
Update on Vascular Cognitive Impairment Associated with Subcortical Small-Vessel Disease
Abstract
Subcortical small-vessel disease (SSVD) is a disorder well characterized from the clinical, imaging, and neuropathological viewpoints. SSVD is considered the most prevalent ischemic brain disorder, increasing in frequency with age. Vascular risk factors include hypertension, diabetes, hyperlipidemia, elevated homocysteine, and obstructive sleep apnea. Ischemic white matter lesions are the hallmark of SSVD; other pathological lesions include arteriolosclerosis, dilatation of perivascular spaces, venous collagenosis, cerebral amyloid angiopathy, microbleeds, microinfarcts, lacunes, and large infarcts. The pathogenesis of SSVD is incompletely understood but includes endothelial changes and blood-brain barrier alterations involving metalloproteinases, vascular endothelial growth factors, angiotensin II, mindin/spondin, and the mammalian target of rapamycin pathway. Metabolic and genetic conditions may also play a role but hitherto there are few conclusive studies. Clinical diagnosis of SSVD includes early executive dysfunction manifested by impaired capacity to use complex information, to formulate strategies, and to exercise self-control. In comparison with Alzheimer’s disease (AD), patients with SSVD show less pronounced episodic memory deficits. Brain imaging has advanced substantially the diagnostic tools for SSVD. With the exception of cortical microinfarcts, all other lesions are well visualized with MRI. Diagnostic biomarkers that separate AD from SSVD include reduction of cerebrospinal fluid amyloid-β (Aβ)42 and of the ratio Aβ42/Aβ40 often with increased total tau levels. However, better markers of small-vessel function of intracerebral blood vessels are needed. The treatment of SSVD remains unsatisfactory other than control of vascular risk factors. There is an urgent need of finding targets to slow down and potentially halt the progression of this prevalent, but often unrecognized, disorder.
INTRODUCTION AND CLASSIFICATION
Since the 17th century, stroke has been recognized as a cause of age-associated cognitive impairment. However, until early in the 20th century, the prevailing concept of age-associated cognitive impairment causation was the impairment of cerebral blood flow (CBF) resulting from partial blockage of the vessels that supply the brain; thus, arteriosclerotic dementia resulting in global brain hypoperfusion was considered the most common form of major cognitive disorder in the elderly [1]. Early neuropathologists recognized subcortical ischemic white matter injury and lacunes as typical lesions resulting from small-vessel disease [2]. During the 1970s, amyloid-β (Aβ) plaques and neurofibrillary tangles were identified as critical components of the neurodegenerative process occurring in demented elderly patients [3]. Alzheimer’s disease (AD) became recognized as the most common form of major cognitive disorder and the notion of chronic brain ischemia as an explanation of progressive cognitive impairment was abandoned. The claim that vascular disease could lead to cognitive disorders not by means of CBF-related energy deficiency but by repeated stroke episodes resulting in cerebral tissue lesions led to the concept of multi-infarct dementia (MID). Although limited by scarce neuropathological support, the diagnosis of MID grew popular and was applied to a larger group of patients than originally proposed. The demonstration that vascular-related white matter damage (Binswanger disease) [4] contributes to cognitive impairment in the elderly began questioning multiple infarcts as the most important cause of vascular cognitive impairment (VCI) [5]. Aging of the population, along with changes in the panorama of cerebrovascular disease (CVD) in terms of reduced stroke mortality [6], have led to reevaluation and renewal of concepts in this area. Moreover, the role of CVD—particularly small-vessel disease—in the clinical expression and pathogenesis of AD has been increasingly recognized [7–9].
AD and VCI are the most common causes of cognitive impairment in the elderly, accounting together for more than 70–75% of cases [7]. They also share common risk factors and mechanisms including atherosclerosis, diabetes, and amyloid angiopathy [10–13]. Vascular comorbidity may be present in 30–60% of AD patients [11, 12], while AD pathology may be present in 40–80% of VCI patients [11, 12]. Indeed, the coexistence of the two disorders, termed mixed dementia (MXD), may be more common than “pure” AD or “pure” VCI [14, 15]; moreover, microscopic cortical infarcts, which are not visualized in neuroimaging, may contribute up to 1/5 to 1/3 in the variability of severity of cognitive impairment [16].
Neuropathology usually provides the final and definite diagnosis in clinical neurology. Therefore, there is unanimous accord that the clinical classification of cerebral small-vessel diseases should be based on the distinct neuropathological lesions [13, 17, 18]. Given the fact that VCI may be the result of a number of cerebrovascular pathologies (Fig. 1) resulting from numerous etiopathogenic causes (Table 1), the occurrence of several clinical forms of VCI (Table 2) is therefore understandable. Among the sporadic forms of VCI, particular attention over the last decades has been paid to post-stroke-associated cognitive impairment or post-stroke dementia (PSD); and, more recently to VCI associated with subcortical small-vessel disease (SSVD), a condition that we last reviewed over a decade ago [19]. Although small-vessel disease may cause cortical atrophy (cortical granular degeneration), and secondary localized atrophy from subcortical lacunar strokes, from the clinical viewpoint, however, most cases of cortical atrophy in elderly patients with cognitive impairment are the result of neurodegenerative conditions, not vascular disease. Thus, when approaching such patients with cognitive loss clinicians rely on the presence of “cortical” versus “subcortical” manifestations that allow a first diagnostic impression. These clinical features are confirmed subsequently by neuropsychological tests, brain imaging, and eventually by neuropathology. Therefore, we decided to maintain the time-honored name “subcortical” in the overall approach to this topic.
Fig.1
Newcastle categorization in six subtypes of different cerebrovascular pathologies associated with VCI. Post-stroke survivors are included in subtypes I–III. Cases with extensive WM disease in the absence of significant other features are included under SVD. Subtype I may result from large vessel occlusion (athero-thromboembolism), artery-to-artery embolism or cardioembolism. Subtype II usually involves arteriosclerosis, lipohyalinosis and hypertensive, arteriosclerotic, amyloid or collagen angiopathy. Subtypes I, II and V may result from aneurysms, arterial dissections, arteriovenous malformations and various forms of arteritis (vasculitis). AD, Alzheimer’s disease; CH, cerebral haemorrhage; CVD, cerebrovascular disease; MI, myocardial infarction; MID, multi-infarct dementia; LVD, large vessel disease; SIVD, subcortical ischemic vascular dementia; SVD, small vessel disease; VCI, vascular cognitive impairment; VaD, vascular dementia. From: Kalaria RN, 2016 [13].
![Newcastle categorization in six subtypes of different cerebrovascular pathologies associated with VCI. Post-stroke survivors are included in subtypes I–III. Cases with extensive WM disease in the absence of significant other features are included under SVD. Subtype I may result from large vessel occlusion (athero-thromboembolism), artery-to-artery embolism or cardioembolism. Subtype II usually involves arteriosclerosis, lipohyalinosis and hypertensive, arteriosclerotic, amyloid or collagen angiopathy. Subtypes I, II and V may result from aneurysms, arterial dissections, arteriovenous malformations and various forms of arteritis (vasculitis). AD, Alzheimer’s disease; CH, cerebral haemorrhage; CVD, cerebrovascular disease; MI, myocardial infarction; MID, multi-infarct dementia; LVD, large vessel disease; SIVD, subcortical ischemic vascular dementia; SVD, small vessel disease; VCI, vascular cognitive impairment; VaD, vascular dementia. From: Kalaria RN, 2016 [13].](https://content.iospress.com:443/media/jad/2018/62-3/jad-62-3-jad170803/jad-62-jad170803-g001.jpg)
Table 1
Pathologic basis of vascular cognitive disorders*
| Parenchymal lesions of vascular etiology |
| (1) Large vessel or atherothromboembolic disease |
| (a) Multiple infarcts |
| (b) Single strategically placed infarct |
| (2) Small vessel disease: |
| (a) Multiple lacunar infarcts in white matter and deep gray matter nuclei |
| (b) Ischemic white matter change |
| (c) Dilatation of perivascular spaces |
| (d) Microinfarcts (cortical and subcortical) and microhemorrhages |
| (3) Hemorrhage |
| (a) Intracerebral hemorrhage |
| (b) Multiple cortical and subcortical microbleeds |
| (c) Subarachnoid hemorrhage |
| (4) Hypoperfusion |
| (a) Hippocampal sclerosis |
| (b) Laminar cortical sclerosis |
| Types of vascular lesions |
| (1) Atherosclerosis |
| (2) Cardiac, atherosclerotic, and systemic emboli |
| (3) Arteriolosclerosis |
| (4) Lipohyalinosis |
| (5) Amyloid angiopathy |
| (6) Vasculitis—infectious and noninfectious |
| (7) Venous collagenosis |
| (8) Arteriovenous fistulae—dural or parenchymal |
| (9) Hereditary angiopathies cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL); cerebral autosomal recessive arteriopathy with subcortical autosomal recessive leukoencephalopathy (CARASIL); etc. |
| (10) Giant cell arteritis |
| (11) Berry aneurysms |
| (12) Miscellaneous vasculopathies—fibromuscular dysplasia, Moya-Moya |
| (13) Systemic microangiopathies without vascular inflammatory cell infiltrates |
| (14) Cerebral venous thrombosis |
*From Vascog [21].
Table 2
VICCCS proposed definitions of major VCI subtypes*
| Post-stroke dementia (PSD): A patient described as having PSD may or may not have presented evidence of mild cognitive impairment before stroke. The patient may exhibit immediate and/or delayed cognitive decline that begins after, but within 6 months, of stroke, that does not recover. PSD results from different vascular causes and changes in brain. It includes cases with multiple cortico-subcortical infarcts, strategic infarcts, subcortical ischemic vascular dementia, and various forms of neurodegenerative pathology, including AD, which develop within 6 months of stroke. This temporal basis for cognitive decline after stroke differentiates PSD from other forms of major VCI. |
| Mixed dementias (MXD): A stand-alone umbrella subgroup termed mixed dementias includes all the phenotypes specified for each combination, that is VCI-AD, VCI-DLB, so forth. It is recommended that a patient is referred to as having “VCI-AD”, according to the phenotypes present, rather than less specific mixed dementia, for example. Where discrimination is possible, the order of terms should reflect the relative contribution of the underlying pathology, that is AD-VCI or VCI-AD. |
| Subcortical ischemic small-vessel disease (SSVD): Small-vessel disease is the main vascular cause of VCI. Lacunar infarct and ischemic white matter lesions are the main type of brain lesions, which are primarily located subcortically. It incorporates the overlapping clinical entities of Binswanger’s disease and the lacunar state. |
| Multi-infarct dementia (MID): MID-relates to the involvement, and likely contribution, of multiple large cortical infarcts in the development of dementia. |
*From VICCCS [22].
The numerous diagnostic criteria proposed for the diagnosis of cognitive disorders resulting from vascular disease reflect the difficulties in the field [20]. The most recently updated definitions and criteria have been proposed by the International Society of Vascular Behavioural and Cognitive Disorders (VASCOG) [21] and by the Vascular Impairment of Cognition Classification Consensus Study (VICCCS) group [22, 23].
Based on ample evidence, there is current consensus on the notion that SSVD in the brain is probably the commonest and most prevalent vascular neurological lesion [24], occurring as part of a systemic dysfunction of arteriolar perfusion affecting mainly heavily perfused tissues such as brain, retina and kidneys [25, 26]. SSVD is ubiquitous in elderly patients with cognitive decline. SSVD presents with a more insidious course than PSD and has been associated with prefrontal symptoms affecting cognition, mood, behavior, gait, and bladder control. Moreover, SSVD has been associated with traditional vascular risk factors (VRFs) such as aging, hypertension, hyperlipidemia, diabetes, smoking, peripheral vascular and heart disease, and with common but frequently overlooked VRFs such as obstructive sleep apnea [27] and hyperhomocysteinemia resulting from dietary deficiencies of vitamin B12 and folate [28], as well as from mutations of the methylenetetrahydrofolate reductase (MTHFR) gene [29]. Also, SSVD provided unexpected links as the terrain that precedes and favors the development of neurodegeneration in AD and other cognitive disorders [7–15].
Finally, the pendulum is swinging back and new developments in brain imaging, in particular arterial spin-labeling (ASL) imaging on magnetic resonance imaging (MRI) – a method that allows noninvasive quantification of CBF [30–32]; as well as novel cerebrospinal fluid (CSF) biomarkers [33, 34], have revitalized the studies on hypoperfusion leading to cortical hypometabolism in some forms of cognitive impairment, by mechanisms different from carotid and intracranial large-vessel stenoses.
PATHOLOGY AND MECHANISM
The pathology of SSVD encompasses tissue injury to the cerebral white matter and to the vessels that supply blood to this tissue [15, 17–19]. One of the most important achievements in the past decade was the implementation of the Vascular Cognitive Impairment Neuropathology Guidelines (VCING), a collaborative study performed in the United Kingdom [35]. The study by Barker et al. [36] and some of the other studies referred to here were carried out on brain samples donated after death from elderly donors who were systematically followed during life with regard to their clinical and psychological state at each yearly visit at the Oxford Study to Investigate Memory and Ageing (OPTIMA). From a total brain archive of over 500 donations, this cohort included subjects who were found after death to have only SSVD and no more than Braak and Braak stage 3-tau pathology. We found that cognitive impairment only affected about a third of subjects, the rest had cognition within normal limits [37], using as instruments to assess cognition the Mini-Mental State Examination and the Cambridge Cognition Examination. The findings may have been different using the Montreal Cognitive Assessment test [38, 39] that has been found to be more sensitive to cognitive changes characteristic of SSVD and VCI; or the Brief Memory and Executive Test [40] for detecting cognitive impairment in small vessel disease.
Pathology
Diffuse damage to the white matter (ischemic leukoencephalopathy) is the most common pathology in SSVD [35]. It is usually most severe in frontal and occipital regions [35, 41–44] and the affected white matter exhibits loss of myelin and axons and a chronic inflammatory infiltrate (Fig. 2A, B). There is also evidence of ongoing axon damage in surviving axons, detected with immunocytochemistry for phosphorylated neurofilament precursor protein [36]. Evidence for axonal depletion in white matter lesions also comes from silver-stained preparations [41, 42], correlated with reduced fractional anisotropy in diffusion tensor imaging [43].
Fig.2
Components of the pathology due to SSVD. A, B) Low power (A) and higher power (B) views of a histological section from a case of SSVD. The section has been stained for myelin (blue) (Luxol fast blue/cresyl violet stain). There is diffuse pallor of staining and, at the top left corner of the section in (A), the tissue is necrotic. B) Damaged white matter at higher power. The nuclei (purple) are chiefly those of infiltrating macrophages. C) Greatly dilated perivascular space (hematoxylin and eosin stain). D) Small artery with a grossly thickened wall in which collagen has replaced smooth muscle (hematoxylin and eosin stain). E) Normal white matter arterioles in which the deeper pink cells are smooth muscle cells. F) Severely fibrotic and stenosed arteriole from a case of SSVD (E and F, hematoxylin and eosin).
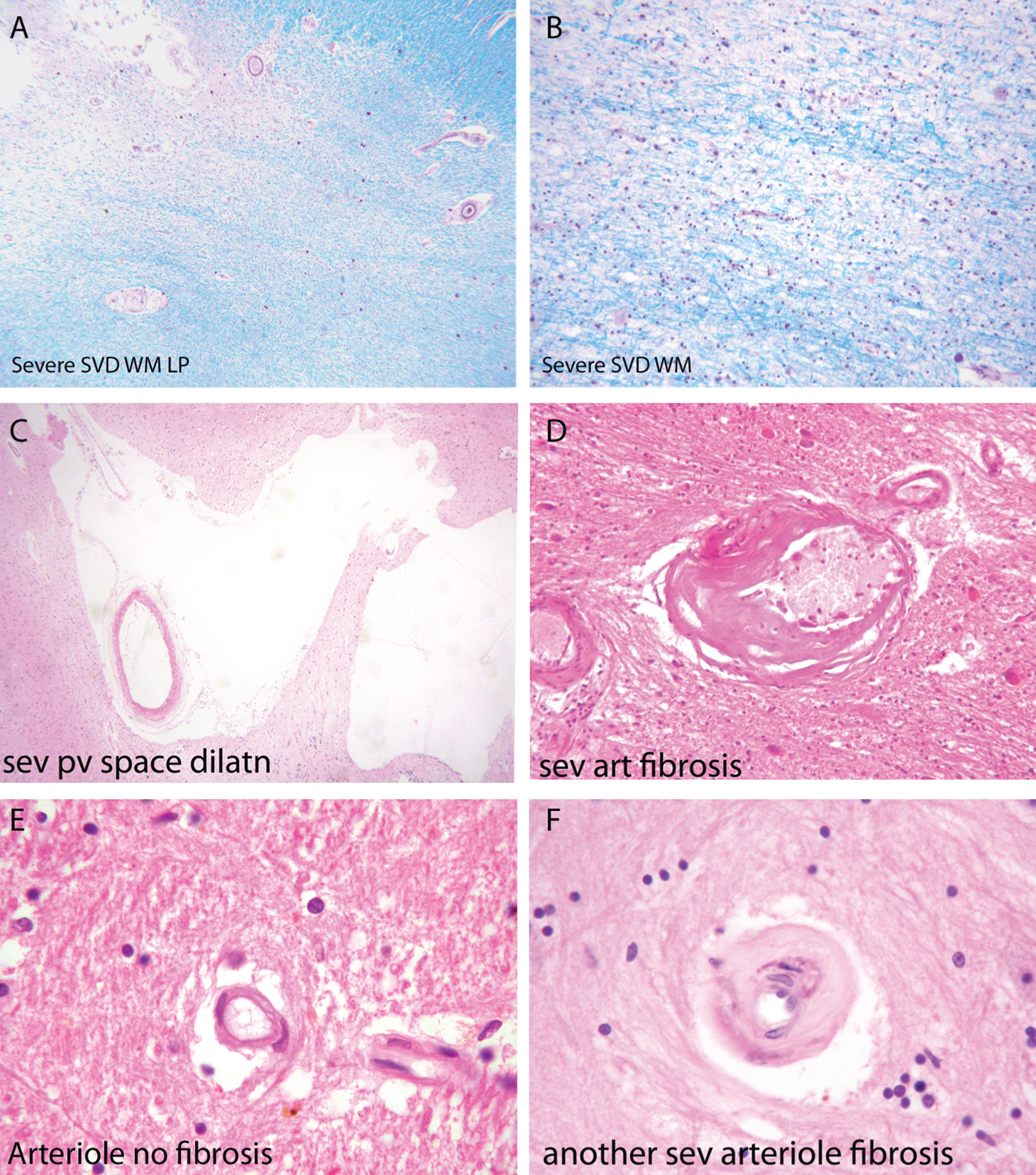
Associated with this diffuse white matter damage, Skrobot et al. [35] found six other pathological features predictive of cognitive impairment: 1) arteriolosclerosis, 2) perivascular space dilatation, 3) leptomeningeal cerebral amyloid angiopathy (CAA), 4) microinfarcts, 5) lacunar infarcts; and 6) large infarcts.
Small arteries and arterioles supplying blood to cerebral white matter in SSVD have thickened walls and basement membranes and diminished luminal spaces (Fig. 2D, F) [44–46]. A thrombus within these vessels is rare but arterioles are sometimes tortuous [47–49]. Smooth muscle in the walls of the vessels is characteristically replaced by collagen, and perivascular spaces are commonly enlarged [50] (Fig. 2C). Perivascular macrophages are frequently present and may contain hemosiderin, indicative of likely earlier small bleeds. A marker of endothelial activation, thrombomodulin, was found to be elevated in vessels affected by SSVD, a change that was interpreted as likely to be a protective response to SSVD rather than causative [47]. Similar changes to those in cerebral white matter are often seen in deep grey matter. Veins adjacent to the lateral ventricles often have thickened walls [48], but the significance of venous pathology in SSVD is only beginning to be systematically studied and related to cognitive state. The same is true of capillaries, although imaging techniques such as Dynamic Susceptibility Contrast (DSC)-MRI perfusion has shown alterations of cortical capillary transit time in AD, consistent with low tissue oxygen tension in white matter hyperintensities (WMH) areas [51].
It is evident that the blood supply to cerebral white matter in SSVD is compromised and there is ischemia and oxidative stress in affected regions [44, 52]. From imaging studies the process appears to progress slowly; the rate of WMH in T2 weighted MRI, indicative of damage due to SSVD, is associated with decline in verbal IQ [53] and executive function [54].
Mechanisms
Epidemiological studies indicate that SSVD is more common with longstanding hypertension in middle age [55–57] and in subjects with diabetes mellitus [58]. Hypertension may damage these small vessels by provoking sheer stresses since these vessels branch out from larger arteries without progressive decrease in size and are therefore liable to experience relatively high pressures. Alternatively, there may be damage inflicted by episodes of hypotension interspersed between episodes of hypertension in people with poorly controlled blood pressure [55]. In the material, we have studied from the OPTIMA cohort we found no relation of SSVD to severity of hypertension or diabetes, but instead we found it to be related to age [59]. Age also had an influence of some SSVD features in the study by Skrobot et al. [35]. Less attention has been paid in the literature of SSVD to the effect of age than of hypertension. This finding emphasizes an interesting similarity between SSVD and sporadic AD for which old age is overwhelmingly the most important risk factor [60, 61]. It is unclear how age may damage small cerebral arteries and arterioles but one change that may be important is stiffening of upstream, larger arteries, reflected in carotid-femoral pulse wave and increased pulse wave velocity which have been linked to WMH [62, 63]. One aspect of vascular smooth muscle that has been shown to increase in expression with age is vascular endothelial growth factor 2 [64]. However, this expression was not related to severity of SSVD in the brains of elderly subjects lacking AD pathology in the OPTIMA cohort [59].
Angiotensin II is a protein capable to damage aging cerebrovascular cells, particularly vascular smooth muscle cells; it shows increased activation with age and is linked to pro-inflammatory effects through activation of leucocytes, cell adhesion molecules, and inflammatory cytokines [65, 66]. It also leads to stimulation of NADPH oxidase, an important source of oxygen species production and downregulation of antioxidant defenses [67, 68].
Though speculative at this stage, it is interesting to note the results of informative parabiosis experiments in animals performed by Wyss-Coray and colleagues. They have found that by engineering a common blood supply between old and young mice they can detect a circulating factor in the blood of young mice that can reduce age-associated cardiac muscle changes in old mice [69]. Clearly, it would be of interest to know if cerebral arterial smooth muscle in elderly mice (and humans) is similarly affected. Another mechanism, which is also worth considering in the pathophysiology of SSVD, is the breakdown of the blood-brain barrier (BBB) [70]. This is common in old age and may at times be severe enough to enable a large molecular weight protein, fibrinogen, to enter the neighboring brain parenchyma from the blood. Bridges et al. [71] examined this feature in material from the OPTIMA cohort. The presence of fibrinogen in cerebral white matter of elderly subjects was readily demonstrable, but its amount was not related to the severity of damage to white matter. This does not exclude a potential role for fibrinogen and its breakdown product fibrin, in the development of white matter damage in SSVD because the damage is likely to have arisen earlier than in the pre-mortem period during which fibrinogen seen in tissue sections is likely to have leaked from blood vessels. It is worth referring to the apparent ability of fibrinogen to damage cerebral cortex in multiple sclerosis [72] and to white matter in EAE, the experimental model of multiple sclerosis [73]. Fibrinogen has also been linked to cortical damage in AD [74]. BBB damage in SSVD may be effected by activation of matrix metalloproteinases [18].
CAA is a factor associated with severity of SSVD and cognitive impairment attributable to SSVD. In a study specifically designed to study the effect of CAA on SSVD [75] we found a complex relationship of CAA to the severity of SSVD-related damage that varied depending on apolipoprotein E (APOE) genotype. There was a modest positive correlation between severity of CAA and severity of SSVD in those who were APOE ɛ4-positive, no effect in those who were APOE ɛ3-positive and a negative effect in those who were APOE ɛ2-positive [75]. CAA has been shown to be associated with microinfarctions in the cerebral cortex. Since the arterioles in the cortex that are affected by CAA also supply subcortical white matter with blood it is plausible that it may promote ischemia in subcortical white matter; Skrobot et al. [35] found that it promoted SSVD in their study. The manner in which APOE genotype may exert this effect is not clear. Nonetheless, Skrobot et al. [35] did not find an effect of the APOE genotype in SSVD.
One further metabolic pathway that would be worth investigating with respect to the effect of age on cerebral arteries and arterioles is the mammalian target of rapamycin (mTOR) pathway [74, 76] which has been shown to have important control over lifespan in animals [77] and to be dysfunctional in AD [78, 79]. The extracellular matrix protein mindin/spondin2 is a protein involved in vascular smooth muscle regulation influenced by the mTOR pathway [80], as is the platelet-derived growth factor-induced control of vascular smooth muscle phenotype [81].
This neuropathology review summarized some of the factors that may contribute to SSVD in the elderly. To be in a position to help those with cognitive impairment due to SSVD, and to help others at risk to avoid it, we need to further clarify mechanisms of damage and find ways to intervene and to combat them.
GENETICS
In 1955, Ludo van Bogaert [82] reported two sisters with a familial form of Binswanger disease and provided the first neuropathological description of the clinical condition that eventually became known as Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL), the first monogenic form of pure SSVD [83]. Other variants of SSVD with a genetic basis were later reported [84, 85]. However, the mechanisms of contribution of risk genes to the development of sporadic SSVD are not fully known, due to the complexity of the pathogenesis caused by the interaction of genetic and environmental factors. The genetic contributions to the monogenic forms of small-vessel diseases are better known [86, 87] and will not be discussed here; these include CADASIL, Cerebral Autosomal Recessive Arteriopathy with Subcortical Infarcts and Leukoencephalopathy, Fabry disease, autosomal dominant Retinal Vasculopathy with Cerebral Leukodystrophy, CAA, and collagen type IV alpha 1 chain-related cerebral small-vessel disease. Although some pathogenic mechanisms may be shared between the monogenic and sporadic forms, they are not identical. Therefore, there is still a need to better understand the genetics of SSVD.
Established diagnostic criteria for SSVD have been insufficiently used in genetic studies and the majority of gene-association studies have focused not on clinical diagnoses but on surrogate markers or disease traits, such as WMH, deep intracerebral hemorrhage and cerebral microbleeds, although these markers are not unique for SSVD. Throughout the years, the association of such surrogate markers and diagnoses with gene variants known as single nucleotide polymorphisms or gene loci has been performed in so-called candidate gene studies. A few of these have also aimed at confirming genetic findings with neuropathological studies, where an accurate diagnosis of SSVD is possible [87]. Many of the candidate gene studies have been performed in small patient groups or have given conflicting results when compared. Therefore, important meta-analyses of these previously reported candidate gene studies have been performed. Moreover, a few genome-wide association studies of VCI have been performed [88, 89] but failed to reproduce the findings from the candidate gene studies and meta-analyses. In these studies, rs290227 of the spleen tyrosine kinase (SYK) gene [89] and rs12007229, located on the X chromosome near the androgen receptor gene were associated with risk of VCI [88], but to our knowledge, no follow-up studies of these gene variants have been published to date.
The gene for apolipoprotein E (APOE), a multifunctional protein involved in lipid and cholesterol transport and Aβ clearance, is the best-known risk gene for AD [90] and CAA [91] where the ɛ4 allele increases the disease risk. Meta-analyses of previously published candidate gene studies of APOE ɛ4 have shown significant association between ɛ4 allele carriers and increased risk of VCI [92–94]. Meta-analyses [94] have also indicated that the APOE ɛ4 allele is associated with an increased risk of cerebral microbleeds [95] but not with WMH [96].
The MTHFR gene codes for an enzyme in the folate cycle, involved in homocysteine metabolism [29]. Although the gene variant C677T (rs1801133) of MTHFR has been shown to be a risk gene for stroke in meta-analyses, it has failed to show associated with WMH and intracerebral hemorrhage in similar studies [96]. In two meta-analyses the T allele of MTHFR was associated with VCI in Asian populations; but this association was not present in Caucasian and Indian populations [93, 94]. A validation study later showed an increased risk of VCI associated with the T allele of rs1801133 in Caucasians [92].
A few genes that have shown associations to VCI have later been confirmed in meta-analyses while some have failed to show associations with disease. The gene variant rs662 for paraoxonase 1 (PON1), a gene involved in homocysteine and lipoprotein metabolism, has failed to be associated with VCI in a published meta-analysis [93] or in a validation study [92]. However, the PON1 L55M (rs854560) variant was found to be associated with VCI, while no association was found with the variant –108C/T (rs705379) of the gene [94]. Variants of two genes related to inflammation, tumor necrosis factor (TNF)-α –850C/T (rs1799724) and transforming growth factor (TGF)-β1 + 29C/T (rs1800470) were found to be associated with risk of VCI in Caucasian and Asians, respectively [94], while the interleukin (IL) gene variants IL-1α –889C/T (rs1800587) and IL-6 –174G/C (rs1800795) showed no significant association in this study. Finally, no significant associations of VCI were found in a meta-analysis [93] and in a validation study [92] of variants of the genes for angiotensin converting enzyme (ACE; rs1799752), alpha-1-antichymotrypsin (ACT/SERPINA3; rs4934) and presenilin (PSEN-1; rs165932). Moreover, ACT A/T (rs4934), APOE promoter–427T/C (rs769446) and PSEN-1 (rs165932) were not associated with VCI in a meta-analysis [94]. The reasons for this could be the reduced number of studies and the limited number of patients included in the meta-analyses, preventing stratification by ethnicity.
The importance of determining exact genetic mechanisms involved in the pathogenesis of SSVD resides in the possibility of prevention and treatment, as recently illustrated with the use of an agonist Notch3 antibody that prevents mural cell loss and modifies plasma proteins associated with Notch3 signaling in an animal model of CADASIL [97]. Therefore, future genetic analyses of definite SSVD cases in large cohorts and subsequent meta-analyses are warranted.
SYMPTOMS PATTERN AND CLINICAL PRESENTATION
Although cognitive impairment in the elderly may affect the entire range of cognitive functions, there is usually an uneven distribution of deficits, i.e., some cognitive functions are more affected than others. The pattern of cognitive impairment reflects the nature of the disease and the distribution of pathological changes in the brain. From the clinical viewpoint, it is useful to simply consider two major types of cognitive syndromes. The first one is characterized by impaired memory, language problems, difficulty with practical tasks, and loss of the capacity to interpret sensory (mainly visual) impressions including recognition ability. The syndrome originates from dysfunction of posterior cortical association regions (posterior brain syndrome) and is typical of AD. The second syndrome results from executive dysfunction and is characterized by mental slowness and personality changes [98]. The executive control function ensures that mental and physical activities achieve the intended goals. It coordinates cognitive functions such as planning, attention, working memory, abstraction capacity, flexibility, and the ability to take action goals. Consequently, when executive dysfunction is present, various types of activity-related cognitive functions are impaired. Memory disorders also occur as a result of loss-of-set but are less pronounced than in the posterior brain syndrome. Recognition and interpretation capabilities remain relatively intact. Gait pattern is often slower, similar to that of Parkinson’s disease. The syndrome originates from dysfunction of frontal subcortical regions of the brain (anterior brain syndrome) and is typical of SSVD [18, 19, 98–101]. Executive dysfunction may appear also in patients with AD but later in the course of the disease, and it is usually associated with impairment of attentional shifting and working memory [102]. In patients with SSVD, the executive dysfunction manifests itself early in the course of the disease causing impairment in the capacity to use complex information, to formulate strategies, and to exercise emotional and behavioral self-control. In comparison with AD, patients with SSVD show less pronounced episodic memory impairment but more depressive symptomatology and greater variability in the speed of progression of the disease [103, 104]. SSVD specifically contributes to deterioration of psychomotor speed, global cognitive function, and impaired executive control. Executive dysfunction causes the loss of functional capacity required to perform activities of daily living, a requisite criterion for the diagnosis of major cognitive disorder. Patients with AD and prominent impairment of memory may remain relatively independent as long as the executive function remains intact; in contrast, SSVD may lead to early and progressive executive dysfunction with considerable risk of developing major cognitive disorder [104]. Executive function is also a determinant in defining the level of institutional care required.
Recent studies have investigated the cognitive profile of SSVD in more detail. In a comprehensive meta-analysis, Vasquez and Zakzanis [105] compared healthy controls with patients with SSVD and mild cognitive impairment (MCI). Patients showed impairments across all cognitive domains but the greatest decline was in mental speed. SSVD patients exhibited more pronounced impairments in executive function and mental speed compared to the non-vascular MCI group. In contrast, the non-vascular MCI group exhibited more pronounced impairment of delayed memory [105]. Although the differences between vascular and non-vascular MCI are in agreement with the results of previous studies, it is essential to notice that there is overlap in cognitive impairment observed in neuropsychological tests between the groups. Based on the recommendations of the 2006 NINDS-CSN consensus conference [106], a neuropsychological battery for MCI-SSVD was developed using a factor analysis approach [107, 108]. The battery was found to be robust and applicable. Attention-executive dysfunction, memory loss and visuospatial difficulties were the most obvious features. However, the Trail Making Test was poorly applicable to older and cognitively impaired patients.
Age at onset appears to be a crucial factor in determining the cognitive impairment profile [109]. Frontal-executive dysfunction was more pronounced in patients with disease onset before age of 65 in comparison with the late-onset group. The early-onset SSVD group had a more severe small-vessel disease burden, whereas the late-onset group exhibited greater amyloid burden. In comparison with AD, the loss of visual memory is less pronounced in SSVD after one year, whereas the opposite is true for loss of physical independence [110].
A number of simplified approaches to the diagnosis of cognitive impairment in SSVD have been proposed. In a study operationalizing MCI criteria, non-amnestic single-domain MCI was unexpectedly common and the authors concluded that it may represent a previously unrecognized MCI subtype associated with SSVD [108]. The Frontal Assessment Battery is a useful tool to differentiate MCI due to SSVD from AD [111]. The Montreal Cognitive Assessment performed better than the Mini-Mental State Examination to detect MCI-SSVD [112]. The Cognitive Assessment Battery appears to be useful for MCI detection of MCI-SSVD [113]. In an autopsy-defined cohort [114], a combined memory and verbal fluency score was able to differentiate between AD and SSVD with a sensitivity of 85%, specificity of 67% and positive likelihood ratio of 2.5. Amnestic memory impairment and lower categorical fluency characterized AD, while the pattern in SSVD exhibited a tendency toward greater impairment on phonemic fluency and better performance on recognition memory [114]. The CLOX test [116] remains a simple and effective tool to detect executive dysfunction (Fig. 3).
Fig.3
Executive Clock-Drawing Task (CLOX): The patient is instructed to draw a clock on a white sheet of paper. The instructions are as follows: “Draw a clock that says 1:45. Set the hands and numbers on the face of the clock so that even a child could read them.” The instructions can be repeated until they are clearly understood, but once the subject begins to draw no further assistance is allowed. This patient scored 7/15 points. The subject’s performance is scored as follows: Does figure resemble a clock? 1 point; Circular face present? 1 point; Dimensions >1 inch? 1 point; All numbers inside the perimeter? 1 point; No sectoring or tic marks? 1 point; Numbers 12, 6, 3, & 9 placed first? 1 point; Spacing Intact? (Symmetry on either side of 12 and 6 o’clock?) 1 point; Only Arabic numerals? 1 point; Only numbers 1 - 12 among the numerals present? 1 point; Sequence 1-12 intact? No omissions or intrusions. 1 point; Only two hands are present? 1 point; All hands represented as arrows? 1 point; Hour hand between 1 and 2 o’clock? 1 point; Minute hand obviously longer than hour? 1 point; 1 point if none of the following are present: 1) Hand pointing to 4 or 5 o’clock? 2) “1:45” present? 3) Any other notation (e.g., “ 9:00”)? 4) Any arrows point inward? 5) Intrusions from “hand” or “face” present? 6) Any letters, words or pictures? From: Royall et al., 1998 [116].
![Executive Clock-Drawing Task (CLOX): The patient is instructed to draw a clock on a white sheet of paper. The instructions are as follows: “Draw a clock that says 1:45. Set the hands and numbers on the face of the clock so that even a child could read them.” The instructions can be repeated until they are clearly understood, but once the subject begins to draw no further assistance is allowed. This patient scored 7/15 points. The subject’s performance is scored as follows: Does figure resemble a clock? 1 point; Circular face present? 1 point; Dimensions >1 inch? 1 point; All numbers inside the perimeter? 1 point; No sectoring or tic marks? 1 point; Numbers 12, 6, 3, & 9 placed first? 1 point; Spacing Intact? (Symmetry on either side of 12 and 6 o’clock?) 1 point; Only Arabic numerals? 1 point; Only numbers 1 - 12 among the numerals present? 1 point; Sequence 1-12 intact? No omissions or intrusions. 1 point; Only two hands are present? 1 point; All hands represented as arrows? 1 point; Hour hand between 1 and 2 o’clock? 1 point; Minute hand obviously longer than hour? 1 point; 1 point if none of the following are present: 1) Hand pointing to 4 or 5 o’clock? 2) “1:45” present? 3) Any other notation (e.g., “ 9:00”)? 4) Any arrows point inward? 5) Intrusions from “hand” or “face” present? 6) Any letters, words or pictures? From: Royall et al., 1998 [116].](https://content.iospress.com:443/media/jad/2018/62-3/jad-62-3-jad170803/jad-62-jad170803-g003.jpg)
Neurological signs have been thoroughly studied in patients with SSVD. A score for focal neurological signs did not correlate with global cognitive performance, but instead with executive dysfunction, as measured by the letter fluency and the Rey-Osterrieth Complex Figure tests [115]. The neurological signs observed more often were the Chaddock sign, stooped posture, decreased arm swing, bradykinesia, and rigidity. Sixty-nine percent exhibited at least one extrapyramidal sign and 58% at least one unilateral lateralizing sign [115]. Motor impersistence and perseveration occurred when cortical areas and underlying white matter tracts associated with the frontoparietal attentional system were affected in patients with SSVD [117]. The LeukoAraiosis and DISability (LADIS) study [118] showed that SSVD was associated with deterioration of gait and balance over time. Also, gait disturbance complaints in non-disabled SSVD patients were, along with age, atrial fibrillation, and the degree of WMH, an independent predictor of disability after 3 years [119]. Interestingly, new studies have found that SSVD plays a role in the development of cognitive impairment in patients with Parkinson’s disease [120, 121].
Neuropsychiatric manifestations such as depressive symptoms, loss of motivation, lack of interest and emotional bluntness are common in patients with SSVD [122]. Small-vessel lesions and amyloid burden independently affect specific neuropsychiatric symptoms [123]. Apathy, depressive symptoms, irritability [123, 124], and sleep disturbances [125] are the most frequent neuropsychiatric symptoms in SSVD. In MCI-associated SSVD both negative and also positive neuropsychiatric symptoms appear; more negative than positive symptoms become more obvious as the disease progresses from MCI to major cognitive disorder [126]. In a pivotal study, both apathy and depressive symptoms were increased in patients with SSVD but typically they did not occur together in the same patient [127]. Diffusion-tensor imaging (DTI) revealed that white matter microstructural changes in SSVD were associated with increased apathy, but not depressive symptoms [127]. These findings have consequences for the formation of treatment strategies for patients with SSVD.
In summary, new studies have confirmed the anterior symptom profile in patients with SSVD [128]. Motor-neurological and neuropsychiatric features have been added to the characteristic frontal-executive pattern of the disorder, which is phenotypically different from that of pure AD [123, 124]. However, the cognitive profile of SSVD is much more complex than typically reported. The results of new studies suggest that overlapping neural networks take part in accomplishing neuropsychological tasks. The field will benefit from further studies on outcome of network pathway disruption [105, 129]. The symptom profiles described here are not sufficiently specific and do not qualify as unique diagnostic markers of SSVD. For diagnostic purposes, other tools such as brain imaging and biochemical markers should be used in combination with the characteristic symptom profile.
NEUROIMAGING
The standard-of-care evaluation in elderly patients with cognitive decline requires at least one brain imaging study [130] to eliminate pathologies different from neurodegeneration and to determine the extent of the vascular lesions [131, 132]. Development of neuroimaging techniques, in particular computerized tomography (CT) and MRI, provided some of the most important tools in the diagnosis of cognitive disorders, allowing accurate demonstration of the location and rate of progression of atrophic changes affecting the brain in AD and other cognitive disorders, as well as the different types of vascular lesions observed in pure forms of vascular cognitive disorders and MXD [133, 134]. Two basic imaging methods are currently used in the clinical evaluation of cognitive disorders: structural brain imaging (CT/MRI) and metabolic imaging (PET/SPECT). CT uses X-rays and the resulting images depend on the electron density and rates of absorption of X-rays by different tissues. MRI uses the changes induced by the magnetic field to alter the rate of spin of the hydrogen atom about its own axis; this change of the electromagnetic properties of protons in water molecules in the tissue added to the small difference in energy produced by the change of spin of the protons is detected and used for the anatomic three-dimensional (3D) reconstruction of the tissue. Positron emission tomography (PET) scans allow measurement of regional cerebral glucose metabolism using 18F-2-fluoro-deoxy-D-glucose (FDG). Cerebral blood flow can be measured with H215O-PET or using single photon emission computerized tomography (SPECT) with technetium hexamethyl-propylene-amine-oxime (99mTc-HMPAO).
Quantification of cortical thickness allows early diagnosis and rate of progression from MCI to major cognitive disorder. White matter involvement can also be quantified with DTI and functional methods including fMRI, functional connectivity, and MR spectroscopy (MRS). Isotope-based techniques include isotope markers for Aβ (11O-PIB, 18F-florbetapir), tau (18FDDNP) and activated microglia (11C-PK11195). Neuroimaging markers are particularly useful at the early symptomatic and preclinical asymptomatic phases of SSVD [131], as well as serving as endpoints in clinical trials. The latter technique allows a significant reduction of the number-needed-to-treat subjects in controlled trials.
According to Thompson and Hakim [24], SSVD is widely recognized as the commonest of all brain lesions. The unusually elevated prevalence of WMH in the elderly was received initially with skepticism, reflected in the name UBOs (unidentified bright objects) used to first describe them [2]. For instance, in 1996, the large population-based brain MRI study on 3301 elderly subjects in the Cardiovascular Health Study [134] found that more than half (64.2%) of all participants had some degree of WMH; only 4.4% had no lesions and 31.4% had minimal lesions. WMH were associated with older age, clinically silent stroke on MRI, hypertension, smoking and lower income indicating limited access to medical services, inadequate diet and living conditions, and poor long-term compliance with antihypertensive treatment [135]; higher grade WMH were accompanied by cognitive impairment and abnormal gait. Correlation studies revealed that the majority of these WMH in the elderly were ischemic in nature [136]. Arne Brun and Elisabet Englund [137] at the University of Lund, Sweden, first called these WMH lesions “incomplete” white matter infarctions. The name “incomplete” indicates that complete ischemic necrosis and cavitation were not present. CT and, more clearly, MRI also revealed the presence of état criblé or dilatation of Virchow-Robin perivascular spaces [138, 139]; lacunes, état lacunaire and Binswanger disease. Joanna Wardlaw and colleagues [140] have extensively reviewed the topic of WMH, including pathogenesis, clinical relevance and imaging features. WMH are not benign lesions since they increase 2-fold the risk of major cognitive disorder and 3-fold the risk of stroke [54, 141]. Recent MRI technical advances [142–144] including manual and automatic volume measurements, DTI, and fMRI should provide further understanding of SSVD lesions. In 2013, Wardlaw and the STandards for ReportIng Vascular changes on nEuroimaging (STRIVE) study group [145] completed a major international effort to standardize neuroimaging acquisition, interpretation and reporting of cerebral small-vessel disease. The criteria include the following SSVD detected on conventional MRI: 1) recent small subcortical infarcts, 2) white matter hyperintensities, 3) lacunes, 4) prominent perivascular spaces, 5) cerebral microbleeds, and 6) atrophy (Fig. 4). Criteria for imaging detection and quantification of cortical cerebral microinfarcts on 3 Tesla MRI have been proposed recently [146].
Fig.4
Examples of different features of small vessel disease, including white matter hyperintensities, as published by the STandards for ReportIng Vascular changes on nEuroimaging (STRIVE) study group. From: Wardlaw et al., 2013 [145].
![Examples of different features of small vessel disease, including white matter hyperintensities, as published by the STandards for ReportIng Vascular changes on nEuroimaging (STRIVE) study group. From: Wardlaw et al., 2013 [145].](https://content.iospress.com:443/media/jad/2018/62-3/jad-62-3-jad170803/jad-62-jad170803-g004.jpg)
Quantification of the total burden of SSVD and the resulting alterations of the BBB leading to decreased CBF has been successfully studied [147, 148]. It has been shown that the severity of the baseline WMH load predates the decrease of CBF after a follow-up of almost 4 years. Cerebrovascular reactivity in SSVD can be measured using blood oxygen level dependent (BOLD) MRI and vasoactive stimulation; preliminary results indicate that cerebrovascular reactivity appears to be decreased in subjects with more severe WMH [147, 148].
Cerebral FDG-PET is widely used in the evaluation of cognitive disorders in the elderly given that cerebral glucose metabolism is an index of neuronal and synaptic function; thus cerebral glucose hypometabolism represents regions of brain destruction such as in stroke, or cortical zones of neurodegeneration in VCI and in most types of degenerative cognitive disorders such as AD [149, 150]. Bloudek et al. [151] performed a meta-analysis to evaluate FDG-PET in AD and found 90% sensitivity (95% CI 84–94%) and 89% specificity (95% CI 81–94%). Kerrouche et al. [152] using voxel-based multivariate analysis of 18FDG-PET separated VCI from AD with 100% accuracy. Hypometabolism in deep gray nuclei, cerebellum, primary cortex, middle temporal gyrus, and anterior cingulate cortex differentiated VCI from AD [152]. In addition to providing a clear diagnostic image of AD and other neurodegenerative cognitive disorders, such as frontotemporal degeneration, FDG-PET is quite useful in the diagnosis of patients with PSD due to multiple strokes and also in determining the contribution of AD to VCI.
The brain is a complex vascular organ; therefore, measurement and understanding of CBF is central to progress in the field of cognition [153]. Neurovascular coupling increases CBF according to local neuronal activity and metabolism [8] serving as the basis for BOLD-MRI and fMRI [150, 151]. A decrease in CBF occurs with normal aging as well as in AD [30–32, 153, 154]. A novel method to measure CBF uses ASL [30, 155], a non-invasive MRI technique that requires neither contrast media nor isotopes [30]. ASL-MRI generates an endogenous contrast by using radiofrequency pulses that “label” water proton spins in blood circulating in carotid and vertebral arteries at the base of the skull. CBF-ASL-MRI images are obtained by subtracting labeled and unlabeled spin exchanges in the brain tissue yielding a quantifiable map of regional CBF (rCBF) [30, 155]. Using 3D ASL-MRI in subjects with SSVD, Sun et al. [156] demonstrated marked decrease in CBF in the cortex of the temporal and frontal lobes, hippocampus, thalamus, and insula that correlated with the degree of cognitive impairment. Also, thinner cortex in frontal, parietal and lateral temporal regions was noted in more severe cases of SSVD.
Excellent correlation of CBF with FDG-PET metabolic rate has been obtained in patients with several forms of cognitive disorders [155, 156], as well as in obstructive sleep apnea [157, 158]. BBB alterations can be detected using a two-stage non-invasive diffusion weighted MRI technique [159] that uses fast and slow diffusion coefficients to differentiate labeled blood in the microvascular compartment (small vessels and capillaries) and in the brain tissue; the ratio of these two signals indicates water exchange rate across the BBB [159]. Large artery integrity can also be determined using flow encoding arterial spin tagging [160].
CBF-ASL in NPH
A promising area of research in SSVD is the use of CBF-ASL is the study of Binswanger disease and normal pressure hydrocephalus (NPH). In 1894, Otto Binswanger described the clinical form of major vascular cognitive disorder that bears his name characterized by extensive subcortical ischemic white matter lesions. Binswanger’s neuropathological report noted the presence of “enormously enlarged ventricles” [4]. Román [2, 4, 161] pointed out the difficulties in clinically separating Binswanger disease from NPH. In fact, both entities are characterized by prominent changes in ambulation, such as small-step gait (marche à petits pas), gait apraxia, frequent falls, and urinary incontinence. A subcortical type of cognitive impairment with changes in mood, behavior, and personality is common in both, as well as pseudobulbar palsy, emotional incontinence, and frontal lobe signs with loss of incentive, drive, and insight. Profound apathy and abulia may be observed in advanced cases of both conditions. Mutism, bradykinesia, rigidity, and dysarthria may lead to confusion with Parkinson disease. Finally, there is frequent occurrence of hypertension and cerebrovascular disease in cases of idiopathic NPH [161], as well as typically noted in Binswanger disease.
Bradley et al. [162] compared the presence and degree of periventricular WMH in patients with treated and untreated NPH and in age-matched control subjects. A highly significant statistical association was found for WMH in NPH when compared with controls. Patients with MRI evidence of WMH but without clinical symptoms of NPH had increased frequency of communicating hydrocephalus. Diffusion of CSF into the periventricular areas and SSVD contribute to periventricular WMH in NPH.
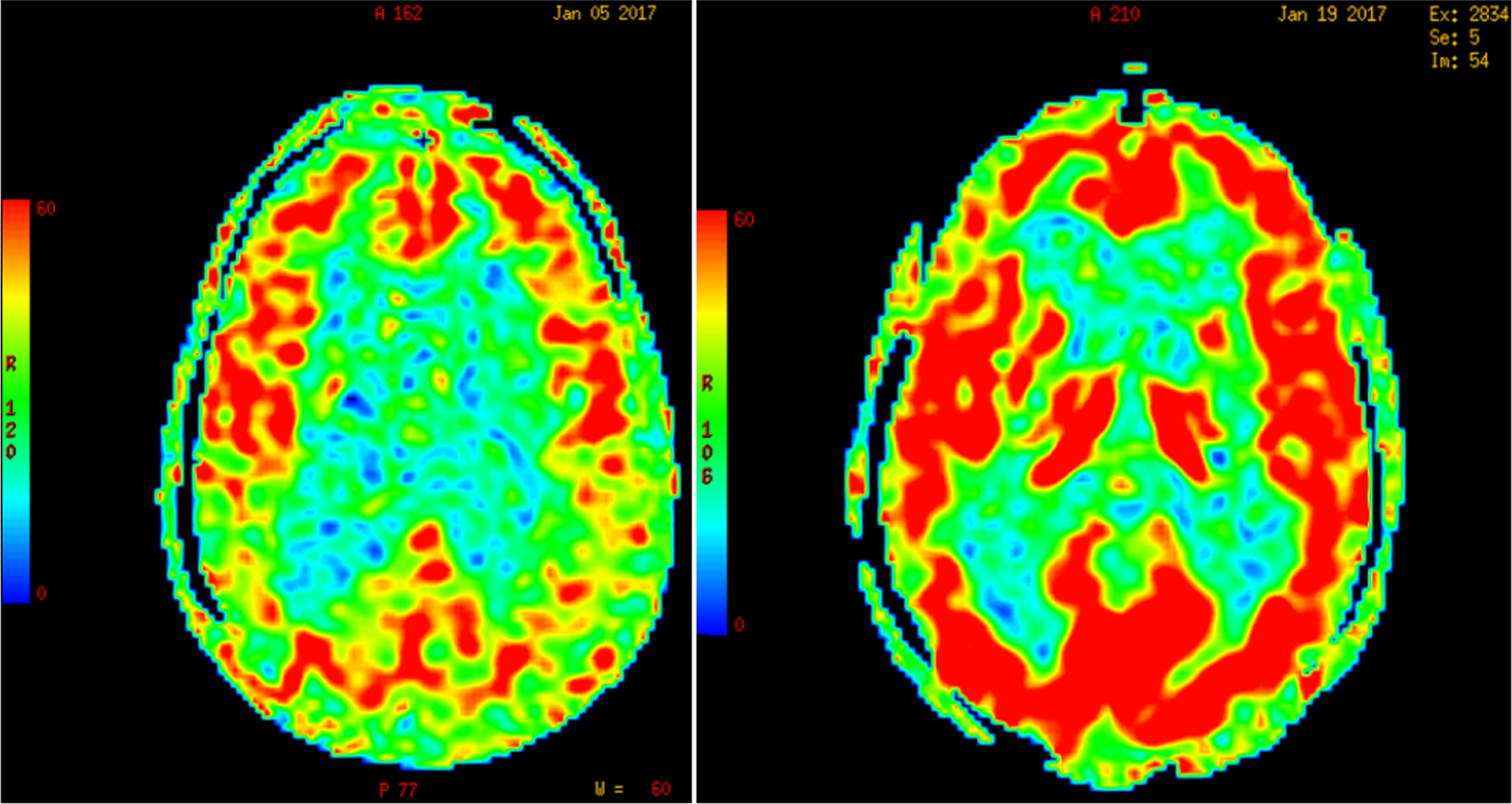
Due to Pascal’s hydrostatic pressure law, the net effect of CSF pressure on the dilated ventricles is an increase of interstitial pressure in the brain parenchyma or transmantle pressure [163]. Perfusion of the periventricular regions proceeds in a centripetal pattern from the surface of the brain toward the ventricles by means of long medullary arteries; therefore, an increase in the intraventricular pressure results in an opposite gradient of centrifugal pressure capable of producing cerebral hypoperfusion [164] and ischemia of the watershed periventricular white matter, particularly in elderly subjects with loss of CBF autoregulation [164], but also in children with hydrocephalus from posterior fossa lesions [165, 166]. As illustrated in Fig. 5, global CBF-ASL perfusion increases post-lumbar puncture (post-LP) in patients with diagnosis of NPH; in contrast, Binswanger disease cases show no change in CBF post-LP.
Fig.5
Increase in global cerebral blood flow determined by arterial spin label MR imaging post-lumbar puncture in a patient with normal-pressure hydrocephalus. (Román GC and Fung S, unpublished data).
In summary, most of the advances in the diagnosis, pathogenesis, epidemiology, and overall understanding of SSVD in the last decade have resulted from the clinical and research application of brain imaging, in particular MRI. Further studies on the vascular mechanisms underlying SSVD could be expected from research using MRI-based CBF-ASL studies and newer imaging modalities. Several groups in Europe recently recommended the implementation of ASL-MRI studies for numerous clinical applications including patients with dementia [30]. Brain imaging should continue to open novel therapeutic paths for these patients.
CEREBROSPINAL FLUID BIOMARKERS
As stated above there are biochemical changes in the brain of patients with SSVD, which may be reflected in the CSF since it is in direct contact with the extracellular space. This assumption and strategy has proved fruitful in the field for AD research. Recently, the index between tau (τ) protein in its total (τT) or phosphorylated form (phospho-tau, τP) and Aβ peptide 1–42 (Aβ42), has been incorporated in the research criteria for AD [33, 167, 168]. The differentiation between AD and VCI, and especially between MXD and “pure” VCI, is not always easy in everyday practice. VCI is a heterogeneous entity with SSVD increasingly recognized as the most common pathological substrate. Here we summarize the diagnostic value of these biomarkers in the differential diagnosis of SSVD [33, 169].
Tau (τ) protein is the major component of intracellular neurofibrillary tangles and is present in a hyperphosphorylated form in AD pathology. Both τT and τP tau are increased in the CSF of patients with AD, as compared to controls. τT is better viewed as a marker of neuronal and/or axonal degeneration, while τP is a more specific marker of tau hyperphosphorylation process and tangle formation in AD pathology [170]. Although the diagnostic accuracy may be reduced to some degree when attempting to differentiate AD from some other types of cognitive disorders, when combined with Aβ42 the above biomarkers achieve sensitivities and specificities approaching or exceeding 90% for the discrimination of AD from normal aging.
CSF τT levels in VCI provide conflicting results ranging from normal [171–173] to increased [174–178], or to intermediate between controls and AD cases but much lower when compared with AD [179, 180]. Some patients with VCI may present with high or even very high τT levels [173, 180–183]. When patients with “pure” VCI and MXD or AD pathology with subcortical white matter lesions are clinically separated, the results were again conflicting with τT in VCI reported as comparable to the controls [173], increased [178] or intermediate but much lower as compared to those of AD [180], while patients with MXD presented with increased τT in all studies. However, normal τT levels are found in patients with lacunar infarcts [171], progressive leukoaraiosis [175], or pure SSVD [184–187]. Phospho-tau (τP) levels have been reported as normal in VCI or SSVD [177, 178, 180, 185–187], while in MXD the levels are increased to those of AD cases [180] or intermediate between controls and AD [186]. Aβ with either 1–40 (Aβ40) or especially 1–42 amino acids (Aβ42) is the major component of amyloid plaques; Aβ is present extracellularly in AD and seems to inversely reflect amyloid pathology, i.e., CSF Aβ42 is 50% reduced in AD as compared to normal aged subjects. CSF-Aβ42 has shown high sensitivity and specificity (over 85%) and is recognized as one of the three “core feasible” CSF markers for AD [170].
Reduction of CSF Aβ42 has been reported in VCI [183] or in pure SSVD [185, 187–189], at levels similar to those of AD or intermediate between controls and AD cases. In other studies, Aβ42 levels in VCI have been comparable to those of controls but higher than in AD [172, 176–179]. Some VCI patients present low Aβ42 levels, but some overlap may be present [180].
In summary, most of the above studies agree that Aβ42 levels in VCI are reduced to a degree comparable to AD. The ratio of Aβ42/Aβ40, which is decreased in AD, may be comparable to the controls in pure VCI [190]. Thus the above index, if normal, may strengthen the diagnosis of SSVD, when used along with clinical imaging and other biomarker data.
Taken together, most of the above studies agree that τP is usually normal in pure SSVD, while some patients may present with low Aβ42 and/or high τT levels. A recent study [191], using NINDS-AIREN criteria [192], showed that only 29% of VCI patients presented with increased τP levels; however, reduced Aβ42 was found in 53% and increased τT levels in 41% of cases [191]. Such deviations from the usual pattern may indicate clinical overlap with AD or MXD, thus reducing sensitivity and specificity of individual biomarkers when used as tools in every day practice; therefore, combination of several biomarkers is required [190].
Overall, for the diagnosis of SSVD and separation from AD—the most common clinical scenario—the use of the above biomarkers as diagnostic tools is of value, but further studies are needed to evaluate sensitivities and specificities in various clinical situations. Combinations of these biomarkers with neurofilament light, metalloproteinase-9, TIMP metallopeptidase inhibitor 1, albumin index and metalloproteinase-2 index may further increase the diagnostic accuracy [178–180].
METABOLIC FACTORS
Risk factors for SSVD include systemic hypertension [193] and conditions causing hypoxia such as smoking [194], chronic obstructive pulmonary disease [195], and sleep apnea syndrome [196]. Hypercoagulation [193], chronic kidney disease [197], and homocysteinemia [198] could worsen the consequences of hypertension and hypoxia. Moreover, the metabolic syndrome due to an adverse lifestyle and aging could also influence the risk of SSVD [199].
Overnutrition and sedentary lifestyle result in excessive adiposity and metabolic disturbances. The risk profile is dependent on the distribution of body fat and the presence of metabolic aberrations [199]. The metabolic syndrome (METS) includes the clustering of abdominal obesity, insulin resistance, dyslipidemia, and elevated blood pressure [199]. In the French Three-City (3C) cohort [200], the presence of METS increased the risk of incident major VCI over 4 years. In 308 healthy older French persons from the longitudinal ESPRIT study [201], those with METS increased two-fold (Odds Ratio [OR] = 2.74) the chance of presenting with high levels of WMH volume, compared with subjects without METS. Furthermore, in 1151 healthy old Japanese people [202], METS was associated with silent brain infarction and WMH, whereas less strong associations were found with increased body mass index alone [202].
In the Japanese study by Bokura et al. [202], dyslipidemia was associated with WMH. Although there have been variable results in terms of the role of hyperlipidemia in WMH/SSVD progression [203], several studies have shown a positive association with hypertriglyceridemia [204]. ApoE is the major transport protein for cholesterol in the central nervous system [205]; ApoE levels in the CSF are significantly lower among patients with AD compared with controls [206], as well as in other forms of cognitive disorders including VCI [207]. ApoE transports cholesterol to neurons via ApoE receptors [208], thereby being involved in the mobilization of lipids for repair, growth and maintenance of myelin and axonal membranes [208, 209]. Thus, the deficiency of ApoE could possibly worsen the consequences of WMH by reducing repair mechanisms. ApoA-I levels in the CSF were reduced in AD but levels were not associated with ApoE ɛ4 allele distribution [207].
Insulin resistance is part of the clustering of risk factors seen in METS [210]. Using transcranial Doppler and MRI in subjects with diabetes mellitus type 2, the estimated WMH volume correlated negatively with CBF velocity [211], which could indicate a possible mechanism underlying WMH development. The results of several studies have suggested the association of impaired glucose homeostasis or diabetes mellitus with VCI or with increased WMH volume [212, 213]; but not all reports are concordant [214]. Moreover, postmortem studies have revealed that in the AD brain, there is resistance to signaling through the insulin receptor and the closely related insulin-like growth factor-I (IGF-I) receptor [215–217]. Neurons resistant to insulin and IGF-I action could lack trophic signals and therefore degenerate [218]; also, insulin and IGF-I are associated with the quantity of oligodendrocytes and myelin production [219–221]. However, it is unclear whether brain insulin resistance is a primary or secondary event in AD and it is unknown if there is resistance to insulin or IGF-I activity in the brains of patients with SSVD.
Subclinical inflammation is associated with both METS and SSVD. Several studies have shown that the inflammatory markers C-reactive protein, IL-6, TNF-α, TGF-β, and vascular endothelial growth factor levels are elevated in SSVD patients [222–225]. Furthermore, some studies have reported stronger associations between inflammatory markers and risk of SSVD/VCI compared to the corresponding associations seen in AD [226, 227]. However, data are not fully conclusive given that unchanged or even decreased levels of inflammatory markers have also been observed [227–229]. An additional hypothesis posits that immunosenescence could be of importance [229]. This means that an aged and defective immune system could be a risk factor for cognitive decline VCI. In conclusion, although inflammation is likely to occur in SSVD development, the exact nature of this involvement is still unclear.
Exposure to chronic stress may result in cognitive decline by activation of the hypothalamic–pituitary–adrenal (HPA) axis [230]. Although glucocorticoids display both neuroprotective and neurodegenerative effects [231], the results of most experimental studies suggest that prolonged or excessive increases in cortisol levels result in neuronal injury and reduced cognitive function [232, 233]. Increased circulating and CSF levels of cortisol have been observed in AD [234, 235], and there are reports of disturbed cortisol secretion also in VCI [236]. In the Lothian Birth Cohort 1936, higher cortisol at the start and the end of a mild cognitive stressor was associated with higher WMH [237]. Chronic stress with activated HPA axis could therefore be a risk factor for the development of SSVD. These concepts will need to be explored in the future.
In summary, sedentary lifestyle combined with overeating, chronic stress, and aging increases the risk of SSVD. However, the underlying mechanisms have not been elucidated in detail, and little is known concerning metabolic changes in the brain. Therefore, further research is needed to explore the role of metabolic factors in WMH development. Lifestyle intervention and medical treatment of metabolic alterations must be studied in clinical trials to determine the potential effect in reducing the risk of SSVD.
CONCLUSIONS
The most important research advance in the field of VCI in the last decade has been the scientific demonstration that age-related white matter involvement is a sign of subcortical small-vessel disease that leads to cognitive failure and impaired functional capacity, i.e., SSVD. Although this is the most homogeneous and common type of VCI and clinical criteria for the disease exist, it is often underdiagnosed both in incipient and manifest stages. Likewise, given the magnitude of the problem, a surprisingly limited number of pharmacological studies on SSVD have been conducted to-date. Interestingly, in a subgroup analysis of the Scandinavian Multi-Infarct Dementia Trial, nimodipine was found to be effective in patients with SSVD [238]; a more recent controlled clinical trial of DL-3-n-butylphthalide (a nutriceutical extracted from celery, Apium graveolens) showed positive results in patients with mild VCI caused by disease of the subcortical small vessels [239]. As mentioned earlier, a therapeutic experiment in an animal model of CADASIL demonstrated that the use of an antibody targeting Notch3 signaling effectively prevents mural cell loss [97]. A similar method could eventually be developed in animal models of SSVD.
One of the most important steps toward providing a greater knowledge about SSVD, and subsequently better care options for the disease, is to accept it as a distinct entity, as has been the case with AD. Indirectly, the lack of success in more than 100 AD trials has facilitated this approach [240, 241]. Recent advances towards that goal have emerged including: 1) increased knowledge of the symptom profile of SSVD, in addition to the characteristic frontal-dysexecutive cognitive profile; 2) new MRI studies have found that white matter involvement is associated with reduced cerebral blood flow; 3) a characteristic non-AD profile of CSF markers reflecting subcortical degeneration, inflammation and extracellular matrix breakdown has been identified; 4) the clinical and pathological criteria for SSVD have been updated; 5) the potential importance of deep vein collagenosis has been studied [242]; and 6) interesting associations between SSVD and NPH have been found. Although several of the new findings need to be replicated, they already now imply that focusing on SSVD using various methods is a promising way of addressing the great challenge of preventing and treating age-related cognitive impairment. Future studies investigating the relationships between sporadic SSVD and genetic factors are needed as well as research on the various mechanisms of causation presented above. These goals aim primarily at finding targets for intervention in patients with SSVD early in the course of the disease but may also have positive consequences for knowledge and treatment of AD [243].
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/17-0803r1).
REFERENCES
[1] | Román GC ((1999) ) A historical review of the concept of vascular dementia: Lessons from the past for the future. Alzheimer Dis Assoc Dis 13: (Suppl 3), S4–S8. |
[2] | Román GC ((1996) ) From UBOs to Binswanger’s disease. Impact of magnetic resonance imaging on vascular dementia research. Stroke 27: , 1269–1273. |
[3] | Blessed G , Tomlinson BE , Roth M ((1968) ) The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J Psychiatry 114: , 797–811. |
[4] | Román GC ((1987) ) Senile dementia of the Binswanger type: A vascular form of dementia in the elderly. JAMA 258: , 1782–1788. |
[5] | Román GC , Sachdev P , Royall DR , Bullock RA , Orgogozo JM , López-Pousa S , Arizaga R , Wallin A ((2004) ) Vascular cognitive disorder: A new diagnostic category updating vascular cognitive impairment and vascular dementia. J Neurol Sci 226: , 81–87. |
[6] | Feigin VL , Forouzanfar MH , Krishnamurthi R , Mensah GA , Connor M , Bennett DA , Moran AE , Sacco RL , Anderson L , Truelsen T , O’Donnell M , Venketasubramanian N , Barker-Collo S , Lawes CM , Wang W , Shinohara Y , Witt E , Ezzati M , Naghavi M , Murray C , Global Burden of Diseases, Injuries, Risk Factors Study 2010 (GBD, 2010), the GBD, Stroke Experts Group ((2014) ) Global and regional burden of stroke during 1990-2010: Findings from the Global Burden of Disease Study 2010. Lancet 383: , 245–254. |
[7] | Gorelick PB , Scuteri A , Black SE , DeCarli C , Greenberg SM , Iadecola C , Launer LJ , Laurent S , Lopez OL , Nyenhuis D , Petersen RC , Schneider JA , Tzourio C , Arnett DK , Bennett DA , Chui HC , Higashida RT , Lindquist R , Nilsson PM , Román GC , Sellke FW , Seshadri S , on behalf of the American Heart Association Stroke Council, Council on Epidemiology and Prevention, Council on Cardiovascular Nursing, Council on Cardiovascular Radiology and Intervention, Council on Cardiovascular Surgery and Anesthesia ((2011) ) Vascular contributions to cognitive impairment and dementia: A statement for healthcare professionals from the American Heart Association / American Stroke Association. Stroke 42: , 2672–2713. |
[8] | Kisler K , Nelson AR , Montagne A , Zlokovic BV ((2017) ) Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nature Rev Neurosci 18: , 419–433. |
[9] | Barnes DE , Yaffe K ((2011) ) The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol 10: , 819–828. |
[10] | Kalback W , Esh C , Castaño EM , Rahman A , Kokjohn T , Luehrs DC , Sue L , Cisneros R , Gerber F , Richardson C , Bohrmann B , Walker DG , Beach TG , Roher AE ((2004) ) Atherosclerosis, vascular amyloidosis and brain hypoperfusion in the pathogenesis of sporadic Alzheimer’s disease. Neurol Res 26: , 527–539. |
[11] | Attems J , Jellinger KA ((2014) ) The overlap between vascular disease and Alzheimer’s disease – Lessons from pathology. BMC Med 12: , 206. |
[12] | Kalaria RN , Ballard C ((1999) ) Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis Assoc Disord 13: (Suppl 3), S115–S123. |
[13] | Kalaria RN ((2016) ) Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer’s disease. Acta Neuropathol 131: , 659–685. |
[14] | Wallin A ((1998) ) The overlap between Alzheimer’s disease and vascular dementia: The role of white matter changes. Dement Geriatr Cogn Disord 1: (Suppl), 30–35. |
[15] | Neuropathology Group. Medical Research Council Cognitive Function and Aging Study ((2001) ) Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Lancet 357: , 169–175. |
[16] | Kövari E , Gold G , Herrmann FR , Canuto A , Hof PR , Michel JP , Bouras C , Giannakopoulos P ((2004) ) Cortical microinfarcts and demyelination significantly affect cognition in brain aging. Stroke 35: , 410–414. |
[17] | Brun A ((1994) ) Pathology and pathophysiology of cerebrovascular dementia: Pure subgroups of obstructive and hypoperfusive etiology. Dementia 5: , 145–147. |
[18] | Rosenberg GA , Wallin A , Wardlaw JM , Markus HS , Montaner J , Wolfson L , Iadecola C , Zlokovic BV , Joutel A , Dichgans M , Duering M , Schmidt R , Korczyn AD , Grinberg LT , Chui HC , Hachinski V ((2016) ) Consensus statement for diagnosis of subcortical small vessel disease. J Cereb Blood Flow Metab 36: , 6–25. |
[19] | Román GC , Erkinjuntti T , Wallin A , Pantoni L , Chui HC ((2002) ) Subcortical ischaemic vascular dementia. Lancet Neurol 1: , 426–436. |
[20] | Perneczky R , Tene O , Attems J , Giannakopoulos P , Ikram MA , Federico A , Sarazin M , Middleton LT ((2016) ) Is the time ripe for new diagnostic criteria of cognitive impairment due to cerebrovascular disease? Consensus report of the International Congress on Vascular Dementia working group. BMC Medicine 14: , 162. |
[21] | Sachdev P , Kalaria R , O’Brien J , Skoog I , Alladi S , Black SE , Blacker D , Blazer DG , Chen C , Chui H , Ganguli M , Jellinger K , Jeste DV , Pasquier F , Paulsen J , Prins N , Rockwood K , Román G , Scheltens P ((2014) ) Diagnostic criteria for vascular cognitive disorders: A VASCOG statement. Alzheimer Dis Assoc Disord 28: , 206–218. |
[22] | Skrobot OA , O’Brien J , Black S , Chen C , DeCarli C , Erkinjuntti T , Ford GA , Kalaria RN , Pantoni L , Pasquier F , Román GC , Wallin A , Sachdev P , Skoog I , VICCCS group, Ben-Shlomo Y , Passmore AP , Love S , Kehoe PG ((2017) ) The Vascular Impairment of Cognition Classification Consensus Study. Alzheimers Dement 13: , 624–633. |
[23] | Kehoe PG , Skrobot OA , O’Brien J , Black S , Chen C , DeCarli C , Erkinjuntti T , Ford GA , Kalaria RN , Pantoni L , Pasquier F , Román GC , Wallin A , Sachdev P , Skoog I , the VICCCS group, Ben-Shlomo Y , Passmore AP , Love S ((2018) ) Vascular Impairment of Cognition Classification Consensus Study (VICCCS). Lancet Neurol, in press. |
[24] | Thompson CS , Hakim AM ((2009) ) Living beyond our physiological means. Small vessel disease of the brain is an expression of a systemic failure in arteriolar function: A unifying hypothesis. Stroke 40: , e322–e330. |
[25] | Grunwald JE , Pistilli M , Ying GS , Daniel E , Maguire MG , Xie D , Whittock-Martin R , Parker Ostroff C , Lo JC , Townsend RR , Gadegbeku CA , Lash JP , Fink JC , Rahman M , Feldman HI , Kusek JW , Chronic Renal Insufficiency Cohort Study Investigators ((2014) ) Retinopathy and progression of CKD: The CRIC study. Clin J Am Soc Nephrol 9: , 1217–1224. |
[26] | Ooi QL , Tow FK , Deva R , Alias MA , Kawasaki R , Wong TY , Mohamad N , Colville D , Hutchinson A , Savige J ((2011) ) The microvasculature in chronic kidney disease. Clin J Am Soc Nephrol 6: , 1872–1878. |
[27] | Ferini-Strambi L , Lombardi GE , Marelli S , Galbiati A ((2017) ) Neurological deficits in obstructive sleep apnea. Curr Treat Options Neurol 19: , 16. |
[28] | Spence JD ((2017) ) Nutrition in stroke prevention. Semin Neurol 37: , 259–266. |
[29] | Román GC ((2015) ) MTHFR gene mutations: A potential marker of late-onset Alzheimer’s disease? J Alzheimers Dis 47: , 323–327. |
[30] | Alsop DC , Detre JA , Golay X , Günther M , Hendrikse J , Hernandez-Garcia L , Lu H , MacIntosh BJ , Parkes LM , Smits M , van Osch MJ , Wang DJ , Wong EC , Zaharchuk G ((2015) ) Recommended implementation of arterial spin-labeled perfusion MRI for clinical applications: A consensus of the ISMRM Perfusion Study Group and the European Consortium for ASL in Dementia. Magn Reson Med 73: , 102–116. |
[31] | Leeuwis AE , Benedictus MR , Kuijer JPA , Binnewijzend MAA , Hooghiemstra AM , Verfaillie SCJ , Koene T , Scheltens P , Barkhof F , Prins ND , van der Flier WM ((2017) ) Lower cerebral blood flow is associated with impairment in multiple cognitive domains in Alzheimer’s disease. Alzheimers Dement 13: , 531–540. |
[32] | Dopper EGP , Chalos V , Ghariq E , den Heijer T , Hafkemeijer A , Jiskoot LC , de Koning I , Seelaar H , van Minkelen R , van Osch MJP , Rombouts SARB , van Swieten JC ((2016) ) Cerebral blood flow in presymptomatic MAPT and GRN mutation carriers: A longitudinal arterial spin labeling study. Neuroimage Clin 12: , 460–465. |
[33] | Wallin A , Kapaki E , Boban M , Engelborghs S , Hermann DM , Huisa B , Jonsson M , Kramberger MG , Lossi L , Malojcic B , Mehrabian S , Merighi A , Mukaetova-Ladinska EB , Paraskevas GP , Popescu BO , Ravid R , Traykov L , Tsivgoulis G , Weinstein G , Korczyn A , Bjerke M , Rosenberg G ((2017) ) Biochemical markers in vascular cognitive impairment associated with subcortical small vessel disease – A consensus report. BMC Neurol 17: , 102. |
[34] | Bocchetta M , Galluzzi S , Kehoe PG , Aguera E , Bernabei R , Bullock R , Ceccaldi M , Dartigues JF , de Mendonça A , Didic M , Eriksdotter M , Félician O , Frölich L , Gertz HJ , Hallikainen M , Hasselbalch SG , Hausner L , Heuser I , Jessen F , Jones RW , Kurz A , Lawlor B , Lleo A , Martinez-Lage P , Mecocci P , Mehrabian S , Monsch A , Nobili F , Nordberg A , Rikkert MO , Orgogozo JM , Pasquier F , Peters O , Salmon E , Sánchez-Castellano C , Santana I , Sarazin M , Traykov L , Tsolaki M , Visser PJ , Wallin ÅK , Wilcock G , Wilkinson D , Wolf H , Yener G , Zekry D , Frisoni GB ((2015) ) The use of biomarkers for the etiologic diagnosis of MCI in Europe: An EADC survey. Alzheimers Dement 11: , 195.e1–206.e1. |
[35] | Skrobot OA , Attems J , Esiri M , Hortobágyi T , Ironside JW , Kalaria RN , King A , Lammie GA , Mann D , Neal J , Ben-Shlomo Y , Kehoe PG , Love S ((2016) ) Vascular Cognitive Impairment Neuropathology Guidelines (VCING): The contribution of cerebrovascular pathology to cognitive impairment. Brain 139: , 2957–2969. |
[36] | Barker MK , Bridges LR , Esiri MM , Hainsworth AH ((2017) ) A vasculo-centric pattern of axonal damage and neurofilament phosphorylation in subcortical white matter in older people. Neuropathology Appl Neurobiol 4: (Suppl 1), 23. |
[37] | Smallwood A , Oulhaj A , Joachim C , Christie S , Smith AD , Esiri M ((2012) ) Cerebral subcortical small vessel disease and its relation to cognition in elderly subjects: A pathological study in the Oxford Project to Investigate Memory and Ageing (OPTIMA) cohort. Neuropathol Appl Neurobiol 38: , 337–343. |
[38] | Nasreddine ZS , Phillips NA , Bedirian V , Charbonneau S , Whitehead V , Colin I , Cummings JL , Chertkow H ((2005) ) The Montreal Cognitive Assessment MOCA: A brief screening tool for mild cognitive impairment. J Amer Geriatr Soc 53: , 695–699. |
[39] | Toglia J , Fitzgerald KA , O’Dell MW , Mastrogiovanni R , Lin C ((2011) ) The Mini-Mental State Examination and Montreal Cognitive Assessment in persons with mild subacute stroke: Relationship to functional outcome. Arch Phys Med Rehabil 92: , 792–798. |
[40] | Brookes RL , Hollocks MJ , Khan U , Morris RG , Markus HS ((2015) ) The Brief Memory and Executive Test (BMET) for detecting vascular cognitive impairment in small vessel disease: A validation study. BMC Med 13: , 51. |
[41] | Jellinger KA ((2007) ) The enigma of vascular cognitive disorder and vascular dementia. Acta Neuropathol 113: , 349–388. |
[42] | Gouw AA , Seewann A , van der Flier WM , Barkhof F , Rozemuller AM , Scheltens P , Geurts JJ ((2011) ) Heterogeneity of small vessel disease: A systematic review of MRI and histopathology correlations. J Neurol Neurosurg Psychiatry 82: , 126–135. |
[43] | Gouw AA , Seewann A , Vrenken H , van der Flier WM , Rozemuller JM , Barkhof F , Scheltens P , Geurts JJ ((2008) ) Heterogeneity of white matter hyperintensities in Alzheimer’s disease: Post-mortem quantitative MRI and neuropathology. Brain 131: , 3286–3298. |
[44] | Fernando M , Simpson J , Matthews F , Brayne C , Lewis C , Barber R , Kalaria RN , Forster G , Esteves F , Wharton SB , Shaw PJ , O’Brien JT , Ince PG ((2006) ) White matter lesions in an unselected cohort of the elderly: Molecular pathology suggests origin from hypoperfusion injury. Stroke 37: , 1391–1398. |
[45] | van Swieten J , van den Hout J , van ketel B , Hijdra A , Wokke J , van Gijn J ((1991) ) Periventricular lesions in the white matter on magnetic resonance imaging in the elderly. A morphometric correlation with arteriolosclerosis and dilated perivascular spaces. Brain 114: , 761–774. |
[46] | Farkas E , Luiten PG ((2001) ) Cerebral microvascular pathology in aging and Alzheimer’s disease. Progr Neurobiol 64: , 575–611. |
[47] | Giwa MO , Williams J , Elderfield K , Jiwa NS , Bridges LR , Kalaria RN , Markus HS , Esiri MM , Hainsworth AH ((2012) ) Neuropathologic evidence of endothelial changes in cerebral small vessel disease. Neurology 78: , 167–174. |
[48] | Brown WR , Moody DM , Challa VR , Thore CR , Anstrom JA ((2002) ) Venous collagenosis and arteriolar tortuosity in leukoaraiosis. J Neurol Sci 203-204: , 159–163. |
[49] | Hassler O ((1965) ) Vascular changes in senile brains. Acta Neuropathol 5: , 40–53. |
[50] | Craggs LJ , Yamamoto Y , Deramecourt V , Kalaria RN ((2014) ) Microvascular pathology and morphometrics of sporadic and hereditary small vessel diseases of the brain. Brain Pathol 24: , 495–509. |
[51] | Eskildsen SF , Gyldensted L , Nagenthiraja K , Nielsen RB , Hansen MB , Dalby RB , Frandsen J , Rodell A , Gyldensted C , Jespersen SN , Lund TE , Mouridsen K , Brændgaard H , Østergaard L ((2017) ) Increased cortical capillary transit time heterogeneity in Alzheimer’s disease: A DSC-MRI perfusion study. Neurobiol Aging 50: , 107–118. |
[52] | Wharton SB , Simpson JE , Brayne C , Ince PG ((2015) ) Age-associated white matter lesions: The MRC Cognitive Function and Ageing Study. Brain Pathol 25: , 35–43. |
[53] | Garde E , Mortenson LE , Rostrup E , Paulson OB ((2005) ) Decline in intelligence is associated with progression in white matter hyperintensity volume. J Neurol Neurosurg Psychiat 76: , 1289–1291. |
[54] | Debette S , Markus HS ((2010) ) The clinical importance of white matter intensities on brain magnetic resonance imaging: A systematic review and meta-analysis. BMJ 341: , c3666. |
[55] | Jeetakathil T , Wolf PA , Beiser A , Massaro J , Seshadri S , D’Agostino RB , DeCarli C ((2004) ) Stroke risk profile predicts white matter hyperintensity volume: The Framingham study. Stroke 35: , 1857–1861. |
[56] | Novak V , Hajjar I ((2010) ) The relationship between blood pressure and cognitive function. Nat Rev Cardiol 7: , 686–698. |
[57] | White WB , Wolfson L , Wakefield DB , Hall CB , Campbell P , Muscufo N , Schmidt J , Kaplan RF , Pearlson G , Guttmann CR ((2011) ) Average daily blood pressure, not office blood pressure, is associated with progression of cerebrovascular disease and cognitive decline in older people. Circulation 124: , 2312–2319. |
[58] | Biessels GJ , Kappelle AC , Bravenboer B , Gispen WH ((1994) ) Cerebral function in diabetes mellitus. Diabetologia 37: , 643–650. |
[59] | Esiri MM , Joachim C , Sloan C , Christie S , Agacinski G , Bridges L , Wilcock GK , Smith AD ((2014) ) Cerebral subcortical small vessel disease in subjects with pathologically confirmed Alzheimer’s disease: A clinicopathological study in the Oxford Project to Investigate Memory and Ageing (OPTIMA). Alzheimer Dis Assoc Disord 28: , 35–40. |
[60] | Katzman R ((1993) ) Clinical and epidemiological aspects of Alzheimer’s disease. Clin Neurosci 1: , 165–170. |
[61] | Kawas C , Katzman R ((1999) ) Epidemiology of dementia and Alzheimer’s disease. In Alzheimer’s disease, Terry R , Katzman R , Bick K , Sisodia S , eds. Lippincott Williams and Wilkins, Philadelphia, pp. 95–116. |
[62] | Dufouil C , Godin O , Chalmers J , Coskun O , MacMahon S , Tzourio-Mazoyer N , Bousser M-G , Anderson C , Mazoyer B , Tzourio C ((2009) ) Severe cerebral white matter hyperintensities predict severe cognitive decline in patients with cerebrovascular disease history. Stroke 40: , 2219–2221. |
[63] | Waldstein SR , Rice SC , Thayer JF , Najjar SS , Scuteri A , Zonderman AB ((2008) ) Pulse pressure and pulse wave velocity are related to cognitive decline in the Baltimore Longitudinal Study of Aging. Hypertension 51: , 99–104. |
[64] | Ahmed-Jushuf F , Jiwa NS , Arwani AS , Foot P , Bridges LR , Kalaria RN , Esiri MM , Hainsworth AH ((2016) ) Age-dependent expression of VEGFR2 in deep brain arteries in small vessel disease, CADASIL and healthy brains. Neurobiol Aging 42: , 110–115. |
[65] | Marchesi C , Paradis P , Schriffin EL ((2008) ) Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol Sci 27: , 367–374. |
[66] | Wang M , Takagi G , Asai K , Resuello RG , Natividad FF , Vatner DE , Vatner SF , Lakatta EG ((2003) ) Age increases aortic MMP-2 activity and angiotensin II in nonhuman primates. Hypertension 41: , 1308–1316. |
[67] | Faraci FM ((2011) ) Protecting against vascular disease in brain. Am J Physiol Circ Physiol 300: , H1566–H1582. |
[68] | Gill R , Tsung A , Billiar T ((2010) ) Linking oxidative stress to inflammation: Toll-like receptors. Free Radical Biol Med 48: , 1121–1132. |
[69] | Loffredo FSA , Steinhauer ML , Jay SM , Gannon J , Pancoast JR , Yalamanshi P , Sinha M , Dall’Osso C , Khong D , Shadrach JL , Miller CM , Singer BS , Stewart A , Psychogios N , Gerszten RE , Hartigan AJ , Kim M-J , Serwold T , Wagers AJ , Lee RT ((2013) ) Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell 153: , 828–839. |
[70] | Farrall AJ , Wardlaw JM ((2009) ) Blood-brain barrier: Ageing and microvascular disease – A systematic review and meta-analysis. Neurobiol Aging 30: , 337–352. |
[71] | Bridges LR , Andoh J , Lawrence AJ , Khoong CH , Poon WW , Esiri MM , Markus HS , Hainsworth AH ((2014) ) Blood-brain barrier dysfunction and cerebral small vessel disease (arteriolosclerosis) in brains of older people. J Neuropathol Exp Neurol 73: , 1026–1033. |
[72] | Yates RL , Esiri MM , Palace J , Jacobs B , Perera R , DeLuca GC ((2017) ) Fibrin(ogen) and neurodegeneration in progressive multiple sclerosis motor cortex. Ann Neurol 82: , 259–270. |
[73] | Davalos D , Ryu JK , Merlini M , beaten KM , Le Moan N , Petersen MA , Deerinck TJ , Smirnoff DS , Bedard C , Hakozaki H , Gonias MS , Ling JB , Lassmann H , Degen JL , Ellisman MK , Akassoglou K ((2012) ) Fibrinogen-induced perivascular clustering is required for development of axonal damage in neuroinflammation. Nat Commun 3: , 1227. |
[74] | O’Neill C ((2013) ) PI3-kinase/Akt/mTOR signalling: Impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp Gerontol 48: , 647–653. |
[75] | Esiri MM , Chance S , Joachim C , Warden D , Smallwood A , Sloan C , Christie S , Wilcock G , Smith AD ((2015) ) Cerebral amyloid angiopathy, subcortical white matter disease and dementia: Literature review and study in OPTIMA. Brain Pathol 25: , 51–62. |
[76] | Lamming DW , Ye L , Katajisto P , Goncalves MD , Saitoh M , Stevens DM , Davis JG , Salmon AB , Sabatini DM , Baur JA ((2012) ) Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335: , 1638–1643. |
[77] | Wu JJ , Liu J , Chen EB , Wang JJ , Cao L , Narayan N , Fergusson MM , Rovira II , Allen M , Springer DA , Lago CU , Zhang S , DuBois W , Ward T , deCabo R , Gavrilova O , Mock B , Finkel T ((2013) ) Increased mammalian lifespan and a segmental and tissue-specific slowing of aging after genetic reduction of MTOR expression. Cell Reports 4: , 913–920. |
[78] | Spilman P , Podlutskaya N , Hart MJ , Debnath J , Gorostiza O , Bredesen D , Richardson A , Strong R , Galvan V ((2010) ) Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-β levels in a mouse model of Alzheimer’s disease. PLoS One 5: , e9979. |
[79] | Yates SC , Zafar A , Hubbard P , Nagy S , Durant S , Bicknell R , Wilcock G , Christie S , Esiri MM , Smith AD , Nagy Z ((2013) ) Dysfunction of the mTOR pathway is a risk factor for Alzheimer’s disease. Acta Neuropath Commun 1: , 3. |
[80] | Zhu L-H , Huang L , Zhang X , Zhang P , Zhang S-M , Guan H , Zhang Y , Zhu X-Y , Tian S , Deng K , Hongliang L ((2015) ) Mindin regulates vascular smooth muscle cell phenotype and prevents neointima formation. Clin Sci 129: , 129–145. |
[81] | Ha JM , Yun SJ , Kim YW , Jin SY , Lee HS , Song SH , Shin HK , Bae SS ((2015) ) Platelet-derived growth factor regulates vascular smooth muscle phenotype via mammalian target of rapamycin complex 1. Biochem Biophys Res Commun 464: , 57–62. |
[82] | van Bogaert L ((1955) ) Encéphalopathie sous-corticale progressive (Binswanger) à évolution rapide chez deux soeurs. Helliniki Iatriki 24: , 3–14. |
[83] | Tournier-Lasserve E , Iba-Zizen MT , Romero N , Bousser MG ((1991) ) Autosomal dominant syndrome with stroke-like episodes and leukoencephalopathy. Stroke 22: , 1297–1302. |
[84] | Sourander P , Wålinder J ((1977) ) Hereditary multi-infarct dementia. Acta Neuropathol (Berl) 39: , 247–254. |
[85] | Zhang WW , Ma KC , Andersen O , Sourander P , Tolesson PO , Olsson Y ((1994) ) The microvascular changes in cases of hereditary multi-infarct disease of the brain. Acta Neuropathol (Berl) 87: , 317–324. |
[86] | Federico A , Di Donato I , Bianchi S , Di Palma C , Taglia I , Dotti MT ((2012) ) Hereditary cerebral small vessel diseases: A review. J Neurol Sci 322: , 25–30. |
[87] | Jones EL , Kalaria RN , Sharp SI , O’Brien JT , Francis PT , Ballard CG ((2011) ) Genetic associations of autopsy-confirmed vascular dementia subtypes. Dement Geriatr Cogn Disord 31: , 247–253. |
[88] | Schrijvers EM , Schurmann B , Koudstaal PJ , van den Bussche H , Van Duijn CM , Hentschel F , Heun R , Hofman A , Jessen F , Kolsch H , Kornhuber J , Peters O , Rivadeneira F , Ruther E , Uitterlinden AG , Riedel-Heller S , Dichgans M , Wiltfang J , Maier W , Breteler MM , Ikram MA ((2012) ) Genome-wide association study of vascular dementia. Stroke 43: , 315–319. |
[89] | Kim Y , Kong M , Lee C ((2013) ) Association of intronic sequence variant in the gene encoding spleen tyrosine kinase with susceptibility to vascular dementia. World J Biol Psychiatry 14: , 220–226. |
[90] | Corder EH , Saunders AM , Strittmatter WJ , Schmechel DE , Gaskell PC , Small GW , Roses AD , Haines JL , Pericak-Vance MA ((1993) ) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261: , 921–923. |
[91] | Greenberg SM , Rebeck GW , Vonsattel JP , Gomez-Isla T , Hyman BT ((1995) ) Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol 38: , 254–259. |
[92] | Skrobot OA , McKnight AJ , Passmore PA , Seripa D , Mecocci P , Panza F , Kalaria R , Wilcock G , Munafo M , Erkinjuntti T , Karhunen P , Pessi T , Martiskainen M , Love S , for the Genetic and Environmental Risk for Alzheimer’s disease Consortium (GERAD1), Kehoe PG ((2016) ) A validation study of vascular cognitive impairment genetics meta-analysis. Findings in an independent collaborative cohort. J Alzheimers Dis 53: , 981–989. |
[93] | Dwyer R , Skrobot OA , Dwyer J , Munafo M , Kehoe PG ((2013) ) Using Alzgene-like approaches to investigate susceptibility genes for vascular cognitive impairment. J Alzheimers Dis 34: , 145–154. |
[94] | Sun JH , Tan L , Wang HF , Tan MS , Tan L , Li JQ , Xu W , Zhu XC , Jiang T , Yu JT ((2015) ) Genetics of vascular dementia: Systematic review and meta-analysis. J Alzheimers Dis 46: , 611–629. |
[95] | Maxwell SS , Jackson CA , Paternoster L , Cordonnier C , Thijs V , Al-Shahi Salman R , Sudlow CL ((2011) ) Genetic associations with brain microbleeds: Systematic review and meta-analyses. Neurology 77: , 158–167. |
[96] | Paternoster L , Chen W , Sudlow CL ((2009) ) Genetic determinants of white matter hyperintensities on brain scans: A systematic assessment of 19 candidate gene polymorphisms in 46 studies in 19,000 subjects. Stroke 40: , 2020–2026. |
[97] | Machuca-Parra AI , Bigger-Allen AA , Sanchez AV , Boutabla A , Cardona-Vélez J , Amarnani D , Saint-Geniez M , Siebel CW , Kim LA , D’Amore PA , Arboleda-Velasquez JF ((2017) ) Therapeutic antibody targeting of Notch3 signaling prevents mural cell loss in CADASIL. J Exp Med 214: , 2271–2282. |
[98] | Román GC , Royall DR ((1999) ) Executive control function: A rational basis for the diagnosis of vascular dementia. Alzheimer Dis Assoc Disord 13: (Suppl 3), 69–80. |
[99] | Cohen RA , Paul RH , Ott BR , Moser DJ , Zawacki TM , Stone W , Gordon N ((2002) ) The relationship of subcortical MRI hyperintensities and brain volume to cognitive function in vascular dementia. J Int Neuropsychol Soc 8: , 743–752. |
[100] | Libon DJ , Price CC , Davis Garrett K , Giovannetti T ((2004) ) From Binswanger’s disease to leuokoaraiosis: What we have learned about subcortical vascular dementia. Clin Neuropsychol 18: , 83–100. |
[101] | Nordlund A , Rolstad S , Klang O , Lind K , Hansen S , Wallin A ((2007) ) Cognitive profiles of mild cognitive impairment with and without vascular disease. Neuropsychology 21: , 706–712. |
[102] | Cannata AP , Alberoni M , Franceschi M , Mariani C ((2002) ) Frontal impairment in subcortical ischemic vascular dementia in comparison to Alzheimer’s disease. Dement Geriatr Cogn Disord 13: , 101–111. |
[103] | Bennett DA , Gilley DW , Lee S , Cochran EJ ((1994) ) White matter changes: Neurobehavioral manifestations of Binswanger’s disease and clinical correlates in Alzheimer’s disease. Dementia 5: , 148–152. |
[104] | Jokinen H , Kalska H , Ylikoski R , Madureira S , Verdelho A , van der Flier WM , Scheltens P , Barkhof F , Visser MC , Fazekas F , Schmidt R , O’Brien J , Waldemar G , Wallin A , Chabriat H , Pantoni L , Inzitari D , Erkinjuntti T , LADIS group ((2009) ) Longitudinal cognitive decline in subcortical ischemic vascular disease–The LADIS Study. Cerebrovasc Dis 27: , 384–391. |
[105] | Vasquez BP , Zakzanis KK ((2015) ) The neuropsychological profile of vascular cognitive impairment not demented: A meta-analysis. J Neuropsychol 9: , 109–136. |
[106] | Hachinski V , Iadecola C , Petersen RC , Breteler MM , Nyenhuis DL , Black SE , Powers WJ , DeCarli C , Merino JG , Kalaria RN , Vinters HV , Holtzman DM , Rosenberg GA , Wallin A , Dichgans M , Marler JR , Leblanc GG ((2006) ) National Institute of Neurological Disorders and Stroke-Canadian Stroke Network vascular cognitive impairment harmonization standards. Stroke 37: , 2220–2241. |
[107] | Salvadori E , Poggesi A , Pracucci G , Inzitari D , Pantoni L , VMCI-Tuscany Study Group ((2015) ) Development and psychometric properties of a neuropsychological battery for mild cognitive impairment with small vessel disease: The VMCI-Tuscany Study. J Alzheimers Dis 43: , 313–323. |
[108] | Salvadori E , Poggesi A , Valenti R , Pracucci G , Pescini F , Pasi M , Nannucci S , Marini S , Del Bene A , Ciolli L , Ginestroni A , Diciotti S , Orlandi G , Di Donato I , De Stefano N , Cosottini M , Chiti A , Federico A , Dotti MT , Bonuccelli U , Inzitari D , Pantoni L , VMCI-Tuscany Study Group ((2016) ) Operationalizing mild cognitive impairment criteria in small vessel disease: The VMCI-Tuscany Study. Alzheimers Dement 12: , 407–418. |
[109] | Jang YK , Kwon H , Kim YJ , Jung NY , Lee JS , Lee J , Chin J , Im K , Jeon S , Lee JM , Seong JK , Kim JH , Kim S , Choe YS , Lee KH , Kim ST , Kim JS , Lee JH , Na DL , Seo SW , Kim HJ ((2016) ) Early- vs. late-onset subcortical vascular cognitive impairment. Neurology 86: , 527–534. |
[110] | Hong YJ , Yoon B , Shim YS , Han IW , Han SH , Park KH , Choi SH , Ku BD , Yang DW ((2014) ) Do Alzheimer’s disease (AD) and subcortical ischemic vascular dementia (SIVD) progress differently? Arch Gerontol Geriatr 58: , 415–419. |
[111] | Hsu YH , Huang CF , Lo CP , Wang TL , Yang CC , Tu MC ((2016) ) Frontal assessment battery as a useful tool to differentiate mild cognitive impairment due to subcortical ischemic vascular disease from Alzheimer disease. Dement Geriatr Cogn Disord 42: , 331–341. |
[112] | Xu Q , Cao WW , Mi JH , Yu L , Lin Y , Li YS ((2014) ) Brief screening for mild cognitive impairment in subcortical ischemic vascular disease: A comparison study of the Montreal Cognitive Assessment with the Mini-Mental State Examination. Eur Neurol 71: , 106–114. |
[113] | Nyström O , Wallin A , Nordlund A ((2015) ) MCI of different etiologies differ on the Cognitive Assessment Battery. Acta Neurol Scand 132: , 31–36. |
[114] | Ramirez-Gomez L , Zheng L , Reed B , Kramer J , Mungas D , Zarow C , Vinters H , Ringman JM , Chui H ((2017) ) Neuropsychological profiles differentiate Alzheimer disease from subcortical ischemic vascular dementia in an autopsy-defined cohort. Dement Geriatr Cogn Disord 44: , 1–11. |
[115] | Choi SH , Kim S , Han SH , Na DL , Kim DK , Cheong HK , Lee JH , Kim SY , Hong CH , Moon SY , Kwon JC , Kim JE , Jeong JH , Na HR , Cha KR , Seo SW , Shim YS , Lee JY , Park KW ((2012) ) Neurologic signs in relation to cognitive function in subcortical ischemic vascular dementia: A CREDOS (Clinical Research Center for Dementia of South Korea) study. Neurol Sci 33: , 839–846. |
[116] | Royall DR , Cordes JA , Polk M ((1998) ) CLOX: An executive clock drawing task. J Neurol Neurosurg Psychiatry 64: , 588–594. |
[117] | Kim GH , Seo SW , Jung K , Kwon OH , Kwon H , Kim JH , Roh JH , Kim MJ , Lee BH , Yoon DS , Hwang JW , Lee JM , Jeong JH , You H , Heilman KM , Na DL ((2016) ) The neural correlates of motor intentional disorders in patients with subcortical vascular cognitive impairment. J Neurol 263: , 89–99. |
[118] | Kreisel SH , Blahak C , Bäzner H , Inzitari D , Pantoni L , Poggesi A , Chabriat H , Erkinjuntti T , Fazekas F , Ferro JM , Langhorne P , O’Brien J , Scheltens P , Visser MC , Wahlund LO , Waldemar G , Wallin A , Hennerici MG ((2013) ) Deterioration of gait and balance over time: The effects of age-related white matter change–The LADIS study. Cerebrovasc Dis 35: , 544–553. |
[119] | Inzitari D , Pracucci G , Poggesi A , Carlucci G , Barkhof F , Chabriat H , Erkinjuntti T , Fazekas F , Ferro JM , Hennerici M , Langhorne P , O’Brien J , Scheltens P , Visser MC , Wahlund LO , Waldemar G , Wallin A , Pantoni L , LADIS Study Group ((2009) ) Changes in white matter as determinant of global functional decline in older independent outpatients: Three year follow-up of LADIS (leukoaraiosis and disability) study cohort. BMJ 339: , b2477. |
[120] | Kandiah N , Zainal NH , Narasimhalu K , Chander RJ , Ng A , Mak E , Au WL , Sitoh YY , Nadkarni N , Tan LC ((2014) ) Hippocampal volume and white matter disease in the prediction of dementia in Parkinson’s disease. Parkinsonism Relat Disord 20: , 1203–1208. |
[121] | Foo H , Mak E , Yong TT , Wen MC , Chander RJ , Au WL , Tan L , Kandiah N ((2016) ) Progression of small vessel disease correlates with cortical thinning in Parkinson’s disease. Parkinsonism Relat Disord 31: , 34–40. |
[122] | Jonsson M , Edman A , Lind K , Rolstad S , Sjögren M , Wallin A ((2010) ) Apathy is a prominent neuropsychiatric feature of radiological white-matter changes in patients with dementia. Int J Geriatr Psychiatry 25: , 588–595. |
[123] | Kim HJ , Kang SJ , Kim C , Kim GH , Jeon S , Lee JM , Oh SJ , Kim JS , Choe YS , Lee KH , Noh Y , Cho H , Yoon CW , Chin J , Cummings JL , Lee JH , Na DL , Seo SW ((2013) ) The effects of small vessel disease and amyloid burden on neuropsychiatric symptoms: A study among patients with subcortical vascular cognitive impairments. Neurobiol Aging 34: , 1913–1920. |
[124] | Staekenborg SS , Gillissen F , Romkes R , Pijnenburg YA , Barkhof F , Scheltens P , van der Flier WM ((2008) ) Behavioural and psychological symptoms are not related to white matter hyperintensities and medial temporal lobe atrophy in Alzheimer’s disease. Int J Geriatr Psychiatry 23: , 387–392. |
[125] | Fuh JL , Wang SJ , Cummings JL ((2005) ) Neuropsychiatric profiles in patients with Alzheimer’s disease and vascular dementia. J Neurol Neurosurg Psychiatry 76: , 1337–1341. |
[126] | Chin J , Seo SW , Kim SH , Park A , Ahn HJ , Lee BH , Kang SJ , Na DL ((2012) ) Neurobehavioral dysfunction in patients with subcortical vascular mild cognitive impairment and subcortical vascular dementia. Clin Neuropsychol 26: , 224–238. |
[127] | Hollocks MJ , Lawrence AJ , Brookes RL , Barrick TR , Morris RG , Husain M , Markus HS ((2015) ) Differential relationships between apathy and depression with white matter microstructural changes and functional outcomes. Brain 138: (Pt 12), 3803–3815. |
[128] | Moretti R , Signori R ((2016) ) Neural correlates for apathy: Frontal-prefrontal and parietal cortical-subcortical circuits. Front Aging Neurosci 8: , 289. |
[129] | Kazui H , Takahashi R , Yamamoto Y , Yoshiyama K , Kanemoto H , Suzuki Y , Sato S , Azuma S , Suehiro T , Shimosegawa E , Ishii K , Tanaka T ((2017) ) Neural basis of apathy in patients with amnestic mild cognitive impairment. J Alzheimers Dis 55: , 1403–1416. |
[130] | Filippi M , Agosta F , Barkhof F , Dubois B , Fox NC , Frisoni GB , Jack CR , Johannsen P , Miller BL , Nestor PJ , Scheltens P , Sorbi S , Teipel S , Thompson PM , Wahlund LO , European Federation of the Neurologic Societies ((2012) ) EFNS task force: The use of neuroimaging in the diagnosis of dementia. Eur J Neurol 19: , 1487–1501. |
[131] | Román G , Pascual B ((2012) ) Contribution of neuroimaging to the diagnosis of Alzheimer’s disease and vascular dementia. Arch Med Res 43: , 671–676. |
[132] | Wahlund LO , Westman E , van Westen D , Wallin A , Shams S , Cavallin L , Larsson EM , for the Imaging Cognitive Impairment Network (ICINET) ((2017) ) Imaging biomarkers of dementia: Recommended visual rating scales with teaching cases. Insights Imaging 8: , 79–90. |
[133] | Schmidt R , Berghold A , Jokinen H , Gouw AA , van der Flier WM , Barkhof F , Scheltens P , Petrovic K , Madureira S , Verdelho A , Ferro JM , Waldemar G , Wallin A , Wahlund LO , Poggesi A , Pantoni L , Inzitari D , Fazekas F , Erkinjuntti T , for the LADIS Study Group ((2012) ) White matter lesion progression in LADIS: Frequency, clinical effects, and sample size calculations. Stroke 43: , 2643–2647. |
[134] | Longstreth WT Jr , Manolio TA , Arnold A , Burke GL , Bryan N , Jungreis CA , Enright PL , O’Leary D , Fried L ((1996) ) Clinical correlates of white matter findings on cranial magnetic resonance imaging of 3301 elderly people. The Cardiovascular Health Study. Stroke 27: , 1274–1282. |
[135] | Braffman BH , Zimmerman RA , Trojanowski JQ , Gonatas NK , Hickey WF , Schlaepfer WW ((1988) ) Brain MR: Pathologic correlation with gross and histopathology. 1. Lacunar infarction and Virchow-Robin spaces. AJR Am J Roentgenol 151: , 551–558. |
[136] | Braffman BH , Zimmerman RA , Trojanowski JQ , Gonatas NK , Hickey WF , Schlaepfer WW ((1988) ) Brain MR: Pathologic correlation with gross and histopathology. 2. Hyperintense white-matter foci in the elderly. AJR Am J Roentgenol 151: , 559–566. |
[137] | Brun A , Englund E ((1986) ) A white matter disorder in dementia of the Alzheimer type: A pathoanatomical study. Ann Neurol 19: , 253–262. |
[138] | Moody DM , Bell MA , Challa VR ((1990) ) Features of the cerebral vascular pattern that predict vulnerability to perfusion or oxygenation deficiency: An anatomic study. AJNR Am J Neuroradiol 11: , 431–439. |
[139] | Spangler KM , Challa VR , Moody DM , Bell MA ((1994) ) Arteriolar tortuosity of the white matter in aging and hypertension. A microradiographic study. J Neuropathol Exp Neurol 53: , 22–26. |
[140] | Wardlaw JM , Valdés Hernández MC , Muñoz-Maniega S ((2015) ) What are white matter hyperintensities made of? Relevance to vascular cognitive impairment. J Am Heart Assoc 4: , 001140. |
[141] | Morris Z , Whiteley WN , Longstreth WT Jr , Weber F , Lee YC , Tsushima Y , Alphs H , Ladd SC , Warlow C , Wardlaw JM , Al-Shahi Salman R ((2009) ) Incidental findings on brain magnetic resonance imaging: Systematic review and meta-analysis. BMJ 339: , b3016. |
[142] | Agosta F , Galantucci S , Filippi M ((2017) ) Advanced magnetic resonance imaging of neurodegenerative diseases. Neurol Sci 38: , 41–51. |
[143] | Blair GW , Hernandez MV , Thrippleton MJ , Doubal FN , Wardlaw JM ((2017) ) Advanced neuroimaging of cerebral small vessel disease. Curr Treat Options Cardiovasc Med 19: , 56. |
[144] | Banerjee G , Wilson D , Jäger HR , Werring DJ ((2016) ) Novel imaging techniques in cerebral small vessel diseases and vascular cognitive impairment. Biochim Biophys Acta 1862: , 926–938. |
[145] | Wardlaw JM , Smith EE , Biessels GJ , Cordonnier C , Fazekas F , Frayne R , Lindley RI , O’Brien JT , Barkhof F , Benavente OR , Black SE , Brayne C , Breteler M , Chabriat H , Decarli C , de Leeuw FE , Doubal F , Duering M , Fox NC , Greenberg S , Hachinski V , Kilimann I , Mok V , Oostenbrugge Rv , Pantoni L , Speck O , Stephan BC , Teipel S , Viswanathan A , Werring D , Chen C , Smith C , van Buchem M , Norrving B , Gorelick PB , Dichgans M , STandards for ReportIng Vascular changes on nEuroimaging (STRIVE, v1) ((2013) ) Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 12: , 822–838. |
[146] | Hilal S , Sikking E , Shaik MA , Chan QL , van Veluw SJ , Vrooman H , Cheng CY , Sabanayagam C , Cheung CY , Wong TY , Venketasubramanian N , Biessels GJ , Chen C , Ikram MK ((2016) ) Cortical cerebral microinfarcts on 3T MRI: A novel marker of cerebrovascular disease. Neurology 87: , 1583–1590. |
[147] | Shi Y , Thrippleton MJ , Makin SD , Marshall I , Geerlings MI , de Craen AJ , van Buchem MA , Wardlaw JM ((2016) ) Cerebral blood flow in small vessel disease: A systematic review and meta-analysis. J Cereb Blood Flow Metab 36: , 1653–1667. |
[148] | Blair GW , Doubal FN , Thrippleton MJ , Marshall I , Wardlaw JM ((2016) ) Magnetic resonance imaging for assessment of cerebrovascular reactivity in cerebral small vessel disease: A systematic review. J Cereb Blood Flow Metab 36: , 833–841. |
[149] | Shivamurthy VK , Tahari AK , Marcus C , Subramaniam RM ((2015) ) Brain FDG PET and the diagnosis of dementia. AJR Am J Roentgenol 204: , .W76–W85. |
[150] | Heiss WD , Rosenberg GA , Thiel A , Berlot R , de Reuck J ((2016) ) Neuroimaging in vascular cognitive impairment: A state-of-the-art review. BMC Med 14: , 174. |
[151] | Bloudek LM , Spackman DE , Blankenburg M , Sullivan SD ((2011) ) Review and meta-analysis of biomarkers and diagnostic imaging in Alzheimer’ disease. J Alzheimers Dis 26: , 627–645. |
[152] | Kerrouche N , Herholz K , Mielke R , Holthoff V , Baron JC ((2006) ) 18FDG PET in vascular dementia: Differentiation from Alzheimer’s disease using voxel-based multivariate analysis. J Cereb Blood Flow Metab 26: , 1213–1221. |
[153] | Zhang N , Gordona ML , Goldberg TE ((2017) ) Cerebral blood flow measured by arterial spin labeling MRI at resting state in normal aging and Alzheimer’s disease. Neurosci Biobehavior Rev 72: , 168–175. |
[154] | Ogoh S ((2017) ) Relationship between cognitive function and regulation of cerebral blood flow. J Physiol Sci 67: , 345–351. |
[155] | Grade M , Hernandez Tamames JA , Pizzini FB , Achten E , Golay X , Smits M ((2015) ) A neuroradiologist’s guide to arterial spin labeling MRI in clinical practice. Neuroradiology 57: , 1181–1202. |
[156] | Sun Y , Cao W , Ding W , Wang Y , Han X , Zhou Y , Xu Q , Zhang Y , Xu J ((2016) ) Cerebral blood flow alterations as assessed by 3D ASL in cognitive impairment in patients with Subcortical Vascular Cognitive Impairment: A marker for disease severity. Front Aging Neurosci 8: , 211. |
[157] | Chen HL , Lin HC , Lu CH , Chen PC , Huang CC , Chou KH , Su MC , Friedman M , Chen YW , Lin WC ((2017) ) Systemic inflammation and alterations to cerebral blood flow in obstructive sleep apnea. J Sleep Res 26: , 789–798. |
[158] | Palomares JA , Tummala S , Wang DJ , Park B , Woo MA , Kang DW , St Lawrence KS , Harper RM , Kumar R ((2015) ) Water exchange across the blood-brain barrier in obstructive sleep apnea: An MRI diffusion-weighted pseudo-continuous arterial spin labeling study. J Neuroimaging 25: , 900–905. |
[159] | Wang J , Fernandez-Seara MA , Wang S , St Lawrence KS ((2007) ) When perfusion meets diffusion: In vivo measurement of water permeability in human brain. J Cereb Blood Flow Metab 27: , 839–849. |
[160] | Wang J , Alsop DC , Song HK , Maldjian JA , Tang K , Salvucci AE , Detre JA ((2003) ) Arterial transit time imaging with flow encoding arterial spin tagging (FEAST). Magn Reson Med 50: , 599–607. |
[161] | Román GC ((1991) ) White matter lesions and normal-pressure hydrocephalus: Binswanger disease or Hakim syndrome? Am J Neuroradiol AJNR 12: , 40–41. |
[162] | Bradley WG Jr , Whittemore AR , Watanabe AS , Davis SJ , Teresi LM , Hornyak M ((1991) ) Association of deep white matter infarction with chronic communicating hydrocephalus: Implications regarding the possible origin of normal-pressure hydrocephalus. AJNR 12: , 31–39. |
[163] | Hakim S , Venegas JG , Burton JD ((1976) ) The physics of the cranial cavity, hydrocephalus and normal pressure hydrocephalus: Mechanical interpretation and mathematical model. Surg Neurol 5: , 187–210. |
[164] | Owler BK , Pickard JD ((2001) ) Normal pressure hydrocephalus and cerebral blood flow: A review. Acta Neurol Scand 104: , 325–342. |
[165] | Yeom KW , Lober RM , Alexander A , Cheshier SH , Edwards MSB ((2014) ) Hydrocephalus decreases arterial spin-labeled cerebral perfusion. AJNR Am J Neuroradiol 35: , 1433–1439. |
[166] | Li MD , Forkert ND , Kundu P , Ambler C , Lober RM , Burns TC , Barnes PD , Gibbs IC , Grant GA , Fisher PG , Cheshier SH , Campen CJ , Monje M , Yeom KW ((2017) ) Brain perfusion and diffusion abnormalities in children treated for posterior fossa brain tumors. J Pediatr 185: , .173.e3–180.e3. |
[167] | McKhann GM , Knopman DS , Chertkow H , Hyman BT , Jack CR Jr , Kawas CH , Klunk WE , Koroshetz WJ , Manly JJ , Mayeux R , Mohs RC , Morris JC , Rossor MN , Scheltens P , Carrillo MC , Thies B , Weintraub S , Phelps CH ((2011) ) The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 263–269. |
[168] | Dubois B , Feldman HH , Jacova C , Hampel H , Molinuevo JL , Blennow K , DeKosky ST , Gauthier S , Selkoe D , Bateman R , Cappa S , Crutch S , Engelborghs S , Frisoni GB , Fox NC , Galasko D , Habert MO , Jicha GA , Nordberg A , Pasquier F , Rabinovici G , Robert P , Rowe C , Salloway S , Sarazin M , Epelbaum S , de Souza LC , Vellas B , Visser PJ , Schneider L , Stern Y , Scheltens P , Cummings JL ((2014) ) Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol 13: , 614–629. |
[169] | Skillbäck T , Delsing L , Synnergren J , Mattsson N , Janelidze S , Nägga K , Kilander L , Hicks R , Wimo A , Winblad B , Hansson O , Blennow K , Eriksdotter M , Zetterberg H ((2017) ) CSF/serum albumin ratio in dementias: A cross-sectional study on 1861 patients. Neurobiol Aging 59: , 1–9. |
[170] | Cavedo E , Lista S , Khachaturian Z , Aisen P , Amouyel P , Herholz K , Jack CR Jr , Sperling R , Cummings J , Blennow K , O’Bryant S , Frisoni GB , Khachaturian A , Kivipelto M , Klunk W , Broich K , Andrieu S , de Schotten MT , Mangin JF , Lammertsma AA , Johnson K , Teipel S , Drzezga A , Bokde A , Colliot O , Bakardjian H , Zetterberg H , Dubois B , Vellas B , Schneider LS , Hampel H ((2014) ) The road ahead to cure Alzheimer’s disease: Development of biological markers and Neuroimaging methods for prevention trials across all stages and target populations. J Prev Alzheimers Dis 1: , 181–202. |
[171] | Mori H , Hosoda K , Matsubara E , Nakamoto T , Furiya Y , Endoh R , Usami M , Shoji M , Maruyama S , Hirai S ((1995) ) Tau in cerebrospinal fluids: Establishment of the sandwich ELISA with antibody specific to the repeat sequence in tau. Neurosci Lett 186: , 181–183. |
[172] | Kapaki E , Paraskevas GP , Zalonis I , Zournas C ((2003) ) CSF tau protein and β-amyloid (1–42) in Alzheimer’s disease diagnosis: Discrimination from normal ageing and other dementias in the Greek population. Eur J Neurol 10: , 119–128. |
[173] | Paraskevas GP , Kapaki E , Liappas I , Theotoka I , Mamali I , Zournas C , Lykouras L ((2005) ) The diagnostic value of cerebrospinal fluid tau protein in dementing and nondementing neuropsychiatric disorders. J Geriatr Psychiatry Neurol 18: , 163–173. |
[174] | Tato RE , Frank A , Hernanz A ((1995) ) Tau protein concentrations in cerebrospinal fluid of patients with dementia of the Alzheimer type. J Neurol Neurosurg Psychiatry 59: , 280–283. |
[175] | Andreasen N , Vanmechelen E , Van de Voorde A , Davidsson P , Hesse C , Tarvonen S , Räihä I , Sourander L , Winblad B , Blennow K ((1998) ) Cerebrospinal fluid tau protein as a biochemical marker for Alzheimer’s disease: A community based follow up study. J Neurol Neurosurg Psychiatry 64: , 298–305. |
[176] | Andreasen N , Minthon L , Davidsson P , Vanmechelen E , Vanderstichele H , Winblad B , Blennow K ((2001) ) Evaluation of CSF-tau and CSF-Abeta42 as diagnostic markers for Alzheimer disease in clinical practice. Arch Neurol 58: , 373–379. |
[177] | Jia JP , Meng R , Sun YX , Sun WJ , Ji XM , Jia LF ((2005) ) Cerebrospinal fluid tau, Abeta1-42 and inflammatory cytokines in patients with Alzheimer’s disease and vascular dementia. Neurosci Lett 383: , 12–16. |
[178] | Stefani A , Bernardini S , Panella M , Pierantozzi M , Nuccetelli M , Koch G , Urbani A , Giordano A , Martorana A , Orlacchio A , Federici G , Bernardi G ((2005) ) AD with subcortical white matter lesions and vascular dementia: CSF markers for differential diagnosis. J Neurol Sci 237: , 83–88. |
[179] | Leszek J , Malyszczak K , Janicka B , Kiejna A , Wiak A ((2003) ) Total tau in cerebrospinal fluid differentiates Alzheimer’s disease from vascular dementia. Med Sci Monit 9: , CR484–CR488. |
[180] | Paraskevas GP , Kapaki E , Papageorgiou SG , Kalfakis N , Andreadou E , Zalonis I , Vassilopoulos D ((2009) ) CSF biomarker profile and diagnostic value in vascular dementia. Eur J Neurol 16: , 205–211. |
[181] | Vandermeeren M , Mercken M , Vanmechelen E , Six J , van de Voorde A , Martin JJ , Cras P ((1993) ) Detection of tau proteins in normal and Alzheimer’s disease cerebrospinal fluid with a sensitive sandwich enzyme-linked immunosorbent assay. J Neurochem 61: , 1828–1834. |
[182] | Vigo-Pelfrey C , Seubert P , Barbour R , Blomquist C , Lee M , Lee D , Coria F , Chang L , Miller B , Lieberburg I , Schenk D ((1995) ) Elevation of microtubule-associated protein tau in the cerebrospinal fluid of patients with Alzheimer’s disease. Neurology 45: , 788–793. |
[183] | Kaerst L , Kuhlmann A , Wedekind D , Stoeck K , Lange P , Zerr I ((2013) ) Cerebrospinal fluid biomarkers in Alzheimer’s disease, vascular dementia and ischemic stroke patients: A critical analysis. J Neurol 260: , 2722–2727. |
[184] | Wallin A , Sjogren M ((2001) ) Cerebrospinal fluid cytoskeleton proteins in patients with subcortical white-matter dementia. Mech Ageing Dev 122: , 1937–1949. |
[185] | Bjerke M , Zetterberg H , Edman A , Blennow K , Wallin A , Andreasson U ((2011) ) Cerebrospinal fluid matrix metalloproteinases and tissue inhibitor of metalloproteinases in combination with subcortical and cortical biomarkers in vascular dementia and Alzheimer’s disease. J Alzheimers Dis 27: , 665–676. |
[186] | Hermann P , Romero C , Schmidt C , Reis C , Zerr I ((2014) ) CSF biomarkers and neuropsychological profiles in patients with cerebral small-vessel disease. PLoS One 9: , e105000. |
[187] | Wallin A , Nordlund A , Jonsson M , Blennow K , Zetterberg H , Öhrfelt A , Stålhammar J , Eckerström M , Carlsson M , Olsson E , Göthlin M , Svensson J , Rolstad S , Eckerström C , Bjerke M ((2016) ) Alzheimer’s disease-subcortical vascular disease spectrum in a hospitalbased setting: Overview of results from the Gothenburg MCI and dementia studies. J Cereb Blood Flow Metab 36: , 95–113. |
[188] | Bjerke M , Andreasson U , Rolstad S , Nordlund A , Lind K , Zetterberg H , Edman A , Blennow K , Wallin A ((2009) ) Subcortical vascular dementia biomarker pattern in mild cognitive impairment. Dement Geriatr Cogn Disord 28: , 348–356. |
[189] | Rosenberg GA , Prestopnik J , Adair JC , Huisa BN , Knoefel J , Caprihan A , Gasparovic C , Thompson J , Erhardt EB , Schrader R ((2015) ) Validation of biomarkers in subcortical ischaemic vascular disease of the Binswanger type: Approach to targeted treatment trials. J Neurol Neurosurg Psychiatry 86: , 1324–1330. |
[190] | Lewczuk P , Esselmann H , Otto M , Maler JM , Henkel AW , Henkel MK , Eikenberg O , Antz C , Krause WR , Reulbach U , Kornhuber J , Wiltfang J ((2004) ) Neurochemical diagnosis of Alzheimer’s dementia by CSF Abeta42, Abeta42/Abeta40 ratio and total tau. Neurobiol Aging 25: , 273–281. |
[191] | Skillbäck T , Farahmand BY , Rosén C , Mattsson N , Nägga K , Kilander L , Religa D , Wimo A , Winblad B , Schott JM , Blennow K , Eriksdotter M , Zetterberg H ((2015) ) Cerebrospinal fluid tau and amyloid-β1-42 in patients with dementia. Brain 138: , 2716–2731. |
[192] | Román GC , Tatemichi TK , Erkinjuntti T , Cummings JL , Masdeu JC , Garcia JH , Amaducci L , Orgogozo JM , Brun A , Hofman A , Moody DM , O’Brien MD , Yamaguchi T , Grafman J , Drayer BP , Bennett DA , Fisher M , Ogata J , Kokmen E , Bermejo F , Wolf PA , Gorelick PB , Bick KL , Pajeau AK , Bell MA , DeCarli C , Culebras A , Korczyn AD , Bogousslavsky J , Hartmann A , Scheinberg P ((1993) ) Vascular dementia: Diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology 43: , 250–260. |
[193] | Abraham H , Wolfson L , Moscufo N , Guttmann C , Kaplan R , White W ((2016) ) Cardiovascular risk factors and small vessel disease of the brain, Bloodressure, white matter lesions, and functional decline in older persons J Cereb Blood Flow Metab 36: , 132–142. |
[194] | Staals J , Makin S , Doubal F , Dennis M , Wardlaw J ((2014) ) Stroke subtype, vascular risk factors, and total MRI brain small-vessel disease burden. Neurology 83: , 1228–1234. |
[195] | Lahousse L , Tiemeier H , Ikram M , Brusselle G ((2015) ) Chronic obstructive pulmonary disease and cerebrovascular disease: A comprehensive review. Resp Med 109: , 1371–1380. |
[196] | Durgan D , Bryan RMJ ((2012) ) Cerebrovascular consequences of obstructive sleep apnea. J Am Heart Assoc 1: , e000091. |
[197] | Toyoda K ((2015) ) Cerebral small vessel disease and chronic kidney disease. J Stroke 17: , 31–37. |
[198] | Hainsworth A , Yeo N , Weekman E , Wilcock D ((2016) ) Homocysteine, hyperhomocysteinemia and vascular contributions to cognitive impairment and dementia (VCID). Biochim Biophys Acta 1862: , 1008–1017. |
[199] | Cornier M , Dabelea D , Hernandez T , Lindstrom R , Steig A , Stob N , Van Pelt R , Wang H , Eckel R ((2008) ) The metabolic syndrome. Endocr Rev 29: , 777–822. |
[200] | Raffaitin C , Gin H , Empana J , Helmer C , Berr C , Tzourio C , Portet F , Dartigues J , Alpérovitch A , Barberger-Gateau P ((2009) ) Metabolic syndrome and risk for incident Alzheimer’s disease or vascular dementia: The Three-City Study. Diabetes Care 32: , 169–174. |
[201] | Portet F , Brickman A , Stern Y , Scarmeas N , Muraskin J , Provenzano F , Berr C , Bonafé A , Artero S , Ritchie K , Akbaraly T ((2012) ) Metabolic syndrome and localization of white matter hyperintensities in the elderly population. Alzheimers Dement 8: (5 Suppl), S88–S95. |
[202] | Bokura H , Yamaguchi S , Iijima K , Nagai A , Oguro H ((2008) ) Metabolic syndrome is associated with silent ischemic brain lesions. Stroke 39: , 1607–1609. |
[203] | Schilling S , Tzourio C , Dufouil C , Zhu Y , Berr C , Alpérovitch A , Crivello F , Mazoyer B , Debette S ((2014) ) Plasma lipids and cerebral small vessel disease. Neurology 83: , 1844–1852. |
[204] | Chang TY , Yamauchi Y , Hasan MT , Chang CC ((2017) ) Cellular cholesterol homeostasis in Alzheimer’s disease. J Lipid Res 58: , 2239–2254. |
[205] | Talwar P , Sinha J , Grover S , Agarwal R , Kushwaha S , Srivastava MV , Kukreti R ((2016) ) Meta-analysis of apolipoprotein E levels in the cerebrospinal fluid of patients with Alzheimer’s disease. J Neurol Sci 360: , 179–187. |
[206] | Landén M , Hesse C , Fredman P , Regland B , Wallin A , Blennow K ((1996) ) Apolipoprotein E in cerebrospinal fluid from patients with Alzheimer’s disease and other forms of dementia is reduced but without any correlation to the ApoE4 isoform. Dementia 7: , 273–278. |
[207] | Johansson P , Almqvist E , Bjerke M , Wallin A , Johansson J , Andreasson U , Blennow K , Zetterberg H , Svensson J ((2017) ) Reduced cerebrospinal fluid concentration of apolipoprotein A-I in patients with Alzheimer’s disease. J Alzheimers Dis 59: , 1017–1026. |
[208] | Liu C , Kanekiyo T , Xu H , Bu G ((2013) ) Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat Rev Neurol 9: , 106–118. |
[209] | Kim J , Basak J , Holtzman D ((2009) ) The role of apolipoprotein E in Alzheimer’s disease. Neuron 63: , 287–303. |
[210] | Stefan N , Schick F , Häring HU ((2017) ) Causes, characteristics, and consequences of metabolically unhealthy normal weight in humans. Cell Metab 26: , 292–300. |
[211] | Novak V , Last D , Alsop D , Abduljalil A , Hu K , Lepicovsky L , Cavallerano J , Lipsitz L ((2006) ) Cerebral blood flow velocity and periventricular white matter hyperintensities in type 2 diabetes. Diabetes Care 29: , 1529–1534. |
[212] | van Harten B , Oosterman J , Potter van Loon B , Scheltens P , Weinstein H ((2007) ) Brain lesions on MRI in elderly patients with type 2 diabetes mellitus. Eur Neurol 57: , 70–74. |
[213] | Sims R , Katzel L , Lefkowitz D , Siegel E , Rosenberger W , Manukyan Z , Whitfield K , Waldstein S ((2014) ) Association of fasting glucose with subclinical cerebrovascular disease in older adults without type 2 diabetes. Diabet Med 31: , 691–698. |
[214] | Weinger K , Jacobson A , Musen G , Lyoo I , Ryan C , Jimerson D , Renshaw P ((2008) ) The effects of type 1 diabetes on cerebral white matter. Diabetologia 51: , 417–425. |
[215] | Rivera E , Goldin A , Fulmer N , Tavares R , Wands J , de la Monte S ((2005) ) Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J Alzheimers Dis 8: , 247–268. |
[216] | Steen E , Terry B , Rivera E , Cannon J , Neely T , Tavares R , Xu X , Wands J , de la Monte S ((2005) ) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease – Is this type 3 diabetes? J Alzheimers Dis 7: , 63–80. |
[217] | Talbot K , Wang H , Kazi H , Han L , Bakshi KP , Stucky A , Fuino R , Kawaguchi K , Samoyedny A , Wilson R , Arvanitakis Z , Schneider J , Wolf B , Bennett D , Trojanowski J , Arnold S ((2012) ) Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest 122: , 1316–1338. |
[218] | Fernandez A , Torres-Alemán I ((2012) ) The many faces of insulin-like peptide signalling in the brain. Nat Rev Neurosci 13: , 225–239. |
[219] | van der Pal R , Koper J , van Golde L , Lopes-Cardozo M ((1988) ) Effects of insulin and insulin-like growth factor (IGF-I) on oligodendrocyte-enriched glial cultures. J Neurosci Res 19: , 483–490. |
[220] | Mozell R , McMorris F ((1991) ) Insulin-like growth factor I stimulates oligodendrocyte development and myelination in rat brain aggregate culture. J Neurosci Res 30: , 382–390. |
[221] | Riikonen R , Turpeinen U ((2005) ) Cerebrospinal fluid insulin-like growth factor 1 is low in acute and chronic white-matter diseases of children. J Child Neurol 20: , 181–184. |
[222] | Tarkowski E , Blennow K , Wallin A , Tarkowski A ((1999) ) Intracerebral production of tumor necrosis factor-alpha, a local neuroprotective agent, in Alzheimer disease and vascular dementia. J Clin Immunol 19: , 223–230. |
[223] | Tarkowski E , Issa R , Sjögren M , Wallin A , Blennow K , Tarkowski A , Kumar P ((2002) ) Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer’s disease and vascular dementia. Neurobiol Aging 23: , 237–243. |
[224] | van Dijk E , Prins N , Vermeer S , Vrooman H , Hofman A , Koudstaal P , Breteler M ((2005) ) C-reactive protein and cerebral small-vessel disease: The Rotterdam Scan Study. Circulation 112: , 900–905. |
[225] | Vilar-Bergua A , Riba-Llena I , Nafría C , Bustamante A , Llombart V , Delgado P , Montaner J ((2016) ) Blood and CSF biomarkers in brain subcortical ischemic vascular disease: Involved pathways and clinical applicability. J Cereb Blood Flow Metab 36: , 55–71. |
[226] | Engelhart M , Geerlings M , Meijer J , Kiliaan A , Ruitenberg A , van Swieten J , Stijnen T , Hofman A , Witteman J , Breteler M ((2004) ) Inflammatory proteins in plasma and the risk of dementia: The Rotterdam study. Arch Neurol 61: , 668–672. |
[227] | Koyama A , O’Brien J , Weuve J , Blacker D , Metti A , Yaffe K ((2013) ) The role of peripheral inflammatory markers in dementia and Alzheimer’s disease: A meta-analysis. J Gerontol A Biol Sci Med Sci 68: , 433–440. |
[228] | Wada M , Nagasawa H , Kurita K , Koyama S , Arawaka S , Kawanami T , Tajima K , Daimon M , Kato T ((2008) ) Cerebral small vessel disease and C-reactive prote, Results of a cross-sectional study in community-based Jaanese elderly J Neurol Sci 264: , 43–49. |
[229] | Chen A , Oakley A , Monteiro M , Tuomela K , Allan L , Mukaetova-Ladinska E , O’Brien J , Kalaria R ((2016) ) Multiplex analyte assays to characterize different dementias: Brain inflammatory cytokines in poststroke and other dementias. Neurobiol Aging 38: , 56–57. |
[230] | Huang C , Lui C , Chang W , Lu C , Wang Y , Chang C ((2009) ) Elevated basal cortisol level predicts lower hippocampal volume and cognitive decline in Alzheimer’s disease. J Clin Neurosci 16: , 1283–1286. |
[231] | Abraham I , Harkany T , Horvath K , Luiten P ((2001) ) Action of glucocorticoids on survival of nerve cells: Promoting neurodegeneration or neuroprotection? J Neuroendocrinol 13: , 749–760. |
[232] | Lowy M , Gault L , Yamamoto B ((1993) ) Adrenalectomy attenuates stress-induced elevations in extracellular glutamate concentrations in the hippocampus. J Neurochem 61: , 1957–1960. |
[233] | De Kloet E , Vreugdenhil E , Oitzl M , Joëls M ((1998) ) Brain corticosteroid receptor balance in health and disease. Endocr Rev 19: , 269–301. |
[234] | Swaab D , Bao A , Lucassen P ((2005) ) The stress system in the human brain in depression and neurodegeneration. Ageing Res Rev 4: , 141–194. |
[235] | Popp J , Schaper K , Kölsch H , Cvetanovska G , Rommel F , Klingmüller D , Dodel R , Ullrich Wüllner U , Jessen F ((2009) ) CSF cortisol in Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 30: , 498–500. |
[236] | Gottfries C , Balldin J , Blennow K , Bråne G , Karlsson I , Regland B , Wallin A ((1994) ) Regulation of the hypothalamic-pituitary-adrenal axis in dementia disorders. Ann N Y Acad Sci 746: , 336–343. |
[237] | Cox S , Bastin M , Ferguson K , Maniega S , MacPherson S , Deary I , Wardlaw J , MacLullich A ((2015) ) Brain white matter integrity and cortisol in older men: The Lothian Birth Cohort 1936. Neurobiol Aging 36: , 257–264. |
[238] | Pantoni L , Rossi R , Inzitari D , Bianchi C , Beneke M , Erkinjuntti T , Wallin A ((2000) ) Efficacy and safety of nimodipine in subcortical vascular dementia: A subgroup analysis of the Scandinavian Multi-Infarct Dementia Trial. J Neurol Sci 175: , 124–134. |
[239] | Jia J , Wei C , Liang J , Zhou A , Zuo X , Song H , Wu L , Chen X , Chen S , Zhang J , Wu J , Wang K , Chu L , Peng D , Lv P , Guo H , Niu X , Chen Y , Dong W , Han X , Fang B , Peng M , Li D , Jia Q , Huang L ((2016) ) The effects of DL-3-n-butylphthalide in patients with vascular cognitive impairment without dementia caused by subcortical ischemic small vessel disease: A multicentre, randomized, double-blind, placebo-controlled trial. Alzheimers Dement 12: , 89–99. |
[240] | Mehta D , Jackson R , Paul G , Shi J , Sabbagh M ((2017) ) Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin Investig Drugs 26: , 735–739. |
[241] | Besser LM , Alosco ML , Ramirez Gomez L , Zhou XH , McKee AC , Stern RA , Gunstad J , Schneider JA , Chui H , Kukull WA ((2016) ) Late-life vascular risk factors and Alzheimer disease neuropathology in individuals with normal cognition. J Neuropathol Exp Neurol 75: , 955–962. |
[242] | Keith J , Gao F , Noor R , Kiss A , Balasubramaniam G , Au K , Rogaeva E , Masellis M , Black SE ((2017) ) Collagenosis of the deep medullary veins: An underrecognized pathologic correlate of white matter hyperintensities and periventricular infarction? J Neuropathol Exp Neurol 76: , 299–312. |
[243] | Chui HC , Ramirez Gomez L ((2017) ) Vascular contributions to cognitive impairment in late life. Neurol Clin 35: , 295–323. |


