki et al. have tested and compared the potential gene transfer efficiency of AAV9 as well as AAV-PHP.B in marmosets using intravenous injection [92]. Their results demonstrate limited transduction of neurons (0–2%) and astrocytes (0.1–2.5%) in both AAV-PHP.B and AAV9 but marked transduction of the peripheral dorsal root ganglia neurons.
Murlidharan et al. have reported on CNS restricted transduction and CRISPR/Cas9-mediated gene deletion with an engineered AAV [93]. They reported the development of a chimeric AAV by replacing the viral protein 3 amino acid residues on AAV9 capsid required for galactose binding on the AAV2 capsid by site directed mutagenesis. The capsid engineered AAV2g9 had a superior CNS transduction efficiency due to preferential neurotropism, CNS restricted biodistribution and minimal transgene expression in off-target peripheral organs. They subsequently demonstrated the utility of AAV2g9 for targeted disruption of the MIR137 locus in CRISPR/Cas9 transgenic mouse. Tervo et al. have used directed evolution approach to develop a designer AAV variant rAAV-retro that exhibits robust retrograde access to projection neurons and has superior genome editing capabilities [94]. This rAAV-retro vector holds significant promise for the therapeutic intervention for neurodegenerative diseases especially AD and Parkinson’s disease wherein multiple AAV injections pose a safety risk and may be insufficient to achieve widespread transduction. This is due to the fact that early intervention by targeting subcortically projecting neurons in the mesocortical regions in the AD and Parkinson’s disease could have the potential to significantly halt or stabilize the disease progression and possible improve or restore cognitive function. It would be interesting to determine whether rAAV-retro vector is capable of efficiently transducing the neural stem cells.
More recently, Choudhury et al. have used directed molecular evolution to construct AAV capsid library by DNA shuffling of AAV1, 2, 4, 5, 6, 8, rh8, rh10, rh39, and AAVrh43 capsid genes [95]. They have developed a novel AAV-B1 capsid, which has superior transduction in the neuronal, glial, and endothelial cell population within the CNS. This AAV-B1 capsid is also highly effective for gene transfer of various peripheral organs including skeletal muscle, heart, lung, pancreas, and retina. Further, AAV-B1 is significantly resistant to neutralization than AAV9 and therefore represents an important improvement over AAV9 for CNS gene therapy. They have also generated a novel AAV-AS vector by insertion of a poly-alanine peptide, which imparts 6 and 15 fold more efficient transduction than AAV9 in the spinal cord and cerebrum, respectively [96]. Kanaan et al. have investigated the efficacy of CNS transduction using a variety of tyrosine and threonine capsid mutants based on AAV2, AAV5, and AAV8 capsids as well as AAV2 mutants incapable of binding heparin sulfate [97]. They found that simultaneously mutating multiple tyrosine residues on the AAV2 capsid significantly enhanced neuronal transduction in the striatum and hippocampus and the ablation of heparan sulfate binding also increased the volumetric spread of the vector in vivo.
CRISPR/Cas9 HOLDS SIGNIFICANT PROMISE FOR THE DEVELOPMENT OF NEW AD MODELS AND PRECISION TARGETED AD THERAPY
Clustered regularly interspaced short palindromic repeat (CRISPR)-Cas nucleases have revolutionized the field of gene editing and have tremendous application in the field of molecular medicine [98–102]. Despite a significant surge in CRISPR/Cas9-mediated genome editing in various disease models, the progress in the field of AD has lagged behind substantially. We believe that genome editing can significantly improve the development of AD models and also create novel opportunities for the development of the next generation precision targeted AD gene and stem cell therapies. Since there are several excellent review articles on CRISPR/Cas9-mediated genome editing, here we will limit our focus on select recent articles that are noteworthy.
CRISPR/Cas9 system can be engineered to either activate transcription (gain-of-function) or achieve gene silencing (Loss-of-function). Dahlman et al. have developed a CRISPR-based system that uses catalytically active Cas9 and distinct single guide (sgRNA) constructs to activate and knockout different genes in the same cell [103]. Konermann et al. have used structure-guided engineering of a CRISPR-Cas9 complex to mediate efficient transcriptional activation at endogenous genomic loci [104]. Using crystallographic studies, they have engineered a combination of sgRNA2.0, NLS-dCas9-VP64 and MS2-p65-HSF1 to develop one of the most effective transcription activation system.
Hu et al. have used phage-assisted continuous evolution (PACE) to evolve an expanded PAM spCas9 variant (xcas9) that can recognize a broad range of PAM sequences including NG, GAA and GAT [105]. The engineered xCas9 has broadest PAM compatibility and much greater specificity than the regular SpCas9 and substantially lower genome-wide off-target activity. Kramer et al. used CRISPR/Cas9 genome-wide gene knockout screens for suppressors and enhancers of C9ORF72 DPR toxicity in human cells [106]. They discovered potent modifiers of DPR toxicity whose gene products function in nucleocytoplasmic transport, endoplasmic reticulum, proteasome, RNA-processing pathways and chromatin modification. Their research findings highlight the potential of CRISPR-Cas9 screens in uncovering novel molecular mechanisms underlying neurodegenerative diseases.
Paquet et al. have used CRISPR/cas9-mediated scarless CORRECT (Consecutive re-guide or re-Cas Steps to Erase CRISPR/Cas-blocked targets) genome editing to develop knock-in model of AD mutations in human iPS cells [107]. Their gene editing approach allows selective introduction of mono- and bi-allelic sequence changes with high efficiency and accuracy. Using this approach, they were successful in generating human iPS cells with heterozygous and homozygous dominant early onset AD causing mutations in amyloid precursor protein (APPSwe)10 and presenilin 1 (PSEN1M146V)11 and derived cortical neurons. This unique strategy has the potential to introduce specific sequence changes using CRISPR/Cas9 to develop disease in a dish model especially for AD using human ES or iPS cells.
Platt et al. have developed a Rosa26-LSL-Cas9 knock-in mice in which a Floxed-STOP polyA stop cassette prevents the expression of the CAG promoter driven downstream bicistronic Cas9 and EGFP [108]. Treatment with Cre recombinase leads to deletion of the STOP cassette thereby resulting in widespread Cas9 and EGFP expression. These Rosa26-LSL-Cas9 knock-in mice can be used to generate either single or multiple specific mutations using an AAV vector expressing Cre under the control of a tissue specific promoter along with single or multiple sgRNAs. To demonstrate the potential utility of the Rosa26-LSL-Cas9 knock-in mice for neuronal gene editing, they developed a chimeric AAV1/2 vector in which a CBh promoter drives the expression of Cre recombinase and U6 promoter drives the expression of RNA-splicing factor NeuN. Stereotactic injection of the AAV into the prefrontal cortex of the Rosa26-LSL-Cas9 knock-in mice followed by the Deep sequencing of the NeuN locus revealed indel formation as well as NeuN protein depletion.
Zheng et al. have utilized dCas9-mediated CRISPR interference-based specific and efficient gene inactivation in the brain [109]. In their studies, they demonstrated the efficiency of CRISPRi in cultured hippocampal neurons to successfully knock down (90%) the expression of Syt1, Vamp2, Stx1a, and Snap25. Further, they modified their lentiviral vectors either to coexpress dCas9-KRAB and 2-3 sgRNAs that were driven by different RNA Pol III promoters in an all-in-one lentiviral vector or used a dual vector CRISPRi system in which one lentiviral vector solely expressed the dCas9-KRAB and another lentiviral vector contained a tandem array of 5 U6:sgRNA cassettes against Syt1, Vamp2, Snap25, Stx1a, and Stx1b. They were successful in knocking down the expression of the five genes in the mouse hippocampus using the multiplexing approach. These studies suggest that multiplex CRISPRi can be successfully used to abrogate gene expression in multigeneic neuronal disorders. Prior studies have shown that genetic fusion between dCas9 and the transcriptional repressor KRAB (dCas9-KRAB) successfully blocks transcription leading to excellent gene knockdown [110–115].
Swiech et al. have utilized a dual AAV system to achieve efficient SpCas9-mediated gene editing of MeCP2 (methyl CpG binding protein) in neurons [116]. They observed significant MeCP2 gene editing 7 days post transduction. Stereotactic injection of a (1:1 ratio) of AAV-SpCas9 and AAV-SpGuide (MeCP2 or LacZ targeting sgrNAs) into the hippocampal dentate gyrus of adult male mice led to ∼70% reduction in MeCP2 nuclei and >60% decrease in MeCP2 protein levels. They further used sgRNAs targeting the family of DNA methyltransferases (DNMTs: Dnmt1, Dnmt3a, and Dnmt3b) which are highly expressed in the adult brain and are required for synaptic plasticity, learning and memory formation. Stereotactic injection of a mixture of AAV-SpCas9 and AAV-SpGuide (targeting Dnmt3a, Dnmt1, and Dnmt3b) into the dentate gyrus of adult mice led to indels in all three loci and revealed ∼75% modification rate in Dnmt1 and Dnmt3a and ∼50% modification in Dnmt3b. CFC behavior tests were used to investigate the effect of SpCas9-mediated knockdown of DNMTs on memory acquisition and consolidation. Their data revealed that triple DNMT knockdown mice had impaired memory formation.
We have recently developed recombinant AAV and lentiviral vectors to achieve CRISPR/Cas9-mediated genome editing of glia maturation factor in microglia [117]. We investigated if CRISPR-Cas9-mediated GMF gene editing leads to inhibition of GMF expression and suppression of microglial activation. Confocal microscopy of murine BV2 microglial cell line transduced with an adeno-associated virus AAV co-expressing Staphylococcus aureus (Sa) Cas9 and a GMF specific guide RNA (GMF-sgRNA) revealed few cells expressing SaCas9 while lacking GMF expression, thereby confirming successful GMF gene editing. To further improve GMF gene editing efficiency we developed lentiviral vectors (LVs) expressing either Streptococcus pyogenes (Sp) Cas9 or GMF-sgRNAs. BV2 cells co-transduced with LVs expressing SpCas9 and GMF-sgRNAs revealed reduced GMF expression and the presence of indels in the exons 2 and 3 of the GMF coding sequence. Lipopolysaccharide (LPS) treatment of GMF-edited cells led to reduced microglial activation as shown by reduced p38 MAPK phosphorylation. We will be testing GMF gene editing in 5xFAD mice. We believe that targeted in vivo GMF gene editing has a significant potential for developing a unique and novel AD therapy.
As compared to the complete gene knockout mouse models, CRISPR/Cas9 based gene editing provides only a partial knockout in a fraction of targeted cells. Therefore, it is important to improve the current gene editing efficiency of the CRISPR/Cas9 systems. Another caveat is that using a ubiquitous promoter to drive the expression of CRISPR/Cas9 can lead to gene editing in the non-target cells. This can be achieved using a highly tissue specific promoter to drive the expression of CRISPR/Cas9. However, despite utilizing a tissue specific promoter, it is still not desirable to have constitutive expression of CRISPR/Cas9 as it may lead to off-target effects. This potential drawback can be overcome by utilizing a conditional expression system wherein the expression levels as well as the duration of CRISPR/Cas9 can be tightly regulated. This has been recently demonstrated by de Solis et al. who have developed a doxycycline-inducible AAV based system for gene editing [118]. Their strategy involved generating two separate AAV/DJ vectors such that the vector harbors a TRE Tight promoter driving the expression of CRISPR/Cas9 while the second vector contains a U6 promoter driving Tet2 sgRNA and a CMV promoter driving the expression of rtTA (Tet-On Advanced and an IRES driven GFP. Surprisingly, their results indicate doxycycline-inducible expression of CRISPR but Tet2 gene editing in a doxycycline independent manner due to leakiness. To overcome the issue of leakiness, they have significantly modified their vectors by utilizing a combination of hybrid H1/TO promoter to drive the expression of Tet2-sgRNA and a CMV promoter controlling the expression of TetR in frame with a self-cleaving P2A sequence followed by a GFP ORF fused to a KASH domain. In this system in the absence of doxycycline, TetR binds to H1/TO promoter and represses the gRNA transcription. However, addition of doxycycline inhibits TetR binding and induces gRNA expression. This system allowed doxycycline dependent genome editing of Tet2 in N2A cells in vitro. Besides, doxycycline inducible system there are several other inducible systems available including rapamycin, mifepristone, tamoxifen, and ecdysone inducible systems that can be engineered to overcome the leakiness of the dinducible system.
The currently used genome editing approaches are inefficient and induce significantly higher random insertions and deletions (indels) at the target locus. Recently, Nishiyama et al. have shown that precise genome editing via homology-directed repair is possible in post mitotic neurons as well as mitotic cells in mouse brain by combining CRISPR-cas9-mediated DNA cleavage and the efficient delivery of donor template with AAV [119]. Using this approach of virus-mediated single-cell labeling of endogenous proteins via HDR (vSLENDER), they were able to achieve highly efficient tagging of endogenous proteins in dissociated and organotypic slice cultures in vitro and various brain areas and cell types in developing and adult brains in vivo.
In a pioneering study, Komor et al. have recently described programmable base editing that enables the direct irreversible conversion of one target DNA base into another in a programmable fashion without requiring dsDNA backbone cleavage or a donor template [120]. They engineered fusions of CRISPR/Cas9 and a cytidine deaminase enzyme that retain the ability to be programmed with a guide RNA, do not induce dsDNA breaks and mediate the direct conversion of cytidine to uridine thereby effecting a C⟶T (or G⟶A) substitution. They successfully demonstrated the potential of programmable base editing to convert a target base in APOE4 into APOE3r in immortalized mouse astrocytes by replacing the endogenous ApoE gene with human APOE4. These exciting new developments in the field of genome editing have the potential to revolutionize the development of next generation patient-specific precision medicine for a wide variety of genetic diseases as well as neurodegenerative diseases especially AD.
EPIGENETIC EDITING
Bustos et al. have analyzed the role of Dlg4 gene which encodes post-synaptic density protein 95 (PSD95), a major synaptic protein that is diminished in neurodegenerative disorders including AD [121]. They engineered a Dlg4/PSD95 zinc finger DNA-binding domain and fused it to the effector domains to either repress (G9a, Suvdel76, SKD) or activate (VP64) transcription thereby generating Herpes simplex viral, lentiviral and AAV-PHP.B vectors expressing artificial transcription factors or epigenetic factors (methylating H3K9). Their in vitro data revealed that PSD95-6ZF fusion constructs induce epigenetic reprogramming and bidirectionally control Dlg4/PSD95 gene expression in N2a cells. Further, in vivo gene transfer led to modulation of Dlg4/PSD95 gene expression, regulation of synapse and spine maturation of hippocampal neurons and improved learning and memory deficits in aged and AD mouse model AβPPswe/PS-1 in vivo. These exciting results suggest that epigenetic editing could be utilized for developing novel gene therapies for the treatment of AD.
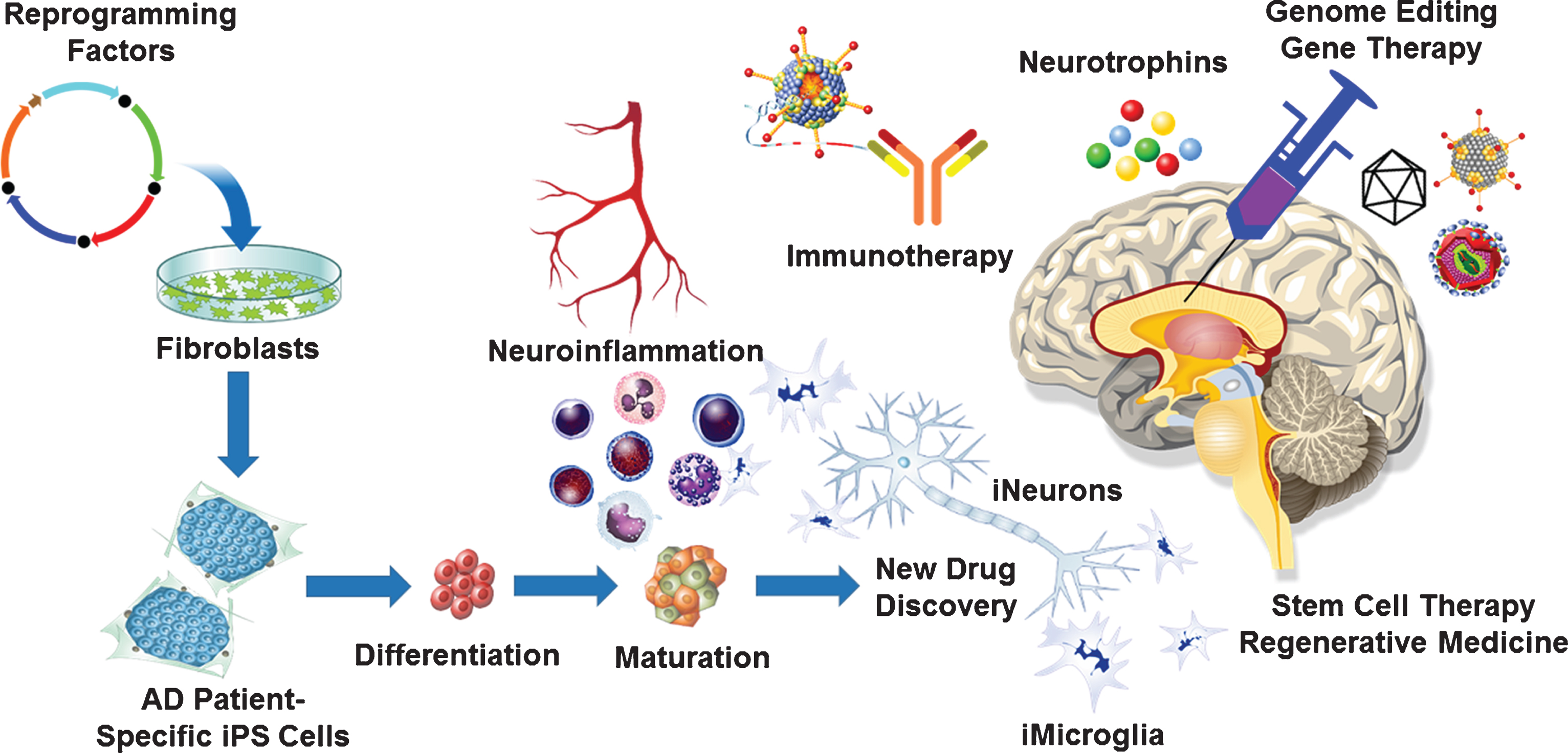
Fig.1
Neuro-immuno-genome-editing-stem-cell-therapy for Alzheimer’s disease (AD). Development of novel AD therapies would involve harnessing the latest technological advancements in the fields of neurology, immunology, molecular biology, virology, molecular medicine as well as stem cell biology. Neuroinflammation plays a significant role in the development and progression of AD. Targeting neuroinflammation using CRISPR/Cas9-mediated gene editing and gene therapy approaches will delay the onset as well as potentially halt the progression of AD. Genetically engineered recombinant viral vectors with enhanced neurotropism due to novel capsid engineering will maximize targeted gene editing and gene therapy efficacy. Latest advancements in the field of stem cell biology and regenerative medicine will enable the development of disease in a dish model using AD patient derived iPS cells to generate 3D organoids, which can be used for the new drug discovery. Such an approach will lead to the development of AD-patient specific neuro-immuno-genome-editing-stem-cell-therapy.
Pu.1
In a large scale genome-wide survival analysis of 14,406 AD cases and 25,849 controls Huang et al. have identified 8 previously reported AD risk loci (CR1, BIN1, SPl1, MS4A, PICALM, SORL1, FERMT2, and APOE) and 14 novel loci (C1orf112, CMC1, SYNPR, PDE6B, LINC00290, PCDHA1, LINC00951, NT5C3A, CSMD1, VLDLR, IPMK, SLC6A15, IQCK, and SUN2) associated with age at onset [122]. They discovered an association with delayed AD onset and lower expression of SPl1 in monocytes and macrophages. The SPl1 gene encodes for the PU.1 transcription factor that plays a critical role in the myeloid and B cell development and function and binds to the cis-regulatory elements of several AD-associated genes. PU.1 was shown to be bound to cis-regulatory elements of many AD-associated genes including ABCA7, CD33, MS4A, MS4A6A, PILRA, PILRB, TREM2, TREML2, and TYROBP but surprisingly not APOE. PU.1 overexpression led to increased phagocytosis while PU.1 knockdown led to diminished phagocytic activity in BV2 cells. Their study revealed that lower SPl1 expression might reduce AD risk potentially by modulating microglial and/or myeloid cell gene expression and function. Overall, these interesting studies highlight the potential of CRISPR/Cas9 mediated SPl1 genome editing as an approach to develop novel therapeutic strategies to treat AD.
STEM CELLS AND REGENERATIVE MEDICINE
There is evidence linking AD related pathology with increased hippocampal neurogenesis in AD patients. Jin et al. examined the expression of neurogenesis marker proteins in hippocampus brains from AD patients and neurologically normal subjects [123]. They found that the expression of several neurogenesis markers was increased in the hippocampus of severely affected AD patients. The upregulated proteins included DCX, PSA-NCAM, TUC-4, and NeuroD. These findings are in contrast to the findings in AD transgenic mice, which show impaired hippocampal neurogenesis [124–126]. Baglietto-Vargas et al. have now investigated the effect of Aβ on neuronal and glial cell proliferation by using APP/PS1 transgenic model and in vitro assays [127]. They showed that neurogenesis is affected early in the APP/PS1 hippocampus due to reduced number of both radial glia-like neural stem cells and intermediate progenitor cells. There is a need to investigate the role of hippocampal neurogenesis in AD pathology as well as cognitive function.
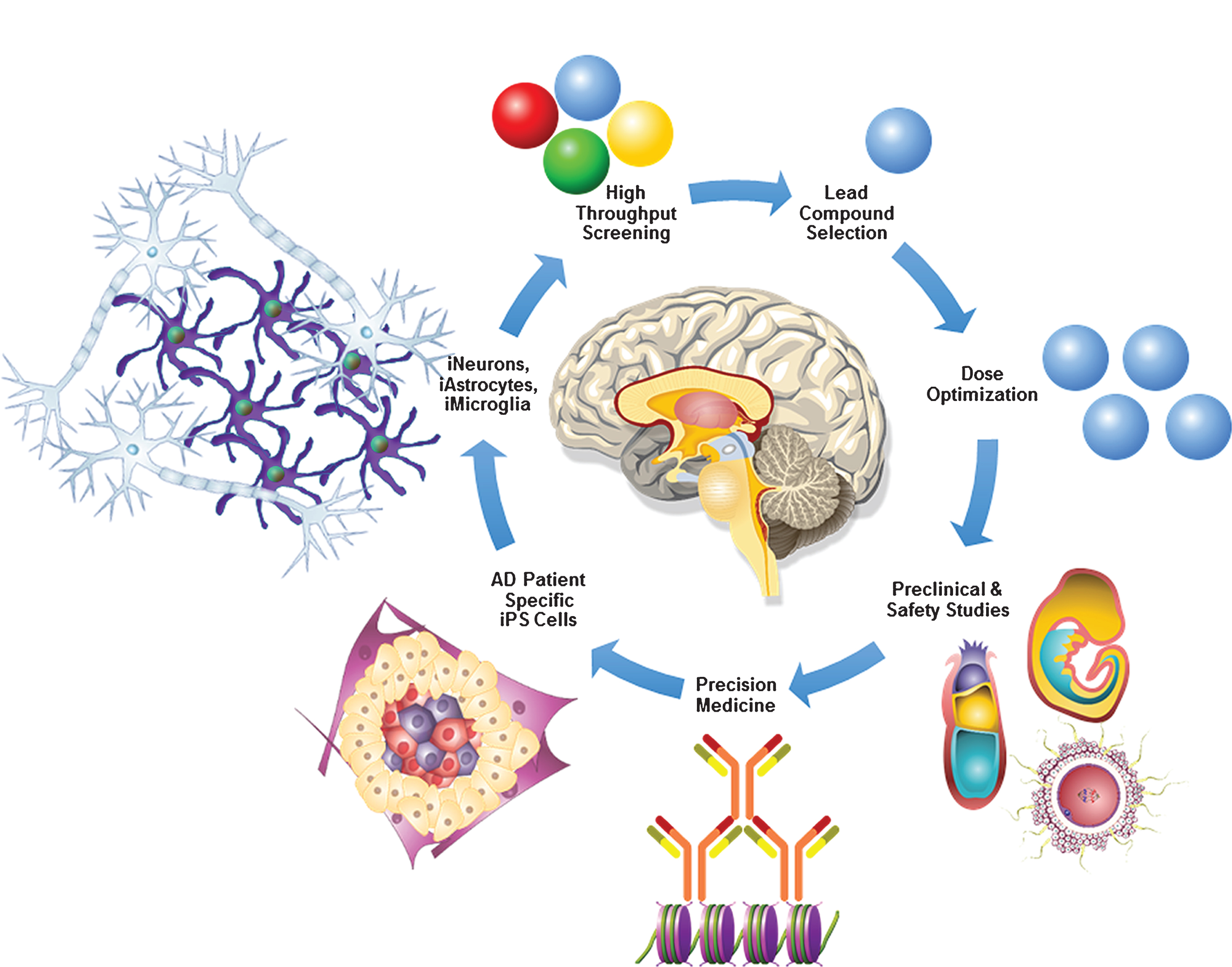
Fig.2
AD in a dish model for precision medicine. AD patient-specific fibroblasts can be reprogrammed into iPS cells. Progressive stepwise differentiation and lineage commitment of AD patient-specific iPS cells will generate iNeurons, iAstrocytes, and iMicroglia which play a crucial role in AD pathogenesis. Advancements in biomedical engineering have made it possible to generate 3D brain-like mini organoids derived from iPS cells, which can be very valuable in robotic high throughput screening of novel AD therapeutic targets. Once a lead compound with significant therapeutic potential is identified it can be used for the dose optimization, preclinical, pharmacokinetic, pharmacodynamic, toxicology, and safety studies in AD murine and non-human primate models. Finally, utilizing crystallography, mass-spectrometry, bioinformatics tools and latest advancements in precision medicine, AD patient-specific novel therapies can be successfully developed.
Han et al. have developed an antibody that induces the differentiation of bone marrow cells into microglia cells that traffic to the brain where they self-organize into typical networks [128]. They created a human ScFv lentiviral intracellular combinatorial antibody library with 108 unique antibody clones. Total bone marrow cells were harvested from mice, transduced with the lentiviral library and then transplanted into lethally irradiated mice. Seven days post transplantation, various organs were harvested and the cells assessed for migration and the DNA was subjected to PCR to amplify and sequence human ScFv sequences. They were able to identify an antibody B1 that induced migration of the cells to the brain. This antibody was also able to differentiate mouse bone marrow or human CD34 + cells into microglia-like cells that expressed mRNA for the microglial markers CX3CR1, IBA1, CD11b, CD68, TMEM119, and HEXB but failed to express mRNA for established oligodendrocyte (Olig1, Olig2, and MOG) and astrocyte (GFAP, SLCA2, and ALDH1LA) gene markers. The induced microglia-like cells exhibited an anti-inflammatory M2-like phenotype and phagocytosed Aβ peptide in vitro. These microglia-like cells when injected in APP/PS1 mice were capable of lowering Aβ deposition in the brain. These exciting results could potentially lead to the development of novel therapeutic strategies to successfully treat AD.
CONCLUSION AND FUTURE PERSPECTIVES
There has been a significant interest in developing novel AD therapies. However, despite significant global efforts, successful AD therapy remains elusive. There are multiple hurdles and roadblocks that need to be overcome before a precision-targeted patient-specific AD therapy becomes a reality. With the latest multi-disciplinary advancements including single-cell whole-genome sequencing [129], transcriptomics [130], metabolomics [131], proteomics [132, 133], CRISPR/Cas-mediated genome and base editing [134, 135], gene therapy [136], stem cell-based brain organoids [137–139], direct neuronal reprogramming and transdifferentiation [140–142], nanotechnology [143], biomolecular 3D organ printing [144], molecular imaging [145, 146], artificial intelligence [147], machine learning [148], robotics [149], and optogenetics [150] there is tremendous excitement and renewed vigor to win the war against AD. Toward our commitment to our long-term goal of developing targeted gene-editing and stem cell-based patient-specific therapies against AD and diabetes, we have very recently proposed a novel paradigm wherein tanycytes have been implicated to play a crucial role in diabetes and AD pathogenesis [151]. Although, the molecular mechanisms linking AD and diabetes are poorly understood, recent studies suggest that elevated glucose and oligomeric Aβ disrupt synapses via S-nitrosylation [152]. Further, we believe that there is critical need to focus our efforts on the deciphering the nexus between traumatic brain injury, post-traumatic stress disorder and AD as it will enable us to uncover potentially novel molecular mechanisms underlying AD initiation and progression [153, 154]. We are cautiously optimistic that, multi-faceted approaches simultaneously targeting neuroinflammation [117, 155, 156], blood-brain barrier [157, 158], robust Aβ processing and clearance by targeted immunotherapies [13], tau phosphorylation [159], APOE4 [160–164], improving mitochondrial function [165–167], iPS cell-based disease modeling [168], development of neural organoids [169, 170], stem cell trials [171, 172], neuroepigenomics [173, 174], microRNAs [175–177], improving CRISPR/Cas9 genome editing [178, 179], designer AAV vectors with enhanced neurotropism [87, 90, 180], improved drug delivery using nanotechnology [181], targeting exosomes [182], development of large animal AD models [183, 184], next generation nutraceuticals [185] as well as advances in microbiome-gut-brain axis [186, 187], neuroimaging [188] and robust, reproducible and reliable biomarker discovery [189, 190] hold significant promise and potential for the development of the next generation patient-specific neuro-immuno-gene and genome-editing as well as exosome and stem cell-based AD therapies (Figs. 1 and 2).
ACKNOWLEDGMENTS
The authors would like to acknowledge the Veteran’s Affairs Merit Award I01BX002477 and the National Institutes of Health Grant AG048205 to AZ.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0422r1).
REFERENCES
[1] | Hardy J , Selkoe DJ ((2002) ) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: , 353–356. |
[2] | Pickford F , Masliah E , Britschgi M , Lucin K , Narasimhan R , Jaeger PA , Small S , Spencer B , Rockenstein E , Levine B , Wyss-Coray T ((2008) ) The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 118: , 2190–2199. |
[3] | Zhang J , Wu X , Qin C , Qi J , Ma S , Zhang H , Kong Q , Chen D , Ba D , He W ((2003) ) A novel recombinant adeno-associated virus vaccine reduces beavihoral impairment and beta-amyloid plaques in a mouse model of Alzheimer’s disease. Neurobiol Dis 14: , 365–379. |
[4] | Fukuchi K , Tahara K , Kim HD , Maxwell JA , Lewis TL , Accavitti-Loper MA , Kim H , Ponnazhagan S , Lalonde R ((2006) ) Anti-Abeta single-chain antibody delivery via adeno-associated virus for treatment of Alzheimer’s disease. Neurobiol Dis 23: , 502–511. |
[5] | Hara H , Monsonego A , Yuasa K , Adachi K , Xiao X , Takeda S , Takahashi K , Weiner HL , Tabira T ((2004) ) Development of a safe oral Abeta vaccine using recombinant adeno-associated virus vector for Alzheimer’s disease. J Alzheimers Dis 6: , 483–488. |
[6] | Mouri A , Noda Y , Hara H , Mizoguchi H , Tabira T , Nabeshima T ((2007) ) Oral vaccination with a viral vector containing Abeta cDNA attenuates age-related Abeta accumulation and memory deficits without causing inflammation in a mouse Alzheimer model. FASEB J 21: , 2135–2148. |
[7] | Levites Y , Jansen K , Smithson LA , Dakin R , Holloway VM , Das P , Golde TE ((2006) ) Intracranial adeno-associated virus-mediated delivery of anti-pan amyloid beta, amyloid beta40, and amyloid beta42 single-chain variable fragments attenuates plaque pathology in amyloid precursor protein mice. J Neurosci 26: , 11923–11928. |
[8] | Carty NC , Nash K , Lee D , Mercer M , Gottschall PE , Meyers C , Muzyczka N , Gordon MN , Morgan D ((2008) ) Adeno-associated Viral (AAV) Serotype 5 vector mediated gene delivery of endothelin-converting enzyme reduces Abeta deposits in APP + PS1 transgenic mice. Mol Ther 16: , 1580–1586. |
[9] | Liu Y , Studzinski C , Beckett T , Guan H , Hersh MA , Murphy MP , Klein R , Hersh LB ((2009) ) Expression of neprilysin in skeletal muscle reduces amyloid burden in a transgenic mouse model of Alzheimer disease. Mol Ther 17: , 1381–1386. |
[10] | Jiang T , Tan L , Zhu XC , Zhang QQ , Cao L , Tan MS , Gu LZ , Wang HF , Ding ZZ , Zhang YD , Yu JT ((2014) ) Upregulation of TREM2 ameliorates neuropathology and rescues spatial cognitive impairment in a transgenic mouse model of Alzheimer’s disease. Neuropsychopharmacology 39: , 2949–2962. |
[11] | Moore BD , Martin J , de Mena L , Sanchez J , Cruz PE , Ceballos-Diaz C , Ladd TB , Ran Y , Levites Y , Kukar TL , Kurian JJ , McKenna R , Koo EH , Borchelt DR , Janus C , Rincon-Limas D , Fernandez-Funez P , Golde TE ((2018) ) Short Abeta peptides attenuate Abeta42 toxicity in vivo. J Exp Med 215: , 283–301. |
[12] | Wang QH , Wang YR , Zhang T , Jiao SS , Liu YH , Zeng F , Li J , Yao XQ , Zhou HD , Zhou XF , Wang YJ ((2016) ) Intramuscular delivery of p75NTR ectodomain by an AAV vector attenuates cognitive deficits and Alzheimer’s disease-like pathologies in APP/PS1 transgenic mice. J Neurochem 138: , 163–173. |
[13] | Sevigny J , Chiao P , Bussiere T , Weinreb PH , Williams L , Maier M , Dunstan R , Salloway S , Chen T , Ling Y , O’Gorman J , Qian F , Arastu M , Li M , Chollate S , Brennan MS , Quintero-Monzon O , Scannevin RH , Arnold HM , Engber T , Rhodes K , Ferrero J , Hang Y , Mikulskis A , Grimm J , Hock C , Nitsch RM , Sandrock A ((2016) ) The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 537: , 50–56. |
[14] | Doody RS , Farlow M , Aisen PS , Alzheimer’s Disease Cooperative Study Data Analysis and Publication Committee ((2014) ) Phase 3 trials of solanezumab and bapineuzumab for Alzheimer’s disease. N Engl J Med 370: , 1460. |
[15] | Doody RS , Thomas RG , Farlow M , Iwatsubo T , Vellas B , Joffe S , Kieburtz K , Raman R , Sun X , Aisen PS , Siemers E , Liu-Seifert H , Mohs R , Alzheimer’s Disease Cooperative Study Steering Committee, Solanezumab Study Group ((2014) ) Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med 370: , 311–321. |
[16] | Honig LS , Vellas B , Woodward M , Boada M , Bullock R , Borrie M , Hager K , Andreasen N , Scarpini E , Liu-Seifert H , Case M , Dean RA , Hake A , Sundell K , Poole Hoffmann V , Carlson C , Khanna R , Mintun M , DeMattos R , Selzler KJ , Siemers E ((2018) ) Trial of solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med 378: , 321–330. |
[17] | Graham WV , Bonito-Oliva A , Sakmar TP ((2017) ) Update on Alzheimer’s disease therapy and prevention strategies. Annu Rev Med 68: , 413–430. |
[18] | Salloway S , Sperling R , Fox NC , Blennow K , Klunk W , Raskind M , Sabbagh M , Honig LS , Porsteinsson AP , Ferris S , Reichert M , Ketter N , Nejadnik B , Guenzler V , Miloslavsky M , Wang D , Lu Y , Lull J , Tudor IC , Liu E , Grundman M , Yuen E , Black R , Brashear HR , Bapineuzumab, Clinical Trial I ((2014) ) Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 370: , 322–333. |
[19] | Spillantini MG , Goedert M ((1998) ) Tau protein pathology in neurodegenerative diseases. Trends Neurosci 21: , 428–433. |
[20] | Goedert M , Crowther RA , Spillantini MG ((1998) ) Tau mutations cause frontotemporal dementias. Neuron 21: , 955–958. |
[21] | Grundke-Iqbal I , Iqbal K , Quinlan M , Tung YC , Zaidi MS , Wisniewski HM ((1986) ) Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem 261: , 6084–6089. |
[22] | Grundke-Iqbal I , Iqbal K , Tung YC , Quinlan M , Wisniewski HM , Binder LI ((1986) ) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 83: , 4913–4917. |
[23] | Ittner LM , Ke YD , Delerue F , Bi M , Gladbach A , van Eersel J , Wolfing H , Chieng BC , Christie MJ , Napier IA , Eckert A , Staufenbiel M , Hardeman E , Gotz J ((2010) ) Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 142: , 387–397. |
[24] | Tan DCS , Yao S , Ittner A , Bertz J , Ke YD , Ittner LM , Delerue F ((2018) ) Generation of a new tau knockout (tauDeltaex1) line using CRISPR/Cas9 genome editing in mice. J Alzheimers Dis 62: , 571–578. |
[25] | Zhou H , Liu J , Zhou C , Gao N , Rao Z , Li H , Hu X , Li C , Yao X , Shen X , Sun Y , Wei Y , Liu F , Ying W , Zhang J , Tang C , Zhang X , Xu H , Shi L , Cheng L , Huang P , Yang H ((2018) ) In vivo simultaneous transcriptional activation of multiple genes in the brain using CRISPR-dCas9-activator transgenic mice. Nat Neurosci 21: , 440–446. |
[26] | Yanamandra K , Kfoury N , Jiang H , Mahan TE , Ma S , Maloney SE , Wozniak DF , Diamond MI , Holtzman DM ((2013) ) Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 80: , 402–414. |
[27] | Ising C , Gallardo G , Leyns CEG , Wong CH , Stewart F , Koscal LJ , Roh J , Robinson GO , Remolina Serrano J , Holtzman DM ((2017) ) AAV-mediated expression of anti-tau scFvs decreases tau accumulation in a mouse model of tauopathy. J Exp Med 214: , 1227–1238. |
[28] | Mullard A ((2017) ) BACE inhibitor bust in Alzheimer trial. Nat Rev Drug Discov 16: , 155. |
[29] | Atwal JK , Chen Y , Chiu C , Mortensen DL , Meilandt WJ , Liu Y , Heise CE , Hoyte K , Luk W , Lu Y , Peng K , Wu P , Rouge L , Zhang Y , Lazarus RA , Scearce-Levie K , Wang W , Wu Y , Tessier-Lavigne M , Watts RJ ((2011) ) A therapeutic antibody targeting BACE1 inhibits amyloid-beta production in vivo. Sci Transl Med 3: , 84ra43. |
[30] | Hu X , Das B , Hou H , He W , Yan R ((2018) ) BACE1 deletion in the adult mouse reverses preformed amyloid deposition and improves cognitive functions. J Exp Med 215: , 927–940. |
[31] | Tuszynski MH , Thal L , Pay M , Salmon DP , U HS , Bakay R , Patel P , Blesch A , Vahlsing HL , Ho G , Tong G , Potkin SG , Fallon J , Hansen L , Mufson EJ , Kordower JH , Gall C , Conner J ((2005) ) A phase 1 clinical trial of nerve growth factor gene therapy for Alzheimer disease. Nat Med 11: , 551–555. |
[32] | Eyjolfsdottir H , Eriksdotter M , Linderoth B , Lind G , Juliusson B , Kusk P , Almkvist O , Andreasen N , Blennow K , Ferreira D , Westman E , Nennesmo I , Karami A , Darreh-Shori T , Kadir A , Nordberg A , Sundstrom E , Wahlund LO , Wall A , Wiberg M , Winblad B , Seiger A , Wahlberg L , Almqvist P ((2016) ) Targeted delivery of nerve growth factor to the cholinergic basal forebrain of Alzheimer’s disease patients: Application of a second-generation encapsulated cell biodelivery device. Alzheimers Res Ther 8: , 30. |
[33] | Ferreira D , Westman E , Eyjolfsdottir H , Almqvist P , Lind G , Linderoth B , Seiger A , Blennow K , Karami A , Darreh-Shori T , Wiberg M , Simmons A , Wahlund LO , Wahlberg L , Eriksdotter M ((2015) ) Brain changes in Alzheimer’s disease patients with implanted encapsulated cells releasing nerve growth factor. J Alzheimers Dis 43: , 1059–1072. |
[34] | Hock C , Heese K , Hulette C , Rosenberg C , Otten U ((2000) ) Region-specific neurotrophin imbalances in Alzheimer disease: Decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch Neurol 57: , 846–851. |
[35] | Nagahara AH , Tuszynski MH ((2011) ) Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov 10: , 209–219. |
[36] | Narisawa-Saito M , Wakabayashi K , Tsuji S , Takahashi H , Nawa H ((1996) ) Regional specificity of alterations in NGF, BDNF and NT-3 levels in Alzheimer’s disease. Neuroreport 7: , 2925–2928. |
[37] | Chen J , Zhang T , Jiao S , Zhou X , Zhong J , Wang Y , Liu J , Deng J , Wang S , Xu Z ((2017) ) proBDNF accelerates brain amyloid-beta deposition and learning and memory impairment in APPswePS1dE9 transgenic mice. J Alzheimers Dis 59: , 941–949. |
[38] | Osborne A , Wang AX , Tassoni A , Widdowson PS , Martin KR ((2018) ) Design of a novel gene therapy construct to achieve sustained brain-derived neurotrophic factor signalling in neurons. Hum Gene Ther 29: , 828–841. |
[39] | Caccamo A , Maldonado MA , Bokov AF , Majumder S , Oddo S ((2010) ) CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 107: , 22687–22692. |
[40] | Nagahara AH , Wilson BR , Ivasyk I , Kovacs I , Rawalji S , Bringas JR , Pivirotto PJ , Sebastian WS , Samaranch L , Bankiewicz KS , Tuszynski MH ((2018) ) MR-guided delivery of AAV2-BDNF into the entorhinal cortex of non-human primates. Gene Ther 25: , 104–114. |
[41] | Mahley RW ((1988) ) Apolipoprotein E: Cholesterol transport protein with expanding role in cell biology. Science 240: , 622–630. |
[42] | Fagan AM , Bu G , Sun Y , Daugherty A , Holtzman DM ((1996) ) Apolipoprotein E-containing high density lipoprotein promotes neurite outgrowth and is a ligand for the low density lipoprotein receptor-related protein. J Biol Chem 271: , 30121–30125. |
[43] | Corder EH , Saunders AM , Risch NJ , Strittmatter WJ , Schmechel DE , Gaskell PC Jr. , Rimmler JB , Locke PA , Conneally PM , Schmader KE , . ((1994) ) Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet 7: , 180–184. |
[44] | Holtzman DM , Bales KR , Tenkova T , Fagan AM , Parsadanian M , Sartorius LJ , Mackey B , Olney J , McKeel D , Wozniak D , Paul SM ((2000) ) Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 97: , 2892–2897. |
[45] | Holtzman DM , Fagan AM , Mackey B , Tenkova T , Sartorius L , Paul SM , Bales K , Ashe KH , Irizarry MC , Hyman BT ((2000) ) Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer’s disease model. Ann Neurol 47: , 739–747. |
[46] | Huang YA , Zhou B , Wernig M , Sudhof TC ((2017) ) ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and Abeta secretion. Cell 168: , 427–441 e421. |
[47] | Shi Y , Yamada K , Liddelow SA , Smith ST , Zhao L , Luo W , Tsai RM , Spina S , Grinberg LT , Rojas JC , Gallardo G , Wang K , Roh J , Robinson G , Finn MB , Jiang H , Sullivan PM , Baufeld C , Wood MW , Sutphen C , McCue L , Xiong C , Del-Aguila JL , Morris JC , Cruchaga C , Alzheimer’s Disease Neuroimaging Initiative, Fagan AM , Miller BL , Boxer AL , Seeley WW , Butovsky O , Barres BA , Paul SM , Holtzman DM ((2017) ) ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549: , 523–527. |
[48] | Dodart JC , Marr RA , Koistinaho M , Gregersen BM , Malkani S , Verma IM , Paul SM ((2005) ) Gene delivery of human apolipoprotein E alters brain Abeta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 102: , 1211–1216. |
[49] | Rosenberg JB , Kaplitt M , De BP , Chen A , Flagiello T , Salami CO , Pey E , Zhao L , Ricart Arbona R , Monette S , Dyke J , Ballon D , Kaminsky SM , Sondhi D , Petsko G , Paul S , Crystal RG ((2018) ) AAVrh.10-mediated APOE2 CNS gene therapy for APOE4-associated Alzheimer’s disease. Hum Gene Ther Clin Dev 29: , 24–47. |
[50] | Kempuraj D , Thangavel R , Natteru PA , Selvakumar GP , Saeed D , Zahoor H , Zaheer S , Iyer SS , Zaheer A ((2016) ) Neuroinflammation induces neurodegeneration. J Neurol Neurosurg Spine 1: , 1003. |
[51] | Ransohoff RM ((2016) ) How neuroinflammation contributes to neurodegeneration. Science 353: , 777–783. |
[52] | Kiyota T , Yamamoto M , Schroder B , Jacobsen MT , Swan RJ , Lambert MP , Klein WL , Gendelman HE , Ransohoff RM , Ikezu T ((2009) ) AAV1/2-mediated CNS gene delivery of dominant-negative CCL2 mutant suppresses gliosis, beta-amyloidosis, and learning impairment of APP/PS1 mice. Mol Ther 17: , 803–809. |
[53] | Kiyota T , Ingraham KL , Swan RJ , Jacobsen MT , Andrews SJ , Ikezu T ((2012) ) AAV serotype 2/1-mediated gene delivery of anti-inflammatory interleukin-10 enhances neurogenesis and cognitive function in APP+PS1 mice. Gene Ther 19: , 724–733. |
[54] | Kiyota T , Okuyama S , Swan RJ , Jacobsen MT , Gendelman HE , Ikezu T ((2010) ) CNS expression of anti-inflammatory cytokine interleukin-4 attenuates Alzheimer’s disease-like pathogenesis in APP+PS1 bigenic mice. FASEB J 24: , 3093–3102. |
[55] | Chakrabarty P , Li A , Ceballos-Diaz C , Eddy JA , Funk CC , Moore B , DiNunno N , Rosario AM , Cruz PE , Verbeeck C , Sacino A , Nix S , Janus C , Price ND , Das P , Golde TE ((2015) ) IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive beavihor. Neuron 85: , 519–533. |
[56] | Petitto JM , McNamara RK , Gendreau PL , Huang Z , Jackson AJ ((1999) ) Impaired learning and memory and altered hippocampal neurodevelopment resulting from interleukin-2 gene deletion. J Neurosci Res 56: , 441–446. |
[57] | Alves S , Churlaud G , Audrain M , Michaelsen-Preusse K , Fol R , Souchet B , Braudeau J , Korte M , Klatzmann D , Cartier N ((2017) ) Interleukin-2 improves amyloid pathology, synaptic failure and memory in Alzheimer’s disease mice. Brain 140: , 826–842. |
[58] | Chakrabarty P , Tianbai L , Herring A , Ceballos-Diaz C , Das P , Golde TE ((2012) ) Hippocampal expression of murine IL-4 results in exacerbation of amyloid deposition. Mol Neurodegener 7: , 36. |
[59] | Vogelaar CF , Mandal S , Lerch S , Birkner K , Birkenstock J , Buhler U , Schnatz A , Raine CS , Bittner S , Vogt J , Kipnis J , Nitsch R , Zipp F ((2018) ) Fast direct neuronal signaling via the IL-4 receptor as therapeutic target in neuroinflammation, Sci Transl Med 10: , eaao2304. |
[60] | Chakrabarty P , Herring A , Ceballos-Diaz C , Das P , Golde TE ((2011) ) Hippocampal expression of murine TNFalpha results in attenuation of amyloid deposition in vivo. Mol Neurodegener 6: , 16. |
[61] | Chakrabarty P , Ceballos-Diaz C , Beccard A , Janus C , Dickson D , Golde TE , Das P ((2010) ) IFN-gamma promotes complement expression and attenuates amyloid plaque deposition in amyloid beta precursor protein transgenic mice. J Immunol 184: , 5333–5343. |
[62] | Chakrabarty P , Jansen-West K , Beccard A , Ceballos-Diaz C , Levites Y , Verbeeck C , Zubair AC , Dickson D , Golde TE , Das P ((2010) ) Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: Evidence against inflammation as a driving force for amyloid deposition. FASEB J 24: , 548–559. |
[63] | Sudol KL , Mastrangelo MA , Narrow WC , Frazer ME , Levites YR , Golde TE , Federoff HJ , Bowers WJ ((2009) ) Generating differentially targeted amyloid-beta specific intrabodies as a passive vaccination strategy for Alzheimer’s disease. Mol Ther 17: , 2031–2040. |
[64] | Borchelt DR , Ratovitski T , van Lare J , Lee MK , Gonzales V , Jenkins NA , Copeland NG , Price DL , Sisodia SS ((1997) ) Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 19: , 939–945. |
[65] | Gordon MN , Holcomb LA , Jantzen PT , DiCarlo G , Wilcock D , Boyett KW , Connor K , Melachrino J , O’Callaghan JP , Morgan D ((2002) ) Time course of the development of Alzheimer-like pathology in the doubly transgenic PS1+APP mouse. Exp Neurol 173: , 183–195. |
[66] | Holcomb L , Gordon MN , McGowan E , Yu X , Benkovic S , Jantzen P , Wright K , Saad I , Mueller R , Morgan D , Sanders S , Zehr C , O’Campo K , Hardy J , Prada CM , Eckman C , Younkin S , Hsiao K , Duff K ((1998) ) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 4: , 97–100. |
[67] | Hsiao K , Chapman P , Nilsen S , Eckman C , Harigaya Y , Younkin S , Yang F , Cole G ((1996) ) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274: , 99–102. |
[68] | Lawlor PA , Bland RJ , Das P , Price RW , Holloway V , Smithson L , Dicker BL , During MJ , Young D , Golde TE ((2007) ) Novel rat Alzheimer’s disease models based on AAV-mediated gene transfer to selectively increase hippocampal Abeta levels. Mol Neurodegener 2: , 11. |
[69] | Kim J , Miller VM , Levites Y , West KJ , Zwizinski CW , Moore BD , Troendle FJ , Bann M , Verbeeck C , Price RW , Smithson L , Sonoda L , Wagg K , Rangachari V , Zou F , Younkin SG , Graff-Radford N , Dickson D , Rosenberry T , Golde TE ((2008) ) BRI2 (ITM2b) inhibits Abeta deposition in vivo. J Neurosci 28: , 6030–6036. |
[70] | Lawlor PA , Bland RJ , Mouravlev A , Young D , During MJ ((2009) ) Efficient gene delivery and selective transduction of glial cells in the mammalian brain by AAV serotypes isolated from nonhuman primates. Mol Ther 17: , 1692–1702. |
[71] | Klein RL , Dayton RD , Leidenheimer NJ , Jansen K , Golde TE , Zweig RM ((2006) ) Efficient neuronal gene transfer with AAV8 leads to neurotoxic levels of tau or green fluorescent proteins. Mol Ther 13: , 517–527. |
[72] | Fu AK , Hung KW , Yuen MY , Zhou X , Mak DS , Chan IC , Cheung TH , Zhang B , Fu WY , Liew FY , Ip NY ((2016) ) IL-33 ameliorates Alzheimer’s disease-like pathology and cognitive decline, Proc Natl Acad Sci U S A 113: , E2705–E2713. |
[73] | Chapuis J , Hot D , Hansmannel F , Kerdraon O , Ferreira S , Hubans C , Maurage CA , Huot L , Bensemain F , Laumet G , Ayral AM , Fievet N , Hauw JJ , DeKosky ST , Lemoine Y , Iwatsubo T , Wavrant-Devrieze F , Dartigues JF , Tzourio C , Buee L , Pasquier F , Berr C , Mann D , Lendon C , Alperovitch A , Kamboh MI , Amouyel P , Lambert JC ((2009) ) Transcriptomic and genetic studies identify IL-33 as a candidate gene for Alzheimer’s disease. Mol Psychiatry 14: , 1004–1016. |
[74] | Mendell JR , Al-Zaidy S , Shell R , Arnold WD , Rodino-Klapac LR , Prior TW , Lowes L , Alfano L , Berry K , Church K , Kissel JT , Nagendran S , L’Italien J , Sproule DM , Wells C , Cardenas JA , Heitzer MD , Kaspar A , Corcoran S , Braun L , Likhite S , Miranda C , Meyer K , Foust KD , Burghes AHM , Kaspar BK ((2017) ) Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med 377: , 1713–1722. |
[75] | Hinderer C , Katz N , Buza EL , Dyer C , Goode T , Bell P , Richman LK , Wilson JM ((2018) ) Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum Gene Ther 29: , 285–298. |
[76] | Hordeaux J , Wang Q , Katz N , Buza EL , Bell P , Wilson JM ((2018) ) The neurotropic properties of AAV-PHP.B are limited to C57BL/6J mice. Mol Ther 26: , 664–668. |
[77] | Hammond SL , Leek AN , Richman EH , Tjalkens RB ((2017) ) Cellular selectivity of AAV serotypes for gene delivery in neurons and astrocytes by neonatal intracerebroventricular injection. PLoS One 12: , e0188830. |
[78] | Aschauer DF , Kreuz S , Rumpel S ((2013) ) Analysis of transduction efficiency, tropism and axonal transport of AAV serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PLoS One 8: , e76310. |
[79] | Choudhury SR , Hudry E , Maguire CA , Sena-Esteves M , Breakefield XO , Grandi P ((2017) ) Viral vectors for therapy of neurologic diseases. Neuropharmacology 120: , 63–80. |
[80] | Hocquemiller M , Giersch L , Audrain M , Parker S , Cartier N ((2016) ) Adeno-associated virus-based gene therapy for CNS diseases. Hum Gene Ther 27: , 478–496. |
[81] | Lau AA , Hemsley KM ((2017) ) Adeno-associated viral gene therapy for mucopolysaccharidoses exhibiting neurodegeneration. J Mol Med (Berl) 95: , 1043–1052. |
[82] | Rosenberg JB , Kaminsky SM , Aubourg P , Crystal RG , Sondhi D ((2016) ) Gene therapy for metachromatic leukodystrophy. J Neurosci Res 94: , 1169–1179. |
[83] | Faust SM , Bell P , Cutler BJ , Ashley SN , Zhu Y , Rabinowitz JE , Wilson JM ((2013) ) CpG-depleted adeno-associated virus vectors evade immune detection. J Clin Invest 123: , 2994–3001. |
[84] | Hudry E , Martin C , Gandhi S , Gyorgy B , Scheffer DI , Mu D , Merkel SF , Mingozzi F , Fitzpatrick Z , Dimant H , Masek M , Ragan T , Tan S , Brisson AR , Ramirez SH , Hyman BT , Maguire CA ((2016) ) Exosome-associated AAV vector as a robust and convenient neuroscience tool. Gene Ther 23: , 380–392. |
[85] | Zhong L , Li B , Jayandharan G , Mah CS , Govindasamy L , Agbandje-McKenna M , Herzog RW , Weigel-Van Aken KA , Hobbs JA , Zolotukhin S , Muzyczka N , Srivastava A ((2008) ) Tyrosine-phosphorylation of AAV2 vectors and its consequences on viral intracellular trafficking and transgene expression. Virology 381: , 194–202. |
[86] | Zhong L , Li B , Mah CS , Govindasamy L , Agbandje-McKenna M , Cooper M , Herzog RW , Zolotukhin I , Warrington KH Jr. , Weigel-Van Aken KA , Hobbs JA , Zolotukhin S , Muzyczka N , Srivastava A ((2008) ) Next generation of adeno-associated virus 2 vectors: Point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc Natl Acad Sci U S A 105: , 7827–7832. |
[87] | Deverman BE , Pravdo PL , Simpson BP , Kumar SR , Chan KY , Banerjee A , Wu WL , Yang B , Huber N , Pasca SP , Gradinaru V ((2016) ) Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol 34: , 204–209. |
[88] | Foust KD , Nurre E , Montgomery CL , Hernandez A , Chan CM , Kaspar BK ((2009) ) Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol 27: , 59–65. |
[89] | Bevan AK , Duque S , Foust KD , Morales PR , Braun L , Schmelzer L , Chan CM , McCrate M , Chicoine LG , Coley BD , Porensky PN , Kolb SJ , Mendell JR , Burghes AH , Kaspar BK ((2011) ) Systemic gene delivery in large species for targeting spinal cord, brain, and peripheral tissues for pediatric disorders. Mol Ther 19: , 1971–1980. |
[90] | Chan KY , Jang MJ , Yoo BB , Greenbaum A , Ravi N , Wu WL , Sanchez-Guardado L , Lois C , Mazmanian SK , Deverman BE , Gradinaru V ((2017) ) Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat Neurosci 20: , 1172–1179. |
[91] | Morabito G , Giannelli SG , Ordazzo G , Bido S , Castoldi V , Indrigo M , Cabassi T , Cattaneo S , Luoni M , Cancellieri C , Sessa A , Bacigaluppi M , Taverna S , Leocani L , Lanciego JL , Broccoli V ((2017) ) AAV-PHP.B-mediated global-scale expression in the mouse nervous system enables GBA1 gene therapy for wide protection from synucleinopathy. Mol Ther 25: , 2727–2742. |
[92] | Matsuzaki Y , Konno A , Mochizuki R , Shinohara Y , Nitta K , Okada Y , Hirai H ((2018) ) Intravenous administration of the adeno-associated virus-PHP.B capsid fails to upregulate transduction efficiency in the marmoset brain. Neurosci Lett 665: , 182–188. |
[93] | Murlidharan G , Sakamoto K , Rao L , Corriher T , Wang D , Gao G , Sullivan P , Asokan A ((2016) ) CNS-restricted transduction and CRISPR/Cas9-mediated gene deletion with an engineered AAV vector. Mol Ther Nucleic Acids 5: , e338. |
[94] | Tervo DG , Hwang BY , Viswanathan S , Gaj T , Lavzin M , Ritola KD , Lindo S , Michael S , Kuleshova E , Ojala D , Huang CC , Gerfen CR , Schiller J , Dudman JT , Hantman AW , Looger LL , Schaffer DV , Karpova AY ((2016) ) A designer AAV variant permits efficient retrograde access to projection neurons. Neuron 92: , 372–382. |
[95] | Choudhury SR , Fitzpatrick Z , Harris AF , Maitland SA , Ferreira JS , Zhang Y , Ma S , Sharma RB , Gray-Edwards HL , Johnson JA , Johnson AK , Alonso LC , Punzo C , Wagner KR , Maguire CA , Kotin RM , Martin DR , Sena-Esteves M ((2016) ) In vivo selection yields AAV-B1 capsid for central nervous system and muscle gene therapy. Mol Ther 24: , 1247–1257. |
[96] | Choudhury SR , Harris AF , Cabral DJ , Keeler AM , Sapp E , Ferreira JS , Gray-Edwards HL , Johnson JA , Johnson AK , Su Q , Stoica L , DiFiglia M , Aronin N , Martin DR , Gao G , Sena-Esteves M ((2016) ) Widespread central nervous system gene transfer and silencing after systemic delivery of novel AAV-AS vector. Mol Ther 24: , 726–735. |
[97] | Kanaan NM , Sellnow RC , Boye SL , Coberly B , Bennett A , Agbandje-McKenna M , Sortwell CE , Hauswirth WW , Boye SE , Manfredsson FP ((2017) ) Rationally engineered AAV capsids improve transduction and volumetric spread in the CNS. Mol Ther Nucleic Acids 8: , 184–197. |
[98] | Cornu TI , Mussolino C , Cathomen T ((2017) ) Refining strategies to translate genome editing to the clinic. Nat Med 23: , 415–423. |
[99] | Barrangou R , Doudna JA ((2016) ) Applications of CRISPR technologies in research and beyond. Nat Biotechnol 34: , 933–941. |
[100] | Doudna JA ((2015) ) Genomic engineering and the future of medicine. JAMA 313: , 791–792. |
[101] | Fellmann C , Gowen BG , Lin PC , Doudna JA , Corn JE ((2017) ) Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat Rev Drug Discov 16: , 89–100. |
[102] | Wright AV , Nunez JK , Doudna JA ((2016) ) Biology and applications of CRISPR systems: Harnessing nature’s toolbox for genome engineering. Cell 164: , 29–44. |
[103] | Dahlman JE , Abudayyeh OO , Joung J , Gootenberg JS , Zhang F , Konermann S ((2015) ) Orthogonal gene knockout and activation with a catalytically active Cas9 nuclease. Nat Biotechnol 33: , 1159–1161. |
[104] | Konermann S , Brigham MD , Trevino AE , Joung J , Abudayyeh OO , Barcena C , Hsu PD , Habib N , Gootenberg JS , Nishimasu H , Nureki O , Zhang F ((2015) ) Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517: , 583–588. |
[105] | Hu JH , Miller SM , Geurts MH , Tang W , Chen L , Sun N , Zeina CM , Gao X , Rees HA , Lin Z , Liu DR ((2018) ) Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556: , 57–63. |
[106] | Kramer NJ , Haney MS , Morgens DW , Jovicic A , Couthouis J , Li A , Ousey J , Ma R , Bieri G , Tsui CK , Shi Y , Hertz NT , Tessier-Lavigne M , Ichida JK , Bassik MC , Gitler AD ((2018) ) CRISPR-Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nat Genet 50: , 603–612. |
[107] | Paquet D , Kwart D , Chen A , Sproul A , Jacob S , Teo S , Olsen KM , Gregg A , Noggle S , Tessier-Lavigne M ((2016) ) Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature 533: , 125–129. |
[108] | Platt RJ , Chen S , Zhou Y , Yim MJ , Swiech L , Kempton HR , Dahlman JE , Parnas O , Eisenhaure TM , Jovanovic M , Graham DB , Jhunjhunwala S , Heidenreich M , Xavier RJ , Langer R , Anderson DG , Hacohen N , Regev A , Feng G , Sharp PA , Zhang F ((2014) ) CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159: , 440–455. |
[109] | Zheng Y , Shen W , Zhang J , Yang B , Liu YN , Qi H , Yu X , Lu SY , Chen Y , Xu YZ , Li Y , Gage FH , Mi S , Yao J ((2018) ) CRISPR interference-based specific and efficient gene inactivation in the brain. Nat Neurosci 21: , 447–454. |
[110] | Gilbert LA , Larson MH , Morsut L , Liu Z , Brar GA , Torres SE , Stern-Ginossar N , Brandman O , Whitehead EH , Doudna JA , Lim WA , Weissman JS , Qi LS ((2013) ) CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154: , 442–451. |
[111] | Gilbert LA , Horlbeck MA , Adamson B , Villalta JE , Chen Y , Whitehead EH , Guimaraes C , Panning B , Ploegh HL , Bassik MC , Qi LS , Kampmann M , Weissman JS ((2014) ) Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159: , 647–661. |
[112] | Fulco CP , Munschauer M , Anyoha R , Munson G , Grossman SR , Perez EM , Kane M , Cleary B , Lander ES , Engreitz JM ((2016) ) Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science 354: , 769–773. |
[113] | Adamson B , Norman TM , Jost M , Cho MY , Nunez JK , Chen Y , Villalta JE , Gilbert LA , Horlbeck MA , Hein MY , Pak RA , Gray AN , Gross CA , Dixit A , Parnas O , Regev A , Weissman JS ((2016) ) A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167: , 1821. |
[114] | Thakore PI , D’Ippolito AM , Song L , Safi A , Shivakumar NK , Kabadi AM , Reddy TE , Crawford GE , Gersbach CA ((2015) ) Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods 12: , 1143–1149. |
[115] | Liu SJ , Horlbeck MA , Cho SW , Birk HS , Malatesta M , He D , Attenello FJ , Villalta JE , Cho MY , Chen Y , Mandegar MA , Olvera MP , Gilbert LA , Conklin BR , Chang HY , Weissman JS , Lim DA ((2017) ) CRISPRi-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 355: , aah7111. |
[116] | Swiech L , Heidenreich M , Banerjee A , Habib N , Li Y , Trombetta J , Sur M , Zhang F ((2015) ) In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat Biotechnol 33: , 102–106. |
[117] | Raikwar SP , Thangavel R , Dubova I , Selvakumar GP , Ahmed ME , Kempuraj D , Zaheer SA , Iyer SS , Zaheer A ((2018) ) Targeted gene editing of glia maturation factor in microglia: A novel Alzheimer’s disease therapeutic target. Mol Neurobiol. doi: 10.1007/s12035-018-1068-y . |
[118] | de Solis CA , Ho A , Holehonnur R , Ploski JE ((2016) ) The development of a viral mediated CRISPR/Cas9 system with doxycycline dependent gRNA expression for inducible in vitro and in vivo genome editing. Front Mol Neurosci 9: , 70. |
[119] | Nishiyama J , Mikuni T , Yasuda R ((2017) ) Virus-mediated genome editing via homology-directed repair in mitotic and postmitotic cells in mammalian brain. Neuron 96: 755–768.e5. |
[120] | Komor AC , Kim YB , Packer MS , Zuris JA , Liu DR ((2016) ) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533: , 420–424. |
[121] | Bustos FJ , Ampuero E , Jury N , Aguilar R , Falahi F , Toledo J , Ahumada J , Lata J , Cubillos P , Henriquez B , Guerra MV , Stehberg J , Neve RL , Inestrosa NC , Wyneken U , Fuenzalida M , Hartel S , Sena-Esteves M , Varela-Nallar L , Rots MG , Montecino M , van Zundert B ((2017) ) Epigenetic editing of the Dlg4/PSD95 gene improves cognition in aged and Alzheimer’s disease mice. Brain 140: , 3252–3268. |
[122] | Huang KL , Marcora E , Pimenova AA , Di Narzo AF , Kapoor M , Jin SC , Harari O , Bertelsen S , Fairfax BP , Czajkowski J , Chouraki V , Grenier-Boley B , Bellenguez C , Deming Y , McKenzie A , Raj T , Renton AE , Budde J , Smith A , Fitzpatrick A , Bis JC , DeStefano A , Adams HHH , Ikram MA , van der Lee S , Del-Aguila JL , Fernandez MV , Ibanez L , International Genomics of Alzheimer’s Project; Alzheimer’s Disease Neuroimaging Initiative, Sims R , Escott-Price V , Mayeux R , Haines JL , Farrer LA , Pericak-Vance MA , Lambert JC , van Duijn C , Launer L , Seshadri S , Williams J , Amouyel P , Schellenberg GD , Zhang B , Borecki I , Kauwe JSK , Cruchaga C , Hao K , Goate AM ((2017) ) A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nat Neurosci 20: , 1052–1061. |
[123] | Jin K , Peel AL , Mao XO , Xie L , Cottrell BA , Henshall DC , Greenberg DA ((2004) ) Increased hippocampal neurogenesis in Alzheimer’s disease. Proc Natl Acad Sci U S A 101: , 343–347. |
[124] | Haughey NJ , Liu D , Nath A , Borchard AC , Mattson MP ((2002) ) Disruption of neurogenesis in the subventricular zone of adult mice, and in human cortical neuronal precursor cells in culture, by amyloid beta-peptide: Implications for the pathogenesis of Alzheimer’s disease. Neuromolecular Med 1: , 125–135. |
[125] | Haughey NJ , Nath A , Chan SL , Borchard AC , Rao MS , Mattson MP ((2002) ) Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer’s disease. J Neurochem 83: , 1509–1524. |
[126] | Wen PH , Shao X , Shao Z , Hof PR , Wisniewski T , Kelley K , Friedrich VL Jr. , Ho L , Pasinetti GM , Shioi J , Robakis NK , Elder GA ((2002) ) Overexpression of wild type but not an FAD mutant presenilin-1 promotes neurogenesis in the hippocampus of adult mice. Neurobiol Dis 10: , 8–19. |
[127] | Baglietto-Vargas D , Sanchez-Mejias E , Navarro V , Jimenez S , Trujillo-Estrada L , Gomez-Arboledas A , Sanchez-Mico M , Sanchez-Varo R , Vizuete M , Davila JC , Garcia-Verdugo JM , Vitorica J , Gutierrez A ((2017) ) Dual roles of Abeta in proliferative processes in an amyloidogenic model of Alzheimer’s disease. Sci Rep 7: , 10085. |
[128] | Han KH , Arlian BM , Macauley MS , Paulson JC , Lerner RA ((2018) ) Migration-based selections of antibodies that convert bone marrow into trafficking microglia-like cells that reduce brain amyloid beta. Proc Natl Acad Sci U S A 115: , E372–E381. |
[129] | Lodato MA , Rodin RE , Bohrson CL , Coulter ME , Barton AR , Kwon M , Sherman MA , Vitzthum CM , Luquette LJ , Yandava CN , Yang P , Chittenden TW , Hatem NE , Ryu SC , Woodworth MB , Park PJ , Walsh CA ((2018) ) Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359: , 555–559. |
[130] | Magistri M , Velmeshev D , Makhmutova M , Faghihi MA ((2015) ) Transcriptomics profiling of Alzheimer’s disease reveal neurovascular defects, altered amyloid-beta homeostasis, and deregulated expression of long noncoding RNAs. J Alzheimers Dis 48: , 647–665. |
[131] | Wilkins JM , Trushina E ((2017) ) Application of metabolomics in Alzheimer’s disease. Front Neurol 8: , 719. |
[132] | Sato C , Barthelemy NR , Mawuenyega KG , Patterson BW , Gordon BA , Jockel-Balsarotti J , Sullivan M , Crisp MJ , Kasten T , Kirmess KM , Kanaan NM , Yarasheski KE , Baker-Nigh A , Benzinger TLS , Miller TM , Karch CM , Bateman RJ ((2018) ) Tau kinetics in neurons and the human central nervous system. Neuron 97: , 1284–1298 e1287. |
[133] | Zhu B , Jiang L , Huang T , Zhao Y , Liu T , Zhong Y , Li X , Campos A , Pomeroy K , Masliah E , Zhang D , Xu H ((2017) ) ER-associated degradation regulates Alzheimer’s amyloid pathology and memory function by modulating gamma-secretase activity. Nat Commun 8: , 1472. |
[134] | Billon P , Bryant EE , Joseph SA , Nambiar TS , Hayward SB , Rothstein R , Ciccia A ((2017) ) CRISPR-mediated base editing enables efficient disruption of eukaryotic genes through induction of STOP codons. Mol Cell 67: , 1068–1079 e1064. |
[135] | Gyorgy B , Loov C , Zaborowski MP , Takeda S , Kleinstiver BP , Commins C , Kastanenka K , Mu D , Volak A , Giedraitis V , Lannfelt L , Maguire CA , Joung JK , Hyman BT , Breakefield XO , Ingelsson M ((2018) ) CRISPR/Cas9 mediated disruption of the Swedish APP allele as a therapeutic approach for early-onset Alzheimer’s disease. Mol Ther Nucleic Acids 11: , 429–440. |
[136] | Rosenberg JB , Kaplitt MG , De BP , Chen A , Flagiello T , Salami C , Pey E , Zhao L , Ricart Arbona RJ , Monette S , Dyke JP , Ballon DJ , Kaminsky SM , Sondhi D , Petsko GA , Paul SM , Crystal RG ((2018) ) AAVrh.10-mediated APOE2 central nervous system gene therapy for APOE4-associated Alzheimer’s disease. Hum Gene Ther Clin Dev 29: , 24–47. |
[137] | Lin YT , Seo J , Gao F , Feldman HM , Wen HL , Penney J , Cam HP , Gjoneska E , Raja WK , Cheng J , Rueda R , Kritskiy O , Abdurrob F , Peng Z , Milo B , Yu CJ , Elmsaouri S , Dey D , Ko T , Yankner BA , Tsai LH ((2018) ) APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types, Neuron 98: ,1141–1154.e7. Erratum in: Neuron 98. 1294, 2018. |
[138] | Mansour AA , Goncalves JT , Bloyd CW , Li H , Fernandes S , Quang D , Johnston S , Parylak SL , Jin X , Gage FH ((2018) ) An in vivo model of functional and vascularized human brain organoids. Nat Biotechnol 36: , 432–441. |
[139] | Karzbrun E , Kshirsagar A , Cohen SR , Hanna JH , Reiner O ((2018) ) Human brain organoids on a chip reveal the physics of folding. Nat Phys 14: , 515–522. |
[140] | Weinberg MS , Criswell HE , Powell SK , Bhatt AP , McCown TJ ((2017) ) Viral vector reprogramming of adult resident striatal oligodendrocytes into functional neurons. Mol Ther 25: , 928–934. |
[141] | Tanabe K , Ang CE , Chanda S , Olmos VH , Haag D , Levinson DF , Sudhof TC , Wernig M ((2018) ) Transdifferentiation of human adult peripheral blood T cells into neurons. Proc Natl Acad Sci U S A 115: , 6470–6475. |
[142] | Gascon S , Masserdotti G , Russo GL , Gotz M ((2017) ) Direct neuronal reprogramming: Achievements, hurdles, and new roads to success. Cell Stem Cell 21: , 18–34. |
[143] | Wen MM , El-Salamouni NS , El-Refaie WM , Hazzah HA , Ali MM , Tosi G , Farid RM , Blanco-Prieto MJ , Billa N , Hanafy AS ((2017) ) Nanotechnology-based drug delivery systems for Alzheimer’s disease management: Technical, industrial, and clinical challenges. J Control Release 245: , 95–107. |
[144] | Haring AP , Sontheimer H , Johnson BN ((2017) ) Microphysiological human brain and neural systems-on-a-chip: Potential alternatives to small animal models and emerging platforms for drug discovery and personalized medicine. Stem Cell Rev 13: , 381–406. |
[145] | Patriarchi T , Cho JR , Merten K , Howe MW , Marley A , Xiong WH , Folk RW , Broussard GJ , Liang R , Jang MJ , Zhong H , Dombeck D , von Zastrow M , Nimmerjahn A , Gradinaru V , Williams JT , Tian L ((2018) ) Ultrafast neuronal imaging of dopamine dynamics with designed genetically encoded sensors., eaat, Science 360: , 4422. |
[146] | Liebmann T , Renier N , Bettayeb K , Greengard P , Tessier-Lavigne M , Flajolet M ((2016) ) Three-dimensional study of Alzheimer’s disease hallmarks using the iDISCO clearing method. Cell Rep 16: , 1138–1152. |
[147] | Romeo-Guitart D , Fores J , Herrando-Grabulosa M , Valls R , Leiva-Rodriguez T , Galea E , Gonzalez-Perez F , Navarro X , Petegnief V , Bosch A , Coma M , Mas JM , Casas C ((2018) ) Neuroprotective drug for nerve trauma revealed using artificial intelligence. Sci Rep 8: , 1879. |
[148] | Bryan RN ((2016) ) Machine learning applied to Alzheimer disease. Radiology 281: , 665–668. |
[149] | Wang RH , Sudhama A , Begum M , Huq R , Mihailidis A ((2017) ) Robots to assist daily activities: Views of older adults with Alzheimer’s disease and their caregivers. Int Psychogeriatr 29: , 67–79. |
[150] | Chen S , Weitemier AZ , Zeng X , He L , Wang X , Tao Y , Huang AJY , Hashimotodani Y , Kano M , Iwasaki H , Parajuli LK , Okabe S , Teh DBL , All AH , Tsutsui-Kimura I , Tanaka KF , Liu X , McHugh TJ ((2018) ) Near-infrared deep brain stimulation via upconversion nanoparticle-mediated optogenetics. Science 359: , 679–684. |
[151] | Raikwar SP , Bhagavan SM , Ramaswamy SB , Thangavel R , Dubova I , Selvakumar GP , Ahmed ME , Kempuraj D , Zaheer S , Iyer S , Zaheer A ((2018) ) Are tanycytes the missing link between type 2 diabetes and Alzheimer’s disease? Mol Neurobiol. doi: 10.1007/s12035-018-1123-8 . |
[152] | Akhtar MW , Sanz-Blasco S , Dolatabadi N , Parker J , Chon K , Lee MS , Soussou W , McKercher SR , Ambasudhan R , Nakamura T , Lipton SA ((2016) ) Elevated glucose and oligomeric beta-amyloid disrupt synapses via a common pathway of aberrant protein S-nitrosylation. Nat Commun 7: , 10242. |
[153] | Weiner MW , Harvey D , Hayes J , Landau SM , Aisen PS , Petersen RC , Tosun D , Veitch DP , Jack CR Jr , Decarli C , Saykin AJ , Grafman J , Neylanthe TC , Department of Defense Alzheimer’s Disease Neuroimaging Initiative ((2017) ) Effects of traumatic brain injury and posttraumatic stress disorder on development of Alzheimer’s disease in Vietnam Veterans using the Alzheimer’s Disease Neuroimaging Initiative: Preliminary report. Alzheimers Dement (N Y) 3: , 177–188. |
[154] | Lou D , Du Y , Huang D , Cai F , Zhang Y , Li T , Zhou W , Gao H , Song W ((2018) ) Traumatic brain injury alters the metabolism and facilitates Alzheimer’s disease in a murine model. Mol Neurobiol 55: , 4928–4939. |
[155] | Wendeln AC , Degenhardt K , Kaurani L , Gertig M , Ulas T , Jain G , Wagner J , Hasler LM , Wild K , Skodras A , Blank T , Staszewski O , Datta M , Centeno TP , Capece V , Islam MR , Kerimoglu C , Staufenbiel M , Schultze JL , Beyer M , Prinz M , Jucker M , Fischer A , Neher JJ ((2018) ) Innate immune memory in the brain shapes neurological disease hallmarks. Nature 556: , 332–338. |
[156] | Steeland S , Gorle N , Vandendriessche C , Balusu S , Brkic M , Van Cauwenberghe C , Van Imschoot G , Van Wonterghem E , De Rycke R , Kremer A , Lippens S , Stopa E , Johanson CE , Libert C , Vandenbroucke RE ((2018) ) Counteracting the effects of TNF receptor-1 has therapeutic potential in Alzheimer’s disease. EMBO Mol Med 10: , e8300. |
[157] | Sweeney MD , Sagare AP , Zlokovic BV ((2018) ) Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol 14: , 133–150. |
[158] | Montagne A , Zhao Z , Zlokovic BV ((2017) ) Alzheimer’s disease: A matter of blood-brain barrier dysfunction? J Exp Med 214: , 3151–3169. |
[159] | Ittner A , Chua SW , Bertz J , Volkerling A , van der Hoven J , Gladbach A , Przybyla M , Bi M , van Hummel A , Stevens CH , Ippati S , Suh LS , Macmillan A , Sutherland G , Kril JJ , Silva AP , Mackay JP , Poljak A , Delerue F , Ke YD , Ittner LM ((2016) ) Site-specific phosphorylation of tau inhibits amyloid-beta toxicity in Alzheimer’s mice. Science 354: , 904–908. |
[160] | Thangavel R , Bhagavan SM , Ramaswamy SB , Surpur S , Govindarajan R , Kempuraj D , Zaheer S , Raikwar S , Ahmed ME , Selvakumar GP , Iyer SS , Zaheer A ((2018) ) Co-expression of glia maturation factor and apolipoprotein E4 in Alzheimer’s disease brain. J Alzheimers Dis 61: , 553–560. |
[161] | Wang C , Najm R , Xu Q , Jeong DE , Walker D , Balestra ME , Yoon SY , Yuan H , Li G , Miller ZA , Miller BL , Malloy MJ , Huang Y ((2018) ) Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med 24: , 647–657. |
[162] | Liao F , Li A , Xiong M , Bien-Ly N , Jiang H , Zhang Y , Finn MB , Hoyle R , Keyser J , Lefton KB , Robinson GO , Serrano JR , Silverman AP , Guo JL , Getz J , Henne K , Leyns CE , Gallardo G , Ulrich JD , Sullivan PM , Lerner EP , Hudry E , Sweeney ZK , Dennis MS , Hyman BT , Watts RJ , Holtzman DM ((2018) ) Targeting of nonlipidated, aggregated apoE with antibodies inhibits amyloid accumulation. J Clin Invest 128: , 2144–2155. |
[163] | Liu CC , Zhao N , Fu Y , Wang N , Linares C , Tsai CW , Bu G ((2017) ) ApoE4 accelerates early seeding of amyloid pathology. Neuron 96: , 1024–1032 e1023. |
[164] | Nuriel T , Angulo SL , Khan U , Ashok A , Chen Q , Figueroa HY , Emrani S , Liu L , Herman M , Barrett G , Savage V , Buitrago L , Cepeda-Prado E , Fung C , Goldberg E , Gross SS , Hussaini SA , Moreno H , Small SA , Duff KE ((2017) ) Neuronal hyperactivity due to loss of inhibitory tone in APOE4 mice lacking Alzheimer’s disease-like pathology. Nat Commun 8: , 1464. |
[165] | Stockburger C , Eckert S , Eckert GP , Friedland-Leuner K , Muller WE ((2018) ) Mitochondrial function, dynamics, and permeability transition: A complex love triangle as a possible target for the treatment of brain aging and Alzheimer’s disease, J Alzheimers Dis 64: (s1), S455–S467. |
[166] | Sorrentino V , Romani M , Mouchiroud L , Beck JS , Zhang H , D’Amico D , Moullan N , Potenza F , Schmid AW , Rietsch S , Counts SE , Auwerx J ((2017) ) Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature 552: , 187–193. |
[167] | Reddy PH , Manczak M , Yin X , Reddy AP ((2018) ) Synergistic protective effects of mitochondrial division inhibitor 1 and mitochondria-targeted small peptide SS31 in Alzheimer’s disease. J Alzheimers Dis 62: , 1549–1565. |
[168] | Kondo T , Imamura K , Funayama M , Tsukita K , Miyake M , Ohta A , Woltjen K , Nakagawa M , Asada T , Arai T , Kawakatsu S , Izumi Y , Kaji R , Iwata N , Inoue H ((2017) ) iPSC-based compound screening and in vitro trials identify a synergistic anti-amyloid beta combination for Alzheimer’s disease. Cell Rep 21: , 2304–2312. |
[169] | Mattei C , Alshawaf A , D’Abaco G , Nayagam B , Dottori M ((2018) ) Generation of neural organoids from human embryonic stem cells using the rotary cell culture system: Effects of microgravity on neural progenitor cell fate. Stem Cells Dev 27: , 848–885. |
[170] | Lee CT , Bendriem RM , Wu WW , Shen RF ((2017) ) 3D brain Organoids derived from pluripotent stem cells: Promising experimental models for brain development and neurodegenerative disorders. J Biomed Sci 24: , 59. |
[171] | Hunsberger JG , Rao M , Kurtzberg J , Bulte JWM , Atala A , LaFerla FM , Greely HT , Sawa A , Gandy S , Schneider LS , Doraiswamy PM ((2016) ) Accelerating stem cell trials for Alzheimer’s disease. Lancet Neurol 15: , 219–230. |
[172] | Duncan T , Valenzuela M ((2017) ) Alzheimer’s disease, dementia, and stem cell therapy. Stem Cell Res Ther 8: , 111. |
[173] | Mano T , Nagata K , Nonaka T , Tarutani A , Imamura T , Hashimoto T , Bannai T , Koshi-Mano K , Tsuchida T , Ohtomo R , Takahashi-Fujigasaki J , Yamashita S , Ohyagi Y , Yamasaki R , Tsuji S , Tamaoka A , Ikeuchi T , Saido TC , Iwatsubo T , Ushijima T , Murayama S , Hasegawa M , Iwata A ((2017) ) Neuron-specific methylome analysis reveals epigenetic regulation and tau-related dysfunction of BRCA1 in Alzheimer’s disease. Proc Natl Acad Sci U S A 114: , E9645–E9654. |
[174] | Maze I , Shen L , Zhang B , Garcia BA , Shao N , Mitchell A , Sun H , Akbarian S , Allis CD , Nestler EJ ((2014) ) Analytical tools and current challenges in the modern era of neuroepigenomics. Nat Neurosci 17: , 1476–1490. |
[175] | Liu D , Tang H , Li XY , Deng MF , Wei N , Wang X , Zhou YF , Wang DQ , Fu P , Wang JZ , Hebert SS , Chen JG , Lu Y , Zhu LQ ((2017) ) Targeting the HDAC2/HNF-4A/miR-101b/AMPK pathway rescues tauopathy and dendritic abnormalities in Alzheimer’s disease. Mol Ther 25: , 752–764. |
[176] | Lee K , Kim H , An K , Kwon OB , Park S , Cha JH , Kim MH , Lee Y , Kim JH , Cho K , Kim HS ((2016) ) Replenishment of microRNA-188-5p restores the synaptic and cognitive deficits in 5XFAD mouse model of Alzheimer’s disease. Sci Rep 6: , 34433. |
[177] | Salta E , Sierksma A , Vanden Eynden E , De Strooper B ((2016) ) miR-132 loss de-represses ITPKB and aggravates amyloid and TAU pathology in Alzheimer’s brain. EMBO Mol Med 8: , 1005–1018. |
[178] | Ihry RJ , Worringer KA , Salick MR , Frias E , Ho D , Theriault K , Kommineni S , Chen J , Sondey M , Ye C , Randhawa R , Kulkarni T , Yang Z , McAllister G , Russ C , Reece-Hoyes J , Forrester W , Hoffman GR , Dolmetsch R , Kaykas A ((2018) ) p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat Med 24: , 939–946. |
[179] | Haapaniemi E , Botla S , Persson J , Schmierer B , Taipale J ((2018) ) CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response. Nat Med. doi: 10.1038/s41591-018-0049-z |
[180] | Rincon MY , de Vin F , Duque SI , Fripont S , Castaldo SA , Bouhuijzen-Wenger J , Holt MG ((2018) ) Widespread transduction of astrocytes and neurons in the mouse central nervous system after systemic delivery of a self-complementary AAV-PHP.B vector. Gene Ther 25: , 83–92. |
[181] | Trompetero A , Gordillo A , Del Pilar MC , Cristina VM , Bustos Cruz RH ((2018) ) Alzheimer’s disease and Parkinson’s disease: A review of current treatment adopting a nanotechnology approach. Curr Pharm Des 24: , 22–45. |
[182] | Asai H , Ikezu S , Tsunoda S , Medalla M , Luebke J , Haydar T , Wolozin B , Butovsky O , Kugler S , Ikezu T ((2015) ) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18: , 1584–1593. |
[183] | Lee SE , Hyun H , Park MR , Choi Y , Son YJ , Park YG , Jeong SG , Shin MY , Ha HJ , Hong HS , Choi MK , Im GS , Park EW , Kim YH , Park C , Kim EY , Park SP ((2017) ) Production of transgenic pig as an Alzheimer’s disease model using a multi-cistronic vector system. PLoS One 12: , e0177933. |
[184] | Jakobsen JE , Johansen MG , Schmidt M , Liu Y , Li R , Callesen H , Melnikova M , Habekost M , Matrone C , Bouter Y , Bayer TA , Nielsen AL , Duthie M , Fraser PE , Holm IE , Jorgensen AL ((2016) ) Expression of the Alzheimer’s disease mutations AbetaPP695sw and PSEN1M146I in double-transgenic Gottingen minipigs. J Alzheimers Dis 53: , 1617–1630. |
[185] | Reddy PH , Manczak M , Yin X , Grady MC , Mitchell A , Tonk S , Kuruva CS , Bhatti JS , Kandimalla R , Vijayan M , Kumar S , Wang R , Pradeepkiran JA , Ogunmokun G , Thamarai K , Quesada K , Boles A , Reddy AP ((2018) ) Protective effects of indian spice curcumin against amyloid-beta in Alzheimer’s disease. J Alzheimers Dis 61: , 843–866. |
[186] | Schmidt C ((2015) ) Mental health: Thinking from the gut. Nature 518: , S12–15. |
[187] | Smith PA ((2015) ) The tantalizing links between gut microbes and the brain. Nature 526: , 312–314. |
[188] | Villemagne VL , Dore V , Burnham SC , Masters CL , Rowe CC ((2018) ) Imaging tau and amyloid-beta proteinopathies in Alzheimer disease and other conditions. Nat Rev Neurol 14: , 225–236. |
[189] | Nakamura A , Kaneko N , Villemagne VL , Kato T , Doecke J , Dore V , Fowler C , Li QX , Martins R , Rowe C , Tomita T , Matsuzaki K , Ishii K , Ishii K , Arahata Y , Iwamoto S , Ito K , Tanaka K , Masters CL , Yanagisawa K ((2018) ) High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature 554: , 249–254. |
[190] | Frisoni GB , Boccardi M , Barkhof F , Blennow K , Cappa S , Chiotis K , Demonet JF , Garibotto V , Giannakopoulos P , Gietl A , Hansson O , Herholz K , Jack CR Jr. , Nobili F , Nordberg A , Snyder HM , Ten Kate M , Varrone A , Albanese E , Becker S , Bossuyt P , Carrillo MC , Cerami C , Dubois B , Gallo V , Giacobini E , Gold G , Hurst S , Lonneborg A , Lovblad KO , Mattsson N , Molinuevo JL , Monsch AU , Mosimann U , Padovani A , Picco A , Porteri C , Ratib O , Saint-Aubert L , Scerri C , Scheltens P , Schott JM , Sonni I , Teipel S , Vineis P , Visser PJ , Yasui Y , Winblad B ((2017) ) Strategic roadmap for an early diagnosis of Alzheimer’s disease based on biomarkers. Lancet Neurol 16: , 661–676. |


