
The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade
Abstract
The amyloid-β oligomer (AβO) hypothesis was introduced in 1998. It proposed that the brain damage leading to Alzheimer’s disease (AD) was instigated by soluble, ligand-like AβOs. This hypothesis was based on the discovery that fibril-free synthetic preparations of AβOs were potent CNS neurotoxins that rapidly inhibited long-term potentiation and, with time, caused selective nerve cell death (Lambert et al., 1998). The mechanism was attributed to disrupted signaling involving the tyrosine-protein kinase Fyn, mediated by an unknown toxin receptor. Over 4,000 articles concerning AβOs have been published since then, including more than 400 reviews. AβOs have been shown to accumulate in an AD-dependent manner in human and animal model brain tissue and, experimentally, to impair learning and memory and instigate major facets of AD neuropathology, including tau pathology, synapse deterioration and loss, inflammation, and oxidative damage. As reviewed by Hayden and Teplow in 2013, the AβO hypothesis “has all but supplanted the amyloid cascade.” Despite the emerging understanding of the role played by AβOs in AD pathogenesis, AβOs have not yet received the clinical attention given to amyloid plaques, which have been at the core of major attempts at therapeutics and diagnostics but are no longer regarded as the most pathogenic form of Aβ. However, if the momentum of AβO research continues, particularly efforts to elucidate key aspects of structure, a clear path to a successful disease modifying therapy can be envisioned. Ensuring that lessons learned from recent, late-stage clinical failures are applied appropriately throughout therapeutic development will further enable the likelihood of a successful therapy in the near-term.
Abbreviations: α7nAChR, alpha 7-nicotinic acetylcholine receptor; 5XFAD, transgenic mouse model of AD carrying 5 AD-related familial mutations; A11, amyloid oligomer polyclonal antibody; Aβ, Amyloid β peptide; Aβ40, Amyloid β peptide 1– 40 sequence; Aβ42, Amyloid β peptide 1– 42 sequence; Aβ43, Amyloid β peptide 1– 43 sequence; AβOs, Aβ oligomers; AD, Alzheimer’s disease; AkT, Protein Kinase B; ALS, Amyotrophic lateral sclerosis; AMPA, α-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor; APOE, Apolipoprotein E gene; ApoE, Apolipoprotein E; APP, Amyloid precursor protein; AFM, atomic force microscopy; BACE, β-secretase; Ca++, calcium ion; CaMKII, Ca++/calmodulin-dependent protein kinase II; cDNA, complementary DNA; CNS, central nervous system; CSF, cerebrospinal fluid; CT, cortex; CTAD, Clinical Trials on Alzheimer’s Disease; CTE, chronic traumatic encephalopathy; DHA, docosahexaenoic acid; DPP4, dipeptidyl peptidase 4; EphB2, Ephrin type B receptor 2; EphA4, Ephrin type A receptor 4; ER, endoplasmic reticulum; ERK, extracellular signal-regulated kinase; Fab, fragment antigen-binding; fAD, Familial Alzheimer’s disease; FAK, focal adhesion kinase; FcγRIIb, Immunoglobulin gamma Fc region receptor II-b; FPR2, N-formyl peptide receptor 2; Fyn, tyrosine-protein kinase Fyn; GSK3β, glycogen synthase kinase 3 β; GTPase Drp-1, GTPase dynamin-related protein 1; HDAC6, histone deacetylase 6; HMW, high molecular weight; HP, hippocampus; i.c.v., intracerebroventricular; IGF-1, insulin-like growth factor 1; iPSC, induced pluripotent stem cells; IR, insulin receptor; IRS-1, insulin receptor substrate 1; kDa, kilodalton; LilRb2, leukocyte immunoglobulin-like receptor subfamily B member 2; LMW, low molecular weight; LRP-1, lipoprotein receptor; LTD, long-term depression; LTP, long-term potentiation; MCI, mild cognitive impairment; mGluR5, metabotropic glutamate receptor 5; MRI, magnetic resonance imaging; NADPH, nicotinamide adenine dinucleotide phosphate; NHPs, non-human primates; NKAα3, Na+/K+ ATPase alpha 3 subunit; nM, nanomolar; NMDARs, N-methyl-D-Aspartate receptors; NO, nitric oxide; NU4, AβO-selective mouse monoclonal antibody; N-VSCCs, N-type voltage-sensitive calcium channels; OC, anti-amyloid fibril antibody; p38 MAPK, p38 mitogen-activated protein kinases; p75NTR, p75 neutrophin receptor; pE, pyroglutamylated; PET, positron emission tomography; PICUP, photo-induced crosslinking of unmodified proteins; POPC/POPS, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC)/1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS); PPAR-γ, peroxisome proliferator-activated receptor gamma; PrPc, cellular prion protein; PS1, presenilin-1; PSEN1, presenilin-1 gene; pTau, phosphorylated tau; Pyk2, protein tyrosine kinase 2; RAGE, receptor for advanced glycation endproducts; ROS, Reactive Oxygen Species; sAD, Sporadic Alzheimer’s disease; SDS, sodium dodecyl sulfate; SDS-PAGE, sodium dodecyl sulfate polyacrylamide gel electrophoresis; SEC, size exclusion chromatography; SEM, standard error of the mean; Sigma-2/PGRMC1, Sigma-2 receptor/progesterone receptor membrane component 1; TBI, traumatic brain injury; TNF, tumor necrosis factor; ThioS, Thioflavin S; Tg, transgenic; TRPM2, transient receptor potential melastatin family subtype 2; VEGF-A, vascular endothelial growth factor A.
INTRODUCTION TO THE AβO HYPOTHESIS
The transition from the amyloid cascade to the AβO hypothesis
The detection of amyloid-β oligomers (AβOs) in human brain parenchyma and vasculature was first reported while the original amyloid cascade hypothesis was being introduced and developed [1–3]. At the time, AβOs were regarded as intermediates en route to generation of amyloid plaques, which were believed to be the pathogenic form of Aβ.
Today, AβOs are widely regarded as the most toxic and pathogenic form of Aβ (Fig.1) [4, 5]. AβOs show an Alzheimer’s disease (AD)-dependent presence in humans and animal models [1, 6–13], and their buildup occurs early, before plaques, evidenced by both immunochemistry [14] and immunohistochemistry [15, 16]. In support of a toxic role for AβOs and not plaques, the Osaka familial AD mutation of Aβ (APP E693Δ) shows extremely low levels of senile plaques [17–21] despite severe cognitive impairment [17, 20], while cerebrospinal fluid (CSF) manifests low levels of overall Aβ, but elevated levels of AβOs [22]. Transgenic (Tg) mice carrying this mutation [19], or a closely related one [23], likewise manifest AβOs and other major forms of AD neuropathology but not plaques. Although historically AD has been defined as dementia with plaques and tangles, replacing plaques with AβOs in this definition may be closer to the pathogenic mechanism.
Fig.1
AβOs, not Aβ monomers or fibrils, instigate the neuron damage leading to dementia. Following cleavage from the membrane, Aβ peptides aggregate to form AβOs, some of which further aggregate to fibrils and some of which instigate the neuron damage leading to dementia. Reprinted with Jannis Productions permissions from the “Progress Report on Alzheimer’s Disease 2004-2005” (ed. AB Rodgers), NIH Publication Number: 05-5724. Digital images produced by Stacy Jannis and Rebekah Fredenburg of Jannis Productions [455].
![AβOs, not Aβ monomers or fibrils, instigate the neuron damage leading to dementia. Following cleavage from the membrane, Aβ peptides aggregate to form AβOs, some of which further aggregate to fibrils and some of which instigate the neuron damage leading to dementia. Reprinted with Jannis Productions permissions from the “Progress Report on Alzheimer’s Disease 2004-2005” (ed. AB Rodgers), NIH Publication Number: 05-5724. Digital images produced by Stacy Jannis and Rebekah Fredenburg of Jannis Productions [455].](https://content.iospress.com:443/media/jad/2018/64-s1/jad-64-s1-jad179941/jad-64-jad179941-g001.jpg)
Synthetic AβOs can assemble at very low concentrations of Aβ42 monomer, in harmony with pre-plaque buildup in brain tissue [24, 25]. In vitro, AβOs form within minutes from low nanomolar concentrations of monomeric Aβ42 [26, 27]. Because Aβ has been found to aggregate in sodium dodecyl sulfate (SDS) [28], some investigators have concluded that the quickly forming AβOs seen in SDS-PAGE are experimental artifacts [29, 30]. However, under full denaturing conditions, SDS-PAGE experiments show monomeric Aβ in the complete absence of AβOs [31]. AβOs can be observed, moreover, in the absence of SDS by atomic force microscopy (AFM) and by size exclusion chromatography [26, 31]. Evidence for structural homology between certain forms of synthetic and brain-derived AβOs has been presented [6]; this is discussed further below.
Besides their presence in brain, AβOs show an AD-dependent buildup in human CSF. An ultrasensitive assay, known as the BioBarcode, which is capable of attomolar measurements, showed median levels of AβOs in CSF from AD patients to be 30-fold higher than from non-demented individuals [32]. This elevation is opposite to the AD-dependent change measured in monomeric Aβ levels, which decrease rather than increase [33]. Levels are so low, however, that for most assays, comparisons of CSF AβO levels are not feasible [12, 32, 34, 35]. Ultrasensitive assays for AβOs in CSF, however, are extremely lengthy and difficult, and their lack of precision requires multiple runs to provide a reliable measurement. These are all factors that preclude their adaptation for the clinic.
Fig.2
AβOs instigate multiple facets of AD-neuropathology. Observed in various culture and animal models. Reprinted by permission from Springer Nature: Acta Neuropathol, 129(2): 183-206, “Amyloid beta oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis” by Viola KL and Klein WL. Copyright 2015 Springer Nature [200].
![AβOs instigate multiple facets of AD-neuropathology. Observed in various culture and animal models. Reprinted by permission from Springer Nature: Acta Neuropathol, 129(2): 183-206, “Amyloid beta oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis” by Viola KL and Klein WL. Copyright 2015 Springer Nature [200].](https://content.iospress.com:443/media/jad/2018/64-s1/jad-64-s1-jad179941/jad-64-jad179941-g002.jpg)
There is extensive evidence that elevated AβO levels in the brain has pathogenic consequences, with results coming from behavioral, neuropathological, and cell biological studies, as discussed below. Memory performance is lost when small quantities of AβOs are injected into the intracerebral ventricle (i.c.v.) of non-Tg animals [36–39]. Long-term potentiation (LTP) and long-term depression, the electrophysiological underpinnings of memory formation, are disrupted by AβOs both ex vivo and in vivo [26, 36, 37, 40, 41]. Synthetic and brain-derived AβOs both exhibit these characteristics. In addition to their cognitive impact, exogenous AβOs instigate multiple facets of AD-neuropathology in culture and animal models, including non-human primates (NHPs) [42–46]. If one assumes an AβO molecular weight in aqueous solution of ∼100 kDa (see below), these effects are elicited at sub-nanomolar AβO concentrations [26, 47–50]. Overall, AβOs have been found to instigate tau pathology [19, 51, 52], loss of neuronal polarity [53–55], impairment of axonal transport [56–58], deterioration of synapses [47, 55], oxidative stress [59–62], endoplasmic reticulum (ER) stress [18, 63, 64], insulin resistance [48, 65–67], neuroinflammation [19, 49, 68, 69], cholinergic impairment [70, 71], loss of trophic factors [45, 72–75], epigenetic changes [74, 76–80], ectopic mitosis [81–83], and selective nerve cell death [26, 84]. A complicating factor is that these various responses were obtained under widely divergent conditions, with different disease models, time-scales, doses, and AβO preparations. Nonetheless, the collective body of evidence offers strong support for a mechanism in which AD neuropathology and cognitive loss are the consequences of the cellular damage instigated by AβOs (Fig.2).
The evidence is strong that AβOs are manifested in AD brain. Experiments in animal models strongly suggest, furthermore, that AβOs are both necessary and sufficient for dementia. Sufficiency, at least vis-á-vis amyloid plaques, is indicated by instances of AD without senile plaque pathology. Highly demented individuals with the Osaka mutation (APP E693Δ) manifest AβOs (and other facets of AD pathology) in the absence of senile plaques [17–21]. This has been experimentally recapitulated in a Tg mouse model harboring this mutation [19]. In addition, Tg mice expressing a different mutation in the same APP residue (Dutch APP E693Q) also exhibit AβO accumulation and altered synaptic structure without plaques [23, 85]. The sufficiency of AβOs for pathogenesis was first indicated in an APP mouse (Indiana APP mutation V717F; outside of Aβ42 sequence) that showed synapse loss despite absence of plaques [86]. In addition, a Tg rat expressing the Indiana mutation also shows pre-plaque AβO-associated cognitive impairment [87]. A later study comparing Tg strains indicated in fact that elevated levels of amyloid plaques likely protected against pathogenic AβO buildup [88].
Fig.3
Single injection (30μg) of an AβO-specific antibody ameliorates cognitive deficits in AD mice for at least 40 days. 5xFAD Tg mice and their wild-type (WT) littermates (6 months of age) were evaluated by Object Recognition Tasks before and after (40 days) a single injection (30μg) of a humanized AβO-specific antibody (anti-AβO) or non-specific human IgG (hIgG). First, locomotor activity was assessed while mice were allowed to habituate to the testing field (Habituation). Assessments were the number of times the mice crossed grids in the field (Crossings, light gray) and the number of times mice put their hind paws on the walls of the field (Rearings, purple), with no differences between WT and 5xFAD mice. Next, the test objects (F1 and F2) were introduced to the mice in the Training session. All mice showed normal exploratory behavior, defined by 50% exploration of each object, as both objects are equal and new to the mice. The ability of mice to remember object placement was then tested 24 hours after the Training session in a hippocampal (HP)-dependent task. Another 24 hours later, the ability of mice to remember the object was tested in a cortical (CT)-dependent task. Only the WT mice were able to recognize the familiar object (F1) from the Training session, as evidenced by >50% exploration of the displaced (D, pink) or new (N, light blue) object. The 5xFAD mice failed to recognize F1 in both tasks. When re-evaluated 40 days post-antibody injection in a HP-dependent task, only the 5xFAD mice that received the AβO antibody recovered their ability to recognize object F1. These data support the hypothesis that AβOs induce memory dysfunction in AD (Bicca and Klein, unpublished).
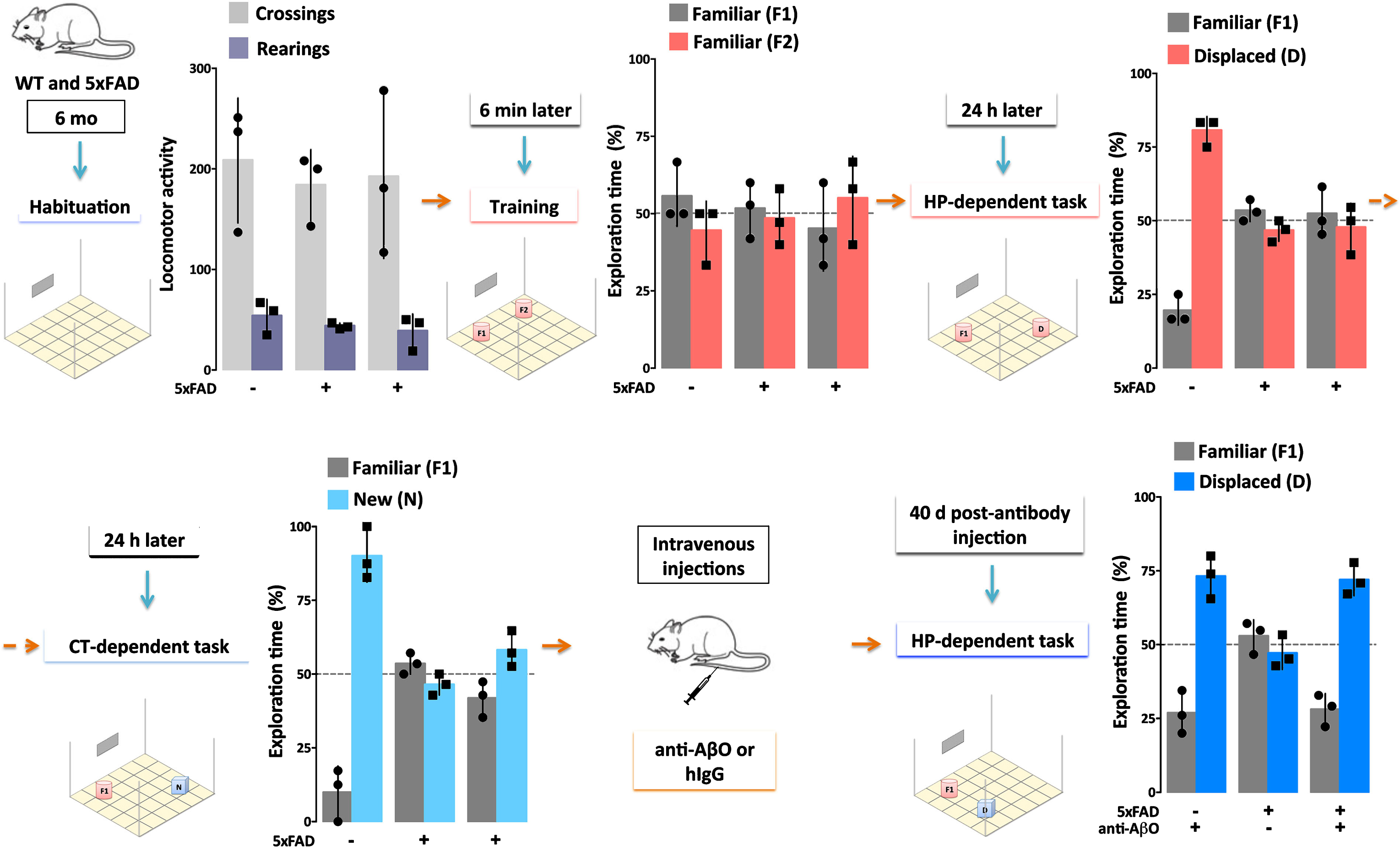
Direct evidence that AβOs are necessary for dementia comes from experiments using AβO-selective antibodies. Such antibodies were first shown to protect cell models against the damage caused by exogenous AβOs [51, 89, 90]. When administered to various Tg AD mice, the antibodies prevent AD-like pathology and rescue memory performance [89–94]. New data from our group indicates that a single injection of an AβO-selective antibody (30μg) can suffice to rescue memory performance in 6-7-month-old Tg 5xFAD mice for at least 40 days (Fig.3; Bicca and Klein, unpublished data). AβOs and plaques in these mice begin to accumulate extensively around 2 months of age [11, 92, 95]. The new data are in harmony with previous evidence that an AβO-selective antibody can reach the parenchyma and engage AβOs [96], but not Thioflavin S (ThioS)-positive amyloid plaques, when injected into 5xFAD mice.
The large body of evidence that AβOs are both necessary and sufficient to trigger AD-associated memory malfunction and neurodegeneration, coupled with the extensive portfolio of documented AβO-triggered cellular and behavioral effects, sets the stage for new AD therapeutic approaches targeting AβOs. As the third decade of the AβO hypothesis begins, the biggest challenge is to mobilize a clinical trial that will validate or invalidate the hypothesis. While “Aβ dyshomeostasis has emerged as the most extensively validated and compelling therapeutic target” [5], the past development of Aβ-based therapeutics has largely concerned plaque elimination, ignoring AβOs. However, the link between plaques and cognitive dysfunction has been tenuous for decades [97–99], and no Aβ-directed therapeutic has yet reached a clinical efficacy endpoint [100–103]. In a potential turning point, an antibody that can engage AβOs, Aducanumab, has recently shown modest therapeutic benefit in early clinical trials [104, 105]. A potential limitation of Aducanumab is that it lacks stringent selectivity for AβOs. Off-target engagement with senile plaques likely accounts for the high dosage-requirement found in trials. Antibodies are needed that target only the most pathogenic configurations of Aβ, i.e., AβOs. Such antibodies will be optimized by a better understanding of AβO structure-toxicity relationships [101, 106–108],
Besides development of AβO-specific antibodies [89, 101, 109], other tactics are likely to improve the prospects of Aβ-directed therapies. Such tactics may be earlier intervention within the disease continuum and better criteria for patient selection [5, 108] and better biomarkers for monitoring of investigational new drugs [103], including inflammation markers to better predict complications [106]. Furthermore, multi-factorial therapies may be needed [106]. Although it has been suggested that Aβ-targeting therapies may only be beneficial in prodromal individuals [110], if AβOs play a role in disease progression, e.g., through promoting propagation of tau pathology (below), there may be a meaningful chance that AβO-immunotherapy would be beneficial even after AD onset. Overall, there is an important call for more rigor in preclinical development. At each phase of the drug discovery process for Aβ-targeting therapies, it has been possible to find significant gaps in data [102]. Target engagement, e.g., was not established for the majority of therapeutic agents analyzed [102, 107]. Furthermore, compounds have been moved into phase III trials on the basis of very limited data [111], premature moves that have had a tendency to poison the well. Consequently, the discouraging track record of Aβ-directed drugs has provided significant impetus to point new drug discovery efforts toward non-Aβ targets, despite the preponderance of evidence that AβOs are the culpable AD neurotoxins [112–114].
STATUS OF THE FIELD
In lieu of clinical efforts based on the AβO hypothesis, there nonetheless have been substantial developments in the last five years regarding more fundamental issues. Of the more than 4,000 publications on Aβ oligomers or oligomeric Aβ, about half were published in the last five years. These fundamental developments regarding AβO pathogenicity are just beginning to be tested clinically and we predict that they will set the stage for therapeutic success. This section will consider major developments regarding: 1) species of AβOs, their assembly, and relation to amyloid plaques, and emerging insights into how to approach molecular structure; 2) mechanisms of how AβOs initiate their impact on neuronal function and structure; 3) downstream pathways resulting in neural damage, and 4) multicellular interactions contributing to AβO pathogenicity.
A multitude of AβO species or just two?
One of the biggest knowledge gaps currently facing the field is the precise identity of the most toxic AβO structures [5, 29, 101, 106, 107, 115–117]. Without this knowledge, it is impossible to know if Aβ-directed therapeutics are engaging the correct target. Characterization of AβO structure has been hindered by AβO metastability and heterogeneity [116, 117]. Consequently, a multitude of AβO species have been identified in the literature [117]. It is not clear which of these AβO species are AD-relevant and which are experimental artifacts. One possibility is that there exist a multitude of pathogenically-relevant AβO species in the AD brain and that their high number correlates with the myriad Aβ-associated toxic pathways identified in the literature [29, 101, 106, 115]. Another possibility is that there are only a few discrete AD-relevant species, and the majority identified in the literature are merely artifacts induced by non-physiological experimental conditions [107, 116, 117]. As stated by Benilova and colleagues, “The lack of a common, agreed-upon experimental description of the toxic Aβ oligomer makes interpretation and direct comparison of data between different research groups impossible [117].”
Fig.4
AβOs can be divided into two classes based on their temporal, spatial, and structural relationships to amyloid plaques as well as their ability to cause memory dysfunction. Type 1 AβOs (aka “peak 1” or HMW) are thought to be associated with memory impairment, while type 2 AβOs (aka “peak 2” or LMW) are not. Only type 2 AβOs are associated with amyloid plaques. Reprinted from “Quaternary Structure Defines a Large Class of Amyloid-beta Oligomers Neutralized by Sequestration” by Liu P, Reed MN, Kotilinek LA, Grant MK, Forster CL, Qiang W, Shapiro SL, Reichl JH, Chiang AC, Jankowsky JL, Wilmot CM, Cleary JP, Zahs KR, and Ashe KH. This was published in Cell Rep, 2015, 11(11): 1760-1771, under the terms of the Creative Commons Attribution Non-Commercial No Derivatives License (CC BY NC ND) https://creativecommons.org/licenses/by-nc-nd/4.0/ [119].
![AβOs can be divided into two classes based on their temporal, spatial, and structural relationships to amyloid plaques as well as their ability to cause memory dysfunction. Type 1 AβOs (aka “peak 1” or HMW) are thought to be associated with memory impairment, while type 2 AβOs (aka “peak 2” or LMW) are not. Only type 2 AβOs are associated with amyloid plaques. Reprinted from “Quaternary Structure Defines a Large Class of Amyloid-beta Oligomers Neutralized by Sequestration” by Liu P, Reed MN, Kotilinek LA, Grant MK, Forster CL, Qiang W, Shapiro SL, Reichl JH, Chiang AC, Jankowsky JL, Wilmot CM, Cleary JP, Zahs KR, and Ashe KH. This was published in Cell Rep, 2015, 11(11): 1760-1771, under the terms of the Creative Commons Attribution Non-Commercial No Derivatives License (CC BY NC ND) https://creativecommons.org/licenses/by-nc-nd/4.0/ [119].](https://content.iospress.com:443/media/jad/2018/64-s1/jad-64-s1-jad179941/jad-64-jad179941-g004.jpg)
Some patterns regarding AβO structure-toxicity relationships are, however, already emerging in the literature. For instance, it appears as if AβOs, whether produced in vitro or present in the brain of animal models or AD patients, can be divided into toxic and non-toxic sub-populations based on simple aspects of their quaternary structure, molecular weight and antibody reactivity, as well as their relationship to amyloid plaques. The toxic AβO species appear to be greater than 50 kDa [16, 55, 118], reactive with the anti-amyloid oligomer antibody A11 [119] and the anti-AβO antibody NU4 [120], and unrelated to amyloid plaques (Fig.4) [118, 119]. On the other hand, the non-toxic AβO species appear to be less than 50 kDa [16, 55, 118], reactive with the anti-fibril antibody OC [119], and related to amyloid plaques temporally, spatially, and structurally [118, 119]. In addition to their convenient immuno-identification, they also can be separated in vitro by size exclusion chromatography [31] or ultrafiltration with a 50 kDa molecular weight cutoff [16, 55, 118]. These populations have been referred to in the literature, respectively, as “peak 1” and “peak 2” [31], high molecular weight (HMW) and low molecular weight (LMW) [16, 55, 115, 118], and “type 1” and “type 2” [119]. Myriad evidence supports a toxic role for type 1 AβOs. In vitro, they bind cultured synapses (Fig.5) [16, 55, 118], inducing production of reactive oxygen species (ROS) [39], while type 2 AβOs cannot. Both species have been implicated in binding PrPc [121–123]. In vivo, type 1 AβOs disrupt memory function [39, 119, 120]. Type 2 AβOs have been found not to be associated with memory dysfunction [119, 120], although in one study, they were [39]. HMW, type 1 AβOs appear to be most prominent AβOs in the AD brain under native conditions [124–126]. The differential toxic impacts of LMW and HMW AβO species has been recently reviewed by Ferreira and colleagues [115].
Fig.5
Only high-molecular weight AβOs are capable of binding cultured hippocampal neurons. Synthetic AβOs were divided into high and low molecular weight populations using 50 kDa molecular weight cutoff ultrafiltration (A-B) or size exclusion chromatography (D-F) and incubated with cultured hippocampal neurons. Only high-molecular weight AβOs bind neurons (A, E); no binding of low-molecular weight AβOs was evident (B, F). Scale bar = 40μm. Reprinted from “Synaptic targeting by Alzheimer’s-related amyloid beta oligomers” by Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, and Klein WL. This was published in J Neurosci, 2004, 24(45): 10191-10200, copyright 2004; permission conveyed through Copyright Clearance Center, Inc. [16].
![Only high-molecular weight AβOs are capable of binding cultured hippocampal neurons. Synthetic AβOs were divided into high and low molecular weight populations using 50 kDa molecular weight cutoff ultrafiltration (A-B) or size exclusion chromatography (D-F) and incubated with cultured hippocampal neurons. Only high-molecular weight AβOs bind neurons (A, E); no binding of low-molecular weight AβOs was evident (B, F). Scale bar = 40μm. Reprinted from “Synaptic targeting by Alzheimer’s-related amyloid beta oligomers” by Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong Y, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, and Klein WL. This was published in J Neurosci, 2004, 24(45): 10191-10200, copyright 2004; permission conveyed through Copyright Clearance Center, Inc. [16].](https://content.iospress.com:443/media/jad/2018/64-s1/jad-64-s1-jad179941/jad-64-jad179941-g005.jpg)
One specific type 1 AβO has been identified, the 56 kDa SDS-stable species sometimes referred to as Aβ*56 [44]. Aβ*56 was first identified as a prominent AβO in AD brain [6] and Tg2576 mice [44] and has been observed more recently in CSF [14]. A recent study by Lesné and colleagues compared the in vitro toxicity of Aβ*56 to two LMW species, dimers and trimers [127]. They found that Aβ*56 interacted with N-methyl-D-aspartate receptors (NMDARs), increased NMDAR-dependent Ca++ influx, and increased the activation of Ca++/calmodulin-dependent kinase IIα (CAMKIIα). The latter was associated with increased site-specific phosphorylation and missorting of tau. Dimers and trimers did not induce any of these effects. On the other hand, trimers were able to induce pathological conformational changes in tau, which was associated with a selective decrease in proteins governing axonal transport [128]. The lack of dimer toxicity is consistent with earlier observations from O’Malley and colleagues utilizing crosslinked dimers [129]. According to their results, they proposed that the contribution of dimers to AD is through their ability to further assemble into larger, more stable synaptotoxic assemblies. It is possible that the toxic response observed with trimers above was similarly due to their ability to assemble into large, more stable synaptotoxic assemblies [106] (i.e., HMW type 1 AβOs). This possibility cannot be discounted since the trimers were not conformationally stabilized in this study.
Many studies of AβO structure and function have been conducted with synthetic AβOs or AβOs derived from Tg mouse brain. Some researchers, however, are calling for analysis to be restricted to AD brain-derived AβOs [107, 130]. Yet, it seems as if there is structural homology between synthetic and brain-derived AβOs. For example, under non-denaturing conditions, synthetic and brain-derived AβOs show structural equivalence in terms of mass, isoelectric point, and immunoreactivity with conformation-sensitive antibodies [6]. Furthermore, as suggested above, at least three identical AβO species can be found in AD brain and the brain of multiple Tg mouse models: Aβ*56, dimers, and trimers [14, 44, 128].
However, one important justification for restricting analysis to AD-derived AβOs is the increasingly apparent presence of Aβ proteoforms in the AD brain as well as the contribution of Aβ proteoforms to Aβ aggregation and toxicity (reviewed in [107, 131]). These proteoforms are not present in synthetic or cell-derived AβOs. It is well known that Aβ40 and Aβ42 are the most abundant Aβ peptides found in AD. However, in addition to these peptides, myriad truncated Aβ peptides also have been found in the AD brain [132] and CSF [133]. Using mass spectrometry, one study identified 26 unique Aβ proteoforms in the AD brain. 73% of these were N-terminal truncations and 30% were C-terminal truncations. The N-terminally truncated peptides were predominately found in the insoluble fraction of the brain, while the C-terminally truncated were predominately found in the soluble fraction. Canonical Aβ42 was a minority of the proteoforms identified and was equally distributed between the insoluble and soluble fractions [132].
Truncated Aβ peptides likely play a role in AD pathogenesis as they can form stable oligomeric complexes with the full-length Aβ42 peptide [133]. In fact, N-terminally truncated Aβ peptides formed through pyroglutamylation of glutamic acid residues are increasingly recognized as very toxic proteoforms of Aβ. Pyroglutamylated (pE) Aβ has been found to drive misfolding of Aβ into more toxic aggregates when present at 5–33% of the total Aβ concentration [134]. Pyroglutamylation also increases the aggregation speed of Aβ [135]. Anti-pE Aβ antibodies have been developed and successfully utilized in Fab form for co-crystallization with pE Aβ [136]. These studies revealed that the pE modification confers a pronounced bulky hydrophobic nature to the N-terminus of Aβ that might explain its enhanced aggregation properties. Interestingly, one group finds that pE AβOs may be the most abundant AβO species in AD brain [137]. Other Aβ proteoforms reported in the past 5 years to increase Aβ toxicity include C-terminally extended Aβ43 [138–140], Aβ peptides with N-terminal extensions up to 40 residues [141, 142], aspartic acid isomerization [143], and phosphorylation [144, 145].
AβO assembly pathways and their relation to amyloid plaques
A preponderance of data now supports the hypothesis that some AβO species are “on-pathway” to fibril formation, while others are “off-pathway”. It is these “off-pathway” oligomeric species that may be the most toxic [146]. This on/off-pathway classification appears to correlate with the HMW/LMW and type 1/2 AβO classifications discussed above. Most data show that LMW, type 2 AβOs are on-pathway to fibril formation, while HMW, type 1 AβOs are off-pathway [118, 119, 147, 148]. High-speed AFM imaging demonstrates that LMW AβOs quickly form fibrils, whereas HMW do not [147]. These aggregation differences between LMW and HMW AβOs are consistent with earlier findings using SDS-PAGE analysis [118]. In fact, it seems as if the only contribution of HMW AβOs to fibril formation may be through their dissociation into LMW AβOs, which then seed fibril formation [147]. Interestingly, differences in the aggregation pathways of these two AβO structures occur as early as the dimer stage [149]. But contrary to the hypothesis of HMW AβOs being more toxic than LMW AβOs, one study has found that HMW AβOs are not as toxic as the LMW AβOs into which they dissociate [150]. And another study utilizing all-atom molecular dynamics simulations observed that compact AβO structures, with an oligomeric order up to 18 (81 kDa), are off-pathway to fibril formation, while larger, elongated AβO structures are on-pathway to fibril formation [149]. Therefore, although there is general agreement in the literature regarding toxicity of AβO species, there is not complete consensus.
An alternate hypothesis to the on/off-pathway model of AβO formation, is the fibril-seeded model [151]. In this model, toxic AβOs are predominantly formed from monomers that dissociate from fibrils only after a small, but critical concentration of fibrils has formed. This model is supported by kinetic experiments, selective radiolabeling experiments, and cell viability assays. Further support for secondary nucleation of AβOs comes from molecular dynamics simulations. These simulations predict that a hydrophobic fibril region causes the structural changes required for catalyzing the formation of AβOs on the fibril surface [152]. However, AβOs can form within minutes in vitro, even at low nanomolar concentrations [26]. Recent AFM studies confirm that AβOs can form within minutes [153]. This quickly forming AβO population is specifically dominated by hexamers and dodecamers and quickly followed by AβO-seeded fibril formation. Therefore, one factor that has led to contrasting conclusions regarding the timing of AβO primary versus secondary nucleation pathways may be the differing time resolutions of the different experimental techniques.
Another hypothesis, the amyloid plaque buffering hypothesis, supports this notion of co-existing primary and secondary nucleation pathways for AβOs. This hypothesis predicts that plaques act as a reservoir or sink for toxic AβOs [107]. AβOs gradually deposit as diffuse plaques, which cause inflammation, but AβOs also can directly cause damage leading to dementia via altered signaling [5, 110]. Evidence for AβOs existing in these diffuse plaques comes from immunofluorescent imaging with anti-AβO antibodies. This has been observed in the AD brain and in the brains of multiple animal models [7, 96, 154, 155]. Over time, this plaque reservoir is saturated or loses capacity and toxic AβOs become free to diffuse and exert toxicity [107, 154, 156, 157]. Overall, it seems as if evidence in the literature converges into the hypothesis that Aβ aggregates into distinct AβO species, with differing toxicities and relationships to fibrils, that can interconvert.
Emerging insights into how to approach molecular structure
A major hurdle to AβO structural characterization is AβO metastability and heterogeneity. One major approach to stabilize and isolate distinct AβO species has been crosslinking. One widely applied crosslinking method for AβO stabilization has been photo-induced crosslinking (PICUP), developed by the Teplow group. Initially, this method was successful in stabilizing only LMW oligomers of the Aβ40 peptide [158]. Recently, PICUP has been improved through use of the mutated Aβ42 peptide [F10, Y42]Aβ42, enabling stabilization of Aβ42 oligomers up to dodecamers [159]. Another crosslinking strategy used for AβO stabilization is dityrosine crosslinking. This method is thought to be AD-relevant as it occurs under conditions of elevated copper and oxidative stress [160]. Copper is relevant to AD as there is some evidence that dyshomeostasis of metals, including copper, may contribute to AD pathogenesis [161]. Furthermore, dityrosine crosslinked proteins are found to be prevalent in AD brain and CSF [160]. Molecular dynamics simulations predict that dityrosine crosslinking promotes Aβ self-assembly, at least up to tetramers [162]. In one study, copper was found to stabilize Aβ in an oligomeric conformation sufficiently to enable 3D structural characterization by small-angle x-ray scattering [163]. The putative mechanism of this copper-mediated stabilization was through copper-induced dityrosine linkage of Aβ peptides [164]. Different copper ratios had different effects on Aβ aggregation, with supra-equimolar ratios favoring formation of ellipsoid oligomers and sub-equimolar ratios favoring formation of fibrils [163]. These ellipsoid AβOs were predicted to contain 38 copies of the Aβ peptide and are therefore consistent with the converging classifications of off-pathway, HMW, type 1 AβOs. Given published findings, it is essential that AD-relevant stabilization techniques continue to evolve to enable direct structure-function comparisons of distinct AβO species under AD-relevant experimental conditions. This will make it possible to properly interpret Aβ-directed clinical findings and make the most informed efforts at rational design of AβO-targeting therapeutics.
What makes AβOs toxic to neurons?
AβOs can be extracellular in vivo, existing in CSF [32, 34, 35, 165] and in interstitial fluid [166]. Some brain cells when exposed experimentally to extracellular AβOs become dysfunctional and deteriorate, as reviewed above. How AβOs instigate pathological changes, and why only some cells are affected, are fundamental questions to which we do not yet have satisfying answers.
The simplest possibility is that cell damage is initiated by physiochemical interactions between AβOs and neuronal membranes. It has been reported that AβOs can insert directly into lipid bilayers, causing disruption by acting as a pore, a phenomenon first observed with artificial membranes [167]. The extensive amount of literature concerning Aβ-lipid membrane interactions and molecular level membrane modeling has recently been reviewed [168], including the possible involvement of metals in the mechanism [169]. AFM was used recently to show structural damage to POPC/POPS lipid bilayers caused by Aβ40 in different aggregation states [170]. Aggregation and lipid interaction properties of Aβ peptide fragments incubated in the absence or presence of total brain lipid extract bilayers indicate that some sequences interact with and disrupt bilayers (e.g., Aβ40) but others do not (e.g., Aβ28 and Aβ12–24) [171]. Some experiments indicate that oligomers of Aβ have more membrane affinity than monomers [172]. Putative oligomers from Aβ that is pyroglutamate-modified also binds neurons and causes a loss of plasma membrane integrity [173]. Ion channel formation in cell membranes [174] has been reported for oligomers of Aβ42, but not Aβ40, and attributed to a pore-forming beta-barrel AβO structure [175]. Individual AβOs larger than trimers reportedly induce Ca++ entry as they cross the cell membrane [176]. Cholesterol enhances formation of an annular octameric channel of Aβ22–35, which induces a zinc-sensitive Ca++ influx [177], suggested as a possible lipid raft association. Recruitment of AβOs to rafts is consistent with findings that depletion of the ganglioside GM1 blocks AβO interactions toxicity [178]. On the other hand, data suggest that a moderate increase in membrane cholesterol content may be protective against AβO toxicity [179]. Protection also is conferred by a pentapeptide from the glycine zipper region of the C-terminal of Aβ, which blocks apparent membrane insertion and abolishes synaptotoxicity [180].
One significant difficulty encountered by the bilayer insertion hypothesis is its inability to account for the specificity of AβO attachment. Two neurons side-by-side can exhibit completely different ability to accumulate AβOs, one showing robust synaptic accumulation and the other showing virtually none [16]. Cell-specific toxicity, measured by tau hyperphosphorylation, correlates with this binding [51]. There also is a difficulty in accounting for binding saturability [16, 96], although it might be argued that AβO insertion into lipid rafts specific to particular synapses could be saturable. It has been hypothesized that AβOs may act through both lipids and proteins, forming pores within membranes while also binding to receptors to induce specific intracellular responses [181].
The receptor hypothesis regards AβOs as gain-of-function pathogenic ligands that bind adventitiously to specific proteins acting as toxin receptors. Overall, the receptor hypothesis provides a mechanism that fits well with salient facets of the cell-based evidence. The hypothesis was introduced to explain toxicity that was cell-specific and dependent on expression of Fyn, and to explain why AβO binding was virtually eliminated by treating cell surfaces with low doses of trypsin [26]. Consistent with the receptor hypothesis, AβO binding shows (A) saturation and high-affinity for cultured neurons and synaptosome preparations; (B) specificity for particular neurons and particular brain regions; (C) targeting of synapses; (D) accumulation at dendritic spines; (E) pathological impact, such as tau hyperphosphorylation, specific to neurons with bound AβOs; (F) sensitivity to low doses of antagonist; (G) binding to trypsin-sensitive proteins; (H) association with small patches of isolatable membranes; and (I) specificity in Far-Western immunoblots [6, 16, 48, 167, 182]. These findings apply generally to brain-derived and synthetic AβOs and support the hypothesis that binding of AβOs is ligand-like and mediated adventitiously by proteins acting as toxin receptors.
Perhaps the most intriguing and well-studied AβO toxin receptor candidate is the cellular prion protein (PrPc). Strittmatter and colleagues in a series of papers have provided strong evidence that PrPc is capable of mediating AβO binding [122, 183–185], starting with their unbiased screening of a cDNA expression library that identified PrPc as a potential high-affinity AβO receptor [186]. Extensive experiments with multiple models support this possibility and connect binding to neural damage [187–190]. It has been reported that binding of AβOs to PrPc is dependent on integrity of cholesterol-rich lipid rafts and that AβOs bound to PrPc accumulate in endosomes after which they are trafficked to lysosomes [191]. Investigations of how externally-oriented PrPc might bring about intracellular damage through transmembrane coupling to Fyn are discussed further below. Coupling of AβO-bound PrPc to Fyn is consistent with the original studies showing that Fyn expression is required for AβO-induced toxicity [26, 192] and evidence showing involvement of Fyn in physiological PrPc signaling [193]. It should be noted that the PrPc hypothesis is still somewhat controversial, and some reports are not in agreement with the role of PrPc as an AβO toxin receptor [29, 194–197].
Another promising candidate receptor is the Na+K+ ATPase alpha3 subunit (NKAα3), recently identified independently by two groups using disparate preparations and identification strategies. It was shown first by Ohnishi and colleagues that NKAα3 can bind both brain-derived and synthetic AβOs [198], each resembling type 1 AβOs with respect to their relatively large size. Verification of NKAα3 as an AβO receptor subsequently was provided by DiChiara et al. [199]. This group used solubilized synaptic membrane proteins reconstituted in nanoscale artificial membranes and an AβO-specific antibody to isolate AβO-bound NKAα3. Co-localization of AβO binding sites with NKAα3 was confirmed in hippocampal cell cultures. As discussed later, down-regulation of NKAα3 could play a significant role in converting AβO binding into cell pathology.
Overall, and rather remarkably, the current list of candidate toxin receptors for AβOs comprises a very large number of membrane proteins besides PrPc and NKAα3. As has been reviewed [115, 200–202], these include the metabotropic glutamate receptor 5 (mGluR5) [182, 184], NMDARs [58, 62], Sigma-2 receptor/progesterone receptor membrane component 1 (PGRMC1) [203, 204], frizzled receptor [205], neuroligin [206], Ephrin type-B receptor 2 (EphB2) [207], Ephrin type-A receptor A (EphA4) [208, 209], p75 neurotrophin receptor (p75NTR) [210], alpha7-nicotinic acetylcholine receptor (α7nAChR) [211, 212], adrenergic receptors [213], the receptor for advanced glycation endproducts (RAGE) [214], calcium channels [215–217], leukocyte immunoglobulin-like receptor subfamily B member 2 (LILRB2)/paired Ig-like receptor B (PirB) [64, 218, 219], N-formyl peptide receptor 2 (FPR2) [220], immunoglobulin gamma Fc region receptor II-b (FcγRIIb) [221], transient receptor potential melastatin 2 (TRPM2) [222], insulin receptor (IR) [48], and the AMPA receptor [223].
It is not known why there are so many candidate receptors. There certainly are different forms of AβOs, which could interact with different membrane proteins. AβO ligands in aqueous buffer are between 100 and 300 kDa (Cline and Klein, unpublished); in this mass range AβOs would comprise 22–66 Aβ monomers. To interact with a binding domain of a toxin receptor, only particular regions of the oligomer surface would be needed. Ligand-like regions could assume multiple configurations influenced by buffer composition. For example, monomeric Aβ in Ham’s F12 media assembles into structures quite different than Aβ in phosphate buffered saline (the former has a high type 1 to type 2 ratio [118], while the latter has a low type 1 to type 2 ratio) (unpublished data). Even within a population of synthetic type 1 AβOs, there is a small subpopulation of synapse-binding AβOs that can be targeted uniquely by a selective single-chain variable fragment antibody [118]. Different targeted binding proteins might nonetheless mediate similar changes downstream. It is known, for instance, that AD-type phosphorylated tau can be induced by oligomers of different proteins such as Aβ [51], α-synuclein [224], and even lysozyme [225].
Receptor transduction mechanisms → how does the initial receptor-AβO interaction on neurons trigger a change that leads to intracellular damage?
The mechanism by which PrPc mediates AβO impact intracellularly has been carefully worked through (Fig.6). It incorporates a number of molecular players previously implicated by multiple laboratories: mGluR5 [182, 184], Fyn [26], tau [51], NDMARs [58, 62] and protein tyrosine kinase 2 (Pyk2) [226]. Both high and low molecular weight AβOs have been implicated in this pathway [121–123, 191]. Data are consistent with a mechanism in which AβOs first bind to PrPc on cell surfaces and stimulate Fyn via mGluR5 activation (reviewed by Nygaard, et al. [227]). Consistent with activation of mGluR5 by AβOs, the ability of glutamate to activate the prion-mGluR5 complex is occluded [228]. Downstream, stimulated Fyn is known to phosphorylate tau [229] and cause tyrosine phosphorylation of the NR2B subunit of NMDARs [183]. It is thought that AβO binding to neurons and AβO neurotoxicity depends on a pre-existing PrPc-mGluR5 complex [230]. However, since PrPc can be removed with full retention of AβO binding [194], it may be that the critical membrane-organizing function of PrPc precedes the ligand binding step.
Fig.6
PrPc mediates AβO toxicity through mGluR5, Fyn kinase, and NMDARs. Downstream consequences of the pathway include calcium dyshomeostasis, tau hyperphosphorylation, and synaptic dysfunction and loss. Reprinted from “Fyn kinase inhibition as a novel therapy for Alzheimer’s disease” by Nygaard HB, van Dyck CH, and Strittmater SM. This was published in Alzheimers Res Ther, 2014, 6(1): 8, under the terms of the Creative Commons Attribution License (CC BY) [227].
![PrPc mediates AβO toxicity through mGluR5, Fyn kinase, and NMDARs. Downstream consequences of the pathway include calcium dyshomeostasis, tau hyperphosphorylation, and synaptic dysfunction and loss. Reprinted from “Fyn kinase inhibition as a novel therapy for Alzheimer’s disease” by Nygaard HB, van Dyck CH, and Strittmater SM. This was published in Alzheimers Res Ther, 2014, 6(1): 8, under the terms of the Creative Commons Attribution License (CC BY) [227].](https://content.iospress.com:443/media/jad/2018/64-s1/jad-64-s1-jad179941/jad-64-jad179941-g006.jpg)
An interesting potential connection exists between this synaptopathic mechanism and Pyk2. Pyk2 has a single nucleotide polymorphism identified as increasing the likelihood of late-onset AD [231]. Functionally, Pyk2 is a focal adhesion kinase (FAK), an enzyme that previously was shown to be stimulated by toxic Aβ preparations and to form complexes with Fyn [226]. Pyk2 normally helps regulate synaptic plasticity [232–235]. It is activated by increased intracellular calcium and by Fyn [236–241]. Ectopic activation of Pyk2 potentially could be an early event in this AβO pathogenic pathway, which would provide a molecular basis for its risk factor status.
Insights also have been obtained into how AβO binding mediated by NKAα3 could be transduced into neuronal damage. As described by Ohnishi et al. [198], AβO binding leads to a slow, time-dependent decrease in ATPase activity. The consequence is Ca++ buildup via N-type voltage-sensitive calcium channels (N-VSCC) and mitochondrial channels and ultimately apoptosis. Decrease in activity was suggested as linked to AβO binding to the ouabain binding site of the NKAα3 [198]. This observation suggests the new hypothesis that, under some circumstances, AβOs could be endogenous ouabain-like physiological regulators of ATPase. The slow, time-dependent decrease in activity, however, could be linked to the observed impact of AβOs on NKAα3 distribution. Following exposure to AβOs, neuronal NKAα3 accumulates in dense clusters along dendrites. These clusters of NKAα3 increase in size and then decrease in abundance (Fig.7) [199]. This presumably occurs at dendritic spines, where AβOs also cluster [16, 55]. Like the NKAα3 redistribution, spines undergo time-dependent changes in morphology and abundance due to AβO exposure. Ultimately, there is a large down-regulation of NKAα3, which could account for decreased ATPase activity (Fig.7).
Fig.7
AβOs induce membrane re-distribution of NKAα3 subunit resulting its downregulation and excessive Ca++ buildup. A hypothesized early event in AβO-induced neuronal damage is binding to NKAα3 on neuronal membranes, causing restructuring of the NKAα3 docking station into toxic clusters of membrane proteins. Ultimately, this results in downregulation of NKAα3 on the neuronal surface and buildup of toxic Ca++. Adapted and reprinted from “Alzheimer’s Toxic Amyloid Beta Oligomers: Unwelcome Visitors to the Na/K ATPase alpha3 Docking Station” by DiChiara T, DiNunno N, Clark J, Bu RL, Cline EN, Rollins MG, Gong Y, Brody DL, Sligar SG, Velasco PT, Viola KL, and Klein WL. This was published in Yale J Biol Med, 2017, 90(1): 45-61, under the terms of the Creative Commons Attribution Non-Commercial No Derivatives License (CC BY NC ND) https://creativecommons.org/licenses/by-nc-nd/4.0/ [199].
![AβOs induce membrane re-distribution of NKAα3 subunit resulting its downregulation and excessive Ca++ buildup. A hypothesized early event in AβO-induced neuronal damage is binding to NKAα3 on neuronal membranes, causing restructuring of the NKAα3 docking station into toxic clusters of membrane proteins. Ultimately, this results in downregulation of NKAα3 on the neuronal surface and buildup of toxic Ca++. Adapted and reprinted from “Alzheimer’s Toxic Amyloid Beta Oligomers: Unwelcome Visitors to the Na/K ATPase alpha3 Docking Station” by DiChiara T, DiNunno N, Clark J, Bu RL, Cline EN, Rollins MG, Gong Y, Brody DL, Sligar SG, Velasco PT, Viola KL, and Klein WL. This was published in Yale J Biol Med, 2017, 90(1): 45-61, under the terms of the Creative Commons Attribution Non-Commercial No Derivatives License (CC BY NC ND) https://creativecommons.org/licenses/by-nc-nd/4.0/ [199].](https://content.iospress.com:443/media/jad/2018/64-s1/jad-64-s1-jad179941/jad-64-jad179941-g007.jpg)
The issue of distribution is a salient one given that NKAα3 acts not only in cation transport, but also as a membrane protein docking station that functions to control signaling pathways [242]. These docking stations organize multiple membrane proteins [8], including neurotransmitter receptors linked to AβO-induced neuron damage [243]. The clustering of NKAα3 is in harmony with the earlier observation that AβOs induce the clustering of mGluR5 [182, 184]. As discussed above, mGluR5 is a Ca++ mobilizing receptor, and it is regarded as a key mediator of AβO-elevated Ca++ buildup and the damage that ensues [184]. The time-dependence of AβO-induced clustering of mGluR5 has been imaged using quantum dots and single-particle tracking in experiments with live neurons [182]. It has been hypothesized that mGluR5 clustering itself is a seminal step for the transduction mechanism [182]. Supporting this possibility, clustering of mGluR5 molecules induced by receptor antibodies mimics the toxic impact of AβOs [182]. Because mGluR5 and NKAα3 each co-localize with cell-surface bound AβOs, they likely are part of the same ectopic clusters. Recently, single-particle tracking experiments have shown that NKAα3 becomes immobilized during exposure of hippocampal neurons to toxic assemblies of synuclein [244]. These results support the hypothesis that NKAα3 has a central role as an immobilizing docking station for toxic oligomers found in multiple proteinopathies. With respect to generation of these clusters, the role of the NKAα3 docking station relative to roles played by mGluR5, or other membrane domain-organizing proteins such as PrPc [245], is not yet clear.
The seminal interactions between AβOs and NKAα3 molecules at the cell surface may be suitable targets for new drug discovery strategies, as suggested by Ohnishi et al. [198]. Attachment of AβOs to NKAα3 is amenable to high-throughput screening for antagonists using Nanodiscs [194, 246]. Results from a preliminary screen showed that AβO binding to spines can be blocked by low doses of a small organic molecule, albeit one with promiscuous binding, precluding its use for therapeutics [194]. Nonetheless, Lee and colleagues have shown that behavior in a Tg AD model could be safely rescued using this same compound [247]. Future investigations of the docking station hypothesis are expected to open the door to therapeutics targeting the first step of a complex pathway that leads to neural damage and dementia.
Whether the NKAα3 acts, as has been suggested [198], as a “death target” for AβOs is not confirmed yet. Most AD-like pathology is evident in cultures containing almost exclusively neurons, but cell death is minimal; neuron death likely requires the presence of factors released by glia [248]. It is possible that the impact of AβOs on NKAα3 may render them more vulnerable to inflammatory cytokines.
TRENDING TOPICS IN AβO RESEARCH
With about 2,000 AβO papers published since 2013, there has been a great deal of progress on many issues. Some of the salient directions are considered briefly in this section.
Toxic effector pathways after initial transduction
Downstream, after the initial transduction steps, the impact of AβOs has been tracked to mitochondrial effects, ER stress, and autophagy/lysosomal dysfunction. These may be the consequences of surface events discussed above, but some studies show that AβOs may themselves enter cells and act directly on these organelles, as discussed below.
AβO-associated NMDAR activation [62] promotes Ca++ release from the ER, which leads to ER stress [249] with subsequent mitochondrial dysfunction [250], astrogliosis [115, 251], and apoptosis [18]. AβOs also have been found to trigger the unfolded protein response, a collection of signaling pathways that respond to ER stress [252]. AβOs further decrease resistance of brain mitochondria to Ca++-induced opening of mitochondrial permeability transition pores [253]. Cytochrome C is released by AβO-activated BAK pores [254]. Voltage-dependent anion channel 1 also interacts with Aβ monomers and oligomers, and the block of mitochondrial pores leads to mitochondrial dysfunction [255]. Morphological effects on mitochondrial fusion and fission dynamics, essential for neuronal function, have been reviewed [256]. AβO targeting of mitochondria promotes mitochondrial fission, disruption of mitochondrial membrane potential, increase of intracellular ROS and activation of mitophagy [257]. AβOs decrease mitochondrial volume [258], and AβO-induced oxidative stress is associated with mitochondrial fission [259]. AβOs activate fragmentation through the GTPase dynamin-related protein 1 (Drp-1) [260] and extracellular signal-regulated kinase (ERK) [259]. Fragmentation also has been associated with AβO-induced mitochondrial transport defects, with histone deacetylase (HDAC6) activation part of the mechanism [260]. AβO-induced mitochondrial damage appears to be restricted to neurons and not microglia or astrocytes [261].
With respect to autophagy, the sole catabolic mechanism for degrading protein aggregates, there is increasing evidence for autophagic dysfunction in AD and other neurodegenerative diseases [262, 263]. The endosomal-lysosomal (autophagy) system is a prominent site of AβPP processing, Aβ uptake, and Aβ production [262]. One study has found that AβOs associate with autophagic vacuoles in AD axons, starting a pathway that impairs retrograde transport, which contributes to autophagic stress [264]. On the other hand, another study found that it is Aβ monomers, and not AβOs, that contribute to autophagy [265]. AD and lysosomal storage disorders share many overlapping pathologies, including neuronal accumulation of lysosomal vesicles, dystrophic axons, ectopic dendrites, cognitive deficits, and neurodegeneration [262]. Lysosomal storage disorder gene variants also have been found to be associated significantly with Parkinson’s disease [266]. Restoration of autophagy function may represent a promising therapeutic target as rifampamycin, a candidate preventative therapeutic thought to restore autophagy function, has been found to inhibit oligomerization of Aβ and tau, tau phosphorylation, synapse loss, and microglial activation in AD mouse models [267].
Intracellular effects of AβOs may be instigated by surface mechanisms but also could be a result of direct interactions between organelles and internalized AβOs. In NHPs, i.c.v.-injected AβOs were observed on the surface and inside neurons [46]. Internalization may involve signaling pathways that affect regulation of receptor-mediated endocytosis. In the human neuroblastoma SH-SY5Y line, AβOs activate p38 mitogen-activated protein kinase (p38 MAPK) and ERK1/2 signaling pathways via the α7nAChR, which in turn results in AβO internalization [268]. Internalized Aβ (monomers and AβOs) localized to all organelles (ER, Golgi complexes, multivesicular bodies/late endosomes, lysosomes, exocytotic vesicles, and mitochondria) and non-membrane-bound cytosolic structures [269, 270]. The uptake of Aβ via endocytosis is rapid and spontaneous. It is retained in lysosomes, where accumulation leads to aggregation [271].
The relationship between neurons, astrocytes, microglia, and AβOs
De Strooper and Karran propose that AD pathogenesis is not simply a neuron-centric, linear cascade initiated by Aβ and leading to dementia, but rather a long, complex cellular phase consisting of feedback and feedforward responses of astrocytes, microglia, and vasculature [202].
Many lines of evidence support this hypothesis. For instance, AβOs have been found to induce astrogliosis [272] and trigger ROS generation in activated astrocytes [273]. AβOs reportedly cause disturbances in the signaling of insulin, protein kinase B (Akt), and excitatory amino acid transporters 1 and 2 [274]. Decreases in the activation and expression of astrocytic glutamate transporters has been linked to impaired synaptic plasticity [275]. AβOs at picomolar levels, within minutes, can increase levels of intracellular Ca++ in astrocytes but not neurons [276]. An increase in ROS production by nicotidamine adenine dinucleotide phosphate (NADPH) oxidase in both neurons and astrocytes has been found to activate caspase-3, also linked to LTP inhibition. In these experiments, only a small fraction of AβOs were impactful and their damage was blocked by clusterin [276]. AβOs and fibrils bind and activate Ca++-sensing receptors, which drives both neurons and astrocytes to secrete Aβ42. While the Aβ-exposed neurons start dying, astrocytes survive, and they keep over-secreting Aβ42, nitric oxide (NO), and vascular endothelial growth factor A (VEGF-A), apparently contributing to the demise of neurons [277]. On the other hand, astrocytes, before they are affected by AβOs, appear to release insulin and insulin-like growth factor (IGF1), whose trophic effects serve to protect neurons from AβO toxicity [73].
Microglia in AD are involved in phagocytosis of Aβ plaques [278–281], a process that is regulated by astrocytes [278]. It is possible that AβOs play a role in attracting microglia to plaques (Bicca and Klein, unpublished data). Besides chemotactic effects, AβOs induce a switch in microglial phenotype to a pro-inflammatory phenotype, leading, e.g., to aberrant tumor necrosis factor (TNF) signaling [282]. Aberrant TNF signaling causes decreased brain serotonin levels and subsequent depression [283]. It also causes insulin receptor substrate (IRS-1) and PKR (dsRNA-dependent protein kinase)-dependent synaptic dysfunction and memory loss [221]. There thus is a link between AβOs, neuroinflammation, mood alterations, metabolic disorders, and memory loss. Microglia also may contribute to AβO formation, by releasing particles that can bind rapidly to Aβ and cross-seed its aggregation, including oligomerization [284]. It should be noted that there also is evidence that microglia may contribute to neuronal loss and memory impairment in a manner independent of Aβ [281]. One potential mechanism is through microglia engulfment of synapses [285].
Tau progression and prion-like action may be instigated and potentiated by AβOs: PART (primary age-related tauopathy)
Most evidence in the literature converges on the hypothesis that AβOs are upstream of tau in AD pathogenesis and not the other way around, as reviewed by Bloom [286]. However, there is currently no consensus in the field, with some studies demonstrating crosstalk between AβOs and tau and some demonstrating that each acts separately [110, 286–290]. In support of the hypothesis that AβOs trigger tau pathology, it was demonstrated in 2008 that AβOs were capable of inducing tau hyperphosphorylation in cultured neurons in the absence of fibrils [51]. Tau distribution in AβO-exposed neurons ectopically redistributes to dendrites [52]. AβOs also have been shown to induce tau-dependent microtubule severing [291], to disrupt tau translocation to excitatory synapses [292], and to stabilize microtubules, the latter leading to tau-dependent loss of spines and tau hyperphosphorylation [293]. AβOs even can seed the formation of tau oligomers, which are thought to be the most toxic form of tau [294]. In the AD brain, synaptic AβOs have been found to precede synaptic phosphorylated tau (pTau), even perhaps driving the synaptic spread of pTau [295]. It is known that spread of tau pathology in a Tg mouse tauopathy model is accelerated by crossing with an APP Tg mouse [296]. Recent data suggests that AβOs may induce neurons to release pTau within exosomes, thereby suggesting a potential mechanism for AβO-induced spread of tau pathology [73]. Interestingly, this release of tau was increased by the presence of insulin. The idea that tau is secreted by neurons is supported by numerous other studies, as reviewed by Pooler et al. [297]. Furthermore, i.c.v. injection of AβOs into NHPs induces tau hyperphosphorylation and formation of neurofibrillary tangles throughout the NHP brain [46].
Many recent studies have attempted to give Aβ and tau an even playing field on which to determine their pathological relationships by crossbreeding mice expressing human tau (wild-type or mutant) and APP/PS1 mutants. However, these studies show inconsistencies in data leading to contrasting conclusions. Co-expression of mutant tau and mutant Aβ appears to support a synergistic action, showing dramatically increased pTau aggregation and spread, inflammation, and synapse loss [296, 298, 299]. Multiple studies utilizing a wild-type human tau instead of mutant tau also support a synergistic model, for example [300, 301]. Although it may be that some aspects of AD pathology are cooperatively affected by Aβ and tau, while others are independently affected [155, 302]. Contrary to these studies, one report found no evidence for pathological interaction between Aβ and tau [303]. These apparently disparate findings may be the result of utilizing different transgenes and/or pathological readouts.
AβOs themselves as prions?
The idea that oligomers of amyloid proteins, including AβOs, may spread from neuron-to-neuron in a prion-like manner has been widely considered. Although there is currently no clear clinical evidence that AD is a transmissible prion-like disease [5], experimental data support the idea that AβOs may spread from cell-to-cell and brain region-to-region in a prion-like manner. A recent review of this hypothesis states that Aβ aggregates have all of the key characteristics of canonical mammalian prions, including a β-sheet rich architecture, the tendency to polymerize into amyloid, templated corruption of like protein molecules, the ability to form structurally and functionally variant strains, the systematic spread by neuronal transport, and resistance to inactivation by heat and formaldehyde [304]. Another review of this concept predicts that small, extracellular oligomers of amyloid proteins would have a high propensity for prion-like spread, while large intracellular oligomers would have a lower propensity for prion-like spread [305]. In support of AβOs acting as prions, one study has found that AβOs can transfer from cell to cell [306]. This transfer was shown to be dependent on insufficient cellular clearance of Aβ peptides and oligomers. The remaining un-degraded Aβ was able to cause seeding and pathology in the receiving cells. Cell transfer was an early event seemingly independent of later toxicity. Aβ can seed its own aggregation in vitro [307, 308] and brain extracts from AD patients and animal models can seed Aβ aggregation in vivo [309, 310]. Furthermore, i.c.v. injections of AβOs to NHPs induced accumulation of AβOs in specific brain regions far from the injection site, suggesting spreading [46]. Hypotheses for the mechanisms of Aβ spread include exosome transfer [311] and spread directed by the limbic connectome [312, 313].
Mechanisms of buildup
Three intriguing new hypotheses for the mechanism of AβO accumulation that have emerged in the literature in the past 5 years are saturated proteostasis, shear-induced amyloid formation, and slowed clearance of AβOs from interstitial fluid. These hypotheses are briefly reviewed below.
Saturated proteostasis
One theory to explain the accumulation of Aβ aggregates is saturated proteostasis [314]. This theory, based on a large body of evidence, states that there may be nothing particularly unique about the Aβ42 peptide that causes it to aggregate into toxic oligomers and amyloid fibrils. In fact, this may be an ability inherent in all proteins if they are placed in the right conditions. There is increasing evidence that many proteins are kinetically, but not thermodynamically, stable in their native states and that they become metastable when their cellular concentrations exceed their critical values. Considering the specific example of Aβ42, one study found that changing the propensity of this peptide to aggregate by only 15% through site-directed mutagenesis resulted in large changes in toxicity [315]. The authors interpret this finding to mean that Aβ42, and other amyloid proteins, may be extremely close to their solubility limit under physiological conditions. Thus, they hypothesize that in AD, or other neurodegenerative diseases associated with misfolded proteins and aging, age-related stress makes the entire proteome susceptible to aggregation, which in turn saturates the protein quality-control system of the cell [314]. Indeed, the majority of proteins implicated in AD were found to be present at supersaturated concentrations in the cell [316]. Therefore, proponents of this saturated proteostasis theory suggest that more effective therapeutics may target the driving forces for whole-proteome aggregation and protein quality-control mechanisms instead of individual disease-related proteins like Aβ [314]. More specifically, the systems that were found to be of importance in maintaining proteostasis in AD were involved in trafficking and clearance mechanisms, including specific branches of the endosomal-lysosomal and ubiquitin-proteasome systems [317].
Shear-induced amyloid formation
Another new hypothesis to explain the etiology of AβO buildup is the shear-induced amyloid formation hypothesis [318]. This hypothesis predicts that Aβ within the slow-flowing interstitial fluid can gain significant shear energy at, or near, the wall of the narrow extracellular spaces of the brain parenchyma. This could cause Aβ to absorb to the brain membrane and form oligomers on the membrane and/or form plaques within the flow pathways of the brain extracellular spaces.
Slowed clearance of AβOs from interstitial fluid
Microdialysis experiments have shown the presence of AβOs in interstitial fluid [166]. These findings are an extension of earlier studies showing a circadian rhythm in interstitial Aβ levels [319]. Clearance through the glymphatic system is inversely correlated with AβO size [166]. Impaired glymphatic functioning is considered to be a likely factor in AβO accumulation [320, 321] (see discussion below).
Etiological factors that trigger AβO buildup in sporadic AD
There is evidence that traumatic brain injury (TBI), atmospheric pollutants, poor quality of diet and sleep, and metabolic diseases (e.g., type 2 diabetes and hypercholesterolemia), may trigger AβO buildup, eventually leading to non-inherited forms of AD (sporadic) (see, e.g., [322]). A hypothesis from De Felice for the contributions of these etiological factors to AβO buildup and AD is illustrated (Fig.8) [322]. Evidence implicating each of these factors in AβO buildup is briefly reviewed below.
Fig.8
A cumulative hypothesis for the development of sporadic AD. From De Felice, sporadic AD is hypothesized to be the result of the cumulative impact over a life-time of injuries to the brain and peripheral organs that results in increased AβO levels. Reprinted from “Alzheimer’s disease and insulin resistance: translating basic science into clinical applications” by De Felice FG. This was published in JClin Invest, 2013, 123(2): 531-539, under Free access [322].
![A cumulative hypothesis for the development of sporadic AD. From De Felice, sporadic AD is hypothesized to be the result of the cumulative impact over a life-time of injuries to the brain and peripheral organs that results in increased AβO levels. Reprinted from “Alzheimer’s disease and insulin resistance: translating basic science into clinical applications” by De Felice FG. This was published in JClin Invest, 2013, 123(2): 531-539, under Free access [322].](https://content.iospress.com:443/media/jad/2018/64-s1/jad-64-s1-jad179941/jad-64-jad179941-g008.jpg)
TBI
TBI is a risk factor for AD [323] with AD developing in 55.5% of TBI patients [324]. Aβ pathology has been found to accumulate in the brain and CSF following TBI, including amyloid plaques [325] and AβOs [199, 323, 326–328]. Soluble Aβ levels, including AβOs, increase with TBI severity [327] and declining patient prognosis [328]. Results are consistent with indications that AβPP expression is injury-related, e.g., in shaken-baby syndrome [329–331]. These observations are supported in TBI animal models, wherein Aβ levels rise within 1 hour after a single mild cortical impact and continue to rise for at least 24 hours [332, 333] and are associated with increased memory impairment [334]. AβO-associated proteins, PrPc and pTau, also are increased in TBI mouse models [335].
Atmospheric pollutants
Recent studies in mice have demonstrated that air pollutants, specifically vehicular-derived airborne nano-sized particulate matter, induce AD-like neuronal damage, including reduced synaptic function [336], altered neuronal differentiation and depression-like responses [337], and reduced neurite outgrowth [338]. Two AD risk factors, age [339] and gender (female) [337], appear to increase susceptibility to these detrimental effects.
Poor quality of diet and sleep
Diets high in sugar, salt, and fat and low in fruits and vegetables are associated with a higher risk of AD [340]. In animal models, diets high in fat increase soluble Aβ without changing plaque burden [341] and diet-induced insulin resistance impairs cognition [342]. In humans, such diets have been shown to perturb the circadian modulation of cortisol secretion, which is associated with poor sleep quality. Poor sleep quality also is associated with dementia and can negatively affect glymphatic system activity, which leads to Aβ accumulation via impaired clearance (see discussion below) [340]. Furthermore, sleep restriction in mice promotes neuroinflammation and synapse loss and potentiates AβO-induced memory deficits [343].
Diabetes
Sporadic AD has been called type 3 diabetes for its molecular and biochemical similarities with type 1 and 2 diabetes [322, 344]. An increasing body of evidence shows that AD is coupled to impaired brain insulin signaling, glucose utilization, and energy metabolism, all of which lead to increased oxidative stress, neuroinflammation, and further increased insulin resistance. Specifically considering AβO buildup, it has been found that glucose concentrations observed in diabetics facilitate Aβ oligomerization [345]. Furthermore, induction of diabetes in rabbits leads to AβO accumulation in the brain and retina [346]. Most recently, type 2 diabetes has been found to be positively associated with Aβ42 in CSF [347]. The mechanism for AβO buildup in diabetes is not known, but it has been hypothesized to be mediated by inflammation [322].
Hypercholesterolemia
Hypercholesterolemia also is an AD risk factor [348, 349]. Many studies have shown that elevated cholesterol levels may contribute to AD pathogenesis, and several cholesterol-related gene polymorphisms are associated with AD, the most well-known of which is APOE [349]. Hypercholesterolemia accelerates AβO accumulation and memory impairment in AD mouse models [350, 351].
It is important to note that even though these factors have been shown experimentally to trigger AβO buildup in sporadic AD, lifestyle and therapeutic interventions aimed at modifying these risk factors in humans have yet to show definitive success. This further highlights the difficulties and challenges associated with developing successful interventions for AD, even when the therapeutic target (AβOs) plays an established role in the disease process.
Endogenous protection and its failure
Astrocyte-mediated clearance of Aβ
A growing body of evidence indicates a role for astrocytes in clearing excess levels of Aβ from the brain [352]. Interestingly, it seems as if astrocytes have differing abilities to clear different aggregation states of Aβ, presumably due to size differences. Not surprisingly, astrocytes seem to have a harder time clearing fibrils compared to AβOs [353]. Astrocyte-mediated clearance of Aβ seems to occur by multiple mechanisms, as recently reviewed, including receptor-mediated uptake, secretion of degrading enzymes, and secretion of ApoE, which acts as a chaperone [352]. A few astrocyte receptors implicated in Aβ clearance are of note: RAGE, which is currently being targeted therapeutically in phase III clinical trials (http://clinicaltrials.gov), and matrix metalloproteinases, which are implicated in AβO-induced disruption of the blood-CSF barrier integrity (see discussion below). Overall, one possibility is that astrocyte protection of the brain from Aβ fails when Aβ accumulation reaches a certain threshold at which astrocyte-mediated clearance is saturated [352]. This hypothesis is consistent with advanced astrocyte pathology in AD brain that is detected by a monoclonal antibody developed against AβOs [89].
It may be the case that astrocytes near amyloid plaques switch from a neuro-supportive role to an inflammatory role. An opposing view is that astrocyte failure in AD is neuroprotective [354]. Another hypothesis is that Aβ, specifically AβOs, can stimulate astrocytes to produce and secrete more Aβ [355, 356]; this mechanism seems to occur through Ca++-sensing receptors expressed on the astrocytes [277]. Other evidence suggests that astrocytic metabolic dysfunction may regulate Aβ production through AβPP processing [354]. And astrocytes may also mediate Aβ clearance through induction of microglial phagocytosis [278]. Thus, more experimentation is needed to fully elucidate the role of astrocytes in Aβ production and clearance within AD. It also was shown recently that healthy astrocytes secrete insulin and IGF1 that act to protect neurons from AβO toxicity [73]. Note that these mechanisms need not be mutually exclusive.
Insulin
Extensive evidence indicates that insulin signaling and AβOs are connected in a vicious cycle of pathogenesis, as recently reviewed [200, 357]. This vicious cycle may be initiated, in some cases, by diabetes, which decreases insulin signaling in the brain. Since insulin signaling protects against AβO accumulation [346] and neurotoxicity [66], this leads to increased AβO accumulation and AβO-associated damage in the brain. AβOs themselves then further disrupt insulin signaling at many levels via pro-inflammatory mechanisms [357], e.g., by downregulating the expression of IRs on the plasma membrane [66]. Thus, a vicious pathogenic cycle is created in which AβOs upregulate their own production and aggregation by disrupting the physiological actions of insulin. Such a mechanism could account in part for AβO buildup in AD brains.
Importantly, the cellular stress and synaptic dysfunction induced by AβOs can be counteracted by stimulating brain insulin signaling [66, 322]. Therefore, either insulin or therapeutics aimed at increasing/repairing insulin signaling may be promising candidates for the treatment of AD [322]. One study exemplifying this promise demonstrated that the anti-diabetes agent exenatide protects against AβO-induced pathologies in cell culture and AβO-induced impaired insulin signaling and cognitive deficits in mice [358]. Furthermore, a recent study testing the effect in Tg mice of a therapeutic targeting multiple receptors involved in insulin signaling found a multitude of benefits including reversal of memory deficits, reduction of apoptotic factors, increase of factors promoting synaptic health, increase in neurogenesis, and reduction in Aβ, neuroinflammation, and oxidative stress [359]. Hopefully the multiple drugs targeting insulin signaling that are currently in clinical trials (see discussion below) also will have such a robust therapeutic effect.
Glymphatic system and impaired AβO clearance
The recently discovered glymphatic system functions to remove metabolic waste, including soluble proteins, from the CNS [360]. The glymphatic system involves CSF inflow to the brain, which drives interstitial fluid to clear waste out of the brain. Recent studies in mice show that glymphatic activity decreases sharply during aging, resulting in decreased Aβ clearance from the brain [320]. Studies in AD mouse models show that this decreased Aβ clearance is due to oligomer formation [321], especially HMW AβOs [166], indicating that the size of larger AβOs may make it more difficult for the glymphatic system to clear them from the brain. Poor sleep quality, which is associated with dementia, might negatively affect the activity of the glymphatic system [340]. CSF levels of Aβ have been found to be increased significantly in insomnia patients [361]. Thus, the role of the glymphatic system in AD with regards to Aβ may be that its decreased activity leads to impaired AβO clearance, which may lead to further aggregation resulting in larger AβOs or insoluble amyloid plaques, both of which the glymphatic system cannot clear. Ultimately, repairing glymphatic activity may be another option for therapeutic targeting in AD treatment.
Blood-CSF barrier
The function of the blood-CSF barrier is to keep undesirable molecules and pathogens out of the brain. Several studies have shown that the integrity of the blood-CSF barrier is disrupted in AD [362], and recently, evidence has been presented that this disruption can be induced by AβOs, seemingly through increased expression and activity of matrix metalloproteinases [363]. Based on their data, the authors of this study hypothesize that AβO-induced breakdown of the blood-CSF barrier might be an early event in AD pathogenesis that would contribute to the enhancement of neuroinflammation. Therefore, early therapeutic inhibition of matrix metalloproteinases may decrease neuroinflammation in AD.
Newer AD models
There is growing consensus in the literature that Tg mice are not ideal models of AD, partly because they are based on genetic mutations present in only <5% of AD patients. Furthermore, although many therapeutics have ameliorated cognitive deficits and/or neuropathology in Tg mouse models, these same therapeutics have failed in clinical trials. The traditional Tg mouse models that express mutant forms of human APP and/or presenilins generally do not develop tau pathology unless they also express mutant forms of tau [364–367]. However, tau mutations are associated with other tauopathies, not AD. It has been argued that Tg mouse models actually simulate the asymptomatic phase of AD and therefore are telling us how to prevent AD, not cure AD [368]. Next-generation Tg mouse models are being developed that accumulate Aβ without phenotypes related to Aβ overexpression, which may be unrelated to AD. It has been recommended that these models be used with the caveat that researchers consider the strengths and limitations of each model against the scientific and therapeutic goal of a prospective preclinical study [369]. There remains a call for more suitable models that recapitulate sporadic AD and more closely model human physiology.
Perhaps one of the most exciting AD model systems recently introduced is the NHP. NHPs have an APP sequence that is completely homologous to that of humans [370] and they develop plaques and tau pathology [370–372]. To speed up development of AD pathology, researchers introduced Aβ preparations containing fibrils into NHPs via i.c.v. injection. This resulted in microglial activation, neuronal loss, and tau phosphorylation [373, 374]. In a major advance for the AβO field, researchers from Brazil and Canada showed that i.c.v. injection of AβO preparations free of fibrils into NHPs induced fundamental features of AD pathology without development of Aβ fibrils and plaques [46]. The pathological features induced by AβOs in these NHPs included synapse loss, tau hyperphosphorylation, and activation of astrocytes and microglia. Most importantly, this research team recently reported that sustaining AβO injections for 12–18 months results in memory deficits and synapse loss [375]. This NHP model shows great promise as a superior AD model for therapeutic testing.
Another potentially powerful new AD model system comprises human induced pluripotent stem cells, or iPSCs. iPSCs derived from both familial AD and sporadic AD patients have been studied, and these AD-derived iPSCs show AβO accumulation, ER stress, oxidative stress, and tau hyperphosphorylation [376, 377]. These studies indicate that familial AD and sporadic AD iPSCs exhibit differential manifestations of ER stress [376] and Aβ40 accumulation [377], indicating that different therapeutics may be effective for patient subsets. In 2015, an organoid human iPSC-derived system was developed, also termed a “3D human neural cell culture system” [378]. This iPSC system developed key events in AD pathogenesis, including extracellular aggregation of Aβ and accumulation of pTau. The De Strooper group recently created a novel chimeric model wherein human iPSCs were studied in a more natural environment, i.e., via transplantation into the brains of APP Tg immunodeficient mice [379]. These human neurons were able to differentiate and integrate into the mouse brain, express 3R/4R tau splice forms, show abnormal phosphorylation and conformational tau changes, and undergo neurodegeneration. Remarkably, transplantation of these human iPSCs altered gene expression, upregulating genes involved in myelination and downregulating genes related to memory and cognition, synaptic transmission, and neuronal projection. Therefore, human iPSC models are attractive AD models for their human origin and ability to integrate into mouse models, which are more easily utilized than NHP models.
Efforts are also being made at developing improved rodent models for AD. Octodon degus, a small rodent endemic to Chile, needs no genetic manipulation as its Aβ sequence differs from human in only 1 amino acid (H13R). Unlike mouse and rat Aβ, this Aβ sequence does form AβOs and this rodent also develops plaques and pTau with age [380, 381]. Tg rats are also being developed as AD models. The TgF344-AD rat expresses mutant APPsw and PS1ΔE9 genes and manifests age-dependent cerebral amyloidosis that precedes tauopathy, gliosis, apoptotic loss of neurons in the cerebral cortex and hippocampus, and cognitive disturbance [367]. These rats also exhibit pathological changes in the retina, including plaques and inflammation, e.g., microglial recruitment and complement activation [382]. This is interesting considering AβOs may also be involved in the pathogenesis of macular degeneration and glaucoma (see discussion below). Another Tg rat was developed using the Tg2576 mouse protocol. These rats exhibit cognitive deficits at 8–12 months, activated astrocytes in the brain, ThioS staining in the hippocampus and cortex, and elevated levels of Aβ38, Aβ40, and Aβ42 [383]. Tg rats expressing the Swedish and Indiana APP mutations also exhibit elevated levels of these Aβ peptides in the CSF [384] as well as pre-plaque intracellular AβO-associated cognitive impairment [87]. There are indications that non-Tg rabbit, which expresses the human Aβ sequence, may also prove valuable for studies of AβO etiology [346].
The use of Drosophila as an AD model was reviewed recently [385]. Drosophila has homologues of human APP and tau. Other advantages of Drosophila for use as an AD model include low genome redundancy, which greatly simplifies the analysis of single gene disruption, short lifespan, and their low cost compared to mammalian models. Drosophila have been used to uncover or validate several pathological pathways or susceptibility genes and have been readily implemented in drug screening pipelines. Interestingly, using a Tg Drosophila model expressing the Artic mutation, it was found that AD-like pathologies affected the circadian system in an age-dependent manner [386]. These Tg flies showed a dramatic degradation of circadian rhythms in tune with their reduced longevity and impaired climbing activity.
The use of Caenorhabditis elegans as an AD model was recently reviewed [387]. C. elegans is useful as an AD model as it has homologs of AD-related genes, including APP, tau, and PSEN1. C. elegans has complex biochemical pathways just like mammals, many of which are conserved. Its neuronal connectivity has already been established, making it a suitable model for learning and memory impairments [387]. Furthermore, C. elegans has a short lifespan, thereby speeding up study of Aβ accumulation, a process that can take months or years in other model organisms. To directly study the impact of the exact human Aβ42 sequence, Tg worms also have been developed and these model organisms have been shown to accumulate AβOs and utilized to study AβO-directed therapeutics [388–391].
Therapeutics
Eliezer Masliah, the current head of the NIA’s Division of Neuroscience, proposed multiple possible strategies for targeting the AβO pathogenic cascade in a 2014 commentary in PNAS (Fig.9) [392]. He and co-author Cassia Overk proposed that therapeutics for AD might involve 1) directly clearing AβOs; 2) blocking AβO surface receptors; 3) interfering with AβO-induced signaling pathways; or 4) decreasing downstream effectors such as tau. The AD therapeutics currently in clinical trials are described in a systematic review of clinicaltrials.gov conducted in January 2017 by Cummings and colleagues [393]. Approximately half of the 105 agents currently in clinical trials are amyloid related. Figure10 illustrates the mechanistic targets of many of the amyloid-targeted therapeutics. Some of the current efforts germane to AβO pathogenic mechanisms are discussed below.
Fig.9
Putative therapeutic targets of the AβO pathogenic cascade. Including: 1) AβOs themselves; 2) AβO receptors; 3) signaling pathways; or 4) downstream effectors such as tau. Reprinted with permission of PNAS from “Toward a unified therapeutics approach targeting putative amyloid-beta oligomer receptors” by Overk CR and Masliah E. This was published in Proc Natl Acad Sci U S A, 2014, 111(38): 13680-13681 [392].
![Putative therapeutic targets of the AβO pathogenic cascade. Including: 1) AβOs themselves; 2) AβO receptors; 3) signaling pathways; or 4) downstream effectors such as tau. Reprinted with permission of PNAS from “Toward a unified therapeutics approach targeting putative amyloid-beta oligomer receptors” by Overk CR and Masliah E. This was published in Proc Natl Acad Sci U S A, 2014, 111(38): 13680-13681 [392].](https://content.iospress.com:443/media/jad/2018/64-s1/jad-64-s1-jad179941/jad-64-jad179941-g009.jpg)
Fig.10
Mechanisms of Aβ-targeting phase III drugs in AD clinical trials. Drugs inhibiting AβO formation (A) or downstream consequences of toxic AβOs (B). Reprinted from “Alzheimer’s disease drug development pipeline: 2017” by Cummings J, Garam L, Mortsdorf T, Ritter A, and Zhong K. This was published in Alzheimers Dementia (N Y), 2017, 3(3): 367-384 [393], under the terms of the Creative Commons Attribution Non-Commercial No Derivatives License (CC BY NC ND) https://creativecommons.org/licenses/by-nc-nd/4.0/) [393].
![Mechanisms of Aβ-targeting phase III drugs in AD clinical trials. Drugs inhibiting AβO formation (A) or downstream consequences of toxic AβOs (B). Reprinted from “Alzheimer’s disease drug development pipeline: 2017” by Cummings J, Garam L, Mortsdorf T, Ritter A, and Zhong K. This was published in Alzheimers Dementia (N Y), 2017, 3(3): 367-384 [393], under the terms of the Creative Commons Attribution Non-Commercial No Derivatives License (CC BY NC ND) https://creativecommons.org/licenses/by-nc-nd/4.0/) [393].](https://content.iospress.com:443/media/jad/2018/64-s1/jad-64-s1-jad179941/jad-64-jad179941-g010.jpg)
1) Directly clearing AβOs or decreasing AβO production. This category comprises anti-Aβ immunotherapies, β-secretase (BACE) inhibitors, and anti-Aβ aggregation agents. About one quarter (27%) of agents in AD phase II clinical trials fall into this category. However, this category comprises more than half (57%) of phase III AD trials [393]. Several recent reviews discuss the progress of these Aβ-centric clinical trials and provide hypotheses for the failures. In 2014, Karran and Hardy reviewed the data reported at each phase of the drug discovery process for Aβ-targeting therapies and found significant gaps in the data in several cases [102]. They observe that target engagement was not established for most therapeutic agents analyzed, an issue also raised by Brody et al. [107]. In 2016, Selkoe presented an updated review of Aβ-targeted phase III clinical trials and concluded that they have failed because of improper patient selection, choice of agent, lack of target engagement, and/or dose or side effects unrelated to target engagement [5].
Anti-Aβ immunotherapies represent 8% of the phase II pipeline and 18% of the phase III pipeline. In 2014, Goure and colleagues of Acumen Pharmaceuticals proposed in their review of immunotherapeutics that current Aβ-directed therapies were failing due to lack of selectivity for AβOs; instead, they bind to monomeric or fibrillar Aβ, or both [101]. Monomers and fibrils are more abundant than AβOs in the AD brain, but less germane to nerve cell damage. The authors suggest that the affinity for monomers and/or fibrils by Aβ immunotherapies in clinical trials is why high doses are required for therapeutic benefit. Acumen, in partnership with Merck, has developed an antibody, known as ACU193, that is unique among Aβ immunotherapeutics in its selectivity for Aβ oligomers. ACU193 has greater than 500-fold selectivity for AβOs over fibrils [394] and greater than 2500-fold selectivity for AβOs over monomers [34]. Success of ACU193 in clinical trials would provide compelling evidence for the hypothesis that soluble AβOs are the primary toxins instigating AD pathogenesis.
What may be two examples supporting AβO-directed immunotherapies are Crenezumab and Aducanumab. Genentech reported at the 2017 Clinical Trials on Alzheimer’s Disease (CTAD) conference that their Aβ immunotherapy Crenezumab had a 10-fold higher affinity for AβOs over Aβ monomers [395]. In immunoprecipitation experiments, its main target was AβOs that are, or dissociate into, SDS-stable dimers. It is of note that dimers themselves are not thought to be toxic [129]. Genentech reported that primary efficacy endpoints were not met for Crenezumab in two phase II trials (ABBY and BLAZE), although subgroup analysis showed greater reduction in cognitive decline in patients with mild AD given the higher dose of Crenezumab. They are currently enrolling patients in two phase III studies (CREAD and CREAD2), which will dose up to 4-fold higher.
Aducanumab (Biogen), an antibody that targets Aβ oligomers and fibrils, has shown in phase Ib trials a reduction of amyloid plaques in a dose- and time-dependent manner and, most importantly, a slowing of cognitive decline. Slowing of cognitive decline required the highest doses tested. However, these doses caused amyloid related imaging abnormalities [105]. While there is optimism that higher doses of these antibodies will result in greater therapeutic efficacy, a fully AβO-selective antibody may be essential for using low doses to avoid complications while still providing disease-modifying efficacy.
BACE inhibitors represent 6% of the phase II pipeline and 18% of the phase III pipeline. Supporting evidence for the therapeutic value of BACE inhibitors comes from the protective A673T APP Icelandic mutation in humans, which reduces BACE processing of AβPP [396, 397]. On the other hand, genetic deletion of BACE in mice causes many side effects, most notably the AD symptoms of neurodegeneration and memory dysfunction [398]. Since the systematic review conducted by Cummings and colleagues in January 2017 [393], Merck has halted its phase II and III trials of the BACE inhibitor verubescestat for lack of efficacy; a leading theory for this failure is the timing of treatment [399]. A recent success in AD mouse models, demonstrating beneficial effects on cellular, long-range circuitry, and memory impairment, has motivated researchers to start another clinical trial with a modified BACE inhibitor [400].
2) Blocking AβO receptors. The AβO receptors currently targeted in clinical trials are RAGE, the Sigma-2 receptor, calcium channels, and IR. Azelirago (vTv Therapeutics), an inhibitor of RAGE, is currently in phase III clinical trials. RAGE has been identified as an AβO-targeted receptor, as reviewed previously [401]. This therapy did show a statistically significant slowing in cognitive decline in phase II trials, although it increased cognitive decline when tested at a higher dose [402]. A Sigma-2 receptor antagonist (CT1812; Cognition Therapeutics) is currently in phase II clinical trials. Sigma-2 receptors have been shown to participate in AβO binding to neurons and synaptotoxicity [204]. The Sigma-2 receptor antagonist blocks AβO binding to cultured neurons and improves cognitive deficits in AD mouse models [203]. In October 2017, the FDA placed CT1812 on fast track and in November, Cognition Therapeutics reported at CTAD that CT1812 was well tolerated at all doses tested [403]. Furthermore, it decreased levels of protein biomarkers including synaptotagmin-1, a marker of synaptic damage [404]. Nilvadipine, a calcium channel blocker (St. James’ Hospital Ireland, Alzheimer Europe, Archer Pharmaceuticals) that is currently indicated to reduce blood pressure, has completed phase III clinical trials for AD, although no results yet have been reported. Nilvadipine has been reported to enhance Aβ clearance from the brain of AD mouse models and improve cognition; its putative mechanisms-of-action being inhibition of BACE, inhibition of RAGE-mediated Aβ brain influx, and/or facilitation of lipoprotein receptor (LRP-1)-mediated Aβ brain efflux [405].
The relationship between insulin resistance and AβOs in AD is discussed above. Therapeutically, insulin signaling may be a convenient target if the many treatments already developed for type 2 diabetes could be repurposed for AD [406, 407]. In hippocampal cell cultures, exogenous as well as astrocyte-secreted insulin and IGF1 displace AβOs bound to cell surfaces [73]. Diabetes drugs fall into five categories: intranasal insulin, incretins, dipeptidyl peptidase 4 (DPP-4) inhibitors, peroxisome proliferator activated receptor (PPAR-γ) agonists, and the common diabetes treatment metformin [406]. Intranasally delivered insulin has been shown to improve memory function in AD patients, although different studies have obtained inconsistent results as to whether this is effective in APOE E4-positive AD patients [406]. The impact of intranasally delivered insulin on AD is currently being investigated in phase I and II clinical trials sponsored by Wake Forest University. Incretins are gastrointestinal hormones that stimulate insulin secretion and inhibit glucagon release in a glucose-dependent manner [406]. Liraglutide, an incretin analog, has been shown to reverse memory impairment, synaptic loss, and reduce plaque load in aged APP/PS1 mice [408]. Liraglutide is currently in phase II clinical trials (Imperial College London). DPP-4 inhibitors block degradation of incretins and have been found to improve memory function, reduce levels of Aβ and pTau, and decrease inflammation in AD rodent models [406]. However, no DPP-4 inhibitors are currently in clinical trials [406]. Activation of PPAR-γ induces the expression of multiple genes involved in the insulin signaling cascade, which improves insulin sensitivity in patients with type 2 diabetes [406]. Metformin, a very widely prescribed drug for diabetes [409], is currently in clinical trials to examine its effect both on aging in general and AD. On the other hand, it has been recently linked to an increased dementia risk in diabetes patients [410].
3) Interfering with AβO-induced signaling path-ways. The 2014 commentary by Overk and Masliah [392] suggests the therapeutic targets in this category are kinases that are activated by the various AβO surface receptors. These kinases include the tyrosine kinases, Tyr K, Fyn, and c-Abl, and also CDK5/GSK3β. Fyn inhibition as a therapeutic strategy is based on the PrPc/mGluR5 pathway discussed earlier and has been reviewed recently [411]. An inhibitor of Src and Abl family kinases, AZD0530, is currently in phase II clinical trials (Yale University). AZD0530 (saracatinib) was previously used to treat cancer. Pre-clinically, it was found to reverse cognitive deficits in AD mice [412]. Another kinase inhibitor repurposed from cancer treatment, nilotinib (Tasigna®), is currently in phase II clinical trials (Georgetown University). Nilotinib targets the tyrosine kinase Abl and may aid in clearance of plaques and tangles through activation of autophagy [413, 414].
4) Decreasing downstream effectors such as tau. Tau-directed therapies, which could block the down-stream effects of AβOs [286, 415], represent 8% of the phase II pipeline and 4% of the phase III pipeline. Similar to Aβ-directed therapies, there are multiple mechanisms of action for therapeutic targeting of tau including inhibiting tau aggregation, decreasing tau hyperphosphorylation, reducing tau levels in the brain, and stabilizing microtubules [415]. LMTM (aka TRx0237; TauRx) is a small molecule derived from the dye methylene blue that has been shown to block tau aggregation in vitro and in tau Tg animal models [416]. Although it initially showed no clinical efficacy when tested as a combination therapy [416], it recently showed an ability to improve cognition and decrease rate of brain atrophy when tested as a monotherapy in phase III clinical trials [417]. No significant efficacy findings have been reported yet for other tau-directed therapies.
Combination treatments
Given the complex neuropathology of AD and the lack of effective biomarkers for sporadic AD, it may be the case that multi-factorial, combination treatments will provide the greatest efficacy in AD treatment. This is a sentiment shared by many in the field [5, 66, 106, 418]. In October 2017, Amylyx Pharmaceuticals received a grant from the Alzheimer’s Association and the Alzheimer’s Drug Discovery Foundation to conduct phase II clinical trials with the combination therapy AMX0035 (Alzheimer’s Association). AMX0035 is a combination of two compounds that synergistically block mitochondrial and ER stress. Preclinical studies show that this combination protects brain cells from inflammation and oxidation in models of amyotrophic lateral sclerosis, AD, and mitochondrial diseases [419]. ALZT-OP1 (AZTherapies) is a combination of two small molecule drugs, cromolyn, an asthma drug, and ibuprofen. Cromolyn was found to inhibit Aβ aggregation in vitro and reduce soluble Aβ in the brain in vivo [420]. The intended effect of ibuprofen in the combination therapy is to reduce neuroinflammation. ALZT-OP1 is currently in phase III clinical trials. It is likely that many more combination therapies will arise in the future.
AD prevention: Diet, exercise, and mental/social engagement
One review article states that although it is difficult to make conclusions regarding diet as an AD risk-factor due to the difficulty in analyzing eating patterns, there does seem to be clear evidence that diet influences AD. Protective foods identified include fish, fruit, coffee, and wine. There is also evidence that a diet high in saturated fats may increase AD risk [421]. In a Tg mouse AD model, high cholesterol promotes earlier buildup of AβOs [350]. A systematic review and meta-analysis by researchers at the Mayo Clinic found that a higher adherence to the Mediterranean diet is associated with a reduced risk of developing mild cognitive impairment (MCI) and AD and a reduced risk of progressing from MCI to AD [422]. Vitamin B may also have a positive effect [423]. To more directly test the impact of diet on AD risk, participants are currently being recruited for a clinical trial to determine the effect of saturated fat and glycemic index on cognition in older individuals with or without an APOE E4 genotype (sponsored by the University of Washington). APOE4 is known to affect the presence and impact of AβOs [424–430]. Recruitment is underway also for clinical trials testing Genistein, a dietary supplement that has been found to have antioxidant and neuroprotective effects on AD and increases PPAR-γ levels, which results in increased APOE expression and Aβ degradation (Fundación para la Investigación del Hospital Clínico de Valencia). The omega-3 fatty acid DHA, which protects against AβO-instigated dendritic spine loss [431], shows potential to decrease AD incidence in APOE4 carriers [423, 432] and is under clinical investigation (http://clinicaltrails.gov). In addition to the potential for diet to modify AD risk, it is well recognized that physical activity also modifies risk [433]. In AD mouse models, exercise decreases AβO levels and increases cognitive performance [434–437]. Furthermore, evidence has been found for mental/social engagement modifying AD risk and AβO accumulation in a mouse model [438] and in humans [439–441]. Therefore, it is encouraging that some cases of sporadic AD may be delayed or even prevented by modifiable lifestyle factors.
AβOs as biomarkers: can AβOs provide metrics to assess experimental drug efficacy and ultimately give a diagnostic to indicate a patient should start AD treatments?
Newly emerging approaches have begun to focus on therapeutic targeting of AβOs, and not amyloid plaques, as AβOs are the form of Aβ that instigates the neural damage leading to AD. A powerful metric for the efficacy of these new approaches to disease-modifying therapeutics would be to monitor a patient’s AβO levels.
There are two big challenges to using AβO levels as a biomarker. First, there is a need for extraordinary sensitivity. Second, there is a need to discriminate oligomeric Aβ from the other, much more abundant but chemically similar forms of the peptide. A uniquely sensitive and specific AβO immunoassay, capable of attomolar quantitation, initially showed that median AβO levels in AD CSF were 10-30-fold higher than in non-AD controls [32]. Although powerful, this assay was not practical, and it has not been explored for clinical use. Since then, other groups have utilized various immunoassay platforms and Aβ-targeting antibodies with varying results. A team at Merck using the AβO-specific antibody ACU193 (see Therapeutics section above), found a significant 3-5-fold increase in AβOs in AD CSF compared with aged-matched controls [34]. On the contrary, Jongbloed and colleagues found that CSF AβO levels decreased from non-dementia to MCI to AD [165]. Another study found no significant difference between AβO levels in AD CSF and controls. However, this study found a small, but significant increase in AβOs levels in MCI compared to controls, suggesting an early rise in AβO levels followed by a later decrease [35]. Most recently, an AβO-specific plasma assay was able to differentiate AD from controls with 78.3% sensitivity and 86.5% specificity, finding AβOs elevated in AD plasma [442]. CSF/plasma measurements of whole-AβO populations do not seem to be diagnostically useful at present due to inconsistent results, but the possibility remains that an assay targeting specific populations of AβOs may be more useful [33, 35, 443–445]. Indeed, a recent study showed that an assay targeting HMW AβOs could be used to monitor efficacy of an anti-amyloid therapeutic [446]. This finding is consistent with immunoassays showing therapeutic efficacy in a mouse model correlated with reduction in a pool of putative type 1 AβOs specifically recognized by the NU4 monoclonal antibody [120].
An alternative to measurements of AβOs in CSF or blood comes from new technologies for AβO imaging. The Pronucleontrademark platform from Adlyfe consists of series of engineered peptides that provide a unique readout when associated with beta-rich fibers and AβOs. There are indications that it can be used for pre-plaque imaging [447]. Another technology is the development of magnetic resonance imaging (MRI) [96] and positron emission tomography (PET) [199] probes using modified antibodies that have high selectivity and affinity for AβOs. These have been shown to discriminate AD Tg mice from non-Tg littermates and to discriminate human AD brain slices from non-AD specimens. The availability of humanized AβO-specific antibodies makes it likely that these probes will soon be ready for clinical testing [101].
In the future, it is foreseeable that scheduled clinical tests of AβO levels could provide an indicator of whether a patient should begin an appropriate AD treatment. Monitoring levels after diagnosis and treatment would provide an initial indicator of how well a patient is responding to a therapeutic. Use of AβO levels as an AD biomarker would thus be akin to monitoring glycated hemoglobin by A1C levels for diabetics.
Other dysfunctions/degenerative neural disorders linked to AβOs?
As discussed above, TBI and diabetes may be etiological factors in the buildup of AβOs. In addition, it may be the case that once accumulated, AβOs may contribute to further pathogenic consequences in these diseases such as insulin resistance (see discussion above), dementia, or chronic traumatic encephalopathy (CTE). AβOs, specifically, have not yet been implicated in CTE, but CTE is associated with repetitive brain injury and amyloid plaques have been observed in CTE brain tissue [448]. Additional disorders that have been linked to AβOs are inclusion body myositis, glaucoma, and macular degeneration. Inclusion body myositis is the most common progressive muscle disease of patients above the age of 50. Forms of Aβ, including AβOs, are known to accumulate in the muscle fibers of diseased patients [449]. Further evidence implicates AβOs in glaucoma and macular degeneration. AβOs have been found to contribute to apoptosis of retinal ganglion cells in glaucoma [450]. Intravitreal injection of AβOs in rat induces molecular changes associated with apoptosis in the rat retina. Apoptosis is hypothesized to take place in macular degeneration according to bioinformatics analysis [451]. AβOs, through RAGE, upregulate VEGF, which stimulates neovascular age-related macular degeneration [452]. Structures reactive with the OC antibody have been found in drusen, a hallmark of eyes affected by macular degeneration [453]. The OC antibody is well characterized and binds to fibrillar type 2 AβOs [148]. In fact, a modulator of Aβ aggregation (MRZ-99030) is neuroprotective with therapeutic treatment in animal models of glaucoma and macular degeneration [454]. However, much more evidence is needed to understand the role of AβOs in these neural disorders.
CONCLUDING REMARKS
As evidenced by the increasing number of publications concerning AβOs in the past 5 years, and the consistency in data supporting a toxic role for AβOs, the AβO hypothesis for AD pathogenesis has garnered considerable support and acceptance. Accordingly, the number of AβO-targeting therapeutics in the AD pipeline has begun to increase. We believe that this emerging interest in AβO targeting will prove beneficial to the treatment and diagnosis of AD. These efforts potentially can extend to a broader proportion of the population, given the evidence for a role for AβOs in other diseases in addition to AD. Ultimately, for these efforts to result in therapeutic and diagnostic benefits, further advances in AβO structure-function studies are needed. Continued investment into this and other research involving AβOs will enable the closing of critical gaps, thereby paving a smoother and shorter path from bench to bedside.
ACKNOWLEDGMENTS
We would like to thank Stephanie Cline, Steven Patrie, and Grant Krafft for editing the manuscript. This work was supported by National Institutes of Health Grants R41AG054337 (WLK) and T32AG020506 (ENC).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/17-9941).
REFERENCES
[1] | Frackowiak J , Zoltowska A , Wisniewski HM ((1994) ) Non-fibrillar beta-amyloid protein is associated with smooth muscle cells of vessel walls in Alzheimer disease, J Neuropathol Exp Neurol 53: , 637–645. |
[2] | Oda T , Pasinetti GM , Osterburg HH , Anderson C , Johnson SA , Finch CE ((1994) ) Purification and characterization of brain clusterin, Biochem Biophys Res Commun 204: , 1131–1136. |
[3] | Oda T , Wals P , Osterburg HH , Johnson SA , Pasinetti GM , Morgan TE , Rozovsky I , Stine WB , Snyder SW , Holzman TF , Krafft GA , Finch CE ((1995) ) Clusterin (Apoj) alters the aggregation of amyloid beta-peptide (Aβ(1-42)) and forms slowly sedimenting Aβ complexes that cause oxidative stress, Exp Neurol 136: , 22–31. |
[4] | Hayden EY , Teplow DB ((2013) ) Amyloid beta-protein oligomers and Alzheimer’s disease, Alzheimers Res Ther 5: , 60. |
[5] | Selkoe DJ , Hardy J ((2016) ) The amyloid hypothesis of Alzheimer’s disease at 25 years, EMBO Mol Med 8: , 595–608. |
[6] | Gong Y , Chang L , Viola KL , Lacor PN , Lambert MP , Finch CE , Krafft GA , Klein WL ((2003) ) Alzheimer’s disease-affected brain: Presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A 100: , 10417–10422. |
[7] | Kayed R , Head E , Thompson JL , McIntire TM , Milton SC , Cotman CW , Glabe CG ((2003) ) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis, Science 300: , 486–489. |
[8] | Noguchi A , Matsumura S , Dezawa M , Tada M , Yanazawa M , Ito A , Akioka M , Kikuchi S , Sato M , Ideno S , Noda M , Fukunari A , Muramatsu S , Itokazu Y , Sato K , Takahashi H , Teplow DB , Nabeshima Y , Kakita A , Imahori K , Hoshi M ((2009) ) Isolation and characterization of patient-derived, toxic, high mass amyloid beta-protein (Abeta) asbly from Alzheimer disease brains, J Biol Chem 284: , 32895–32905 sem. |
[9] | Mc Donald JM , Savva GM , Brayne C , Welzel AT , Forster G , Shankar GM , Selkoe DJ , Ince PG , Walsh D ; Medical Research Council Cognitive Function and Ageing Study ((2010) ) The presence of sodium dodecyl sulphate-stable Abeta dimers is strongly associated with Alzheimer-type dementia, Brain 133: , 1328–1341. |
[10] | Pham E , Crews L , Ubhi K , Hansen L , Adame A , Cartier A , Salmon D , Galasko D , Michael S , Savas JN , Yates JR , Glabe C , Masliah E ((2010) ) Progressive accumulation of amyloid-beta oligomers in Alzheimer’s disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins, FEBS J 277: , 3051–3067. |
[11] | Ohno M , Chang L , Tseng W , Oakley H , Citron M , Klein WL , Vassar R , Disterhoft JF ((2006) ) Temporal memory deficits in Alzheimer’s mouse models: Rescue by genetic deletion of BACE1, Eur J Neurosci 23: , 251–260. |
[12] | Esparza TJ , Zhao H , Cirrito JR , Cairns NJ , Bateman RJ , Holtzman DM , Brody DL ((2013) ) Amyloid-beta oligomerization in Alzheimer dementia versus high-pathology controls, Ann Neurol 73: , 104–119. |
[13] | Masters CL , Simms G , Weinman NA , Multhaup G , McDonald BL , Beyreuther K ((1985) ) Amyloid plaque core protein in Alzheimer disease and Down syndrome, Proc Natl Acad Sci U S A 82: , 4245–4249. |
[14] | Lesne SE , Sherman MA , Grant M , Kuskowski M , Schneider JA , Bennett DA , Ashe KH ((2013) ) Brain amyloid-beta oligomers in ageing and Alzheimer’s disease, Brain 136: , 1383–1398. |
[15] | Gyure KA , Durham R , Stewart WF , Smialek JE , Troncoso JC ((2001) ) Intraneuronal abeta-amyloid precedes development of amyloid plaques in Down syndrome, Arch Pathol Lab Med 125: , 489–492. |
[16] | Lacor PN , Buniel MC , Chang L , Fernandez SJ , Gong Y , Viola KL , Lambert MP , Velasco PT , Bigio EH , Finch CE , Krafft GA , Klein WL ((2004) ) Synaptic targeting by Alzheimer’s-related amyloid beta oligomers, J Neurosci 24: , 10191–10200. |
[17] | Tomiyama T , Nagata T , Shimada H , Teraoka R , Fukushima A , Kanemitsu H , Takuma H , Kuwano R , Imagawa M , Ataka S , Wada Y , Yoshioka E , Nishizaki T , Watanabe Y , Mori H ((2008) ) A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia, Ann Neurol 63: , 377–387. |
[18] | Nishitsuji K , Tomiyama T , Ishibashi K , Ito K , Teraoka R , Lambert MP , Klein WL , Mori H ((2009) ) The E693Delta mutation in amyloid precursor protein increases intracellular accumulation of amyloid beta oligomers and causes endoplasmic reticulum stress-induced apoptosis in cultured cells, Am J Pathol 174: , 957–969. |
[19] | Tomiyama T , Matsuyama S , Iso H , Umeda T , Takuma H , Ohnishi K , Ishibashi K , Teraoka R , Sakama N , Yamashita T , Nishitsuji K , Ito K , Shimada H , Lambert MP , Klein WL , Mori H ((2010) ) A mouse model of amyloid beta oligomers: Their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo, J Neurosci 30: , 4845–4856. |
[20] | Shimada H , Ataka S , Tomiyama T , Takechi H , Mori H , Miki T ((2011) ) Clinical course of patients with familial early-onset Alzheimer’s disease potentially lacking senile plaques bearing the E693Delta mutation in amyloid precursor protein, Dement Geriatr Cogn Disord 32: , 45–54. |
[21] | Inayathullah M , Teplow DB ((2011) ) Structural dynamics of the DeltaE22 (Osaka) familial Alzheimer’s disease-linked amyloid beta-protein, Amyloid 18: , 98–107. |
[22] | Kutoku Y , Ohsawa Y , Kuwano R , Ikeuchi T , Inoue H , Ataka S , Shimada H , Mori H , Sunada Y ((2015) ) A second pedigree with amyloid-less familial Alzheimer’s disease harboring an identical mutation in the amyloid precursor protein gene (E693delta), Intern Med 54: , 205–208. |
[23] | Gandy S , Simon AJ , Steele JW , Lublin AL , Lah JJ , Walker LC , Levey AI , Krafft GA , Levy E , Checler F , Glabe C , Bilker WB , Abel T , Schmeidler J , Ehrlich ME ((2010) ) Days to criterion as an indicator of toxicity associated with human Alzheimer amyloid-beta oligomers, Ann Neurol 68: , 220–230. |
[24] | Podlisny MB , Walsh DM , Amarante P , Ostaszewski BL , Stimson ER , Maggio JE , Teplow DB , Selkoe DJ ((1998) ) Oligomerization of endogenous and synthetic amyloid beta-protein at nanomolar levels in cell culture and stabilization of monomer by Congo red, Biochemistry 37: , 3602–3611. |
[25] | LeVine H 3rd ((2004) ) Alzheimer’s beta-peptide oligomer formation at physiologic concentrations, Anal Biochem 335: , 81–90. |
[26] | Lambert MP , Barlow AK , Chromy BA , Edwards C , Freed R , Liosatos M , Morgan TE , Rozovsky I , Trommer B , Viola KL , Wals P , Zhang C , Finch CE , Krafft GA , Klein WL ((1998) ) Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins, Proc Natl Acad Sci U S A 95: , 6448–6453. |
[27] | Chang L , Bakhos L , Wang Z , Venton DL , Klein WL ((2003) ) Femtomole immunodetection of synthetic and endogenous amyloid-beta oligomers and its application to Alzheimer’s disease drug candidate screening, J Mol Neurosci 20: , 305–313. |
[28] | Barghorn S , Nimmrich V , Striebinger A , Krantz C , Keller P , Janson B , Bahr M , Schmidt M , Bitner RS , Harlan J , Barlow E , Ebert U , Hillen H ((2005) ) Globular amyloid beta-peptide oligomer - a homogenous and stable neuropathological protein in Alzheimer’s disease, J Neurochem 95: , 834–847. |
[29] | Lesne SE ((2013) ) Breaking the code of amyloid-beta oligomers, Int J Cell Biol 2013: , 950783. |
[30] | Bitan G , Fradinger EA , Spring SM , Teplow DB ((2005) ) Neurotoxic protein oligomers–what you see is not always what you get, Amyloid 12: , 88–95. |
[31] | Chromy BA , Nowak RJ , Lambert MP , Viola KL , Chang L , Velasco PT , Jones BW , Fernandez SJ , Lacor PN , Horowitz P , Finch CE , Krafft GA , Klein WL ((2003) ) Self-assembly of Abeta(1-42) into globular neurotoxins, Biochemistry 42: , 12749–12760. |
[32] | Georganopoulou DG , Chang L , Nam JM , Thaxton CS , Mufson EJ , Klein WL , Mirkin CA ((2005) ) Nanoparticle-based detection in cerebral spinal fluid of a soluble pathogenic biomarker for Alzheimer’s disease, Proc Natl Acad Sci U S A 102: , 2273–2276. |
[33] | Blennow K , Mattsson N , Scholl M , Hansson O , Zetterberg H ((2015) ) Amyloid biomarkers in Alzheimer’s disease, Trends Pharmacol Sci 36: , 297–309. |
[34] | Savage MJ , Kalinina J , Wolfe A , Tugusheva K , Korn R , Cash-Mason T , Maxwell JW , Hatcher NG , Haugabook SJ , Wu G , Howell BJ , Renger JJ , Shughrue PJ , McCampbell A ((2014) ) A sensitive abeta oligomer assay discriminates Alzheimer’s and aged control cerebrospinal fluid, J Neurosci 34: , 2884–2897. |
[35] | Yang T , O’Malley TT , Kanmert D , Jerecic J , Zieske LR , Zetterberg H , Hyman BT , Walsh DM , Selkoe DJ ((2015) ) A highly sensitive novel immunoassay specifically detects low levels of soluble Abeta oligomers in human cerebrospinal fluid, Alzheimers Res Ther 7: , 14. |
[36] | Walsh DM , Klyubin I , Shankar GM , Townsend M , Fadeeva JV , Betts V , Podlisny MB , Cleary JP , Ashe KH , Rowan MJ , Selkoe DJ ((2005) ) The role of cell-derived oligomers of Abeta in Alzheimer’s disease and avenues for therapeutic intervention, Biochem Soc Trans 33: , 1087–1090. |
[37] | Shankar GM , Li S , Mehta TH , Garcia-Munoz A , Shepardson NE , Smith I , Brett FM , Farrell MA , Rowan MJ , Lemere CA , Regan CM , Walsh DM , Sabatini BL , Selkoe DJ ((2008) ) Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory, Nat Med 14: , 837–842. |
[38] | Reed MN , Hofmeister JJ , Jungbauer L , Welzel AT , Yu C , Sherman MA , Lesne S , LaDu MJ , Walsh DM , Ashe KH , Cleary JP ((2011) ) Cognitive effects of cell-derived and synthetically derived Abeta oligomers, Neurobiol Aging 32: , 1784–1794. |
[39] | Figueiredo CP , Clarke JR , Ledo JH , Ribeiro FC , Costa CV , Melo HM , Mota-Sales AP , Saraiva LM , Klein WL , Sebollela A , De Felice FG , Ferreira ST ((2013) ) Memantine rescues transient cognitive impairment caused by high-molecular-weight abeta oligomers but not the persistent impairment induced by low-molecular-weight oligomers, J Neurosci 33: , 9626–9634. |
[40] | Townsend M , Shankar GM , Mehta T , Walsh DM , Selkoe DJ ((2006) ) Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: A potent role for trimers, J Physiol 572: , 477–492. |
[41] | Klyubin I , Betts V , Welzel AT , Blennow K , Zetterberg H , Wallin A , Lemere CA , Cullen WK , Peng Y , Wisniewski T , Selkoe DJ , Anwyl R , Walsh DM , Rowan MJ ((2008) ) Amyloid beta protein dimer-containing human CSF disrupts synaptic plasticity: Prevention by systemic passive immunization, J Neurosci 28: , 4231–4237. |
[42] | Walsh DM , Klyubin I , Fadeeva JV , Cullen WK , Anwyl R , Wolfe MS , Rowan MJ , Selkoe DJ ((2002) ) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo, Nature 416: , 535–539. |
[43] | Cleary JP , Walsh DM , Hofmeister JJ , Shankar GM , Kuskowski MA , Selkoe DJ , Ashe KH ((2005) ) Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function, Nat Neurosci 8: , 79–84. |
[44] | Lesne S , Koh MT , Kotilinek L , Kayed R , Glabe CG , Yang A , Gallagher M , Ashe KH ((2006) ) A specific amyloid-beta protein assembly in the brain impairs memory, Nature 440: , 352–357. |
[45] | Zussy C , Brureau A , Keller E , Marchal S , Blayo C , Delair B , Ixart G , Maurice T , Givalois L ((2013) ) Alzheimer’s disease related markers, cellular toxicity and behavioral deficits induced six weeks after oligomeric amyloid-beta peptide injection in rats, PLoS One 8: , e53117. |
[46] | Forny-Germano L , Lyra e Silva NM , Batista AF , Brito-Moreira J , Gralle M , Boehnke SE , Coe BC , Lablans A , Marques SA , Martinez AM , Klein WL , Houzel JC , Ferreira ST , Munoz DP , De Felice FG ((2014) ) Alzheimer’s disease-like pathology induced by amyloid-beta oligomers in nonhuman primates, J Neurosci 34: , 13629–13643. |
[47] | Shankar GM , Bloodgood BL , Townsend M , Walsh DM , Selkoe DJ , Sabatini BL ((2007) ) Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway, J Neurosci 27: , 2866–2875. |
[48] | Zhao WQ , De Felice FG , Fernandez S , Chen H , Lambert MP , Quon MJ , Krafft GA , Klein WL ((2008) ) Amyloid beta oligomers induce impairment of neuronal insulin receptors, FASEB J 22: , 246–260. |
[49] | Maezawa I , Zimin PI , Wulff H , Jin LW ((2011) ) Amyloid-beta protein oligomer at low nanomolar concentrations activates microglia and induces microglial neurotoxicity, J Biol Chem 286: , 3693–3706. |
[50] | Larson M , Sherman MA , Amar F , Nuvolone M , Schneider JA , Bennett DA , Aguzzi A , Lesne SE ((2012) ) The complex PrP(c)-Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer’s disease, J Neurosci 32: , 16857–16871a. |
[51] | De Felice FG , Wu D , Lambert MP , Fernandez SJ , Velasco PT , Lacor PN , Bigio EH , Jerecic J , Acton PJ , Shughrue PJ , Chen-Dodson E , Kinney GG , Klein WL ((2008) ) Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by A beta oligomers, Neurobiol Aging 29: , 1334–1347. |
[52] | Zempel H , Thies E , Mandelkow E , Mandelkow EM ((2010) ) Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines, JNeurosci 30: , 11938–11950. |
[53] | Snyder EM , Nong Y , Almeida CG , Paul S , Moran T , Choi EY , Nairn AC , Salter MW , Lombroso PJ , Gouras GK , Greengard P ((2005) ) Regulation of NMDA receptor trafficking by amyloid-beta, Nat Neurosci 8: , 1051–1058. |
[54] | Roselli F , Tirard M , Lu J , Hutzler P , Lamberti P , Livrea P , Morabito M , Almeida OF ((2005) ) Soluble beta-amyloid1-40 induces NMDA-dependent degradation of postsynaptic density-95 at glutamatergic synapses, J Neurosci 25: , 11061–11070. |
[55] | Lacor PN , Buniel MC , Furlow PW , Clemente AS , Velasco PT , Wood M , Viola KL , Klein WL ((2007) ) Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease, J Neurosci 27: , 796–807. |
[56] | Pigino G , Morfini G , Atagi Y , Deshpande A , Yu C , Jungbauer L , LaDu M , Busciglio J , Brady S ((2009) ) Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta, Proc Natl Acad Sci U S A 106: , 5907–5912. |
[57] | Poon WW , Blurton-Jones M , Tu CH , Feinberg LM , Chabrier MA , Harris JW , Jeon NL , Cotman CW ((2011) ) beta-Amyloid impairs axonal BDNF retrograde trafficking, Neurobiol Aging 32: , 821–833. |
[58] | Decker H , Jurgensen S , Adrover MF , Brito-Moreira J , Bomfim TR , Klein WL , Epstein AL , De Felice FG , Jerusalinsky D , Ferreira ST ((2010) ) N-methyl-D-aspartate receptors are required for synaptic targeting of Alzheimer’s toxic amyloid-beta peptide oligomers, J Neurochem 115: , 1520–1529. |
[59] | Longo VD , Viola KL , Klein WL , Finch CE ((2000) ) Reversible inactivation of superoxide-sensitive aconitase in Abeta1-42-treated neuronal cell lines, J Neurochem 75: , 1977–1985. |
[60] | Sponne I , Fifre A , Drouet B , Klein C , Koziel V , Pincon-Raymond M , Olivier JL , Chambaz J , Pillot T ((2003) ) Apoptotic neuronal cell death induced by the non-fibrillar amyloid-beta peptide proceeds through an early reactive oxygen species-dependent cytoskeleton perturbation, JBiol Chem 278: , 3437–3445. |
[61] | Tabner BJ , El-Agnaf OM , Turnbull S , German MJ , Paleologou KE , Hayashi Y , Cooper LJ , Fullwood NJ , Allsop D ((2005) ) Hydrogen peroxide is generated during the very early stages of aggregation of the amyloid peptides implicated in Alzheimer disease and familial British dementia, J Biol Chem 280: , 35789–35792. |
[62] | De Felice FG , Velasco PT , Lambert MP , Viola K , Fernandez SJ , Ferreira ST , Klein WL ((2007) ) Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine, J Biol Chem 282: , 11590–11601. |
[63] | Umeda T , Tomiyama T , Sakama N , Tanaka S , Lambert MP , Klein WL , Mori H ((2011) ) Intraneuronal amyloid beta oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo, J Neurosci Res 89: , 1031–1042. |
[64] | Resende R , Ferreiro E , Pereira C , Oliveira CR ((2008) ) ER stress is involved in Abeta-induced GSK-3beta activation and tau phosphorylation, J Neurosci Res 86: , 2091–2099. |
[65] | Zhao WQ , Lacor PN , Chen H , Lambert MP , Quon MJ , Krafft GA , Klein WL ((2009) ) Insulin receptor dysfunction impairs cellular clearance of neurotoxic oligomeric a{beta}, J Biol Chem 284: , 18742–18753. |
[66] | De Felice FG , Vieira MN , Bomfim TR , Decker H , Velasco PT , Lambert MP , Viola KL , Zhao WQ , Ferreira ST , Klein WL ((2009) ) Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Abeta oligomers, Proc Natl Acad Sci U S A 106: , 1971–1976. |
[67] | Ma QL , Yang F , Rosario ER , Ubeda OJ , Beech W , Gant DJ , Chen PP , Hudspeth B , Chen C , Zhao Y , Vinters HV , Frautschy SA , Cole GM ((2009) ) Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: Suppression by omega-3 fatty acids and curcumin, J Neurosci 29: , 9078–9089. |
[68] | Hu J , Akama KT , Krafft GA , Chromy BA , Van Eldik LJ ((1998) ) Amyloid-beta peptide activates cultured astrocytes: Morphological alterations, cytokine induction and nitric oxide release, Brain Res 785: , 195–206. |
[69] | Ferretti MT , Bruno MA , Ducatenzeiler A , Klein WL , Cuello AC ((2012) ) Intracellular Abeta-oligomers and early inflammation in a model of Alzheimer’s disease, Neurobiol Aging 33: , 1329–1342. |
[70] | Heinitz K , Beck M , Schliebs R , Perez-Polo JR ((2006) ) Toxicity mediated by soluble oligomers of beta-amyloid(1-42) on cholinergic SN56.B5.G4 cells, J Neurochem 98: , 1930–1945. |
[71] | Nunes-Tavares N , Santos LE , Stutz B , Brito-Moreira J , Klein WL , Ferreira ST , de Mello FG ((2012) ) Inhibition of choline acetyltransferase as a mechanism for cholinergic dysfunction induced by amyloid-beta peptide oligomers, J Biol Chem 287: , 19377–19385. |
[72] | Kitiyanant N , Kitiyanant Y , Svendsen CN , Thangnipon W ((2012) ) BDNF-, IGF-1- and GDNF-secreting human neural progenitor cells rescue amyloid beta-induced toxicity in cultured rat septal neurons, Neurochem Res 37: , 143–152. |
[73] | Pitt J , Wilcox KC , Tortelli V , Diniz LP , Oliveira MS , Dobbins C , Yu XW , Nandamuri S , Gomes FCA , DiNunno N , Viola KL , De Felice FG , Ferreira ST , Klein WL ((2017) ) Neuroprotective astrocyte-derived insulin/insulin-like growth factor 1 stimulates endocytic processing and extracellular release of neuron-bound Abeta oligomers, Mol Biol Cell 28: , 2623–2636. |
[74] | Sen A , Nelson TJ , Alkon DL ((2015) ) ApoE4 and Abeta oligomers reduce BDNF expression via HDAC nuclear translocation, J Neurosci 35: , 7538–7551. |
[75] | Poon WW , Carlos AJ , Aguilar BL , Berchtold NC , Kawano CK , Zograbyan V , Yaopruke T , Shelanski M , Cotman CW ((2013) ) beta-Amyloid (Abeta) oligomers impair brain-derived neurotrophic factor retrograde trafficking by down-regulating ubiquitin C-terminal hydrolase, UCH-L1, J Biol Chem 288: , 16937–16948. |
[76] | Sharma M , Dierkes T , Sajikumar S ((2017) ) Epigenetic regulation by G9a/GLP complex ameliorates amyloid-beta 1-42 induced deficits in long-term plasticity and synaptic tagging/capture in hippocampal pyramidal neurons, Aging Cell 16: , 1062–1072. |
[77] | Krishna K , Behnisch T , Sajikumar S ((2016) ) Inhibition of histone deacetylase 3 restores amyloid-beta oligomer-induced plasticity deficit in hippocampal CA1 pyramidal neurons, J Alzheimers Dis 51: , 783–791. |
[78] | Mastroeni D , Khdour OM , Arce PM , Hecht SM , Coleman PD ((2015) ) Novel antioxidants protect mitochondria from the effects of oligomeric amyloid beta and contribute to the maintenance of epigenome function, ACS Chem Neurosci 6: , 588–598. |
[79] | Hodgson N , Trivedi M , Muratore C , Li S , Deth R ((2013) ) Soluble oligomers of amyloid-beta cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake, J Alzheimers Dis 36: , 197–209. |
[80] | Lithner CU , Lacor PN , Zhao WQ , Mustafiz T , Klein WL , Sweatt JD , Hernandez CM ((2013) ) Disruption of neocortical histone H3 homeostasis by soluble Abeta: Implications for Alzheimer’s disease, Neurobiol Aging 34: , 2081–2090. |
[81] | Varvel NH , Bhaskar K , Patil AR , Pimplikar SW , Herrup K , Lamb BT ((2008) ) Abeta oligomers induce neuronal cell cycle events in Alzheimer’s disease, J Neurosci 28: , 10786–10793. |
[82] | Varvel NH , Bhaskar K , Kounnas MZ , Wagner SL , Yang Y , Lamb BT , Herrup K ((2009) ) NSAIDs prevent, but do not reverse, neuronal cell cycle reentry in a mouse model of Alzheimer disease, J Clin Invest 119: , 3692–3702. |
[83] | Bhaskar K , Miller M , Chludzinski A , Herrup K , Zagorski M , Lamb BT ((2009) ) The PI3K-Akt-mTOR pathway regulates Abeta oligomer induced neuronal cell cycle events, Mol Neurodegener 4: , 14. |
[84] | Kim HJ , Chae SC , Lee DK , Chromy B , Lee SC , Park YC , Klein WL , Krafft GA , Hong ST ((2003) ) Selective neuronal degeneration induced by soluble oligomeric amyloid beta protein, FASEB J 17: , 118–120. |
[85] | Price KA , Varghese M , Sowa A , Yuk F , Brautigam H , Ehrlich ME , Dickstein DL ((2014) ) Altered synaptic structure in the hippocampus in a mouse model of Alzheimer’s disease with soluble amyloid-beta oligomers and no plaque pathology, Mol Neurodegener 9: , 41. |
[86] | Mucke L , Masliah E , Yu GQ , Mallory M , Rockenstein EM , Tatsuno G , Hu K , Kholodenko D , Johnson-Wood K , McConlogue L ((2000) ) High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation, J Neurosci 20: , 4050–4058. |
[87] | Leon WC , Canneva F , Partridge V , Allard S , Ferretti MT , DeWilde A , Vercauteren F , Atifeh R , Ducatenzeiler A , Klein W , Szyf M , Alhonen L , Cuello AC ((2010) ) A novel transgenic rat model with a full Alzheimer’s-like amyloid pathology displays pre-plaque intracellular amyloid-beta-associated cognitive impairment, J Alzheimers Dis 20: , 113–126. |
[88] | Cheng IH , Scearce-Levie K , Legleiter J , Palop JJ , Gerstein H , Bien-Ly N , Puolivali J , Lesne S , Ashe KH , Muchowski PJ , Mucke L ((2007) ) Accelerating amyloid-beta fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models, J Biol Chem 282: , 23818–23828. |
[89] | Lambert MP , Velasco PT , Chang L , Viola KL , Fernandez S , Lacor PN , Khuon D , Gong Y , Bigio EH , Shaw P , De Felice FG , Krafft GA , Klein WL ((2007) ) Monoclonal antibodies that target pathological assemblies of Abeta, J Neurochem 100: , 23–35. |
[90] | Zameer A , Kasturirangan S , Emadi S , Nimmagadda SV , Sierks MR ((2008) ) Anti-oligomeric Abeta single-chain variable domain antibody blocks Abeta-induced toxicity against human neuroblastoma cells, J Mol Biol 384: , 917–928. |
[91] | Klyubin I , Walsh DM , Lemere CA , Cullen WK , Shankar GM , Betts V , Spooner ET , Jiang L , Anwyl R , Selkoe DJ , Rowan MJ ((2005) ) Amyloid beta protein immunotherapy neutralizes Abeta oligomers that disrupt synaptic plasticity in vivo, Nat Med 11: , 556–561. |
[92] | Xiao C , Davis FJ , Chauhan BC , Viola KL , Lacor PN , Velasco PT , Klein WL , Chauhan NB ((2013) ) Brain transit and ameliorative effects of intranasally delivered anti-amyloid-beta oligomer antibody in 5XFAD mice, JAlzheimers Dis 35: , 777–788. |
[93] | Zhao M , Wang SW , Wang YJ , Zhang R , Li YN , Su YJ , Zhou WW , Yu XL , Liu RT ((2014) ) Pan-amyloid oligomer specific scFv antibody attenuates memory deficits and brain amyloid burden in mice with Alzheimer’s disease, Curr Alzheimer Res 11: , 69–78. |
[94] | Sebollela A , Cline EN , Popova I , Luo K , Sun X , Ahn J , Barcelos MA , Bezerra VN , Lyra E Silva NM , Patel J , Pinheiro NR , Qin LA , Kamel JM , Weng A , DiNunno N , Bebenek AM , Velasco PT , Viola KL , Lacor PN , Ferreira ST , Klein WL ((2017) ) A human scFv antibody that targets and neutralizes high molecular weight pathogenic amyloid-beta oligomers, J Neurochem 142: , 934–947. |
[95] | Oakley H , Cole SL , Logan S , Maus E , Shao P , Craft J , Guillozet-Bongaarts A , Ohno M , Disterhoft J , Van Eldik L , Berry R , Vassar R ((2006) ) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: Potential factors in amyloid plaque formation, J Neurosci 26: , 10129–10140. |
[96] | Viola KL , Sbarboro J , Sureka R , De M , Bicca MA , Wang J , Vasavada S , Satpathy S , Wu S , Joshi H , Velasco PT , MacRenaris K , Waters EA , Lu C , Phan J , Lacor P , Prasad P , Dravid VP , Klein WL ((2015) ) Towards non-invasive diagnostic imaging of early-stage Alzheimer’s disease, Nat Nanotechnol 10: , 91–98. |
[97] | Terry RD , Masliah E , Salmon DP , Butters N , DeTeresa R , Hill R , Hansen LA , Katzman R ((1991) ) Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment, Ann Neurol 30: , 572–580. |
[98] | Arriagada PV , Growdon JH , Hedley-Whyte ET , Hyman BT ((1992) ) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease, Neurology 42: , 631–639. |
[99] | Crystal H , Dickson D , Fuld P , Masur D , Scott R , Mehler M , Masdeu J , Kawas C , Aronson M , Wolfson L ((1988) ) Clinico-pathologic studies in dementia: Nondemented subjects with pathologically confirmed Alzheimer’s disease, Neurology 38: , 1682–1687. |
[100] | Cummings JL , Morstorf T , Zhong K ((2014) ) Alzheimer’s disease drug-development pipeline: Few candidates, frequent failures, Alzheimers Res Ther 6: , 37. |
[101] | Goure WF , Krafft GA , Jerecic J , Hefti F ((2014) ) Targeting the proper amyloid-beta neuronal toxins: A path forward for Alzheimer’s disease immunotherapeutics, Alzheimers Res Ther 6: , 42. |
[102] | Karran E , Hardy J ((2014) ) A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease, Ann Neurol 76: , 185–205. |
[103] | Karran E , Hardy J ((2014) ) Antiamyloid therapy for Alzheimer’s disease–are we on the right road? N Engl J Med 370: , 377–378. |
[104] | Siemers ER , Sundell KL , Carlson C , Case M , Sethuraman G , Liu-Seifert H , Dowsett SA , Pontecorvo MJ , Dean RA , Demattos R ((2016) ) Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer’s disease patients, Alzheimers Dement 12: , 110–120. |
[105] | Sevigny J , Chiao P , Bussiere T , Weinreb PH , Williams L , Maier M , Dunstan R , Salloway S , Chen T , Ling Y , O’Gorman J , Qian F , Arastu M , Li M , Chollate S , Brennan MS , Quintero-Monzon O , Scannevin RH , Arnold HM , Engber T , Rhodes K , Ferrero J , Hang Y , Mikulskis A , Grimm J , Hock C , Nitsch RM , Sandrock A ((2016) ) The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease, Nature 537: , 50–56. |
[106] | Sengupta U , Nilson AN , Kayed R ((2016) ) The role of amyloid-beta oligomers in toxicity, propagation, and immunotherapy, EBioMedicine 6: , 42–49. |
[107] | Brody DL , Jiang H , Wildburger N , Esparza TJ ((2017) ) Non-canonical soluble amyloid-beta aggregates and plaque buffering: Controversies and future directions for target discovery in Alzheimer’s disease, Alzheimers Res Ther 9: , 62. |
[108] | van Dyck CH ((2017) ) Anti-amyloid-beta monoclonal antibodies for Alzheimer’s disease: Pitfalls and promise, Biol Psychiatry 83: , 311–319. |
[109] | Klein WL , Krafft GA , Finch CE ((2001) ) Targeting small Abeta oligomers: The solution to an Alzheimer’s disease conundrum? Trends Neurosci 24: , 219–224. |
[110] | Karran E , De Strooper B ((2016) ) The amyloid cascade hypothesis: Are we poised for success or failure? J Neurochem 139: (Suppl 2), 237–252. |
[111] | Gold M ((2017) ) Phase II clinical trials of anti-amyloid beta antibodies: When is enough, enough? Alzheimers Dement (N Y) 3: , 402–409. |
[112] | Morris GP , Clark IA , Vissel B ((2014) ) Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease, Acta Neuropathol Commun 2: , 135. |
[113] | Le Couteur DG , Hunter S , Brayne C ((2016) ) Solanezumab and the amyloid hypothesis for Alzheimer’s disease, BMJ 355: . |
[114] | Abbott A , Dolgin E ((2016) ) Failed Alzheimer’s trial does not kill leading theory of disease, Nature 540: , 15–16. |
[115] | Ferreira ST , Lourenco MV , Oliveira MM , De Felice FG ((2015) ) Soluble amyloid-beta oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease, Front Cell Neurosci 9: , 191. |
[116] | Teplow DB ((2013) ) On the subject of rigor in the study of amyloid beta-protein assembly, Alzheimers Res Ther 5: , 39. |
[117] | Benilova I , Karran E , De Strooper B ((2012) ) The toxic Abeta oligomer and Alzheimer’s disease: An emperor in need of clothes, Nat Neurosci 15: , 349–357. |
[118] | Velasco PT , Heffern MC , Sebollela A , Popova IA , Lacor PN , Lee KB , Sun X , Tiano BN , Viola KL , Eckermann AL , Meade TJ , Klein WL ((2012) ) Synapse-binding subpopulations of Abeta oligomers sensitive to peptide assembly blockers and scFv antibodies, ACS Chem Neurosci 3: , 972–981. |
[119] | Liu P , Reed MN , Kotilinek LA , Grant MK , Forster CL , Qiang W , Shapiro SL , Reichl JH , Chiang AC , Jankowsky JL , Wilmot CM , Cleary JP , Zahs KR , Ashe KH ((2015) ) Quaternary structure defines a large class of amyloid-beta oligomers neutralized by sequestration, Cell Rep 11: , 1760–1771. |
[120] | Knight EM , Kim SH , Kottwitz JC , Hatami A , Albay R , Suzuki A , Lublin A , Alberini CM , Klein WL , Szabo P , Relkin NR , Ehrlich M , Glabe CG , Gandy S , Steele JW ((2016) ) Effective anti-Alzheimer Abeta therapy involves depletion of specific Abeta oligomer subtypes, Neurol Neuroimmunol Neuroinflamm 3: , e237. |
[121] | Dohler F , Sepulveda-Falla D , Krasemann S , Altmeppen H , Schluter H , Hildebrand D , Zerr I , Matschke J , Glatzel M ((2014) ) High molecular mass assemblies of amyloid-beta oligomers bind prion protein in patients with Alzheimer’s disease, Brain 137: , 873–886. |
[122] | Kostylev MA , Kaufman AC , Nygaard HB , Patel P , Haas LT , Gunther EC , Vortmeyer A , Strittmatter SM ((2015) ) Prion-protein-interacting amyloid-beta oligomers of high molecular weight are tightly correlated with memory impairment in multiple Alzheimer mouse models, J Biol Chem 290: , 17415–17438. |
[123] | Williams TL , Choi JK , Surewicz K , Surewicz WK ((2015) ) Soluble prion protein binds isolated low molecular weight amyloid-beta oligomers causing cytotoxicity inhibition, ACS Chem Neurosci 6: , 1972–1980. |
[124] | Mc Donald JM , O’Malley TT , Liu W , Mably AJ , Brinkmalm G , Portelius E , Wittbold WM 3rd , Frosch MP , Walsh DM ((2015) ) The aqueous phase of Alzheimer’s disease brain contains assemblies built from approximately 4 and approximately 7 kDa Abeta species, Alzheimers Dement 11: , 1286–1305. |
[125] | Upadhaya AR , Lungrin I , Yamaguchi H , Fandrich M , Thal DR ((2012) ) High-molecular weight Abeta oligomers and protofibrils are the predominant Abeta species in the native soluble protein fraction of the AD brain, J Cell Mol Med 16: , 287–295. |
[126] | Savioz A , Giannakopoulos P , Herrmann FR , Klein WL , Kovari E , Bouras C , Giacobini E ((2016) ) A study of Abeta oligomers in the temporal cortex and cerebellum of patients with neuropathologically confirmed Alzheimer’s disease compared to aged controls, Neurodegener Dis 16: , 398–406. |
[127] | Amar F , Sherman MA , Rush T , Larson M , Boyle G , Chang L , Gotz J , Buisson A , Lesne SE ((2017) ) The amyloid-beta oligomer Abeta*56 induces specific alterations in neuronal signaling that lead to tau phosphorylation and aggregation, Sci Signal 10: , eaal2021. |
[128] | Sherman MA , LaCroix M , Amar F , Larson ME , Forster C , Aguzzi A , Bennett DA , Ramsden M , Lesne SE ((2016) ) Soluble conformers of Abeta and tau alter selective proteins governing axonal transport, J Neurosci 36: , 9647–9658. |
[129] | O’Malley TT , Oktaviani NA , Zhang D , Lomakin A , O’Nuallain B , Linse S , Benedek GB , Rowan MJ , Mulder FA , Walsh DM ((2014) ) Abeta dimers differ from monomers in structural propensity, aggregation paths and population of synaptotoxic assemblies, Biochem J 461: , 413–426. |
[130] | Walsh DM , Teplow DB ((2012) ) Alzheimer’s disease and the amyloid beta-protein, Prog Mol Biol Transl Sci 107: , 101–124. |
[131] | Naylor R , Hill AF , Barnham KJ ((2008) ) Is covalently crosslinked Abeta responsible for synaptotoxicity in Alzheimer’s disease? Curr Alzheimer Res 5: , 533–539. |
[132] | Wildburger NC , Esparza TJ , LeDuc RD , Fellers RT , Thomas PM , Cairns NJ , Kelleher NL , Bateman RJ , Brody DL ((2017) ) Diversity of amyloid-beta proteoforms in the Alzheimer’s disease brain, Sci Rep 7: , 9520. |
[133] | Rudinskiy N , Fuerer C , Demurtas D , Zamorano S , De Piano C , Herrmann AG , Spires-Jones TL , Oeckl P , Otto M , Frosch MP , Moniatte M , Hyman BT , Schmid AW ((2016) ) Amyloid-beta oligomerization is associated with the generation of a typical peptide fragment fingerprint, Alzheimers Dement 12: , 996–1013. |
[134] | Galante D , Ruggeri FS , Dietler G , Pellistri F , Gatta E , Corsaro A , Florio T , Perico A , D’Arrigo C ((2016) ) A critical concentration of N-terminal pyroglutamylated amyloid beta drives the misfolding of Ab1-42 into more toxic aggregates, Int J Biochem Cell Biol 79: , 261–270. |
[135] | Lee J , Gillman AL , Jang H , Ramachandran S , Kagan BL , Nussinov R , Teran Arce F ((2014) ) Role of the fast kinetics of pyroglutamate-modified amyloid-beta oligomers in membrane binding and membrane permeability, Biochemistry 53: , 4704–4714. |
[136] | Piechotta A , Parthier C , Kleinschmidt M , Gnoth K , Pillot T , Lues I , Demuth HU , Schilling S , Rahfeld JU , Stubbs MT ((2017) ) Structural and functional analyses of pyroglutamate-amyloid-beta-specific antibodies as a basis for Alzheimer immunotherapy, J Biol Chem 292: , 12713–12724. |
[137] | Saido TC , Iwatsubo T , Mann DM , Shimada H , Ihara Y , Kawashima S ((1995) ) Dominant and differential deposition of distinct beta-amyloid peptide species, A beta N3(pE), in senile plaques, Neuron 14: , 457–466. |
[138] | Zou K , Liu J , Watanabe A , Hiraga S , Liu S , Tanabe C , Maeda T , Terayama Y , Takahashi S , Michikawa M , Komano H ((2013) ) Abeta43 is the earliest-depositing Abeta species in APP transgenic mouse brain and is converted to Abeta41 by two active domains of ACE, Am J Pathol 182: , 2322–2331. |
[139] | Conicella AE , Fawzi NL ((2014) ) The C-terminal threonine of Abeta43 nucleates toxic aggregation via structural and dynamical changes in monomers and protofibrils, Biochemistry 53: , 3095–3105. |
[140] | Kretner B , Trambauer J , Fukumori A , Mielke J , Kuhn PH , Kremmer E , Giese A , Lichtenthaler SF , Haass C , Arzberger T , Steiner H ((2016) ) Generation and deposition of Abeta43 by the virtually inactive presenilin-1 L435F mutant contradicts the presenilin loss-of-function hypothesis of Alzheimer’s disease, EMBO Mol Med 8: , 458–465. |
[141] | Welzel AT , Maggio JE , Shankar GM , Walker DE , Ostaszewski BL , Li S , Klyubin I , Rowan MJ , Seubert P , Walsh DM , Selkoe DJ ((2014) ) Secreted amyloid beta-proteins in a cell culture model include N-terminally extended peptides that impair synaptic plasticity, Biochemistry 53: , 3908–3921. |
[142] | Szczepankiewicz O , Linse B , Meisl G , Thulin E , Frohm B , Sala Frigerio C , Colvin MT , Jacavone AC , Griffin RG , Knowles TP , Walsh DM , Linse S ((2015) ) N-terminal extensions retard Abeta42 fibril formation but allow cross-seeding and co-aggregation with Abeta42, J Am Chem Soc 137: , 14673–14685. |
[143] | Sugiki T , Utsunomiya-Tate N ((2013) ) Site-specific aspartic acid isomerization regulates self-assembly and neurotoxicity of amyloid-beta, Biochem Biophys Res Commun 441: , 493–498. |
[144] | Kumar S , Wirths O , Stuber K , Wunderlich P , Koch P , Theil S , Rezaei-Ghaleh N , Zweckstetter M , Bayer TA , Brustle O , Thal DR , Walter J ((2016) ) Phosphorylation of the amyloid beta-peptide at Ser26 stabilizes oligomeric assembly and increases neurotoxicity, Acta Neuropathol 131: , 525–537. |
[145] | Rijal Upadhaya A , Kosterin I , Kumar S , von Arnim CA , Yamaguchi H , Fandrich M , Walter J , Thal DR ((2014) ) Biochemical stages of amyloid-beta peptide aggregation and accumulation in the human brain and their association with symptomatic and pathologically preclinical Alzheimer’s disease, Brain 137: , 887–903. |
[146] | Matsumura S , Shinoda K , Yamada M , Yokojima S , Inoue M , Ohnishi T , Shimada T , Kikuchi K , Masui D , Hashimoto S , Sato M , Ito A , Akioka M , Takagi S , Nakamura Y , Nemoto K , Hasegawa Y , Takamoto H , Inoue H , Nakamura S , Nabeshima Y , Teplow DB , Kinjo M , Hoshi M ((2011) ) Two distinct amyloid beta-protein (Abeta) assembly pathways leading to oligomers and fibrils identified by combined fluorescence correlation spectroscopy, morphology, and toxicity analyses, J Biol Chem 286: , 11555–11562. |
[147] | Watanabe-Nakayama T , Ono K , Itami M , Takahashi R , Teplow DB , Yamada M ((2016) ) High-speed atomic force microscopy reveals structural dynamics of amyloid beta1-42 aggregates, Proc Natl Acad Sci U S A 113: , 5835–5840. |
[148] | Breydo L , Kurouski D , Rasool S , Milton S , Wu JW , Uversky VN , Lednev IK , Glabe CG ((2016) ) Structural differences between amyloid beta oligomers, Biochem Biophys Res Commun 477: , 700–705. |
[149] | Barz B , Liao Q , Strodel B ((2018) ) Pathways of amyloid-beta aggregation depend on oligomer shape, J Am Chem Soc 140: , 319–327. |
[150] | Yang T , Li S , Xu H , Walsh DM , Selkoe DJ ((2017) ) Large soluble oligomers of amyloid beta-protein from Alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate, J Neurosci 37: , 152–163. |
[151] | Cohen SI , Linse S , Luheshi LM , Hellstrand E , White DA , Rajah L , Otzen DE , Vendruscolo M , Dobson CM , Knowles TP ((2013) ) Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism, Proc Natl Acad Sci U S A 110: , 9758–9763. |
[152] | Barz B , Strodel B ((2016) ) Understanding amyloid-beta oligomerization at the molecular level: The role of the fibril surface, Chemistry 22: , 8768–8772. |
[153] | Economou NJ , Giammona MJ , Do TD , Zheng X , Teplow DB , Buratto SK , Bowers MT ((2016) ) Amyloid beta-protein assembly and Alzheimer’s disease: Dodecamers of Abeta42, but not of Abeta40, seed fibril formation, JAm Chem Soc 138: , 1772–1775. |
[154] | Koffie RM , Meyer-Luehmann M , Hashimoto T , Adams KW , Mielke ML , Garcia-Alloza M , Micheva KD , Smith SJ , Kim ML , Lee VM , Hyman BT , Spires-Jones TL ((2009) ) Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques, Proc Natl Acad Sci U S A 106: , 4012–4017. |
[155] | Jackson RJ , Rudinskiy N , Herrmann AG , Croft S , Kim JM , Petrova V , Ramos-Rodriguez JJ , Pitstick R , Wegmann S , Garcia-Alloza M , Carlson GA , Hyman BT , Spires-Jones TL ((2016) ) Human tau increases amyloid beta plaque size but not amyloid beta-mediated synapse loss in a novel mouse model of Alzheimer’s disease, Eur J Neurosci 44: , 3056–3066. |
[156] | Koffie RM , Hashimoto T , Tai HC , Kay KR , Serrano-Pozo A , Joyner D , Hou S , Kopeikina KJ , Frosch MP , Lee VM , Holtzman DM , Hyman BT , Spires-Jones TL ((2012) ) Apolipoprotein E4 effects in Alzheimer’s disease are mediated by synaptotoxic oligomeric amyloid-beta, Brain 135: , 2155–2168. |
[157] | Sadleir KR , Kandalepas PC , Buggia-Prevot V , Nicholson DA , Thinakaran G , Vassar R ((2016) ) Presynaptic dystrophic neurites surrounding amyloid plaques are sites of microtubule disruption, BACE1 elevation, and increased Abeta generation in Alzheimer’s disease, Acta Neuropathol 132: , 235–256. |
[158] | Ono K , Condron MM , Teplow DB ((2009) ) Structure-neurotoxicity relationships of amyloid beta-protein oligomers, Proc Natl Acad Sci U S A 106: , 14745–14750. |
[159] | Hayden EY , Conovaloff JL , Mason A , Bitan G , Teplow DB ((2017) ) Preparation of pure populations of covalently stabilized amyloid beta-protein oligomers of specific sizes, Anal Biochem 518: , 78–85. |
[160] | Al-Hilaly YK , Williams TL , Stewart-Parker M , Ford L , Skaria E , Cole M , Bucher WG , Morris KL , Sada AA , Thorpe JR , Serpell LC ((2013) ) A central role for dityrosine crosslinking of Amyloid-beta in Alzheimer’s disease, Acta Neuropathol Commun 1: , 83. |
[161] | Bush AI ((2013) ) The metal theory of Alzheimer’s disease, J Alzheimers Dis 33: (Suppl 1), S277–281. |
[162] | Zhang S , Fox DM , Urbanc B ((2017) ) Insights into formation and structure of Abeta oligomers cross-linked via tyrosines, J Phys Chem B 121: , 5523–5535. |
[163] | Ryan TM , Kirby N , Mertens HD , Roberts B , Barnham KJ , Cappai R , Pham CL , Masters CL , Curtain CC ((2015) ) Small angle X-ray scattering analysis of Cu-induced oligomers of the Alzheimer’s amyloid beta peptide, Metallomics 7: , 536–543. |
[164] | Smith DP , Ciccotosto GD , Tew DJ , Fodero-Tavoletti MT , Johanssen T , Masters CL , Barnham KJ , Cappai R ((2007) ) Concentration dependent Cu2+ induced aggregation and dityrosine formation of the Alzheimer’s disease amyloid-beta peptide, Biochemistry 46: , 2881–2891. |
[165] | Jongbloed W , Bruggink KA , Kester MI , Visser PJ , Scheltens P , Blankenstein MA , Verbeek MM , Teunissen CE , Veerhuis R ((2015) ) Amyloid-beta oligomers relate to cognitive decline in Alzheimer’s disease, J Alzheimers Dis 45: , 35–43. |
[166] | Takeda S , Hashimoto T , Roe AD , Hori Y , Spires-Jones TL , Hyman BT ((2013) ) Brain interstitial oligomeric amyloid beta increases with age and is resistant to clearance from brain in a mouse model of Alzheimer’s disease, FASEB J 27: , 3239–3248. |
[167] | Arispe N ((2004) ) Architecture of the Alzheimer’s A beta P ion channel pore, J Membr Biol 197: , 33–48. |
[168] | Drolle E , Hane F , Lee B , Leonenko Z ((2014) ) Atomic force microscopy to study molecular mechanisms of amyloid fibril formation and toxicity in Alzheimer’s disease, Drug Metab Rev 46: , 207–223. |
[169] | Watt AD , Villemagne VL , Barnham KJ ((2013) ) Metals, membranes, and amyloid-beta oligomers: Key pieces in the Alzheimer’s disease puzzle?S, J Alzheimers Dis 33: (Suppl 1), 283–293. |
[170] | Canale C , Seghezza S , Vilasi S , Carrotta R , Bulone D , Diaspro A , San Biagio PL , Dante S ((2013) ) Different effects of Alzheimer’s peptide Abeta(1-40) oligomers and fibrils on supported lipid membranes, Biophys Chem 182: , 23–29. |
[171] | Yates EA , Owens SL , Lynch MF , Cucco EM , Umbaugh CS , Legleiter J ((2013) ) Specific domains of Abeta facilitate aggregation on and association with lipid bilayers, J Mol Biol 425: , 1915–1933. |
[172] | Sarkar B , Das AK , Maiti S ((2013) ) Thermodynamically stable amyloid-beta monomers have much lower membrane affinity than the small oligomers, Front Physiol 4: , 84. |
[173] | Gunn AP , Wong BX , Johanssen T , Griffith JC , Masters CL , Bush AI , Barnham KJ , Duce JA , Cherny RA ((2016) ) Amyloid-beta peptide Abeta3pE-42 induces lipid peroxidation, membrane permeabilization, and calcium influx in neurons, J Biol Chem 291: , 6134–6145. |
[174] | Bode DC , Baker MD , Viles JH ((2017) ) Ion channel formation by amyloid-beta42 oligomers but not amyloid-beta40 in cellular membranes, J Biol Chem 292: , 1404–1413. |
[175] | Serra-Batiste M , Ninot-Pedrosa M , Bayoumi M , Gairi M , Maglia G , Carulla N ((2016) ) Abeta42 assembles into specific beta-barrel pore-forming oligomers in membrane-mimicking environments, Proc Natl Acad Sci U S A 113: , 10866–10871. |
[176] | Drews A , Flint J , Shivji N , Jonsson P , Wirthensohn D , De Genst E , Vincke C , Muyldermans S , Dobson C , Klenerman D ((2016) ) Individual aggregates of amyloid beta induce temporary calcium influx through the cell membrane of neuronal cells, Sci Rep 6: , 31910. |
[177] | Di Scala C , Troadec JD , Lelievre C , Garmy N , Fantini J , Chahinian H ((2014) ) Mechanism of cholesterol-assisted oligomeric channel formation by a short Alzheimer beta-amyloid peptide, J Neurochem 128: , 186–195. |
[178] | Evangelisti E , Wright D , Zampagni M , Cascella R , Fiorillo C , Bagnoli S , Relini A , Nichino D , Scartabelli T , Nacmias B , Sorbi S , Cecchi C ((2013) ) Lipid rafts mediate amyloid-induced calcium dyshomeostasis and oxidative stress in Alzheimer’s disease, Curr Alzheimer Res 10: , 143–153. |
[179] | Evangelisti E , Zampagni M , Cascella R , Becatti M , Fiorillo C , Caselli A , Bagnoli S , Nacmias B , Cecchi C ((2014) ) Plasma membrane injury depends on bilayer lipid composition in Alzheimer’s disease, J Alzheimers Dis 41: , 289–300. |
[180] | Peters C , Fernandez-Perez EJ , Burgos CF , Espinoza MP , Castillo C , Urrutia JC , Streltsov VA , Opazo C , Aguayo LG ((2013) ) Inhibition of amyloid beta-induced synaptotoxicity by a pentapeptide derived from the glycine zipper region of the neurotoxic peptide, Neurobiol Aging 34: , 2805–2814. |
[181] | Jang H , Connelly L , Arce FT , Ramachandran S , Kagan BL , Lal R , Nussinov R ((2013) ) Mechanisms for the insertion of toxic, fibril-like beta-amyloid oligomers into the membrane, J Chem Theory Comput 9: , 822–833. |
[182] | Renner M , Lacor PN , Velasco PT , Xu J , Contractor A , Klein WL , Triller A ((2010) ) Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5, Neuron 66: , 739–754. |
[183] | Haas LT , Strittmatter SM ((2016) ) Oligomers of amyloid beta prevent physiological activation of the cellular prion protein-metabotropic glutamate receptor 5 complex by glutamate in Alzheimer disease, J Biol Chem 291: , 17112–17121. |
[184] | Um JW , Kaufman AC , Kostylev M , Heiss JK , Stagi M , Takahashi H , Kerrisk ME , Vortmeyer A , Wisniewski T , Koleske AJ , Gunther EC , Nygaard HB , Strittmatter SM ((2013) ) Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein, Neuron 79: , 887–902. |
[185] | Um JW , Nygaard HB , Heiss JK , Kostylev MA , Stagi M , Vortmeyer A , Wisniewski T , Gunther EC , Strittmatter SM ((2012) ) Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons, Nat Neurosci 15: , 1227–1235. |
[186] | Lauren J , Gimbel DA , Nygaard HB , Gilbert JW , Strittmatter SM ((2009) ) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers, Nature 457: , 1128–1132. |
[187] | Caetano FA , Beraldo FH , Hajj GN , Guimaraes AL , Jurgensen S , Wasilewska-Sampaio AP , Hirata PH , Souza I , Machado CF , Wong DY , De Felice FG , Ferreira ST , Prado VF , Rylett RJ , Martins VR , Prado MA ((2011) ) Amyloid-beta oligomers increase the localization of prion protein at the cell surface, J Neurochem 117: , 538–553. |
[188] | Ganzinger KA , Narayan P , Qamar SS , Weimann L , Ranasinghe RT , Aguzzi A , Dobson CM , McColl J , St George-Hyslop P , Klenerman D ((2014) ) Single-molecule imaging reveals that small amyloid-beta(1-42) oligomers interact with the cellular prion protein (PrPC), Chembiochem 15: , 2515–2521. |
[189] | Peters C , Espinoza MP , Gallegos S , Opazo C , Aguayo LG ((2015) ) Alzheimer’s Abeta interacts with cellular prion protein inducing neuronal membrane damage and synaptotoxicity, Neurobiol Aging 36: , 1369–1377. |
[190] | Aimi T , Suzuki K , Hoshino T , Mizushima T ((2015) ) Dextran sulfate sodium inhibits amyloid-beta oligomer binding to cellular prion protein, J Neurochem 134: , 611–617. |
[191] | Rushworth JV , Griffiths HH , Watt NT , Hooper NM ((2013) ) Prion protein-mediated toxicity of amyloid-beta oligomers requires lipid rafts and the transmembrane LRP1, J Biol Chem 288: , 8935–8951. |
[192] | Chin J , Palop JJ , Yu GQ , Kojima N , Masliah E , Mucke L ((2004) ) Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice, J Neurosci 24: , 4692–4697. |
[193] | Mouillet-Richard S , Ermonval M , Chebassier C , Laplanche JL , Lehmann S , Launay JM , Kellermann O ((2000) ) Signal transduction through prion protein, Science 289: , 1925–1928. |
[194] | Wilcox KC , Marunde MR , Das A , Velasco PT , Kuhns BD , Marty MT , Jiang H , Luan CH , Sligar SG , Klein WL ((2015) ) Nanoscale synaptic membrane mimetic allows unbiased high throughput screen that targets binding sites for Alzheimer’s-associated Abeta oligomers, PLoS One 10: , e0125263. |
[195] | Benilova I , De Strooper B ((2010) ) Prion protein in Alzheimer’s pathogenesis: A hot and controversial issue, EMBO Mol Med 2: , 289–290. |
[196] | Balducci C , Beeg M , Stravalaci M , Bastone A , Sclip A , Biasini E , Tapella L , Colombo L , Manzoni C , Borsello T , Chiesa R , Gobbi M , Salmona M , Forloni G ((2010) ) Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein, Proc Natl Acad Sci U S A 107: , 2295–2300. |
[197] | Rial D , Piermartiri TC , Duarte FS , Tasca CI , Walz R , Prediger RD ((2012) ) Overexpression of cellular prion protein (PrP(C)) prevents cognitive dysfunction and apoptotic neuronal cell death induced by amyloid-beta (Abeta(1)(-)(4)(0)) administration in mice, Neuroscience 215: , 79–89. |
[198] | Ohnishi T , Yanazawa M , Sasahara T , Kitamura Y , Hiroaki H , Fukazawa Y , Kii I , Nishiyama T , Kakita A , Takeda H , Takeuchi A , Arai Y , Ito A , Komura H , Hirao H , Satomura K , Inoue M , Muramatsu S , Matsui K , Tada M , Sato M , Saijo E , Shigemitsu Y , Sakai S , Umetsu Y , Goda N , Takino N , Takahashi H , Hagiwara M , Sawasaki T , Iwasaki G , Nakamura Y , Nabeshima Y , Teplow DB , Hoshi M ((2015) ) Na, K-ATPase alpha3 is a death target of Alzheimer patient amyloid-beta assembly, Proc Natl Acad Sci U S A 112: , E4465–4474. |
[199] | DiChiara T , DiNunno N , Clark J , Bu RL , Cline EN , Rollins MG , Gong Y , Brody DL , Sligar SG , Velasco PT , Viola KL , Klein WL ((2017) ) Alzheimer’s toxic amyloid beta oligomers: Unwelcome visitors to the Na/K ATPase alpha3 docking station, Yale J Biol Med 90: , 45–61. |
[200] | Viola KL , Klein WL ((2015) ) Amyloid beta oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis, Acta Neuropathol 129: , 183–206. |
[201] | Jarosz-Griffiths HH , Noble E , Rushworth JV , Hooper NM ((2016) ) Amyloid-beta receptors: The good, the bad, and the prion protein, J Biol Chem 291: , 3174–3183. |
[202] | De Strooper B , Karran E ((2016) ) The cellular phase of Alzheimer’s disease, Cell 164: , 603–615. |
[203] | Izzo NJ , Staniszewski A , To L , Fa M , Teich AF , Saeed F , Wostein H , Walko T 3rd , Vaswani A , Wardius M , Syed Z , Ravenscroft J , Mozzoni K , Silky C , Rehak C , Yurko R , Finn P , Look G , Rishton G , Safferstein H , Miller M , Johanson C , Stopa E , Windisch M , Hutter-Paier B , Shamloo M , Arancio O , LeVine H 3rd , Catalano SM ((2014) ) Alzheimer’s therapeutics targeting amyloid beta 1-42 oligomers I: Abeta 42 oligomer binding to specific neuronal receptors is displaced by drug candidates that improve cognitive deficits, PLoS One 9: , e111898. |
[204] | Izzo NJ , Xu J , Zeng C , Kirk MJ , Mozzoni K , Silky C , Rehak C , Yurko R , Look G , Rishton G , Safferstein H , Cruchaga C , Goate A , Cahill MA , Arancio O , Mach RH , Craven R , Head E , LeVine H 3rd , Spires-Jones TL , Catalano SM ((2014) ) Alzheimer’s therapeutics targeting amyloid beta 1-42 oligomers II: Sigma-2/PGRMC1 receptors mediate Abeta 42 oligomer binding and synaptotoxicity, PLoS One 9: , e111899. |
[205] | Magdesian MH , Carvalho MM , Mendes FA , Saraiva LM , Juliano MA , Juliano L , Garcia-Abreu J , Ferreira ST ((2008) ) Amyloid-beta binds to the extracellular cysteine-rich domain of Frizzled and inhibits Wnt/beta-catenin signaling, J Biol Chem 283: , 9359–9368. |
[206] | Brito-Moreira J , Lourenco MV , Oliveira MM , Ribeiro FC , Ledo JH , Diniz LP , Vital JFS , Magdesian MH , Melo HM , Barros-Aragao F , de Souza JM , Alves-Leon SV , Gomes FCA , Clarke JR , Figueiredo CP , De Felice FG , Ferreira ST ((2017) ) Interaction of amyloid-beta (Abeta) oligomers with neurexin 2alpha and neuroligin 1 mediates synapse damage and memory loss in mice, J Biol Chem 292: , 7327–7337. |
[207] | Suzuki K , Aimi T , Ishihara T , Mizushima T ((2016) ) Identification of approved drugs that inhibit the binding of amyloid beta oligomers to ephrin type-B receptor 2, FEBS Open Bio 6: , 461–468. |
[208] | Fu AK , Hung KW , Huang H , Gu S , Shen Y , Cheng EY , Ip FC , Huang X , Fu WY , Ip NY ((2014) ) Blockade of EphA4 signaling ameliorates hippocampal synaptic dysfunctions in mouse models of Alzheimer’s disease, Proc Natl Acad Sci U S A 111: , 9959–9964. |
[209] | Vargas JY , Fuenzalida M , Inestrosa NC ((2014) ) In vivo activation of Wnt signaling pathway enhances cognitive function of adult mice and reverses cognitive deficits in an Alzheimer’s disease model, J Neurosci 34: , 2191–2202. |
[210] | Takamura A , Sato Y , Watabe D , Okamoto Y , Nakata T , Kawarabayashi T , Oddo S , Laferla FM , Shoji M , Matsubara E ((2012) ) Sortilin is required for toxic action of Abeta oligomers (AbetaOs): Extracellular AbetaOs trigger apoptosis, and intraneuronal AbetaOs impair degradation pathways, Life Sci 91: , 1177–1186. |
[211] | Dineley KT , Westerman M , Bui D , Bell K , Ashe KH , Sweatt JD ((2001) ) Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer’s disease, J Neurosci 21: , 4125–4133. |
[212] | Wang HY , Lee DH , D’Andrea MR , Peterson PA , Shank RP , Reitz AB ((2000) ) Beta-Amyloid(1-42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology, J Biol Chem 275: , 5626–5632. |
[213] | Wang D , Govindaiah G , Liu R , De Arcangelis V , Cox CL , Xiang YK ((2010) ) Binding of amyloid beta peptide to beta2 adrenergic receptor induces PKA-dependent AMPA receptor hyperactivity, FASEB J 24: , 3511–3521. |
[214] | Patel AN , Jhamandas JH ((2012) ) Neuronal receptors as targets for the action of amyloid-beta protein (Abeta) in the brain, Expert Rev Mol Med 14: , e2. |
[215] | Arispe N , Pollard HB , Rojas E ((1996) ) Zn2+ interaction with Alzheimer amyloid beta protein calcium channels, Proc Natl Acad Sci U S A 93: , 1710–1715. |
[216] | Nimmrich V , Grimm C , Draguhn A , Barghorn S , Lehmann A , Schoemaker H , Hillen H , Gross G , Ebert U , Bruehl C ((2008) ) Amyloid beta oligomers (A beta(1-42) globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents, J Neurosci 28: , 788–797. |
[217] | Bobich JA , Zheng Q , Campbell A ((2004) ) Incubation of nerve endings with a physiological concentration of Abeta1-42 activates CaV2.2(N-Type)-voltage operated calcium channels and acutely increases glutamate and noradrenaline release, J Alzheimers Dis 6: , 243–255. |
[218] | Hu T , Wang S , Chen C , Sun J , Yang X ((2017) ) Real-time analysis of binding events between different Abeta1-42 species and human Lilrb2 by dual polarization interferometry, Anal Chem 89: , 2606–2612. |
[219] | Kim T , Vidal GS , Djurisic M , William CM , Birnbaum ME , Garcia KC , Hyman BT , Shatz CJ ((2013) ) Human LilrB2 is a beta-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model, Science 341: , 1399–1404. |
[220] | Heurtaux T , Michelucci A , Losciuto S , Gallotti C , Felten P , Dorban G , Grandbarbe L , Morga E , Heuschling P ((2010) ) Microglial activation depends on beta-amyloid conformation: Role of the formylpeptide receptor 2, J Neurochem 114: , 576–586. |
[221] | Kam TI , Song S , Gwon Y , Park H , Yan JJ , Im I , Choi JW , Choi TY , Kim J , Song DK , Takai T , Kim YC , Kim KS , Choi SY , Choi S , Klein WL , Yuan J , Jung YK ((2013) ) FcgammaRIIb mediates amyloid-beta neurotoxicity and memory impairment in Alzheimer’s disease, J Clin Invest 123: , 2791–2802. |
[222] | Ostapchenko VG , Chen M , Guzman MS , Xie YF , Lavine N , Fan J , Beraldo FH , Martyn AC , Belrose JC , Mori Y , MacDonald JF , Prado VF , Prado MA , Jackson MF ((2015) ) The transient receptor potential melastatin 2 (TRPM2) channel contributes to beta-amyloid oligomer-related neurotoxicity and memory impairment, J Neurosci 35: , 15157–15169. |
[223] | Zhao WQ , Santini F , Breese R , Ross D , Zhang XD , Stone DJ , Ferrer M , Townsend M , Wolfe AL , Seager MA , Kinney GG , Shughrue PJ , Ray WJ ((2010) ) Inhibition of calcineurin-mediated endocytosis and alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors prevents amyloid beta oligomer-induced synaptic disruption, J Biol Chem 285: , 7619–7632. |
[224] | Wills J , Credle J , Haggerty T , Lee JH , Oaks AW , Sidhu A ((2011) ) Tauopathic changes in the striatum of A53T alpha-synuclein mutant mouse model of Parkinson’s disease, PLoS One 6: , e17953. |
[225] | Vieira MN , Forny-Germano L , Saraiva LM , Sebollela A , Martinez AM , Houzel JC , De Felice FG , Ferreira ST ((2007) ) Soluble oligomers from a non-disease related protein mimic Abeta-induced tau hyperphosphorylation and neurodegeneration, J Neurochem 103: , 736–748. |
[226] | Zhang C , Qiu HE , Krafft GA , Klein WL ((1996) ) A beta peptide enhances focal adhesion kinase/Fyn association in a rat CNS nerve cell line, Neurosci Lett 211: , 187–190. |
[227] | Nygaard HB , van Dyck CH , Strittmatter SM ((2014) ) Fyn kinase inhibition as a novel therapy for Alzheimer’s disease, Alzheimers Res Ther 6: , 8. |
[228] | Folch J , Petrov D , Ettcheto M , Abad S , Sanchez-Lopez E , Garcia ML , Olloquequi J , Beas-Zarate C , Auladell C , Camins A ((2016) ) Current research therapeutic strategies for Alzheimer’s disease treatment, Neural Plast 2016: , 8501693. |
[229] | Lee G , Thangavel R , Sharma VM , Litersky JM , Bhaskar K , Fang SM , Do LH , Andreadis A , Van Hoesen G , Ksiezak-Reding H ((2004) ) Phosphorylation of tau by fyn: Implications for Alzheimer’s disease, J Neurosci 24: , 2304–2312. |
[230] | Beraldo FH , Ostapchenko VG , Caetano FA , Guimaraes AL , Ferretti GD , Daude N , Bertram L , Nogueira KO , Silva JL , Westaway D , Cashman NR , Martins VR , Prado VF , Prado MA ((2016) ) Regulation of amyloid beta oligomer binding to neurons and neurotoxicity by the prion protein-mGluR5 complex, J Biol Chem 291: , 21945–21955. |
[231] | Lambert JC , Ibrahim-Verbaas CA , Harold D , Naj AC , Sims R , Bellenguez C , DeStafano AL , Bis JC , Beecham GW , Grenier-Boley B , Russo G , Thorton-Wells TA , Jones N , Smith AV , Chouraki V , Thomas C , Ikram MA , Zelenika D , Vardarajan BN , Kamatani Y , Lin CF , Gerrish A , Schmidt H , Kunkle B , Dunstan ML , Ruiz A , Bihoreau MT , Choi SH , Reitz C , Pasquier F , Cruchaga C , Craig D , Amin N , Berr C , Lopez OL , De Jager PL , Deramecourt V , Johnston JA , Evans D , Lovestone S , Letenneur L , Moron FJ , Rubinsztein DC , Eiriksdottir G , Sleegers K , Goate AM , Fievet N , Huentelman MW , Gill M , Brown K , Kamboh MI , Keller L , Barberger-Gateau P , McGuiness B , Larson EB , Green R , Myers AJ , Dufouil C , Todd S , Wallon D , Love S , Rogaeva E , Gallacher J , St George-Hyslop P , Clarimon J , Lleo A , Bayer A , Tsuang DW , Yu L , Tsolaki M , Bossu P , Spalletta G , Proitsi P , Collinge J , Sorbi S , Sanchez-Garcia F , Fox NC , Hardy J , Deniz Naranjo MC , Bosco P , Clarke R , Brayne C , Galimberti D , Mancuso M , Matthews F , Moebus S , Mecocci P , Del Zompo M , Maier W , Hampel H , Pilotto A , Bullido M , Panza F , Caffarra P , Nacmias B , Gilbert JR , Mayhaus M , Lannefelt L , Hakonarson H , Pichler S , Carrasquillo MM , Ingelsson M , Beekly D , Alvarez V , Zou F , Valladares O , Younkin SG , Coto E , Hamilton-Nelson KL , Gu W , Razquin C , Pastor P , Mateo I , Owen MJ , Faber KM , Jonsson PV , Combarros O , O’Donovan MC , Cantwell LB , Soininen H , Blacker D , Mead S , Mosley TH Jr , Bennett DA , Harris TB , Fratiglioni L , Holmes C , de Bruijn RF , Passmore P , Montine TJ , Bettens K , Rotter JI , Brice A , Morgan K , Foroud TM , Kukull WA , Hannequin D , Powell JF , Nalls MA , Ritchie K , Lunetta KL , Kauwe JS , Boerwinkle E , Riemenschneider M , Boada M , Hiltuenen M , Martin ER , Schmidt R , Rujescu D , Wang LS , Dartigues JF , Mayeux R , Tzourio C , Hofman A , Nothen MM , Graff C , Psaty BM , Jones L , Haines JL , Holmans PA , Lathrop M , Pericak-Vance MA , Launer LJ , Farrer LA , van Duijn CM , Van Broeckhoven C , Moskvina V , Seshadri S , Williams J , Schellenberg GD , Amouyel P ((2013) ) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease, Nat Genet 45: , 1452–1458. |
[232] | Huang Y , Lu W , Ali DW , Pelkey KA , Pitcher GM , Lu YM , Aoto H , Roder JC , Sasaki T , Salter MW , MacDonald JF ((2001) ) CAKbeta/Pyk2 kinase is a signaling link for induction of long-term potentiation in CA1 hippocampus, Neuron 29: , 485–496. |
[233] | Hsin H , Kim MJ , Wang CF , Sheng M ((2010) ) Proline-rich tyrosine kinase 2 regulates hippocampal long-term depression, J Neurosci 30: , 11983–11993. |
[234] | Bartos JA , Ulrich JD , Li H , Beazely MA , Chen Y , Macdonald JF , Hell JW ((2010) ) Postsynaptic clustering and activation of Pyk2 by PSD-95, J Neurosci 30: , 449–463. |
[235] | Giralt A , Brito V , Chevy Q , Simonnet C , Otsu Y , Cifuentes-Diaz C , de Pins B , Coura R , Alberch J , Gines S , Poncer JC , Girault JA ((2017) ) Pyk2 modulates hippocampal excitatory synapses and contributes to cognitive deficits in a Huntington’s disease model, Nat Commun 8: , 15592. |
[236] | Andreev J , Galisteo ML , Kranenburg O , Logan SK , Chiu ES , Okigaki M , Cary LA , Moolenaar WH , Schlessinger J ((2001) ) Src and Pyk2 mediate G-protein-coupled receptor activation of epidermal growth factor receptor (EGFR) but are not required for coupling to the mitogen-activated protein (MAP) kinase signaling cascade, J Biol Chem 276: , 20130–20135. |
[237] | Dikic I , Tokiwa G , Lev S , Courtneidge SA , Schlessinger J ((1996) ) A role for Pyk2 and Src in linking G-protein-coupled receptors with MAP kinase activation, Nature 383: , 547–550. |
[238] | Qian D , Lev S , van Oers NS , Dikic I , Schlessinger J , Weiss A ((1997) ) Tyrosine phosphorylation of Pyk2 is selectively regulated by Fyn during TCR signaling, J Exp Med 185: , 1253–1259. |
[239] | Collins M , Bartelt RR , Houtman JC ((2010) ) T cell receptor activation leads to two distinct phases of Pyk2 activation and actin cytoskeletal rearrangement in human T cells, Mol Immunol 47: , 1665–1674. |
[240] | Collins M , Tremblay M , Chapman N , Curtiss M , Rothman PB , Houtman JC ((2010) ) The T cell receptor-mediated phosphorylation of Pyk2 tyrosines 402 and 580 occurs via a distinct mechanism than other receptor systems, J Leukoc Biol 87: , 691–701. |
[241] | Park SY , Avraham HK , Avraham S ((2004) ) RAFTK/Pyk2 activation is mediated by trans-acting autophosphorylation in a Src-independent manner, J Biol Chem 279: , 33315–33322. |
[242] | Reinhard L , Tidow H , Clausen MJ , Nissen P ((2013) ) Na(+),K (+)-ATPase as a docking station: Protein-protein complexes of the Na(+),K (+)-ATPase, Cell Mol Life Sci 70: , 205–222. |
[243] | Bayburt TH , Sligar SG ((2010) ) Membrane protein assembly into Nanodiscs, FEBS Lett 584: , 1721–1727. |
[244] | Xie Z , Cai T ((2003) ) Na+-K+–ATPase-mediated signal transduction: From protein interaction to cellular function, Mol Interv 3: , 157–168. |
[245] | Akkuratov EE , Lopacheva OM , Kruusmagi M , Lopachev AV , Shah ZA , Boldyrev AA , Liu L ((2015) ) Functional interaction between Na/K-ATPase and NMDA receptor in cerebellar neurons, Mol Neurobiol 52: , 1726–1734. |
[246] | Schuler MA , Denisov IG , Sligar SG ((2013) ) Nanodiscs as a new tool to examine lipid-protein interactions, Methods Mol Biol 974: , 415–433. |
[247] | Lee M , Guo JP , Schwab C , McGeer EG , McGeer PL ((2012) ) Selective inhibition of the membrane attack complex of complement by low molecular weight components of the aurin tricarboxylic acid synthetic complex, Neurobiol Aging 33: , 2237–2246. |
[248] | Lourenco MV , Clarke JR , Frozza RL , Bomfim TR , Forny-Germano L , Batista AF , Sathler LB , Brito-Moreira J , Amaral OB , Silva CA , Freitas-Correa L , Espirito-Santo S , Campello-Costa P , Houzel JC , Klein WL , Holscher C , Carvalheira JB , Silva AM , Velloso LA , Munoz DP , Ferreira ST , De Felice FG ((2013) ) TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys, Cell Metab 18: , 831–843. |
[249] | Costa RO , Lacor PN , Ferreira IL , Resende R , Auberson YP , Klein WL , Oliveira CR , Rego AC , Pereira CM ((2012) ) Endoplasmic reticulum stress occurs downstream of GluN2B subunit of N-methyl-d-aspartate receptor in mature hippocampal cultures treated with amyloid-beta oligomers, Aging Cell 11: , 823–833. |
[250] | Ferreira IL , Ferreiro E , Schmidt J , Cardoso JM , Pereira CM , Carvalho AL , Oliveira CR , Rego AC ((2015) ) Abeta and NMDAR activation cause mitochondrial dysfunction involving ER calcium release, Neurobiol Aging 36: , 680–692. |
[251] | Alberdi E , Wyssenbach A , Alberdi M , Sanchez-Gomez MV , Cavaliere F , Rodriguez JJ , Verkhratsky A , Matute C ((2013) ) Ca(2+)-dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid beta-treated astrocytes and in a model of Alzheimer’s disease, Aging Cell 12: , 292–302. |
[252] | De Felice FG , Lourenco MV ((2015) ) Brain metabolic stress and neuroinflammation at the basis of cognitive impairment in Alzheimer’s disease, Front Aging Neurosci 7: , 94. |
[253] | Morkuniene R , Cizas P , Jankeviciute S , Petrolis R , Arandarcikaite O , Krisciukaitis A , Borutaite V ((2015) ) Small Abeta1-42 oligomer-induced membrane depolarization of neuronal and microglial cells: Role of N-methyl-D-aspartate receptors, J Neurosci Res 93: , 475–486. |
[254] | Kim J , Yang Y , Song SS , Na JH , Oh KJ , Jeong C , Yu YG , Shin YK ((2014) ) Beta-amyloid oligomers activate apoptotic BAK pore for cytochrome c release, Biophys J 107: , 1601–1608. |
[255] | Manczak M , Reddy PH ((2012) ) Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau causes mitochondrial dysfunction in Alzheimer’s disease, Hum Mol Genet 21: , 5131–5146. |
[256] | Arrázola MS , Silva-Alvarez C , Inestrosa NC ((2015) ) How the Wnt signaling pathway protects from neurodegeneration: The mitochondrial scenario, Front Cell Neurosci 9: , 166. |
[257] | Han XJ , Hu YY , Yang ZJ , Jiang LP , Shi SL , Li YR , Guo MY , Wu HL , Wan YY ((2017) ) Amyloid beta-42 induces neuronal apoptosis by targeting mitochondria, Mol Med Rep 16: , 4521–4528. |
[258] | Chang PK , Boridy S , McKinney RA , Maysinger D ((2013) ) Letrozole potentiates mitochondrial and dendritic spine impairments induced by beta amyloid, J Aging Res 2013: , 538979. |
[259] | Kim B , Park J , Chang KT , Lee DS ((2016) ) Peroxiredoxin 5 prevents amyloid-beta oligomer-induced neuronal cell death by inhibiting ERK-Drp1-mediated mitochondrial fragmentation, Free Radic Biol Med 90: , 184–194. |
[260] | Rui Y , Zheng JQ ((2016) ) Amyloid beta oligomers elicit mitochondrial transport defects and fragmentation in a time-dependent and pathway-specific manner, Mol Brain 9: , 79. |
[261] | Mastroeni D , Nolz J , Khdour OM , Sekar S , Delvaux E , Cuyugan L , Liang WS , Hecht SM , Coleman PD ((2018) ) Oligomeric amyloid β preferentially targets neuronal and not glial mitochondrial-encoded mRNAs, Alzheimers Dement, doi: 10.1016/j.jalz.2017.12.005 |
[262] | Orr ME , Oddo S ((2013) ) Autophagic/lysosomal dysfunction in Alzheimer’s disease, Alzheimers Res Therapy 5: , 53–53. |
[263] | Onyenwoke RU , Brenman JE ((2015) ) Lysosomal storage diseases—regulating neurodegeneration, J Exp Neurosci 9: , 81–91. |
[264] | Tammineni P , Ye X , Feng T , Aikal D , Cai Q ((2017) ) Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons, Elife 6: , e21776. |
[265] | Guglielmotto M , Monteleone D , Piras A , Valsecchi V , Tropiano M , Ariano S , Fornaro M , Vercelli A , Puyal J , Arancio O , Tabaton M , Tamagno E ((2014) ) Abeta1-42 monomers or oligomers have different effects on autophagy and apoptosis, Autophagy 10: , 1827–1843. |
[266] | Robak LA , Jansen IE , van Rooij J , Uitterlinden AG , Kraaij R , Jankovic J , Heutink P , Shulman JM ((2017) ) Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease, Brain 140: , 3191–3203. |
[267] | Umeda T , Ono K , Sakai A , Yamashita M , Mizuguchi M , Klein WL , Yamada M , Mori H , Tomiyama T ((2016) ) Rifampicin is a candidate preventive medicine against amyloid-beta and tau oligomers, Brain 139: , 1568–1586. |
[268] | Yang WN , Ma KG , Chen XL , Shi LL , Bu G , Hu XD , Han H , Liu Y , Qian YH ((2014) ) Mitogen-activated protein kinase signaling pathways are involved in regulating alpha7 nicotinic acetylcholine receptor-mediated amyloid-beta uptake in SH-SY5Y cells, Neuroscience 278: , 276–290. |
[269] | Zheng L , Calvo-Garrido J , Hallbeck M , Hultenby K , Marcusson J , Cedazo-Minguez A , Terman A ((2013) ) Intracellular localization of amyloid-beta peptide in SH-SY5Y neuroblastoma cells, J Alzheimers Dis 37: , 713–733. |
[270] | Kaminsky YG , Tikhonova LA , Kosenko EA ((2015) ) Critical analysis of Alzheimer’s amyloid-beta toxicity to mitochondria, Front Biosci (Landmark Ed) 20: , 173–197. |
[271] | Esbjorner EK , Chan F , Rees E , Erdelyi M , Luheshi LM , Bertoncini CW , Kaminski CF , Dobson CM , Kaminski Schierle GS ((2014) ) Direct observations of amyloid beta self-assembly in live cells provide insights into differences in the kinetics of Abeta(1-40) and Abeta(1-42) aggregation, Chem Biol 21: , 732–742. |
[272] | Wyssenbach A , Quintela T , Llavero F , Zugaza JL , Matute C , Alberdi E ((2016) ) Amyloid beta-induced astrogliosis is mediated by beta1-integrin via NADPH oxidase 2 in Alzheimer’s disease, Aging Cell 15: , 1140–1152. |
[273] | Diniz LP , Tortelli V , Matias I , Morgado J , Bergamo Araujo AP , Melo HM , Seixas da Silva GS , Alves-Leon SV , de Souza JM , Ferreira ST , De Felice FG , Gomes FCA ((2017) ) Astrocyte transforming growth factor beta 1 protects synapses against Abeta oligomers in Alzheimer’s disease model, J Neurosci 37: , 6797–6809. |
[274] | Han X , Yang L , Du H , Sun Q , Wang X , Cong L , Liu X , Yin L , Li S , Du Y ((2016) ) Insulin attenuates beta-amyloid-associated insulin/Akt/EAAT signaling perturbations in human astrocytes, Cell Mol Neurobiol 36: , 851–864. |
[275] | Huang S , Tong H , Lei M , Zhou M , Guo W , Li G , Tang X , Li Z , Mo M , Zhang X , Chen X , Cen L , Wei L , Xiao Y , Li K , Huang Q , Yang X , Liu W , Zhang L , Qu S , Li S , Xu P ((2018) ) Astrocytic glutamatergic transporters are involved in Abeta-induced synaptic dysfunction, Brain Res 1678: , 129–137. |
[276] | Narayan P , Holmstrom KM , Kim DH , Whitcomb DJ , Wilson MR , St George-Hyslop P , Wood NW , Dobson CM , Cho K , Abramov AY , Klenerman D ((2014) ) Rare individual amyloid-beta oligomers act on astrocytes to initiate neuronal damage, Biochemistry 53: , 2442–2453. |
[277] | Dal Pra I , Chiarini A , Pacchiana R , Gardenal E , Chakravarthy B , Whitfield JF , Armato U ((2014) ) Calcium-sensing receptors of human astrocyte-neuron teams: Amyloid-beta-driven mediators and therapeutic targets of Alzheimer’s disease, Curr Neuropharmacol 12: , 353–364. |
[278] | DeWitt DA , Perry G , Cohen M , Doller C , Silver J ((1998) ) Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer’s disease, Exp Neurol 149: , 329–340. |
[279] | Jimenez S , Baglietto-Vargas D , Caballero C , Moreno-Gonzalez I , Torres M , Sanchez-Varo R , Ruano D , Vizuete M , Gutierrez A , Vitorica J ((2008) ) Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: Age-dependent switch in the microglial phenotype from alternative to classic, JNeurosci 28: , 11650–11661. |
[280] | Lee S , Varvel NH , Konerth ME , Xu G , Cardona AE , Ransohoff RM , Lamb BT ((2010) ) CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models, Am J Pathol 177: , 2549–2562. |
[281] | Spangenberg EE , Lee RJ , Najafi AR , Rice RA , Elmore MR , Blurton-Jones M , West BL , Green KN ((2016) ) Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology, Brain 139: , 1265–1281. |
[282] | El-Shimy IA , Heikal OA , Hamdi N ((2015) ) Minocycline attenuates Abeta oligomers-induced pro-inflammatory phenotype in primary microglia while enhancing Abeta fibrils phagocytosis, Neurosci Lett 609: , 36–41. |
[283] | Ledo JH , Azevedo EP , Beckman D , Ribeiro FC , Santos LE , Razolli DS , Kincheski GC , Melo HM , Bellio M , Teixeira AL , Velloso LA , Foguel D , De Felice FG , Ferreira ST ((2016) ) Cross talk between brain innate immunity and serotonin signaling underlies depressive-like behavior induced by Alzheimer’s amyloid-beta oligomers in mice, J Neurosci 36: , 12106–12116. |
[284] | Venegas C , Kumar S , Franklin BS , Dierkes T , Brinkschulte R , Tejera D , Vieira-Saecker A , Schwartz S , Santarelli F , Kummer MP , Griep A , Gelpi E , Beilharz M , Riedel D , Golenbock DT , Geyer M , Walter J , Latz E , Heneka MT ((2017) ) Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease, Nature 552: , 355–361. |
[285] | Schafer Dorothy P , Lehrman Emily K , Kautzman Amanda G , Koyama R , Mardinly Alan R , Yamasaki R , Ransohoff Richard M , Greenberg Michael E , Barres Ben A , Stevens B ((2012) ) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner, Neuron 74: , 691–705. |
[286] | Bloom GS ((2014) ) Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis, JAMA Neurol 71: , 505–508. |
[287] | Nisbet RM , Polanco JC , Ittner LM , Götz J ((2015) ) Tau aggregation and its interplay with amyloid-β, Acta Neuropathol 129: , 207–220. |
[288] | Spires-Jones TL , Attems J , Thal DR ((2017) ) Interactions of pathological proteins in neurodegenerative diseases, Acta Neuropathol 134: , 187–205. |
[289] | Guerrero-Munoz MJ , Gerson J , Castillo-Carranza DL ((2015) ) Tau oligomers: The toxic player at synapses in Alzheimer’s disease, Front Cell Neurosci 9: , 464. |
[290] | Ittner LM , Gotz J ((2011) ) Amyloid-beta and tau–a toxic pas de deux in Alzheimer’s disease, Nat Rev Neurosci 12: , 65–72. |
[291] | Zempel H , Luedtke J , Kumar Y , Biernat J , Dawson H , Mandelkow E , Mandelkow EM ((2013) ) Amyloid-beta oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin, EMBO J 32: , 2920–2937. |
[292] | Frandemiche ML , De Seranno S , Rush T , Borel E , Elie A , Arnal I , Lante F , Buisson A ((2014) ) Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers, J Neurosci 34: , 6084–6097. |
[293] | Qu X , Yuan FN , Corona C , Pasini S , Pero ME , Gundersen GG , Shelanski ML , Bartolini F ((2017) ) Stabilization of dynamic microtubules by mDia1 drives Tau-dependent Abeta1-42 synaptotoxicity, J Cell Biol 216: , 3161–3178. |
[294] | Lasagna-Reeves CA , Castillo-Carranza DL , Guerrero-Muoz MJ , Jackson GR , Kayed R ((2010) ) Preparation and characterization of neurotoxic tau oligomers, Biochemistry 49: , 10039–10041. |
[295] | Bilousova T , Miller CA , Poon WW , Vinters HV , Corrada M , Kawas C , Hayden EY , Teplow DB , Glabe C , Albay R 3rd , Cole GM , Teng E , Gylys KH ((2016) ) Synaptic amyloid-beta oligomers precede p-Tau and differentiate high pathology control cases, Am J Pathol 186: , 185–198. |
[296] | Pooler AM , Polydoro M , Maury EA , Nicholls SB , Reddy SM , Wegmann S , William C , Saqran L , Cagsal-Getkin O , Pitstick R , Beier DR , Carlson GA , Spires-Jones TL , Hyman BT ((2015) ) Amyloid accelerates tau propagation and toxicity in a model of early Alzheimer’s disease, Acta Neuropathol Commun 3: , 14. |
[297] | Pooler AM , Polydoro M , Wegmann S , Nicholls SB , Spires-Jones TL , Hyman BT ((2013) ) Propagation of tau pathology in Alzheimer’s disease: Identification of novel therapeutic targets, Alzheimers Res Therapy 5: , 49–49. |
[298] | Saul A , Sprenger F , Bayer TA , Wirths O ((2013) ) Accelerated tau pathology with synaptic and neuronal loss in a novel triple transgenic mouse model of Alzheimer’s disease, Neurobiol Aging 34: , 2564–2573. |
[299] | Heraud C , Goufak D , Ando K , Leroy K , Suain V , Yilmaz Z , De Decker R , Authelet M , Laporte V , Octave JN , Brion JP ((2014) ) Increased misfolding and truncation of tau in APP/PS1/tau transgenic mice compared to mutant tau mice, Neurobiol Dis 62: , 100–112. |
[300] | Umeda T , Maekawa S , Kimura T , Takashima A , Tomiyama T , Mori H ((2014) ) Neurofibrillary tangle formation by introducing wild-type human tau into APP transgenic mice, Acta Neuropathol 127: , 685–698. |
[301] | Chabrier MA , Cheng D , Castello NA , Green KN , LaFerla FM ((2014) ) Synergistic effects of amyloid-beta and wild-type human tau on dendritic spine loss in a floxed double transgenic model of Alzheimer’s disease, Neurobiol Dis 64: , 107–117. |
[302] | Guo Q , Li H , Cole AL , Hur JY , Li Y , Zheng H ((2013) ) Modeling Alzheimer’s disease in mouse without mutant protein overexpression: Cooperative and independent effects of Abeta and tau, PLoS One 8: , e80706. |
[303] | Yetman MJ , Fowler SW , Jankowsky JL ((2016) ) Humanized tau mice with regionalized amyloid exhibit behavioral deficits but no pathological interaction, PLoS One 11: , e0153724. |
[304] | Rasmussen J , Jucker M , Walker LC ((2017) ) Abeta seeds and prions: How close the fit? Prion 11: , 215–225. |
[305] | Espargaro A , Busquets MA , Estelrich J , Sabate R ((2016) ) Key Points Concerning Amyloid Infectivity and Prion-Like Neuronal Invasion, Front Mol Neurosci 9: , 29. |
[306] | Domert J , Rao SB , Agholme L , Brorsson AC , Marcusson J , Hallbeck M , Nath S ((2014) ) Spreading of amyloid-beta peptides via neuritic cell-to-cell transfer is dependent on insufficient cellular clearance, Neurobiol Dis 65: , 82–92. |
[307] | Jarrett JT , Lansbury PT Jr ((1993) ) Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 73: , 1055–1058. |
[308] | Marzesco AM , Flotenmeyer M , Buhler A , Obermuller U , Staufenbiel M , Jucker M , Baumann F ((2016) ) Highly potent intracellular membrane-associated Abeta seeds, Sci Rep 6: , 28125. |
[309] | Ridley RM , Baker HF , Windle CP , Cummings RM ((2006) ) Very long term studies of the seeding of beta-amyloidosis in primates, J Neural Transm (Vienna) 113: , 1243–1251. |
[310] | Ye L , Fritschi SK , Schelle J , Obermuller U , Degenhardt K , Kaeser SA , Eisele YS , Walker LC , Baumann F , Staufenbiel M , Jucker M ((2015) ) Persistence of Abeta seeds in APP null mouse brain, Nat Neurosci 18: , 1559–1561. |
[311] | Vella LJ , Hill AF , Cheng L ((2016) ) Focus on extracellular vesicles: Exosomes and their role in protein trafficking and biomarker potential in Alzheimer’s and Parkinson’s disease, Int J Mol Sci 17: , 173. |
[312] | Ye L , Hamaguchi T , Fritschi SK , Eisele YS , Obermuller U , Jucker M , Walker LC ((2015) ) Progression of seed-induced Abeta deposition within the limbic connectome, Brain Pathol 25: , 743–752. |
[313] | Iturria-Medina Y , Sotero RC , Toussaint PJ , Evans AC ((2014) ) Epidemic spreading model to characterize misfolded proteins propagation in aging and associated neurodegenerative disorders, PLoS Comput Biol 10: , e1003956. |
[314] | Ciryam P , Kundra R , Morimoto RI , Dobson CM , Vendruscolo M ((2015) ) Supersaturation is a major driving force for protein aggregation in neurodegenerative diseases, Trends Pharmacol Sci 36: , 72–77. |
[315] | Luheshi LM , Tartaglia GG , Brorsson AC , Pawar AP , Watson IE , Chiti F , Vendruscolo M , Lomas DA , Dobson CM , Crowther DC ((2007) ) Systematic in vivo analysis of the intrinsic determinants of amyloid Beta pathogenicity, PLoS Biol 5: , e290. |
[316] | Ciryam P , Tartaglia GG , Morimoto RI , Dobson CM , Vendruscolo M ((2013) ) Widespread aggregation and neurodegenerative diseases are associated with supersaturated proteins, Cell Rep 5: , 781–790. |
[317] | Kundra R , Ciryam P , Morimoto RI , Dobson CM , Vendruscolo M ((2017) ) Protein homeostasis of a metastable subproteome associated with Alzheimer’s disease, Proc Natl Acad Sci U S A 114: , E5703–e5711. |
[318] | Trumbore CN ((2016) ) Shear-induced amyloid formation in the brain: I. Potential vascular and parenchymal processes. J Alzheimers Dis 54: , 457–470 . |
[319] | Roh JH , Huang Y , Bero AW , Kasten T , Stewart FR , Bateman RJ , Holtzman DM ((2012) ) Disruption of the sleep-wake cycle and diurnal fluctuation of beta-amyloid in mice with Alzheimer’s disease pathology, Sci Transl Med 4: , 150ra122. |
[320] | Kress BT , Iliff JJ , Xia M , Wang M , Wei HS , Zeppenfeld D , Xie L , Kang H , Xu Q , Liew JA , Plog BA , Ding F , Deane R , Nedergaard M ((2014) ) Impairment of paravascular clearance pathways in the aging brain, Ann Neurol 76: , 845–861. |
[321] | Peng W , Achariyar TM , Li B , Liao Y , Mestre H , Hitomi E , Regan S , Kasper T , Peng S , Ding F , Benveniste H , Nedergaard M , Deane R ((2016) ) Suppression of glymphatic fluid transport in a mouse model of Alzheimer’s disease, Neurobiol Dis 93: , 215–225. |
[322] | De Felice FG ((2013) ) Alzheimer’s disease and insulin resistance: Translating basic science into clinical applications, J Clin Invest 123: , 531–539. |
[323] | Ikonomovic MD , Mi Z , Abrahamson EE ((2017) ) Disordered APP metabolism and neurovasculature in trauma and aging: Combined risks for chronic neurodegenerative disorders, Ageing Res Rev 34: , 51–63. |
[324] | Julien J , Joubert S , Ferland MC , Frenette LC , Boudreau-Duhaime MM , Malo-Veronneau L , de Guise E ((2017) ) Association of traumatic brain injury and Alzheimer disease onset: A systematic review, Ann Phys Rehabil Med 60: , 347–356. |
[325] | Johnson VE , Stewart W , Smith DH ((2012) ) Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans, Brain Pathol 22: , 142–149. |
[326] | Vincent AS , Roebuck-Spencer TM , Cernich A ((2014) ) Cognitive changes and dementia risk after traumatic brain injury: Implications for aging military personnel.S, Alzheimers Dement 10: , 174–187. |
[327] | Abu Hamdeh S , Waara ER , Moller C , Soderberg L , Basun H , Alafuzoff I , Hillered L , Lannfelt L , Ingelsson M , Marklund N ((2017) ) Rapid amyloid-beta oligomer and protofibril accumulation in traumatic brain injury, Brain Pathol. doi: 10.1111/bpa.12532 |
[328] | Gatson JW , Warren V , Abdelfattah K , Wolf S , Hynan LS , Moore C , Diaz-Arrastia R , Minei JP , Madden C , Wigginton JG ((2013) ) Detection of beta-amyloid oligomers as a predictor of neurological outcome after brain injury, JNeurosurg 118: , 1336–1342. |
[329] | Finnie JW , Manavis J , Blumbergs PC ((2010) ) Diffuse neuronal perikaryal amyloid precursor protein immunoreactivity in an ovine model of non-accidental head injury (the shaken baby syndrome), J Clin Neurosci 17: , 237–240. |
[330] | Gleckman AM , Evans RJ , Bell MD , Smith TW ((2000) ) Optic nerve damage in shaken baby syndrome: Detection by beta-amyloid precursor protein immunohistochemistry, Arch Pathol Lab Med 124: , 251–256. |
[331] | Gleckman AM , Bell MD , Evans RJ , Smith TW ((1999) ) Diffuse axonal injury in infants with nonaccidental craniocerebral trauma: Enhanced detection by beta-amyloid precursor protein immunohistochemical staining, Arch Pathol Lab Med 123: , 146–151. |
[332] | Tran HT , LaFerla FM , Holtzman DM , Brody DL ((2011) ) Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-beta accumulation and independently accelerates the development of tau abnormalities, J Neurosci 31: , 9513–9525. |
[333] | Washington PM , Morffy N , Parsadanian M , Zapple DN , Burns MP ((2014) ) Experimental traumatic brain injury induces rapid aggregation and oligomerization of amyloid-beta in an Alzheimer’s disease mouse model, JNeurotrauma 31: , 125–134. |
[334] | Brody DL , Holtzman DM ((2006) ) Morris water maze search strategy analysis in PDAPP mice before and after experimental traumatic brain injury, Exp Neurol 197: , 330–340. |
[335] | Rubenstein R , Chang B , Grinkina N , Drummond E , Davies P , Ruditzky M , Sharma D , Wang K , Wisniewski T ((2017) ) Tau phosphorylation induced by severe closed head traumatic brain injury is linked to the cellular prion protein, Acta Neuropathol Commun 5: , 30. |
[336] | Davis DA , Akopian G , Walsh JP , Sioutas C , Morgan TE , Finch CE ((2013) ) Urban air pollutants reduce synaptic function of CA1 neurons via an NMDA/NO pathway in vitro, J Neurochem 127: , 509–519. |
[337] | Davis DA , Bortolato M , Godar SC , Sander TK , Iwata N , Pakbin P , Shih JC , Berhane K , McConnell R , Sioutas C , Finch CE , Morgan TE ((2013) ) Prenatal exposure to urban air nanoparticles in mice causes altered neuronal differentiation and depression-like responses, PLoS One 8: , e64128. |
[338] | Cheng H , Davis DA , Hasheminassab S , Sioutas C , Morgan TE , Finch CE ((2016) ) Urban traffic-derived nanoparticulate matter reduces neurite outgrowth via TNFalpha in vitro, JNeuroinflammation 13: , 19. |
[339] | Zhang H , Liu H , Davies KJ , Sioutas C , Finch CE , Morgan TE , Forman HJ ((2012) ) Nrf2-regulated phase II enzymes are induced by chronic ambient nanoparticle exposure in young mice with age-related impairments, Free Radic Biol Med 52: , 2038–2046. |
[340] | Pistollato F , Sumalla Cano S , Elio I , Masias Vergara M , Giampieri F , Battino M ((2016) ) Associations between sleep, cortisol regulation, and diet: Possible implications for the risk of Alzheimer disease, Adv Nutr 7: , 679–689. |
[341] | Pedrini S , Thomas C , Brautigam H , Schmeidler J , Ho L , Fraser P , Westaway D , Hyslop PS , Martins RN , Buxbaum JD , Pasinetti GM , Dickstein DL , Hof PR , Ehrlich ME , Gandy S ((2009) ) Dietary composition modulates brain mass and solubilizable Abeta levels in a mouse model of aggressive Alzheimer’s amyloid pathology, Mol Neurodegener 4: , 40. |
[342] | Stranahan AM , Norman ED , Lee K , Cutler RG , Telljohann RS , Egan JM , Mattson MP ((2008) ) Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats, Hippocampus 18: , 1085–1088. |
[343] | Kincheski GC , Valentim IS , Clarke JR , Cozachenco D , Castelo-Branco MTL , Ramos-Lobo AM , Rumjanek V , Donato J Jr , De Felice FG , Ferreira ST ((2017) ) Chronic sleep restriction promotes brain inflammation and synapse loss, and potentiates memory impairment induced by amyloid-beta oligomers in mice, Brain Behav Immun 64: , 140–151. |
[344] | de la Monte SM ((2014) ) Type 3 diabetes is sporadic Alzheimers disease: Mini-review, Eur Neuropsychopharmacol 24: , 1954–1960. |
[345] | Kedia N , Almisry M , Bieschke J ((2017) ) Glucose directs amyloid-beta into membrane-active oligomers, Phys Chem Chem Phys 19: , 18036–18046. |
[346] | Bitel CL , Kasinathan C , Kaswala RH , Klein WL , Frederikse PH ((2012) ) Amyloid-beta and tau pathology of Alzheimer’s disease induced by diabetes in a rabbit animal model, J Alzheimers Dis 32: , 291–305. |
[347] | Li W , Risacher SL , Gao S , Boehm SL 2nd , Elmendorf JS , Saykin AJ ((2018) ) Type 2 diabetes mellitus and cerebrospinal fluid Alzheimer’s disease biomarker amyloid beta1-42 in Alzheimer’s Disease Neuroimaging Initiative participants, Alzheimers Dement (Amst) 10: , 94–98. |
[348] | Chakrabarti S , Khemka VK , Banerjee A , Chatterjee G , Ganguly A , Biswas A ((2015) ) Metabolic risk factors of sporadic Alzheimer’s disease: Implications in the pathology, pathogenesis and treatment, Aging Dis 6: , 282–299. |
[349] | Xue-Shan Z , Juan P , Qi W , Zhong R , Li-Hong P , Zhi-Han T , Zhi-Sheng J , Gui-Xue W , Lu-Shan L ((2016) ) Imbalanced cholesterol metabolism in Alzheimer’s disease, Clin Chim Acta 456: , 107–114. |
[350] | Umeda T , Tomiyama T , Kitajima E , Idomoto T , Nomura S , Lambert MP , Klein WL , Mori H ((2012) ) Hypercholesterolemia accelerates intraneuronal accumulation of Abeta oligomers resulting in memory impairment in Alzheimer’s disease model mice, Life Sci 91: , 1169–1176. |
[351] | Fitz NF , Cronican A , Pham T , Fogg A , Fauq AH , Chapman R , Lefterov I , Koldamova R ((2010) ) Liver X receptor agonist treatment ameliorates amyloid pathology and memory deficits caused by high-fat diet in APP23 mice, J Neurosci 30: , 6862–6872. |
[352] | Batarseh YS , Duong QV , Mousa YM , Al Rihani SB , Elfakhri K , Kaddoumi A ((2016) ) Amyloid-beta and astrocytes interplay in amyloid-beta related disorders, Int J Mol Sci 17: , 338. |
[353] | Nielsen HM , Mulder SD , Belien JA , Musters RJ , Eikelenboom P , Veerhuis R ((2010) ) Astrocytic A beta 1-42 uptake is determined by A beta-aggregation state and the presence of amyloid-associated proteins, Glia 58: , 1235–1246. |
[354] | Yan LJ , Xiao M , Chen R , Cai Z ((2013) ) Metabolic dysfunction of astrocyte: An initiating factor in beta-amyloid pathology? Aging Neurodegener 1: , 7–14. |
[355] | Perez JL , Carrero I , Gonzalo P , Arevalo-Serrano J , Sanz-Anquela JM , Ortega J , Rodriguez M , Gonzalo-Ruiz A ((2010) ) Soluble oligomeric forms of beta-amyloid (Abeta) peptide stimulate Abeta production via astrogliosis in the rat brain, Exp Neurol 223: , 410–421. |
[356] | Dal Pra I , Chiarini A , Gui L , Chakravarthy B , Pacchiana R , Gardenal E , Whitfield JF , Armato U ((2015) ) Do astrocytes collaborate with neurons in spreading the “infectious” abeta and Tau drivers of Alzheimer’s disease? Neuroscientist 21: , 9–29. |
[357] | De Felice FG , Lourenco MV , Ferreira ST ((2014) ) How does brain insulin resistance develop in Alzheimer’s disease? Alzheimers Dement 10: , S26–S32. |
[358] | Bomfim TR , Forny-Germano L , Sathler LB , Brito-Moreira J , Houzel JC , Decker H , Silverman MA , Kazi H , Melo HM , McClean PL , Holscher C , Arnold SE , Talbot K , Klein WL , Munoz DP , Ferreira ST , De Felice FG ((2012) ) An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease-associated Abeta oligomers, J Clin Invest 122: , 1339–1353. |
[359] | Tai J , Liu W , Li Y , Li L , Holscher C ((2018) ) Neuroprotective effects of a triple GLP-1/GIP/glucagon receptor agonist in the APP/PS1 transgenic mouse model of Alzheimer’s disease, Brain Res 1678: , 64–74. |
[360] | Jessen NA , Munk ASF , Lundgaard I , Nedergaard M ((2015) ) The glymphatic system: A beginner’s guide, Neurochem Res 40: , 2583–2599. |
[361] | Chen DW , Wang J , Zhang LL , Wang YJ , Gao CY ((2018) ) Cerebrospinal fluid amyloid-beta levels are increased in patients with insomnia, J Alzheimers Dis 61: , 645–651. |
[362] | Balusu S , Brkic M , Libert C , Vandenbroucke RE ((2016) ) The choroid plexus-cerebrospinal fluid interface in Alzheimer’s disease: More than just a barrier, Neural Regen Res 11: , 534–537. |
[363] | Brkic M , Balusu S , Van Wonterghem E , Gorle N , Benilova I , Kremer A , Van Hove I , Moons L , De Strooper B , Kanazir S , Libert C , Vandenbroucke RE ((2015) ) Amyloid beta oligomers disrupt blood-CSF barrier integrity by activating matrix metalloproteinases, J Neurosci 35: , 12766–12778. |
[364] | Oddo S , Caccamo A , Kitazawa M , Tseng BP , LaFerla FM ((2003) ) Amyloid deposition precedes tangle formation in a triple transgenic model of Alzheimer’s disease, Neurobiol Aging 24: , 1063–1070. |
[365] | Oddo S , Caccamo A , Shepherd JD , Murphy MP , Golde TE , Kayed R , Metherate R , Mattson MP , Akbari Y , LaFerla FM ((2003) ) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction, Neuron 39: , 409–421. |
[366] | Chabrier MA , Blurton-Jones M , Agazaryan AA , Nerhus JL , Martinez-Coria H , LaFerla FM ((2012) ) Soluble abeta promotes wild-type tau pathology in vivo, J Neurosci 32: , 17345–17350. |
[367] | Cohen RM , Rezai-Zadeh K , Weitz TM , Rentsendorj A , Gate D , Spivak I , Bholat Y , Vasilevko V , Glabe CG , Breunig JJ , Rakic P , Davtyan H , Agadjanyan MG , Kepe V , Barrio JR , Bannykh S , Szekely CA , Pechnick RN , Town T ((2013) ) A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric abeta, and frank neuronal loss, J Neurosci 33: , 6245–6256. |
[368] | Zahs KR , Ashe KH ((2010) ) ‘Too much good news’ - are Alzheimer mouse models trying to tell us how to prevent, not cure, Alzheimer’s disease? Trends Neurosci 33: , 381–389. |
[369] | Sasaguri H , Nilsson P , Hashimoto S , Nagata K , Saito T , De Strooper B , Hardy J , Vassar R , Winblad B , Saido TC ((2017) ) APP mouse models for Alzheimer’s disease preclinical studies, EMBO J 36: , 2473–2487. |
[370] | Podlisny MB , Tolan DR , Selkoe DJ ((1991) ) Homology of the amyloid beta protein precursor in monkey and human supports a primate model for beta amyloidosis in Alzheimer’s disease, Am J Pathol 138: , 1423–1435. |
[371] | Oikawa N , Kimura N , Yanagisawa K ((2010) ) Alzheimer-type tau pathology in advanced aged nonhuman primate brains harboring substantial amyloid deposition, Brain Res 1315: , 137–149. |
[372] | Schultz C , Hubbard GB , Rub U , Braak E , Braak H ((2000) ) Age-related progression of tau pathology in brains of baboons, Neurobiol Aging 21: , 905–912. |
[373] | Geula C , Wu CK , Saroff D , Lorenzo A , Yuan M , Yankner BA ((1998) ) Aging renders the brain vulnerable to amyloid beta-protein neurotoxicity, Nat Med 4: , 827–831. |
[374] | Leung E , Guo L , Bu J , Maloof M , El Khoury J , Geula C ((2011) ) Microglia activation mediates fibrillar amyloid-beta toxicity in the aged primate cortex, Neurobiol Aging 32: , 387–397. |
[375] | Boehnke SE , Wither RG , Nashed JY , Cook DJ , Levy R , De Felice FG , Munoz DP ((2017) ) Changes in cerebrospinal fluid and neuroimaging biomarkers in a non-human primate model of Alzheimer’s Disease, Society for Neuroscience, Washington, DC, Presentation 356.04. |
[376] | Kondo T , Asai M , Tsukita K , Kutoku Y , Ohsawa Y , Sunada Y , Imamura K , Egawa N , Yahata N , Okita K , Takahashi K , Asaka I , Aoi T , Watanabe A , Watanabe K , Kadoya C , Nakano R , Watanabe D , Maruyama K , Hori O , Hibino S , Choshi T , Nakahata T , Hioki H , Kaneko T , Naitoh M , Yoshikawa K , Yamawaki S , Suzuki S , Hata R , Ueno S , Seki T , Kobayashi K , Toda T , Murakami K , Irie K , Klein WL , Mori H , Asada T , Takahashi R , Iwata N , Yamanaka S , Inoue H ((2013) ) Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness, Cell Stem Cell 12: , 487–496. |
[377] | Ochalek A , Mihalik B , Avci HX , Chandrasekaran A , Teglasi A , Bock I , Giudice ML , Tancos Z , Molnar K , Laszlo L , Nielsen JE , Holst B , Freude K , Hyttel P , Kobolak J , Dinnyes A ((2017) ) Neurons derived from sporadic Alzheimer’s disease iPSCs reveal elevated TAU hyperphosphorylation, increased amyloid levels, and GSK3B activation, Alzheimers Res Ther 9: , 90. |
[378] | Kim YH , Choi SH , D’Avanzo C , Hebisch M , Sliwinski C , Bylykbashi E , Washicosky KJ , Klee JB , Brüstle O , Tanzi RE , Kim DY ((2015) ) A 3D human neural cell culture system for modeling Alzheimer’s disease, Nat Protoc 10: , 985–1006. |
[379] | Espuny-Camacho I , Arranz AM , Fiers M , Snellinx A , Ando K , Munck S , Bonnefont J , Lambot L , Corthout N , Omodho L , Vanden Eynden E , Radaelli E , Tesseur I , Wray S , Ebneth A , Hardy J , Leroy K , Brion JP , Vanderhaeghen P , De Strooper B ((2017) ) Hallmarks of Alzheimer’s disease in stem-cell-derived human neurons transplanted into mouse brain, Neuron 93: , 1066–1081.e1068. |
[380] | Castro-Fuentes R , Socas-Pérez R ((2013) ) Octodon degus: A strong attractor for Alzheimer research, Basic Clin Neurosci 4: , 91–96. |
[381] | Tarragon E , Lopez D , Estrada C , Ana GC , Schenker E , Pifferi F , Bordet R , Richardson JC , Herrero MT ((2013) ) Octodon degus: A model for the cognitive impairment associated with Alzheimer’s disease, CNS Neurosci Ther 19: , 643–648. |
[382] | Tsai Y , Lu B , Ljubimov AV , Girman S , Ross-Cisneros FN , Sadun AA , Svendsen CN , Cohen RM , Wang S ((2014) ) Ocular changes in TgF344-AD rat model of Alzheimer’s disease, Invest Ophthalmol Vis Sci 55: , 523–534. |
[383] | O’Hare E , Ardis T , Page D , Scopes DI , Kim EM ((2013) ) AbetaPP-overexpressing transgenic rat model of Alzheimer’s disease utilizing the Tg2576 mouse protocol, J Alzheimers Dis 37: , 77–88. |
[384] | Iulita MF , Allard S , Richter L , Munter LM , Ducatenzeiler A , Weise C , Do Carmo S , Klein WL , Multhaup G , Cuello AC ((2014) )Intracellular Abeta pathology and early cognitive impairments in a transgenic rat overexpressing human amyloid precursor protein: A multidimensional study. Acta Neuropathol Commun 2: , 61. |
[385] | Bouleau S , Tricoire H ((2015) ) Drosophila models of Alzheimer’s disease: Advances, limits, and perspectives, J Alzheimers Dis 45: , 1015–1038. |
[386] | Long DM , Blake MR , Dutta S , Holbrook SD , Kotwica-Rolinska J , Kretzschmar D , Giebultowicz JM ((2014) ) Relationships between the circadian system and Alzheimer’s disease-like symptoms in Drosophila, PLoS One 9: , e106068. |
[387] | Alexander AG , Marfil V , Li C ((2014) ) Use of Caenorhabditis elegans as a model to study Alzheimer’s disease and other neurodegenerative diseases, Front Genet 5: , 279. |
[388] | Diomede L , Cassata G , Fiordaliso F , Salio M , Ami D , Natalello A , Doglia SM , De Luigi A , Salmona M ((2010) ) Tetracycline and its analogues protect Caenorhabditis elegans from beta amyloid-induced toxicity by targeting oligomers, Neurobiol Dis 40: , 424–431. |
[389] | Wu Y , Cao Z , Klein WL , Luo Y ((2010) ) Heat shock treatment reduces beta amyloid toxicity in vivo by diminishing oligomers, Neurobiol Aging 31: , 1055–1058. |
[390] | Ryan TM , Roberts BR , McColl G , Hare DJ , Doble PA , Li QX , Lind M , Roberts AM , Mertens HD , Kirby N , Pham CL , Hinds MG , Adlard PA , Barnham KJ , Curtain CC , Masters CL ((2015) ) Stabilization of nontoxic Abeta-oligomers: Insights into the mechanism of action of hydroxyquinolines in Alzheimer’s disease, J Neurosci 35: , 2871–2884. |
[391] | McColl G , Roberts BR , Pukala TL , Kenche VB , Roberts CM , Link CD , Ryan TM , Masters CL , Barnham KJ , Bush AI , Cherny RA ((2012) ) Utility of an improved model of amyloid-beta (Abeta(1)(-)(4)(2)) toxicity in Caenorhabditis elegans for drug screening for Alzheimer’s disease, Mol Neurodegener 7: , 57. |
[392] | Overk CR , Masliah E ((2014) ) Toward a unified therapeutics approach targeting putative amyloid-beta oligomer receptors, Proc Natl Acad Sci U S A 111: , 13680–13681. |
[393] | Cummings J , Lee G , Mortsdorf T , Ritter A , Zhong K ((2017) ) Alzheimer’s disease drug development pipeline: 2017, Alzheimers Dement (N Y) 3: , 367–384. |
[394] | Krafft GA , Hefti F , Goure WF , Jerecic J , Iverson KS , Walicke PA ((2013) ) ACU-193: A candidate therapeutic antibody that selectively targets soluble beta-amyloid oligomers, Alzheimers Dement 9: , P326. |
[395] | Meilandt WJ , Maloney JA , Imperio J , Bainbridge TW , Reichelt M , Mandikian D , Lu Y , Ernst JA , Fuji RN , Atwal JK ((2017) ) Characterization of the selective in vivo and in vitro binding properties of crenezumab: Insights into crenezumab’s unique mechanism of action, J Prev Alzheimers Dis 4: , 293. |
[396] | Jonsson T , Atwal JK , Steinberg S , Snaedal J , Jonsson PV , Bjornsson S , Stefansson H , Sulem P , Gudbjartsson D , Maloney J , Hoyte K , Gustafson A , Liu Y , Lu Y , Bhangale T , Graham RR , Huttenlocher J , Bjornsdottir G , Andreassen OA , Jonsson EG , Palotie A , Behrens TW , Magnusson OT , Kong A , Thorsteinsdottir U , Watts RJ , Stefansson K ((2012) ) A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline, Nature 488: , 96–99. |
[397] | Maloney JA , Bainbridge T , Gustafson A , Zhang S , Kyauk R , Steiner P , van der Brug M , Liu Y , Ernst JA , Watts RJ , Atwal JK ((2014) ) Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein, J Biol Chem 289: , 30990–31000. |
[398] | Vassar R ((2014) ) BACE1 inhibitor drugs in clinical trials for Alzheimer’s disease, Alzheimers Res Ther 6: , 89. |
[399] | Mullard A ((2017) ) BACE inhibitor bust in Alzheimer trial, Nat Rev Drug Discov 16: , 155. |
[400] | Keskin AD , Kekušs M , Adelsberger H , Neumann U , Shimshek DR , Song B , Zott B , Peng T , Förstl H , Staufenbiel M , Nelken I , Sakmann B , Konnerth A , Busche MA ((2017) ) BACE inhibition-dependent repair of Alzheimer’s pathophysiology, Proc Natl Acad Sci U S A 114: , 8631–8636. |
[401] | Ferreira ST , Klein WL ((2011) ) The Abeta oligomer hypothesis for synapse failure and memory loss in Alzheimer’s disease, Neurobiol Learn Mem 96: , 529–543. |
[402] | Galasko D , Bell J , Mancuso JY , Kupiec JW , Sabbagh MN , van Dyck C , Thomas RG , Aisen PS ((2014) ) Clinical trial of an inhibitor of RAGE-Abeta interactions in Alzheimer disease, Neurology 82: , 1536–1542. |
[403] | Schneider LS , Grundman M , DeKosky ST , Morgan R , Guttendorf R , Higgin M , Pribyl J , Mozzoni K , Izzo NJ , Safferstein H , Houser C , Woodward M , Catalano SM ((2017) ) The anti-AB oligomer drug CT1812 for Alzheimer’s: Phase 1B/2A safety trial outcomes, J Prev Alzheimers Dis 4: , 317. |
[404] | Catalano SM , Schachner LS , DeKosky S , Morgan R , Rehak C , Mozzoni K , Izzo NJ , Grundman M , Schirm M , Guilbaud R , Watson M , Chelsky D ((2017) ) Biomarker outcomes from the phase 1b/2a safety trial of the anti-AB oligomer drug CT1812 in Alzheimer’s patients, J Prev Alzheimers Dis 4: , 401. |
[405] | Paris D , Bachmeier C , Patel N , Quadros A , Volmar CH , Laporte V , Ganey J , Beaulieu-Abdelahad D , Ait-Ghezala G , Crawford F , Mullan MJ ((2011) ) Selective antihypertensive dihydropyridines lower Abeta accumulation by targeting both the production and the clearance of Abeta across the blood-brain barrier, Mol Med 17: , 149–162. |
[406] | Chen Y , Zhang J , Zhang B , Gong CX ((2016) ) Targeting insulin signaling for the treatment of Alzheimer’s disease, Curr Top Med Chem 16: , 485–492. |
[407] | Yarchoan M , Arnold SE ((2014) ) Repurposing diabetes drugs for brain insulin resistance in Alzheimer disease, Diabetes 63: , 2253–2261. |
[408] | McClean PL , Holscher C ((2014) ) Liraglutide can reverse memory impairment, synaptic loss and reduce plaque load in aged APP/PS1 mice, a model of Alzheimer’s disease, Neuropharmacology 76: (Pt A), 57–67. |
[409] | Cheng M , Santich BH , Xu H , Ahmed M , Huse M , Cheung NK ((2016) ) Successful engineering of a highly potent single-chain variable-fragment (scFv) bispecific antibody to target disialoganglioside (GD2) positive tumors, Oncoimmunology 5: , e1168557. |
[410] | Kuan YC , Huang KW , Lin CL , Hu CJ , Kao CH ((2017) ) Effects of metformin exposure on neurodegenerative diseases in elderly patients with type 2 diabetes mellitus, Prog Neuropsychopharmacol Biol Psychiatry 79: , 77–83. |
[411] | Nygaard HB ((2018) ) Targeting fyn kinase in Alzheimer’s disease, Biol Psychiatry 83: , 369–376. |
[412] | Kaufman AC , Salazar SV , Haas LT , Yang J , Kostylev MA , Jeng AT , Robinson SA , Gunther EC , van Dyck CH , Nygaard HB , Strittmatter SM ((2015) ) Fyn inhibition rescues established memory and synapse loss in Alzheimer mice, Ann Neurol 77: , 953–971. |
[413] | Lonskaya I , Hebron ML , Desforges NM , Schachter JB , Moussa CE ((2014) ) Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance, J Mol Med (Berl) 92: , 373–386. |
[414] | Pagan F , Hebron M , Valadez EH , Torres-Yaghi Y , Huang X , Mills RR , Wilmarth BM , Howard H , Dunn C , Carlson A , Lawler A , Rogers SL , Falconer RA , Ahn J , Li Z , Moussa C ((2016) ) Nilotinib effects in Parkinson’s disease and dementia with Lewy bodies, J Parkinsons Dis 6: , 503–517. |
[415] | Bakota L , Brandt R ((2016) ) Tau biology and tau-directed therapies for Alzheimer’s disease, Drugs 76: , 301–313. |
[416] | Gauthier S , Feldman HH , Schneider LS , Wilcock GK , Frisoni GB , Hardlund JH , Moebius HJ , Bentham P , Kook KA , Wischik DJ , Schelter BO , Davis CS , Staff RT , Bracoud L , Shamsi K , Storey JM , Harrington CR , Wischik CM ((2016) ) Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: A randomised, controlled, double-blind, parallel-arm, phase 3 trial, Lancet 388: , 2873–2884. |
[417] | Wilcock GK , Gauthier S , Frisoni GB , Jia J , Hardlund JH , Moebius HJ , Bentham P , Kook KA , Schelter BO , Wischik DJ , Davis CS , Staff RT , Vuksanovic V , Ahearn T , Bracoud L , Shamsi K , Marek K , Seibyl J , Riedel G , Storey JMD , Harrington CR , Wischik CM ((2018) ) Potential of low dose leuco-methylthioninium bis(hydromethanesulphonate) (LMTM) monotherapy for treatment of mild Alzheimer’s disease: Cohort analysis as modified primary outcome in a phase III clinical trial, JAlzheimers Dis 61: , 435–457. |
[418] | Hendrix JA , Bateman RJ , Brashear HR , Duggan C , Carrillo MC , Bain LJ , DeMattos R , Katz RG , Ostrowitzki S , Siemers E , Sperling R , Vitolo OV ((2016) ) Challenges, solutions, and recommendations for Alzheimer’s disease combination therapy, Alzheimers Dement 12: , 623–630. |
[419] | Leslie K , Cohen J , Klee J , Arnold SE ((2017) ) Phase II study to assess safety and target engagement of AMX0035, a novel combination therapy, in Alzheimer’s disease, Neurotherapeutics 14: (3), 825–826. |
[420] | Hori Y , Takeda S , Cho H , Wegmann S , Shoup TM , Takahashi K , Irimia D , Elmaleh DR , Hyman BT , Hudry E ((2015) ) A Food and Drug Administration-approved asthma therapeutic agent impacts amyloid beta in the brain in a transgenic model of Alzheimer disease, J Biol Chem 290: , 1966–1978. |
[421] | Yusufov M , Weyandt LL , Piryatinsky I ((2017) ) Alzheimer’s disease and diet: A systematic review, Int J Neurosci 127: , 161–175. |
[422] | Singh B , Parsaik AK , Mielke MM , Erwin PJ , Knopman DS , Petersen RC , Roberts RO ((2014) ) Association of mediterranean diet with mild cognitive impairment and Alzheimer’s disease: A systematic review and meta-analysis, J Alzheimers Dis 39: , 271–282. |
[423] | D’Cunha NM , Georgousopoulou EN , Dadigamuwage L , Kellett J , Panagiotakos DB , Thomas J , McKune AJ , Mellor DD , Naumovski N ((2018) ) Effect of long-term nutraceutical and dietary supplement use on cognition in the elderly: A 10-year systematic review of randomised controlled trials, Br J Nutr 119: , 280–298. |
[424] | Cerf E , Gustot A , Goormaghtigh E , Ruysschaert JM , Raussens V ((2011) ) High ability of apolipoprotein E4 to stabilize amyloid-beta peptide oligomers, the pathological entities responsible for Alzheimer’s disease, FASEB J 25: , 1585–1595. |
[425] | Garai K , Verghese PB , Baban B , Holtzman DM , Frieden C ((2014) ) The binding of apolipoprotein E to oligomers and fibrils of amyloid-beta alters the kinetics of amyloid aggregation, Biochemistry 53: , 6323–6331. |
[426] | Kang MK , Lee J , Nguyen AH , Sim SJ ((2015) ) Label-free detection of ApoE4-mediated beta-amyloid aggregation on single nanoparticle uncovering Alzheimer’s disease, Biosens Bioelectron 72: , 197–204. |
[427] | Bien-Ly N , Andrews-Zwilling Y , Xu Q , Bernardo A , Wang C , Huang Y ((2011) ) C-terminal-truncated apolipoprotein (apo) E4 inefficiently clears amyloid-beta (Abeta) and acts in concert with Abeta to elicit neuronal and behavioral deficits in mice, Proc Natl Acad Sci U S A 108: , 4236–4241. |
[428] | Manelli AM , Bulfinch LC , Sullivan PM , LaDu MJ ((2007) ) Abeta42 neurotoxicity in primary co-cultures: Effect of apoE isoform and Abeta conformation, Neurobiol Aging 28: , 1139–1147. |
[429] | Pankiewicz JE , Guridi M , Kim J , Asuni AA , Sanchez S , Sullivan PM , Holtzman DM , Sadowski MJ ((2014) ) Blocking the apoE/Abeta interaction ameliorates Abeta-related pathology in APOE epsilon2 and epsilon4 targeted replacement Alzheimer model mice, Acta Neuropathol Commun 2: , 75. |
[430] | Hashimoto T , Serrano-Pozo A , Hori Y , Adams KW , Takeda S , Banerji AO , Mitani A , Joyner D , Thyssen DH , Bacskai BJ , Frosch MP , Spires-Jones TL , Finn MB , Holtzman DM , Hyman BT ((2012) ) Apolipoprotein E, especially apolipoprotein E4, increases the oligomerization of amyloid beta peptide, J Neurosci 32: , 15181–15192. |
[431] | Cole GM , Frautschy SA ((2006) ) Docosahexaenoic acid protects from amyloid and dendritic pathology in an Alzheimer’s disease mouse model, Nutr Health 18: , 249–259. |
[432] | Yassine HN , Braskie MN , Mack WJ , Castor KJ , Fonteh AN , Schneider LS , Harrington MG , Chui HC ((2017) ) Association of docosahexaenoic acid supplementation with Alzheimer disease stage in apolipoprotein E epsilon4 carriers: A review, JAMA Neurol 74: , 339–347. |
[433] | Stephen R , Hongisto K , Solomon A , Lonnroos E ((2017) ) Physical activity and Alzheimer’s disease: A systematic review, J Gerontol A Biol Sci Med Sci 72: , 733–739. |
[434] | Nichol KE , Poon WW , Parachikova AI , Cribbs DH , Glabe CG , Cotman CW ((2008) ) Exercise alters the immune profile in Tg2576 Alzheimer mice toward a response coincident with improved cognitive performance and decreased amyloid, J Neuroinflammation 5: , 13. |
[435] | Adlard PA , Perreau VM , Pop V , Cotman CW ((2005) ) Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer’s disease, J Neurosci 25: , 4217–4221. |
[436] | Rocchi A , Yamamoto S , Ting T , Fan Y , Sadleir K , Wang Y , Zhang W , Huang S , Levine B , Vassar R , He C ((2017) ) A Becn1 mutation mediates hyperactive autophagic sequestration of amyloid oligomers and improved cognition in Alzheimer’s disease, PLoS Genet 13: , e1006962. |
[437] | Tapia-Rojas C , Aranguiz F , Varela-Nallar L , Inestrosa NC ((2016) ) Voluntary running attenuates memory loss, decreases neuropathological changes and induces neurogenesis in a mouse model of Alzheimer’s disease, Brain Pathol 26: , 62–74. |
[438] | Mainardi M , Di Garbo A , Caleo M , Berardi N , Sale A , Maffei L ((2014) ) Environmental enrichment strengthens corticocortical interactions and reduces amyloid-beta oligomers in aged mice, Front Aging Neurosci 6: , 1. |
[439] | Fratiglioni L , Paillard-Borg S , Winblad B ((2004) ) An active and socially integrated lifestyle in late life might protect against dementia, Lancet Neurol 3: , 343–353. |
[440] | Wang HX , Karp A , Winblad B , Fratiglioni L ((2002) ) Late-life engagement in social and leisure activities is associated with a decreased risk of dementia: A longitudinal study from the Kungsholmen project, Am J Epidemiol 155: , 1081–1087. |
[441] | Karp A , Paillard-Borg S , Wang HX , Silverstein M , Winblad B , Fratiglioni L ((2006) ) Mental, physical and social components in leisure activities equally contribute to decrease dementia risk, Dement Geriatr Cogn Disord 21: , 65–73. |
[442] | Wang MJ , Yi S , Han JY , Park SY , Jang JW , Chun IK , Kim SE , Lee BS , Kim GJ , Yu JS , Lim K , Kang SM , Park YH , Youn YC , An SSA , Kim S ((2017) ) Oligomeric forms of amyloid-beta protein in plasma as a potential blood-based biomarker for Alzheimer’s disease, Alzheimers Res Ther 9: , 98. |
[443] | Blennow K , Zetterberg H ((2015) ) The past and the future of Alzheimer’s disease CSF biomarkers-a journey toward validated biochemical tests covering the whole spectrum of molecular events, Front Neurosci 9: , 345. |
[444] | Chiang ACA , Fowler SW , Reddy R , Pletnikova O , Troncoso JC , Sherman MA , Lesne SE , Jankowsky JL ((2018) ) Discrete pools of oligomeric amyloid-beta track with spatial learning deficits in a mouse model of Alzheimer amyloidosis, Am J Pathol 188: , 739–756. |
[445] | Bao F , Wicklund L , Lacor PN , Klein WL , Nordberg A , Marutle A ((2012) ) Different beta-amyloid oligomer assemblies in Alzheimer brains correlate with age of disease onset and impaired cholinergic activity, Neurobiol Aging 33: , 825 e821–813. |
[446] | Kasai T , Kondo M , Ishii R , Tanaka A , Ataka S , Shimada H , Tomiyama T , Mori H , Taylor M , Allsop D , Nakagawa M , Mizuno T , Tokuda T ((2017) ) Abeta levels in the jugular vein and high molecular weight Abeta oligomer levels in CSF can be used as biomarkers to indicate the anti-amyloid effect of IVIg for Alzheimer’s disease.e, PLoS One 12: , 0174630. |
[447] | Nyborg AC , Moll JR , Wegrzyn RD , Havas D , Hutter-Paier B , Feuerstein GG , Rudolph AS ((2013) ) In vivo and ex vivo imaging of amyloid-beta cascade aggregates with a Pronucleon peptide, J Alzheimers Dis 34: , 957–967. |
[448] | Stein TD , Montenigro PH , Alvarez VE , Xia W , Crary JF , Tripodis Y , Daneshvar DH , Mez J , Solomon T , Meng G , Kubilus CA , Cormier KA , Meng S , Babcock K , Kiernan P , Murphy L , Nowinski CJ , Martin B , Dixon D , Stern RA , Cantu RC , Kowall NW , McKee AC ((2015) ) Beta-amyloid deposition in chronic traumatic encephalopathy, Acta Neuropathol 130: , 21–34. |
[449] | Nogalska A , D’Agostino C , Engel WK , Klein WL , Askanas V ((2010) ) Novel demonstration of amyloid-beta oligomers in sporadic inclusion-body myositis muscle fibers, Acta Neuropathol 120: , 661–666. |
[450] | Yin H , Chen L , Chen X , Liu X ((2008) ) Soluble amyloid beta oligomers may contribute to apoptosis of retinal ganglion cells in glaucoma, Med Hypotheses 71: , 77–80. |
[451] | Fisichella V , Giurdanella G , Platania CB , Romano GL , Leggio GM , Salomone S , Drago F , Caraci F , Bucolo C ((2016) ) TGF-beta1 prevents rat retinal insult induced by amyloid-beta (1-42) oligomers, Eur J Pharmacol 787: , 72–77. |
[452] | Ma W , Lee SE , Guo J , Qu W , Hudson BI , Schmidt AM , Barile GR ((2007) ) RAGE ligand upregulation of VEGF secretion in ARPE-19 cells, Invest Ophthalmol Vis Sci 48: , 1355–1361. |
[453] | Isas JM , Luibl V , Johnson LV , Kayed R , Wetzel R , Glabe CG , Langen R , Chen J ((2010) ) Soluble and mature amyloid fibrils in drusen deposits, Invest Ophthalmol Vis Sci 51: , 1304–1310. |
[454] | Parsons CG , Ruitenberg M , Freitag CE , Sroka-Saidi K , Russ H , Rammes G ((2015) ) MRZ-99030 - A novel modulator of Abeta aggregation: I - Mechanism of action (MoA) underlying the potential neuroprotective treatment of Alzheimer’s disease, glaucoma and age-related macular degeneration (AMD), Neuropharmacology 92: , 158–169. |
[455] | Rodgers AB ((2005) ) Progress report on Alzheimer’s disease 2004-2005. U.S. Department of Health and Human Services; National Institutes on Aging; National Institutes of Health. http://www.alzheimers.org/pr04-05/Progress_Report_on_Alzheimers_Disease_2004-2005-small.pdf |


