
Influence of Lewy Pathology on Alzheimer’s Disease Phenotype: A Retrospective Clinico-Pathological Study
Abstract
Background:
Studies have shown the frequent coexistence of Lewy pathology (LP) in Alzheimer’s Disease (AD).
Objective:
The aim of this study was to determine the influence of LP on the clinical and cognitive phenotype in a cohort of patients with a neuropathological diagnosis of AD.
Methods:
We reviewed neuropathologically proven AD cases, reaching Braak stages V and VI in the brain banks of Lille and Paris between 1993 and 2016, and classified them according to LP extension (amygdala, brainstem, limbic, or neocortical). We then searched patient files for all available clinical and neuropsychiatric features and neuropsychological data.
Results:
Thirty-three subjects were selected for this study, among which 16 were devoid of LP and 17 presented AD with concomitant LP. The latter were stratified into two subgroups according to LP distribution: 7 were AD with amygdala LP and 10 were AD with ‘classical’ (brainstem, limbic or neocortical) LP. When analyzing the incidence of each clinical feature at any point during the disease course, we found no significant difference in symptom frequency between the three groups. However, fluctuations appeared significantly earlier in patients with classical LP (2±3.5 years) than in patients without LP (7±1.7 years) or with amygdala LP (8±2.8 years; p < 0.01). There was no significant difference in cognitive profiles.
Conclusion:
Our findings suggest that the influence of LP on the clinical phenotype of AD is subtle. Core features of dementia with Lewy bodies do not allow clinical diagnosis of a concomitant LP on a patient-to-patient basis.
INTRODUCTION
Alzheimer’s disease (AD) and dementia with Lewy bodies (DLB) are two leading causes of degenerative dementia in the elderly. AD, on the one hand, is pathologically characterized by intraneuronal inclusions of tau protein forming neurofibrillary tangles, and by extracellular deposits of amyloid-β peptide (Aβ) in senile plaques [1]. While senile plaques are widespread early in the course of AD, neurofibrillary tangles progress according to Braak stages from the transentorhinal cortex (stage 1) to the neocortex (stages 5-6), where AD becomes fully symptomatic [1, 2]. Lewy pathology (LP) on the other hand are intraneuronal inclusions of α-synuclein forming Lewy bodies and Lewy neurites. The anatomical extension of LP has been divided into three main subtypes (brainstem, limbic [transitional], and diffuse neocortical) [3, 4]. DLB is mostly diagnosed when LP reaches this latest stage. Additionally, LP can be concentrated in the amygdala without significant involvement of the brainstem, limbic areas or the neocortex [3].
However, AD and DLB are often intertwined. Studies have shown the frequent coexistence of LP in AD [5–7], a situation referred to as LB variant of AD, AD with DLB or AD with LBs in the literature. Furthermore, up to one third of AD cases without significant LP in the brainstem, limbic areas or neocortex present with LP in the amygdala [8, 9], a condition termed AD with amygdala Lewy bodies.
Several studies have tried to determine whether LP had an influence on the clinical phenotype of AD. Findings have been inconsistent so far. For instance, Stern et al. did not observe any relation between LP and AD clinical phenotype [10]. In a much broader retrospective clinic-pathological cohort, Chung et al. showed that patients who had AD with LBs had higher Neuropsychiatric Inventory Questionnaire (NPI-Q) and Unified Parkinson Disease Rating Scale (UPDRS) motor scores as compared with those without [11]. However, there were no data on specific LP subtypes. In particular, the clinical correlates of AD with amygdala LP remain notoriously elusive.
Identification of concomitant LP may however be crucial for AD management. DLB patients have a cognitive profile characterized by fluctuations, prominent attentional deficits, and executive and visuospatial dysfunction, which requires specific cognitive training and caregiver education. Therapeutic approaches must take into account the potential reduction of neuropsychiatric symptoms with cholinesterase inhibitors and the sensitivity to serious adverse events with antipsychotics. Furthermore, acknowledgement of a concomitant pathology contributing to cognitive dysfunction is paramount for clinical trials targeting AD pathology.
The aim of this retrospective clinic-pathological study was to determine the influence of LP according to its anatomical extension in AD. Clinical and neuropsychological characteristics of patients with a neuropathological diagnosis of AD were compared in different LP subtypes.
MATERIALS AND METHODS
Subjects
We reviewed cases with autopsy-proven AD in Lille and Paris brain banks between 1993 and 2016. To avoid any ambiguity as to whether some cases should have been requalified as probable DLB, AD diagnosis was retained only if National Institute on Aging– Alzheimer’s Association (NIA-AA) pathology score was high and Braak neurofibrillary pathology score was V-VI [2, 12]. Depending on the presence and extent of concomitant LP, this situation corresponds to a zero, low or intermediate likelihood of DLB [4]. Additionally, we excluded the subjects with confounding diagnoses, including significant cerebrovascular disease. Subjects had to have been followed up by an expert memory clinic with all clinical and neuropsychological data available.
Neuropathological assessment
Autopsies were performed in Lille, Paris, Marseille, Nice, and Montpellier University Hospitals and brains were recovered in the departments of neuropathology of Lille (n = 22) and Paris (n = 11). AD diagnosis was established according to the NIA-AA criteria [12]. LP was screened according to current protocols using a monoclonal antibody against α-synuclein. The extension of LP was classified into one of the aforementioned subtypes [3].
Informed consent for autopsy was obtained before death from the patient or at the time of death from the next of kin.
Clinical and neuropsychological assessment
Demographic data included sex, age at onset, age at the initial visit, age at death and education. Disease duration was calculated based on age at onset and age at death.
For each patient, all the available reports were searched for clinical features (parkinsonism, fluctuations) and neuropsychiatric symptoms (hallucinations, delusion, anxiety, apathy, irritability, disinhibition, depression and agitation). Each symptom was placed on a timeline from disease onset until death, to report their appearance and duration during the course of AD. We collected Mini-Mental Status Examination (MMSE), Mattis Dementia Rating Scale (MDRS), and Frontal Assessment Battery (FAB) scores when they were informed and placed them on the time line [13–15]. The clinical diagnoses were collected as well at each visit.
Statistical analysis
The results of quantitative variables are presented as a mean or median±standard deviation (SD). For dichotomous variables, numbers and calculated percentages are presented. The comparative analyses of clinical and demographic data were performed using Fisher’s and chi-square tests for dichotomous variables and one-way analysis of variance for quantitative variables. Results were deemed significant if p < 0.05.
RESULTS
Study population
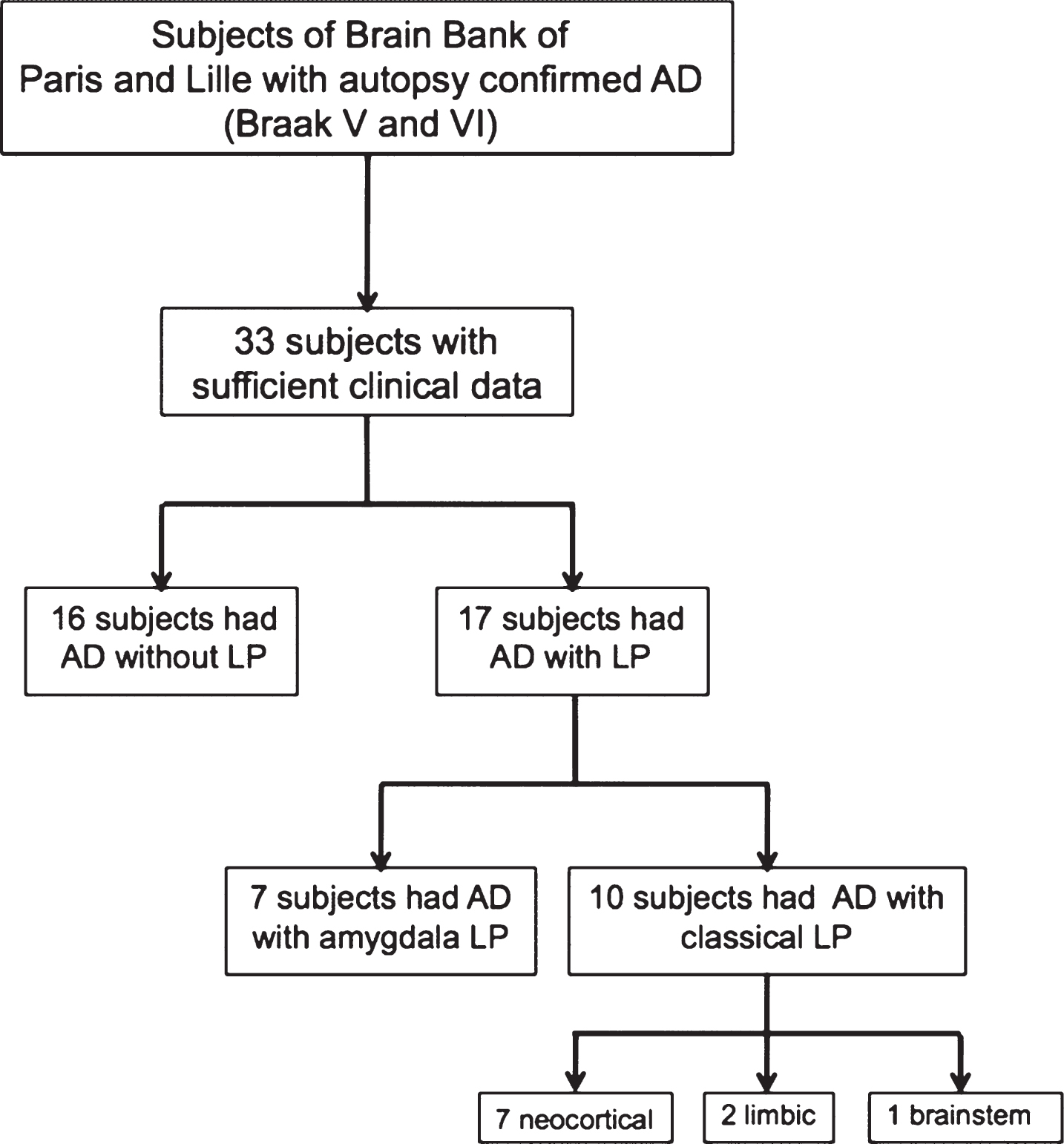
Thirty-three subjects were selected for this study, among which 16 were AD without LP and 17 were AD with LP. The 17 subjects with LP were stratified into 2 groups based on LP distribution. Seven subjects had AD with amygdala LP and 10 had AD with limbic, brainstem or diffuse neocortical LP, hereafter referred to as ‘classical’ LP (Fig. 1).
Fig.1
Flow chart. AD, Alzheimer disease; LP, Lewy pathology.
Demographic characteristics
The characteristics of patients with pure AD, AD with amygdala LP and AD with extra-amygdala LP are summarized in Table 1. Disease duration, education and sex distribution did not significantly differ between groups. The mean age at onset was significantly lower for subjects who had AD with amygdala LP (53.6±4.4 years) than for subjects who had AD without LP (61.9±8.4 years) or AD with classical LP (66.2±9.6 years; p = 0.01).
Table 1
Demographic characteristics of patients with pure AD, AD with amygdala LP and AD with classical LP
| Subjects who had AD | ||||
| Without LP (n = 16) | Amygdala LP (n = 7) | Classical* LP (n = 10) | p | |
| Characteristics | Mean (SD) | Mean (SD) | Mean (SD) | |
| Age of onset (y) | 61.9 (8.4) | 53.6 (4.4) | 66.2 (9.6) | 0.01 |
| Age of initial visit (y) | 66.6 (7.9) | 56.7 (4.8) | 68.6 (6.8) | 0.004 |
| Age of death (y) | 72.0 (8.2) | 65.0 (3.4) | 73.5 (6.8) | 0.08 |
| Disease duration (y) | 9.9 (4.0) | 11.4 (1.7) | 9.5 (3.1) | 0.50 |
| Education (y) | 14.6 (4.0) | 13.9 (4.3) | 15.4 (4.3) | 0.75 |
| Female sex, No. (%) | 7 (43.8) | 1 (14.3) | 7 (70.0) | 0.08 |
*neocortical, limbic and brainstem Lewy bodies. AD, Alzheimer disease; LP, Lewy pathology.
Antemortem clinical diagnosis
Of the 16 subjects without LP, 14 (88%) were diagnosed with AD clinically diagnosed, among which one was diagnosed with AD with DLB. The remainder was diagnosed with DLB (n = 1) and semantic dementia (n = 1).
Of the 17 subjects with LP, 15 (88%) were diagnosed with AD before death, among which one was correctly diagnosed with AD with DLB. The remainder was diagnosed with DLB (n = 1) and corticobasal degeneration (n = 1). All 7 subjects with amygdala LP were diagnosed with AD.
Overall, of the 33 pure or combined AD patients included in the study, 29 (88%) were correctly diagnosed with AD antemortem.
Clinical characteristics
Comparisons of the clinical features between patients with pure AD, AD with amygdala LP and AD with classical LP is summarized in Tables 2 and 3.
Table 2
Clinical characteristics: Frequency of clinical symptoms of patients with pure AD, AD with amygdala LP and AD with classical LP
| Subjects who had AD | ||||
| Without LP | Amygdala LP | Classical* LP | p | |
| Clinical symptoms | (n = 16) n (%) | (n = 7) n (%) | (n = 10) n (%) | |
| Parkinsonism | 8 (44) | 3 (43) | 8 (80) | 0.13 |
| Fluctuations | 6 (38) | 2 (29) | 4 (40) | 1 |
| Hallucinations | 8 (50) | 5 (71) | 5 (50) | 0.66 |
| Delusions | 4 (25) | 1 (14) | 2 (20) | 1 |
| Anxiety | 6 (38) | 6 (86) | 4 (40) | 0.11 |
| Apathy | 5 (31) | 4 (57) | 1 (10) | 0.13 |
| Irritability | 6 (38) | 1 (14) | 0 (0) | 0.06 |
| Disinhibition | 3 (19) | 2 (29) | 2 (20) | 0.87 |
| Depression | 6 (38) | 3 (43) | 3 (30) | 0.90 |
| Aggressive behavior | 5 (31) | 4 (57) | 5 (50) | 0.47 |
*neocortical, limbic and brainstem Lewy bodies. AD, Alzheimer disease; LP, Lewy pathology.
Table 3
Clinical characteristics: Delay of clinical symptoms of patients with pure AD, AD with amygdala LP and AD with classical LP
| Subjects who had AD | ||||
| Without LP (n = 16) | Amygdala LP (n = 7) | Classical* LP (n = 10) | p | |
| Clinical symptoms delay (y) | Mean (SD) | Mean (SD) | Mean (SD) | |
| Parkinsonism | 6.4 (4.5) | 6.7 (3.5) | 4.6 (3.5) | 0.61 |
| Fluctuations | 7.0 (1.7) | 8.0 (2.8) | 2.0 (3.5) | <0.01 |
| Hallucinations | 8.8 (3.9) | 7.6 (3.4) | 4.6 (3.9) | 0.18 |
| Delusions | 9.3 (4.0) | 8.0 (0) | 6.0 (7.1) | NC |
| Anxiety | 8.3 (4.9) | 5.5 (2.4) | 3.3 (2.9) | 0.13 |
| Apathy | 5.8 (5.7) | 6.3 (2.6) | 3.0 (0) | NC |
| Irritability | 4.8 (3.8) | 9.0 (0) | 0 (0) | NC |
| Disinhibition | 8.7 (5.7) | 8.0 (4.2) | 10.0 (1.4) | 0.54 |
| Depression | 4.2 (4.0) | 2.7 (3.1) | 2.0 (1.0) | 0.63 |
| Aggressive behavior | 9.2 (3.9) | 5.8 (3.4) | 6.6 (3.8) | 0.37 |
*neocortical, limbic and brainstem Lewy bodies. AD, Alzheimer disease; LP, Lewy pathology; NC, non calculable.
When analyzing the incidence of each clinical feature at any point during the course of the disease, we found no significant difference between the three groups. Parkinsonism was more frequent in AD with classical LP (80% versus 44% in AD without LP and 43% in AD with amygdala LP) but results failed to reach significance. Hallucinations were not specific to AD with classical LP (50%) but were also common in AD without LP (50%) and in AD with amygdala LP (71%). Anxiety and apathy appeared to be more frequent in AD with amygdala LP (86% and 57%) than in AD without LP (38% and 31%) and AD with classical LP (40% and 10%, respectively) but then again, results were not significant.
We then looked for the delay of onset of each symptom from the start of the disease. Although there was no difference in the frequency of fluctuations between groups, fluctuations appeared significantly earlier in patients with classical LP (2±3.5 years) than in patients with AD without LP (7±1.7 years) or with amygdala LP (8±2.8 years; p < 0.01). Hallucinations and anxiety tended to appear earlier in AD with classical LP but results were not significant.
Neuropsychological tests
Comparison of the MMSE, FAB and MDRS scores between the three groups is summarized in Table 4. Analysis of covariance revealed no significant differences. Analysis of FAB subscores (conceptualization, mental flexibility, programming, sensitivity to interference, inhibitory control and environmental autonomy) as well as analysis of MDRS subscores (attention, initiation, construction, conceptualization and memory) revealed no significant differences.
Table 4
Neuropsychological data of patients with pure AD, AD with amygdala LP and AD with classical LP
| Subjects who had AD | ||||
| Without LP | Amygdala LP | Classical* LP | p | |
| Neuropsychological data | Mean (SD) | Mean (SD) | Mean (SD) | |
| First MMSE score | n = 16 | n = 7 | n = 10 | 0.81 |
| Score/30 | 18.6 (7.1) | 16.6 (6.7) | 17.8 (6.9) | |
| FAB | n = 10 | n = 5 | n = 7 | 0.52 |
| Score/18 | 9.6 (3.7) | 7.2 (2.9) | 8.6 (4.5) | |
| MDRS | n = 7 | n = 4 | n = 6 | 0.62 |
| Score/144 | 107.7 (17.7) | 111.3 (12.9) | 101.8 (13.3) | |
*neocortical, limbic and brainstem Lewy bodies. AD, Alzheimer disease; LP, Lewy pathology; MMSE, Mini-Mental State Examination; FAB, Frontal Assessment Battery; MDRS, Mattis Dementia Rating Scale.
DISCUSSION
The main finding of this retrospective clinico-pathological study is that concomitant LP in AD is not reliably diagnosed in expert memory centers. We found that LP was correctly predicted in only 2 of 17 AD cases, one of which was thought to be ‘pure’ DLB. Sensitivity of the clinical diagnosis of LP was especially low (2/17 cases with any kind of LP and 2/8 cases with classical LP). Interestingly, positive predictive value was low as well, since only 2/5 cases with a predicted Lewy body disease did have LP at autopsy.
Noticeably, fluctuations appeared significantly earlier in AD with classical LP. The importance of fluctuations in DLB diagnosis has been confirmed in the latest clinical criteria [4]. Of the three ‘historical’ core clinical features, i.e., parkinsonism, hallucinations, and fluctuations, it is the most counter-intuitive feature. Accurate diagnosis of fluctuations requires training, and the use of semi-structured questionnaires provides a valuable help [16]. Yet fluctuations stand among the most specific features of DLB. Like other core features, fluctuations can be part of ‘pure’ AD in later stages [17]. However, our results show that it might be the feature that best differentiates ‘pure’ AD from AD with classical LB when it is present early in the course of the disease. In contrast to our findings, a recent clinico-pathological study by Thomas et al. showed that the emergence of complex visual hallucinations in the course of AD suggested the presence of LP. However fluctuations at baseline or in the course of the disease was not associated with LP [18]. The reasons for this discrepancy are unclear and might be due to the younger age of our patients, the simpler characterization of hallucinations and the better characterization of fluctuations and chronological appearance of symptoms in our work.
DLB is classically characterized by cortical and sub-cortical impairments with attentional deficits and executive and visuospatial dysfunction [4]. However, this distinctive cognitive profile seems to wane when AD pathology is present. We did not find any significant differences between subgroups in FAB or MDRS scores and subscores. Consistent with our findings, previous studies showed that patients with AD with LP did not differ significantly from ‘pure’ AD patients on neuropsychological tests [11, 19]. Only patients with ‘pure’ DLB performed worse on measures of executive function and attention [19]. Likewise, we did not show any difference in behavioral and psychological symptom beyond hallucinations. We did not confirm in our sample the greater prevalence of agitation and delusion previously reported in the in LB variant of AD [11, 20, 21]. Last, disease duration was not significantly different between subgroups in our sample. Literature generally supports a faster decline in the Lewy variant of AD. However results are inconsistent when it comes to disease duration ([11, 19, 22], reviewed in [23]). Overall, our findings are similar to that of other comparative studies [24, 25] suggesting that the Lewy variant of AD is hard to distinguish from pure AD on the basis of cognitive profile and core clinical features. There is a complex paradoxical interplay between AD and LP. Indeed cognitive decline in DLB depends on AD pathology, but AD pathology alters the clinical phenotype and masks LP-specific symptoms [18, 26, 27].
Interestingly, we found a younger age at onset in AD with amygdala LP. With the noticeable exception of Chung et al. [11], most studies have found no significant differences in age at onset in AD with or without LP [21, 25, 28, 29]. However, to our knowledge, there was no specific data on the amygdala variant. It may be that concomitant pathologies on the medial temporal lobe lower the threshold for AD to become symptomatic and precipitate disease onset, as was shown for argyrophilic grain disease [30].
AD with amygdala LP is generally thought to be an incidental finding without clinical correlate [8, 19]. Yet amygdala is a major component of the limbic system and has a major role in memory, learning, motivation, emotional regulation and autonomic control [31]. Interestingly we found that anxiety and apathy were more frequent in AD with amygdala LP, but results fell short to be significant. Likewise, Lopez et al. showed that AD with amygdala LP was associated with a higher prevalence of major depression [32], a finding that is not universal [8]. Future studies should continue to decipher the clinical phenotype associated with amygdala LP.
Our study has several weaknesses. First, the lack of standardized scales to evaluate behavioral and psychological symptoms, fluctuation, parkinsonism or sleep disorders has to be acknowledged. Second, scarce or absent data on dysautonomia, olfactory impairment, and sleep disorders may have precluded identification of specific clinical markers of LP that could resist AD pathology [33–35]. Last, the low sample size is obviously the main limitation of our work as compared with others [11, 36]. However, we performed in-depth analysis of clinical reports from three specialized memory clinics. Data search was unbiased, blinded to neuropathological results, looking for clinical features that could allow prediction of LP in AD. We posit that although limited, the sample size would have allowed for detection of clinically relevant differences that could be of use for diagnosis.
In conclusion, our findings suggest that LP does not sufficiently alter the clinical presentation of AD to be diagnosed in a patient-to-patient basis. While bigger cohorts could identify differences, the clinical overlap with AD precludes DLB core features to be used as reliable diagnostic markers of LP in AD. Early fluctuation is perhaps the best clinical clue of LP in AD. Last, our study contributes to decipher the clinical phenotype associated with LP in the amygdala.
ACKNOWLEDGMENTS
The authors wish to thank Lille NeuroBank, the Association pour la Recherche en Neuroimagerie et Neuropsychologie (ARNN), the Fondation pour la Recherche sur Alzheimer (FRA) and the Neuro-CEB for providing brains and medical reports.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/17-0914r1).
REFERENCES
[1] | Duyckaerts C , Delatour B , Potier M-C ((2009) ) Classification and basic pathology of Alzheimer disease. Acta Neuropathol 118: , 5–36. |
[2] | Braak H , Alafuzoff I , Arzberger T , Kretzschmar H , Del Tredici K ((2006) ) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112: , 389–404. |
[3] | Alafuzoff I , Ince PG , Arzberger T , Al-Sarraj S , Bell J , Bodi I , Bogdanovic N , Bugiani O , Ferrer I , Gelpi E , Gentleman S , Giaccone G , Ironside JW , Kavantzas N , King A , Korkolopoulou P , Kovács GG , Meyronet D , Monoranu C , Parchi P , Parkkinen L , Patsouris E , Roggendorf W , Rozemuller A , Stadelmann-Nessler C , Streichenberger N , Thal DR , Kretzschmar H ((2009) ) Staging/typing of Lewy body related α-synuclein pathology: A study of the BrainNet Europe Consortium. Acta Neuropathol 117: , 635–652. |
[4] | McKeith IG , Boeve BF , Dickson DW , Halliday G , Taylor J-P , Weintraub D , Aarsland D , Galvin J , Attems J , Ballard CG , Bayston A , Beach TG , Blanc F , Bohnen N , Bonanni L , Bras J , Brundin P , Burn D , Chen-Plotkin A , Duda JE , El-Agnaf O , Feldman H , Ferman TJ , Ffytche D , Fujishiro H , Galasko D , Goldman JG , Gomperts SN , Graff-Radford NR , Honig LS , Iranzo A , Kantarci K , Kaufer D , Kukull W , Lee VMY , Leverenz JB , Lewis S , Lippa C , Lunde A , Masellis M , Masliah E , McLean P , Mollenhauer B , Montine TJ , Moreno E , Mori E , Murray M , O’Brien JT , Orimo S , Postuma RB , Ramaswamy S , Ross OA , Salmon DP , Singleton A , Taylor A , Thomas A , Tiraboschi P , Toledo JB , Trojanowski JQ , Tsuang D , Walker Z , Yamada M , Kosaka K ((2017) ) Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 89: , 88–100. |
[5] | Hamilton RL ((2000) ) Lewy bodies in Alzheimer’s disease: A neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol 10: , 378–384. |
[6] | Parkkinen L , Soininen H , Alafuzoff I ((2003) ) Regional distribution of alpha-synuclein pathology in unimpaired aging and Alzheimer disease. J Neuropathol Exp Neurol 62: , 363–367. |
[7] | Murray ME , Graff-Radford NR , Ross OA , Petersen RC , Duara R , Dickson DW ((2011) ) Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: A retrospective study. Lancet Neurol 10: , 785–796. |
[8] | Uchikado H , Lin W-L , DeLucia MW , Dickson DW ((2006) ) Alzheimer disease with amygdala Lewy bodies: A distinct form of alpha-synucleinopathy. J Neuropathol Exp Neurol 65: , 685–697. |
[9] | Popescu A , Lippa CF , Lee VMY , Trojanowski JQ ((2004) ) Lewy bodies in the amygdala: Increase of alpha-synuclein aggregates in neurodegenerative diseases with tau-based inclusions. Arch Neurol 61: , 1915–1919. |
[10] | Stern Y , Jacobs D , Goldman J , Gomez-Tortosa E , Hyman BT , Liu Y , Troncoso J , Marder K , Tang MX , Brandt J , Albert M ((2001) ) An investigation of clinical correlates of Lewy bodies in autopsy-proven Alzheimer disease. Arch Neurol 58: , 460–465. |
[11] | Chung EJ , Babulal GM , Monsell SE , Cairns NJ , Roe CM , Morris JC ((2015) ) Clinical features of Alzheimer disease with and without Lewy bodies. JAMA Neurol 72: , 789–12. |
[12] | Hyman BT , Phelps CH , Beach TG , Bigio EH , Cairns NJ , Carrillo MC , Dickson DW , Duyckaerts C , Frosch MP , Masliah E , Mirra SS , Nelson PT , Schneider JA , Thal DR , Thies B , Trojanowski JQ , Vinters HV , Montine TJ ((2012) ) National Institute on Aging– Alzheimer“s Association guidelines for the neuropathologic assessment of Alzheimer”s disease. Alzheimers Dement 8: , 1–13. |
[13] | Folstein MF , Folstein SE , McHugh PR ((1975) ) “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12: , 189–198. |
[14] | Schmidt R , Freidl W , Fazekas F , Reinhart B , Grieshofer P , Koch M , Eber B , Schumacher M , Polmin K , Lechner H ((1994) ) The Mattis Dementia Rating Scale: Normative data from 1,001 healthy volunteers. Neurology 44: , 964–964. |
[15] | Dubois B , Slachevsky A , Litvan I , Pillon B ((2000) ) The FAB: A frontal assessment battery at bedside. Neurology 55: , 1621–1626. |
[16] | Ferman TJ , Smith GE , Boeve BF , Ivnik RJ , Petersen RC , Knopman D , Graff-Radford N , Parisi J , Dickson DW ((2004) ) DLB fluctuations: Specific features that reliably differentiate DLB from AD and normal aging. Neurology 62: , 181–187. |
[17] | Escandon A , Al-Hammadi N , Galvin JE ((2010) ) Effect of cognitive fluctuation on neuropsychological performance in aging and dementia. Neurology 74: , 210–217. |
[18] | Thomas AJ , Mahin-Babaei F , Saidi M , Lett D , Taylor J-P , Walker L , Attems J ((2018) ) Improving the identification of dementia with Lewy bodies in the context of an Alzheimer’s-type dementia. Alzheimers Res Ther 10: , 27. |
[19] | Kraybill ML , Larson EB , Tsuang DW , Teri L , McCormick WC , Bowen JD , Kukull WA , Leverenz JB , Cherrier MM ((2005) ) Cognitive differences in dementia patients with autopsy-verified AD, Lewy body pathology, or both. Neurology 64: , 2069–2073. |
[20] | Hansen L , Salmon D , Galasko D , Masliah E , Katzman R , DeTeresa R , Thal L , Pay MM , Hofstetter R , Klauber M ((1990) ) The Lewy body variant of Alzheimer’s disease: A clinical and pathologic entity. Neurology 40: , 1–8. |
[21] | Weiner MF , Risser RC , Cullum CM , Honig L , White C , Speciale S , Rosenberg RN ((1996) ) Alzheimer’s disease and its Lewy body variant: A clinical analysis of postmortem verified cases. Am J Psychiatry 153: , 1269–1273. |
[22] | Olichney JM , Galasko D , Salmon DP , Hofstetter CR , Hansen LA , Katzman R , Thal LJ ((1998) ) Cognitive decline is faster in Lewy body variant than in Alzheimer’s disease. Neurology 51: , 351–357. |
[23] | Breitve MH , Chwiszczuk LJ , Hynninen MJ , Rongve A , Brønnick K , Janvin C , Aarsland D ((2014) ) A systematic review of cognitive decline in dementia with Lewy bodies versus Alzheimer’s disease. Alzheimers Res Ther 6: , 445–13. |
[24] | Lopez OL , Hamilton RL , Becker JT , Wisniewski S , Kaufer DI , DeKosky ST ((2000) ) Severity of cognitive impairment and the clinical diagnosis of AD with Lewy bodies. Neurology 54: , 1780–1787. |
[25] | Yoshizawa H , Vonsattel JPG , Honig LS ((2013) ) Early neuropsychological discriminants for Lewy body disease: An autopsy series. J Neurol Neurosurg Psychiatry 84: , 1326–1330. |
[26] | Merdes AR , Hansen LA , Jeste DV , Galasko D , Hofstetter CR , Ho GJ , Thal LJ , Corey-Bloom J ((2003) ) Influence of Alzheimer pathology on clinical diagnostic accuracy in dementia with Lewy bodies. Neurology 60: , 1586–1590. |
[27] | Walker L , McAleese KE , Thomas AJ , Johnson M , Martin-Ruiz C , Parker C , Colloby SJ , Jellinger K , Attems J ((2015) ) Neuropathologically mixed Alzheimer’s and Lewy body disease: Burden of pathological protein aggregates differs between clinical phenotypes. Acta Neuropathol 129: , 729–748. |
[28] | Samuel W , Galasko D , Masliah E , Hansen LA ((1996) ) Neocortical lewy body counts correlate with dementia in the Lewy body variant of Alzheimer’s disease. J Neuropathol Exp Neurol 55: , 44–52. |
[29] | Connor DJ , Salmon DP , Sandy TJ , Galasko D , Hansen LA , Thal LJ ((1998) ) Cognitive profiles of autopsy-confirmed Lewy body variant vs pure Alzheimer disease. Arch Neurol 55: , 994–1000. |
[30] | Thal DR , Arnim von CAF , Griffin WST , Mrak RE , Walker L , Attems J , Arzberger T ((2015) ) Frontotemporal lobar degeneration FTLD-tau: Preclinical lesions, vascular, and Alzheimer-related co-pathologies. J Neural Transm (Vienna) 122: , 1007–1018. |
[31] | Murray EA ((2007) ) The amygdala, reward and emotion. Trends Cogn Sci 11: , 489–497. |
[32] | Lopez OL , Becker JT , Sweet RA , Martin-Sanchez FJ , Hamilton RL ((2006) ) Lewy bodies in the amygdala increase risk for major depression in subjects with Alzheimer disease. Neurology 67: , 660–665. |
[33] | Allan LM , Ballard CG , Allen J , Murray A , Davidson AW , McKeith IG , Kenny RA ((2007) ) Autonomic dysfunction in dementia. J Neurol Neurosurg Psychiatry 78: , 671–677. |
[34] | Ferman TJ , Boeve BF , Smith GE , Lin SC , Silber MH , Pedraza O , Wszolek Z , Graff-Radford NR , Uitti R , Van Gerpen J , Pao W , Knopman D , Pankratz VS , Kantarci K , Boot B , Parisi JE , Dugger BN , Fujishiro H , Petersen RC , Dickson DW ((2011) ) Inclusion of RBD improves the diagnostic classification of dementia with Lewy bodies. Neurology 77: , 875–882. |
[35] | Williams SS , Williams J , Combrinck M , Christie S , Smith AD , McShane R ((2009) ) Olfactory impairment is more marked in patients with mild dementia with Lewy bodies than those with mild Alzheimer disease. J Neurol Neurosurg Psychiatry 80: , 667–670. |
[36] | Schneider JA , Arvanitakis Z , Yu L , Boyle PA , Leurgans SE , Bennett DA ((2012) ) Cognitive impairment, decline and fluctuations in older community-dwelling subjects with Lewy bodies. Brain 135: , 3005–3014. |


