
Moringa Oleifera Alleviates Homocysteine-Induced Alzheimer’s Disease-Like Pathology and Cognitive Impairments
Abstract
Alzheimer’s disease (AD) is multifactorial with unclear etiopathology. Due to the complexity of AD, many attempted single therapy treatments, like Aβ immunization, have generally failed. Therefore, there is a need for drugs with multiple benefits. Naturally occurring phytochemicals with neuroprotective, anti-amyloidogenic, antioxidative, and anti-inflammatory properties could be a possible way out. In this study, the effect of Moringa oleifera (MO), a naturally occurring plant with high antioxidative, anti-inflammatory, and neuroprotective effects, was evaluated on hyperhomocysteinemia (HHcy) induced AD-like pathology in rats. Homocysteine (Hcy) injection for 14 days was used to induce AD-like pathology. Simultaneous MO extract gavage followed the injection as a preventive treatment or, after injection completion, MO gavage was performed for another 14 days as a curative treatment. MO was found to not only prevent but also rescue the oxidative stress and cognitive impairments induced by Hcy treatment. Moreover, MO recovered the decreased synaptic proteins PSD93, PSD95, Synapsin 1 and Synaptophysin, and improved neurodegeneration. Interestingly, MO decreased the Hyc-induced tau hyperphosphorylation at different sites including S-199, T-231, S-396, and S-404, and at the same time decreased Aβ production through downregulation of BACE1. These effects in HHcy rats were accompanied by a decrease in calpain activity under MO treatment, supporting that calpain activation might be involved in AD pathogenesis in HHcy rats. Taken together, our data, for the first time, provided evidence that MO alleviates tau hyperphosphorylation and Aβ pathology in a HHcy AD rat model. This and previous other studies support MO as a good candidate for, and could provide new insights into, the treatment of AD and other tauopathies.
INTRODUCTION
Alzheimer’s disease (AD) is currently the most widely prevalent neurodegenerative disease affecting global health, leading to the deterioration of behavioral and cognitive capacities in aged individuals [1, 2]. The histopathology of the disease is marked by extracellular senile plaques and intracellular neurofibrillary tangles (NFTs) [3, 4], which are predominantly made up of amyloid-β (Aβ) peptides and hyperphosphorylated tau [5] respectively. Aβ peptides result from the successive cleavage of the amyloid-β protein precursor (AβPP) by the beta-amyloid precursor protein cleaving enzyme 1 (BACE1), or β-secretase, and the γ-secretase [6, 7] to form aggregates into plaques, while the hyperphosphorylated tau protein in the NFTs are due to the imbalance in the kinase/phosphatase system, with glycogen synthase kinase 3β (GSK3β) and protein phosphatase 2A (PP2A) mostly involved in this process [1, 8, 9]. Oxidative stress seen as lipids, proteins, and nucleic acids oxidations, is an incontestable player in the pathogenesis of AD as evidenced by multiple studies through causing PP2A inactivation and GSK3β activation [10– 12]. Oxidative stress, neuroinflammation, and calcium homeostasis disturbances, that are components of AD brains lead to increased Aβ production by activating BACE1 via activation of CDK5 by calpain [13]. Owing to the strong implication of oxidative stress in the pathogenesis of AD, agents with multiple antioxidative components could be a possible therapeutic option for this disease.
Homocysteine (Hcy), an intermediate product of the methionine cycle, is one of the major risk factors of AD [14]. It plays a critical role in the development of the disease through inducing oxidative stress, DNA damage, NMDA receptor activation [15– 20] and modifying both Aβ and tau pathways [21, 22]. In triple transgenic mice (3×Tg) model of the AD, Hcy was reported to exacerbate Aβ and tau lesions by increasing γ-secretase and CDK5 activities, respectively [21], and also through Aβ/fibrinogen interaction inducing its oligomerization and clots formation, compromising cerebral blood flow, thus exacerbating AD [22]. Hcy was also reported to induce tau hyperphosphorylation by modulating related tau kinases and phosphatases [23, 24]. The observed AD neuropathological lesions, including Aβ accumulation, tau hyperphosphorylation, synaptic protein loss and Wnt signaling and memory impairments in high methionine diet treated mice might also be associated with the resulting high plasma Hcy level [25].
Moringa oleifera (MO) belongs to the family of Moringaceae and is believed to be indigenous of the Indian subcontinent but is now distributed widely in many African and Asian countries [26]. The bioactive compounds, including vitamins, carotenoids, polyphenols, phenolic acids, flavonoids, alkaloids, glucosinolates, isothiocyanates, tannins and saponins, from various parts of the plant including leaves, roots, bark, gum, flowers, fruits, seeds, and seeds oil have been reported to have high nutritional and medicinal effects [27, 28]. MO has been reported to have many pharmacological qualities like antimicrobial, antihypercholesterolemic, antitumor, antidiabetic, anti-inflammatory, and antioxidant [29– 34]. MO was found to protect against focal cerebral ischemia in rats [35] as well as against oxidative DNA damage [36]. In AD, MO was reported to have a nootropic effect by improving colchicine-induced dysregulated lipid peroxidation, reduced glutathione, catalase, superoxide dismutase (SOD), acetylcholine, and choline acetyltransferase levels and activities [31, 37]. It also restored the disturbed brain monoamines to almost the control level [26], enhanced memory and protected from neurodegeneration [31]. Interestingly, toxicological studies have demonstrated that MO is safe even at higher doses. The leaves extract was found to be safe at a dose of 1000 mg/kg/body weight [38] and even as high as 2000 mg/kg where it was observed to enhance learning and memory and protect against pentylenetetrazol-induced convulsion at doses of 250– 2000 mg/kg [39]. No visible adverse reactions nor pathological changes were found in a single dose of 5000 mg/kg or a 14 days dose of 1000 mg/kg of aqueous MO extract [40]. It was also reported that the LD50 of oral ethanolic extract is as high as 6400 mg/kg [39] while the LD50 of acute oral, intraperitoneal toxicity study of aqueous extract was found to be 1585 mg/kg and no death was observed at an oral dose of as high as 6400 mg/kg/body weight [41].
Aging is the major risk factor associated with AD. Increasing life span through improved quality of life has contributed to a rising proportion of elderly population globally, translating to an increasing prevalence of AD. The complex etiology of the AD led to the failure of many attempted single therapy treatments [42– 44]. Thus, there is an urgent need for drugs with multiple effects and therefore naturally occurring phytochemicals with neuroprotective, anti-amyloidogenic, antioxidative and anti-inflammatory properties, which are characteristics of MO as well, could be a possible way out [45]. Extracts like EGb761 and HSS-888 have already shown antioxidative, anti-tau hyperphosphorylation and anti-Aβ potentials [24, 46]. Moreover, phytoconstituents like heptanol, Trans-linaloloxide and Linalool oxide in MO, have a good permeability of blood-brain barrier with low toxicity [47]. All of these make MO a strong therapeutic option for oxidative stress-related neurodegenerative diseases including AD. Moreover, till now no effect of MO has been reported on the two main characteristics of the AD: tau hyperphosphorylation and Aβ peptides production and aggregation. Therefore, in the present study, we investigated the effect of MO on these two main features of AD and their underlying mechanisms.
MATERIALS AND METHODS
Animals
84 male Sprague-Dawley rats (8 weeks old, 250±25 g) were purchased from the Experimental Animal Center of Tongji Medical College, Huazhong University of Science and Technology, and were housed six animals per cage with access to food and water under a 12:12 h reversed light-dark cycle. The rats were divided into 7 groups of 12 in each as: control, Hcy, preventive low, preventive high, treatment low, treatment high and positive control and treated (Table 1). All animal experiments were performed according to the ‘Policies on the Use of Animals and Humans in Neuroscience Research’ revised and approved by the Society for Neuroscience in 1995.
Table 1
Grouping and treatment of rats used in this study
| Groups | Injection | Gavage | |
| Control | 0.25 ml saline (0.9%) | then | 1 ml saline (0.9%) |
| Homocysteine | 400 μg/kg/day Hcy | then | 1 ml saline (0.9%) |
| Preventive low | 400 μg/kg/day Hcy | simultaneous | 200 mg/kg/day MO |
| Preventive high | 400 μg/kg/day Hcy | simultaneous | 400 mg/kg/day MO |
| Treatment low | 400 μg/kg/day Hcy | then | 200 mg/kg/day MO |
| Treatment high | 400 μg/kg/day Hcy | then | 400 mg/kg/day MO |
| Positive control | 400 μg/kg/day Hcy | then | 1 mg/kg/day SCR1693 |
Hcy, homocysteine; MO, Moringa oleifera.
Homocysteine
DL-Homocysteine (Hcy) (Sigma Chemical COSt. Louis, MO, USA) was dissolved in saline (0.9% NaCl) to a final concentration of 400μg/ml immediately before injection. 400μg/kg/day Hcy was injected for 14 days through vena caudalis every day from 9:00 am to 2:00 pm. The control group was injected with 0.25 ml of saline.
Moringa oleifera (MO) extraction
MO leaf powder was sourced from Moringa Smart (China). The powder was macerated exhaustively in 80% methanol. Briefly, 200 g of the MO powder was soaked in 1000 mL of 80% methanol and allowed at room temperature with continuous shaking for 2 days. The obtained extract was later filtered through Whatman filter paper and concentrated using a vacuum rotary evaporator at 40°C. The condensed residue was of slurry nature and dark green. This extract was kept in a deep freezer at – 80°C until use.
Oral gavage
The rats were divided into 7 groups as stated earlier. The MO extract was weighted and dissolved in saline (0.9% NaCl) to a final concentration of 200 mg/ml immediately before gavage. Using a stainless steel oral gavage needle, the animals were given intra-gastric appropriate doses (see Table 1). For the first 14 days, the preventive low and high groups did not receive any treatment. Only in the last 14 days, they were given the simultaneous Hcy injection and MO gavage. At the same time (last 14 days) the control and Hcy groups were given oral normal saline, the treatment low and high were given oral MO while the positive control was given oral SCR1693 following the first 14 days of injection. The doses were selected according to guidelines from current literature [29, 31, 33]. The training for the water maze was started six days before the last treatment day, and the test was carried out one day after the last treatment day and animals were sacrificed immediately after the water maze test for further experiment.
Reagents
The primary antibodies employed in this study and their properties are listed in Table 2. Secondary antibodies for western blotting were from Amersham Pharmacia Biotech (Little Chalfont, Buckinghamshire, UK). Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody (A10040) use for IF was from Invitrogen. Hoechst, Thioflavin-S were from Sigma Chemical Co (St. Louis, MO, USA). Cresyl violet for Nissl staining was from Beyotime (China).
Table 2
Antibodies employed in this study
| Antibody | Specific | Cat Number | Type | Dilution | Source |
| pS199 | Phosphorylated tau at Ser199 | 44734G | pAb | 1:1000 for WB | Thermo Fisher |
| pT231 | Phosphorylated tau at Thr231 | 11110 | pAb | 1:1000 for WB | Signalway Antibody CollegePark, MD, USA |
| pS396 | Phosphorylated tau at Ser396 | 11102 | pAb | 1:1000 for WB | Signalway Antibody CollegePark, MD, USA |
| pS404 | Phosphorylated tau at Ser404 | 11112 | pAb | 1:1000 for WB | Signalway Antibody CollegePark, MD, USA |
| Tau-1 | Non-phosphorylated tau at Ser198/199/202 | MAB3420-KC | mAb | 1:1000 for WB | Millipore, Temecula, CA, USA |
| Tau-5 | Total tau | ab80579 | mAb | 1:1000 for WB | Abcam, Cambridge, MA, USA |
| PP2Ac | PP2A catalytic subunit | 2038 | pAb | 1:1000 for WB | Cell Signaling Danvers, MA, USA |
| PP2Ac-mL309 | Methylated PP2Ac at Lue309 | MA518072 | mAb | 1:1000 for WB | Millipore, Temecula, CA, USA |
| GSK3β | Total GSK3β | 12456 | mAb | 1:1000 for WB | Cell Signaling Danvers, MA, USA |
| GSK3β-pS9 | Phosphorylated GSK3β at Ser9 | 9323 | pAb | 1:1000 for WB | Cell Signaling Danvers, MA, USA |
| CaMKII | Total CaMKII | 3362 | pAb | 1:1000 for WB | Cell Signaling Danvers, MA, USA |
| P-CaMKII | Phosphorylated CaMKII at Thr286 | 3361 | pAb | 1:1000 for WB | Cell Signaling Danvers, MA, USA |
| CDK5 | Total CDK5 | sc-6247 | mAb | 1:1000 for WB | Santa Cruz, CA, USA |
| p35 | Total p35 | sc-820 | pAb | 1:1000 for WB | Santa Cruz, CA, USA |
| Calpain | Anti calpain1 | MA112434 | mAb | 1:1000 for WB | Millipore, Temecula, CA, USA |
| Spectrin | Anti spectrin alpha chain | MAB1622 | mAb | 1:1000 for WB | Millipore, Temecula, CA, USA |
| AβPP | Full length of total AβPP | 2452S | pAb | 1:1000 for WB | Cell Signaling Danvers, MA, USA |
| AβPPβ | Anti-human sAβPPβ antibodies | 18957 | pAb | 1:1000 for WB | IBL |
| BACE1 | Total BACE1 | 5606S | pAb | 1:1000 for WB | Cell Signaling Danvers, MA, USA |
| PSD93 | Post Synaptic Density 93 aa 352– 366 | ab2930 | pAb | 1:1000 for WB | Abcam, Cambridge, MA, USA |
| PSD95 | PSD95 N-terminal | 507 | pAb | 1:1000 for WB | Cell Signaling Danvers, MA, USA |
| Synapsin1 | Synapsin-1 C-term | AB1543 | pAb | 1:1000 for WB | Millipore, Temecula, CA, USA |
| Synaptophysin | Synaptophysin aa 250– 350 (C terminal) | 701503 | pAb | 1:1000 for WB | Millipore, Temecula, CA, USA |
| MAP2 | Total MAP2 | AB5622 | pAb | 1:200 for IF | Millipore, Temecula, CA, USA |
| β-actin | Total actin | ab6276 | mAb | 1:1000 for WB | Abcam, Cambridge, MA, USA |
mAb, monoclonal antibody; pAb, polyclonal antibody; WB, western blot; IF, immunofluorescence.
Tissue preparation
After anesthesia with chloral hydrate the rats were perfused transcardially with 400 ml of 0.9% filtered saline then the brain was dissected and the hippocampus removed and frozen at – 80°C for further use. For Golgi staining, the brain was removed and placed in the Golgi solution (1 g potassium chromate, 1 g mercuric chloride, 0.8 g potassium chloride and 100 ml double-distilled water) for three weeks while changing the solution every two days. For Nissl staining and immunofluorescence, the saline perfusion was followed by 400 ml phosphate buffer containing 4% paraformaldehyde and the brains were dissected out and post-fixed in the same 4% paraformaldehyde solution overnight at 4°C. After that, the brains were transferred sequentially into 30% and 40% sucrose solutions. Then coronal sections were cut using vibratome (VT 1000 s, Leica, Germany).
Measurement of superoxide dismutase and malondialdehyde
After anesthesia, blood samples were collected from the orbital arteries and immediately centrifuged at 5000×g for 10 min at 4°C. The supernatants (serum) were used for SOD and malondialdehyde (MDA) tests. For brain samples, hippocampi were immediately removed, washed with PBS and homogenized in saline (0.9% NaCl) at (tissue weight (g): saline volume (mL) = 1:9) and centrifuged at 10,000×g for 15 min at 4°C and the supernatants were used for SOD and MDA tests. Total superoxide dismutase activity in the serum was determined with a commercial kit from Jiancheng Bioengineering Institute (Central Road, Nanjing, China), based on the ability of the xanthine-xanthine oxidase system to inhibit the oxidation of hydroxylamine. The mauve product (nitrite (N)) produced by the oxidation of hydroxylamine has an absorbance at 550 nm. One unit of SOD activity was defined as the amount that reduces the absorbance at 550 nm by 50%. MDA, a metabolite of lipid peroxides, was used as an indicator of lipids peroxidation. MDA was determined with a commercial kit from Jiancheng Bioengineering Institute (Central Road, Nanjing, China) by measuring the malondialdehyde formed by the thiobarbituric acid reaction. N-butanol was used for extraction during this process. An equal amount of N-butanol was added into each tube of a mixed solution prepared according to the instructions, and then centrifuged the newly mixed solution at 10,000×g for 10 min at 4°C, and the supernatants were measured at 532 nm.
ELISA assay for Aβ1 - 42 measurement
The amount of Aβ1 - 42 was selectively detected in protein soluble fractions by using a sandwich ELISA kit according to manufacturer’s instructions (Elabscience Inc.). Briefly, hippocampi were immediately removed and weighed, cut in small pieces and rinsed in ice-cold PBS (0.01 M, pH = 7.4) homogenized in the same PBS at (tissue weight (g): PBS volume (mL) = 1:9) and centrifuged for 5 min at 5000×g at 4°C and the supernatants were used for the assay. The protein concentration was measured by bicinchoninic acid (BCA) (Pierce, Rockford, IL, USA) method, and 100μl containing 300μg of proteins from the soluble fractions were incubated in the microplate pre-coated with the anti-Aβ1 - 42 antibody for 90 min at 37°C. Wells were washed and then incubated with the biotinylated detection antibody for 60 min at 37°C. Samples were washed then incubated with an HRP-labeled conjugate for 30 min at 37°C, then washed again and incubated with a substrate reagent for 20 min at 37°C and finally the stop solution was added. Plates were read with a microplate reader at 450 nm.
Western blotting
The hippocampi were homogenized in a buffer containing 50 mM Tris, pH 7.4, 40 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 1.5 mM Na3VO4, 50 mM NaF, 10 mM sodium pyrophosphate, 10 mM sodium β-glycerophosphate, supplemented with protease inhibitors cocktail. The homogenates were mixed with one-third of sample buffer (200 mM Tris–HCl, 8% sodium dodecyl sulfate and 40% glycerol), boiled for 10 min, sonicated and centrifuged at 12,000×g for 10 min. The supernatants were transferred into new tubes, and protein concentrations were measured using the bicinchoninic acid (BCA) (Pierce, Rockford, IL, USA) method. The same amount of proteins was separated by 10% SDS-polyacrylamide gel electrophoresis gel and then transferred to nitrocellulose membrane. After blocking in 3% non-fat milk for 1 h at 25°C, the membranes were then incubated with primary antibodies at 4°C overnight. Then after three washes with TBST, the membranes were incubated with anti-rabbit or anti-mouse IgG conjugated to IRDyeTM (800CW) for 1 h at 25°C followed by another three washes with TBST and visualized using the Odyssey Infrared Imaging System (Licor Biosciences, Lincoln, NE, USA).
Combined immunofluorescence and Thioflavin-S staining
Sections were cut at 30μm using vibratome (VT 1000 s, Leica, Germany). The sections were permeated for 30 min with a mixture of 0.5% Triton X-100 in PBS and blocked in 5% BSA in PBS for 1 h at room temperature. Then the brain slices were incubated with the MAP2 primary antibody overnight at 4°C. Slices were washed with PBS and incubated with Rabbit IgG (H + L) Highly Cross-Adsorbed Secondary Antibody in PBS for 2 h at 25°C. Then after washing with PBS, cell nuclei were visualized by incubation with Hoechst for 10 mn at 25°C. Slices were washed 3 times with PBS and mounted onto glass slides, allowed to dry in the dark and then incubated with Thioflavin-S staining solution for 8 mn at 25°C. The slices were then successively passed through 2 changes of 50% ethanol and 2 changes of double distilled water and finally sealed using 50% glycerin in PBS. Images were obtained using a 710-laser scanning confocal microscope (Zeiss).
Nissl staining
After 30μm coronal sections were cut, sections were mounted on gelatin-coated slides and were incubated in Cresyl violet for 10 min at 25°C, dehydrated through 50%, 75%, 95%, and 100% alcohol, then cleared in xylene and cover-slipped with neutral balsam. Pictures were taken using a light microscope.
Golgi-cox staining
Sections of 80μm thickness were cut and placed on gelatin-coated glass slides. After rinsing with double-distilled water, slides were incubated in ammonium hydroxide for 50 min. The slides were rinsed again with distilled water, then incubated for 30 min in a black and white film developer, diluted 1:9 with water, in the dark. Following another rinse in distilled water, the slides were dehydrated in subsequent concentrations of alcohol 50%, 70% and 95% for 1 min each, then in 3 changes of 100% alcohol for 5 min each. The slides were then cleared for 15 min in CXA (chloroform, xylene, alcohol at 1:1:1) solution. Slides were mounted with neutral balsam and cover-slipped and allowed to dry for 24 h then visualized under a light microscope.
Morris water maze test
The maze consisted of a circular black painted pool, filled with water colored with non-toxic black ink. A platform, 12 cm in diameter, was submerged 1.5 cm below the surface of the water in one of the 4 imaginary quadrants. The rats were trained in water maze to find the hidden platform for 6 consecutive days, 3 trials per day with a 30 min interval from 8 am to 2 pm. For each trial, the rat started from the middle of one of the other three quadrants facing the wall of the pool and ended when the animal climbed on the platform. The rats were allowed to search for the platform for a maximum of 60 s after which if they did not find the platform they were gently guided to the platform and the rats were allowed to remain on the platform for 30 s. The swimming pathway and escape latency of the rats to find the hidden platform were recorded by Noldus video tracking system (Ethovision, Noldus Information Technology, Holland). The learning and memory were tested on the seventh day, the platform removed from the pool and the rats were allowed to search for 60 s. The longer a rat stayed in the target quadrant, the better it scored in the spatial memory.
Statistical analysis
Data were expressed as mean±SD and analyzed using GraphPad Prism 6.05 for Windows statistical software (GraphPad Software, La Jolla California USA, http://www.graphpad.com). Statistical significance was determined by one-way ANOVA procedure followed by Student-Newman-Keuls post hoc test with 95% confidence and Student’s two-tailed t-test. A level of p < 0.05 was accepted as statistically significant.
RESULTS
MO improved the homocysteine-induced oxidative stress
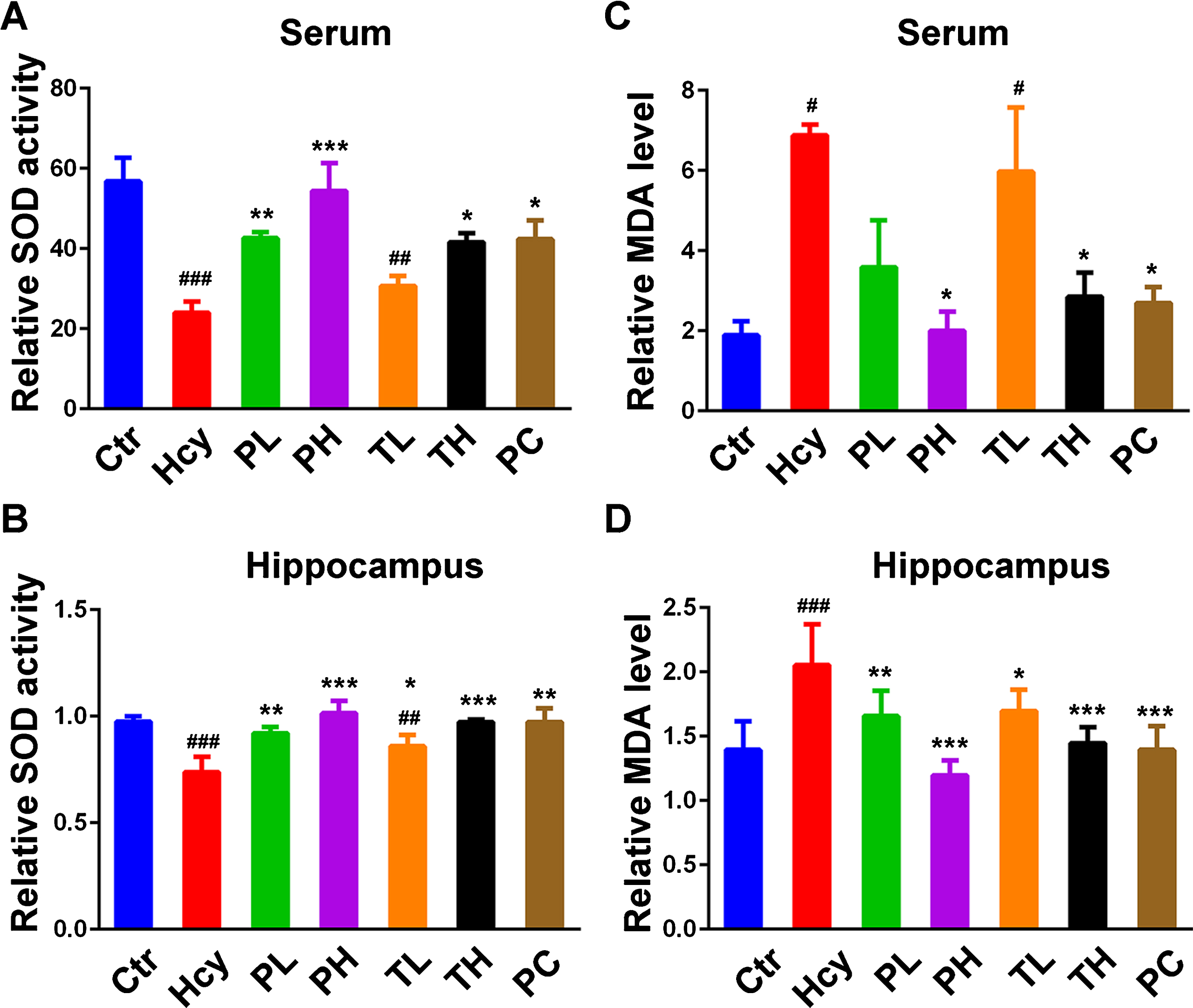
HHcy has been reported to induce increase oxidative stress [15] while MO reduced it [30, 32]. To evaluate the HHcy induced oxidative stress and the effect of MO in this study, the levels of oxidative markers SOD and MDA were measured in the blood and hippocampal lysates from the experimental animals (Fig. 1A-D). Hcy treatment significantly decreased SOD activity (Fig. 1A, B) and increased MDA level (Fig. 1C, D) when compared with the control. However, treatment with MO significantly prevented and rescued the Hcy induced decrease in SOD activity in both serum (Fig. 1A) and hippocampal lysate (Fig. 1B) in a dose-dependent manner, with the preventive group even having more effect than the positive control (SCR1693). At the same time, MO treatment decreased the level of serum (Fig. 1C) and hippocampal (Fig. 1D) MDA elevated by the Hcy injection in the rats. The preventive MO treatment particularly the high dose one, had better restoration effects on the Hcy disturbed oxidative stress markers SOD and MDA. These results confirm the antioxidative effect of MO treatment in the Hcy injected rats.
Fig.1
MO attenuated HHcy-induced oxidative damage. 84 male SD rats were divided into Control (Ctr) (saline), Homocysteine (Hcy), Preventive Low (PL), Preventive High (PH), Treatment Low (TL), Treatment High (TH) and Positive Control (PC) (SCR1693) groups. A, B) Relative SOD activity in serum and hippocampal lysates. C, D) Relative level of MDA in serum and hippocampal lysates. Compared with control group, HHcy rats showed decreased activity of SOD and increased level of MDA. Both prevention and curative treatments with MO markedly increased SOD activity and decreased MDA level. The data were expressed as mean±SD. Data were from 3 different animals in each group (n = 3). #p < 0.05, # #p < 0.01, # # #p < 0.001 versus control; *p < 0.05, **p < 0.01, ***p < 0.001 versus Hcy.
MO attenuated the cognitive impairments induced by homocysteine
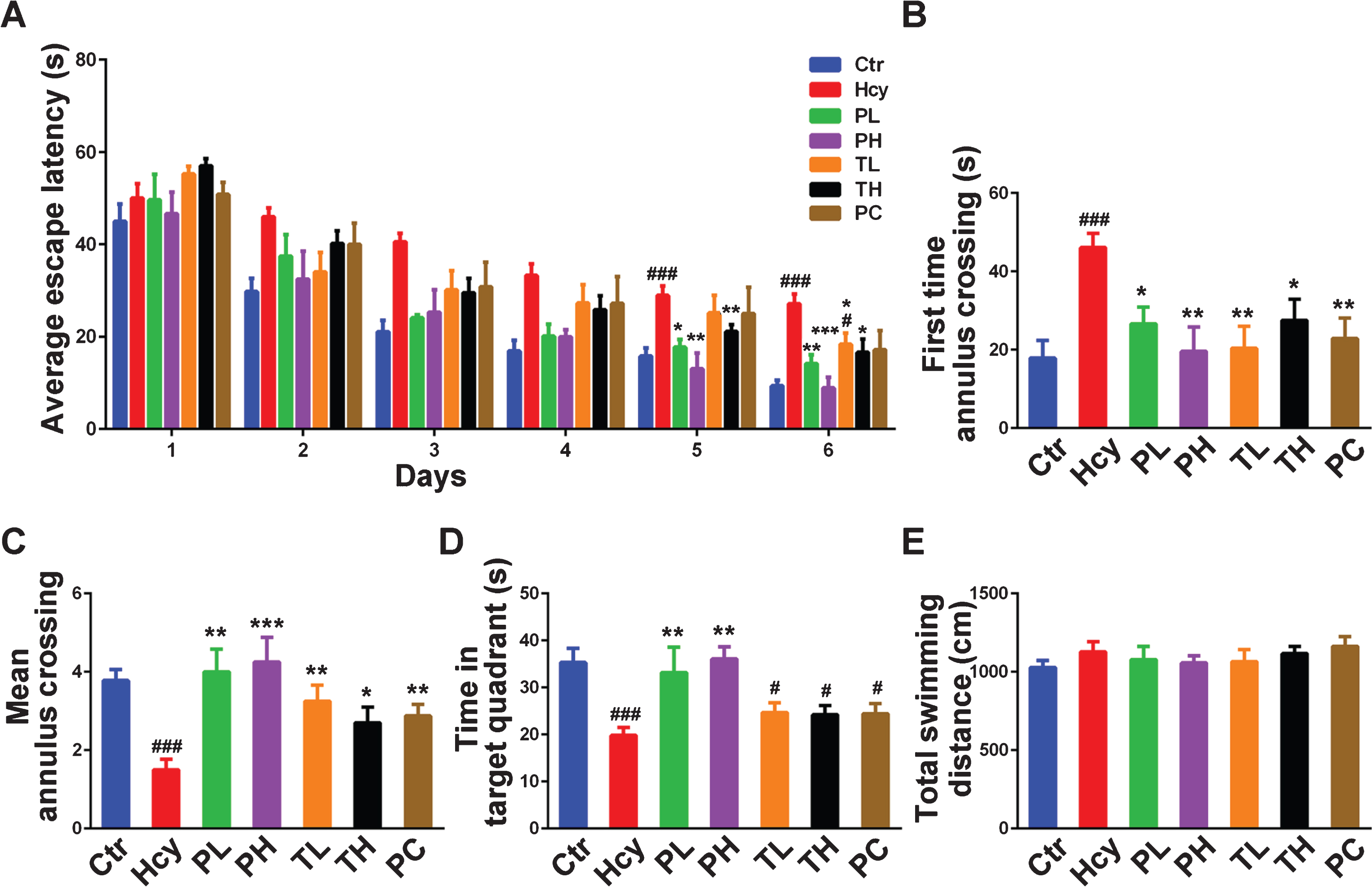
Accumulating evidence showed the deleterious effects of HHcy through inducing oxidative stress and its downstream effects, thus leading to the development or potentiation of AD-like pathologies including memory impairments [23–25, 48]. HHcy could also be a potential diagnostic marker in AD [49]. To evaluate the effect of MO intervention on the Hcy induced cognitive impairments, Morris Water Maze (MWM) test was carried out. This tests the hippocampus-dependent spatial learning and memory. Hcy injection led to significant learning and memory disabilities in the injected rats when compared with the control (Fig. 2A-D). The Hcy injected animals showed an increase in the escape latency to find the platform while the MO treatment decreased it to a level similar to the control and positive control (Fig. 2A, B). The mean annulus crossing, or number of platform position crossing, was significantly reduced by the Hcy injection and MO intervention increased it to control level with better effects seen with preventive doses (Fig. 2C). Similarly, the time spent in the target quadrant was also decreased by Hcy and brought to a level equivalent to the control by the preventive MO treatment (Fig. 2D). However, although increased compared to Hcy group, the curative MO treatment and the positive control showed no significant differences (Fig. 2D). Moreover, neither Hcy nor MO did affect the motor activity because the total swimming distance remained comparable among all groups (Fig. 2E). These results indicate that MO treatment significantly prevented and improved Hcy induced learning and memory deficits.
Fig.2
MO treatment improved HHcy-induced spatial memory impairments. A) Escape latency to find the hidden platform in Morris water maze for the six training days. B) Escape latency to find the position of the platform during the test. C-E) Mean number of platform crossing, the time spent in the platform quadrant and the total swimming distance during the 60-s test. HHcy rats showed impaired spatial learning and memory and either preventive or curative treatment with MO rescued this. The data were expressed as mean±SD (n = 12). #p < 0.05, # # #p < 0.001 versus control; *p < 0.05, **p < 0.01, ***p < 0.001 versus Hcy.
MO decreased tau hyperphosphorylation in hyperhomocysteinemia rats
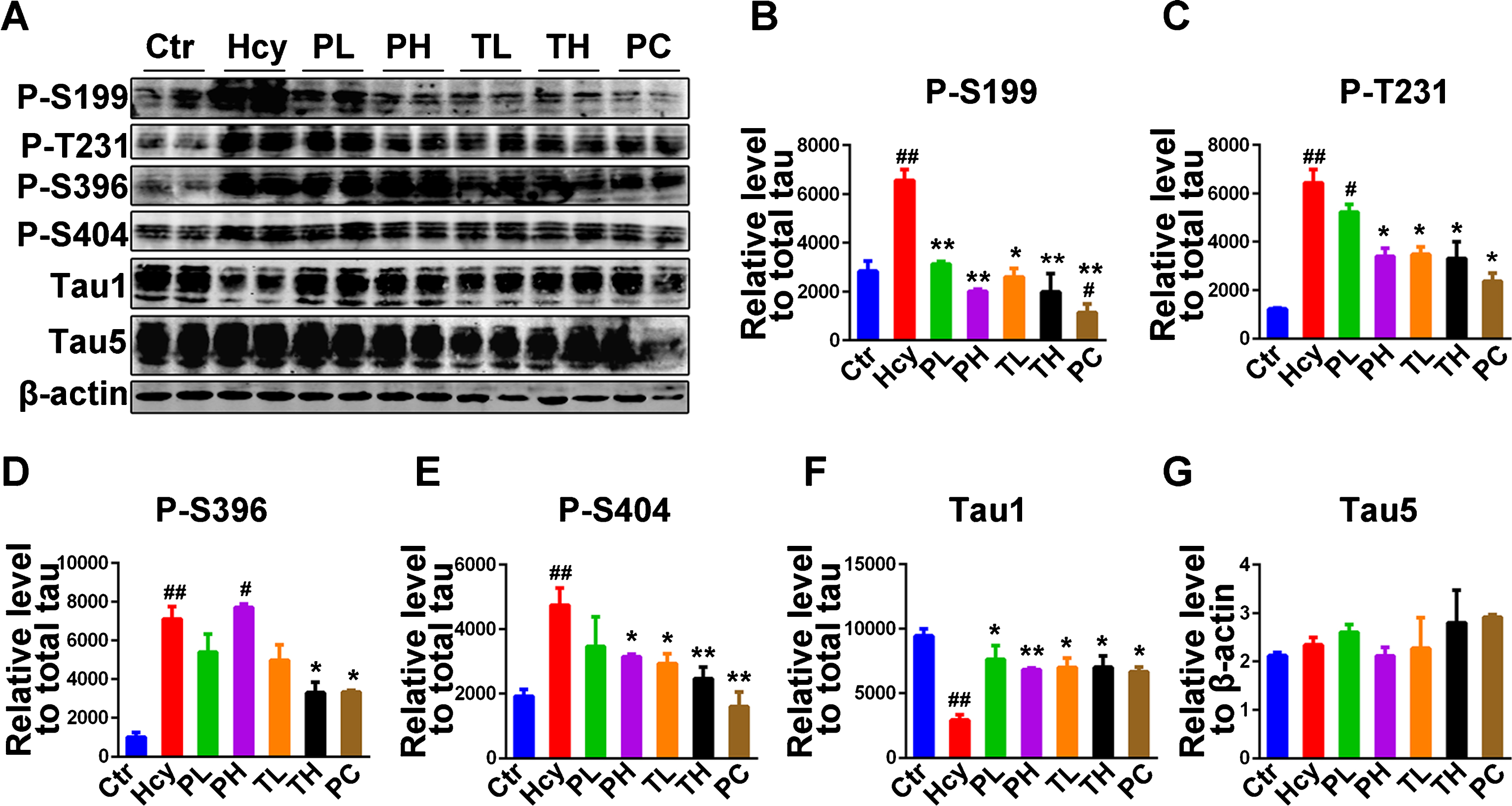
Tau phosphorylation is one of the two hallmarks of AD. Hcy was reported to induce tau hyperphosphorylation in rats [24] as well as exacerbating tau and Aβ pathology in AD animal model [21]. As an antioxidant MO might modulate the Hcy induced tau hyperphosphorylation. To answer this question, the phosphorylation levels of tau were evaluated by western blot in hippocampal lysates from treated animals. Different tau phosphorylation sites were investigated including Ser199 (p-S199), Thr231 (p-T231), Ser396 (p-S396), and Ser404 (p-S404). Hcy injection induced tau hyperphosphorylation at S199, T231, S396 and S404 in the hippocampi of injected rats when compared to the control (Fig. 3A-E). As expected, MO treatment prevented and rescued the tau hyperphosphorylation at all these sites. The low preventive dose of MO showed no significant difference with the Hcy group at T231 (Fig. 3A, C), S396 (Fig. 3A, D), and S404 (Fig. 3A, E). At the same time Tau1, recognizing non-phosphorylated tau at Ser198/199/202 sites, immunoreactivity was significantly lower in the Hcy treated rats when compared with the control and MO treatment increased it to a level similar with the control (Fig. 3A, F). Moreover, no significant difference was found, among all groups, in the immunoreactivity of Tau5 epitope recognizing the total tau (Fig. 3A, G). Overall, our data suggest that MO treatment effectively mitigated the Hcy induced tau hyperphosphorylation in the injected rats.
Fig.3
Treatment with MO mitigated tau phosphorylation induced by HHcy. A) Levels of tau phosphorylation were measured by western blotting in the hippocampus after treatment (see method). B-E) Quantitative analysis of the blots of phosphorylated tau probed with several phosphorylated-tau antibodies, and (F) non-phosphorylated tau probed with Tau1 antibody, all normalized with total tau (Tau5) level. G) Total tau was normalized to β-actin. The significant increase in tau phosphorylation at several studied sites was decreased by MO administration. The data were expressed as mean±SD (n = 6). #p < 0.05, # #p < 0.01 versus control; *p < 0.05, **p < 0.01 versus Hcy.
MO decreased tau phosphorylation by modulating tau-related kinases and phosphatase
The phosphorylation status of the tau protein is regulated by the kinases/phosphatases system which was found to be disrupted in AD [50]. Hcy was also reported to decrease PP2A activity, the main phosphatase of tau, by increasing its phosphorylation at Tyr307, and increase GSK3β activity by decreasing its Ser9 phosphorylation [23, 48]. PP2A methylation at Leu309 is also indicative of its activity [51, 52]. To investigate the effects of MO on the underlying mechanism of the Hcy induced tau phosphorylation, western blot was performed to check the protein levels and activation status of different kinases and phosphatase (Fig. 4A-J). Recently we found PP2A is inactivated by Hcy through increased Try307 phosphorylation [24]. Here in, the total PP2A protein level was not affected (Fig. 4A, B). However, its methylation at Leu309 was significantly decreased in the Hcy injected rats when compared with the control while MO treatment efficiently prevented and rescued it to levels comparable to the control with high preventive dose significantly higher than the control (Fig. 4A, C).
Fig.4
Treatment with MO modulated tau related phosphatase and kinases. A) Western blotting of total protein phosphatase 2A (PP2Ac), methylated PP2Ac at Leu309 (M-L309), total glucose synthase kinase 3β (GSK3β), phosphorylated GSK3β at Ser9 (P-S9), total cyclin dependent kinase 5 (CDK5), activators of CDK5, p35 and p25, the total Ca2 +/calmodulin-dependent protein kinase (CaMKII) and the phosphorylated CaMKII at Thr286 (P-CaMKII) from hippocampal lysates. B-J) Quantitative analysis of the blots, total PP2A, GSK3β, CDK5, p35, p25, and CaMKII were normalized to β-actin and phosphorylated GSK3β and CaMKII and methylated PP2Ac were normalized to their total proteins. The data were expressed as mean±SD (n = 6). #p < 0.05, # #p < 0.01, # # #p < 0.001, # # # #p < 0.0001 versus control; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus Hcy.
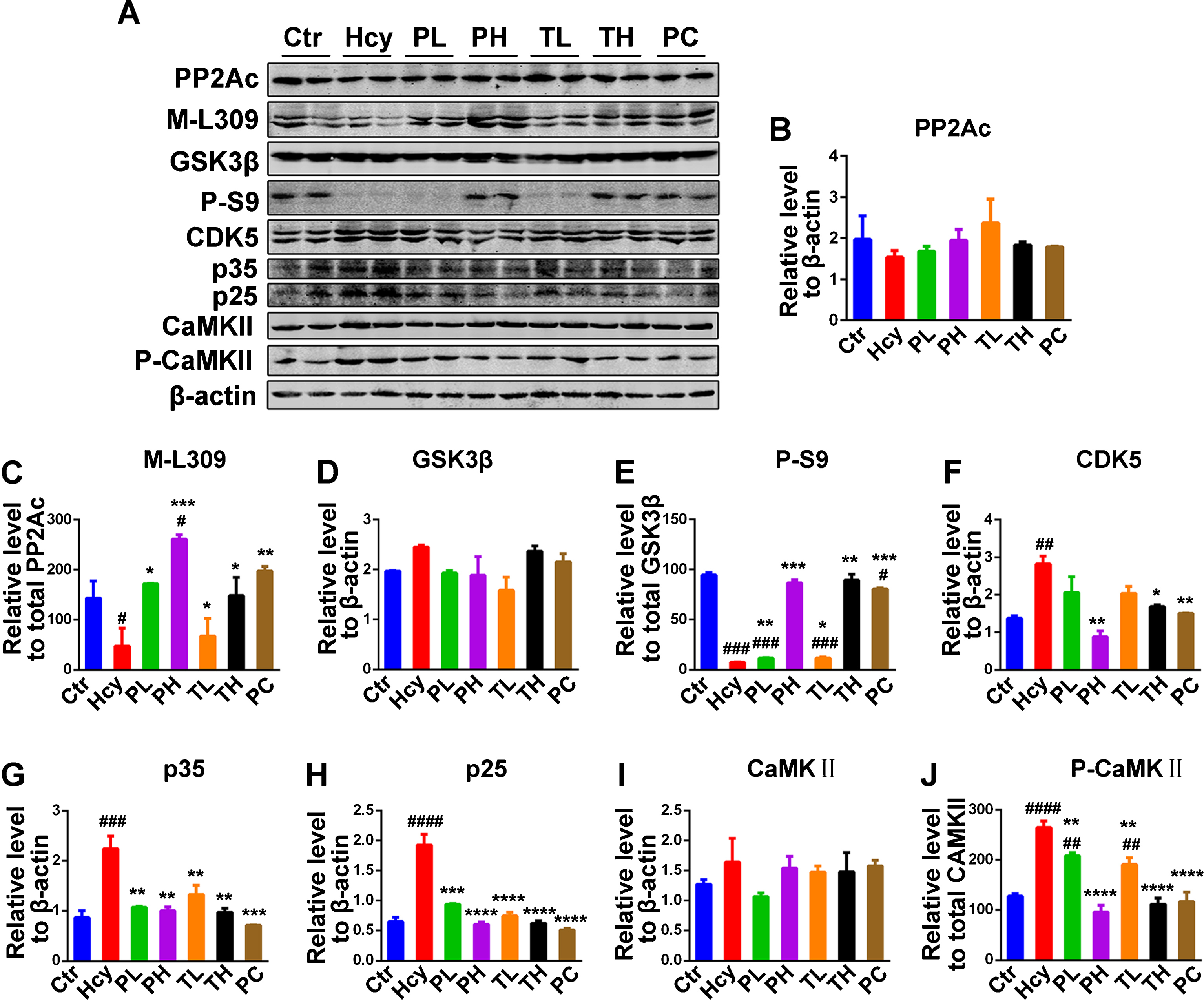
GSK3β is one of the key kinase related with tau phosphorylation [53] and associated with oxidative stress [10]. Thus, the protein level and Ser9 phosphorylation (P-S9), as a measure of its activity, were evaluated. The GSK3β level was unchanged among the groups, however, its phosphorylation at Ser9 was significantly reduced in the Hcy injected rats compared to the control (Fig. 4A, D, E). The decreased level of phosphorylated GSK3β at Ser9 was recovered to control level in the high doses treatments while low doses groups, even though significantly higher than the Hcy, are significantly lower compared to the control (Fig. 4A, E). Another way the Hcy induces its deleterious effect in the brain is through activation of NMDA receptors leading to calcium influx into the cells [54–56]. Therefore, the calcium-dependent kinases CaMKII and CDK5 were also evaluated. The preventive and curative treatments of MO significantly attenuated the increase in the levels of CDK5 and its activators p35 and p25 similar to the control level (Fig. 4A, F-H). The phosphorylated-CaMKII (P-CaMKII), the active form of this kinase was also increased in Hcy group and recovered to control level with high doses of MO but also significantly decreased in the lower doses when compared with the Hcy group (Fig. 4A, J). The total level of CaMKII remained unchanged (Fig. 4A, I). Taken together, these results strongly suggest that MO treatment decreased tau phosphorylation by significantly modulating tau-related phosphatase and kinases.
MO mitigated the induced Aβ pathology in homocysteine injected rats
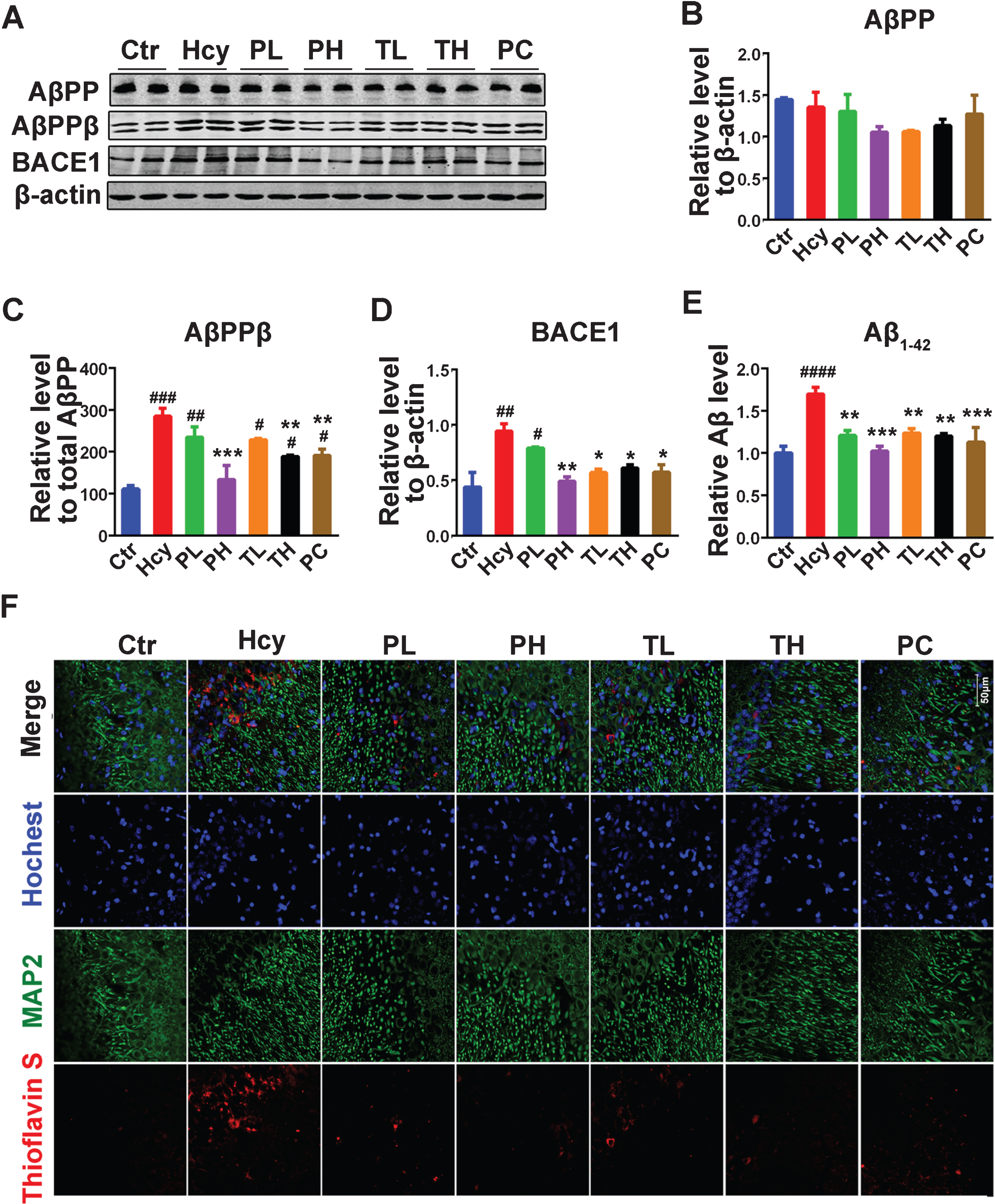
Aβ is the other hallmark of AD and is believed to be the trigger in AD pathogenesis [57, 58]. Hcy was reported to exacerbate Aβ pathology but also induce it in animal models of AD [21, 25]. We therefore evaluated the effect of MO treatment on the Hcy induced Aβ pathology. Aβ peptides result from the action of BACE1 on AβPP leading to AβPPβ, which is then cleaved by γ-secretase to produce Aβ peptides [6]. In this study the levels of the full-length AβPP, AβPPβ and BACE1 were estimated by western blotting with hippocampal lysates (Fig. 5A-D). No significant difference was found in the level of the full-length AβPP (Fig. 5A, B). However, the protein level of AβPPβ was significantly increased in the Hcy treated animals compared with the control (Fig. 5A, C). Although only preventive high dose MO decreased it to control level, preventive low, curative low and high doses also decreased it compared to the Hcy with significant decrease observed with the curative high dose (Fig. 5A, C). Consistent with the increase in AβPPβ level, the protein level of BACE1 was also increased in the Hcy group compared with control, and MO treatment significantly reduced it to control level except for the preventive low dose of MO (Fig. 5A, D). To corroborate these findings, we measured the level of Aβ1 - 42 in the hippocampal lysates and by immunofluorescence (Fig. 5E, F). As expected, the ELISA result showed a significant increase in the amount of Aβ1 - 42 in the Hcy treated animals when compared with the control while no significant difference was observed when the MO treatment groups were compared with the control (Fig. 5E). Interestingly, when compared with the control group, the Hcy injected rats brain slices had an increase in the Aβ aggregate fluorescence as shown by the combined immunofluorescence/Thioflavin-S staining (Fig. 5F), while the fluorescence in the MO treatment groups was similar to that of the control and the positive control groups. These data collectively suggest that MO effectively attenuated the Aβ induced pathology in the treated animals.
Fig.5
MO attenuated Aβ pathology induced by HHcy. A) Levels of total amyloid-β protein precursor (AβPP), the beta AβPP cleaving enzyme 1 (BACE1) cleaved AβPP (AβPPβ) and the total BACE1 were estimated through western blotting. B-D) Quantification of the western blots with AβPP and BACE1 normalized to β-actin and AβPPβ normalized to total AβPP (n = 6). E) ELISA assay for Aβ1 - 42. F) Representative confocal microscopy images of combined immunofluorescence/Thioflavin-S staining of brain slices of rats, Channels: Thioflavin S (red), MAP2 (green), nuclear dye Hoechst 33342 (blue), chat bar = 50μm. The data were expressed as mean±SD (n = 3) #p < 0.05, # #p < 0.01, # # #p < 0.001, # # # #p < 0.0001 versus control; *p < 0.05, **p < 0.01, ***p < 0.001 versus Hcy.
MO decreased the homocysteine-induced elevated calpain activity in the rats
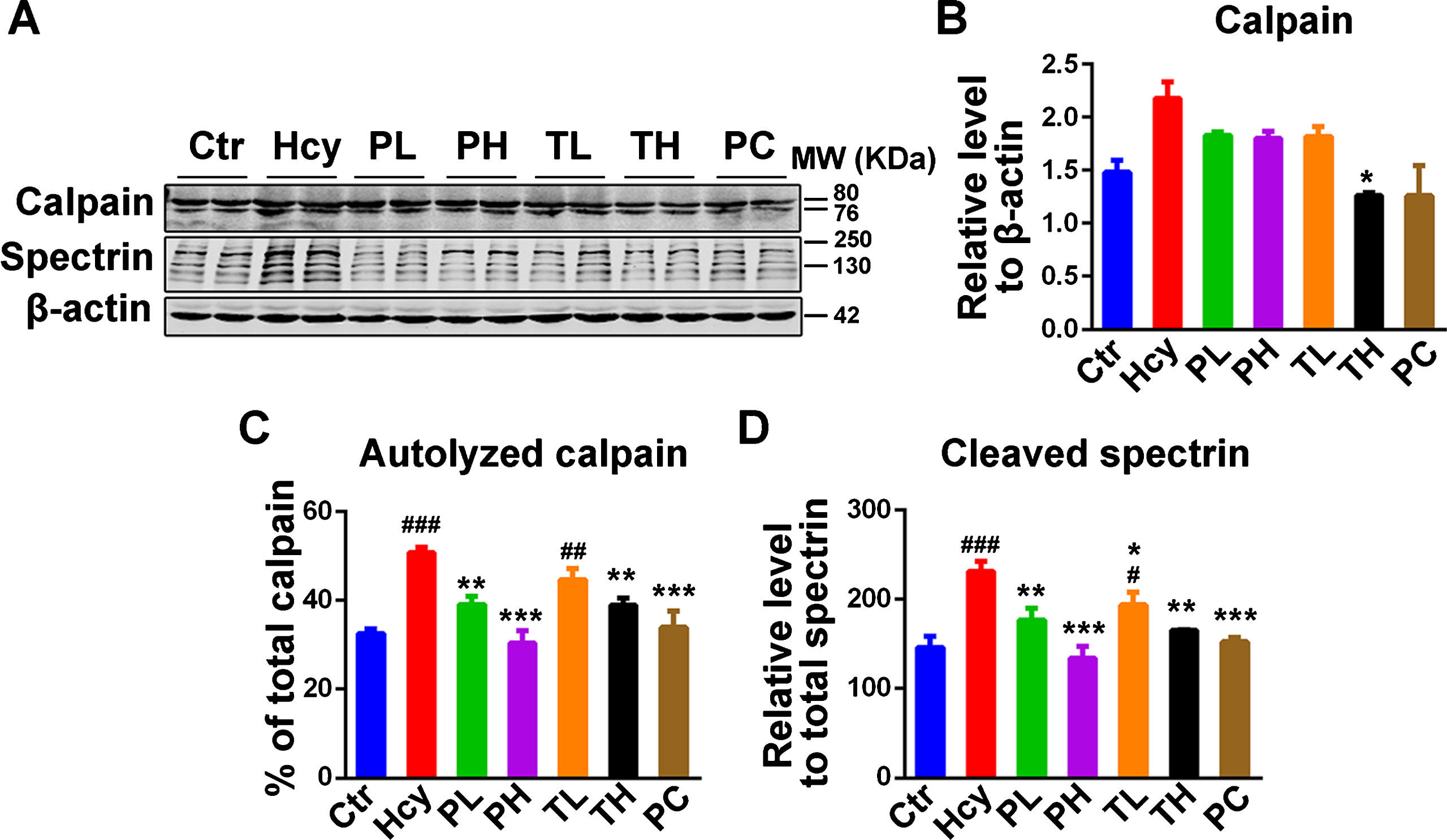
Calpain is a cysteine protease that is activated calcium-dependently in the cell [59] and is ubiquitously expressed in cells of human and other organisms. This enzyme is found to be implicated in AD where oxidative stress and activation of NMDA receptor lead to calcium influx thus hyperactivating this enzyme. Upon activation, calpain mediates the cleavage of p35 to p25 (the most potent form of these CDK5 activators), DARPP-32, promotes BACE1 expression, AβPP processing, and increases GSK3β activity by cleaving its inhibitory domain [60–62]. All of these result in increased Aβ plaques formation and tau hyperphosphorylation in AD. In the present model of HHcy, we assumed that the increased oxidative stress might induce calpain activation and therefore driving the downstream AD pathology including increased tau phosphorylation and Aβ aggregation. Thus, the calpain protein level and its activity were evaluated. On one hand, no significant difference in the calpain protein level was seen among the groups and even though it is apparently high in the Hcy group, it does not reach significance (Fig. 6A, B). On the other, the calpain activity as measured by the level of its autolyzed fragment (76 kDa) and the cleavage of its substrate spectrin, was significantly increased in the hippocampal lysates of Hcy injected rats compared to the control ones, while MO significantly prevented and rescued it (Fig. 6A, C, D). This indicates that the MO could significantly downregulate the increased calpain activity resulting from the Hcy induced oxidative stress.
Fig.6
MO treatment attenuated the increase in calpain activity due to HHcy. A) Western blotting of total calpain and its cleaved substrate spectrin. The increased cleaved spectrin as measure of calpain activity was reduced by MO treatments. B-D) Quantification of western blots, calpain was normalized to β-actin and cleaved spectrin to total spectrin. The data were expressed as mean±SD (n = 6). #p < 0.05, # #p < 0.01, # # #p < 0.001 versus control; *p < 0.05, **p < 0.01, ***p < 0.001 versus Hcy.
MO ameliorated neurodegeneration induced by homocysteine injection in rats
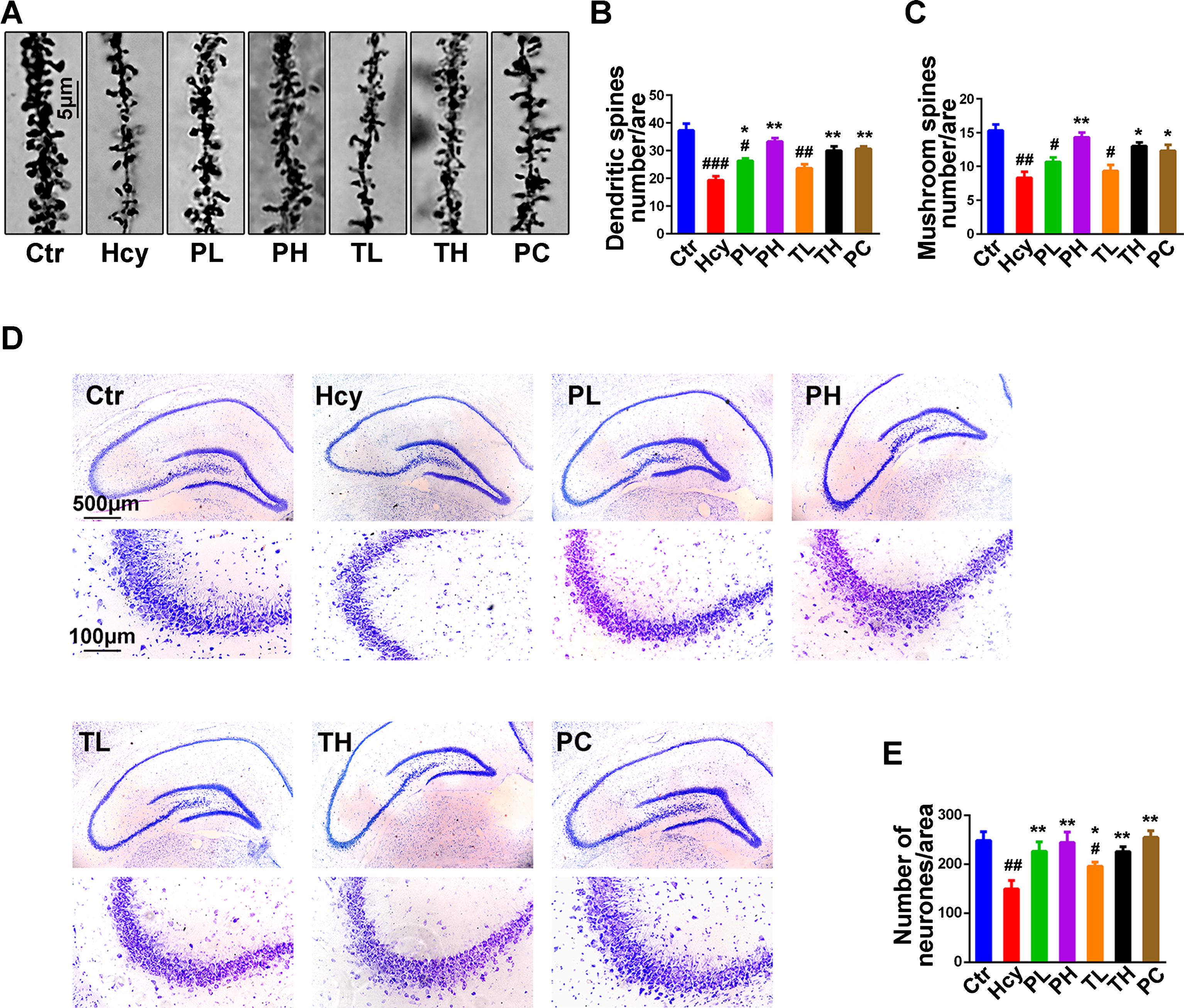
Dendritic spines are essential for memory as they encapsulate postsynaptic elements and undergo dynamic changes in response to synaptic activity, changing their morphology to accommodate the incoming stimulus. This is the basis of synaptic plasticity which is central to learning and memory [63, 64]. Furthermore, Hcy injection is associated with dendritic spines and neuronal loss leading to memory impairments [23]. To investigate on how MO administration ameliorated the Hcy induced memory impairments in the injected animals, Golgi staining and Nissl staining were performed (Fig. 7). Hcy induced a significant loss in the total number of dendritic spines and the mushroom type spines when compared with the control (Fig. 7A-C), and also led to a decrease in the number of neurons (Fig. 7D, E). The treatment with MO significantly improved these impairments especially at higher doses. These results indicate that MO significantly prevented and rescued the Hcy induced synaptic impairments and neuronal cell loss.
Fig.7
Treatment with MO reversed dendritic spine and neuronal loss. After two weeks of Hcy (400μg/kg/day) treatment, with simultaneous or post-injection two weeks MO treatments, the rats were sacrificed following behavior test. A) Representative Golgi staining of hippocampal neurons, chat bar = 5μm. B, C) Quantification of total dendritic spines and the mushroom type spines densities from randomly selected dendritic segments of randomly selected hippocampal neurons. D, E) Representative Nissl staining images and the quantification of neuron density, chart bar = 500 and 100μm for low and high magnifications respectively. The data were expressed as mean±SD (n = 3), #p < 0.05, # #p < 0.01, # # #p < 0.001 versus control; *p < 0.05, **p < 0.01 versus Hcy.
MO recovered a panel of synaptic proteins in homocysteine injected rats
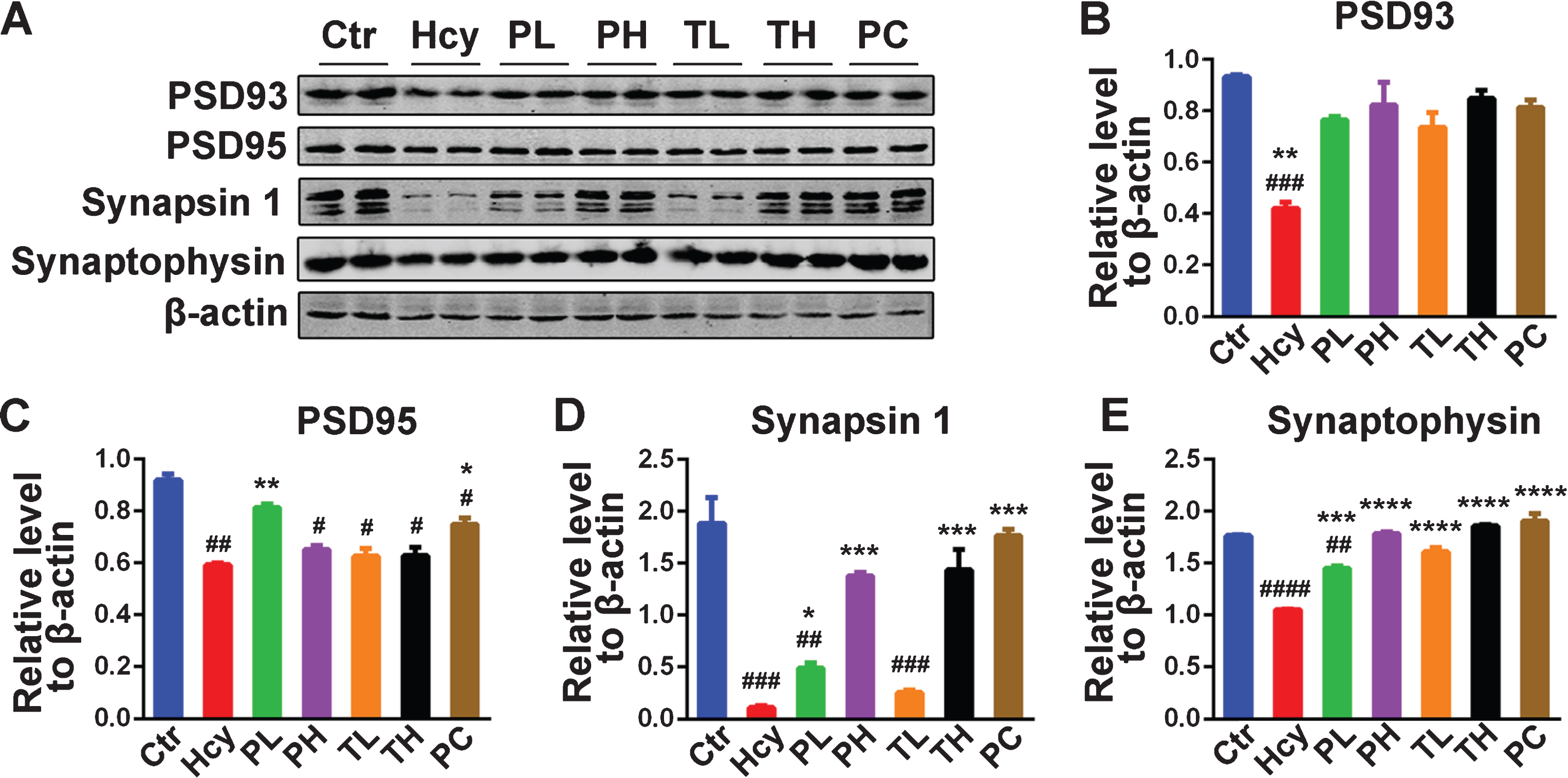
Synaptic plasticity plays a central role in learning and memory, and synaptic integrity is dependent upon stably expressed synaptic proteins. To understand the molecular basis of MO-mediated memory improvement in this study, the levels of some synaptic proteins were investigated by mean of western blot (Fig. 8). Hcy injection induced a tremendous deterioration in the amount of synaptic proteins including PSD93, PSD95, Synapsin 1 and Synaptophysin in the hippocampus of the injected rats when compared with that of the control animals (Fig. 8A-E). The administration of MO, as preventive or curative doses, significantly ameliorated this effect, for PSD93 (Fig. 8A, B) and Synaptophysin (Fig. 8A, E), to levels comparable with the control. However, only high doses of MO rescued Synapsin 1 to control level. This altogether implies that MO had potential in preventing and restoring synaptic proteins loss induced by Hcy in rats.
Fig.8
MO recovered memory-related proteins levels. A) Levels of PSD93, PSD95, Synapsin 1 and Synaptophysin were detected by western blotting in the hippocampus and β-actin was used as loading control. B-E) Quantitative analysis of the blots showed that Hcy dramatically decreased the expression of these synaptic proteins in the hippocampus and either preventive or curative treatment with MO reversed these effects. The data were expressed as mean±SD (n = 6). #p < 0.05, # #p < 0.01, # # #p < 0.001, # # # #p < 0.0001 versus control; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus Hcy.
DISCUSSION
Neurodegenerative diseases are mostly age related, and AD as the most common one is a characteristic illustration. With Aβ plaques, NFTs, neuronal and synaptic loss as histopathological signatures, AD is clinically characterized by gradual loss of short-term memory, a progressive deterioration of cognition and behavior which may affect thinking, planning, judgment and social skills, and finally overt dementia culminating into an inability to carry out daily life [65]. Oxidative stress plays a critical role in the development of the disease [10–13], and HHcy is an established environmental AD risk factor [14–20] by inducing oxidative stress. As previously reported, we induced AD-like pathology by Hcy injection in rats in order to study the effect of a medicinal plant, MO, with strong synergic antioxidant, anti-inflammatory and neuroprotective properties [31, 66–68]. In line with a previous study [31], we found that MO decreased the level of oxidative stress and neurodegeneration induced by Hcy. Most importantly MO decreased the Hcy induced tau hyperphosphorylation and Aβ pathology. To have a line of comparison for the effect of MO in this setting we make use of SCR1693, a synthesized hybrid compound composed of an acetylcholinesterase inhibitor and a calcium channel blocker. SCR1693 was found to improve memory impairments [23, 69, 70], cell death and hippocampal neuron loss [70], inhibited Aβ production and induced tau dephosphorylation [71]. Moreover, this compound has provided some positive effects in HHcy setting, including improved memory deficits, neurodegeneration, synaptic loss and tau hyperphosphorylation [23]. Therefore, we used this compound as a positive control.
In our study, we found that Hcy injection induced an increase in MDA and a decrease in SOD indicating an increase in oxidative stress. MO reversed these effects as preventive or curative treatments in both serum and hippocampal lysates, with more effect observed with the high doses. This could be attributed to the high content of antioxidants in MO as reported previously [28].
Increase in oxidative stress markers correlates with a decline in cognitive ability [72]. Oxidative stress triggers the inactivation of PP2A [73]. Activation of NMDA receptors by Hcy leads to a myriad of events that eventually lead to neurodegeneration and memory impairments [19, 20, 56]. For example, calcium influx due to activation of NMDA receptors may lead to calpain activation which leads to the cleavage of the inhibitory domain (Ser9) of GSK3β and therefore increasing its activity and resulting in tau hyperphosphorylation [10]. Calpain can also cleave the activator of CDK5 p35 into its most active form p25 also leading to tau hyperphosphorylation [21]. Hcy was also reported to increase tau phosphorylation via inhibiting PP2A through decreased methylation and increased phosphorylation [74]. In the present study, Hcy induced cognitive impairments which were prevented and recovered by the MO.
Tau hyperphosphorylation results in NFTs which is a histological mark of AD, and the level of NFTs significantly correlates with the progression of the disease [75, 76]. Here we found that tau phosphorylation was increased by the Hcy and significantly reduced by the MO. Tau hyperphosphorylation in the AD is due to the imbalance between the kinases and phosphatases. As previously reported Hcy and oxidative stress both lead to inactivation of PP2A and activation of GSK3β. We also found a decrease in the methylation of PP2A as well as an increase in the activity of GSK3β which were both recovered by MO administration. However, we also observed an increase in the protein level of CDK5, recovered by MO. This may be due to impaired degradation or increased synthesis, which are not evaluated here, and therefore need further investigations. The increase in CDK5 activators p35 and p25 might also be related to calpain activity as previously mentioned. P25/CDK5 inhibition has been reported to attenuate tauopathy in a model of frontotemporal dementia [77]. CaMKII a calcium-dependent kinase can be auto-phosphorylated in the presence of calcium [78]. In this study, no difference was observed in the total protein level of CaMKII. However, its phosphorylation at Thr286 (most active form) was increased by Hcy and rescued or recovered by MO.
Aβ is believed to be the trigger in the pathogenesis of AD. It has been reported that elevated levels of Aβ1 - 40 and Aβ1 - 42 were associated with increased levels of oxidation products from proteins, lipids and nucleic acids in AD hippocampus and cortex [79], and oxidative stress increased the expression of presenilin 1 and BACE1 and its activity [80, 81]. Oxidative stress and increased activation of CDK5 were also reported to increase the expression of BACE1 through increasing its transcription factors [13]. Here we observed an increase in the protein level of AβPPβ which is justified by the increased in the BACE1 protein level leading to an increase in the Aβ1 - 42 protein level and Aβ fluorescence in the Hcy injected animals which were reduced by MO treatments. From our result, it could be speculated that MO might decrease Aβ processing and increase its clearance. It has been reported that hyperhomocysteinemia increases Aβ production by the activation of γ-secretase [82] while GSK3β is known to promote the amyloidogenic processing of AβPP by forming a complex with presenilins and promoting their γ-secretase activities [83, 84]. Our present study showed that supplementation of MO could inhibit GSK3β. Therefore, MO may attenuate Aβ production through the inhibition of GSK3β activity and subsequently reducing the γ-secretase activities. Moreover, it is known that BACE1 is the rate-limiting enzyme in processing AβPP to Aβ and we reported an increase in BACE1 and AβPPβ protein levels in the Hcy treated animal which were significantly reduced following preventive or curative treatment with MO. MO might also influence Aβ clearance as suggested by the result where MO decreased Aβ protein level in the brain lysates of the treated animals. More especially, the immunofluorescence result showed a decrease in Aβ aggregates in the brain of MO treated animals. However, we have not evaluated Aβ clearance markers like ATP-binding cassette transporter A1 (ABCA1), an increase of which is suggestive of increased Aβ clearance [85].
In normal physiological conditions, calpain plays a biomodulator role in Ca2 +-regulated processes including signal transduction, cell proliferation, cell cycle progression, differentiation, apoptosis [86] and its enzymatic activity is tightly regulated by Ca2 + that activates it and the endogenous protein calpastatin that inhibits it [59, 87]. However, in pathological conditions, including AD, an increase in calpain protein level [88–90] and its activity as measured by the level of its autolyzed fragments [91] and its cleaved substrate spectrin [61, 92–95] were reported. Here, although the protein level of calpain was found to be increased in the HHcy rats compared to the control, no significant difference was observed among all groups. However, in accordance with previous reports, its activity, assessed by the level of its autolyzed fragments and cleaved spectrin, was significantly increased. MO administration recovered this increased activity. This could explain the increase in the CDK5 activators p35 and p25 and the increase in the level of BACE1 reported in the present study.
Normal functional synapses require a precise pre- and postsynaptic communications and are essential for learning and memory [63, 96]. Here we found a decrease in the pre- and postsynaptic proteins PSD93, PSD95, Synapsin 1 and Synaptophysin, following Hcy injection. MO treatments significantly rescued these impairments. This decrease could be attributed to the increased oxidative stress induced by the Hcy. Oxidative stress and increased activation of CDK5 were reported to increase the expression of BACE1 through increasing its transcription factors [13]. A previous study also found that synaptic loss was recovered by decreasing BACE1 in a p25/CDK5 model of neurodegeneration [97]. We also reported an increase in BACE1 protein level which was rescued by MO. This could explain the recovery effect of MO on the synaptic proteins. Altogether, our findings are consistent with the fact that Hcy induces AD-like pathology including behavioral deficits, tau hyperphosphorylation, Aβ accumulation and synaptic and neuronal loss. In our study, we found that MO more especially at high doses recovered these impairments and we also provided the underlying mechanisms. This could be attributed to the previously reported antioxidative, anti-inflammatory and neuroprotective properties of MO. Downregulation of calpain activity following MO treatment might play a central role in this process. However further researches are needed to investigate whether some molecules in the MO that have the ability to pass through the blood-brain barrier might have a direct effect. To the best of our knowledge, this is the first report about the effect of MO on the two hallmarks of AD. This study provides a new insight into the treatment of AD by naturally occurring phytochemicals.
ACKNOWLEDGMENTS
This work was supported in part by grants from National Natural Science Foundation of China (81571255, 31771114, 31528010, and 81660223), Innovative Research Groups of the National Natural Science Foundation of China (81721005), grant from the Ministry of Science and Technology of China (2016YFC1305800) and the Academic Frontier Youth Team Project to Xiaochuan Wang from Huazhong University of Science and Technology.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0091r1).
REFERENCES
[1] | Wang JZ , Wang ZH , Tian Q ((2014) ) Tau hyperphosphorylation induces apoptotic escape and triggers neurodegeneration in Alzheimer’s disease. Neurosci Bull 30: , 359–366. |
[2] | Association AS ((2013) ) 2013 Alzheimer’s disease facts and figures. Alzheimers Dement 9: , 208–245. |
[3] | Iqbal K , Alonso AD , Gondal JA , Gong CX , Haque N , Khatoon S , Sengupta A , Wang JZ , Grundke-Iqbal I ((2000) ) Mechanism of neurofibrillary degeneration and pharmacologic therapeutic approach. J Neural Transm Suppl 59: , 213–222. |
[4] | Geylis V , Steinitz M ((2006) ) Immunotherapy of Alzheimer’s disease (AD): From murine models to anti-amyloid beta (Abeta) human monoclonal antibodies. Autoimmun Rev 5: , 33–39. |
[5] | Braak H , Braak E , Grundke-Iqbal I , Iqbal K ((1986) ) Occurrence of neuropil threads in the senile human brain and in Alzheimer’s disease: A third location of paired helical filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci Lett 65: , 351–355. |
[6] | Reitz C , Brayne C , Mayeux R ((2011) ) Epidemiology of Alzheimer disease. Nat Rev Neurol 7: , 137–152. |
[7] | Li YM , Xu M , Lai MT , Huang Q , Castro JL , DiMuzio-Mower J , Harrison T , Lellis C , Nadin A , Neduvelil JG , Register RB , Sardana MK , Shearman MS , Smith AL , Shi XP , Yin KC , Shafer JA , Gardell SJ ((2000) ) Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405: , 689–694. |
[8] | Wang JZ , Grundke-Iqbal I , Iqbal K ((1996) ) Restoration of biological activity of Alzheimer abnormally phosphorylated tau by dephosphorylation with protein phosphatase-2A, -2B and -1. Brain Res Mol Brain Res 38: , 200–208. |
[9] | Wang JZ , Wu Q , Smith A , Grundke-Iqbal I , Iqbal K ((1998) ) Tau is phosphorylated by GSK-3 at several sites found in Alzheimer disease and its biological activity markedly inhibited only after it is prephosphorylated by A-kinase. FEBS Lett 436: , 28–34. |
[10] | Feng Y , Xia Y , Yu G , Shu X , Ge H , Zeng K , Wang J , Wang X ((2013) ) Cleavage of GSK-3beta by calpain counteracts the inhibitory effect of Ser9 phosphorylation on GSK-3beta activity induced by H(2)O(2). J Neurochem 126: , 234–242. |
[11] | Garcia L , Garcia F , Llorens F , Unzeta M , Itarte E , Gomez N ((2002) ) PP1/PP2A phosphatases inhibitors okadaic acid and calyculin A block ERK5 activation by growth factors and oxidative stress. FEBS Lett 523: , 90–94. |
[12] | Mondragón-Rodríguez S , Perry G , Zhu X , Moreira PI , Acevedo-Aquino MC , Williams S ((2013) ) Phosphorylation of tau protein as the link between oxidative stress, mitochondrial dysfunction, and connectivity failure: Implications for Alzheimer’s disease. Oxid Med Cell Longev 2013: , 1–6. |
[13] | Chami L , Checler F ((2012) ) BACE1 is at the crossroad of a toxic vicious cycle involving cellular stress and beta-amyloid production in Alzheimer’s disease. Mol Neurodegener 7: , 52. |
[14] | Drazen JM ((2012) ) Transparency for clinical trials–the TEST Act. N Engl J Med 367: , 863–864. |
[15] | Seshadri S ((2006) ) Elevated plasma homocysteine levels: Risk factor or risk marker for the development of dementia and Alzheimer’s disease? J Alzheimers Dis 9: , 393–398. |
[16] | Perna AF , Ingrosso D , De Santo NG ((2003) ) Homocysteine and oxidative stress. Amino Acids 25: , 409–417. |
[17] | Kuszczyk M , Gordon-Krajcer W , Lazarewicz JW ((2009) ) Homocysteine-induced acute excitotoxicity in cerebellar granule cells in vitro is accompanied by PP2A-mediated dephosphorylation of tau. Neurochem Int 55: , 174–180. |
[18] | Boldyrev AA , Johnson P ((2007) ) Homocysteine and its derivatives as possible modulators of neuronal and non-neuronal cell glutamate receptors in Alzheimer’s disease. J Alzheimers Dis 11: , 219–228. |
[19] | Ganapathy PS , White RE , Ha Y , Bozard BR , McNeil PL , Caldwell RW , Kumar S , Black SM , Smith SB ((2011) ) The role of N-methyl-D-aspartate receptor activation in homocysteine-induced death of retinal ganglion cells. Invest Ophthalmol Vis Sci 52: , 5515–5524. |
[20] | Poddar R , Paul S ((2009) ) Homocysteine-NMDA receptor-mediated activation of extracellular signal-regulated kinase leads to neuronal cell death. J Neurochem 110: , 1095–1106. |
[21] | Li JG , Chu J , Barrero C , Merali S , Pratico D ((2014) ) Homocysteine exacerbates beta-amyloid pathology, tau pathology, and cognitive deficit in a mouse model of Alzheimer disease with plaques and tangles. Ann Neurol 75: , 851–863. |
[22] | Chung YC , Kruyer A , Yao Y , Feierman E , Richards A , Strickland S , Norris EH ((2016) ) Hyperhomocysteinemia exacerbates Alzheimer’s disease pathology by way of the beta-amyloid fibrinogen interaction. J Thromb Haemost 14: , 1442–1452. |
[23] | Xia Y , Liu R , Chen R , Tian Q , Zeng K , Hu J , Liu X , Wang Q , Wang P , Wang XC , Wang JZ ((2014) ) Novel multipotent AChEI-CCB attenuates hyperhomocysteinemia-induced memory deficits and Neuropathologies in rats. J Alzheimers Dis 42: , 1029–1039. |
[24] | Zeng K , Li M , Hu J , Mahaman YAR , Bao J , Huang F , Xia Y , Liu X , Wang Q , Wang JZ , Yang Y , Liu R , Wang X ((2018) ) Ginkgo biloba extract EGb761 attenuates Hyperhomocysteinemia-induced AD like tau hyperphosphorylation and cognitive impairment in rats. Curr Alzheimer Res 15: , 89–99. |
[25] | Tapia-Rojas C , Lindsay CB , Montecinos-Oliva C , Arrazola MS , Retamales RM , Bunout D , Hirsch S , Inestrosa NC ((2015) ) Is L-methionine a trigger factor for Alzheimer’s-like neurodegeneration? Changes in Abeta oligomers, tau phosphorylation, synaptic proteins, Wnt signaling and behavioral impairment in wild-type mice. Mol Neurodegener 10: , 62. |
[26] | Ganguly R , Guha D ((2008) ) Alteration of brain monoamines & EEG wave pattern in rat model of Alzheimer’s disease & protection by Moringa oleifera. Indian J Med Res 128: , 744–751. |
[27] | Mbikay M ((2012) ) Therapeutic potential of Moringa oleifera leaves in chronic hyperglycemia and dyslipidemia: A review. Front Pharmacol 3: , 24. |
[28] | Leone A , Spada A , Battezzati A , Schiraldi A , Aristil J , Bertoli S ((2015) ) Cultivation, genetic, ethnopharmacology, phytochemistry and pharmacology of moringa oleifera leaves: An overview. Int J Mol Sci 16: , 12791–12835. |
[29] | Fakurazi S , Sharifudin SA , Arulselvan P ((2012) ) Moringa oleifera hydroethanolic extracts effectively alleviate acetaminophen-induced hepatotoxicity in experimental rats through their antioxidant nature. Molecules 17: , 8334–8350. |
[30] | Tan WS , Arulselvan P , Karthivashan G , Fakurazi S ((2015) ) Moringa oleifera flower extract suppresses the activation of inflammatory mediators in lipopolysaccharide-stimulated RAW 264.7 macrophages via NF-kappaB pathway. Mediators Inflamm 2015: , 720171. |
[31] | Sutalangka C , Wattanathorn J , Muchimapura S , Thukham-mee W ((2013) ) Moringa oleifera mitigates memory impairment and neurodegeneration in animal model of age-related dementia. Oxid Med Cell Longev 2013: , 695936. |
[32] | Karthivashan G , Kura AU , Arulselvan P , Md Isa N , Fakurazi S ((2016) ) The modulatory effect of Moringa oleifera leaf extract on endogenous antioxidant systems and inflammatory markers in an acetaminophen-induced nephrotoxic mice model. PeerJ 4: , e2127. |
[33] | Gupta R , Mathur M , Bajaj VK , Katariya P , Yadav S , Kamal R , Gupta RS ((2012) ) Evaluation of antidiabetic and antioxidant activity of Moringa oleifera in experimental diabetes. J Diabetes 4: , 164–171. |
[34] | Tiloke C , Phulukdaree A , Chuturgoon AA ((2013) ) The antiproliferative effect of Moringa oleifera crude aqueous leaf extract on cancerous human alveolar epithelial cells. BMC Complement Altern Med 13: , 226. |
[35] | Kirisattayakul W , Wattanathorn J , Tong-Un T , Muchimapura S , Wannanon P , Jittiwat J ((2013) ) Cerebroprotective effect of Moringa oleifera against focal ischemic stroke induced by middle cerebral artery occlusion. Oxid Med Cell Longev 2013: , 951415. |
[36] | Singh BN , Singh BR , Singh RL , Prakash D , Dhakarey R , Upadhyay G , Singh HB ((2009) ) Oxidative DNA damage protective activity, antioxidant and anti-quorum sensing potentials of Moringa oleifera. Food Chem Toxicol 47: , 1109–1116. |
[37] | C R ((2014) ) A nootropic effect of Moringa oleifera on Ach and ChAT activity in colchicine induced experimental rat model of Alzheimer’s disease: Possible involvement of antioxidants. Al Ameen J Med Sci 7: , 125–133. |
[38] | Asare GA , Gyan B , Bugyei K , Adjei S , Mahama R , Addo P , Otu-Nyarko L , Wiredu EK , Nyarko A ((2012) ) Toxicity potentials of the nutraceutical Moringa oleifera at supra-supplementation levels. J Ethnopharmacol 139: , 265–272. |
[39] | Bakre AG , Aderibigbe AO , Ademowo OG ((2013) ) Studies on neuropharmacological profile of ethanol extract of Moringa oleifera leaves in mice. J Ethnopharmacol 149: , 783–789. |
[40] | Asiedu-Gyekye IJ , Frimpong-Manso S , Awortwe C , Antwi DA , Nyarko AK ((2014) ) Micro- and macroelemental composition and safety evaluation of the nutraceutical Moringa oleifera leaves. J Toxicol 2014: , 786979. |
[41] | Awodele O , Oreagba IA , Odoma S , da Silva JA , Osunkalu VO ((2012) ) Toxicological evaluation of the aqueous leaf extract of Moringa oleifera Lam. (Moringaceae). J Ethnopharmacol 139: , 330–336. |
[42] | Extance A ((2010) ) Alzheimer’s failure raises questions about disease-modifying strategies. Nat Rev Drug Discov 9: , 749–751. |
[43] | Holmes C , Boche D , Wilkinson D , Yadegarfar G , Hopkins V , Bayer A , Jones RW , Bullock R , Love S , Neal JW , Zotova E , Nicoll JAR ((2008) ) Long-term effects of Aβ42 immunisation in Alzheimer’s disease: Follow-up of a randomised, placebo-controlled phase I trial. Lancet 372: , 216–223. |
[44] | Rinne JO , Brooks DJ , Rossor MN , Fox NC , Bullock R , Klunk WE , Mathis CA , Blennow K , Barakos J , Okello AA , Rodriguez Martinez de Liano S , Liu E , Koller M , Gregg KM , Schenk D , Black R , Grundman M ((2010) ) 11C-PiB PET assessment of change in fibrillar amyloid-beta load in patients with Alzheimer’s disease treated with bapineuzumab: A phase 2, double-blind, placebo-controlled, ascending-dose study. Lancet Neurol 9: , 363–372. |
[45] | Kim J , Lee HJ , Lee KW ((2010) ) Naturally occurring phytochemicals for the prevention of Alzheimer’s disease. J Neurochem 112: , 1415–1430. |
[46] | Shytle RD , Tan J , Bickford PC , Rezai-Zadeh K , Hou L , Zeng J , Sanberg PR , Sanberg CD , Alberte RS , Fink RC , Roschek B Jr. ((2012) ) Optimized turmeric extract reduces beta-Amyloid and phosphorylated Tau protein burden in Alzheimer’s transgenic mice. Curr Alzheimer Res 9: , 500–506. |
[47] | Samant LR , Sangar VC , Rege A , Chowdhary S , Pawar S , Chowdhary A ((2015) ) In silico ADME and bioactivity prediction of phytoconstituents of Moringa oleifera against Alzheimer’s disease. Indo Am J Pharm Res 5: , 2682–2689. |
[48] | Chai GS , Jiang X , Ni ZF , Ma ZW , Xie AJ , Cheng XS , Wang Q , Wang JZ , Liu GP ((2013) ) Betaine attenuates Alzheimer-like pathological changes and memory deficits induced by homocysteine. J Neurochem 124: , 388–396. |
[49] | Janel N , Alexopoulos P , Badel A , Lamari F , Camproux AC , Lagarde J , Simon S , Feraudet-Tarisse C , Lamourette P , Arbones M , Paul JL , Dubois B , Potier MC , Sarazin M , Delabar JM ((2017) ) Combined assessment of DYRK1A, BDNF and homocysteine levels as diagnostic marker for Alzheimer’s disease. Transl Psychiatry 7: , e1154. |
[50] | Wang JZ , Grundke-Iqbal I , Iqbal K ((2007) ) Kinases and phosphatases and tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci 25: , 59–68. |
[51] | Nicholls RE , Sontag JM , Zhang H , Staniszewski A , Yan S , Kim CY , Yim M , Woodruff CM , Arning E , Wasek B , Yin D , Bottiglieri T , Sontag E , Kandel ER , Arancio O ((2016) ) PP2A methylation controls sensitivity and resistance to beta-amyloid-induced cognitive and electrophysiological impairments. Proc Natl Acad Sci U S A 113: , 3347–3352. |
[52] | Sontag JM , Nunbhakdi-Craig V , Sontag E ((2013) ) Leucine carboxyl methyltransferase 1 (LCMT1)-dependent methylation regulates the association of protein phosphatase 2A and Tau protein with plasma membrane microdomains in neuroblastoma cells. J Biol Chem 288: , 27396–27405. |
[53] | Chu J , Lauretti E , Pratico D ((2017) ) Caspase-3-dependent cleavage of Akt modulates tau phosphorylation via GSK3beta kinase: Implications for Alzheimer’s disease. Mol Psychiatry 22: , 1002–1008. |
[54] | Choudhury S , Borah A ((2015) ) Activation of NMDA receptor by elevated homocysteine in chronic liver disease contributes to encephalopathy. Med Hypotheses 85: , 64–67. |
[55] | Sibarov DA , Abushik PA , Giniatullin R , Antonov SM ((2016) ) GluN2A subunit-containing NMDA receptors are the preferential neuronal targets of homocysteine. Front Cell Neurosci 10: , 246. |
[56] | Abushik PA , Niittykoski M , Giniatullina R , Shakirzyanova A , Bart G , Fayuk D , Sibarov DA , Antonov SM , Giniatullin R ((2014) ) The role of NMDA and mGluR5 receptors in calcium mobilization and neurotoxicity of homocysteine in trigeminal and cortical neurons and glial cells. J Neurochem 129: , 264–274. |
[57] | Bennett RE , DeVos SL , Dujardin S , Corjuc B , Gor R , Gonzalez J , Roe AD , Frosch MP , Pitstick R , Carlson GA , Hyman BT ((2017) ) Enhanced tau aggregation in the presence of amyloid beta. Am J Pathol 187: , 1601–1612. |
[58] | Audrain M , Souchet B , Alves S , Fol R , Viode A , Haddjeri A , Tada S , Orefice NS , Josephine C , Bemelmans AP , Delzescaux T , Deglon N , Hantraye P , Akwa Y , Becher F , Billard JM , Potier B , Dutar P , Cartier N , Braudeau J ((2017) ) betaAPP processing drives gradual tau pathology in an age-dependent amyloid rat model of Alzheimer’s disease. Cereb Cortex, doi: 10.1093/cercor/bhx260. |
[59] | Ono Y , Sorimachi H ((2012) ) Calpains: An elaborate proteolytic system. Biochim Biophys Acta 1824: , 224–236. |
[60] | Cho K , Cho MH , Seo JH , Peak J , Kong KH , Yoon SY , Kim DH ((2015) ) Calpain-mediated cleavage of DARPP-32 in Alzheimer’s disease. Aging Cell 14: , 878–886. |
[61] | Goni-Oliver P , Lucas JJ , Avila J , Hernandez F ((2007) ) N-terminal cleavage of GSK-3 by calpain: A new form of GSK-3 regulation. J Biol Chem 282: , 22406–22413 . |
[62] | Liang B , Duan BY , Zhou XP , Gong JX , Luo ZG ((2010) ) Calpain activation promotes BACE1 expression, amyloid precursor protein processing, and amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J Biol Chem 285: , 27737–27744. |
[63] | Roberts TF , Tschida KA , Klein ME , Mooney R ((2010) ) Rapid spine stabilization and synaptic enhancement at the onset of behavioural learning. Nature 463: , 948–952. |
[64] | Cho C , MacDonald R , Shang J , Cho MJ , Chalifour LE , Paudel HK ((2017) ) Early growth response-1-mediated down-regulation of drebrin correlates with loss of dendritic spines. J Neurochem 142: , 56–73. |
[65] | Iulita MF , Cuello AC ((2014) ) Nerve growth factor metabolic dysfunction in Alzheimer’s disease and Down syndrome. Trends Pharmacol Sci 35: , 338–348. |
[66] | Giacoppo S , Rajan TS , Iori R , Rollin P , Bramanti P , Mazzon E ((2017) ) The alpha-cyclodextrin complex of the Moringa isothiocyanate suppresses lipopolysaccharide-induced inflammation in RAW 264.7 macrophage cells through Akt and p38 inhibition. Inflamm Res 66: , 487–503. |
[67] | Omodanisi EI , Aboua YG , Oguntibeju OO ((2017) ) Assessment of the anti-hyperglycaemic, anti-inflammatory and antioxidant activities of the methanol extract of Moringa oleifera in diabetes-induced nephrotoxic male Wistar rats. Molecules 22: , E439. |
[68] | Randriamboavonjy JI , Rio M , Pacaud P , Loirand G , Tesse A ((2017) ) Moringa oleifera seeds attenuate vascular oxidative and nitrosative stresses in spontaneously hypertensive rats. Oxid Med Cell Longev 2017: , 4129459. |
[69] | Zhu X , Tian J , Sun S , Dong Q , Zhang F , Zhang X ((2016) ) (-)-SCR1693 protects against memory impairment and hippocampal damage in a chronic cerebral hypoperfusion rat model. Sci Rep 6: , 28908. |
[70] | Wang XL , Xiong Y , Yang Y , Tuo QZ , Wang XC , Chen R , Tian Q , Zhang ZP , Yan X , Yang ZY , Wang JZ , Liu R ((2015) ) A novel tacrine-dihydropyridine hybrid (-)SCR1693 induces tau dephosphorylation and inhibits Abeta generation in cells. Eur J Pharmacol 754: , 134–139. |
[71] | Zhang Z , Chen R , An W , Wang C , Liao G , Dong X , Bi A , Yin Z , Luo L ((2016) ) A novel acetylcholinesterase inhibitor and calcium channel blocker SCR-1693 improves Abeta25-35-impaired mouse cognitive function. Psychopharmacology (Berl) 233: , 599–613. |
[72] | Torres LL , Quaglio NB , de Souza GT , Garcia RT , Dati LM , Moreira WL , Loureiro AP , de Souza-Talarico JN , Smid J , Porto CS , Bottino CM , Nitrini R , Barros SB , Camarini R , Marcourakis T ((2011) ) Peripheral oxidative stress biomarkers in mild cognitive impairment and Alzheimer’s disease. J Alzheimers Dis 26: , 59–68. |
[73] | Jin Jung K , Hyun Kim D , Kyeong Lee E , Woo Song C , Pal Yu B , Young Chung H ((2013) ) Oxidative stress induces inactivation of protein phosphatase 2A, promoting proinflammatory NF-kappaB in aged rat kidney. Free Radic Biol Med 61: , 206–217. |
[74] | Zhang CE , Tian Q , Wei W , Peng JH , Liu GP , Zhou XW , Wang Q , Wang DW , Wang JZ ((2008) ) Homocysteine induces tau phosphorylation by inactivating protein phosphatase 2A in rat hippocampus. Neurobiol Aging 29: , 1654–1665. |
[75] | Wu XL , Pina-Crespo J , Zhang YW , Chen XC , Xu HX ((2017) ) Tau-mediated neurodegeneration and potential implications in diagnosis and treatment of Alzheimer’s disease. Chin Med J (Engl) 130: , 2978–2990. |
[76] | Kantarci K , Murray ME , Schwarz CG , Reid RI , Przybelski SA , Lesnick T , Zuk SM , Raman MR , Senjem ML , Gunter JL , Boeve BF , Knopman DS , Parisi JE , Petersen RC , Jack CR Jr , Dickson DW ((2017) ) White-matter integrity on DTI and the pathologic staging of Alzheimer’s disease. Neurobiol Aging 56: , 172–179. |
[77] | Seo J , Kritskiy O , Watson LA , Barker SJ , Dey D , Raja WK , Lin YT , Ko T , Cho S , Penney J , Silva MC , Sheridan SD , Lucente D , Gusella JF , Dickerson BC , Haggarty SJ , Tsai LH ((2017) ) Inhibition of p25/Cdk5 attenuates tauopathy in mouse and iPSC models of frontotemporal dementia. J Neurosci 37: , 9917–9924. |
[78] | Lucic V , Greif GJ , Kennedy MB ((2008) ) Detailed state model of CaMKII activation and autophosphorylation. Eur Biophys J 38: , 83–98. |
[79] | Butterfield DA , Lauderback CM ((2002) ) Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: Potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med 32: , 1050–1060. |
[80] | Oda A , Tamaoka A , Araki W ((2010) ) Oxidative stress up-regulates presenilin 1 in lipid rafts in neuronal cells. J Neurosci Res 88: , 1137–1145. |
[81] | Tamagno E , Bardini P , Obbili A , Vitali A , Borghi R , Zaccheo D , Pronzato MA , Danni O , Smith MA , Perry G , Tabaton M ((2002) ) Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiol Dis 10: , 279–288. |
[82] | Zhang CE , Wei W , Liu YH , Peng JH , Tian Q , Liu GP , Zhang Y , Wang JZ ((2009) ) Hyperhomocysteinemia increases beta-amyloid by enhancing expression of gamma-secretase and phosphorylation of amyloid precursor protein in rat brain. Am J Pathol 174: , 1481–1491. |
[83] | Gantier R , Gilbert D , Dumanchin C , Campion D , Davoust D , Toma F , Frebourg T ((2000) ) The pathogenic L392V mutation of presenilin 1 decreases the affinity to glycogen synthase kinase-3 beta. Neurosci Lett 283: , 217–220. |
[84] | Michel G , Mercken M , Murayama M , Noguchi K , Ishiguro K , Imahori K , Takashima A ((1998) ) Characterization of tau phosphorylation in glycogen synthase kinase-3beta and cyclin dependent kinase-5 activator (p23) transfected cells. Biochim Biophys Acta 1380: , 177–182. |
[85] | Medeiros R , Kitazawa M , Chabrier MA , Cheng D , Baglietto-Vargas D , Kling A , Moeller A , Green KN , LaFerla FM ((2012) ) Calpain inhibitor A-705253 mitigates Alzheimer’s disease-like pathology and cognitive decline in aged 3xTgAD mice. Am J Pathol 181: , 616–625. |
[86] | Goll DE , Thompson VF , Li H , Wei W , Cong J ((2003) ) The calpain system. Physiol Rev 83: , 731–801. |
[87] | Saido TC , Nagao S , Shiramine M , Tsukaguchi M , Sorimachi H , Murofushi H , Tsuchiya T , Ito H , Suzuki K ((1992) ) Autolytic transition of mu-calpain upon activation as resolved by antibodies distinguishing between the pre- and post-autolysis forms. J Biochem 111: , 81–86. |
[88] | Saito K , Elce JS , Hamos JE , Nixon RA ((1993) ) Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: A potential molecular basis for neuronal degeneration. Proc Natl Acad Sci U S A 90: , 2628–2632. |
[89] | Nixon RA , Saito KI , Grynspan F , Griffin WR , Katayama S , Honda T , Mohan PS , Shea TB , Beermann M ((1994) ) Calcium-activated neutral proteinase (calpain) system in aging and Alzheimer’s disease. Ann N Y Acad Sci 747: , 77–91. |
[90] | Gao L , Tian S , Gao H , Xu Y ((2013) ) Hypoxia increases Abeta-induced tau phosphorylation by calpain and promotes behavioral consequences in AD transgenic mice. J Mol Neurosci 51: , 138–147. |
[91] | Vaisid T , Kosower NS , Katzav A , Chapman J , Barnoy S ((2007) ) Calpastatin levels affect calpain activation and calpain proteolytic activity in APP transgenic mouse model of Alzheimer’s disease. Neurochem Int 51: , 391–397. |
[92] | Ferreira A , Bigio EH ((2011) ) Calpain-mediated tau cleavage: A mechanism leading to neurodegeneration shared by multiple tauopathies. Mol Med 17: , 676–685. |
[93] | Jin N , Yin X , Gu J , Zhang X , Shi J , Qian W , Ji Y , Cao M , Gu X , Ding F , Iqbal K , Gong CX , Liu F ((2015) ) Truncation and activation of dual specificity tyrosine phosphorylation-regulated kinase 1A by calpain I: A molecular mechanism linked to tau pathology in Alzheimer disease. J Biol Chem 290: , 15219–15237. |
[94] | Jin N , Yin X , Yu D , Cao M , Gong CX , Iqbal K , Ding F , Gu X , Liu F ((2015) ) Truncation and activation of GSK-3beta by calpain I: A molecular mechanism links to tau hyperphosphorylation in Alzheimer’s disease. Sci Rep 5: , 8187. |
[95] | Kurbatskaya K , Phillips EC , Croft CL , Dentoni G , Hughes MM , Wade MA , Al-Sarraj S , Troakes C , O’Neill MJ , Perez-Nievas BG , Hanger DP , Noble W ((2016) ) Upregulation of calpain activity precedes tau phosphorylation and loss of synaptic proteins in Alzheimer’s disease brain. Acta Neuropathol Commun 4: , 34. |
[96] | El-Husseini AE , Schnell E , Chetkovich DM , Nicoll RA , Bredt DS ((2000) ) PSD-95 involvement in maturation of excitatory synapses. Science 290: , 1364–1368. |
[97] | Giusti-Rodriguez P , Gao J , Graff J , Rei D , Soda T , Tsai LH ((2011) ) Synaptic deficits are rescued in the p25/Cdk5model of neurodegeneration by the reduction of beta-secretase (BACE1). J Neurosci 31: , 15751–15756. |

