
Involvement of Activation of Asparaginyl Endopeptidase in Tau Hyperphosphorylation in Repetitive Mild Traumatic Brain Injury
Abstract
Traumatic brain injury (TBI) is an established risk factor for the development of neurodegeneration and dementia late in life. Repetitive mild TBI (r-mTBI) is directly associated with chronic traumatic encephalopathy (CTE), a progressive neurodegenerative disorder characterized by focal perivascular to widespread Alzheimer-type neurofibrillary pathology of hyperphosphorylated tau. Studies in animal models have shown hyperphosphorylation of tau after TBI. However, the molecular mechanisms by which TBI leads to tau pathology are not understood. In this study, we employed western blots and immunohistochemistry to test, in triple-transgenic mouse model of Alzheimer’s disease (3xTg-AD), the effect of r-mTBI on tau hyperphosphorylation and activation of asparaginyl endopeptidase (AEP), a cysteine proteinase which is known to be involved in tau hyperphosphorylation. We found that the level of active AEP was increased and correlated with the level of tau hyperphosphorylation following r-mTBI, and that fimbria showed increased immunoreactivity to phospho-tau. In addition, inhibitor 2 of protein phosphatase 2A (I2PP2A) was translocated from neuronal nucleus to the cytoplasm and colocalized with hyperphosphorylated tau. These data suggest the involvement of AEP-I2PP2A-PP2A-ptau pathway in tau pathology in TBI.
INTRODUCTION
Traumatic brain injury (TBI) is a leading cause of death and morbidity worldwide. The annual incidence of TBI with varying severity can be as high as 790 per 100,000 persons, according to a recent population-based study in New Zealand [1]; in the US alone, ∼1.7 million people suffer from a TBI annually with 52,000 deaths as a result [2], and ∼5.3 million people live with a TBI-related disability [1, 3, 4]. TBI is an established risk factor for the development of neurodegeneration and dementia late in life [5, 6]. Among different types of TBI, repetitive mild brain traumas, as seen in professional athletes involved in contact sports and military veterans deployed in wars, often lead to chronic traumatic encephalopathy (CTE), a unique pattern of neurodegeneration characterized by focal perivascular to later widespread neurofibrillary tangles (NFTs) with less overt extracellular deposition of amyloid-β (Aβ) protein [7–9].
Neurofibrillary pathology in CTE, as that seen in other neurodegenerative tauopathies including Alzheimer’s disease (AD), is made up of abnormally hyperphosphorylated tau protein [10–13]. Hyperphosphorylation of tau has been reported in a variety of TBI models, including repetitive mild TBI (r-mTBI), controlled cortical impact (CCI), and fluid percussion injury [14–17], and is thus thought to play a role in etiopathogenesis of CTE [18]. Moderate CCI has been shown to significantly increase, as measured by immunohistochemistry and western blots, the level of phospho-tau at multiple sites in ipsilateral hippocampus and fimbria in young adult 3xTg-AD mice [14], which carry presenilin-1 knock-in mutation and overexpress human amyloid precursor protein with Swedish mutation and human tau with P301L mutation [19]. In aged hTau transgenic mice which express all six isoforms of non-mutated human tau on a murine tau null background, close-head r-mTBI, a total of 5 impacts over 9 days, augments phospho-tau immunoreactivity in the somatodendritic compartment of neurons [20]. Tau hyperphosphorylation was also observed in adult C57BL/6 mouse brain that sustained repetitive closed-head rotation/acceleration injury [21], and in the brain of wild-type rats following fluid percussion injury [18, 22]. However, the molecular mechanism underlying the pathogenesis of neurofibrillary pathology of abnormal hyperphosphorylation of tau after TBI remains elusive.
The phosphorylation state of tau is physiologically orchestrated in a balanced manner by activities of protein kinases and protein phosphatases [23, 24] which, after TBI, are likely altered during the secondary damage initiated by direct mechanical tissue deformation [25]. Given that protein phosphatase 2A (PP2A) accounts for about 70% of total tau phosphatase activity in adult human brain [26], inhibition of PP2A may shift the balance between phosphorylation and dephosphorylation, leading to a net increase in tau phosphorylation. The SET protein, also known as template-activating factor 1β (TAF-1β) or inhibitor 2 of protein phosphatase 2A (I2PP2A), was first characterized as a nuclear phosphoprotein of the set gene that is associated with myeloid leukemogenesis [27, 28]. SET is a multifunctional protein. It has been shown to be involved in gene transcription, control of cell cycle, apoptosis, and cell migration [29–33]. SET is significantly upregulated in AD brain [34]. Notably, SET potently inhibits PP2A activity by acting with its catalytic subunit PP2Ac and thus is known as I2PP2A [35].
Despite its predominant localization in the nucleus, I2PP2A/SET translocates to the cell cytoplasm following its cleavage at N175 into N- and C-terminal fragments; both fragments potently inhibit PP2A and thereby promote hyperphosphorylation of tau [36]. Asparaginyl endopeptidase (AEP), a lysosomal cysteine proteinase [37] which is also known as legumain cleaves both I2PP2A and tau and thereby indirectly leads to hyperphosphorylation of tau in AD brain [38, 39]. Thus, acidosis of the brain tissue could lead to hyperphosphorylation of tau through activation of AEP. TBI can lead to increase in lactate in the brain tissue [25], which may facilitate the activation of AEP. However, it remains uncertain whether AEP plays a role in hyperphosphorylation of tau after TBI.
In the present study, we investigated the effect of r-mTBI on tau hyperphosphorylation and activation of AEP in 3xTg-AD mice. We found that AEP activation was associated with tau hyperphosphorylation following r-mTBI, probably via cleavage of I2PP2A and its translocation from neuronal nucleus to the cytoplasm.
MATERIALS AND METHODS
Animals
Adult female 3xTg-AD mice, 4–6 months of age, were used in this study; female 3xTg-AD mice start showing Aβ pathology at ∼9 months and tau pathology at ∼12 months of age [19]. Mice were purchased from the Jackson Laboratory (Bar Harbor, ME), bred in our animal colony, and housed under a 12-h light/dark cycle, with ad libitum access to food and water. All animal procedures and use were as per the protocol approved by our Institutional Animal Care and Use Committee, in accordance with the PHS Policy on Humane Care and Use of Laboratory Animals (revised February 15, 2015).
Protocols for traumatic brain injury
All TBI surgeries were performed by using the Impact OneTM stereotaxic impactor (Leica Biosystems, Richmond, IL). The r-mTBI was performed as described previously [40]. Briefly, mice were anesthetized by intraperitoneal injection of 2,2,2-tribromoethanol (Sigma, St. Louis, MO) solution (250 mg/kg). The mouse head was shaved and then restrained using a stereotaxic frame and closed-head injury was delivered by a blunt metal tip, which was 5 mm in diameter, centered at midway of the sagittal suture and zeroed at the scalp. The scalp was mildly moisturized with saline and an electronic contact sensor was utilized to help optimize the vertical zero position on the scalp. The strike was set at 5.0 m/s velocity, 200 ms dwell time and 1.0 mm depth. Mice were subjected to a total of 5 impacts with inter-impact interval of 48 h. The sham control mice received anesthesia and stereotaxic restraint only. Mice were kept warm on a soft heating pad during surgery and until fully awake.
Brain tissue sampling
Given that the tau protein is rapidly dephosphorylated at many sites during postmortem interval at room temperature, as shown by us recently [41], mice were sacrificed by cervical dislocation at 24 h after the fifth brain impact, and the brain was quickly dissected out and cooled down in a large volume of ice-cold phosphate-buffered saline (PBS), in which the brain regions from the left hemisphere were further dissected for western blots. The right hemisphere was immersion fixed in buffered 4% paraformaldehyde at room temperature for 48 h for immunohistochemistry.
For western blots, the forebrain was first separated from other brain regions, and then further dissected into two portions, i.e., the hippocampus and the remaining forebrain. These two brain parts were instantly frozen on dry ice and stored in –80°C until used.
Western blot analysis
The brain tissue was homogenized in pre-cooled buffer containing 50 mMTris-HCl, pH 7.4, 2 mM EDTA, 2 mM Na3VO4, 50 mM NaF, 1 mM 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF), and 10μg/ml of each of aprotinin, leupeptin, and pepstatin A. The resulting 10% (w/v) homogenate was mixed with equal volume of 2×Lemaeli buffer, followed by boiling for 5 min. Protein concentration was measured by using A660 kit (Pierce, Rockford, IL). The samples were subjected to 10% SDS-PAGE and electro-transferred onto Immobilon-P membrane (Milipore, Bedford, MA). The blots were then probed with primary antibodies against total tau, phospho-taus, or AEP (Table 1), and developed with species-matched peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA) and ECL kit (Pierce). The Multi Gauge V3.0 densitometry software (Fuji Photo Film Co., Ltd.) was utilized to quantify the density of protein bands in western blots.
Table 1
Primary antibodies employed in the present study
| Antibody | Type | Species | Specificity | Site(s) recognized | Working dilution | Source (catalog and/or reference) | ||
|---|---|---|---|---|---|---|---|---|
| Anti-AEP | Poly- | S | Mouse AEP | / | 1:500 | R&D Systems (AF2058) | ||
| Anti-pS199tau | Poly- | R | P-tau | pS199 | 1:1000 | Invitrogen (44734G) | ||
| Anti-pT205tau | Poly- | R | P-tau | pT205 | 1:1000 | Invitrogen (44738G) | ||
| Anti-pT212tau | Poly- | R | P-tau | pT212 | 1:1000 | Invitrogen (44740G) | ||
| Anti-pS262tau | Poly- | R | P-tau | pS262 | 1:1000 | Invitrogen (44750G) | ||
| PHF-1 | Mono- | M | P-tau | pS396/404 | 1:500 | Dr. P. Davies | ||
| AT8 | Mono- | M | P-tau | pS202 | / | pT205 | 1:1,000 | ThermoFisher Scientific (MN1020) |
| Anti-pS396tau | Poly- | R | P-tau | pS396 | 1:1,000 | Invitrogen (44752G) | ||
| 134D | Poly- | R | Total tau; murine &human tau | / | 1:1,000 | [80] | ||
| Anti-MAP2 | Mono- | M | MAP2 | / | 1:2,000 | Steinberger MonoclonalsIncorpoerated (SMI-52) | ||
| Anti-Syn1 | Poly- | R | Synapsin-1 | / | 1:2,000 | Stressgen Biotechnologies (VAP-SV060) | ||
| 10E7 | Mono- | M | Anti-I2PP2A | / | 1:1,000 | [38] | ||
| Anti GAPDH | Poly- | R | GAPDH | / | 1:3,000 | Santa-Cruz (sc-25778) |
Poly-, polyclonal; Mono-, monoclonal; S, sheep; R, rabbit; M, mouse; P-tau, phosphorylated tau
Immunohistochemistry
The right brain hemispheres of r-mTBI and sham control mice were embedded in paraffin. Adjacent serial sagittal sections, 6μm in thickness and positioned at ∼1 mm lateral to the midline, were immunostained for phospho-tau at Ser202/Thr205 (AT8), Thr212, or Ser396/404 (PHF-1) separately. Briefly, sections were deparaffinized, rehydrated and subjected to antigen retrieval by boiling in pH 6.0 citrate buffer in a water bath for 30 min. Sections were then washed in PBS, subjected to permeabilization and removal of endogenous peroxidase activity, blocked in normal goat serum, and incubated with primary antibodies (see Table 1) overnight at 4°C. After washing in PBS, the sections were incubated with species-matched horseradish peroxidase-conjugated secondary antibodies at room temperature for 2 h, washed and simultaneously developed in 0.05% diaminobenzidine (Sigma) plus 0.015% hydrogen peroxide (Fisher Scientific, Fair Lawn, NJ) to guarantee an identical development condition. Sections were counterstained with Mayer’s hematoxylin, dehydrated in ascending concentrations of ethanol, cleared in Histoclear (National Diagnostics, Atlanta, GA), and covered with coverslips.
Photomicrogaphs were taken on a Nikon 90i digital microscope by using identical imaging settings for quantitation of immnunoreactivity with NIH’s ImageJ software package. For comparison of staining between groups, signals were extracted from images by using the color threshold function with identical settings.
Immunofluorescence
Sagittal paraffin-embedded sections at ∼1 mm lateral to the midline were deparaffinized, rehydrated and subjected to immunofluorescence staining. Briefly, sections were blocked in normal goat serum, and incubated overnight at 4°C with primary antibodies against phospho-tau, I2PP2A, microtubule-associated protein 2 (MAP-2), synapsin-1 or AEP (Table 1), or a combination of two for double immunofluorescence staining. After washing in PBS, the sections were incubated with species-matched fluorophore-conjugated secondary antibodies (1:1,000 each, Life Technologies, Rockford, IL) plus TO-PRO 3 iodide (1:1,000, Thermo Fisher Scientific) for nuclear staining at room temperature for 2 h. Sections were washed in PBS and covered with anti-fade mounting medium.
Fluorescence photomicrographs were taken using a TCS-SP2 laser scanning confocal microscope (Leica Microsystems GmbH, Wetzlar, Germany). For quantification of immunofluorescent staining, images were taken with a 100× objective lens; 4–6 randomly selected visual fields in each of at least two sections per animal were employed and the measures averaged for individual animal. Quantification of fluorescence immunoreactivity was performed using ImageJ, and the criteria for signal thresholding, Moments in most cases or Yen’s arithmetic when suitable, were kept consistent across groups. Percent area occupied by a particular marker was calculated for each animal using the sum of immunopositive areas and that of the total areas.
Statistical analysis
Quantitative data were analyzed using the GraphPad Prism 5.0 software package (GraphPad Software, Inc., La Jolla, CA). Data were plotted as scattered dot plots with mean±SEM. Unpaired t test, with Welch’s correction in the case of unequal variance, was used for comparison between two groups. Pearson correlation was used to analyze the relation of tau phosphorylation level with the level of active AEP. p < 0.05 was considered statistically significant.
RESULTS
Tau is hyperphosphorylated in mouse brain after r-mTBI
To identify the effect of r-mTBI on tau hyperphosphorylation in 3xTg-AD mice, we first examined levels of phosphorylations at multiple sites of tau using western blots. We found that the level of phospho-tau, when normalized with total tau was increased at Thr205 and pSer262 in hippocampus and at PHF-1 (pSer396/404) site in both the hippocampus and forebrain (Fig. 1A, B). In forebrains of mice with r-mTBI, levels of Thr205 and pSer262 showed a trend of increase (Fig. 1B). Immunohistochemical staining showed over 27% increase in phospho-tau immunoreactivity in fimbria in brains with r-mTBI when compared to that in the sham control, as detected by PHF-1 and by antibody against pSer396-tau (Fig. 2A, B). The pSer396-tau immunoreactivity was also visible in subiculum as clusters of dots but with a markedly lower density than the clusters seen in fimbria (Fig. 2B). However, neither fimbria nor other brain regions showed AT8 (pSer202/pThr205) or anti-pThr212 tau immunoreactivity (Fig. 2A). These results indicate that tau is hyperphosphorylated at selected sites in mouse brain after r-mTBI.
Fig. 1
Tau phosphorylation increases at multiple sites in the brain following r-mTBI in 3xTg-AD mice. Adult female 3xTg-AD mice, 4— months of age, were subjected to repetitive mild closed-head brain impacts, totaling five with 48-h inter-impact intervals, and sacrificed 24 h after the final impact. A) Representative western blots showing the level of indicated proteins/phosphorylation sites. B) Densitometric quantification of the blots. Among the phosphorylation sites examined, tau was hyperphosphorylated at pThr205, pSer262 and PHF-1 (pSer396/404) sites. Data are presented as scatter dot plots with mean±SEM (n = 6-7 mice each) and analyzed using unpaired t test, with Welch’s correction in the case of unequal variance.
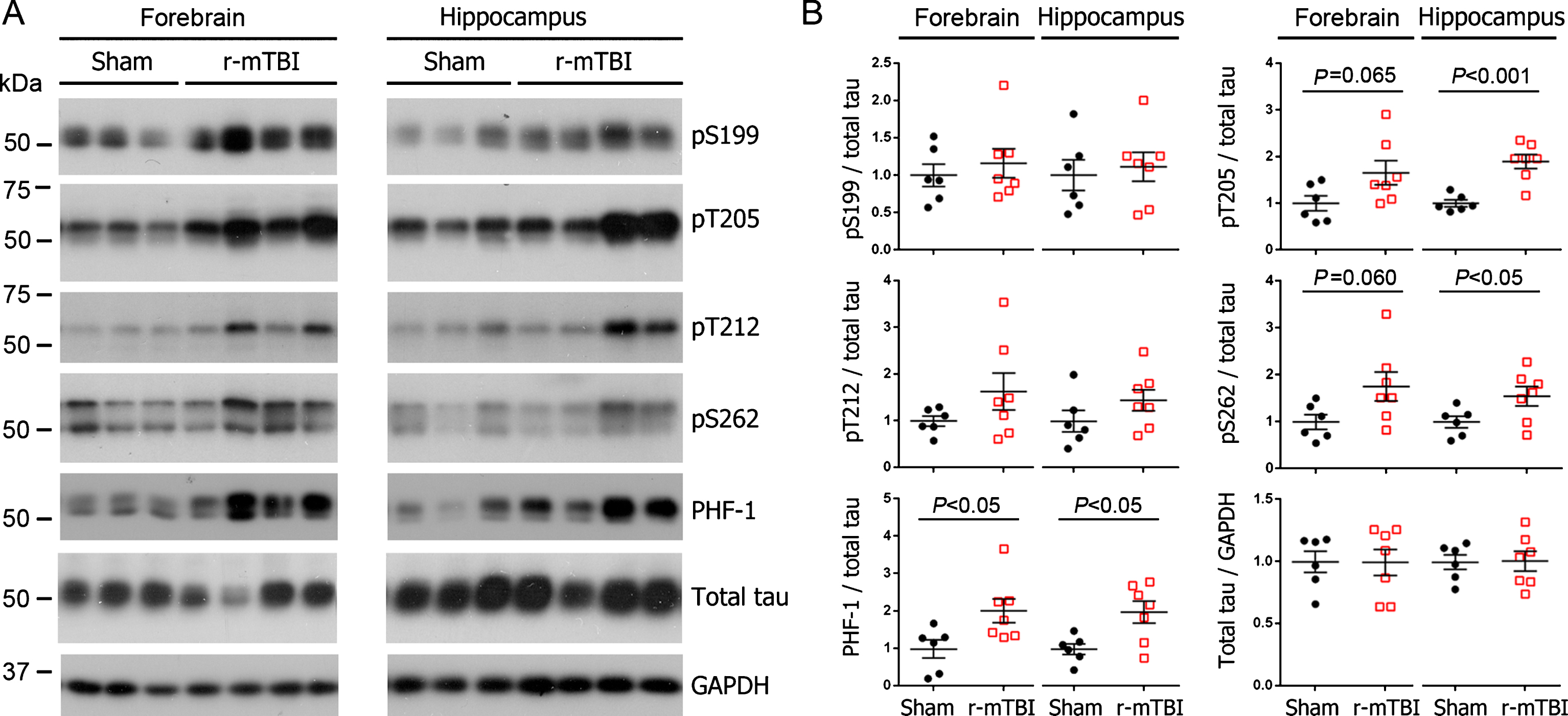
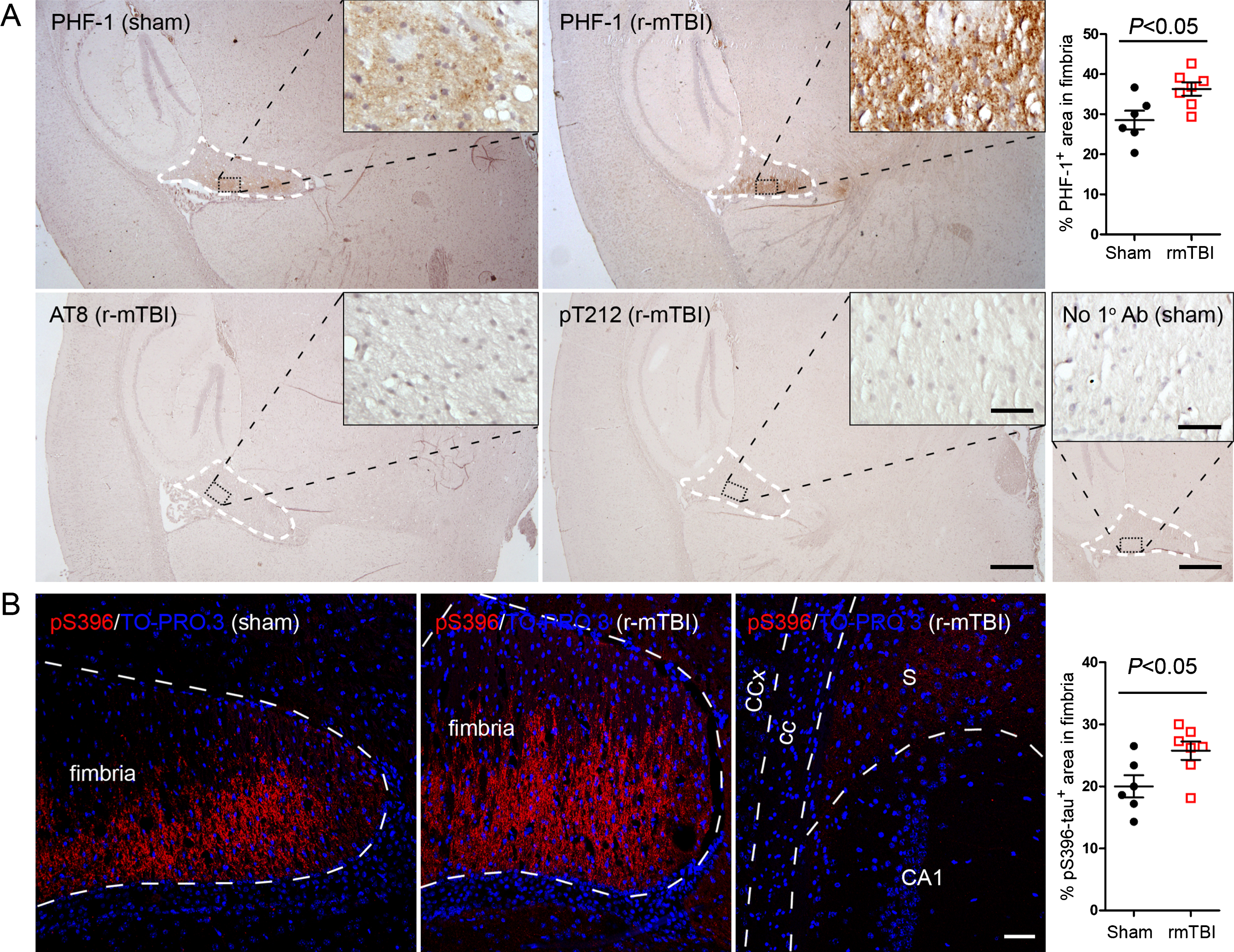
Fig. 2
Tau is hyperphosphorylated in fimbria of the hippocampus in 3xTg-AD mice after r-mTBI. A) Representative images showing immunohistochemical staining of sagittal brain sections with antibodies PHF-1, AT8 (pSer202/pThr205) or anti-pThr212-tau. Fimbria showed easily visible PHF-1 immunoreactivity, and quantification revealed significantly more immunoreactivity in r-mTBI group than sham control group. No staining was detected by AT8 or anti-pThr212-tau. No primary antibody control for PHF-1 staining exhibited no staining. B) Immunofluorescence staining with anti-pSer396-tau. Immunoreactivity was mostly seen in fimbria, though subiculum also showed sparse staining. In addition, quantification which was performed by using images taken with a 20× objective lens showed more immunoreactivity in fimbria in r-mTBI group than in sham controls. Data are expressed as scattered dot plots with mean±SEM (n = 6-7 mice/group) and analyzed with unpaired Student t test. Scale bar = 500μm (A), 50μm (insert and B).
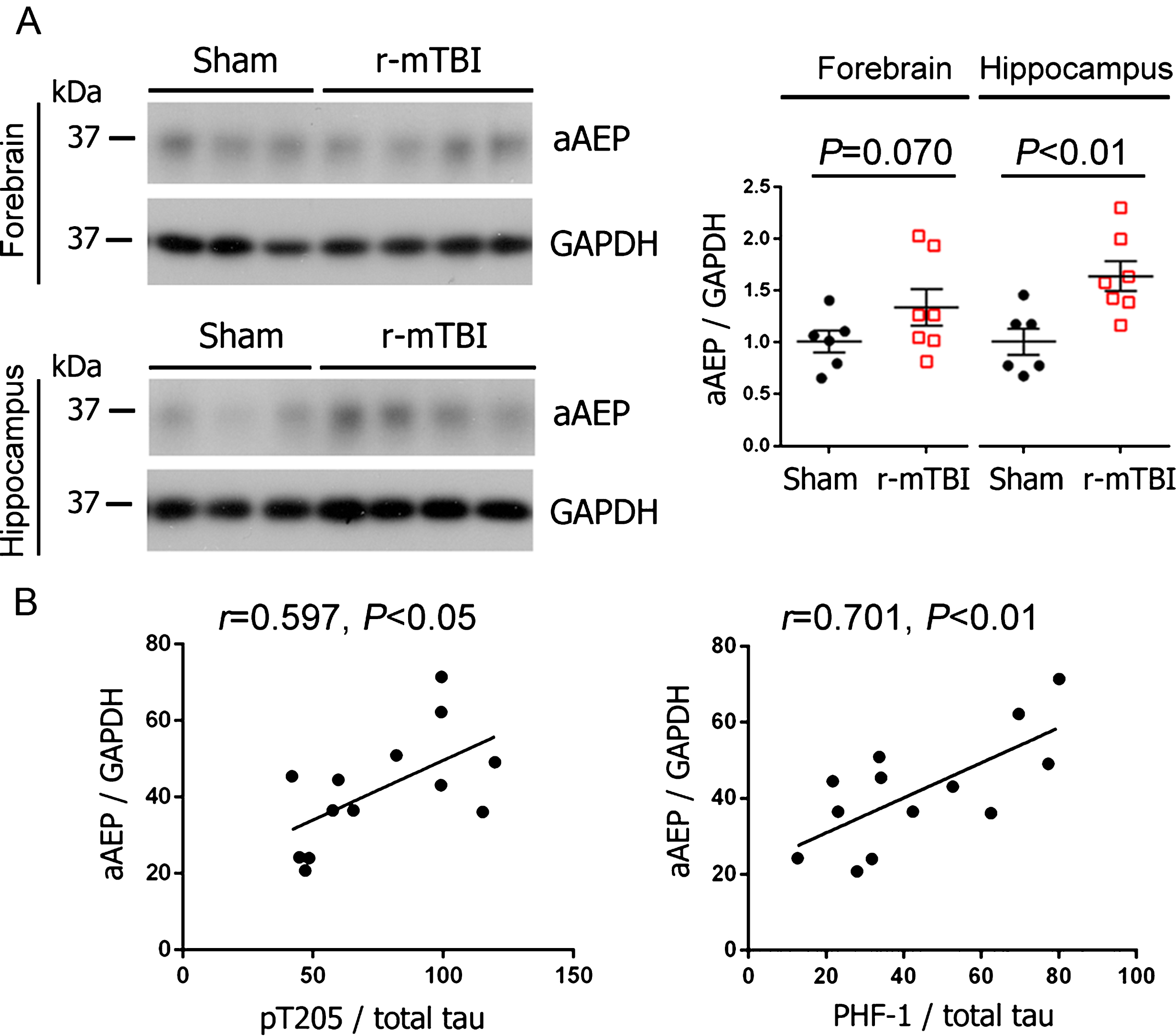
Level of activated AEP increases and correlates with tau hyperphosphorylation after r-mTBI
AEP is synthesized as a 56 kDa zymogen (pro-AEP) and is autocatalytically cleaved to produce 36 kDa active enzyme (active AEP) under acidic conditions [42, 43]. To study the role of AEP in tau pathogenesis after TBI, we analyzed the level of active AEP (36 kDa) by western blots. We found that AEP was significantly more activated in the hippocampi of r-mTBI mice than sham control animals (Fig. 3A); in forebrain, though the increase in the level of activated AEP did not reach statistical significance, it showed a trend (Fig. 3A). Intriguingly, the level of active AEP was positively correlated with the level of tau phosphorylated at Thr205 and PHF-1 sites (Fig. 3B). These data suggest the involvement of AEP in tau hyperphosphorylation in r-mTBI.
Fig. 3
Activation of AEP is increased in the brain and correlated with the level of hyperphosphorylated tau following r-mTBI in 3xTg-AD mice. A) Representative western blots and quantification showing the level of active AEP (aAEP). Data are expressed as scattered dot plots with mean±SEM (n = 6-7 mice/group) and analyzed with unpaired Student t test. B) Pearson correlation of aAEP level to the level of indicated phospho-tau in the hippocampus.
Translocation of I2PP2A to the cytoplasm links AEP activation to tau hyperphosphorylation after r-mTBI
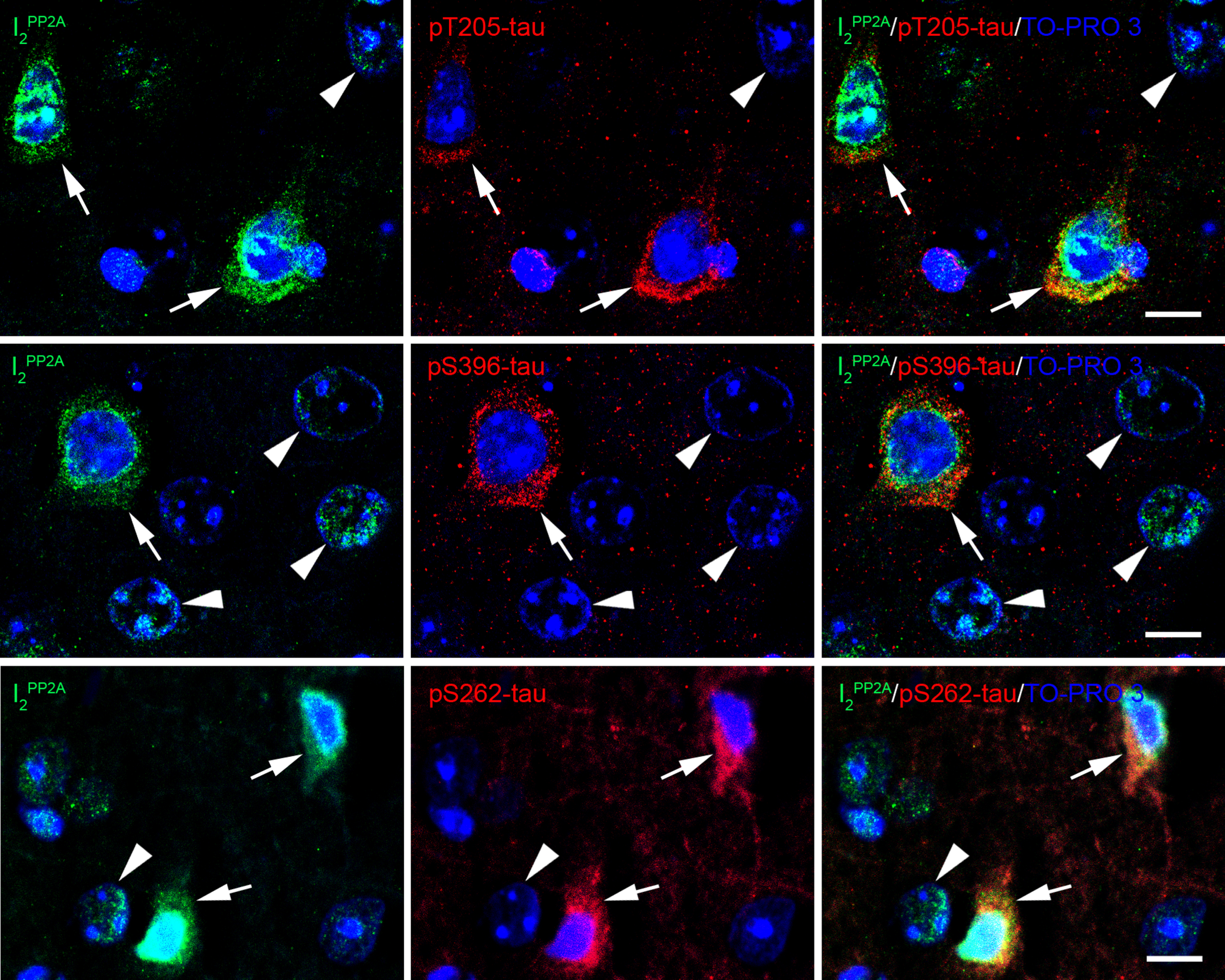
Immunofluorescent staining showed that translocation of I2PP2A to the neuronal cytoplasm in cerebral cortex was significantly increased after r-mTBI whereas sham control animals exhibited I2PP2A immunoreactivity mostly in the neuronal nuclei (Fig. 4A, B). Double immunofluorescent staining revealed that increase in translocation of I2PP2A was associated with increase in the expression of AEP in the neuronal cytoplasm (Fig. 4C), and that cytoplasmic I2PP2A was colocalized with tau hyperphosphorylated at Thr205, pSer262 and pSer396 (Fig. 5). These data suggest that AEP could be involved in translocation of I2PP2A to the neuronal cytoplasm and consequently tau hyperphosphorylation in r-mTBI.
Fig. 4
r-mTBI promotes translocation of I2PP2A/SET from neuronal nucleus to the cytoplasm in 3xTg-AD mice. A) Representative immunofluorescence staining showing translocation of inhibitor 2 of protein phosphatase 2A (I2PP2A or SET) to neuronal cytoplasm in cerebral cortex. B) Quantification of I2PP2A translocation, as percentage of cytoplasmic over total I2PP2A immunoreactivity within the same visual field. Data are expressed as scattered dot plots with mean±SEM (n = 6-7 mice/group) and analyzed with unpaired Student t test. C) Double immunofluorescent staining of I2PP2A and AEP in brain sections. Lysosome-like AEP-positive puncta and I2PP2A coexisted in the cytoplasm of neurons (arrows) in brains with r-mTBI; neurons in sham control brains showed fewer AEP puncta (hollow arrowhead). Scale bar = 10μm for all images.
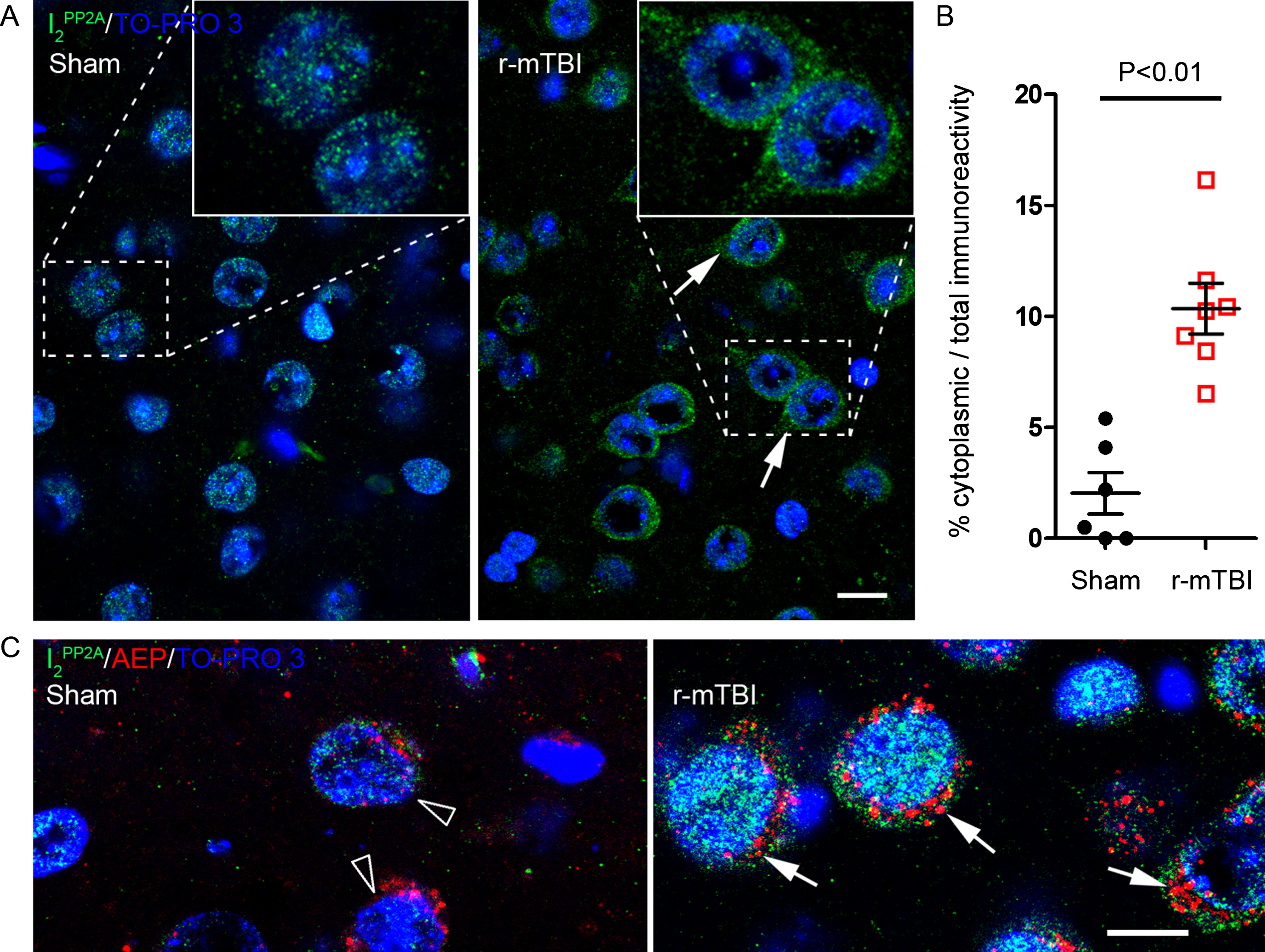
Fig. 5
Translocated I2PP2A/SET is colocalized with hyperphosphorylated tau in the neuronal cytoplasm in cerebral cortex of 3xTg-AD mice with r-mTBI. Brain sections were fluorescently dual immuno-stained for I2PP2A and phospho-tau. Translocated I2PP2A colocalized with pThr205-tau, pSer396-tau and with pSer262-tau in the cytoplasm of neurons (arrows). However, there was no somatodendritic phospho-tau staining in neurons where cytoplasmic I2PP2A was absent (filled arrowheads). Scale bar = 10μm for all images.
Mice with r-mTBI show neurodegeneration
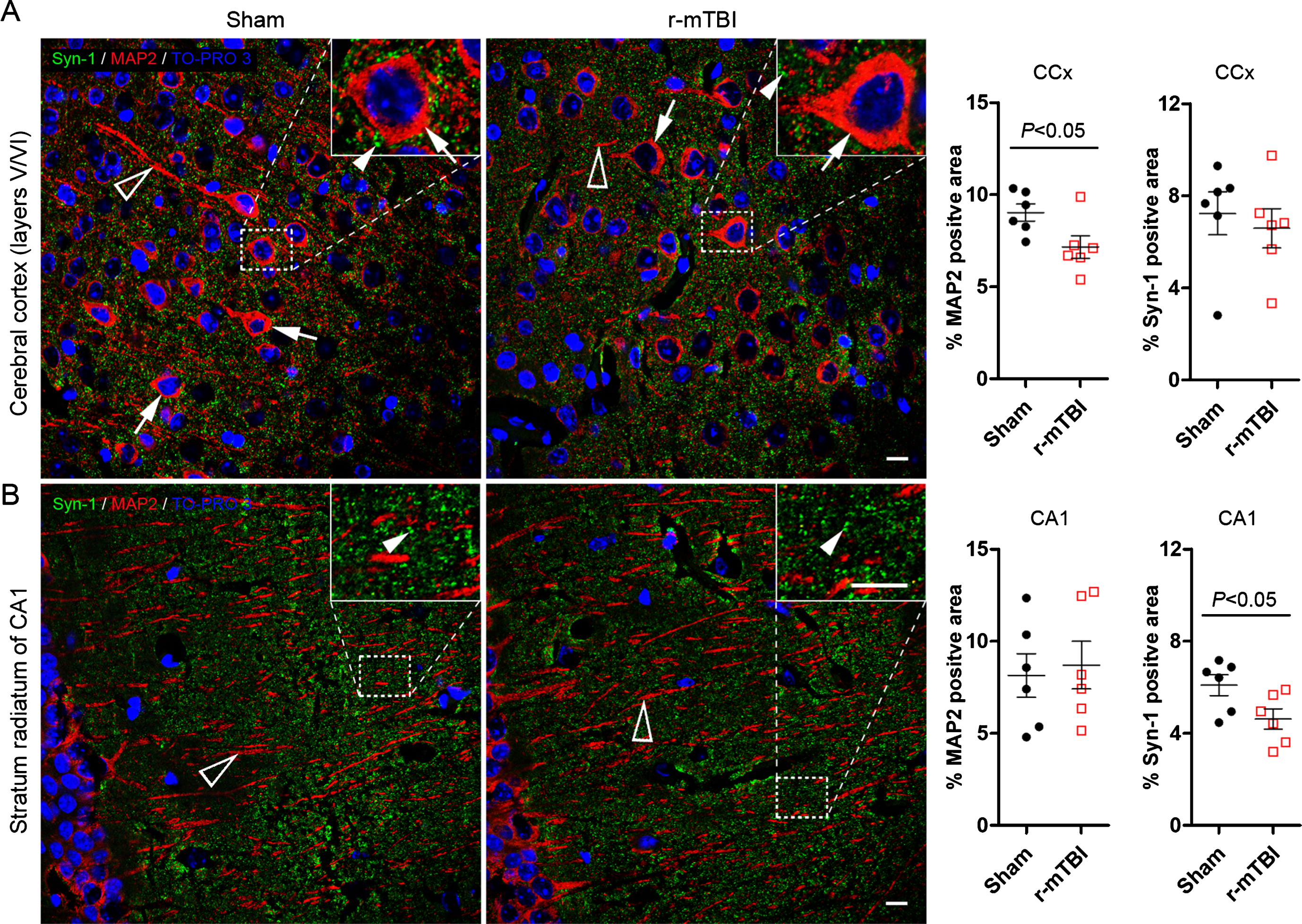
To study neurodegeneration induced by r-mTBI, we performed double-immunofluorescent staining of brain sections for MAP-2 and synapsin-1. MAP-2 is a neuronal somatodendritic marker, and synapsin-1 is a presynaptic marker. The mice with r-mTBI exhibited 21% decrease in MAP-2 immuno-positive area in the cerebral cortex and 24% reduction in density of synapsin-1 positive presynaptic puncta in the hippocampus as compared to sham control animals (Fig. 6A, B). These data indicate neurodegeneration after r-mTBI in mice.
Fig. 6
Repetitive mild TBI induces dendritic and synaptic degeneration in 3xTg-AD mice. Brain sections were double immunofluorescent stained for microtubule-associated protein 2 (MAP2) and synapsin-1 (Syn-1), as somatodendritic and presynaptic markers, respectively. Representative photomicrographs show the staining and quantification in cerebral cortex (A) and the striatum radiatum of hippocampal CA1 subfield (B). Data are expressed as scattered dot plots with mean±SEM (n = 6-7 mice/group) and analyzed with unpaired Student t test. Scale bar = 10μm for all.
DISCUSSION
Repetitive mild TBI in mice in which diffuse shearing and tearing of axons and vessels occur similar to that in mTBI in humans [15, 16, 25, 44], is a suitable model to study the pathophysiology of CTE. Although CTE is proposed to represent a progressive neurodegeneration associated with alterations of tau, TDP-43 and Aβ proteins that occur as a consequence of r-mTBI [6, 8, 45], it is histopathologically characterized by hyperphosphorylated tau inclusions ranging in severity from focal perivascular NFTs to severe tau pathology affecting widespread brain regions [8, 45]. Approximately one-third of individuals who sustain a single moderate to severe TBI show abundant and widely distributed NFTs many years after the injury [46]. Although majority of TBI cases belong to mild TBI [1], repetitive mild TBI has been associated with profound neurofibrillary pathology in the brain in retired professional American football players [9]. The nature of TBI-induced neurodegeneration has led to the belief that hyperphosphorylation of tau represents a pivotal biochemical event in pathogenesis of CTE [45].
Murine tau is less prone to hyperphosphorylation and aggregation than human tau after TBI [45]. Although r-mTBI in aged hTau transgenic mice, which overexpress all six isoforms of wild-type human tau, showed overtly augmented tau immunoreactivity [20], the same r-mTBI model in C57BL/6 mice showed neither elevated tau immunohistochemical staining nor biochemically increased level of phospho-tau compared to sham control animals in the chronic phase [15]. In the present study, we employed 3xTg-AD mice which overexpress mutated human tau (P301L tau) as an animal model, and we detected biochemically an increased level of tau phosphorylation at Thr205, Ser262, and PHF-1 (Ser396/404) sites 24 h after the fifth of a total of five mild closed-head impacts in these animals. However, we detected no significant change in total tau level in r-mTBI group as compared to sham control animals by western blots. Immunohistochemical staining confirmed increased level of phosphorylated tau (pSer396/404) despite that the immunoreactivity was seen mainly in fimbria of the hippocampus. This is consistent, at least in part, with the findings of Tran and colleagues who showed substantial increase of phospho-tau immunoreactivity, including pSer396, in ipsilateral fimbria after moderate CCI in 3xTg-AD mice [14]. We observed occasional hyperphosphorylation of tau at Thr205, Ser262 or pSer396 site by immunofluorescent staining in the neuronal somatodendritic compartment; nevertheless, we only detected immunohistochemically a marked increase in tau hyperphosphorylated at pSer396/404 site in fimbria, an exclusively axonal area. This may be due to aggregation of pSer396/404-tau [47] which is enriched in axons and/or less rapid postmortem dephosphorylation of tau at PHF-1 than at Ser262 and Thr205 [41], two more preferred sites for PP2A [26, 48].
Molecular mechanisms underlying the development of tau pathology after TBI remain to be unraveled. Since the phosphorylation state of tau is governed by balanced regulation of tau protein kinases and phosphatases, one possible mechanism of etiopathogenesis of tau in TBI could be the dysregulation of proteases, such as calpain and AEP that can modulate tau kinases/phosphatases, as a result of initial mechanical insult to the brain and/or secondary damage that follows [25]. Both calpain and AEP have been shown to play a role in tau hyperphosphorylation in AD and related tauopathies [38, 49–51]. Although the role of calpain activation in TBI-induced neurodegeneration has been extensively studied [52–56], no previous study has explored the role of brain AEP in TBI.
The activity of PP2A, the major tau phosphatase in the brain [26], is inhibited by full length [57] and also N-and C-terminal fragments of I2PP2A which are known to induce Alzheimer-like pathology in vivo. AEP is the main cysteine proteinase involved in the cleavage of I2PP2A and may thus contribute to tau hyperphosphorylation in AD brain [38]. Calcium overload and release of excitatory amino acids, two cardinal initiating events in secondary damage after TBI, can lead to ischemia, hypoxia and increase in lactate [25] and thereby acidosis of the brain tissue [59–64] and lysosomal abnormalities [65], conditions in which AEP can be activated and facilitated to cleave I2PP2A [66, 67]. In the present study, we found a significant increase in the level of active AEP which was correlated to tau hyperphosphorylation after r-mTB in mice. Furthermore, we found a marked cytoplasmic translocation of I2PP2A, and more importantly its colocalization with hyperphosphorylated tau, suggesting a potential increase in inhibition of PP2A activity which favors tau hyperphosphorylation in the neuronal cytoplasm [68]. Taken together, these data suggest a role of AEP-I2PP2A-PP2A pathway in hyperphosphorylation of tau in TBI.
Neurofibrillary pathology correlates with the severity of dementia in AD [69, 70], and hyperphosphorylated oligomeric tau has been shown to represent neurotoxic [71] and seeding-competent [72–75] tau species. Given that the development of neurofibrillary pathology and neurodegeneration are long-term phenomena in neurodegenerative diseases [76], persistent neuronal load of hyperphosphorylated tau may thus possibly represent a pathophysiological event that causes neural degeneration after TBI. Using MAP-2 as a marker for dendritic damage or plasticity [77] and synapsin-1 as a presynaptic marker, in the present study we found that dendritc and synaptic degeneration was evident 24 h after the final impact of a total of five brain impacts. Based on the indicated role of AEP activation and subsequent cleavage of I2PP2A in hyperphosphorylation of tau in cell models and in AD brain [38, 58] and the findings of the present study, we propose that activation of AEP as a consequence of primary/secondary brain damage represents a mechanism by which TBI leads to tau hyperphosphorylation that may eventually contribute to CTE pathology (Fig. 7).
Fig. 7
Proposed model demonstrating the involvement of AEP activation in tau hyperphosphorylation in TBI. TBI induces acidosis of the brain tissue, which increases the level of active AEP and its translocation from neuronal lysosomes to the cytoplasm and the nucleus. Active AEP cleaves I2PP2A into amino- and carboxy-terminal fragments [38], both of which are translocated to the cytoplasm and inhibit PP2A activity [36], leading to hyperphosphorylation of tau. I2PP2A, inhibitor 2 of protein phosphatase 2A; I2NTF, amino-terminal fragment of I2PP2A; I2CTF, carboxy-terminal fragment of I2PP2A; P-tau, hyperphosphorylated tau.
![Proposed model demonstrating the involvement of AEP activation in tau hyperphosphorylation in TBI. TBI induces acidosis of the brain tissue, which increases the level of active AEP and its translocation from neuronal lysosomes to the cytoplasm and the nucleus. Active AEP cleaves I2PP2A into amino- and carboxy-terminal fragments [38], both of which are translocated to the cytoplasm and inhibit PP2A activity [36], leading to hyperphosphorylation of tau. I2PP2A, inhibitor 2 of protein phosphatase 2A; I2NTF, amino-terminal fragment of I2PP2A; I2CTF, carboxy-terminal fragment of I2PP2A; P-tau, hyperphosphorylated tau.](https://content.iospress.com:443/media/jad/2018/64-3/jad-64-3-jad180177/jad-64-jad180177-g007.jpg)
A characteristic pattern of tau pathology in human CTE is perivascular foci of neurofibrillary tangles at the depth of sulci, which are thought to represent the early accumulation of tau aggregates in CTE brains [8, 9]. This pattern of neurofibrillary pathology is not recapitulated in rodent models [45] because the rodent brain differs significantly from the human brain in anatomical architecture. Importantly, the ratio of white matter versus gray matter in rodent brain is only 10% as compared to 60% in human brain; and the rodent cerebral cortex does not have convoluted folds, in contrast to extensive sulci in the human cerebral cortex. Recent studies employing postmortem CTE brains suggest that disruption of blood-brain barrier [78] and/or axons [79] underlying the sulci, which are vulnerable to damage by shearing and tearing forces at brain concussion [25], may account for the start of tau pathology in perivascular foci at the depth of sulci. In the present study, we observed significantly increased accumulation of phospho-tau in fimbria and occasional cytoplasmic accumulation of phospho-tau in the cerebral cortex in mice with r-mTBI, a pattern different from that seen in human CTE. This distinction may be a consequence of different anatomical and physiological features of the mouse brain from the human brain. The role of AEP in downstream pathophysiological events of blood-brain barrier damage and/or axonal disruption could be explored in future studies.
In short, in the present study we found that the level of active AEP was increased in brains of 3xTg-AD mice with r-mTBI, and tau hyperphosphorylation correlated well with the level of active AEP and it was colocalized with I2PP2A that was translocated from neuronal nucleus to the cytoplasm. In addition, the sites of tau which showed hyperphosphorylation and colocalized with cytoplasmic I2PP2A, i.e., Thr205, Ser262 and PHF-1 (Ser396/404), are known to be sensitive sites for dephosphorylation by PP2A [41]. These data suggest that AEP may potentially play an important role in pathogenesis of tau in CTE. The role of AEP in TBI could be investigated in future studies by observing the dynamic change of tau hyperphosphorylation and neurofibrillary pathology on a chronic basis.
ACKNOWLEDGMENTS
We are grateful to Dr. Peter Davies from The Feinstein Institute for Medical Research, Manhasset, NY, USA for generously providing antibody PHF-1. This work was funded in part by the New York State Office for People with Developmental Disabilities (OPWDD) and by grants from the Alzheimer’s Association, USA (ZEN-120241433, DSAD-15-363172 and 2016-NIRG-397030) and from National Natural Science Foundation of China (31671046 and 81773713).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0177r1).
REFERENCES
[1] | Feigin VL , Theadom A , Barker-Collo S , Starkey NJ , McPherson K , Kahan M , Dowell A , Brown P , Parag V , Kydd R , Jones K , Jones A , Ameratunga S , Group BS ((2013) ) Incidence of traumatic brain injury in New Zealand: A population-based study. Lancet Neurol 12: , 53–64. |
[2] | US Department of Health and Human Sevice, Centers for Disease Control and Prevention, National Center for Injury Prevention and Control. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002-2006, http://www.cdc.gov/traumaticbraininjury/pdf/blue_book.pdf |
[3] | Selassie AW , Wilson DA , Pickelsimer EE , Voronca DC , Williams NR , Edwards JC ((2013) ) Incidence of sport-related traumatic brain injury and risk factors of severity: A population-based epidemiologic study. Ann Epidemiol 23: , 750–756. |
[4] | Langlois JA , Rutland-Brown W , Wald MM ((2006) ) The epidemiology and impact of traumatic brain injury: A brief overview. J Head Trauma Rehabil 21: , 375–378. |
[5] | Moretti L , Cristofori I , Weaver SM , Chau A , Portelli JN , Grafman J ((2012) ) Cognitive decline in older adults with a history of traumatic brain injury. Lancet Neurol 11: , 1103–1112. |
[6] | Smith DH , Johnson VE , Stewart W ((2013) ) Chronic neuropathologies of single and repetitive TBI: Substrates of dementia?. Nat Rev Neurol 9: , 211–221. |
[7] | Montenigro PH , Baugh CM , Daneshvar DH , Mez J , Budson AE , Au R , Katz DI , Cantu RC , Stern RA ((2014) ) Clinical subtypes of chronic traumatic encephalopathy: Literature review and proposed research diagnostic criteria for traumatic encephalopathy syndrome. Alzheimers Res Ther 6: , 68. |
[8] | McKee AC , Stern RA , Nowinski CJ , Stein TD , Alvarez VE , Daneshvar DH , Lee HS , Wojtowicz SM , Hall G , Baugh CM , Riley DO , Kubilus CA , Cormier KA , Jacobs MA , Martin BR , Abraham CR , Ikezu T , Reichard RR , Wolozin BL , Budson AE , Goldstein LE , Kowall NW , Cantu RC ((2013) ) The of disease in chronic traumatic encephalopathy. Brain 136: , 43–64. |
[9] | Mez J , Daneshvar DH , Kiernan PT , Abdolmohammadi B , Alvarez VE , Huber BR , Alosco ML , Solomon TM , Nowinski CJ , McHale L , Cormier KA , Kubilus CA , Martin BM , Murphy L , Baugh CM , Montenigro PH , Chaisson CE , Tripodis Y , Kowall NW , Weuve J , McClean MD , Cantu RC , Goldstein LE , Katz DI , Stern RA , Stein TD , McKee AC ((2017) ) Clinicopathological evaluation of chronic traumatic encephalopathy in players of American Football. JAMA 318: , 360–370. |
[10] | Grundke-Iqbal I , Iqbal K , Tung YC , Quinlan M , Wisniewski HM , Binder LI ((1986) ) Abnormal phosphorylation of the microtubule-associated protein τ (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci USA 83: , 4913–4917. |
[11] | Lee VM , Goedert M , Trojanowski JQ ((2001) ) Neurodegenerative tauopathies. Annu Rev Neurosci 24: , 1121–1159. |
[12] | Tolnay M , Probst A ((1999) ) REVIEW: Tau protein pathology in Alzheimer’s disease and related disorders. Neuropathol Appl Neurobiol 25: , 171–187. |
[13] | Iqbal K , Liu F , Gong CX ((2016) ) Tau and neurodegenerative disease: The story so far. Nat Rev Neurol 12: , 15–27. |
[14] | Tran HT , LaFerla FM , Holtzman DM , Brody DL ((2011) ) Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-beta accumulation and independently accelerates the development of tau abnormalities. J Neurosci 31: , 9513–9525. |
[15] | Mouzon BC , Bachmeier C , Ferro A , Ojo JO , Crynen G , Acker CM , Davies P , Mullan M , Stewart W , Crawford F ((2014) ) Chronic neuropathological and neurobehoral changes in a repetitive mild traumatic brain injury model. Ann Neurol 75: , 241–254. |
[16] | Kane MJ , Angoa-Perez M , Briggs DI , Viano DC , Kreipke CW , Kuhn DM ((2012) ) A mouse model of human repetitive mild traumatic brain injury. J Neurosci Methods 203: , 41–49. |
[17] | Cheng JS , Craft R , Yu GQ , Ho K , Wang X , Mohan G , Mangnitsky S , Ponnusamy R , Mucke L ((2014) ) Tau reduction diminishes spatial learning and memory deficits after mild repetitive traumatic brain injury in mice. PLoS One 9: , e115765. |
[18] | Shultz SR , Wright DK , Zheng P , Stuchbery R , Liu SJ , Sashindranath M , Medcalf RL , Johnston LA , Hovens CM , Jones NC , O’Brien TJ ((2015) ) Sodium selenate reduces hyperphosphorylated tau and improves outcomes after traumatic brain injury. Brain 138: , 1297–1313. |
[19] | Oddo S , Caccamo A , Shepherd JD , Murphy MP , Golde TE , Kayed R , Metherate R , Mattson MP , Akbari Y , LaFerla FM ((2003) ) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 39: , 409–421. |
[20] | Ojo JO , Mouzon B , Greenberg MB , Bachmeier C , Mullan M , Crawford F ((2013) ) Repetitive mild traumatic brain injury augments tau pathology and glial activation in aged hTau mice. J Neuropathol Exp Neurol 72: , 137–151. |
[21] | Namjoshi DR , Cheng WH , McInnes KA , Martens KM , Carr M , Wilkinson A , Fan J , Robert J , Hayat A , Cripton PA , Wellington CL ((2014) ) Merging pathology with biomechanics using CHIMERA (Closed-Head Impact Model of Engineered Rotational Acceleration): A novel, surgery-free model of traumatic brain injury. Mol Neurodegener 9: , 55. |
[22] | Hoshino S , Tamaoka A , Takahashi M , Kobayashi S , Furukawa T , Oaki Y , Mori O , Matsuno S , Shoji S , Inomata M , Teramoto A ((1998) ) Emergence of immunoreactivities for phosphorylated tau and amyloid-beta protein in chronic stage of fluid percussion injury in rat brain. Neuroreport 9: , 1879–1883. |
[23] | Wang Y , Mandelkow E ((2016) ) Tau in physiology and pathology. Nat Rev Neurosci 17: , 22–35. |
[24] | Martin L , Latypova X , Wilson CM , Magnaudeix A , Perrin ML , Yardin C , Terro F ((2013) ) Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res Rev 12: , 289–309. |
[25] | Xiong Y , Mahmood A , Chopp M ((2013) ) Animal models of traumatic brain injury. Nat Rev Neurosci 14: , 128–142. |
[26] | Liu F , Grundke-Iqbal I , Iqbal K , Gong CX ((2005) ) Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci 22: , 1942–1950. |
[27] | Adachi Y , Pavlakis GN , Copeland TD ((1994) ) Identification and characterization of SET, a nuclear phosphoprotein encoded by the translocation break point in acute undifferentiated leukemia. J Biol Chem 269: , 2258–2262. |
[28] | von Lindern M , van Baal S , Wiegant J , Raap A , Hagemeijer A , Grosveld G ((1992) ) Can, a putative oncogene associated with myeloid leukemogenesis, may be activated by fusion of its 3’ half to different genes: Characterization of the set gene. Mol Cell Biol 12: , 3346–3355. |
[29] | Compagnone NA , Zhang P , Vigne JL , Mellon SH ((2000) ) Novel role for the nuclear phosphoprotein SET in transcriptional activation of P450c17 and initiation of neurosteroidogenesis. Mol Endocrinol 14: , 875–888. |
[30] | Canela N , Rodriguez-Vilarrupla A , Estanyol JM , Diaz C , Pujol MJ , Agell N , Bachs O ((2003) ) The SET protein regulates G2/M transition by modulating cyclin B-cyclin-dependent kinase 1 activity. J Biol Chem 278: , 1158–1164. |
[31] | Carujo S , Estanyol JM , Ejarque A , Agell N , Bachs O , Pujol MJ ((2006) ) Glyceraldehyde 3-phosphate dehydrogenase is a SET-binding protein and regulates cyclin B-cdk1 activity. Oncogene 25: , 4033–4042. |
[32] | Madeira A , Pommet JM , Prochiantz A , Allinquant B ((2005) ) SET protein (TAF1beta, I2PP2A) is involved in neuronal apoptosis induced by an amyloid precursor protein cytoplasmic subdomain. FASEB J 19: , 1905–1907. |
[33] | ten Klooster JP , Leeuwen I , Scheres N , Anthony EC , Hordijk PL ((2007) ) Rac1-induced cell migration requires membrane recruitment of the nuclear oncogene SET. EMBO J 26: , 336–345. |
[34] | Tanimukai H , Grundke-Iqbal I , Iqbal K ((2005) ) Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer’s disease. Am J Pathol 166: , 1761–1771. |
[35] | Li M , Makkinje A , Damuni Z ((1996) ) The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J Biol Chem 271: , 11059–11062. |
[36] | Arnaud L , Chen S , Liu F , Li B , Khatoon S , Grundke-Iqbal I , Iqbal K ((2011) ) Mechanism of inhibition of PP2A activity and abnormal hyperphosphorylation of tau by I2(PP2A)/SET. FEBS Lett 585: , 2653–2659. |
[37] | Dall E , Brandstetter H ((2016) ) Structure and function of legumain in health and disease. Biochimie 122: , 126–150. |
[38] | Basurto-Islas G , Grundke-Iqbal I , Tung YC , Liu F , Iqbal K ((2013) ) Activation of asparaginyl endopeptidase leads to Tau hyperphosphorylation in Alzheimer disease. J Biol Chem 288: , 17495–17507. |
[39] | Zhang Z , Song M , Liu X , Kang SS , Kwon IS , Duong DM , Seyfried NT , Hu WT , Liu Z , Wang JZ , Cheng L , Sun YE , Yu SP , Levey AI , Ye K ((2014) ) Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat Med 20: , 1254–1262. |
[40] | Mouzon B , Chaytow H , Crynen G , Bachmeier C , Stewart J , Mullan M , Stewart W , Crawford F ((2012) ) Repetitive mild traumatic brain injury in a mouse model produces learning and memory deficits accompanied by histological changes. J Neurotrauma 29: , 2761–2773. |
[41] | Wang Y , Zhang Y , Hu W , Xie S , Gong CX , Iqbal K , Liu F ((2015) ) Rapid alteration of protein phosphorylation during postmortem: Implication in the study of protein phosphorylation. Sci Rep 5: , 15709. |
[42] | Halfon S , Patel S , Vega F , Zurawski S , Zurawski G ((1998) ) Autocatalytic activation of human legumain at aspartic acid residues. FEBS Lett 438: , 114–118. |
[43] | Li DN , Matthews SP , Antoniou AN , Mazzeo D , Watts C ((2003) ) Multistep autoactivation of asparaginyl endopeptidase in vitro and in vivo . J Biol Chem 278: , 38980–38990. |
[44] | Dixon CE , Clifton GL , Lighthall JW , Yaghmai AA , Hayes RL ((1991) ) A controlled cortical impact model of traumatic brain injury in the rat. J Neurosci Methods 39: , 253–262. |
[45] | Ojo JO , Mouzon BC , Crawford F ((2016) ) Repetitive head trauma, chronic traumatic encephalopathy and tau: Challenges in translating from mice to men. Exp Neurol 275 Pt 3: , 389–404. |
[46] | Johnson VE , Stewart W , Smith DH ((2012) ) Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol 22: , 142–149. |
[47] | Liu F , Li B , Tung EJ , Grundke-Iqbal I , Iqbal K , Gong CX ((2007) ) Site-specific effects of tau phosphorylation on its microtubule asbly activity and self-aggregation. Eur J Neurosci 26: , 3429–3436. |
[48] | Gong CX , Lidsky T , Wegiel J , Zuck L , Grundke-Iqbal I , Iqbal K ((2000) ) Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain - Implications for neurofibrillary degeneration in Alzheimer’s disease. J Biol Chem 275: , 5535–5544. |
[49] | Jin N , Yin X , Gu J , Zhang X , Shi J , Qian W , Ji Y , Cao M , Gu X , Ding F , Iqbal K , Gong CX , Liu F ((2015) ) Truncation and activation of dual specificity tyrosine phosphorylation-regulated kinase 1A by calpain I: A molecular mechanism linked to tau pathology in Alzheimer disease. J Biol Chem 290: , 15219–15237. |
[50] | Patrick GN , Zukerberg L , Nikolic M , de la Monte S , Dikkes P , Tsai LH ((1999) ) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402: , 615–622. |
[51] | Jin N , Yin X , Yu D , Cao M , Gong CX , Iqbal K , Ding F , Gu X , Liu F ((2015) ) Truncation and activation of GSK-3beta by calpain I: A molecular mechanism links to tau hyperphosphorylation in Alzheimer’s disease. Sci Rep 5: , 8187. |
[52] | Saatman KE , Bozyczko-Coyne D , Marcy V , Siman R , McIntosh TK ((1996) ) Prolonged calpain-mediated spectrin breakdown occurs regionally following experimental brain injury in the rat. J Neuropathol Exp Neurol 55: , 850–860. |
[53] | Kampfl A , Posmantur R , Nixon R , Grynspan F , Zhao X , Liu SJ , Newcomb JK , Clifton GL , Hayes RL ((1996) ) mu-calpain activation and calpain-mediated cytoskeletal proteolysis following traumatic brain injury. J Neurochem 67: , 1575–1583. |
[54] | Zhao X , Posmantur R , Kampfl A , Liu SJ , Wang KK , Newcomb JK , Pike BR , Clifton GL , Hayes RL ((1998) ) Subcellular localization and duration of mu-calpain and m-calpain activity after traumatic brain injury in the rat: A casein zymography study. J Cereb Blood Flow Metab 18: , 161–167. |
[55] | Kupina NC , Nath R , Bernath EE , Inoue J , Mitsuyoshi A , Yuen PW , Wang KK , Hall ED ((2001) ) The novel calpain inhibitor SJA6017 improves functional outcome after delayed administration in a mouse model of diffuse brain injury. J Neurotrauma 18: , 1229–1240. |
[56] | Saatman KE , Abai B , Grosvenor A , Vorwerk CK , Smith DH , Meaney DF ((2003) ) Traumatic al injury results in biphasic calpain activation and retrograde transport impairment in mice. J Cereb Blood Flow Metab 23: , 34–42. |
[57] | Li M , Guo H , Damuni Z ((1995) ) Purification and characterization of two potent heat-stable protein inhibitors of protein phosphatase 2A from bovine kidney. Biochemistry 34: , 1988–1996. |
[58] | Wang X , Blanchard J , Kohlbrenner E , Clement N , Linden RM , Radu A , Grundke-Iqbal I , Iqbal K ((2010) ) The carboxy-terminal fragment of inhibitor-2 of protein phosphatase-2A induces Alzheimer disease pathology and cognitive impairment. FASEB J 24: , 4420–4432. |
[59] | Yin T , Lindley TE , Albert GW , Ahmed R , Schmeiser PB , Grady MS , Howard MA , Welsh MJ ((2013) ) Loss of Acid sensing ion channel-1a and bicarbonate administration attenuate the severity of traumatic brain injury. PLoS One 8: , 72379. |
[60] | Timofeev I , Nortje J , Al-Rawi PG , Hutchinson PJ , Gupta AK ((2013) ) Extracellular brain pH with or without hypoxia is a marker of profound metabolic derangement and increased mortality after traumatic brain injury. J Cereb Blood Flow Metab 33: , 422–427. |
[61] | Clausen T , Khaldi A , Zauner A , Reinert M , Doppenberg E , Menzel M , Soukup J , Alves OL , Bullock MR ((2005) ) Cerebral acid-base homeostasis after severe traumatic brain injury. J Neurosurg 103: , 597–607. |
[62] | DeSalles AA , Kontos HA , Becker DP , Yang MS , Ward JD , Moulton R , Gruemer HD , Lutz H , Maset AL , Jenkins L , Marmarou A , Muizelaar P ((1986) ) Prognostic significance of ventricular CSF lactic acidosis in severe head injury. J Neurosurg 65: , 615–624. |
[63] | Gupta AK , Zygun DA , Johnston AJ , Steiner LA , Al-Rawi PG , Chatfield D , Shepherd E , Kirkpatrick PJ , Hutchinson PJ , Menon DK ((2004) ) Extracellular brain pH and outcome following severe traumatic brain injury. J Neurotrauma 21: , 678–684. |
[64] | Enevoldsen EM , Cold G , Jensen FT , Malmros R ((1976) ) Dynamic changes in regional CBF, intraventricular pressure, CSF pH and lte levels during the acute phase of head injury. J Neurosurg 44: , 191–214. |
[65] | Castejon OJ ((2004) ) Lysosome abnormalities and lipofucsin content of nerve cells of oedematous human cerebral cortex. J Submicrosc Cytol Pathol 36: , 263–271. |
[66] | Liu Z , Jang SW , Liu X , Cheng D , Peng J , Yepes M , Li XJ , Matthews S , Watts C , Asano M , Hara-Nishimura I , Luo HR , Ye K ((2008) ) Neuroprotective actions of PIKE-L by inhibition of SET proteolytic degradation by asparagine endopeptidase. Mol Cell 29: , 665–678. |
[67] | Ishizaki T , Erickson A , Kuric E , Shamloo M , Hara-Nishimura I , Inacio AR , Wieloch T , Ruscher K ((2010) ) The asparaginyl endopeptidase legumain after experimental stroke. J Cereb Blood Flow Metab 30: , 1756–1766. |
[68] | Arif M , Wei J , Zhang Q , Liu F , Basurto-Islas G , Grundke-Iqbal I , Iqbal K ((2014) ) Cytoplasmic retention of protein phosphatase 2A inhibitor 2 (I2PP2A) induces Alzheimer-like abnormal hyperphosphorylation of Tau. J Biol Chem 289: , 27677–27691. |
[69] | Bancher C , Braak H , Fischer P , Jellinger KA ((1993) ) Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer’s and Parkinson’s disease patients. Neurosci Lett 162: , 179–182. |
[70] | Thal DR , Holzer M , Rub U , Waldmann G , Gunzel S , Zedlick D , Schober R ((2000) ) Alzheimer-related tau-pathology in the perforant path target zone and in the hippocampal stratum oriens and radiatum correlates with onset and degree of dementia. Exp Neurol 163: , 98–110. |
[71] | Takashima A ((2008) ) Hyperphosphorylated tau is a cause of neuronal dysfunction in tauopathy. J Alzheimers Dis 14: , 371–375. |
[72] | Alonso AC , Grundke-Iqbal I , Iqbal K ((1996) ) Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disasbles microtubules. Nat Med 2: , 783–787. |
[73] | Hu W , Zhang X , Tung YC , Xie S , Liu F , Iqbal K ((2016) ) Hyperphosphorylation determines both the spread and the morphology of tau pathology. Alzheimers Dement 12: , 1066–1077. |
[74] | Takeda S , Wegmann S , Cho H , DeVos SL , Commins C , Roe AD , Nicholls SB , Carlson GA , Pitstick R , Nobuhara CK , Costantino I , Frosch MP , Muller DJ , Irimia D , Hyman BT ((2015) ) Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat Commun 6: , 8490. |
[75] | Dai CL , Hu W , Tung YC , Liu F , Gong CX , Iqbal K ((2018) ) Tau passive immunization blocks seeding and spread of Alzheimer hyperphosphorylated Tau-induced pathology in 3xTg-AD mice. Alzheimers Res Ther 10: , 13. |
[76] | Iqbal K , Liu F , Gong C-X ((2016) ) Tau and neurodegenerative disease: The story so far. Nat Rev Neurol 12: , 15–27. |
[77] | Johnson GV , Jope RS ((1992) ) The role of microtubule-associated protein 2 (MAP-2) in neuronal growth, plasticity, and degeneration. J Neurosci Res 33: , 505–512. |
[78] | Doherty CP , O’Keefe E , Wallace E , Loftus T , Keaney J , Kealy J , Humphries MM , Molloy MG , Meaney JF , Farrell M , Campbell M ((2016) ) Blood-brain barrier dysfunction as a hallmark pathology in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 75: , 656–662. |
[79] | Holleran L , Kim JH , Gangolli M , Stein T , Alvarez V , McKee A , Brody DL ((2017) ) Axonal disruption in white matter underlying cortical sulcus tau pathology in chronic traumatic encephalopathy. Acta Neuropathol 133: , 367–380. |
[80] | Gong CX , Lidsky T , Wegiel J , Zuck L , Grundke-Iqbal I , Iqbal K ((2000) ) Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer’s disease. J Biol Chem 275: , 5535–5544. |


