
Pyk2 is a Novel Tau Tyrosine Kinase that is Regulated by the Tyrosine Kinase Fyn
Abstract
The protein tyrosine kinase Pyk2 is encoded by PTK2B, a novel Alzheimer’s disease (AD) susceptibility variant, with the PTK2B risk allele being associated with increased mRNA levels, suggestive of increased Pyk2 levels. However, the role of Pyk2, a member of the focal adhesion kinase (FAK) family, in AD pathology and its regulation are largely unknown. To address this, we generated mice with neuronal expression of human Pyk2. Because we had previously reported an association of Pyk2 and hyperphosphorylated tau (a hallmark feature of AD) in human tau transgenic pR5 mice, we also generated Pyk2/tau double-transgenic mice, which exhibit increased tyrosine phosphorylation and accumulation of tau. We further demonstrated that Pyk2 colocalizes, interacts with, and phosphorylates tau in vivo and in vitro. Importantly, although Pyk2 interacts with the established tyrosine-directed tau kinase Fyn, we identified an increased Pyk2 activity in mice which constitutively overexpress Fyn (FynCA), and a decreased activity in mice lacking Fyn (FynKO). Together, our study reveals a novel role for Pyk2 as a direct tyrosine kinase of tau that is active downstream of Fyn. Our analysis may enhance the understanding of how the PTK2B risk allele contributes to tauopathy.
INTRODUCTION
Familial Alzheimer’s disease (AD) is caused by autosomal dominant genes, whereas the more frequent sporadic forms of AD are due to a combination of environmental and genetic risk factors. More than 20 years ago, APOE has been identified as a susceptibility gene, using large-scale genetic approaches [1]. Since then, additional risk genes have been identified, most prominently TREM2 [2, 3]. This finding was accompanied by a wake of large genome-wide association studies (GWAS) that identified additional AD susceptibility loci including the PTK2B (protein tyrosine kinase 2 beta) gene [4–6]. Importantly, the PTK2B risk allele rs28834970C was found to be associated with increased PTK2B mRNA expression [7], suggesting elevated protein levels under disease conditions.
A histopathological hallmark of AD are aggregates of the microtubule-associated protein tau, that are also a defining feature of several other neurodegenerative diseases that are collectively termed tauopathies [8, 9]. Tau is best described as a scaffolding protein that is mainly found in neurons, where it is enriched in axons. Under pathological conditions, tau undergoes many posttranslational modifications, most notably phosphorylation at serine, threonine and tyrosine residues [8–11]. In the course of disease, tau forms oligomers and eventually fibrils that constitute the so-called neurofibrillary tangles that fill up the entire soma of degenerating neurons. In this process, tau accumulates in part because of increased tau synthesis, a process that also entails increased phosphorylation at multiple residues [12]. Pathologically accumulating tau impairs many neuronal functions, including axonal transport, synaptic transmission, action potential generation, oxidative phosphorylation, and chromatin structure [13–19].
Previously, we found that the PTK2B-encoded protein Pyk2 colocalizes with pathologically phosphorylated tau in neurons of P301L tau transgenic pR5 mice [20], at a stage when the mice undergo pronounced changes in tau phosphorylation and aggregation [21]. Consistent with this, a modifier screen in Drosophila combined with a validation in human tissue revealed that Pyk2 was present in neurofibrillary tangles and neuritic processes in human AD brain [22]. In spite of these findings, not much mechanistic evidence has been provided and to date, the role of Pyk2 in tau pathology remained elusive.
Pyk2 belongs to the focal adhesion kinase (FAK) family. Under physiological conditions, the enzyme is involved in the calcium-induced regulation of ion channels, and in the activation of the mitogen-activated protein kinase signaling pathway in response to various stimuli. In non-neuronal cells, Pyk2 is involved in broad cellular functions, including the organization of the actin cytoskeleton to facilitate changes in cellular polarity, migration, and adhesion; it also has a role in bone remodeling and immune functions [23]. In the central nervous system, Pyk2 is found in the postsynaptic density (PSD), a morphological and functional specialization of the postsynapse [24, 25], where Fyn (another tyrosine kinase) is also localized [26, 27]. Of note, Fyn is a Src family kinase (SFK) member and an established tyrosine kinase of tau that mediates excitotoxicity [12, 28–30]. Pyk2 has also been implicated in long-term potentiation [24, 31] and long-term depression [32], two measures of synaptic plasticity, contributing to cognitive deficits in neurodegenerative disease [31].
To better address the interplay of tau, Fyn, and Pyk2, we generated a novel Pyk2 transgenic mouse strain and crossed the mice with pR5 mice, complemented by in vitro and cellular studies. We also examined transgenic mice that express a constitutive active form of Fyn (FynCA Tg) [33] and Fyn knockout (FynKO) mice that together support the notion that Pyk2 is a tau kinase that is regulated by Fyn.
MATERIALS AND METHODS
Animals
Wild-type (WT) C57BL/6 mice, Fyn knockout mice (FynKO) [33], Fyn transgenic mice (FynCA Tg) expressing a constitutively active Y531F mutant form of human Fyn [33], and P301L-tau (pR5) transgenic mice [34] were used in this study. In addition, Pyk2 transgenic mice were generated by pronuclear microinjection as previously described [35]. Mice were maintained on a 12 h light/dark cycle with ad libitum access to food and water and were housed 3–5 mice per cage. All experiments were carried out with ethical approval from the University of Queensland Animal Ethics Committee (approval numbers QBI/027/12/NHMRC and QBI/412/14/NHMRC).
Vectors and small interfering RNAs (siRNAs)
Tau-V5 and Fyn-Myc expression constructs were generated using pENTR donor vectors that contained full-length human 2N4R tau and human Fyn [26]. pDONR223-PTK2B was a generous gift from William Hahn and David Root (Addgene plasmid # 23898). The human Pyk2 cDNA was derived from the pDONR223-PTK2B vector and subcloned into an mThy1.2 expression vector to generate Pyk2 transgenic mice. Pyk2-V5 and Pyk2-GFP vectors were generated by cloning the Pyk2 cDNA into pcDNA6.2/V5-DEST and pcDNA6.2/N-EmGFP-DEST vectors (Gateway cloning, Thermo Fisher Invitrogen). Fyn-GFP was constructed in a similar manner. Mutagenesis of Pyk2 to generate Pyk2Y402F, Tau to generate TauY18A (Y18 is a Fyn phosho-epitope) and Fyn to generate FynKD (K299A, kinase-dead), FynCA (Y531F, constitutively active), FynΔSH2 (SH2 domain (149-246 aa)-deleted) and FynΔSH3 (SH3 domain (82-143 aa)-deleted) constructs was performed following the manufacturer’s instructions (Q5 site-directed mutagenesis kit, New England Biolabs, E0554S). A DNA purification kit (NucleoBond Xtra Midi-EF, MACHEREY-NAGEL) was used for plasmid preparation. ON-TARGET plus human PTK2B siRNA (L-003165-00-0005) smart pool and non-targeting pool control siRNAs were purchased from Dharmocon. Co-transfection of the siRNA and the Tau-V5 plasmid was performed using the DharmaFECT 1 transfection reagent.
In vitro phosphorylation and dephosphorylation assays
Freshly thawed aliquots of recombinant proteins were briefly mixed and incubated in the appropriate kinase incubation buffer (20 mM Tris-HCl, 10 mM MgCl2, 5 mM dithiothreitol, 0.5 mM EDTA, pH 7.5) depending on the assay. For instance, to test if phosphorylation was ATP-dependent, recombinant tau and Pyk2 were incubated with or without 2.5 mM ATP. The phosphorylation reaction was carried out at 30°C for 1 h, except for the time course analysis for which samples were taken at various time points throughout the reaction. For the in vitro dephosphorylation assay, a cell lysate from HEK cells co-transfected with Tau-V5 and Fyn-Myc and 50–100μg of a lysate taken from pR5 brains were included as positive controls to detect phospho-tau epitopes. Lambda phosphatase, alkaline phosphatase and Antarctic phosphatase (New England BioLabs) were used following standard protocols. Phosphorylation and dephosphorylation reactions were terminated by adding SDS sample buffer, and phosphorylation was determined by electrophoresis on 8% polyacrylamide gels followed by western blotting with phospho-tau specific antibodies.
Sequential extraction
Isolated hippocampal slices were homogenized with metal beads in RAB buffer (G-Bioscience, Cat# 786-91) using TissueLyser LT (Qiagen) in a 2 ml tube for 4 min at 4°C and maximum speed. The supernatant was removed and centrifuged at 21,000 g for 90 min at 4°C. The resultant supernatant (RAB-soluble proteins) was collected, and the pellet homogenized in the same volume of RIPA buffer (Cell Signaling Technology, #9806) and centrifuged at 21,000 g for 90 min at 4°C. The supernatant (RIPA-soluble proteins) was then collected. RAB/RIPA fractions were aliquoted and stored at – 80°C for further analysis.
Co-immunoprecipitation
Co-immunoprecipitation was performed following standard procedures. In brief, cells were washed in PBS and lysed in cold NP-40 buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 1% NP-40) supplemented with a protease/phosphatase inhibitor cocktail (Cell Signaling Technology). The lysate was briefly sonicated and centrifuged at 14,000 rpm for 10 min at 4°C. The supernatant was then collected and 1 mg of total protein lysate was incubated with protein G-conjugated sepharose beads (GE Healthcare) with primary antibodies (Table 1) overnight at 4°C. For brain lysates, 500μg total protein lysate was precleared with protein A-conjugated sepharose beads (GE Healthcare) for 1 h before incubation with the antibody-sepharose complex for 4 h at 4°C. The beads were spun down, washed with lysis buffer or PBS buffer, and eluted with 2×SDS sample buffer, followed by western blotting.
Table 1
Antibody applications
| Application | Antibodies | Manufacture | Catalog No. | Dilution |
| WB | GFP (rb) | Thermo Fisher | A-11122 | 1:2,000 |
| WB | MAP2 (ms) | Sigma Aldrich | M1406 | 1:1,000 |
| WB | Tau5 (ms) | Merck Millipore | MAB361 | 1:5,000 |
| WB | Fyn (ms) | BD Bioscience | 610163 | 1:1,000 |
| WB | p-SFK(rb) | Cell Signaling Techn. | #2101 | 1:1,000 |
| WB | V5 (rb) | Sigma Aldrich | V8137 | 1:5,000 |
| WB | DakoTau (rb) | DAKO | A0024 | 1:10,000 |
| WB | Myc (ms) | Sigma Aldrich | M4439 | 1:5,000 |
| WB | Actin (ms) | Merck Millipore | #04-1116 | 1:10,000 |
| WB | pTyr18-Tau (ms) | Medimabs | NM-0194-100 | 1:1,000 |
| WB | HnRNP A2/B1 (ms) | Cell Signaling Techn. | #9304 | 1:2,000 |
| WB | PSD95 (ms) | Merck Millipore | MABN68 | 1:5,000 |
| WB | pY402-Pyk2 (rb) | Cell Signaling Techn. | #3291 | 1:2,000 |
| WB | Pyk2 (ms, C-ter) | Cell Signaling Techn. | #3480 | 1:2,000 |
| WB | Pyk2 (rb, N-ter) | Abcam | Ab32571 | 1:2,000 |
| WB | HT7 (ms) | Thermo Fisher | MN1000 | 1:5,000 |
| WB | AT8 (ms) | Thermo Fisher | MN1020 | 1:1,000 |
| WB | AT180 (ms) | Thermo Fisher | MN1040 | 1:1,000 |
| WB | pS422-tau | GenTex | GTX8147 | 1:1000 |
| WB | Gapdh (rb) | Merck Millipore | ABS16 | 1:5,000 |
| IP | Pyk2 (ms) | Cell Signaling Techn. | #3480 | 1:200 |
| IP | FynAC | Santa Cruz | SC-16AC | 1:200 |
| IP | Myc (ms) | Sigma Aldrich | M4439 | 1:200 |
| IP | DakoTau (rb) | DAKO | A0024 | 1:1000 |
| IP | Rabbit IgG | Cell Signaling Techn. | #2729 | 1:100 |
| IF | Pyk2 (rb) | Abcam | Ab32571 | 1:500 |
| IF | pY402-pyk2 (rb) | Thermo Fisher | 44-168G | 1:500 |
| IF | Tau5 (ms) | Merck Millipore | MAB361 | 1:1,000 |
| IF | Tau (ck) | Abcam | Ab75714 | 1:1,000 |
| IP | DakoTau (rb) | DAKO | A0024 | 1:10,000 |
| IF | MAP2(ck) | Abcam | Ab92434 | 1:1,000 |
| IF | PSD95 (ms) | Merck Millipore | MABN68 | 1:1,000 |
| IF | FLAG-Alexa 488 | Cell Signaling Techn. | #5407 | 1:200 |
| IF | pTyr18-Tau (ms) | Medimabs | NM-0194-100 | 1:1,000 |
WB, western blotting; IF, immunofluorescence staining; IP, immunoprecipitation; rb, rabbit; ms, mouse; ck, chicken; N-ter, amino-terminal epitope of Pyk2; C-ter, at carboxy-terminal epitope of Pyk2; FynAC, Fyn antibody agarose-conjugated; FLAG-Alexa 488, Alexa Fluor 488-conjugated FLAG antibody.
Western blotting
Equal amounts of protein were loaded (10–50μg depending on the assay) and resolved by SDS-PAGE in Tris-glycine-SDS buffer (Bio-Rad), followed by transfer onto low fluorescence polyvinylidene fluoride membranes (Bio-Rad). The membranes were blocked with Odyssey blocking buffer TBS (LICOR) for 1 h, and then reacted with primary antibodies in Odyssey blocking buffer (LICOR) overnight at 4°C under gentle rocking. After washing, the corresponding IR Dye 680RD/800CW secondary antibodies (LICOR, 1:10,000 dilution) were added for 1 h at room temperature under constant agitation. After washing, the membrane was scanned in the Odyssey Fc Imaging system (LICOR) for detection of an infrared signal.
Immunofluorescence
Cells grown on coverslips or microfluidic chamber devices were washed gently with PBS and fixed in 4% paraformaldehyde, followed by permeabilization with 0.3% Triton X-100. Paraffin-embedded brain sections were deparaffinized in xylene and then rehydrated prior to microwave-mediated antigen retrieval in citrate buffer. The cells or sections were then blocked for 1 h in 5% goat serum with 1% bovine serum albumin (BSA), followed by incubation overnight with primary antibodies at 4°C. Free-floating slices were rinsed three times in PBS for 10 min each, incubated in blocking solution (10% normal goat serum in PBS supplemented with 0.2% Triton X-100), and placed on an orbital shaker for 2 h. Slices were incubated with primary antibodies at 4°C for 24 h. Secondary antibodies were Alexa Fluor (405, 488, 555, 647)-labeled goat anti-mouse, goat anti-rabbit and goat anti-chicken IgG antibodies (Thermo Fisher, 1:500 dilution). DAPI was used to visualize the nuclei. Fluorescence images were taken with a 20×or 63×objective on a Zeiss LSM710 laser scanning confocal microscope or Diskovery spinning disk confocal microscopy at the Queensland Brain Institute’s Advanced Microscopy Facility.
Image analysis
For western blotting, band intensities from immunoblots were quantified using Image J software. They were first normalized to the intensity of an internal control protein and then normalized to the control group as indicated, except that phosphorylated proteins were generally normalized to the amount of total protein instead of the internal control. For the analysis of fluorescence intensities by Image J, all images were converted to 8-bit images and thresholded before the hippocampal area was selected as region of interest and the percentage area of positivity was determined. The sample size for each group is indicated in the figure legends.
Statistical analysis
The molecular and biochemical analysis was performed using a minimum of three biological replicates per condition. The statistical analyses were done using the Prism 7 software (GraphPad). The data distribution was assumed to be normal with similar variance between groups; however, datasets were first assessed with normality tests. Two groups were analyzed with the two-tailed unpaired Student’s t test when the datasets were normally distributed. Three or four groups were assessed with one-way ANOVA followed by a post hoc test (Dunnett’s test, or Sidak’s test) for multiple comparisons. Values are presented as the mean±standard error of mean (s.e.m). The sample size is indicated in the figure legends.
Data availability
Additional data that support the findings of this study are available from the corresponding author upon request. Additional methods are available in the Supplementary Material.
RESULTS
Pyk2 enhances tau pathology in vivo
We previously reported that neurons bearing pathologically phosphorylated tau are enriched for the activated form of Pyk2 [20], a tyrosine kinase encoded by a recently identified AD risk gene PTK2B. To address whether Pyk2 can enhance tau pathology in vivo, we established a novel transgenic mouse strain that overexpresses FLAG-tagged human full-length Pyk2 driven by the neuronal mThy1.2 promoter, and crossed this strain with tau transgenic pR5 mice (a model of tauopathy) that overexpress human full-length P301L tau under control of the same promoter (Fig. 1A) [34]. Both the Pyk2 and pR5 transgenic mice showed a significantly lower weight compared to WT, that was more pronounced for double transgenic mice compared to pR5 (p = 0.0732; Supplementary Figure 1A), reflecting a role of Pyk2 in regulating body weight. In contrast, only Pyk2 transgenic mice exhibited a significantly lower brain weight than WT mice (p < 0.001; Supplementary Figure 1B), whereas the brain weight of pR5/Pyk2 double transgenic and pR5 mice was relatively normal (Supplementary Figure 1B). A histological analysis of the double transgenic mice revealed extensive human Pyk2 expression in neuronal cell bodies and neurites in areas such as the hippocampus and cortex, as indicated by immunostaining with the FLAG antibody (Supplementary Figures 1C and 2A). Co-staining for the activated form of Pyk2 (pY402-Pyk2) suggested that human Pyk2 (detected with the FLAG antibody) was widely activated (Fig. 1B, C and Supplementary Figure 1C).
Fig.1
Pyk2 enhances tau pathology in vivo. A) The murine Thy1.2 promoter drives human P301L tau and FLAG-tagged Pyk2 expression in pR5 and Pyk2 transgenic mice, respectively. Representative immunofluorescence staining of brain sections from 12–15-month-old pR5 and double transgenic pR5/Pyk2 animals shown in (B) with magnified view in (C). Scale bars represent 500μm (B) and 50μm (C). D) Quantification of the pY18-tau positive area in the hippocampus of transgenic brains. Mean±s.e.m, n = 7 mice per group, two-tailed t test, **p < 0.01. E) Validation of pY18-tau antibody using immunoblots of a lysate from human tau-expressing HEK cells. WT, wild-type; Y18A, tyrosine to alanine mutant. F) Representative immunoblots of sequentially (RAB/RIPA) fractionated hippocampal homogenates from 12–15-month-old pR5 and pR5/Pyk2 mice. Both pY18-tau and HT7 antibodies react with transgenic human tau but not endogenous mouse tau. ‘h’ indicates human transgenic tau, and ‘m’ indicates endogenous murine tau. G) Quantification of immunoblots in (F). Fold changes are relative to the pR5 group. Mean±s.e.m, n = 4–6 mice per group, two-tailed t test, #p = 0.075, *p < 0.05.
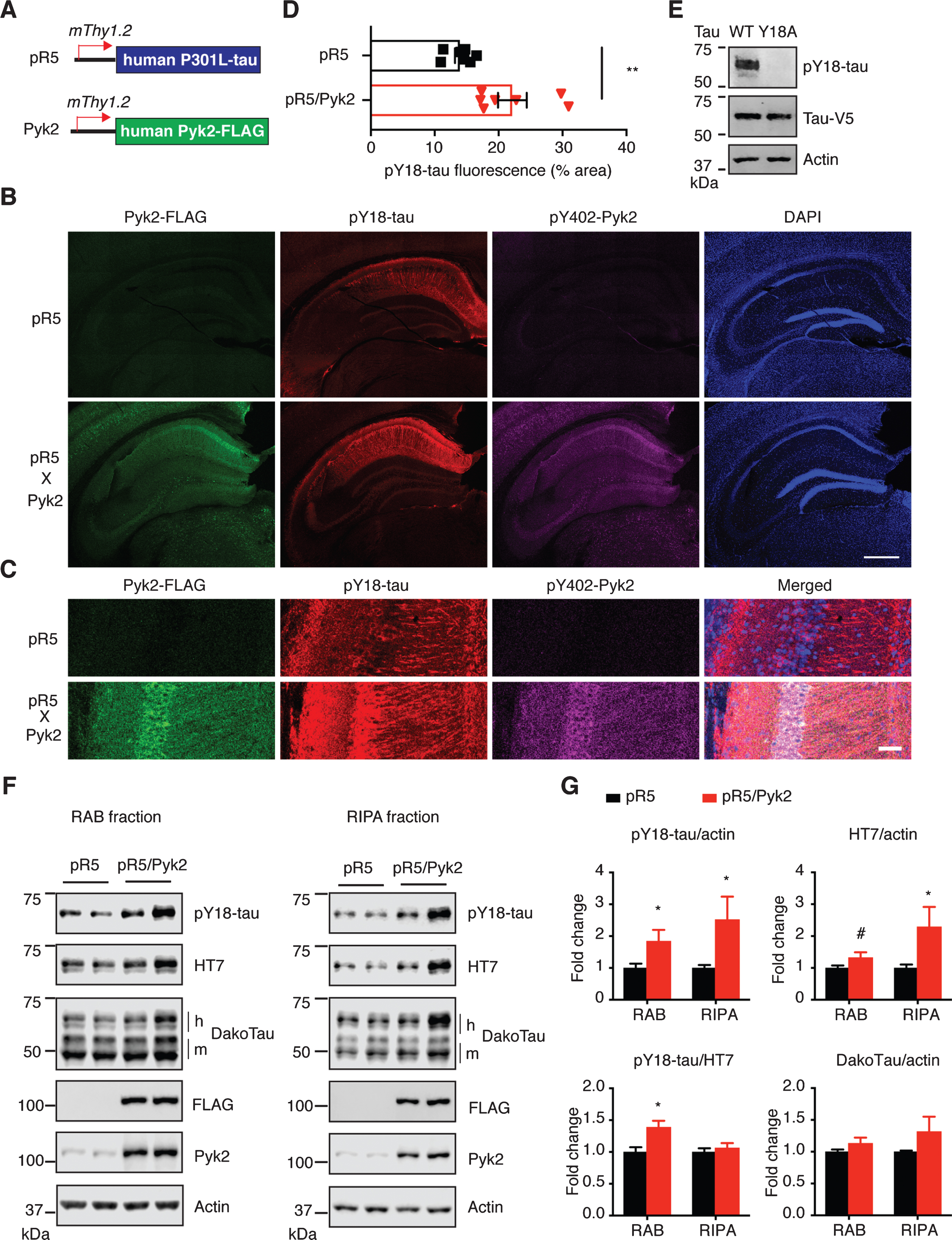
Since Pyk2 is a protein tyrosine kinase, we next investigated phosphorylation of tau at Y18, the major tyrosine phospho-epitope [36, 37]. Interestingly, whereas Pyk2 was expressed throughout the cortex and hippocampus (Supplementary Figures 1C and 2A), most of the pY18-tau signal was confined to hippocampal neurons (Fig. 1B, Supplementary Figures 1C and 2A), to which total tau could also be localized using the DakoTau antibody (Supplementary Figure 2B). As shown for the CA1 region, Pyk2 colocalized with pY18-tau not only in the cell bodies, but also in the dendrites (Fig. 1C and Supplementary Figure 2A). More importantly, hippocampal immunoreactivity for pY18-tau was significantly higher in pR5/Pyk2 compared to pR5 mice (Fig. 1B-D, p < 0.01).
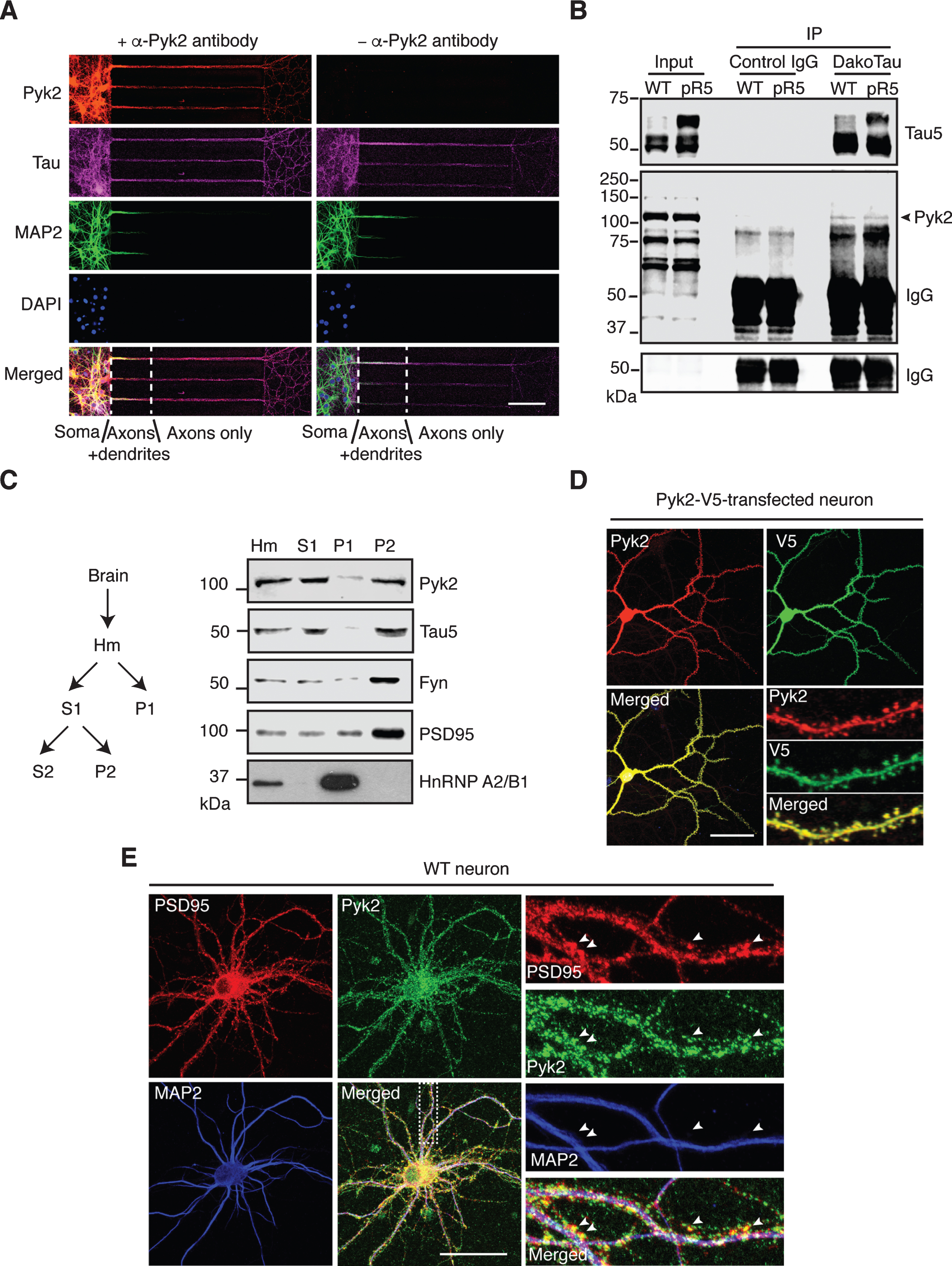
Fig.2
Localization and interaction of Pyk2 and tau. A) Representative immunofluorescence staining of Pyk2 in primary hippocampal neurons (DIV (days in vitro) 10) cultured in microfluidic chambers. Cells were counterstained for tau (all compartments including axons) and MAP2 (all compartments excluding axons). DAPI labels the nuclei. Scale bar: 100μm. B) Co-immunoprecipitation of Pyk2 and tau in RAB fractions of brain lysates obtained from WT and pR5 mice reveals weak interaction of endogenous Pyk2 and tau in vivo. Note that cleaved forms of endogenous Pyk2 were detected in the input lysate. IP, immunoprecipitation. Three independent experiments were performed. C) Endogenous Pyk2 is present in the crude synaptosomal fraction, together with tau. WT brain tissue was processed to obtain fractions including total homogenate (Hm), supernatant S1, pellet P1 and pellet P2, followed by immunoblotting with the indicated antibodies. D) Pyk2-V5-transfected hippocampal neurons (DIV16) stained with Pyk2 and V5, revealing strong localization of Pyk2 in dendritic spines. Scale bar: 50μm. E) WT hippocampal neurons (DIV16) stained with PSD95 (postsynaptic marker) and MAP2. Colocalization of endogenous Pyk2 and PSD95 shown in spines of a WT neuron indicated by arrowheads. Scale bar: 50μm.
The pR5 strain is characterized by the accumulation of hyperphosphorylated tau in many brain areas, and sequential extraction in RAB (more soluble) and then in RIPA buffer (less soluble) is routinely employed to demonstrate increased insolubility of tau. We therefore carried out a sequential RAB/RIPA extraction of hippocampal tissue from brains of 12-month-old pR5 and pR5/Pyk2 mice since tau pathology in the pR5 strain is pronounced in this brain area [21]. pY18-tau and HT7 antibodies were used to detect exogenous human tau, the former to detect tyrosine-phosphorylated and the latter to detect total tau in a phosphorylation-independent manner. Epitope specificity of the pY18-tau antibody was validated in HEK cells expressing WT or a tyrosine-to-alanine (Y18A) mutated tau (Fig. 1E). (Note: Whereas the Y18 epitope is conserved in human and murine tau, the sequence flanking this residue differs and therefore the antibody specific for pY18 only reacts with phosphorylated human and not murine tau.)
Consistent with what we had observed by immunohistochemistry (Fig. 1B, C), in both the RAB and RIPA fractions, levels of pY18-tau were significantly increased by Pyk2 overexpression. Similarly, human tau levels in the RAB fraction were moderately increased (1.3-fold, p = 0.075), being 2.3 times (p < 0.05) higher than for the pR5 RIPA fraction (Fig. 1E, F). In contrast, phosphorylation at Ser/Thr epitopes of tau (AT8, AT180 and pS422) did not differ between the two strains in either fraction (Supplementary Figure 3A, B). We then asked whether Pyk2 activity was increased due to the presence of transgenic tau. Therefore, we determined Pyk2 activation in both strains; however, Pyk2 activity was not changed when tau was overexpressed (Supplementary Figure 3C, D). Together, our in vivo data suggest that Pyk2 significantly augments tau pathology by both tyrosine phosphorylation of tau and increases in total tau.
Fig.3
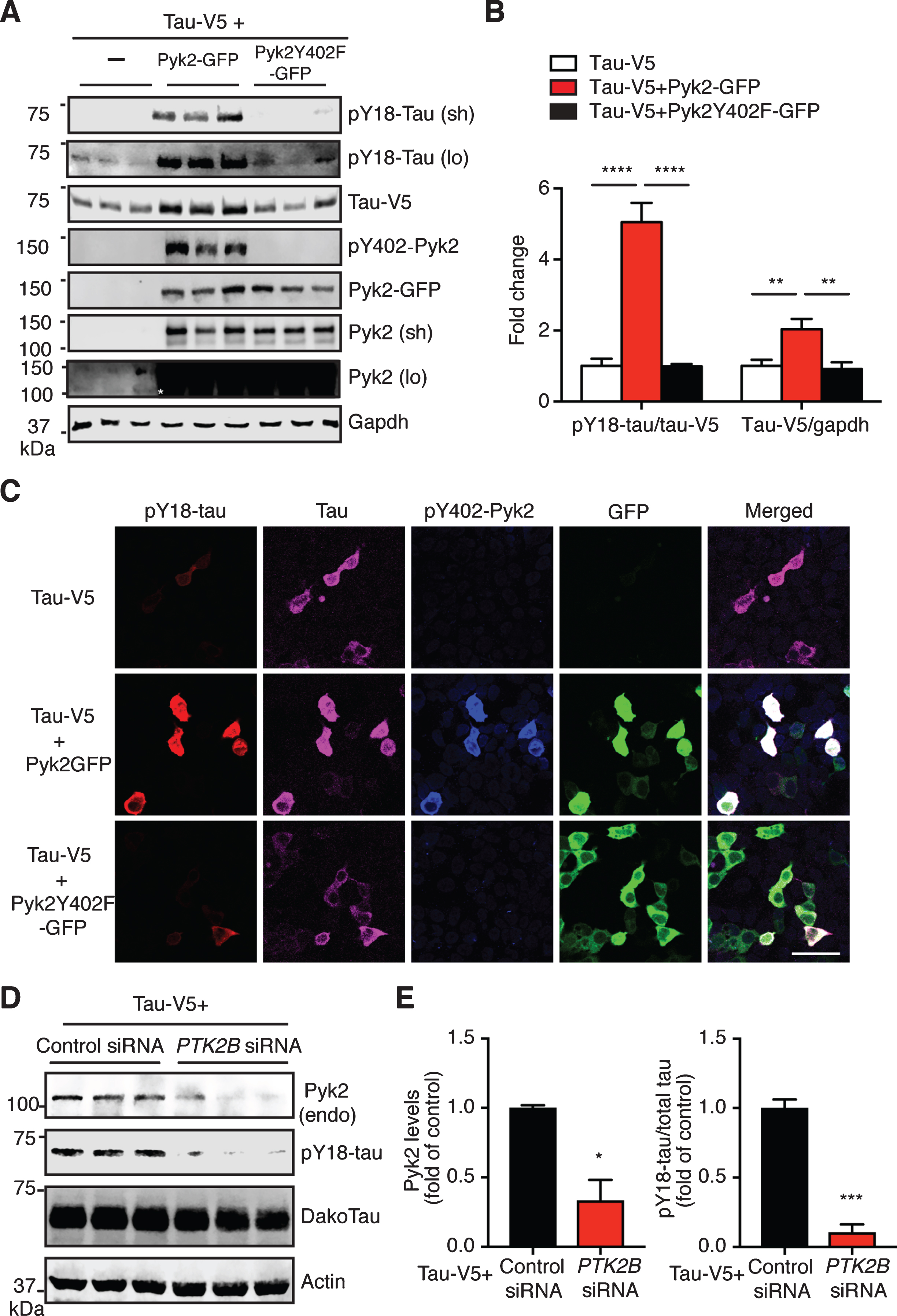
Pyk2 phosphorylates tau in HEK293T cells. A) Representative immunoblots of lysates from HEK293T cells transfected with V5-tagged tau and GFP-tagged Pyk2. Validation of the Y402F mutant form of Pyk2 by immunoblotting using a phospho-Y402-Pyk2 specific antibody. Sh, short exposure; lo, long exposure. Asterisk denotes the endogenous expression of Pyk2 in HEK cells using a longer exposure of the immunoblot in the absence of Pyk2-GFP overexpression. B) Quantification of immunoblots in (A) showing relative tau and pY18-tau levels. Mean±s.e.m, n = 6 per group, one-way ANOVA, Dunnett’s multiple comparisons test, **p < 0.01, ****p < 0.0001. C) Representative immunofluorescence staining of HEK293T cells transfected with V5-tagged tau and GFP-tagged Pyk2 indicating a strong positive correlation of pY18-tau immunoreactivity and expression of the active form of Pyk2. Scale bar: 50μm. D) Immunoblots of lysates from HEK293T cells co-transfected with V5-tagged tau and PTK2B or control siRNA. E) Quantification of immunoblots in (D). Mean±s.e.m, n = 3 per group, two-tailed t test, *p < 0.05, ***p < 0.001.
Pyk2 colocalizes and interacts with tau in neurons
The in vivo evidence of a role of Pyk2 in tau pathology led us to investigate the association of Pyk2 and tau in more detail, including under physiological conditions. We first observed immunoreactivity for both Pyk2 and its activated form in glial cells that were negative for the neuronal marker tau (Supplementary Figure 4A, B), reflecting the nature of Pyk2 as a prevalent focal adhesion kinase. In mature WT primary hippocampal neurons, Pyk2 was strongly present in cell bodies and tau-positive neurites, regardless of whether they were MAP2-positive or not (with MAP2 serving as dendritic marker) (Supplementary Figure 4C), indicating pronounced coexistence of Pyk2 and tau in dendrites and axons. To further confirm that Pyk2 is also localized to axons (where tau is enriched under physiological conditions), we used microfluidic chambers that allow for a better discrimination of dendrites and axons based on their length. Counterstaining for Pyk2, tau and MAP2 confirmed that Pyk2 expression extended into axons (Fig. 2A). Interestingly, Pyk2 and tau were detectable together in axons as early as neurons started to differentiate (Supplementary Figure 5A, B).
Fig.4
Pyk2 phosphorylates tau in vitro. A) Coomassie staining and immunoblots of purified recombinant His-tagged Tau-V5 and Pyk2-V5 proteins. The full-length forms of the recombinant proteins are indicated at the expected molecular weight. Carboxy-terminally cleaved Pyk2 was detected using both anti-V5 and anti-C-terminal Pyk2 antibodies. N-terminal and C-terminal Pyk2 antibodies were raised against an amino- and carboxy-terminal epitope of human Pyk2, respectively. B) Immunoblots of pY18-tau after incubation of recombinant tau with or without recombinant Pyk2 in the in vitro phosphorylation reaction. C) Immunoblots of pY18-tau after incubation of recombinant tau and Pyk2 with or without ATP in the in vitro phosphorylation reaction. D) Quantification of immunoblots in (C). Mean±s.e.m, n = 3 per group, two-tailed t test, ***p < 0.001. E) Representative immunoblots of a time course assay of Pyk2-catalyzed tau phosphorylation in vitro. Reactions were carried out at 30°C in the presence of 0.5μM recombinant Pyk2 and 1.9μM recombinant tau. F) Normalized pY18-tau levels in (E) were plotted as a function of incubation time, with fold-changes relative to time point ‘0 s’. The curve was generated by nonlinear regression. Three independent experiments were performed.
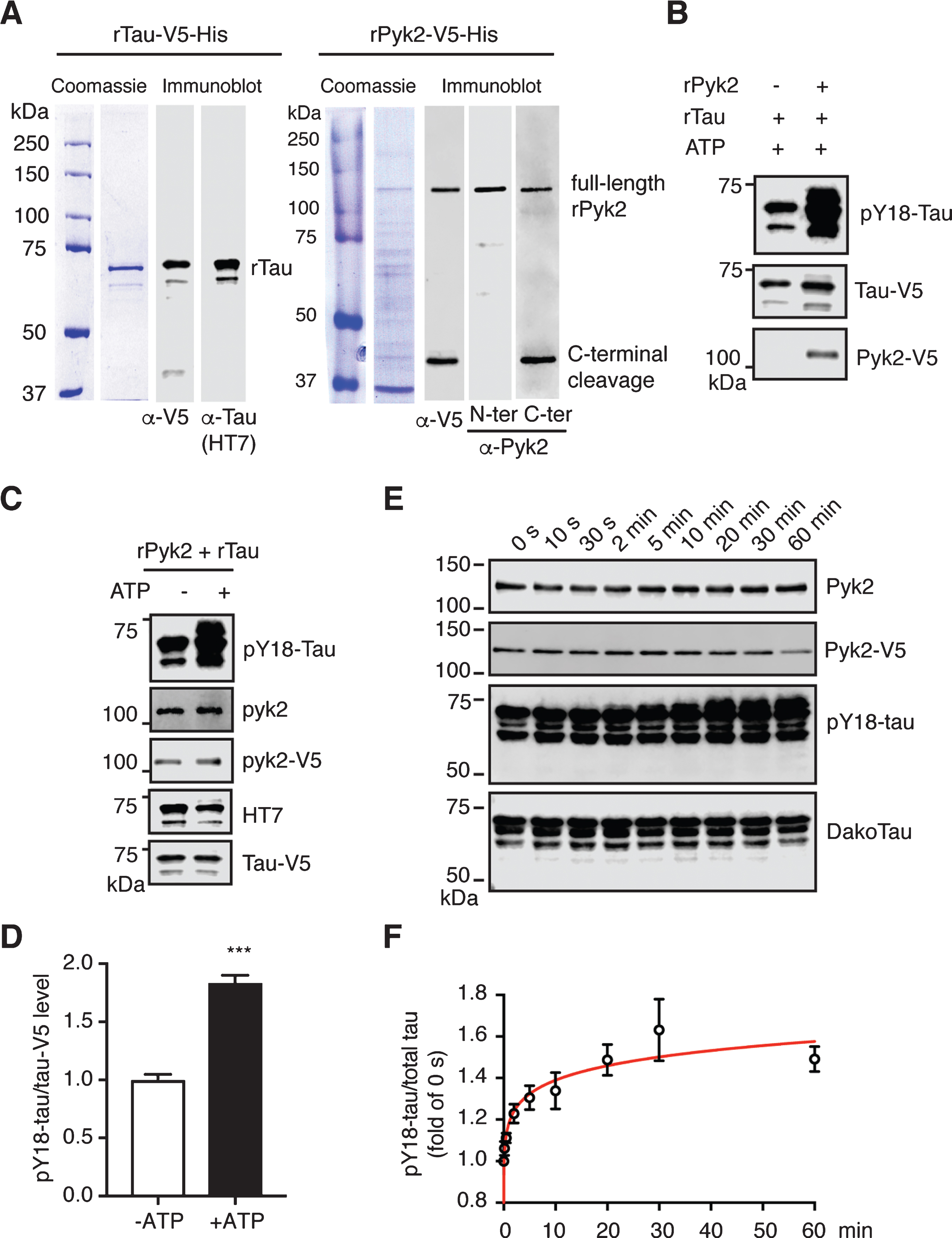
Fig.5
Pyk2 interacts with the tau tyrosine kinase Fyn. A) Immunoblots of cell lysates from HEK293T transfected with V5-tagged tau and Myc-tagged Fyn, indicating that Fyn robustly phosphorylates tau at Tyr18. Sh, short exposure; lo, long exposure. B) Quantification of immunoblots in (A) indicating elevation of pY18-tau levels by Fyn overexpression. Mean±s.e.m, n = 4 per group, two-tailed t test, ****p < 0.0001. C) Representative fluorescence image of HEK293T cells overexpressing Fyn-RFP and Pyk2-GFP. DAPI labels the nuclei. Scale bar: 10μm. D) Immunoprecipitation of Pyk2 and Fyn expressed in HEK293T cells reveals their interaction. E) Immunoprecipitation of Pyk2 and truncated forms of Fyn (FynΔSH2-GFP, SH2 domain-deleted Fyn; FynΔSH3-GFP, and SH3 domain-deleted Fyn) reveals the role of SH2 domain in the interaction. F) Immunoprecipitation of Fyn together with Pyk2 or a phosphorylation-deficient (inactive) Y402F mutant form of Pyk2. G) Quantification of immunoblots (F) indicating a reduced interaction between Fyn and inactive Pyk2. Mean±s.e.m, n = 4 per group, two-tailed t test, ***p < 0.001.
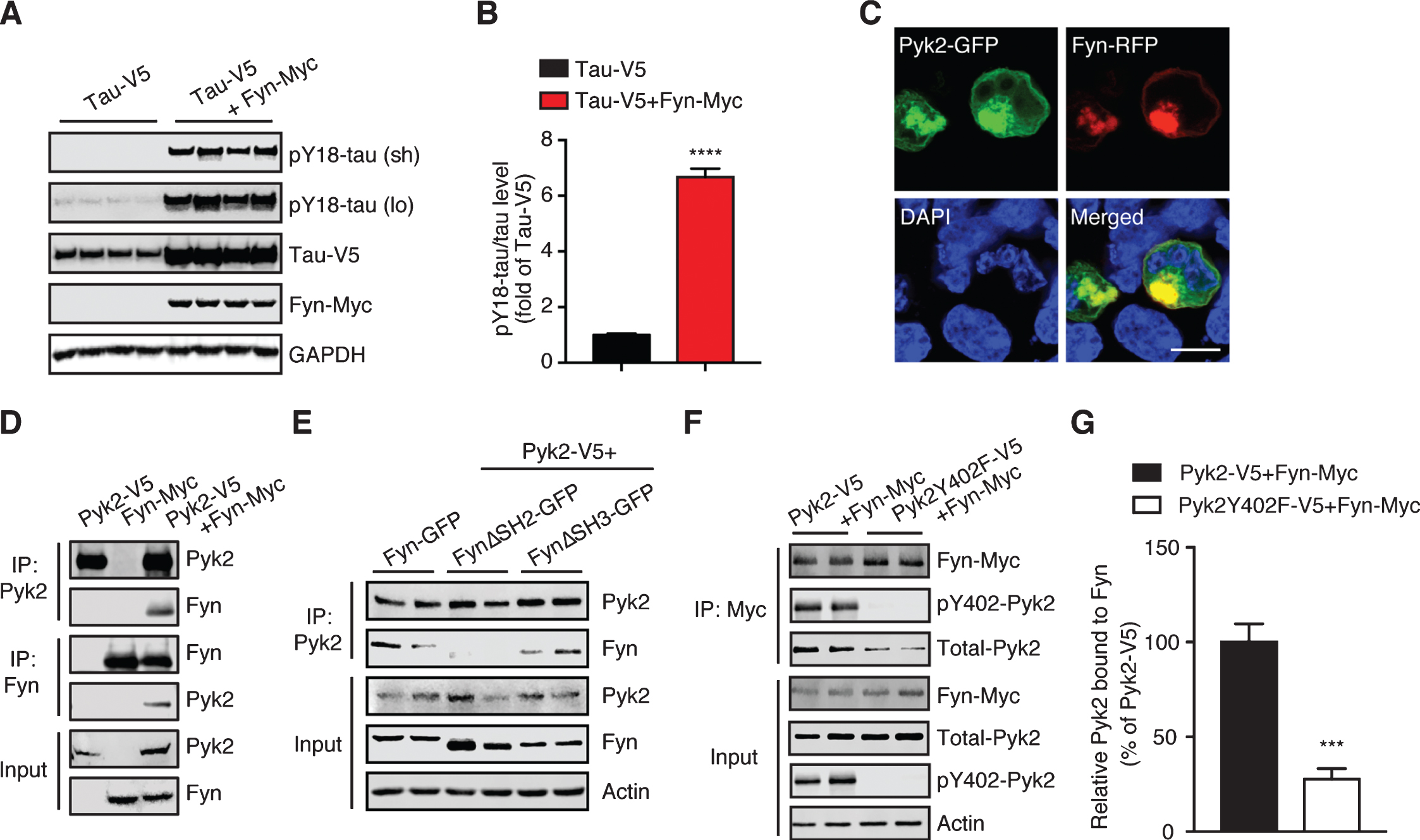
We next examined whether there was a physical interaction between endogenous Pyk2 and tau. Using co-immunoprecipitation, we found that Pyk2 was able to interact with tau in WT brain lysates, although the interaction appeared not to be very strong; a similar interaction (with respect to co-immunoprecipitated full-length Pyk2) was found in brain lysates from pR5 mice that overexpress tau (Fig. 2B). In addition, we observed cleaved forms of endogenous Pyk2 in the input lysate, in agreement with what we had found for recombinant Pyk2 protein (see below).
Pyk2 has also been shown to be localized to the PSD, a scaffolding structure within dendritic spines [24, 25], where tau is also present [28]. We therefore performed fractionations of mouse brain tissue and obtained a synaptosomal (P2) fraction in which Pyk2 was present together with tau, as well as other PSD components such as PSD95 and Fyn (Fig. 2C). Furthermore, when Pyk2 was fused with the green fluorescent protein (Pyk2-GFP) for visualization and then expressed in cultured hippocampal neurons, we observed a colocalization with PSD95 in dendritic spines (Supplementary Figure 5C). In support, by tagging Pyk2 with the much smaller V5-tag (in order to rule out potential leakage of GFP), the same localization pattern was obtained, irrespective of whether an anti-V5 or anti-Pyk2 antibody was used (Fig. 2D). Moreover, labelling for endogenous Pyk2 consistently demonstrated its presence in dendritic spines (Fig. 2E). Together, these data demonstrate that Pyk2 colocalizes and interacts with tau in neurons.
Pyk2 phosphorylates tau in HEK293T cells
We next determined whether Pyk2 is a tau kinase. We therefore transfected HEK293T cells with a tau expression vector either alone or together with a Pyk2-GFP expression vector. As expected, Pyk2 expression significantly increased the amount of tau phosphorylated at Y18 (>5-fold, after normalization to total tau levels) compared to the tau-only transfection (Fig. 3A, B). Surprisingly, the level of total tau was also increased in the co-transfected group (∼ 2-fold). However, these increases of pY18-tau and total tau in the co-transfected group were completely abolished when a phosphorylation-deficient mutant form of Pyk2 (Pyk2Y402F-GFP) was used (Fig. 3A, B). Of note: Only a long exposure time reveals Y18 phosphorylation and endogenous Pyk2 expression (Fig. 3A). In agreement with the biochemical analysis, immunofluorescence staining supports Y18 phosphorylation of tau by Pyk2 unless an inactive kinase is expressed (Fig. 3C).
We next examined the loss-of-function effect of Pyk2 on tau phosphorylation. Since the pY18-tau antibody only detects human tau, we overexpressed a human tau construct in HEK cells that express endogenous Pyk2 protein (albeit at a relatively low level). Using siRNAs against human PTK2B gene, we successfully reduced endogenous Pyk2 levels by 66% (Fig. 3D, E; p < 0.05). This significantly reduced pY18-tau levels by 90% compared to the control (p < 0.001). Together, these data strongly suggest that Pyk2 is a tyrosine kinase of tau.
Pyk2 phosphorylates tau in vitro
To exclude the possibility of indirect phosphorylation, we attempted to validate the tyrosine kinase activity of Pyk2 for tau more directly. We therefore cloned His-tagged Pyk2 and tau cDNAs into bacterial expression vectors followed by affinity chromatography and size exclusion chromatography. Recombinant proteins were validated with Coomassie staining and immunoblotting using antibodies against the V5-tag, tau (HT7) and Pyk2 (Fig. 4A). As revealed with a C- and N-terminal anti-Pyk2 antibody, we observed more C- than N-terminally cleaved products, consistent with the pattern for V5, as the V5-tag was fused C-terminally. As expected, levels of pY18-tau were massively increased when tau was incubated with Pyk2 protein (Fig. 4B).
Of note, we consistently observed a basal level of pY18 phosphorylation for recombinant tau (Supplementary Figure 6A). In order to remove the basal phospho-groups from tyrosine residues, we incubated tau with multiple phosphatases but failed to achieve basal Y18 dephosphorylation, which may or may not be related to the conformation of tau, similar to what is known for its dephosphorylation by protein phosphatase 2A (Supplementary Figure B-D).
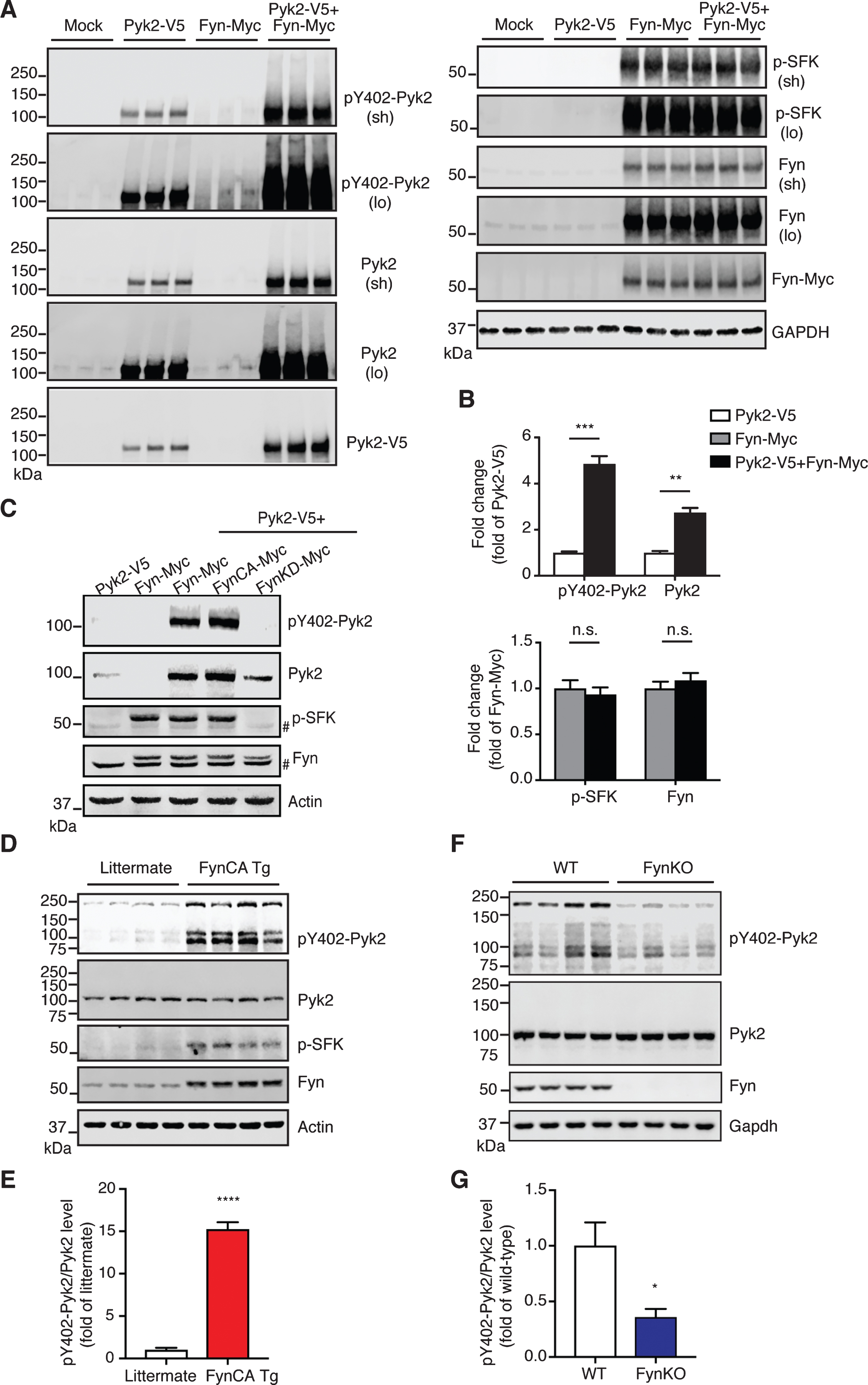
Fig.6
Pyk2 is activated by Fyn in vivo. A) Immunoblots of lysates obtained from Pyk2-V5- and Fyn-Myc-transfected HEK293T cells. Mock, transfection with Lipofectamine only. Sh, short exposure; lo, long exposure. B) Quantification of immunoblots in (A) suggests that increased activity of Pyk2 is mediated by Fyn. Mean±s.e.m, n = 3 per group, two-tailed t test, n.s., not significant; **p < 0.01; ***p < 0.001. C) Immunoblots of HEK293T cell lysates demonstrating that Pyk2 activation is Fyn activity-dependent. FynCA-Myc, constitutively active mutant Fyn (Y531F); FynKD-Myc, kinase-dead mutant Fyn (K299A). # denotes endogenous Fyn. D) Immunoblots of whole brain lysates obtained from 3-week-old Fyn CA transgenic mice and littermate controls. FynCA Tg, transgenic mice neuronally overexpress a constitutively active mutant Fyn (Y531F). E) Quantification of immunoblots in (D) suggests that the increased activity of Pyk2 is mediated by Fyn. Mean±s.e.m, n = 4 per group, two-tailed t test, ****p < 0.0001. F) Immunoblots of whole brain lysates obtained from 6-month-old Fyn knockout (FynKO) mice and WT controls. G) Quantification of immunoblots in (F). Mean±s.e.m, n = 4 per group, two-tailed t test, *p < 0.05.
We next carried out a kinase assay by co-incubating Pyk2 and tau protein with or without ATP. Y18 phosphorylation by Pyk2 was increased >1.8-fold in the presence of ATP, indicating that this reaction is ATP-dependent (Fig. 4C, D). Furthermore, although basal pY18-tau levels were significant, we were able to establish the time course of the Pyk2-catalyzed tau phosphorylation reaction over a 60 min timeframe, showing a time-dependent increase of the pY18-tau signal that was accompanied by a slight molecular weight shift (Fig. 4E, F). A typical enzyme-substrate reaction dynamics was indicated by fitting a curve generated by nonlinear regression to the data (Fig. 4F). Together, our in vitro findings suggest that Pyk2 is a direct tyrosine kinase of tau.
Pyk2 interacts with the tau kinase Fyn
Src family kinases are the most thoroughly studied kinases linked to tau tyrosine phosphorylation [38–40]. Of these, Fyn is one of the most significant kinases in phosphorylating tau under pathological conditions [26, 28, 30]. We therefore sought to determine whether Fyn is involved in Pyk2-mediated tau phosphorylation. We first confirmed the tyrosine kinase activity of Fyn in relation to tau by co-transfecting tau and Fyn expression constructs in HEK293T cells, revealing that pY18-tau levels were induced >6.6-fold by Fyn (Fig. 5A, B). Increased tau levels (2.5-fold) were also observed when Fyn was co-transfected.
We next addressed the relationship of Pyk2 and Fyn, considering that both enzymes can phosphorylate tau at residue Y18. Co-transfection of GFP-tagged Pyk2 and Fyn revealed significant colocalization (Fig. 5C), which was validated by immunoprecipitation (Fig. 5D). Fyn, like any Src family kinase member, engages its conserved SH2 and SH3 domains to mediate protein interactions [41]. We therefore used Fyn constructs that lacked either domain and established that the SH2 domain was responsible for the Fyn/Pyk2 interaction (Fig. 5E). Given that a motif containing phosphorylated tyrosine is a general binding substrate for SH2, we also investigated whether the phosphorylated Y402 epitope of Pyk2 was involved in this interaction. As expected, interaction between the Pyk2 mutant Y402F and Fyn was reduced by more than 72% relative to that of control Pyk2 (Fig. 5F, G; p < 0.001). Together, this demonstrates that Pyk2 interacts with Fyn via the autophosphorylation epitope pY402 of Pyk2 and the SH2 domain of Fyn.
Pyk2 is strongly activated by Fyn in vivo
Having demonstrated that both Pyk2 and Fyn phosphorylate tau and that they are closely coupled, we next investigated how their activities are regulated. Surprisingly, when Pyk2 and Fyn were co-transfected in HEK293T cells, the level of both pY402-Pyk2 and Pyk2 was significantly increased (4.9-fold and 2.7-fold, respectively) compared to Pyk2 only-transfected cells (Fig. 6A, B). In contrast, the signal for neither activated Fyn (p-SFK) nor total Fyn was changed in the co-transfection group relative to cells that were only transfected with Fyn (Fig. 6A, B). Notably, in Fyn-transfected cells, pronounced activation of endogenous Pyk2 was observed compared to the mock control (third group in Fig. 6A), whereas no endogenous Fyn was activated by Pyk2 in Pyk2-transfected cells. We wondered how Fyn regulates the increase of Pyk2 levels and performed a real-time quantitative PCR analysis of transcript levels. An increase of Pyk2 by Fyn was confirmed 16 h after transfection, and a 2.7-fold increase of total Pyk2 transcripts was observed in Fyn-overexpressing cells (Supplementary Figure 7 A, B), indicating that the Fyn-mediated Pyk2 increase is at least partially a result of enhanced transcription. These results strongly indicate that Pyk2 is activated by Fyn, but not vice versa.
To confirm this finding further, we used a constitutively active (FynCA) and a kinase-dead (FynKD) Fyn mutant, mutants we have previously used to address Fyn function [12], and found that Y402 phosphorylation of Pyk2 was Fyn activity-dependent (Fig. 6C).
To further address this regulation in vivo, we analyzed brain lysates from transgenic mice that overexpress constitutively active mutant Fyn in neurons (FynCA Tg) [33]. Owing to the severe premature lethality (lifespan < 7 weeks) of these mice, we had to collect their brains and that of littermate controls already at the young age of 3 weeks. By analyzing brain lysates, we found a remarkable 15.2-fold increase in the active form of Pyk2 (pY402-Pyk2/Pyk2) in FynCA Tg mice compared to the control (****p < 0.0001, Fig. 6D, E), whereas total Pyk2 levels remained unchanged. To determine whether Fyn equally regulates Pyk2 activation in adult animals, we assessed brain lysates from 6-month-old FynKO and WT mice for both total and activated Pyk2. We found that Pyk2 activity was decreased by 64% in FynKO brains, whereas total levels were not altered (*p < 0.05, Fig. 6F, G). Furthermore, we analyzed brain lysates from 15-month-old Pyk2 transgenic mice. Although the p-SFK signal in the Pyk2 transgenic mice was relatively weak, the increase was significant compared to the controls (3.1-fold, Supplementary Figure 7 C, D, **p < 0.01). Together, our in vivo data show that Pyk2 is preferentially activated by Fyn, but not vice versa, supporting the notion of a direct tyrosine phosphorylation of tau mediated by Pyk2.
DISCUSSION
In our study, we addressed the role of the tyrosine kinase Pyk2 in tau pathology, using a novel in vivo model, that was complemented by in vitro and cell culture experiments. Our study was spurred by the recent identification of the PTK2B gene encoding Pyk2 as a susceptibility locus for AD in multiple GWAS studies [4–6]. Its risk allele rs28834970C was subsequently found to be associated with increased PTK2B mRNA expression [7], suggesting elevated protein levels under disease conditions. Given that our earlier work had demonstrated that activated Pyk2 colocalized with pathologically phosphorylated tau in pR5 mice [20], these findings encouraged us to explore the role of Pyk2 in an AD context. In the present study, we demonstrate for the first time that tau pathology is augmented by Pyk2 overexpression in vivo. We also found that Pyk2 phosphorylates tau in mammalian cells and in vitro supporting the notion that Pyk2 contributes to tau pathology.
First, we generated a novel transgenic strain overexpressing Pyk2 in neurons. Besides of a reduced total weight, this strain is overtly normal, displaying only mild motor deficits (unpublished data). After crossing this line with tau transgenic pR5 mice, this revealed consistently and significantly elevated pY18-tau levels in the soluble RAB and less soluble RIPA fractions of double transgenic compared to pR5 hippocampi. The pR5 strain is characterized by predominant tau pathology in the hippocampus, even though the human P301L tau transgene is driven by a forebrain-specific neuronal promoter. Different from the pR5 phospho-tau pattern, the Pyk2 transgene is expressed in both the cerebral cortex and hippocampus, but in the double transgenic strain, a more severe tau pathology was found in the hippocampus. We speculate that this may be due to more availability of the Pyk2 substrate tau in the hippocampus compared with the cortex. Phosphorylation is a critical driver in tau accumulation and aggregation, which further leads to a shift of tau solubility from the RAB to the RIPA fraction, resulting in increased human tau levels in the latter. In contrast to the phospho-Tyr changes, we did not observe any changes for the three phospho-Ser/Thr epitopes analyzed, implying a role of the phospho-Tyr epitope in shifting the tau status, possibly independent of phospho-Ser/Thr epitopes.
For various reasons, Tyr phosphorylation of tau has been less explored than that of Ser/Thr epitopes. There are overwhelmingly more Ser/Thr than Tyr residues in tau (ratio of 80/5), and most studies into tau phosphorylation assessed Ser/Thr sites, that have also been targeted for therapeutic intervention [42]. With the availability of a vast range of phospho-Ser/Thr tau-specific antibodies, the development of antibodies for the phospho-Tyr epitopes is lagging behind [36]. Of those, pY18-tau is the most studied. Besides the binding of tau phosphorylated at Y18 to the SH2 domain of Fyn, a recent study demonstrated that this epitope is also required for the modulation of NMDA receptor-dependent Ca2 + influx and subsequent excitotoxicity [37], highlighting a vital role of this specific phosphorylation site in synaptic transmission. To date, given that tau is enriched in the axon of mature neurons and is mainly expressed in the central nervous system, only a few tyrosine kinases have been identified for tau, including c-Abl, Syk and the SFK Fyn [38–40]. Our study identified the FAK family member Pyk2 as a novel tau tyrosine kinase; however, whether other FAK family members also phosphorylate tau remains to be determined.
The presence of Pyk2 and tau together in multiple neuronal compartments, that extends into both axons and spines, provided the basis for our observation of a direct interaction between endogenous Pyk2 and tau. In agreement with earlier studies, we found an enrichment of Pyk2 in the PSD fraction of hippocampal neurons [24, 25]. Neuronal activation has been shown to lead to Pyk2 clusters and an interaction with the synaptic scaffolding protein PSD95 at the postsynapse [25]. Because a somatodendritic toxic function of tau has been identified in AD [28], with tau mediating synaptic loss when the protein is hyperphosphorylated in dendritic spines [26, 43], this supports the notion that Pyk2 may act as a regulator of tau physiology in this compartment.
We also addressed whether Pyk2 on its own can phosphorylate tau, a hypothesis supported by our data that were obtained in both transfected mammalian cells and in vitro. We found that autophosphorylation of Pyk2 at Y402 was required for full Pyk2 phosphorylation and activation. Phosphorylation of tau at Y18 was massively abolished when the Y402 of Pyk2 was mutated into a phosphorylation-deficient phenylalanine, revealing that this process is dependent on Pyk2 activation. Furthermore, knocking down endogenous Pyk2 expression revealed decreased Y18 phosphorylation of tau, again supporting a role for Pyk2 as tau kinase. However, a recent study in Drosophila reported no role for Pyk2 in tau phosphorylation at either residue [22]. In contrast to this study in flies, our results provide evidence for a function of Pyk2 as a tau tyrosine kinase both in vitro and in vivo. The discrepancy may be due to the lack of homology of tau in flies compared to mammals, and also the possible loss of protein function in Drosophila resulting from differences in the post-translational modification system in vertebrates compared to invertebrates.
Fyn is intimately linked to tau pathology and tau-mediated Aβ toxicity [27, 28, 30]; on the one hand because Fyn is a direct tyrosine kinase of tau, and on the other hand due to the fact that the SH3 and SH2 domains of Fyn bind to PXXP motifs and the pY18 epitope of tau, respectively [28, 38, 44]. We have recently shown that Fyn regulates tau pathology, not only by acting as a tau tyrosine kinase, but also by mediating a signaling pathway involving the kinases ERK and S6 that controls the de novo protein translation of tau in the somatodendritic compartment [12]. We had also shown that increased Fyn-mediated tau translation occurs with concomitant tau phosphorylation at both Y18 and serine and threonine residues, further contributing to tau pathology in AD [12].
Although a number of links have been established between Pyk2 and SFKs in both neuronal and non-neuronal systems, the interaction of Pyk2 and Fyn has been underexplored [45–47]. Here, we found that both tau tyrosine kinases, Pyk2 and Fyn, are tightly associated, and that this association is dependent on the binding of the SH2 domain of Fyn and the pY402 epitope of Pyk2, a prerequisite for subsequent Y579 phosphorylation and full kinase activation [46]. These findings are supported by a previous study which revealed that Pyk2 autophosphorylation, but not its kinase activity, is necessary for its binding to c-Src [48].
Interestingly, both endogenous and exogenous Pyk2 are phospho-activated at the Y402 site by Fyn, and this is further supported by our in vivo evidence that Pyk2 is intensely activated in brains from 3-week-old FynCA transgenic mice expressing a constitutively active form of Fyn. In contrast, we observed a comparatively weak Fyn activation in 15-month-old Pyk2 transgenic mice. More importantly, complementing the finding of Fyn-mediated Pyk2 activation in young FynCA mice with that of a massively decreased Pyk2 activation in adult mice lacking Fyn expression (FynKO) identifies a role for Fyn in activating Pyk2.
Together, our data strongly suggest that Pyk2 is preferentially activated by Fyn; however, a role for Fyn downstream of Pyk2 in tauopathy is not fully ruled out. Consequently, it is possible that not only Fyn itself directly phosphorylates tau, but also that Fyn, by activating downstream Pyk2, further contributes to tyrosine phosphorylation of tau. Given the presence of Pyk2, tau and Fyn in synaptosomal fractions, and the previous evidence of their dendritic function, it is conceivable to assume that the interplay of Pyk2, tau and Fyn is critical for tau pathology in dendritic spines in a physiological and pathological context.
Conclusions
In summary, our data reveal that Pyk2 is a novel non-SFK tyrosine kinase of tau, enhancing tau pathology in vitro and also in vivo, as shown by establishing a novel Pyk2 transgenic mouse model and crossing this strain with a tau transgenic mouse model. We further demonstrated an association of Pyk2 and the established tau tyrosine kinase, Fyn, that is closely coupled to tau pathology. These findings not only offer a better understanding of how Pyk2 is involved in the development of tau pathology, but also shed light on the interplay of Pyk2, tau and Fyn as targets for therapeutic intervention in neurodegeneration.
ACKNOWLEDGMENTS
We thank Dr. Di Xia for assistance in making overexpression constructs, and Rowan Tweedale for critically reading the manuscript. This study was supported by the Estate of Dr. Clem Jones AO, the State Government of Queensland, the Federal Government of Australia (ACT900116), and by grants from the Australian Research Council [DP160103812, LE130100078] and the National Health and Medical Research Council of Australia [GNT1127999, GNT1037746] to JG.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0054r1).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-180054.
REFERENCES
[1] | Strittmatter WJ , Saunders AM , Schmechel D , Pericak-Vance M , Enghild J , Salvesen GS , Roses AD ((1993) ) Apolipoprotein E: High-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A 90: , 1977–1981. |
[2] | Guerreiro R , Wojtas A , Bras J , Carrasquillo M , Rogaeva E , Majounie E , Cruchaga C , Sassi C , Kauwe JS , Younkin S , Hazrati L , Collinge J , Pocock J , Lashley T , Williams J , Lambert JC , Amouyel P , Goate A , Rademakers R , Morgan K , Powell J , St George-Hyslop P , Singleton A , Hardy J , Alzheimer Genetic Analysis Group ((2013) ) TREM2 variants in Alzheimer’s disease. N Engl J Med 368: , 117–127. |
[3] | Ulrich JD , Ulland TK , Colonna M , Holtzman DM ((2017) ) Elucidating the role of TREM2 in Alzheimer’s disease. Neuron 94: , 237–248. |
[4] | Lambert JC , Ibrahim-Verbaas CA , Harold D , Naj AC , Sims R , Bellenguez C , DeStafano AL , Bis JC , Beecham GW , Grenier-Boley B , Russo G , Thorton-Wells TA , Jones N , Smith AV , Chouraki V , Thomas C , Ikram MA , Zelenika D , Vardarajan BN , Kamatani Y , Lin CF , Gerrish A , Schmidt H , Kunkle B , Dunstan ML , Ruiz A , Bihoreau MT , Choi SH , Reitz C , Pasquier F , Cruchaga C , Craig D , Amin N , Berr C , Lopez OL , De Jager PL , Deramecourt V , Johnston JA , Evans D , Lovestone S , Letenneur L , Morón FJ , Rubinsztein DC , Eiriksdottir G , Sleegers K , Goate AM , Fiévet N , Huentelman MW , Gill M , Brown K , Kamboh MI , Keller L , Barberger-Gateau P , McGuiness B , Larson EB , Green R , Myers AJ , Dufouil C , Todd S , Wallon D , Love S , Rogaeva E , Gallacher J , St George-Hyslop P , Clarimon J , Lleo A , Bayer A , Tsuang DW , Yu L , Tsolaki M , Bossú P , Spalletta G , Proitsi P , Collinge J , Sorbi S , Sanchez-Garcia F , Fox NC , Hardy J , Deniz Naranjo MC , Bosco P , Clarke R , Brayne C , Galimberti D , Mancuso M , Matthews F European Alzheimer’s Disease Initiative (EADI); Genetic and Environmental Risk in Alzheimer’s Disease; Alzheimer’s Disease Genetic Consortium; Cohorts for Heart and Aging Research in Genomic Epidemiology , Moebus S , Mecocci P , Del Zompo M , Maier W , Hampel H , Pilotto A , Bullido M , Panza F , Caffarra P , Nacmias B , Gilbert JR , Mayhaus M , Lannefelt L , Hakonarson H , Pichler S , Carrasquillo MM , Ingelsson M , Beekly D , Alvarez V , Zou F , Valladares O , Younkin SG , Coto E , Hamilton-Nelson KL , Gu W , Razquin C , Pastor P , Mateo I , Owen MJ , Faber KM , Jonsson PV , Combarros O , O’Donovan MC , Cantwell LB , Soininen H , Blacker D , Mead S , Mosley THJr , Bennett DA , Harris TB , Fratiglioni L , Holmes C , de Bruijn RF , Passmore P , Montine TJ , Bettens K , Rotter JI , Brice A , Morgan K , Foroud TM , Kukull WA , Hannequin D , Powell JF , Nalls MA , Ritchie K , Lunetta KL , Kauwe JS , Boerwinkle E , Riemenschneider M , Boada M , Hiltuenen M , Martin ER , Schmidt R , Rujescu D , Wang LS , Dartigues JF , Mayeux R , Tzourio C , Hofman A , Nöthen MM , Graff C , Psaty BM , Jones L , Haines JL , Holmans PA , Lathrop M , Pericak-Vance MA , Launer LJ , Farrer LA , van Duijn CM , Van Broeckhoven C , Moskvina V , Seshadri S , Williams J , Schellenberg GD , Amouyel P ((2013) ) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45: , 1452–1458. |
[5] | Beecham GW , Hamilton K , Naj AC , Martin ER , Huentelman M , Myers AJ , Corneveaux JJ , Hardy J , Vonsattel JP , Younkin SG , Bennett DA , De Jager PL , Larson EB , Crane PK , Kamboh MI , Kofler JK , Mash DC , Duque L , Gilbert JR , Gwirtsman H , Buxbaum JD , Kramer P , Dickson DW , Farrer LA , Frosch MP , Ghetti B , Haines JL , Hyman BT , Kukull WA , Mayeux RP , Pericak-Vance MA , Schneider JA , Trojanowski JQ , Reiman EM , Alzheimer’s Disease Genetics Consortium , Schellenberg GD , Montine TJ ((2014) ) Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet 10: , e1004606. |
[6] | De Jager PL , Srivastava G , Lunnon K , Burgess J , Schalkwyk LC , Yu L , Eaton ML , Keenan BT , Ernst J , McCabe C , Tang A , Raj T , Replogle J , Brodeur W , Gabriel S , Chai HS , Younkin C , Younkin SG , Zou F , Szyf M , Epstein CB , Schneider JA , Bernstein BE , Meissner A , Ertekin-Taner N , Chibnik LB , Kellis M , Mill J , Bennett DA ((2014) ) Alzheimer’s disease: Early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci 17: , 1156–1163. |
[7] | Chan G , White CC , Winn PA , Cimpean M , Replogle JM , Glick LR , Cuerdon NE , Ryan KJ , Johnson KA , Schneider JA , Bennett DA , Chibnik LB , Sperling RA , Bradshaw EM , De Jager PL ((2015) ) CD33 modulates TREM2: Convergence of Alzheimer loci. Nat Neurosci 18: , 1556–1558. |
[8] | Bakota L , Brandt R ((2016) ) Tau biology and tau-directed therapies for Alzheimer’s disease. Drugs 76: , 301–313. |
[9] | Guo T , Noble W , Hanger DP ((2017) ) Roles of tau protein in health and disease. Acta Neuropathol 133: , 665–704. |
[10] | Chen F , David D , Ferrari A , Götz J ((2004) ) Posttranslational modifications of tau–role in human tauopathies and modeling in transgenic animals. Curr Drug Targets 5: , 503–515. |
[11] | Berger Z , Roder H , Hanna A , Carlson A , Rangachari V , Yue M , Wszolek Z , Ashe K , Knight J , Dickson D , Andorfer C , Rosenberry TL , Lewis J , Hutton M , Janus C ((2007) ) Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J Neurosci 27: , 3650–3662. |
[12] | Li C , Götz J ((2017) ) Somatodendritic accumulation of Tau in Alzheimer’s disease is promoted by Fyn-mediated local protein translation. EMBO J 36: , 3120–3138. |
[13] | Hatch RJ , Wei Y , Xia D , Götz J ((2017) ) Hyperphosphorylated tau causes reduced hippocampal CA1 excitability by relocating the axon initial segment. Acta Neuropathol 133: , 717–730. |
[14] | Violet M , Chauderlier A , Delattre L , Tardivel M , Chouala MS , Sultan A , Marciniak E , Humez S , Binder L , Kayed R , Lefebvre B , Bonnefoy E , Buee L , Galas MC ((2015) ) Prefibrillar tau oligomers alter the nucleic acid protective function of tau in hippocampal neurons in vivo. Neurobiol Dis 82: , 540–551. |
[15] | Friedland-Leuner K , Stockburger C , Denzer I , Eckert GP , Muller WE ((2014) ) Mitochondrial dysfunction: Cause and consequence of Alzheimer’s disease. Prog Mol Biol Transl Sci 127: , 183–210. |
[16] | Pickett EK , Henstridge CM , Allison E , Pitstick R , Pooler A , Wegmann S , Carlson G , Hyman BT , Spires-Jones TL ((2017) ) Spread of tau down neural circuits precedes synapse and neuronal loss in the rTgTauEC mouse model of early Alzheimer’s disease. Synapse 71: , e21965. |
[17] | Gilley J , Seereeram A , Ando K , Mosely S , Andrews S , Kerschensteiner M , Misgeld T , Brion JP , Anderton B , Hanger DP , Coleman MP ((2012) ) Age-dependent axonal transport and locomotor changes and tau hypophosphorylation in a “P301L” tau knockin mouse. Neurobiol Aging 33: , 621 e621–621 e615. |
[18] | Frost B , Hemberg M , Lewis J , Feany MB ((2014) ) Tau promotes neurodegeneration through global chromatin relaxation. Nat Neurosci 17: , 357–366. |
[19] | Kandimalla R , Manczak M , Fry D , Suneetha Y , Sesaki H , Reddy PH ((2016) ) Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum Mol Genet 25: , 4881–4897. |
[20] | Köhler C , Dinekov M , Götz J ((2013) ) Active glycogen synthase kinase-3 and tau pathology-related tyrosine phosphorylation in pR5 human tau transgenic mice. Neurobiol Aging 34: , 1369–1379. |
[21] | Deters N , Ittner LM , Götz J ((2008) ) Divergent phosphorylation pattern of tau in P301L tau transgenic mice. Eur J Neurosci 28: , 137–147. |
[22] | Dourlen P , Fernandez-Gomez FJ , Dupont C , Grenier-Boley B , Bellenguez C , Obriot H , Caillierez R , Sottejeau Y , Chapuis J , Bretteville A , Abdelfettah F , Delay C , Malmanche N , Soininen H , Hiltunen M , Galas MC , Amouyel P , Sergeant N , Buee L , Lambert JC , Dermaut B ((2017) ) Functional screening of Alzheimer risk loci identifies PTK2B as an in vivo modulator and early marker of Tau pathology. Mol Psychiatry 22: , 874–883. |
[23] | Lipinski CA , Loftus JC ((2010) ) Targeting Pyk2 for therapeutic intervention. Expert Opin Ther Targets 14: , 95–108. |
[24] | Huang Y , Lu W , Ali DW , Pelkey KA , Pitcher GM , Lu YM , Aoto H , Roder JC , Sasaki T , Salter MW , MacDonald JF ((2001) ) CAKβ/Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus. Neuron 29: , 485–496. |
[25] | Bartos JA , Ulrich JD , Li H , Beazely MA , Chen Y , Macdonald JF , Hell JW ((2010) ) Postsynaptic clustering and activation of Pyk2 by PSD-95. J Neurosci 30: , 449–463. |
[26] | Xia D , Li C , Götz J ((2015) ) Pseudophosphorylation of Tau at distinct epitopes or the presence of the P301L mutation targets the microtubule-associated protein Tau to dendritic spines. Biochim Biophys Acta 1852: , 913–924. |
[27] | Um JW , Nygaard HB , Heiss JK , Kostylev MA , Stagi M , Vortmeyer A , Wisniewski T , Gunther EC , Strittmatter SM ((2012) ) Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci 15: , 1227–1235. |
[28] | Ittner LM , Ke YD , Delerue F , Bi M , Gladbach A , van Eersel J , Wolfing H , Chieng BC , Christie MJ , Napier IA , Eckert A , Staufenbiel M , Hardeman E , Götz J ((2010) ) Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell 142: , 387–397. |
[29] | Roberson ED , Halabisky B , Yoo JW , Yao J , Chin J , Yan F , Wu T , Hamto P , Devidze N , Yu GQ , Palop JJ , Noebels JL , Mucke L ((2011) ) Amyloid-β/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J Neurosci 31: , 700–711. |
[30] | Larson M , Sherman MA , Amar F , Nuvolone M , Schneider JA , Bennett DA , Aguzzi A , Lesné SE ((2012) ) The complex PrP(c)-Fyn couples human oligomeric Aβ with pathological tau changes in Alzheimer’s disease. J Neurosci 32: 16857–16871a. |
[31] | Giralt A , Brito V , Chevy Q , Simonnet C , Otsu Y , Cifuentes-Diaz C , de Pins B , Coura R , Alberch J , Gines S , Poncer JC , Girault JA ((2017) ) Pyk2 modulates hippocampal excitatory synapses and contributes to cognitive deficits in a Huntington’s disease model. Nat Commun 8: , 15592. |
[32] | Hsin H , Kim MJ , Wang CF , Sheng M ((2010) ) Proline-rich tyrosine kinase 2 regulates hippocampal long-term depression. J Neurosci 30: , 11983–11993. |
[33] | Xia D , Götz J ((2014) ) Premature lethality, hyperactivity, and aberrant phosphorylation in transgenic mice expressing a constitutively active form of Fyn. Front Mol Neurosci 7: , 40. |
[34] | Götz J , Chen F , van Dorpe J , Nitsch RM ((2001) ) Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Aβ 42 fibrils. Science 293: , 1491–1495. |
[35] | Ittner LM , Götz J ((2007) ) Pronuclear injection for the production of transgenic mice. Nat Protoc 2: , 1206–1215. |
[36] | Lee G , Thangavel R , Sharma VM , Litersky JM , Bhaskar K , Fang SM , Do LH , Andreadis A , Van Hoesen G , Ksiezak-Reding H ((2004) ) Phosphorylation of tau by fyn: Implications for Alzheimer’s disease. J Neurosci 24: , 2304–2312. |
[37] | Miyamoto T , Stein L , Thomas R , Djukic B , Taneja P , Knox J , Vossel K , Mucke L ((2017) ) Phosphorylation of tau at Y18, but not tau-fyn binding, is required for tau to modulate NMDA receptor-dependent excitotoxicity in primary neuronal culture. Mol Neurodegener 12: , 41. |
[38] | Lee G , Newman ST , Gard DL , Band H , Panchamoorthy G ((1998) ) Tau interacts with src-family non-receptor tyrosine kinases. J Cell Sci 111 (Pt 21): , 3167–3177. |
[39] | Lebouvier T , Scales TM , Hanger DP , Geahlen RL , Lardeux B , Reynolds CH , Anderton BH , Derkinderen P ((2008) ) The microtubule-associated protein tau is phosphorylated by Syk. Biochim Biophys Acta 1783: , 188–192. |
[40] | Derkinderen P , Scales TM , Hanger DP , Leung KY , Byers HL , Ward MA , Lenz C , Price C , Bird IN , Perera T , Kellie S , Williamson R , Noble W , Van Etten RA , Leroy K , Brion JP , Reynolds CH , Anderton BH ((2005) ) Tyrosine 394 is phosphorylated in Alzheimer’s paired helical filament tau and in fetal tau with c-Abl as the candidate tyrosine kinase. J Neurosci 25: , 6584–6593. |
[41] | Saito YD , Jensen AR , Salgia R , Posadas EM ((2010) ) Fyn: A novel molecular target in cancer. Cancer 116: , 1629–1637. |
[42] | Li C , Götz J ((2017) ) Tau-based therapies in neurodegeneration: Opportunities and challenges. Nat Rev Drug Discov 16: , 863–883. |
[43] | Hoover BR , Reed MN , Su J , Penrod RD , Kotilinek LA , Grant MK , Pitstick R , Carlson GA , Lanier LM , Yuan LL , Ashe KH , Liao D ((2010) ) Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68: , 1067–1081. |
[44] | Bhaskar K , Yen SH , Lee G ((2005) ) Disease-related modifications in tau affect the interaction between Fyn and Tau. J Biol Chem 280: , 35119–35125. |
[45] | Canobbio I , Cipolla L , Guidetti GF , Manganaro D , Visconte C , Kim S , Okigaki M , Falasca M , Kunapuli SP , Torti M ((2015) ) The focal adhesion kinase Pyk2 links Ca2+signalling to Src family kinase activation and protein tyrosine phosphorylation in thrombin-stimulated platelets. Biochem J 469: , 199–210. |
[46] | Zhao M , Finlay D , Zharkikh I , Vuori K ((2016) ) Novel role of Src in priming Pyk2 phosphorylation. PLoS One 11: , e0149231. |
[47] | Dikic I , Tokiwa G , Lev S , Courtneidge SA , Schlessinger J ((1996) ) A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation. Nature 383: , 547–550. |
[48] | Lakkakorpi PT , Bett AJ , Lipfert L , Rodan GA , Duong LT ((2003) ) PYK2 autophosphorylation, but not kinase activity, is necessary for adhesion-induced association with c-Src, osteoclast spreading, and bone resorption. J Biol Chem 278: , 11502–11512. |


