
Expression Profiling of Cytokine, Cholinergic Markers, and Amyloid-β Deposition in the APPSWE/PS1dE9 Mouse Model of Alzheimer’s Disease Pathology
Abstract
Background:
Alzheimer’s disease (AD), a neurodegenerative disease, is associated with dysfunction of the olfactory and the entorhinal cortex of the brain that control memory and cognitive functions and other daily activities. Pro-inflammatory cytokines, amyloid-β (Aβ), and the cholinergic system play vital roles in the pathophysiology of AD. However, the role of changes in cholinergic system components, Aβ accumulation, and cytokines in both the olfactory and entorhinal cortex is not known clearly.
Objective:
The present study is aimed to evaluate the changes of cholinergic system components, Aβ accumulation, and cytokines in both the olfactory bulb (OB) and entorhinal cortex (EC) of young and aged APPSWE/PS1dE9 transgenic (Tg) mice.
Methods:
We have explored the changes of cholinergic system components, Aβ accumulation, and expression profiling of cytokines in the OB and EC of aged APPswe transgenic mice and age-matched wild type mice using quantitative Real-Time PCR assays and immunohistochemistry techniques.
Results:
In aged Tg mice, a significant increase of expression of interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and chemokine MCP1 (p < 0.001, p < 0.001, and p = 0.001, respectively) and a significant reduction of nAChRα4 (p = 0.048) and AChE (p = 0.023) was observed when compared with age-matched wild type mice. Higher levels of AChE and BuChE are expressed in OB and EC of the APPSWE/PS1dE9 of Tg mice. Aβ accumulation was observed in OB and EC of the APPSWE/PS1dE9 of Tg mice.
Conclusion:
The study demonstrates the expression profiling of pro-inflammatory cytokines and cholinergic markers as well as Aβ accumulation in OB and EC of the APPSWE/PS1dE9 Tg mice. Moreover, the study also demonstrated that the APPSWE/PS1dE9 Tg mice can be useful as a mouse model to understand the role of pro-inflammatory cytokines and cholinergic markers in pathophysiology of AD.
INTRODUCTION
Alzheimer’s disease (AD) is a neurodegenerative disease affecting regions of the brain that control memory and cognitive functions, and reduces the ability to learn, communicate, and carry out daily activities. Current diagnosis of AD is largely based on exclusion of other causes for dementia and on the major neuropathologic hallmarks such as the deposition of amyloid-β (Aβ) peptide in the form of plaques, accumulation of neurofibrillary tangles composed of hyperphosphorylated tau, cytokine dysregulation, and loss or reduction of cholinergic neurons in the basal forebrain. Multiple experimental, genetic, and in vivo studies have supported the role of inflammation in the pathogenesis of AD [1, 2]. Inflammation occurs in AD brain, with increased expression of acute phase proteins and proinflammatory cytokines [3]. In AD, brain amyloid plaques and tangles are related to local stimulation of innate immune and inflammatory responses, and astrocytes, microglia, and neurons are responsible for the inflammatory reaction [4]. A similar intercellular and intracellular signaling process induced by cytokines occurres in microglia, astrocytes, and peripheral cells[5–7].
The pro-inflammatory cytokines may induce the synthesis of Aβ that, by positive feedback mechanism, causes the expression of these cytokines in astrocytes and microglia. Further, chemokines may drive the entry of activated leukocytes into the CNS, and effector mechanisms such as macrophage activation and glial-glial and glial-neuronal interaction contribute to the perivascular inflammatory infiltratesformation.
Several studies in AD demonstrated that impaired olfactory function appears earlier than memory loss, reduced intelligence, personality modification, and dementia. Many AD patients suffer from olfactory dysfunction [8–11], so evaluation of olfactory function may help to identify the onset and early stages of neurodegenerative disorders[12–14].
Cholinergic activity, including nicotinic and muscarinic receptors, in the olfactory system modulates discrimination between similar odors in rats [15]; therefore, performance of olfactory behavioral tasks may be affected during cholinergic neuromodulation, and lesions of cholinergic inputs to the cortex reduced the differentiation of similar odorants [16]. Also, functions such as attention, arousal, learning, and memory are modulated by the cholinergic system. An impaired cholinergic fibers function, in the basal forebrain, hippocampus, and cortex is related to memory loss, altered cognition, and neurodegenerative alterations characterizing aging and AD [17]. Therapies based on cholinergic replacement by acetylcholinesterase (AChE) inhibitors showed amelioration of cholinergic deficit at very early stages of dementia and slow down its progression.
Mutations of genes coding for amyloid precursor protein (APP) and the presenilins (PSs) are responsible for amyloid peptide formation and amyloid plaques deposition in brain, and then for neuroinflammation. Various animal models are used to evaluate factors implicated in the pathogenesis of AD. Although the Aβ peptide sequence of mouse is slightly different to its human counterpart, transgenic mice with mutation of APPswe and PSEN1 that develops Aβ deposition and cognitive deficits such as memory, language, thinking, spatial learning and judgment, represent an effective model for the study of AD [18–28].
Dysfunction of olfactory and entorhinal cortex is associated with AD [29, 30]. Pro-inflammatory cytokines, Aβ, and the cholinergic system play vital roles in the pathophysiology of AD. However, changes in expression levels of cholinergic system components, Aβ accumulation, and cytokines in both the olfactory and entorhinal cortex are not known clearly. In this background, the aim of the present study was to analyze the changes of cholinergic system components and Aβ accumulation as well cytokines in both the olfactory bulb (OB) and entorhinal cortex (EC) of young and aged APPswe transgenic (Tg) mice.
MATERIALS AND METHODS
Transgenic animals
B6.Cg-Tg (APPswe, PSEN1dE9) 85Dbo/J strain(common name APPswe/PS1dE9) expresses achimeric mouse/human amyloid precursor protein(Mo/HuAPP695swe) and a mutant human presenilin1 (PS1-dE9). Mice of either sex were bred and housedin ventilated racks, in groups of 5 mice under 12 hday/night cycle with a half-hour transition as sunriseand sunset, 50% HR and ad libitum food and water(irradiated global diet 2918 Harlan® and water autoclaved), at the Cajal Institute Animal House Facility.Experimental procedures were conducted per theCouncil of Europe (2010/63/UE) and Spanish guidelines, and approved by the Ethical Committee ofCSIC, Bioethical Committee at the Animals ResearchSpanish Council (CSIC) and the local authorities ofthe Community of Madrid. These mice were genotyped by PCR [31]. Genotype determination wasdone with tail biopsies using the following primer todetect the mutant human APP gene product (400 pb):forward, 5’-CCGAGATCTCTGAAGTGAAGATGGATG-3’. The presence of a 1300 bp PS1 gene product was confirmed with the following primers: 5’-CAGGTGGTGGAGCAAGATG-3’ and the PrP internal control product (750pb): forward, 5’-CCTCTTTGTGACTATGTGGACTGATGTCGG-3’, and reverse 5’-GTGGATACCCCCTCCCCCAGCCTAGACC-3’.
RNA extraction, reverse transcription, and Real-Time PCR (qPCR)
Mice were sacrificed by cervical dislocation; brains were removed and both OB and EC were dissected out on ice [32, 33]. The samples were stored in RNAlater™ at 4°C until used.
For cytokines and cholinergic markers quantification, total RNA from the Tg and wild type (WT) mice was isolated using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer’s recommendations.
The concentration of total RNA was measured with NanoDrop 2000 UV-Vis Spectrophotometer (Thermo Scientific, Waltham, MA, USA) at 260 nm and the purity was assessed considering the absorbance ratio at 260 and 280 nm (A260/A280) and A260/A230. Then, 1μg of total RNA was transcribed to cDNA using the QuantiTec® Revers Transcription Kit with integrated removal of genomic DNA contamination (Qiagen, Hilden, Germany) according to the manufacturer’s instructions.
Next, quantitative Real-Time PCR assays were performed in triplicate using GoTaq® qPCR Master Mix (Promega, Madison, USA), and specific mouse primer pairs (given below).
Relative expression of each gene was normalized by HPRT using the ΔCT method, where ΔCT = CT (BuChE,AChE,nAChRα7, nAChRα4,nAChRβ2,IL1β,TNFα, MCP1) – CT (HPRT) . Relative fold changes in gene expression were determined by the 2–ΔΔCT method, where ΔΔCt =ΔCt experimental sample-ΔCt calibrator. Experimental cDNAs of Tg mice were prepared in parallel with the cDNAs of WT mice.
Immunohistochemistry
Mice (n = 5) were deeply anesthetized with pentobarbital (30–50 mg/kg) intraperitoneally and transcardially perfused with paraformaldehyde 4% (PF), pH 7.4. Brains were carefully dissected out, post-fixed in PF overnight at 4°C. Brains were removed from skulls and 50μm coronal sections were vibratome sectioned, and then transferred to phosphate buffer saline (PBS) containing 0.1% Triton X-100 (PBS-T) for 5 min and then blocked for 60 min in 5% NGS in PBS-T. Sections were incubated in the following primary antibodies: β-amyloid antibody (1 : 250; Cell signaling), mouse anti-GFAP (1 : 1000; Millipore), and mouse anti-NeuN (1 : 1000; Chemicon) in the same blocking solution for 48 h at 4°C. After further rinsing, the sections were incubated for 2 h with Alexa Fluor® 488 and Alexa Fluor® 568 conjugated goat anti-rabbit, and goat anti-mouse (1 : 1000; Invitrogen, Carlsbad, CA, USA). Finally, sections were rinsed, counterstained with bismencimide (Hoechst 33342, 1 : 50000), mounted onto slides in Mowiol. Images were obtained on a Leica TCS-SP5 confocal microscope, acquiring two or three different channels simultaneously.
Statistical methods
To give an indication of the precision of the fold change, calculated with 2–ΔΔCt method, the 95% confidence interval (95% CI) was determined. Student t-test for one sample was applied to evaluate statistical differences in mRNA expression levels between young (6 months old) and aged Tg (24 months old) and their age-matched WT with predetermined value equal to 1. Student t-test for unpaired data was applied to evaluate the differences in mRNA expression levels between young and aged mice. Spearman Rho correlation coefficients were performed to evaluate statistically significant correlation between AChE, butyrylcholinesterase (BuChE), and cytokines. In all statistical tests the threshold of statistical significance will be assumed equal to p = 0.05. Data will be analyzed by SPSS® Advanced StatisticalTM 13 (Chicago, IL, USA) and R open source software.
RESULTS
Aβ deposits distribution in the OB and EC at different ages
In the Tg mice brain, anti-Aβ expression was mainly detected in the granular layer of the OB from three months old (data not shown). Figure 1 shows Aβ deposits throughout the different layers of OB. At 6 months old (Fig. 1B), Aβ deposits were distributed within the glomerular and granular layers. At 24 months, the granular layer still shows the largest number of Aβ deposits (Fig. 1C), while the mitral cell layer shows the lowest number of Aβ deposits. Aβ deposits were also distributed along different cortical areas (Fig. 2), increasing in number and size with the age. At 6 months, Aβ deposits were restricted to EC, frontal cortex, and hippocampus (Fig. 2A). At 24 months, those Aβ deposits were widely distributed throughout all the brain (Fig. 2B).
| Gene | Mouse PCR primer pairs [5’-3’] | |
| Forward | Revers | |
| HPRT | TTGGATACAGGCCAGACTTTG | TGGCAACATCAACAGGACTC |
| BuChE | TAGCACAATGTGGCCTGTCT | ATTGCTCCAGCGATGAAATC |
| AChE | ATTTTGCCCGCACAGGGGAC | CGCCTCGTCCAGAGTATCGGT |
| nAChRα7 | TGATTCCGTGCCCTTGATAG | GAATGATCCTGGTCCACTTAGG |
| nAChRα4 | GTAGAAGGCGTCCAGTACATTG | AGATCATACCAGCCAACCATG |
| nAChRβ2 | GCTTCATTGCGGACCATATG | CCAAAGACACAGACAAAGACAAAG |
| IL1β | TTGACGGACCCCAAAAGATG | AGAAGGTGCTCATGTCCTCA |
| TNFα | TGGAGTCATTGCTCTGTGAAG | CCTGAGCCATAATCCCCTTTC |
| MCP1 | GGTCCCTGTCATGCTTCTGG | CCTGCTGCTGGTGATCCTCT |
Fig.1
Distribution Aβ-deposits in APPswe/PS1dE9 mice in olfactory bulb (OB) at different ages. A) Localization of Aβ-deposits throughout the different layers (GL, glomerular layer; EPL, external plexiform layer; GCL, granular Cell layer) of the OB in an old mouse. OB in 6-month-old (B) and 24-month-old mice (C). Anti-Aβ in green, and DAPI nuclear staining in blue. Bars, 50μm. D) t-test number of Aβ deposits in OB young (111.467±14.934) versus old (1510.733±72.113) ***p < 0.001. E) t-test % area young (0.658±0.151) versus old (10.063±0.555) ***p < 0.001.
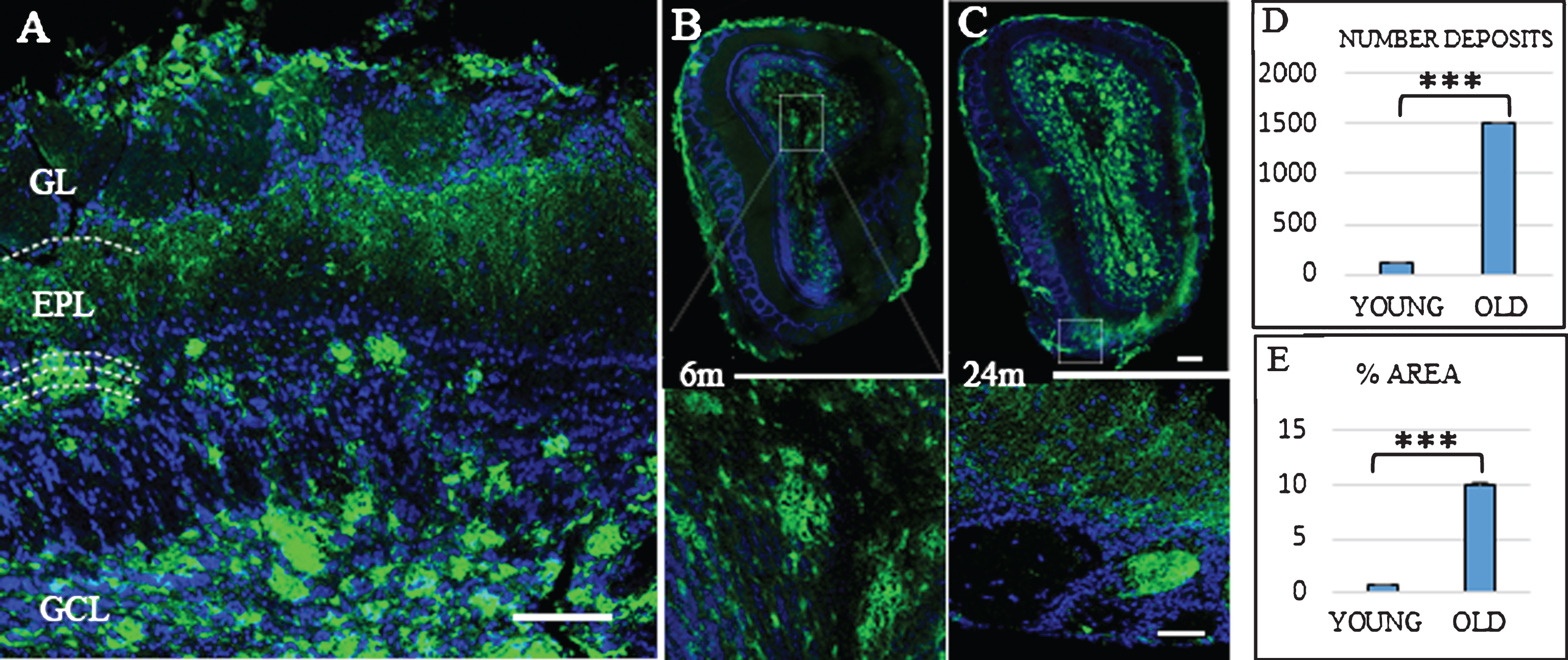
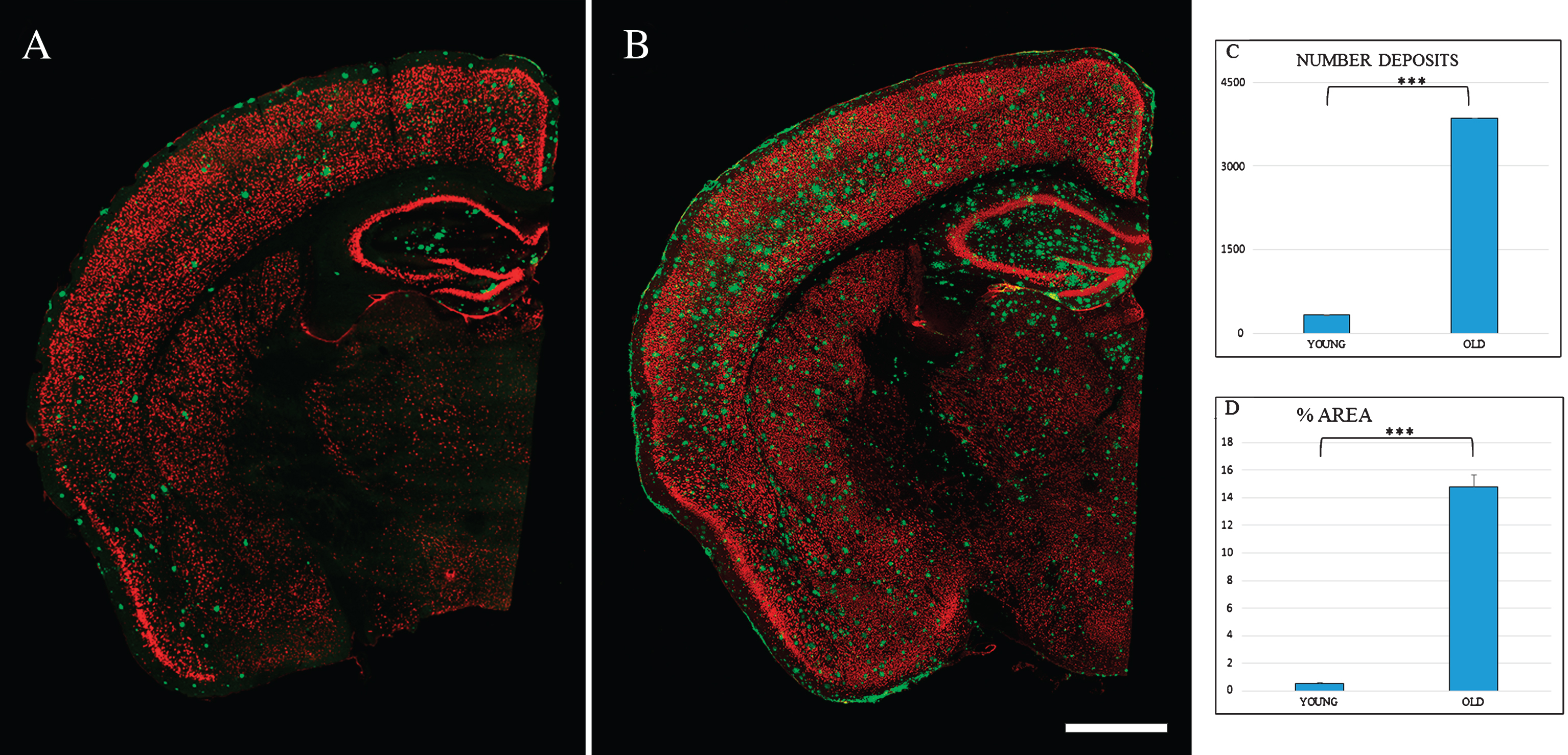
Fig.2
Spatial distribution Aβ-deposits in entorhinal cortex in APPswe/PS1dE9 mice at different ages. Expression of NeuN (red) and antiAβ (green) antibodies. 6-month-old mice (A), 24-month- old mice (B). Coronal section, bars 50μm. C) t-test number of Aβ-deposits in entorhinal cortex in young (324.733±28.747) versus old (3854.867±252.844) ***p < 0.001. D) t-test % area in young (0.539±0.069) versus old (14.801±0.866) ***p < 0.001.
AChE and BuChE expression in OB and EC brain regions
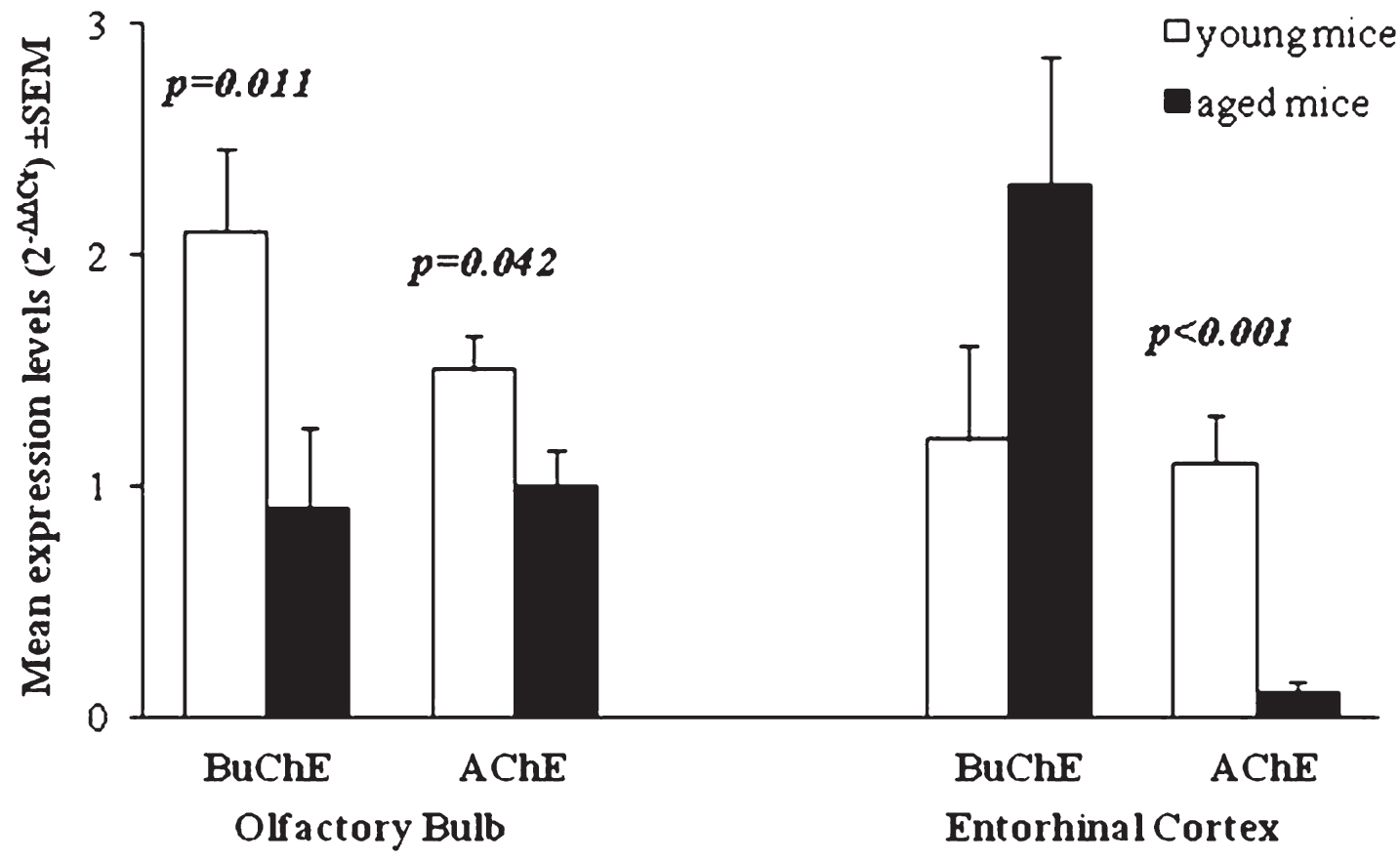
In OB of young Tg mice, AChE and BuChE expression levels are significantly increased with respect to expression in OB of WT mice (p < 0.001). When we compared expression of ChE enzymes in young versus aged Tg mice, BuChE and AChE expression is significantly reduced over time (p = 0.011 and p = 0.042, respectively) (Table 1). These results are in accord with studies on individuals with mild cognitive impairment showing that at the early stages of disease pathogenesis, the cholinergic system could be more active than normal (i.e., the levels of cholinergic enzymes and receptors could beincreased) [34].
Table 1
Mean and 95% confidence interval of AChE and BuChE expression levels (2–ΔΔCt) in OB of young and aged Tg mice respect to age-matched WT
| Olfactory Bulb | |||
| Young mice | Aged mice | p-valuea | |
| BuChE | 2.1 (1.6–2.8) | 0.9 (0.6–1.6) | 0.011 |
| AChE | 1.5 (1.2–1.8) | 1.0 (0.7–1.3) | 0.042 |
at-test for unpaired data Young mice versus Aged mice. Statistically significant comparisons respect to WT are shown in bold character (p < 0.05).
The expression of BuChE in EC of aged Tg mice was significantly increased respect to WT (p = 0.008). An increase of BuChE was also observed between young and aged Tg. In EC of aged Tg mice, the expression of AChE is significantly reduced with respect to WT (p < 0.001) and with respect to young Tg (p < 0.001) (Table 2). Comparison of BuChE and AChE expression in both OB and EC of young and aged Tg mice is shown in Fig. 3.
Table 2
Mean and 95% confidence interval of AChE and BuChE expression (2–ΔΔCt) in EC of young and aged Tg mice respect to age-matched WT
| Entorhinal Cortex | |||
| Young mice | Aged mice | p-valuea | |
| BuChE | 1.2 (0.7–2.0) | 2.3 (1.3–4.0) | 0.131 |
| AChE | 1.1 (0.8–1.5) | 0.1 (0.1–0.2) | <0.001 |
at-test for unpaired data Young mice versus Aged mice. Statistically significant comparisons respect to WT are shown in bold character (p < 0.05).
Fig.3
Mean of ChE enzymes’ expression levels (2-ΔΔCt) in young and aged Tg mice compared to age-matched WT. p-values reported in figure are relative to comparison between age groups. Error bars represent standard error of mean (SEM).
Cholinergic receptors expression
In OB and EC, an expression study performed by RT-PCR revealed that nAChR subunits expression were higher in Tg than in WT mice. In the OB of young Tg mice, the levels of nAChRα7 were 5-fold higher than in age-matched WT mice (p < 0.001) and the levels of nAChRα4 and nAChRβ2 were about 4-fold higher than in age-matched WT mice (p < 0.001). Also in EC of both young and aged Tg mice, all nAChR subunits are more expressed than in WT, but the increase was statistically significant for nAChRα7 in both age group (p = 0.045), and for nAChRβ2 only in the younger one (p = 0.017). Interestingly, when we compared over-time nAChRs expression in OB of Tg mice, we observed a significant reduction for nAChRα7 (p = 0.016) and nAChRβ2 (p = 0.002) (Table 3).
Table 3
Mean and 95% confidence interval of nAChRs expression (2-ΔΔCt) in OB and EC of young and aged Tg mice respect to age-matched WT
| Olfactory Bulb | Entorhinal Cortex | |||||
| Young mice | Aged mice | p-valuea | Young mice | Aged mice | p-valuea | |
| nAChRα7 | 5.3 (3.5–8.1) | 1.7 (0.8–3.7) | 0.016 | 1.9 (1.1–3.2) | 2.2 (1.1–4.7) | 0.759 |
| nAChRα4 | 3.8 (2.7–5.3) | 2.0 (1.1–3.5) | 0.067 | 1.4 (0.9–2.3) | 1.1 (0.7–1.8) | 0.453 |
| nAChRβ2 | 4.3 (3.5–5.2) | 1.4 (0.8–2.6) | 0.002 | 1.6 (1.1–2.4) | 1.5 (0.8–2.9) | 0.836 |
at-test for unpaired data Young mice versus Aged mice. Statistically significant comparisons respect to WT are shown in bold character (p < 0.05).
Proinflammatory cytokines expression
In order to reveal the expression profile of proinflammatory cytokines TNFα and IL1β as well as chemokine MCP1, OB and EC brain sections from young and aged Tg and WT mice were subjected to RT-PCR assay. Here we reported that IL1β, TNFα, and MCP1 mRNA expression was significantly increased in OB of young Tg mice, compared with the expression in OB of age-matched WT mice (respectively p = 0.001, p = 0.008, and p < 0.001). In EC, IL1β significantly increased in young Tg mice (p < 0.001), while MCP1 expression detected in young Tg mice was reduced with respect to age-matched WT mice.
The over-time statistically significant increase of TNFα was observed in OB (p = 0.041). The observed over-time decrease of IL1β and TNFα in EC was not significantly, as reported in Table 4.
Table 4
Mean and 95% confidence interval of IL1β, TNFα, and MCP1 expression (2–ΔΔCt) in OB and EC of young and aged Tg mice respect to age-matched WT
| Olfactory Bulb | Entorhinal Cortex | |||||
| Young mice | Aged mice | p-valuea | Young mice | Aged mice | p-valuea | |
| IL1β | 3.8 (1.8–7.9) | 3.9 (1.3–12.0) | 0.960 | 7.9 (4.5–13.8) | 2.3 (0.7–7.9) | 0.085 |
| TNFα | 1.9 (1.2–3.1) | 7.0 (2.4–20.5) | 0.041 | 1.3 (0.8–2.2) | 1.1 (0.4–2.8) | 0.755 |
| MCP1 | 2.1 (1.4–3.0) | 1.1 (0.5–2.5) | 0.179 | 0.7 (0.5–1.0) | 1.5 (0.6–3.7) | 0.143 |
at-test for unpaired data Young mice versus Aged mice. Statistically significant comparisons respect to WT are shown in bold character (p < 0.05).
Different regional expression of cholinergic markers and cytokines
In young Tg mice, the expression of cholinergic markers and cytokines is differently higher (with respect to WT) in OB and EC. In fact, while nAChR subunits, AChE, BuChE, TNFα, and MCP1 are higher expressed in OB of young Tg mice, IL1β is higher expressed in EC (Tables 1–4). In aged Tg mice, the expression of α7 and nAChRβ2 subunits and BuChE is higher expressed in EC than OB, and nAChRα4 and AChE is higher expressed in OB versus EC (Tables 1–3).
Also, cytokines are differently expressed in OB and EC of young and aged Tg mice. In OB, higher levels of IL1β and TNFα were observed in aged mice, while the same cytokines are higher expressed in EC of young Tg mice.
In OB of young mice, a statistically significant correlation between AChE, BuChE, and IL1β (Rho = 0.766, p = 0.027 and Rho = 0.738, p = 0.037, respectively) was observed. A similar relationship was found for AChE (Rho = 0.905, p = 0.002) in aged mice, and a significant correlation between AChE and TNFα (Rho = 0.786, p = 0.021) was observed in OB of aged mice.
DISCUSSION
The brain is significantly affected by aging and the time-dependent accumulation of cellular damage is widely considered to be the general cause of aging [35–37]. Understanding the molecular mechanisms that characterize the aging-related changes in normal and diseased brains is a constant challenge. Recently a link between aging and the risk of amyloidosis was found; in fact, the time of Aβ clearance in the brain is strongly correlated with age. In healthy and young brain, Aβ production and elimination is an equilibrium, while in aging or pathologic conditions such as AD, there is accumulation of Aβ due to impaired formation and clearance [38] that result in plaques formation, characteristic of AD.
A large number of studies demonstrated that at an early stage of AD and cognitive impairment, cholinergic enzymes and receptors were more active than normal with subsequent decline with increasing severity [39, 40]. Studies on central cholinergic function in mice with double mutations in APP and PS1 genes [41] gave contradictory results. Thus, in Tg2576 mice, vesicular acetylcholine transporter was found both unchanged and increased [42, 43]. Keeping in mind that in the APPswe/PS1dE9 mouse model of AD, amyloid plaques are associated with inflammatory response, we have characterized the relationship between amyloid deposition, inflammatory mediators, and the cholinergic system in the brain and in OB, a primary region associated with the development of neurodegenerative pathologies [44] and in EC, implicated in the stages of AD characterized by changes in the tau protein and in the cleaved fragments of APP.
Decrease in olfactory functions including olfactory sensitivity, perceived intensity of supra-threshold odorants, odor quality discrimination, and identification has been well documented in aging [45].
nAChR is a ligand-gated ion channel constituted by five different α subunits (α2-α9) and three different β subunits (β2-β4) that belong to a large family and have been implicated in complex diseases affecting the nervous system, including aging-associated neurodegenerative diseases. α7 and α4β2 nAChRs are the most abundant subunits in the brain [46, 47], and several authors have also suggested that α7nAChR may accelerate the progression of AD [48, 49], and its altered expression may be responsible for the impaired cholinergic neurotransmission, and contribute to the initiation and development of amyloid AD brain pathology [50]. Western blot and autoradiographic analyses have indicated that the α7nAChR subunit protein was upregulated in human brain samples from AD patients, as well as in animal models of AD [51, 52]. Aβ interacts with α7nAChR that might be involved in nicotine mediated reduction of Aβ1–42 deposition [53], although the nature of this relationship remains not well defined. The α4β2-nAChRs are predominant nAChR subtype in the brain, implicated in perception, cognition, and emotion. Exposure of telencephalic neurons to Aβ has revealed a massive reduction of α4-expressing neurons, and in accord with the α4β2 subtype reduction in the cortex of patients with AD, our results showed that also in OB of Tg mice both α4 and β2 subunits were reduced. This finding points to a possible impact of Aβ on the expression of the nAChR α4β2 subunit in OB.
Aβ peptides are potent activators of glial cells. Once activated, microglia and astrocytes release a variety of cytokines, chemokines, and free radical oxygen species, which can contribute to neuronal dysfunction and death, and some specified glia-derived cytokines may also increase Aβ generation. IL1 plays a key role in the pathogenesis of AD inflammation by starting up a cytokine cycle, and MCP1 may amplify subsequent tissue reactions by regulation of microglia migration and recruitment of astrocytes to the area of neuroinflammation. Although the increased expression of inflammatory cytokines, such as TNFα, IL1β, and IL6, have been identified in amyloid plaques and/or glial cells in brain tissues of AD patients, only a limited number of studies have addressed the progression of neuroinflammation in transgenic AD mouse models with variable results.
Olfactory dysfunction has been reported in several neurodegenerative diseases, including AD. Kovacs’s study suggested that olfactory dysfunction is an early event of AD [13], preceding the appearance of typical AD symptoms. Therefore, the underlying changes on gene expression levels might help identify potential therapeutic targets for AD.
In this study, examination was performed to detect the change of cholinergic markers in B6C3-Tg AD mice brain with the growth of age and to analyze the correlation between the expression of nAChR, ChEnzyme, and inflammatory cytokines. Judging from the results, we can figure out that the brain BuChE expression of AD mice at the age of 6 and over 24 months were slowly higher than the WT mice. Our results are in accord with previous studies that reported unchanged AChE activity in APP695SWE transgenic mice despite extensive Aβ plaques [43, 54], and with observation that this AD Tg model did not exhibit AChE activity, in contrast with AD [55, 56]. Monica et al. [57] characterized amyloid deposition in the APPswe/PS1dE9 mouse model of AD. In this study, accordingly with studies that showed the association between BuChE activity and Aβ plaques, we have found a statistically significant increase of BuChE and Aβ deposition in OB of aged Tg mice with respect to young Tg mice or WT.
We propose that the Aβ deposits may induce nAChRs, which in turn induce increased choline-sterase (ChE) expression very early in the OB of APP SW Tg2576 mice suggesting that Aβ, ChE, and nAChR may be coordinately regulated and involved in OB dysfunction. Since inflammatory cytokines including IL-1β and TNFα contribute to Aβ deposition, interestingly we observed an increase of these cytokines in OB of APP SW Tg2576 mouse related to accumulation of Aβ and increase of ChE and nAChR expression. Alteration of the cholinergic system is a robust finding in AD and correlates with cognitive impairment [58–60]. An inflammatory response invariably accompanies elevated Aβ levels in the brain, and our study showed elevated brain inflammatory cytokine levels in APPSWE/PS1dE9 mice, signifying early and persistent activation of inflammatory processes. An age-related rise in MCP1 levels was also detected in Tg mice. MCP1 induces astrocyte chemotaxis and contributes to the recruitment of astrocytes around Aβ plaques. A similar elevation in TNFα levels was observed in older WT and Tg mice compared to the younger group. It should be noted that the results obtained by qPCR cannot be extrapolated to protein levels, so further studies will be required to confirm these results.
Conclusion
AD is a multifactorial disease that is not dependent on only one gene, or inciting insult. But there are a variety of aberrant genes present in AD patients that could be responsible for cognitive loss andphysical functions. In AD, the pro-inflammatory molecules and cholinergic system are involved and could represent a target for therapeutic intervention. The present study demonstrates the expression profiling of pro-inflammatory cytokines and cholinergic markers as well as Aβ in OB and EC of the APPSWE/PS1dE9 of Tg mice. Moreover, the study also demonstrates that APPSWE/PS1dE9 Tg mice can be useful as a mouse model to understand the role of pro-inflammatory cytokines and cholinergic markers in the pathophysiology of AD. Further investigations are in progress to demonstrate the molecular link between pro-inflammatory cytokines and cholinergic markers as well as Aβ in OB and EC of the APPSWE/PS1dE9 Tg mice.
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/17-0999r1).
REFERENCES
[1] | Reale M , Kamal MA , Velluto L , Gamb D , Di Nicola M , Greig NH ((2012) ) Relationship between inflammatory mediators, Aβ levels and ApoE genotype in Alzheimer disease. Curr Alzheimer Res 9: , 447–457. |
[2] | Reale M , Di Nicola M , Velluto L , D’Angelo C , Costantini E , Lahiri DK , Kamal MA , Yu Q-S , Greig NH ((2014) ) Selective acetyl- and butyrylcholinesterase inhibitors reduce amyloid-β ex vivo activation of peripheral chemo-cytokines from Alzheimer’s disease subjects: exploring the cholinergic anti-inflammatory pathway. Curr Alzheimer Res 11,: , 608–622. |
[3] | Cacquevel M , Lebeurrier N , Cheenne S , Vivien D ((2004) ) Cytokines in neuroinflammation and Alzheimer’s disease. Curr Drug Targets 6: , 529–534. |
[4] | Akiyama H , Barger S , Barnum S , Bradt B , Bauer J , Cole GM , Cooper NR , Eikelenboom P , Emmerling M , Fiebich BL , Finch CE , Frautschy S , Griffin WS , Hampel H , Hull M , Landreth G , Lue L , Mrak R , Mackenzie IR , McGeer PL , O’Banion MK , Pachter J , Pasinetti G , Plata-Salaman C , Rogers J , Rydel R , Shen Y , Streit W , Strohmeyer R , Tooyoma I , Van Muiswinkel FL , Veerhuis R , Walker D , Webster S , Wegrzyniak B , Wenk G , Wyss-Coray T ((2000) ) Inflammation and Alzheimer’s disease. Neurobiol Aging 21: , 383–421. |
[5] | Ekaterina S , Tarja M , Henna K , Riikka HH , Cindy G-T , Sara W , Rashid G , Jari K , Johanna M ((2016) ) Aβ and inflammatory stimulus activate diverse signaling pathways in monocytic cells: Implications in retaining phagocytosis in Aβ-laden environment. Front Cell Neurosci 10: , 279. |
[6] | Turola E , Furlan R , Bianco F , Matteoli M , Verderio C ((2012) ) Microglial microvesicle secretion and intercellular signaling. Front Physiol 3: , 149. |
[7] | Sawikr Y , Yarla NS , Peluso I , Kamal MA , Aliev G , Bishayee A ((2017) ) Neuroinflammation in Alzheimer’s disease: The preventive and therapeutic potential of polyphenolic nutraceuticals. Adv Protein Chem Struct Biol 108: , 33–57. |
[8] | Doty RL , Reyes P , Gregor T ((1987) ) Presence of both odor identification and detection deficits in Alzheimer’s disease. Brain Res Bull 18: , 597–600. |
[9] | Knupfer L , Spiegel R ((1986) ) Differences in olfactory test performance between normal aged, Alzheimer and vascular type dementia individuals. Int J Geriatr Psychiatry 1: , 3–14. |
[10] | Warner MD , Peabody CA , Flattery JJ , Tinklenberg JR ((1986) ) Olfactory deficits and Alzheimer’s disease. Biol Psychiatry 21: , 116–118. |
[11] | Devanand DP , Lee S , Manly J , Andrews H , Schupf N , Doty RL , Stern Y , Zahodne LB , Louis ED , Mayeux R ((2015) ) Olfactory deficits predict cognitive decline and Alzheimer dementia in an urban community. Neurology 84: , 182–189. |
[12] | Albers MW , Tabert MH , Devanand DP ((2006) ) Olfactory dysfunction as a predictor of neurodegenerative disease. Curr Neurol Neurosci. Rep 6: , 379–386. |
[13] | Kovacs T , Cairns NJ , Lantos PL ((2001) ) Olfactory centres in Alzheimer’s disease: Olfactory bulb is involved in early Braak’s stages. Neuroreport 12: , 285–288. |
[14] | Mesholam RI , Moberg PJ , Mahr RN , Doty RL ((1998) ) Olfaction in neurodegenerative disease: A meta-analysis of olfactory functioning in Alzheimer’s and Parkinson’s diseases. Arch Neurol 55: , 84–90. |
[15] | Doty RL , Bagla R , Kim N ((1999) ) Physostigmine enhances performance on an odor mixture discrimination test. Physiol Behav 65: , 801–804. |
[16] | Linster C , Cleland TA ((2002) ) Cholinergic modulation of sensory representations in the olfactory bulb. Neural Netw 15: , 709–717. |
[17] | Morris JC ((2002) ) Challenging assumptions about Alzheimer’s disease: Mild cognitive impairment and the cholinergic hypothesis. Ann Neurol 51: , 143–144. |
[18] | Yamada K , Nabeshima T ((2000) ) Animal models of Alzheimer’s disease and evaluation of anti-dementia drugs. Pharmacol Therapeut 88: , 93–113. |
[19] | Arendash GW , King DL , Gordon MN , Morgan D , Hatcher JM , Hope CE , Diamond DM ((2001) ) Progressive, age-related behavioral impairments intransgenic mice carrying both mutant amyloid precursor protein and presenilin-1 transgenes. Brain Res 891: , 42–53. |
[20] | Hsiao K , Chapman P , Nilsen S , Eckman C , Harigaya Y , Younkin S , Yang F , Cole G ((1996) ) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274: , 99–102. |
[21] | Kelly PH , Bondolfi L , Hunziker D , Schlecht HP , Carver K , Maguire E , Abramowski D , Wiederhold KH , Sturchler-Pierrat C , Jucker M , Bergmann R , Staufenbiel M , Sommer B ((2003) ) Progressive age-related impairment of cognitive behavior in APP23 transgenic mice. Neurobiol Aging 24: , 365–378. |
[22] | Allué JA , Sarasa L , Izco M , Pérez-Grijalba V , Fandos N , Pascual-Lucas M , Ogueta S , Pesini P , Sarasa M ((2016) ) Outstanding phenotypic differences in the profile of amyloid-β between Tg2576 and APPswe/PS1dE9 transgenic mouse models of Alzheimer’s disease. J Alzheimers Dis 53: , 773–785. |
[23] | Pistell PJ , Zhu M , Ingram DK ((2008) ) Acquisition of conditioned taste aversion is impaired in the amyloid precursor protein/presenilin 1 mouse model of Alzheimer’s disease. Neuroscience 52: , 594–600. |
[24] | Kilgore M , Miller CA , Fass DM , Hennig KM , Haggarty SJ , Sweatt JD , Rumbaugh G ((2010) ) Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 35: , 870–880. |
[25] | Reiserer RS , Harrison FE , Syverud DC , McDonald MP ((2007) ) Impaired spatial learning in the APPSwe + PSEN1DeltaE9 bigenic mouse model of Alzheimer’s disease. Genes Brain Behav 6: , 54–65. |
[26] | Zhang W , Hao J , Liu R , Zhang Z , Lei G , Su C , Miao J , Li Z ((2011) ) Soluble Abeta levels correlate with cognitive deficits in the 12-month-old APPswe/PS1dE9 mouse model of Alzheimer’s disease. Behav Brain Res 222: , 342–350. |
[27] | Finnie GS , Gunnarsson R , Manavis J , Blumbergs PC , Mander KA , Edwards S , Van den Heuvel C , Finnie JW ((2017) ) Characterization of an ‘amyloid only’ transgenic (B6C3 Tg(APPswe, PSEN1dE9)85Dbo/Mmjax) mouse model of Alzheimer’s disease. J Comp Pathol 156: , 389–399. |
[28] | Yan L , Deng Y , Gao J , Liu Y , Li F , Shi J , Gong Q ((2017) ) Icariside II effectively reduces spatial learning and memory impairments in Alzheimer’s disease model mice targeting beta-amyloid production. Front Pharmacol 8: , 106. |
[29] | Ryo Y , Takeuchi M , Ueda N , Ohi K , Kihara H , Shimada T , Uehara T , Kawasaki Y ((2017) ) Olfactory function in neuropsychiatricdisorders. Psychiatry Res 252: , 175–179. |
[30] | Khan UA , Liu L , Provenzano FA , Berman DE , Profaci CP , Sloan R , Mayeux R , Duff KE , Small SA ((2014) ) Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease. Nat Neurosci 17: , 304–311. |
[31] | Jankowsky JL , Slunt HH , Ratovitski T , Jenkins NA , Copeland NG , Borchelt DR ((2001) ) Co-expression of multiple transgenes in mouse CNS: A comparison of strategies. Biomol Eng 17: , 157–165. |
[32] | Paxinos G , Franklin KBJ ((2004) ) The Mouse Brain in Stereotaxic Coordinates, second edition. Gulf Professional Publishing, Elsevier, USA. |
[33] | Canto CB , Wouterlood FG , Witter MP ((2008) ) What does the anatomical organization of the entorhinal cortex tell us? Neural Plast 2008: , 381243. |
[34] | Small DH ((2004) ) Do acetylcholinesterase inhibitors boost synaptic scaling in Alzheimer’s disease? Trends Neurosci 27: , 245–249. |
[35] | Gems D , Partridge L ((2013) ) Genetics of longevity in model organisms: Debates and paradigm shifts. Annu Rev Physiol 75: , 621–644. |
[36] | Kirkwood TB ((2005) ) Understanding the odd science of aging. Cell 120: , 437–447. |
[37] | Vijg J , Campisi J ((2008) ) Puzzles, promises and a cure for ageing. Nature 454: , 1065–1071. |
[38] | Harkany T , Abraham I , Timmerman W , Laskay G , Toth B , Sasvari M , Kónya C , Sebens JB , Korf J , Nyakas C , Zarándi M , Soós K , Penke B , Luiten PG ((2000) ) Beta-amyloid neurotoxicity is mediated by a glutamate-triggered excitotoxic cascade in rat nucleus basalis. Eur J Neurosci 12: , 2735–2745. |
[39] | Frolich L ((2002) ) The cholinergic pathology in Alzheimer’s disease–discrepancies between clinical experience and pathophysiological findings. J Neural Transm (Vienna) 109: , 1003–1013. |
[40] | DeKosky ST ((2002) ) Neurobiology and molecular biology of Alzheimer’s disease. Rev Neurol 35: , 752–760. |
[41] | Kar S , Quirion R ((2004) ) Amyloid beta peptides and central cholinergic neurons: Functional interrelationship and relevance to Alzheimer’s disease pathology. Prog Brain Res 145: , 261–274. |
[42] | Gau D , Lemberger T , von Gall C , Kretz O , Le Minh N , Gass P , Schmid W , Schibler U , Korf HW , Schütz G ((2002) ) Phosphorylation of CREB Ser142 regulates light-induced phase shifts of the circadian clock. Neuron 34: , 245–253. |
[43] | Apelt J , Kumar A , Schliebs R ((2002) ) Impairment of cholinergic neurotransmission in adult and aged transgenic Tg2576 mouse brain expressing the Swedish mutation of human beta-amyloid precursor protein. Brain Res 953: , 17–30. |
[44] | Doty RL ((2011) ) Olfaction in Parkinson’s disease and related disorders. Neurobiol Dis 46: , 527–552. |
[45] | Peters JM , Hummel T , Kratzsch T , Lötsch J , Skarke C , Frölich L ((2003) ) Olfactory function in mild cognitive impairment and Alzheimer’s disease: An investigation using psychophysical and electrophysiological techniques. Am J Psychiatry 60: , 1995–2002. |
[46] | Buisson B , Bertrand D ((2002) ) Nicotine addiction: The possible role of functional upregulation. Trends Pharmacol Sci 23: , 130–136. |
[47] | Taly A , Corringer PJ , Guedin D , Lestage P , Changeux JP ((2009) ) Nicotinic receptors: Allosteric transitions and therapeutic targets in the nervous system. Nat Rev Drug Discov 8: , 733–750. |
[48] | Wallace TL , Porter RH ((2011) ) Targeting the nicotinic alpha7 acetylcholine receptor to enhance cognition in disease. Biochem Pharmacol 82: , 891–903. |
[49] | Barrantes FJ , Borroni V , Valles S ((2010) ) Neuronal nicotinic acetylcholine receptor-cholesterol crosstalk in Alzheimer’s disease. FEBS Lett 584: , 1856–1863. |
[50] | Yu W , Mechawar N , Krantic S , Chabot JG , Quirion R ((2012) ) Upregulation of astrocytic α7 nicotinic receptors in Alzheimer’s disease brain- possible relevant to amyloid pathology. Mol Neurodegen 7: , O7. |
[51] | Dineley KT , Westerman M , Bui D , Bell K , Ashe KH , Sweatt JD ((2001) ) Beta-amyloid activates the mitogen-activated protein kinase cascade via hippocampal alpha7 nicotinic acetylcholine receptors: In vitro and in vivo mechanisms related to Alzheimer’s disease. J Neurosci 21: , 4125–4133. |
[52] | Bednar I , Paterson D , Marutle A , Pham TM , Svedberg M , Hellström-Lindahl E , Mousavi M , Court J , Morris C , Perry E , Mohammed A , Zhang X , Nordberg A ((2002) ) Selective nicotinic receptor consequences in APP(SWE) transgenic mice. Mol Cell Neurosci 20: , 354–365. |
[53] | Jones IW , Westmacott A , Chan E , Jones RW , Dineley K , O’Neill MJ , Wonnacott S ((2006) ) Alpha7 nicotinic acetylcholine receptor expression in Alzheimer’s disease: Receptor densities in brain regions of the APP(SWE) mouse model and in human peripheral blood lymphocytes. J Mol Neurosci 30: , 83–84. |
[54] | Boncristiano S , Calhoun ME , Kelly PH , Pfeifer M , Bondolfi L , Stalder M , Phinney AL , Abramowski D , Sturchler-Pierrat C , Enz A , Sommer B , Staufenbiel M , Jucker M ((2002) ) Cholinergic changes in the APP23 transgenic mouse model of cerebral amyloidosis. J Neurosci 22: , 3234–3243. |
[55] | Morán MA , Mufson EJ , Gomez-Ramos P ((1993) ) Colocalization of cholinesterases with beta amyloid protein in aged and Alzheimer’s brains. Acta Neuropathol (Berl) 85: , 362–369. |
[56] | Guillozet AL , Smiley JF , Marsh DC , Mesulam MM ((1997) ) Butyrylcholinesterase in the life cycle of amyloid plaques. Ann Neurol 42: , 909–918. |
[57] | Garcia-Alloza M , Robbins EM , Zhang-Nunes SX , Purcell SM , Betensky RA , Raju S , Prada C , Greenberg SM , Bacskai BJ , Frosch MP ((2006) ) Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis 24: , 516–524. |
[58] | Collerton D ((1986) ) Cholinergic function and intellectual decline in Alzheimer’s disease. Neurosci 19: , 1–28. |
[59] | DeKosky ST , Harbaugh RE , Schmitt FA , Bakay RA , Chui HC , Knopman DS , Reeder TM , Shetter AG , Senter HJ , Markesbery WR ((1992) ) Cortical biopsy in Alzheimer’s disease: Diagnostic accuracy and neurochemical, neuropathological, and cognitive correlations. Intraventricular Bethanecol Study Group. Ann Neurol 32: , 625–632. |
[60] | Bierer LM , Haroutunian V , Gabriel S , Knott PJ , Carlin LS , Purohit DP , Perl DP , Schmeidler J , Kanof P , Davis KL ((1995) ) Neurochemical correlates of dementia severity in Alzheimer’s disease: Relative importance of the cholinergic deficits. J Neurochem 64: , 749–760. |


