
Increased Albumin Oxidation in Cerebrospinal Fluid and Plasma from Alzheimer’s Disease Patients
Abstract
Background:
Oxidative stress in the brain and peripheral systems is considered a major player in Alzheimer’s disease (AD). Albumin is the main transporter and the main extracellular antioxidant in the human body.
Objective:
Here we explore for the first time the oxidation status of cerebrospinal fluid (CSF) and plasma albumin in AD in comparison to healthy subjects.
Methods:
Plasma and CSF samples were obtained from mild-moderate AD patients and control healthy age-matched donors. Albumin redox state forms (reduced: HMA; reversibly oxidized: HNA1; irreversibly oxidized: HNA2) were determined by HPLC. Albumin post-translational modifications (PTM) analysis was performed by mass spectrometry.
Results:
HPLC showed less HMA in AD plasma than in controls (54.1% versus 65.2% ; p < 0.0001), mainly at expense of HNA1 (42.8% versus 32.5% ; p < 0.0001). In AD CSF, HMA was drastically decreased compared to controls (9.6% versus 77.4% ; p < 0.0001), while HNA2 was increased (52.8% versus 7.4% ; p < 0.0001). In AD patients but not in healthy controls, CSF albumin was much more irreversibly oxidized than in plasma (close to 20-fold increase in HNA2). PTM analysis showed that AD CSF albumin samples behave as a differentiated cluster, thus confirming the albumin oxidative pattern observed by HPLC.
Conclusion:
CSF albumin oxidation in AD patients was dramatically increased comparing to healthy controls, while in plasma this increase was smaller. CSF albumin in AD patients was much more oxidized than in plasma, but this effect was not observed in healthy controls. These results suggest that albumin oxidation, especially in CSF, and its role in AD deserves further investigation.
INTRODUCTION
Alzheimer’s disease (AD) is the most frequent cause of neurodegenerative dementia worldwide. The neuropathology hallmarks of AD include the characteristic formation of senile plaques (due to abnormal extracellular deposition of amyloid-β (Aβ) protein), neurofibrillary tangles (due to intraneuronal accumulation of hyperphosphorylated tau protein), cerebral neuroinflammation mediated by microglia activation, and development of Lewy bodies (in 50–60% of AD cases) [1].
Additionally, oxidative stress in the brain and peripheral nervous system has been associated with AD pathogenesis, which is related to the excess Aβ and hyperphosphorylated tau [2]. Oxidative stress on nervous tissue involves the generation of excess reactive oxygen species (ROS)—free radicals deriving from molecular oxygen—that may seriously damage the brain, especially when there is an imbalance between this reactive species and antioxidant defenses. ROS can react with a variety of biomolecules and may damage their structures and functions [3].
In AD, oxidative stress triggers oxidative modification of different proteins in the brain, with the dysfunction of these proteins likely related to the pathology [3–5]. Consistently, in cerebrospinal fluid (CSF) obtained from AD patients, different oxidative stress markers (protein carbonylation and nitration, and lipid peroxidation) have increased [4]. By proteomic approaches, oxidized forms of specific proteins have been identified in CSF from AD (i.e., apolipoprotein E and alpha-1-antitrypsin among others) [4, 6, 7]. Similarly, in plasma from AD patients, elevated levels of protein oxidation markers have been also observed, including albumin [4, 8–10].
Albumin is the most abundant protein in the human body, including both plasma and CSF, and beyond its role as volume expander and transporter, albumin is the main extracellular antioxidant [11]. Such antioxidant capacity mainly relies on its Cys34 residue that can be transformed into more oxidized forms, preventing the oxidation of other entities [12]. According to the oxidation status of the thiol group (-SH) at the Cys34 residue, 3 forms of albumin with decreasing antioxidant capacity can be distinguished: reduced (human mercaptoalbumin; HMA), reversibly oxidized (human nonmercaptalbumin 1; HNA1) and irreversibly oxidized (human nonmercaptalbumin 2; HNA2) [13]. In addition to this scavenger activity, metal-binding properties of albumin also contribute to its antioxidant activity.
In some diseases, such as acute-on-chronic liver failure, an impairment of albumin non-oncotic capacities has been described (i.e., antioxidant and binding capacity). This led to the concept of ‘effective albumin concentration’ which is dependent upon the functional characteristics of albumin rather than its quantity [14]. Likewise, during aging and in some pathological conditions such as mild cognitive impairment and AD where oxidative stress especially in brain is thought to be central, plasma albumin has been found to be more oxidized [9, 11]. Nevertheless, the redox state of albumin in CSF has never been addressed so far.
In the current study, the status of CSF and plasma albumin oxidation in AD has been explored for the first time by conducting a compared characterization in AD patients and age-matched healthy donors.
MATERIAL AND METHODS
Sampling
Samples were baseline specimens provided by the following centers participating in the clinical trial EudraCT #2007-000414-36 (which results have been published elsewhere [15, 16]): ACE Foundation – Catalan Institute of Applied Neurosciences (Barcelona, Spain), University Hospital Gregorio Marañón (Madrid, Spain), Alzheimer’s Research Corporation – Mid Atlantic Geriatric Association (Manchester, NJ, USA) and Howard University (Washington, DC, USA).
Plasma (n = 37) and CSF (n = 34) samples were obtained from 39 mild-moderate AD patients (aged 53–78; with NINCDS-ADRDA criteria for probable AD, and Mini-Mental State Examination (MMSE) scores between ≥18 and ≤26; and CSF Aβ 42 levels within the reference values for AD diagnosis [17, 18]). In 32 cases, plasma and CSF samples came from the same subject (5 plasma samples had no paired CSF; 2 CSF samples had no paired plasma).
Control samples of plasma (n = 31, aged 48–81) and CSF (n = 16, aged 65–81) from healthy (not diagnosed with AD) age-matched subjects were kindly supplied by ACE Foundation – Catalan Institute of Applied Neurosciences (Barcelona, Spain) from their sample repository or purchased from Sera Laboratories International Ltd (West Sussex, UK). A subset of these control samples consisting of plasma and CSF paired specimens both obtained from the same subjects (n = 6, aged 66–81) were therefore used to compare plasma and CSF results in controls.
All donors gave their informed consent in compliance with the Code of Ethics of the World Medical Association (Declaration of Helsinki) for studies involving humans.
Plasma and CSF collection procedures
The drawing of blood and lumbar puncture were carried out following the standard techniques of each center, with minor variations among them. Briefly, plasma samples of up to 10 mL were obtained after collecting peripheral blood in EDTA tubes. After gently mixing, the tubes were centrifuged at >1000 g for 15 min to separate the plasma that was aliquoted in cryotubes at a maximum rate of 1 mL and immediately stored at ≤–70°C until analysis. CSF samples of up to 15 mL were collected on poly-propylene tubes by using a routine lumbar puncture. The tubes were centrifuged if necessary at 2000 g for 10 min at 4°C and aliquoted at a maximum rate of 1 mL in cryotubes. Once collected, CSF samples were immediately stored at ≤– 70°C until analysis.
Experimental design
The concentration of albumin in plasma and CSF of all samples was measured by standard laboratory tests and verified to be similar in controls and AD patients. In addition, the albumin quotient (CSF albumin / plasma albumin) was calculated to confirm the correct function and integrity of the blood-brain barrier (BBB) to be below the threshold of 8, considered adequate for patients older than 40 years [19–21].
The redox state of albumin in the study samples was analyzed within the following comparisons: 1) Plasma: AD patients versus controls (unpaired samples); 2) CSF: AD patients versus controls (unpaired samples); 3) AD patients: Plasma versus CSF (paired samples); and 4) Controls: Plasma versus CSF (paired samples).
The different albumin redox state forms were determined in plasma and CSF by anionic exchange high performance liquid chromatography (HPLC), while the determination of albumin post-translational modifications (PTMs) in terms of their identification and quantification were performed by means of mass spectrometry technique (MS).
Anion exchange high performance liquid chromatography (HPLC)
To assess the redox state of albumin in plasma and CSF, anionic exchange HPLC (Waters, Milford, MA) with the Shodex-Asahipak ES-502N column (Showa Denko Europe GmbH, Munich, Germany) coupled to a fluorescent detector (Waters) was used [22, 23]. Thawed samples were filtered through 0.22μm and were immediately placed in the carousel of the HPLC apparatus. All the samples were processed in parallel to the albumin oxidation controls to ensure sample stability during analysis.
Three albumin fractions were identified according to the oxidation status of the thiol group at the Cys34 albumin residue: HMA, HNA1 and HNA2 forms. Each form was normalized to the total amount of albumin in each sample; their percentage was quantified based on peak heights (dividing the peak height of each form by the sum of peaks height of all the albumin forms and multiplying them by 100) using the Empower software (Waters).
Liquid chromatography-mass spectrometry analysis
Albumin PTMs were assessed on the intact mass protein (top-down) by using ultra high-performance liquid chromatography coupled to electrospray ionization mass spectrometer (LC-ESI-qTOF-MS) [24].
For plasma, albumin-enriched samples were obtained by Cibacron Blue dye (Pierce Albumin Depletion Kit; Thermo Fisher Scientific Inc., Waltham, MA, USA) diluted with ultra-pure water (0.1% formic acid) and separated by liquid chromatography (Agilent 1290 Infinity UHPLC, Agilent Technologies, Santa Clara, CA, USA). In the case of CSF, samples were dialyzed just before chromatography. MS analysis was subsequently carried out on a Quadrupole Time of Flight analyzer with electrospray ionization source (ESI) (Agilent qTOF 6550 Jet Stream, Agilent Technologies). Albumin oxidation controls were processed in parallel to samples to ensure sample stability during analysis. Mass chromatograms were recorded as total ion current (TIC) within the mass range 1180–1500 m/z (mass/charge). ESI mass spectra were deconvoluted by using Agilent BioConfirm 7.0 software (Agilent Technologies), normalized and finally aligned with GeneSpring 13.1 software (Agilent Technologies). Isoform relative intensity (% ) was calculated by dividing the isoform absolute intensity by the sum of all isoform absolute intensities, expressed as percentage [25]. For every particular analysis, a filter of frequency was applied, thus considering only those isoforms presented in all samples in at least one group.
The identification of albumin PTMs was performed as previously described [25, 26] by analyzing 60,000–75,000 Da mass range and comparing the theoretical mass with the experimental value, allowing a mass drift of 10 Da (Agilent BioConfirm software).
Statistical analysis
Variables are expressed as the mean and standard deviation (SD) or median and interquartile range (IQR) as appropriate. Paired or unpaired Student’s t-tests (or Wilcoxon / Mann-Whitney tests, respectively) were used for the comparisons. Benjamini-Hochberg’s correction for multiple comparisons was applied when comparing differences in albumin PTMs relative abundance between groups.
Principal Component Analysis (PCA) and unsupervised hierarchical cluster analysis (HCA) multivariant techniques were used to identify a pattern between samples based on aligned, normalized and log2 transformed MS data. HCA of all PTMs was performed with Euclidian correlation distance, followed by dendrogram formation using Ward’s linkage. Analyses were conducted using Mass Profiler Professional (v. 14.5) from Agilent Technologies.
In order to examine the magnitude of the differences in albumin oxidation status between AD patients and healthy controls, the effect sizes based on standardized differences between mean scores were calculated (Cohen’s delta, [27]). As previously described [27], effect sizes of 0.20, 0.50, and 0.80 were considered small, medium, and large, respectively.
Software used for charting and calculations was Microsoft Excel 2010 (Microsoft Corporation, Redmond, WA, USA), Graph-Pad Prism v6 (San Diego, CA, USA) and GeneSpring 13.1 (Agilent Technologies).
RESULTS
Albumin oxidation assessed by HPLC
The concentration of total albumin in controls and AD patients was similar in plasma (36.9±5.6 mg/mL and 39.1±5.7 mg/mL, respectively), as well as in CSF (0.230±0.173 mg/mL and 0.260±0.190 mg/mL, respectively). Percentages of the three forms of albumin redox state (HMA, HNA1 and HNA2) assessed by HPLC are summarized in Fig. 1. Globally, it can be observed that albumin was maximally oxidized in AD CSF (lowest HMA and highest HNA2).
Fig.1
Albumin oxidation in plasma and cerebrospinal fluid (CSF) from controls (healthy age-matched donors) and Alzheimer’s disease (AD) patients. The subfigures show HPLC analysis of the reduced albumin form HMA, the reversible oxidized form HNA1 and the irreversible oxidized form HNA2 in plasma (a) and CSF (b) samples. Data are shown as median±interquartile range. Unpaired t test ( ***p < 0.001; ****p < 0.0001; AD versus control).
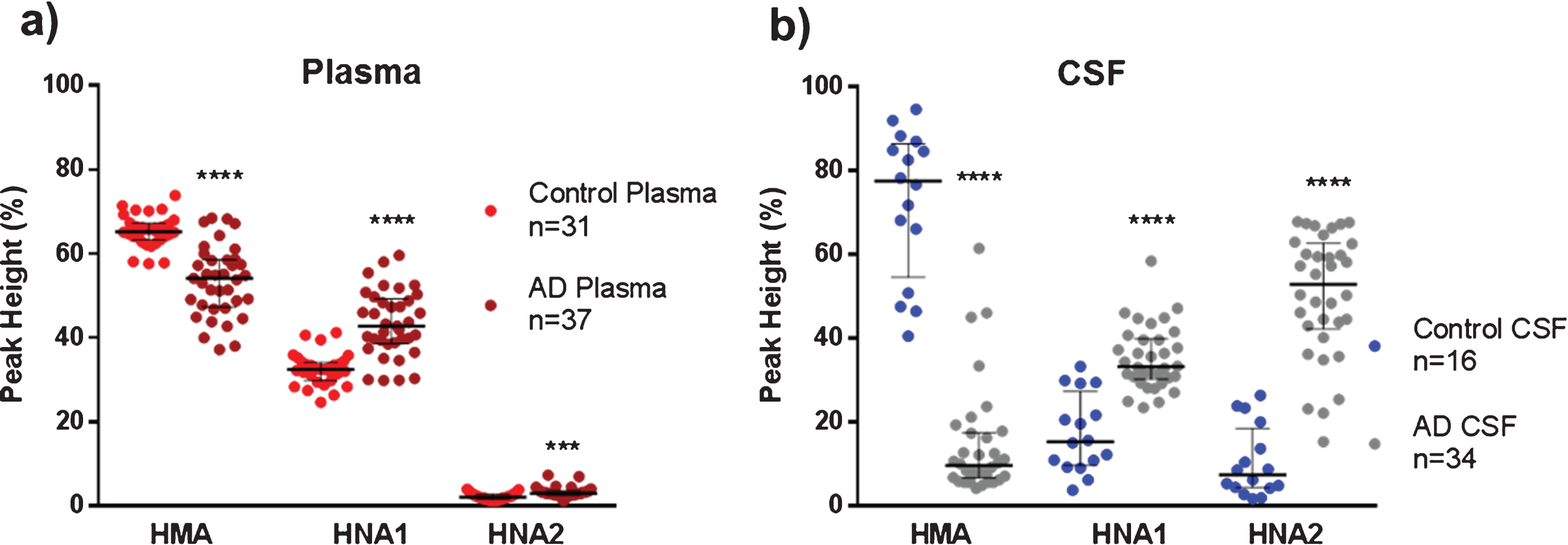
Plasma albumin from AD patients showed a moderate but significant reduced form HMA than healthy controls (54.1% versus 65.2% , p < 0.0001, Cohen’s delta: 1.8, Fig. 1a), mainly at the expense of the increased reversible oxidation form HNA1 (42.8% versus 32.5% , p < 0.0001, Cohen’s delta: 1.7, Fig. 1a). Interestingly, CSF albumin from AD patients showed a marked decrease in the reduced form HMA in comparison to controls (Fig. 1b), with both the reversible and the irreversible oxidation forms increased (Fig. 1b). Of note, in AD CSF, HMA was around 8-fold lower (9.6% versus 77.4% , p < 0.0001, Cohen’s delta: 3.9) and HNA2 was around 7-fold higher (52.8% versus 7.4% , p < 0.0001, Cohen’s delta: 3.1) when compared to controls (Fig. 1b).
When comparing paired plasma and CSF albumin oxidation (from the same subject) within groups, CSF albumin from AD patients was found much more oxidized than in plasma (Table 1). A marked decrease in the reduced fraction (HMA) and a close to 20-fold increase in the irreversible oxidized form HNA2 can be observed (Table 1). Conversely, this effect was not found in healthy controls, where the reduced albumin form was increased in CSF with respect to plasma (Table 1).
Table 1
Albumin oxidation in paired plasma and CSF from controls (healthy age-matched donors) and Alzheimer’s disease (AD) patients assessed by HPLC analysis. The percentages of the reduced (HMA), reversible oxidized (HNA1), and irreversible oxidized (HNA2) forms of albumin, in plasma and cerebrospinal fluid (CSF) samples from the same subject, are shown (controls n = 6 and AD n = 32; median± interquartile range)
| Plasma | CSF | p value | Cohen’s delta | |
| Controls | ||||
| HMA | 65.5% [63.3% – 66.2%] | 86.4% [79.8% – 92.6%] | 0.0313 | 3.4 |
| HNA1 | 32.2% [31.4% – 34.3%] | 10.0% [5.6% – 14.1%] | 0.0313 | 5.1 |
| HNA2 | 2.6% [2.2% – 2.8%] | 3.8% [1.9% – 6.2%] | 0.4375 | 0.9 |
| AD | ||||
| HMA | 54.0% [48.1% – 58.5%] | 9.6% [6.3% – 17.0%] | <0.0001 | 4.3 |
| HNA1 | 43.1% [39.0% – 48.7%] | 33.2% [30.6% – 40.4%] | 0.0003 | 1.0 |
| HNA2 | 2.9% [2.5% – 3.3%] | 52.8% [43.2% – 62.8%] | <0.0001 | 5.0 |
Global MS data analysis
Figure 2 shows representative mass spectra of plasma and CSF samples from AD patients and controls. The deconvoluted ESI mass spectra of albumin were obtained from the multicharged mass spectra to enable the identification of different proteinisoforms.
As shown in Fig. 2, the following PTMs were identified: Native albumin (Alb-SH; a representative form of HMA fraction with the reduced thiol group, 66,440 Da), Alb+Cys (a representative form of HNA1 fraction, with cysteinilation of Cys34 through a reversible disulphide bond, +119 Da), and Alb+SO2H (a representative form of HNA2 fraction involving the oxidation of Cys34 to sulfinic acid, +32 Da). In addition to the forms explained above, the method allowed the tentative identification of different modifications such as other oxidized forms (i.e., Alb+SO3H), glycated (Alb+Glyc) and truncated (Alb-DA) isoforms.
Fig.2
Representative deconvoluted mass spectra from controls (healthy age-matched donors) and Alzheimer’s disease (AD) patients (in both plasma and cerebrospinal fluid [CSF]). In addition to the native albumin (Alb-SH), cysteinylation of the Cys34 residue (Alb+Cys) and sulfinylation of the Cys34 residue (Alb+SO2H); the following structural alterations were detected: truncation of the last two aminoacid residues at the N-terminal (Alb-DA); glycation (Alb+Glyc) and the sulfonylation of the Cys34 residue (Alb+SO3H).
![Representative deconvoluted mass spectra from controls (healthy age-matched donors) and Alzheimer’s disease (AD) patients (in both plasma and cerebrospinal fluid [CSF]). In addition to the native albumin (Alb-SH), cysteinylation of the Cys34 residue (Alb+Cys) and sulfinylation of the Cys34 residue (Alb+SO2H); the following structural alterations were detected: truncation of the last two aminoacid residues at the N-terminal (Alb-DA); glycation (Alb+Glyc) and the sulfonylation of the Cys34 residue (Alb+SO3H).](https://content.iospress.com:443/media/jad/2018/63-4/jad-63-4-jad180243/jad-63-jad180243-g002.jpg)
Importantly, the MS analysis of albumin PTMs in AD CSF revealed a distinctive mass spectra profile, which was mainly related to those peaks corresponding to albumin oxidation isoforms (Alb+Cys, Alb+SO2H and Alb+SO3H) (Fig. 2).
Principal component analysis (PCA) scores based on 17 albumin PTMs are shown in Fig. 3a. The Component 1 versus Component 2 plot showed that samples from the same group clustered together, showing a good separation between CSF and plasma albumin from AD patients as well as from CSF albumin from controls, while plasma albumins from both AD and controls would present some similarities (Fig. 3a). The Component 1 and the Component 2 accounted for 35.67% and the 25.79% of the original observation variance, respectively. An additional PCA analysis only based on 4 albumin oxidation related PTMs (Alb-SH, Alb+Cys, Alb+SO2H and Alb+SO3H) showed similar separation, its two principal components accounting for a total variance of 93.42% (data not shown).
Fig.3
Scores from Principal Component Analysis (PCA) decomposition (a) and hierarchical clustering analysis (b) of mass spectra of albumin post-translational modifications (PTMs) to compare within- and between-group variation datasets.
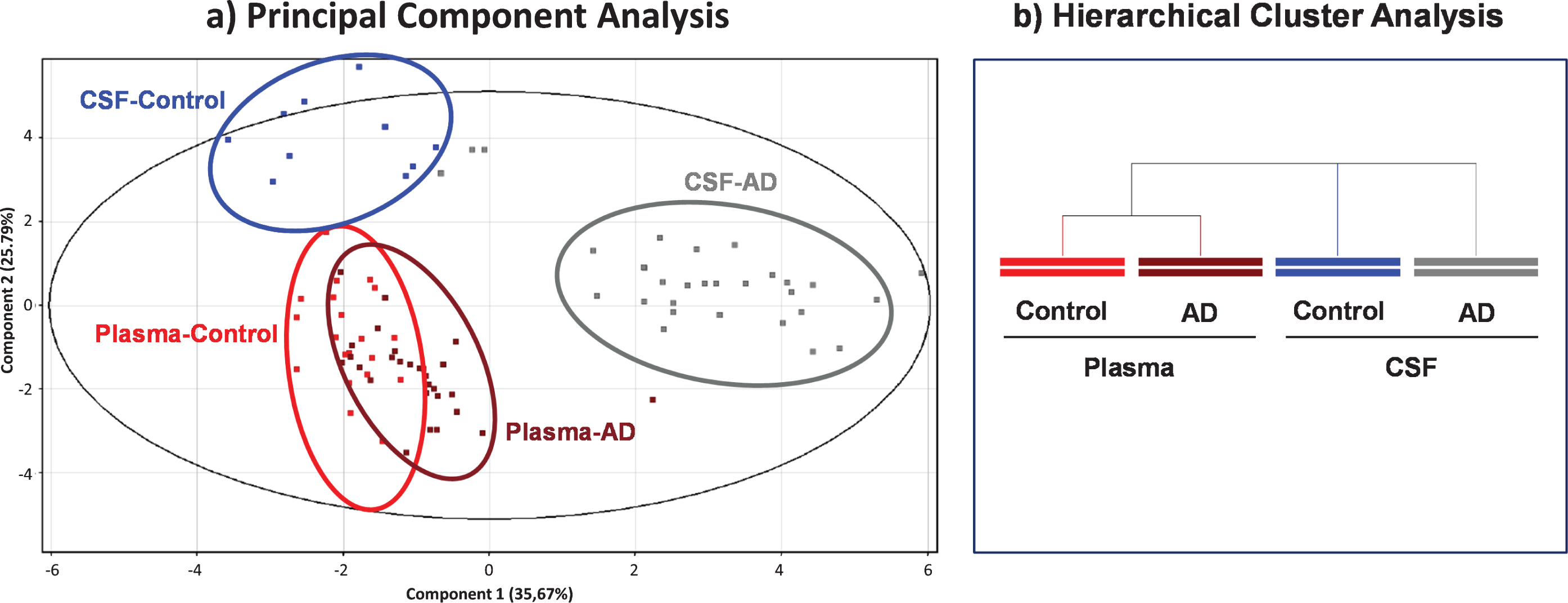
Consistently, hierarchical clustering (Fig. 3b) highlighted that plasma samples from AD and controls are more similar than CSF samples, either from AD or controls, whose PTM profile would differ much more.
Albumin oxidation assessed by MS
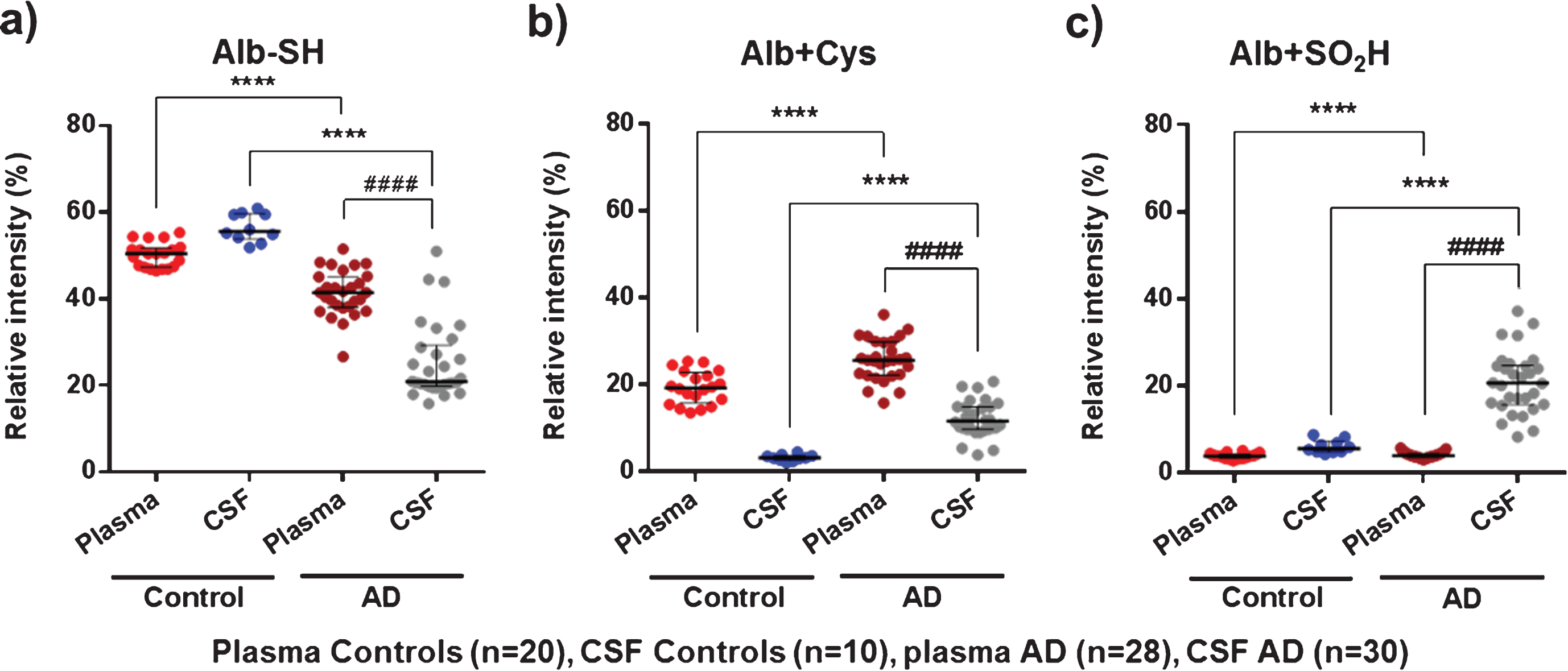
When focusing on the representative albumin oxidation forms obtained by MS (Alb-SH, Alb+Cys and Alb+SO2H), the analysis of these modifications confirmed the albumin oxidative pattern observed by HPLC analysis (see Fig. 4).
Plasma AD albumin showed a significantly higher oxidation profile than plasma albumin from controls (lower Alb-SH and higher Alb+Cys; 41.6% versus 50.5% , p < 0.0001, Cohen’s delta: 2.0, and 25.8% versus 19.4% , p < 0.0001, Cohen’s delta: 1.4 (respectively); Fig. 4a, b). In CSF, AD albumin is much more oxidized than in controls. Specifically, Alb+Cys (11.8% versus 3.3% , p < 0.0001, Cohen’s delta: 2.5) and especially Alb+SO2H (20.7% versus 5.7, p < 0.001, Cohen’s delta: 2.4) are markedly increased at the expense of Alb-SH form (21.0% versus 55.6% , p < 0.0001, Cohen’s delta: 4.0) (Fig. 4). Again, comparisons between CSF and plasma albumin within the same group of AD patients showed increased albumin oxidation modifications (lower Alb-SH and increased Alb+SO2H) in CSF(Fig. 4a, c).
Fig.4
MS results of representative albumin oxidation forms (Alb-SH, Alb+Cys, and Alb+SO2H) in plasma and cerebrospinal fluid (CSF) from controls (healthy age-matched donors) and Alzheimer’s disease (AD) patients. The subfigures show relative intensities (% ) of the native albumin (Alb-SH) (a), the cysteinylation of the Cys34 residue (Alb+Cys) (b), and sulfinylation of the Cys34 residue (Alb+SO2H) (c). Data are shown as median±interquartile range. Unpaired t test: ( ****p < 0.0001; AD versus Control); Paired t test: ( # # # # p < 0.0001; AD CSF versus AD Plasma).
DISCUSSION
Oxidative stress in AD patients induces the dysfunction of a variety of proteins in the brain and blood, including albumin [4, 6, 8, 9, 11]. Albumin is the main extracellular antioxidant in the human body as well as the most abundant protein in CSF and plasma; therefore, characterization of albumin oxidation status in these body compartments may provide key insights to the understanding of AD pathogenesis and involvement of oxidative stress. In our study, we found that globally, albumin was more oxidized in AD patients than in healthy controls, and this effect was much stronger in CSF than in plasma.
Different studies regarding protein oxidation in AD patients have been conducted over the past several years. Most of these studies found that in CSF, there is an increase in protein modifications by oxidation, such as in coenzyme Q-10 [6, 7]. By contrast, in other studies the protein studied, such as transthyretin, is less oxidized in AD patients [28]. However, all the proteins described in these papers are present in lower levels than albumin. When examining AD patients versus healthy controls in our study, we observed that plasma albumin was more oxidized in AD patients than in controls in around 10 percentage points, mainly due to an increase in the reversible oxidized albumin form HNA1. These results confirm those described previously by Greilbelger et al. [9], showing an increase in plasma albumin disulphide (Alb-SSR, also called HNA1) in patients with AD and mild-cognitive impairment, based on a similar HPLC approach. Oxidative damage to plasma lipids and proteins has been described as an important event in the pathogenesis of neurodegenerative diseases such as AD. Although plasma Alb-SSR can be a useful indicator of oxidative stress, it is not considered a specific marker for neurodegenerative disease such as AD [9].
In CSF, albumin was also more oxidized in AD patients than in controls, but this time much more evident; the irreversibly oxidized albumin (HNA2) was predominant in AD patients, whereas in controls the predominant form is the reduced one (HMA). Previous studies in subjects of different ages, including an older group with a similar age range than our subjects (mean±SD: 65.4±8.3), reported results of albumin oxidation similar to those observed in our controls, using an HPLC technique [29]. This would support the validity of our control measurements. In addition, to our knowledge no previous data exists on CSF albumin oxidation status in AD patients, where oxidative stress seems to play a key role especially in the brain. Nevertheless, an increase of CSF albumin oxidation has been described in patients undergoing ischemic stroke, a known trigger of oxidative stress [30].
This is the first study in which albumin intact mass LC-MS analysis is applied to AD patients. The global MS analysis (based on PCA and hierarchical clustering) of albumin post-translational modifications confirmed that CSF from AD patients and controls formed clearly differentiated clusters. In addition, the analysis of discrete post-translational modifications related to albumin oxidation (Alb-SH, Alb+Cys and Alb+SO2H) confirmed the albumin oxidation status shown by HPLC analysis, both in plasma and CSF of AD patients and controls.
When focusing on the albumin oxidation status of CSF versus plasma within each group (AD patients and healthy controls), the most striking point was the significant decrease observed in albumin reduced form HMA within AD patients, which was not shown in healthy controls. This different degree of oxidation in both fluids from AD patients suggests that the communication of both compartments is restricted, with a higher oxidative environment present in the brain and peripheral nervous system than in plasma.
Regarding study limitations, the cohorts of subjects studied were relatively small (particularly in the paired plasma-CSF comparison of healthy controls), between 6 and 37 individuals. This could be especially relevant in PCA analysis where the combination of multiple variables and a reduced sample size might cause an overfitting; however, it is important to note that we have been able to reproduce the results considering only four variables. Nevertheless, a larger sample should be tested to confirm our results, as well as the analysis of samples from other neurodegenerative and non-neurodegenerative disorders.
Aside from these limitations, these results may be of particular interest since one of the major concerns in AD is the need for a test or procedure with diagnostic or theragnostic power. In fact, albumin oxidized forms (HNA1 and HNA2, alone or in combination) have been described as markers of systemic oxidative stress in different pathologies [22, 31, 32].
In patients with probable AD, a pathophysiologic imbalance in favor of oxidants against antioxidants has been shown in blood/plasma [8–10, 33–36] as well as in CSF/brain [4, 6, 7]. Due to is closeness to the central nervous system, CSF is the most logical source to look for biomarkers directly related to AD pathology, although recognizing that its collection is invasive. Even accepting that the CSF volume is relatively small and with a low protein concentration, albumin is the most abundant protein in CSF (around 0.2 mg/ml) [37] and the main extracellular antioxidant. Nevertheless, studies of albumin PTMs, oxidation status or functional roles in CSF of AD patients are lacking. In our study, the analysis of albumin oxidation status as well as the study of PTMs showed a very marked oxidation of albumin in CSF. If this albumin oxidation status is a marker of oxidative stress in AD patients, that could in turn reflect either disease stages, or has theragnostic potential, it deserves further investigation. Albumin is the main extracellular antioxidant in both plasma and CSF, and our results indicate that in these two compartments albumin from AD patients has a reduced antioxidant activity, being much more altered in the CSF.
CSF albumin comes mainly from plasma, where it is synthesized by the liver in its native form, although it may also be directly originated in the microglia [38, 39]. After secretion and under an oxidative environment, albumin can be transformed into more oxidized forms, having an impact on albumin distribution and metabolism: albumin transporters mainly recognize native albumin whereas an increased catabolism has been described for modified albumins via specific scavenger receptors [39]. Therefore, it can be deduced that the oxidation process observed in AD CSF albumin is undergone inside the brain. To our knowledge, it is uncertain if this CSF oxidized albumin in the brain has a pathogenic function in AD, apart from those derived from the oxidative stress unbalance. Likewise, an increase in the reversible oxidized albumin form, both in CSF and plasma, has been observed in our study, which deserves further investigation taking into account that some pro-inflammatory effects of the reversible oxidized albumin have been recently described [40]. All in all, understanding the role of albumin in the pathobiology of AD, not only in context of oxidative stress and oxidation imbalance but also taking into account its role as a carrier protein, may be useful for the design of new therapies.
Also, it is worth mentioning that different approaches, including plasma-based therapies, are under investigation in AD. Among them, a new therapeutic approach including plasma exchange (removal of the patient’s plasma that may carry known and unknown pathological factors such as the amyloid-β bound to albumin) and replacement with therapeutic albumin (Albutein ®, Grifols) is being evaluated. Previous Pilot/Phase II studies using this approach showed encouraging results [15, 16] that lead to the initiation of a Phase IIB/III study (AMBAR trial, NCT01561053).
In conclusion, results from the present study have shown for the first time that CSF albumin of AD patients was significantly more oxidized than in healthy subjects. Such effect was less evident in plasma albumin. These data confirm that oxidative stress is crucially involved in AD, thus supporting the need for further research to better understand the role of albumin in this pathology.
ACKNOWLEDGMENTS
The investigators thank patients and donors for their indispensable contribution. Araclon Biotech S.L. (Zaragoza, Spain) is acknowledged for scientific support. Pol Herrero and Núria Canela (Centre for Omic Sciences, Eurecat, Universitat Rovira i Virgili, Reus, Spain) are acknowledged for the MS analysis support. Carlota Gelabert, Eva Vior, Jordi Vidal, Santiago Garcia, Aida Raventós and Francisca Doncel (Grifols) are acknowledged for their expert technical assistance. Jordi Bozzo PhD, CMPP is acknowledged for medical writing and editorial assistance in the preparation of the manuscript.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0243).
REFERENCES
[1] | Mrak RE ((2009) ) Neuropathology and the neuroinflammation idea. J Alzheimers Dis 18: , 473–481. |
[2] | Melov S , Adlard PA , Morten K , Johnson F , Golden TR , Hinerfeld D , Schilling B , Mavros C , Masters CL , Volitakis I , Li QX , Laughton K , Hubbard A , Cherny RA , Gibson B , Bush AI ((2007) ) Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS One 2: , e536. |
[3] | Barnham KJ , Masters CL , Bush AI ((2004) ) Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov 3: , 205–214. |
[4] | Di Domenico F , Coccia R , Butterfield DA , Perluigi M ((2011) ) Circulating biomarkers of protein oxidation for Alzheimer disease: Expectations within limits. Biochim Biophys Acta 1814: , 1785–1795. |
[5] | Zhao Y , Zhao B ((2013) ) Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid Med Cell Longev 2013: , 316523. |
[6] | Di Domenico F , Pupo G , Giraldo E , Badia MC , Monllor P , Lloret A , Schinina ME , Giorgi A , Cini C , Tramutola A , Butterfield DA , Vina J , Perluigi M ((2016) ) Oxidative signature of cerebrospinal fluid from mild cognitive impairment and Alzheimer disease patients. Free Radic Biol Med 91: , 1–9. |
[7] | Isobe C , Abe T , Terayama Y ((2009) ) Increase in the oxidized/total coenzyme Q-10 ratio in the cerebrospinal fluid of Alzheimer’s disease patients. Dement Geriatr Cogn Disord 28: , 449–454. |
[8] | Altunoglu E , Guntas G , Erdenen F , Akkaya E , Topac I , Irmak H , Derici H , Yavuzer H , Gelisgen R , Uzun H ((2015) ) Ischemia-modified albumin and advanced oxidation protein products as potential biomarkers of protein oxidation in Alzheimer’s disease. Geriatr Gerontol Int 15: , 872–880. |
[9] | Greilberger J , Koidl C , Greilberger M , Lamprecht M , Schroecksnadel K , Leblhuber F , Fuchs D , Oettl K ((2008) ) Malondialdehyde, carbonyl proteins and albumin-disulphide as useful oxidative markers in mild cognitive impairment and Alzheimer’s disease. Free Radic Res 42: , 633–638. |
[10] | Puertas MC , Martinez-Martos JM , Cobo MP , Carrera MP , Mayas MD , Ramirez-Exposito MJ ((2012) ) Plasma oxidative stress parameters in men and women with early stage Alzheimer type dementia. Exp Gerontol 47: , 625–630. |
[11] | Colombo G , Clerici M , Giustarini D , Rossi R , Milzani A , Dalle-Donne I ((2012) ) Redox albuminomics: Oxidized albumin in human diseases. Antioxid Redox Signal 17: , 1515–1527. |
[12] | Cha MK , Kim IH ((1996) )Biochem Biophys Res Commun 222: , 619–625 Glutathione-linked thiol peroxidase activity of human serum album, Aossible antioxidant role of serum albumin in blood plasma. |
[13] | Kawai K , Hayashi T , Matsuyama Y , Minami T , Era S ((2010) ) Difference in redox status of serum and aqueous humor in senile cataract patients as monitored via the albumin thiol-redox state. Jpn J Ophthalmol 54: , 584–588. |
[14] | Garcia-Martinez R , Caraceni P , Bernardi M , Gines P , Arroyo V , Jalan R ((2013) )Hepatology 58: , 1836–1846 Album, athophysiologic basis of its role in the treatment of cirrhosis and its complications. |
[15] | Boada M , Anaya F , Ortiz P , Olazaran J , Shua-Haim JR , Obisesan TO , Hernandez I , Munoz J , Buendia M , Alegret M , Lafuente A , Tarraga L , Nunez L , Torres M , Grifols JR , Ferrer I , Lopez OL , Paez A ((2017) ) Efficacy and safety of plasma exchange with 5% albumin to modify cerebrospinal fluid and plasma amyloid-beta concentrations and cognition outcomes in Alzheimer’s disease patients: A multicenter, randomized, controlled clinical trial. J Alzheimers Dis 56: , 129–143. |
[16] | Cuberas-Borrós G , Roca I , Boada M , Tárraga L , Hernández I , Buendia M , Rubio L , Torres G , Bittini A , Guzmán de Villoria J , Pujadas F , Torres M , Núñez L , Castell J , Páez A ((2018) ) Longitudinal neuroimaging analysis in mild-moderate Alzheimer’s disease patients treated with plasma exchange with 5% human albumin. J Alzheimers Dis 61: , 321–332. |
[17] | Sjogren M , Davidsson P , Gottfries J , Vanderstichele H , Edman A , Vanmechelen E , Wallin A , Blennow K ((2001) ) The cerebrospinal fluid levels of tau, growth-associated protein-43 and soluble amyloid precursor protein correlate in Alzheimer’s disease, reflecting a common pathophysiological process. Dement Geriatr Cogn Disord 12: , 257–264. |
[18] | Hulstaert F , Blennow K , Ivanoiu A , Schoonderwaldt HC , Riemenschneider M , De Deyn PP , Bancher C , Cras P , Wiltfang J , Mehta PD , Iqbal K , Pottel H , Vanmechelen E , Vanderstichele H ((1999) ) Improved discrimination of AD patients using beta-amyloid(1-42) and tau levels in CSF. Neurology 52: , 1555–1562. |
[19] | Andersson M , Alvarez-Cermeno J , Bernardi G , Cogato I , Fredman P , Frederiksen J , Fredrikson S , Gallo P , Grimaldi LM , Grønning M , Keir G , Lamers K , Link H , Magalhães A , Massaro AR , Öhman S , Reiber H , Rönnbäck L , Schluep M , Schuller E , Sindic CJM , Thompson EJ , Trojano M , Wurster U ((1994) ) Cerebrospinal fluid in the diagnosis of multiple sclerosis: A consensus report. J Neurol Neurosurg Psychiatry 57: , 897–902. |
[20] | Blennow K , Fredman P , Wallin A , Gottfries CG , Karlsson I , Langstrom G , Skoog I , Svennerholm L , Wikkelso C ((1993) ) Protein analysis in cerebrospinal fluid. II. Reference values derived from healthy individuals 18-88 years of age. Eur Neurol 33: , 129–133. |
[21] | Tibbling G , Link H , Ohman S ((1977) ) Principles of albumin and IgG analyses in neurological disorders. I. Establishment of reference values. Scand J Clin Lab Invest 37: , 385–390. |
[22] | Oettl K , Marsche G ((2010) ) Redox state of human serum albumin in terms of cysteine-34 in health and disease. Methods Enzymol 474: , 181–195. |
[23] | Soejima A , Matsuzawa N , Hayashi T , Kimura R , Ootsuka T , Fukuoka K , Yamada A , Nagasawa T , Era S ((2004) ) Alteration of redox state of human serum albumin before and after hemodialysis. Blood Purif 22: , 525–529. |
[24] | Kawakami A , Kubota K , Yamada N , Tagami U , Takehana K , Sonaka I , Suzuki E , Hirayama K ((2006) ) Identification and characterization of oxidized human serum albumin. A slight structural change impairs its ligand-binding and antioxidant functions. FEBS J 273: , 3346–3357. |
[25] | Naldi M , Giannone FA , Baldassarre M , Domenicali M , Caraceni P , Bernardi M , Bertucci C ((2013) ) A fast and validated mass spectrometry method for the evaluation of human serum albumin structural modifications in the clinical field. Eur J Mass Spectrom (Chichester) 19: , 491–496. |
[26] | Domenicali M , Baldassarre M , Giannone FA , Naldi M , Mastroroberto M , Biselli M , Laggetta M , Patrono D , Bertucci C , Bernardi M , Caraceni P ((2014) )Hepatology 60: , 1851–1860 Posttranscriptional changes of serum album, Clinical andrognostic significance in hospitalized patients with cirrhosis. |
[27] | Cohen J ((1988) ) Statistical Power Analysis for the Behavioral Sciences, Lawrence Erlbaum Associates, Hillsdale, N.J. |
[28] | Biroccio A , Del Boccio P , Panella M , Bernardini S , Di Ilio C , Gambi D , Stanzione P , Sacchetta P , Bernardi G , Martorana A , Federici G , Stefani A , Urbani A ((2006) ) Differential post-translational modifications of transthyretin in Alzheimer’s disease: A study of the cerebral spinal fluid. Proteomics 6: , 2305–2313. |
[29] | Matsuyama Y , Hayashi T , Terawaki H , Negawa T , Terada T , Okano Y , Era S ((2009) ) Human astrocytes and aortic endothelial cells actively convert the oxidized form of albumin to the reduced form: Reduced albumin might participate in redox regulation of nerve and blood vessel systems. J Physiol Sci 59: , 207–215. |
[30] | Moon GJ , Shin DH , Im DS , Bang OY , Nam HS , Lee JH , Joo IS , Huh K , Gwag BJ ((2011) ) Identification of oxidized serum albumin in the cerebrospinal fluid of ischaemic stroke patients. Eur J Neurol 18: , 1151–1158. |
[31] | Oettl K , Stauber RE ((2007) ) Physiological and pathological changes in the redox state of human serum albumin critically influence its binding properties. Br J Pharmacol 151: , 580–590. |
[32] | Stauber RE , Spindelboeck W , Haas J , Putz-Bankuti C , Stadlbauer V , Lackner C , Oettl K ((2014) ) Human nonmercaptalbumin-2: A novel prognostic marker in chronic liver failure. Ther Apher Dial 18: , 74–78. |
[33] | Casado A , Encarnacion Lopez-Fernandez M , Concepcion Casado M , de La Torre R ((2008) ) Lipid peroxidation and antioxidant enzyme activities in vascular and Alzheimer dementias. Neurochem Res 33: , 450–458. |
[34] | Guidi I , Galimberti D , Lonati S , Novembrino C , Bamonti F , Tiriticco M , Fenoglio C , Venturelli E , Baron P , Bresolin N , Scarpini E ((2006) ) Oxidative imbalance in patients with mild cognitive impairment and Alzheimer’s disease. Neurobiol Aging 27: , 262–269. |
[35] | Ihara Y , Hayabara T , Sasaki K , Fujisawa Y , Kawada R , Yamamoto T , Nakashima Y , Yoshimune S , Kawai M , Kibata M , Kuroda S ((1997) ) Free radicals and superoxide dismutase in blood of patients with Alzheimer’s disease and vascular dementia. J Neurol Sci 153: , 76–81. |
[36] | Sinclair AJ , Bayer AJ , Johnston J , Warner C , Maxwell SR ((1998) ) Altered plasma antioxidant status in subjects with Alzheimer’s disease and vascular dementia. Int J Geriatr Psychiatry 13: , 840–845. |
[37] | Thompson EJ ((2005) ) The roster of CSF proteins. In Proteins of the Cerebrospinal Fluid: Analisys and interpretation in the Diagnosis and Treatment of Neurological Disease, Academic Press, London, UK. |
[38] | Ahn SM , Byun K , Cho K , Kim JY , Yoo JS , Kim D , Paek SH , Kim SU , Simpson RJ , Lee B ((2008) ) Human microglial cells synthesize albumin in brain. PLoS One 3: , e2829. |
[39] | Merlot AM , Kalinowski DS , Richardson DR ((2014) ) Unraveling the mysteries of serum albumin-more than just a serum protein. Front Physiol 5: , 299. |
[40] | Magzal F , Sela S , Szuchman-Sapir A , Tamir S , Michelis R , Kristal B ((2017) ) In-vivo oxidized albumin- a pro-inflammatory agent in hypoalbuminemia. PLoS One 12: , e0177799. |


