
Nature of Tau-Associated Neurodegeneration and the Molecular Mechanisms
Abstract
Neurodegeneration is defined as the progressive loss of structure or function of the neurons. As the nature of degenerative cell loss is currently not clear, there is no specific molecular marker to measure neurodegeneration. Therefore, researchers have been using apoptotic markers to measure neurodegeneration. However, neurodegeneration is completely different from apoptosis by morphology and time course. Lacking specific molecular marker has been the major hindrance in research of neurodegenerative disorders. Alzheimer’s disease (AD) is the most common neurodegenerative disorder, and tau accumulation forming neurofibrillary tangles is a hallmark pathology in the AD brains, suggesting that tau must play a critical role in AD neurodegeneration. Here we review part of our published papers on tau-related studies, and share our thoughts on the nature of tau-associated neurodegeneration in AD.
THE NEUROPROTECTIVE ROLE OF TAU PHOSPHORYLATION
Hyperphosphorylation of wild-type tau proteins forming neurofibrillary tangles is one of the hallmark pathologies in the brains of sporadic Alzheimer’s disease (AD) patients, which accounts for over 95% of the AD populations. As hyperphosphorylated tau is the major protein component of the neurofibrillary tangles in the degenerated neurons of AD brains, neuroscientists have been taken it for granted that tau phosphorylation must promote cell apoptosis. However, initiated from unexpected results, our systemic studies demonstrate that tau phosphorylation not only not promotes cell death, but also renders the cells more resistant to apoptosis induced by various pro-apoptotic factors.
Tau phosphorylation renders cells anti-apoptosis
We first established HEK293 cell lines with stable expression of wild-type full-length human tau or the empty vector, or selected N2a cell lines that express low level or high level of tau protein (to be noted that different N2a cell lines show variable expression levels of endogenous tau proteins, from scarce expression to high level of endogenous tau; therefore, it is important to identify which type of cell lines you are using during tau studies). Then we treated the cells with different apoptosis inducers, including staurosporine, tunicamycin, thapsigargin, camptothecin, amyloid-β (Aβ), and hydrogen peroxide, respectively, and analyzed the relationship of tau phosphorylation and cell apoptosis. We found unexpectedly that the cells or neurons expressing hyperphosphorylated tau exhibit marked resistance to apoptosis induced by the aforementioned apoptotic stimuli [1, 2]. We also demonstrated that activating glycogen synthase kinase 3β (GSK-3β) induces apoptosis, while simultaneous expression of tau attenuates GSK-3β-induced cell apoptosis [1].
Phosphotyrosyl phosphatase activator (PTPA) is decreased in the AD brains. We found that PTPA is located in the integral membrane of mitochondria, and downregulating PTPA induces cell apoptosis by decreasing mitochondrial membrane potential, which leads to translocation of Bax and a simultaneous release of cytochrome C. Overexpression of tau rescues PTPA deficit-induced cell apoptosis [3]. Phosphorylation of tau by death-associated protein kinase 1 also antagonizes kinase-induced cell apoptosis [4]. On the other hand, dephosphorylation of tau promotes cell apoptosis [5]. These data together support the role of tau phosphorylation in anti-apoptosis.
Tau phosphorylation plays an essential role in neurogenesis
We observed that immature neurons were only immunoreactive to phosphorylated tau but not to the non-phosphorylated tau in adult rat brain and human AD brain slices. A correlative increase of immature neuron markers and tau phosphorylation was induced in rat hippocampal dentate gyrus (DG), a niche enriched with progenitor cells, by upregulating GSK-3, and the increased neurogenesis was due to an enhanced proliferation but not survival or differentiation of the newborn neurons [6]. These data imply that spatial/temporal manipulation of tau phosphorylation may be compensatory for the neuron loss in AD. The molecular mechanisms underlying tau phosphorylation-regulated neurogenesis deserve further investigation.
Mechanisms underlying the anti-apoptotic effect of tau
As a cytoskeleton protein, how tau phosphorylation affects cell apoptosis is currently not fully understood. Our studies showed that tau hyperphosphorylation was accompanied by an increased nuclear translocation of β-catenin, a survival-priming molecule. We found that tau hyperphosphorylation inhibits competitively phosphorylation and proteolysis of β-catenin in the cytoplasm. Increasing cytoplasmic β-catenin leads to a significant increase in the level of nuclear β-catenin. Thus, we speculate that increased tau phosphorylation may prevent β-catenin from phosphorylation/proteolysis, and nuclear translocation of the preserved β-catenin primes survival signaling pathway to lead the cells to escape apoptosis; or in other words, the substrate competitive phosphorylation of tau and β-catenin renders the cells anti-apoptosis [1, 7]. We also observed that overexpression of tau attenuates endoplasmic reticulum (ER) stress-induced apoptosis with upregulated unfolded protein response and immunoglobulin-binding protein (Bip) [8, 9], which indicates the involvement of ER in the anti-apoptosis of tau. The anti-apoptotic effects of tau also involve activation of Akt and modulation of the mitochondrial pathway, such as preservation of Bcl-2 and suppression of Bax, of cytochrome C release, and of caspase-3 activation [2, 5]. Furthermore, expression of 1N3R-tau isoform inhibits cell proliferation by inducing S phase arrest in N2a cells, which maybe also involved in the anti-apoptotic effect of tau [10].
In addition to tau phosphorylation, other anti-apoptotic machineries also deserve investigation during aging and in the AD brains. For instance, we found that protein phosphatase-2A inhibitor-2 (I2PP2A) could simultaneously activate Akt and p53. Akt is actively involved in the survival-promoting signaling, while p53 mainly participates in apoptotic pathways. Therefore, activation of Akt can counteract the hyperactivated p53-related cell apoptosis. In the brains of AD patients and transgenic mice, the levels of I2PP2A, Akt, and p53 were all increased, suggesting that increased I2PP2A can trigger apoptosis by p53 upregulation, but simultaneous activation of Akt can lead to neurons aborting from apoptosis [11].
During the long course of AD, the brain must be soaked in a pro-apoptotic environment. However, the majority of neurons, especially those enriched with neurofibrillary tangles, do not seem to go into apoptosis. How can this happen? Our studies suggest that tau phosphorylation can lead the neurons to escape from apoptosis.
THE DELETERIOUS EFFECTS OF TAU ACCUMULATION
As mentioned above, tau phosphorylation can lead the neurons to escape apoptosis. Then, is tau phosphorylation eventually beneficial to the neurons? Our observations suggest that tau phosphorylation plays a dual role in protecting the neurons from acute apoptosis, and simultaneously triggering chronic neurodegeneration. We speculate that the turning point from protection to damage may be due to the increasing intracellular accumulation of tau proteins.
Factors promoting tau accumulation
In AD brains, abnormally hyperphosphorylated tau is the major protein component of neurofibrillary tangles [12, 13], suggesting a critical role of tau accumulation in tangle formation and neurodegeneration. It has been shown that abnormal hyperphosphorylation of tau promotes self-assembly into tangles of paired helical filaments and straight filaments [14]. To further explore why the hyperphosphorylated tau proteins are more prone to the aggregation, we studied the interaction of tau phosphorylation with other posttranslational modifications, such as SUMOylation. We observed that SUMOylation of tau at K340 mutually promotes tau phosphorylation at multiple AD-associated sites, which in turn inhibits tau ubiquitination and its proteolytic degradation with a reduced solubility. These data suggest that a mutual stimulation of tau phosphorylation and SUMOylation induces its accumulation [15]. As tau has much less putative sites for SUMOylation than phosphorylation, targeting SUMOylation may be more promising in preventing tau accumulation.
We also observed that inhibiting proteasome activity by lactacystin increases total and insoluble tau levels [16]. A moderate tau phosphorylation activates proteasome, while a profound tau phosphorylation/accumulation in turn inhibits the protease activity. These results suggest a mutual stimulation of tau phosphorylation and proteasome inhibition [17]. Additionally, we observed that O-glycosylation attenuates tau phosphorylation [18], while N-glycosylation may aggravate tau phosphorylation and accumulation [19]. In future studies, it will be interesting to clarify that how these different post-translational modifications of tau interact each other to affect the structural and biological activity of tau proteins.
Tau accumulation impairs synaptic function
Accumulation of hyperphosphorylated tau has been employed as readout in AD studies. We speculate that intracellular tau accumulation may in turn serve as an upstream causative factor to affect neuronal metabolisms and the functions, i.e., the cause-and-effect alternating vicious circle (Fig. 1). Series of studies have been carried out to test this point. We observed that tau accumulation causes remarkable dephosphorylation of cAMP response element binding protein (CREB) in the nuclear fraction. The mechanisms involve activation of calcium-dependent protein phosphatase calcineurin, which mediates tau-induced CREB dephosphorylation [20]. As CREB is a critical transcriptional factor involved in synapse and cognitive functions, our data reveal a novel mechanism underlying tau-induced synapse impairments and cognitive deficits. Additionally, tau accumulation can mutually activate GSK-3β [1] and disrupts axonal transport in cholinergic neurons [21].
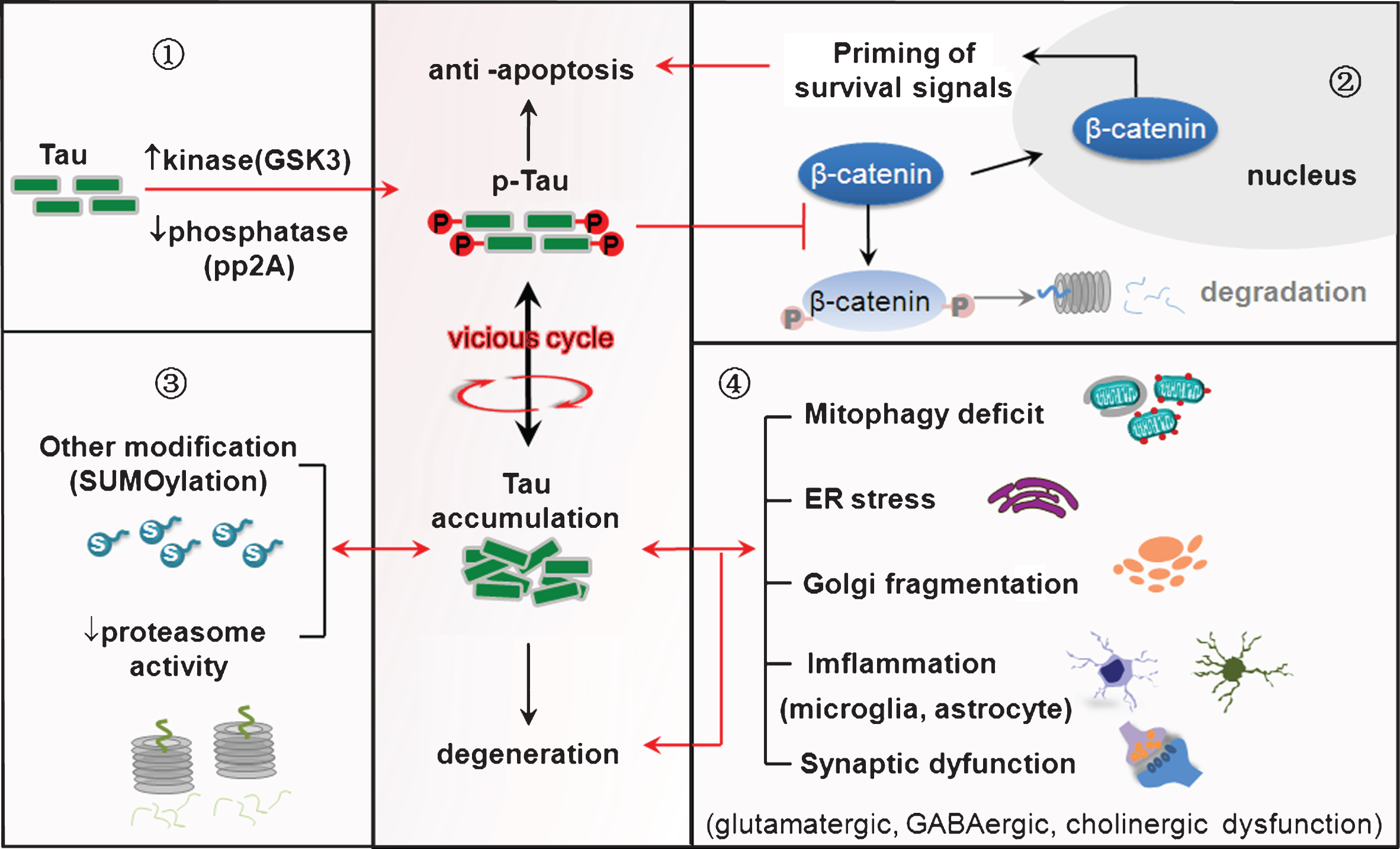
Fig.1
Tau hyperphosphorylation plays a dual role in anti-apoptosis and neurodegeneration. ➀Imbalance of protein kinases and protein phosphatases induces tau hyperphosphorylation. ➁Tau hyperphosphorylation leads the neurons abort acute apoptosis with the mechanisms involving preservation of β-catenin. ➂Tau phosphorylation mutually promotes its accumulation by inducing other posttranslational modifications (such as SUMOylation) and inhibiting proteasome activity. ➃Tau accumulation induces mitochondrial dysfunction, ER stress, inflammation and synaptic impairments, eventually leading to neurodegeneration.
In HEK293 cells, the exogenously transfected tau proteins were expressed with the hyperphosphorylated form and a large proportion of the phosphorylated tau was detected in the nuclear fraction, in which the Thr205 and Ser214 phosphorylated tau proteins were almost exclusively detected in the nuclear fraction when proper amount of protein was loaded. Furthermore, accumulation of the hyperphosphorylated tau induces calcium/calmodulin-dependent protein kinase-IV (CaMKIV) activation that aggravates nuclear tau hyperphosphorylation [22]. These data suggest that HEK293 cells with transient or stable expression of human tau may serve as an ideal model for studying tau hyperphosphorylation and its nuclear effects and as well as drug screening.
Tau accumulation impairs mitochondrial dynamics and induces mitophagy deficit
Mitochondrion is the power station of the cells that directly influence cell survival or death. To understand how tau proteins may regulate cell viability, we measured the effects of tau accumulation on mitochondrial dynamics and the functions. We found that tau proteins were allocated dose-dependently into the mitochondria along with its cytoplasmic accumulation. The mitochondrial insertion of tau enhances mitochondrial fusion and their perinuclear aggregation, increases mitochondrial membrane potential which disrupts mitochondrial residence of PTEN-induced kinase 1 (PINK1) and Parkin. The mitochondrial accumulation of tau also induces mitophagy deficits shown by the increased levels of COX IV, TOMM20, and the increased ratio of mtDNA to genomic DNA (mt-Atp6/Rpl13). At a later stage, tau accumulation decreases ATP level and complex I activity. Most interestingly, we observed that the mitophagy deficit was only shown in the AD patients who had an increased total tau level [23, 24].
Tau accumulation mutually induces ER stress
ER stress plays a crucial role in AD pathogenesis, but the role of tau was not understood. We observed that overexpression of tau induces ER stress shown by the upregulation of unfolded protein response, i.e., the increased phosphorylation of PERK, eIF2, and IRE1 with an increased cleavage of ATF6 and ATF4 [8]. Furthermore, upregulation of Bip during ER stress promotes association of GSK-3β with tau, which promotes tau hyperphosphorylation [9], suggesting a mutual stimulation of ER stress and tau hyperphosphorylation/accumulation. We also found that the cerebellum was more resistant to tunicamycin-induced ER stress compared with the temporal cortex, frontal cortex, and hippocampus. These observations partially explain why the AD-like neuropathologies, such as neurofibrillary tangles, have been scarcely seen in cerebellum [25]. Additionally, Golgi fragmentation induces tau hyperphosphorylation by activating cyclin-dependent kinase-5 and extracellular signal-regulated kinase [26]. It is currently not known whether and how intracellular tau accumulation may impair Golgi apparatus.
Tau accumulation activates microglia and induces inflammation
Microglia activation and inflammation have been observed in the AD brains. Tau was observed in microglia, but its role was not illustrated. We observed that microglia was activated in rodents’ brain during aging with a significantly enhanced tau phosphorylation in the activated microglia. We demonstrated that transient expression of tau in the cultured rat microglia increased Iba1 expression with elevation of interleukin (IL)-1β, IL-6, IL-10, tumor necrosis factor-α, and nitric oxide. The exogenously expressed human tau promotes microglial migration and phagocytosis without affecting its proliferation. These data suggest a novel role of tau in microglial activation [27]. The in vivo role of different tau species on glial cells and the influence on the neurons or vice versa deserve further investigations.
Tau accumulation impairs GABAergic and cholinergic functions
Patients with AD commonly show psychotic disorders, such as depression, and anxiety behaviors, and so on. We found that overexpression of human tau in mouse hippocampus significantly decreases the extracellular γ-aminobutyric acid (GABA) level with inhibition of γ-oscillation and the evoked inhibitory postsynaptic potential, and the mice show age-dependent anxiety behaviors. Further studies demonstrated that tau accumulation selectively suppresses expression of the intracellular vesicular GABA transporter (vGAT), among the factors responsible for GABA synthesis, release, uptake, and transport. The mechanism involves an increased miR92a, which targets vGAT mRNA 3’ UTR and inhibits vGAT translation [28]. These data suggest that the AD-like tau accumulation induces anxiety by disrupting miR92a-vGAT-GABA signaling.
We also observed that tau accumulation inhibits α4 subunit of nicotinic acetylcholine receptors (nAChRs) with an increased cleavage of the receptor producing a ∼55 kDa fragment in primary hippocampal neurons and in the rat brains. Meanwhile, the α4 nAChR currents decreased, suggesting the impairment of cholinergic functions [29]. The molecular and neural-circuit mechanisms need further investigation.
Taken together, our studies strongly suggest that tau phosphorylation plays a dual role in AD neurodegeneration, i.e., transient tau phosphorylation helps cells abort from an acute apoptosis with the mechanisms involving preservation of β-catenin, while persistent tau hyperphosphorylation/accumulation induces a chronic neurodegeneration by impairing multiple cellular functions (Fig. 1). We believe that the nature of “AD neurodegeneration” represents a novel type of tau-regulated chronic neuron death, termed neurodegenerasis [7].
We propose that during the long chronic course of AD pathogenesis, the neurons lack of tau hyperphosphorylation machinery die of apoptosis in the early phases when challenged by pro-apoptotic stimuli, while the neurons with tau hyperphosphorylation escape from the acute apoptosis. With increasing accumulation of hyperphosphorylated tau and formation of tangles, the neurons degenerate. Therefore, the degenerated neurons bearing tangles observed in AD brain autopsy must be those that survived the tau-induced apoptosis escape. As the turning point between anti-apoptosis and deterioration is the increasing intracellular tau accumulation, and targeting tau accumulation may prohibit neurodegeneration in AD and other tauopathies (Fig. 2).
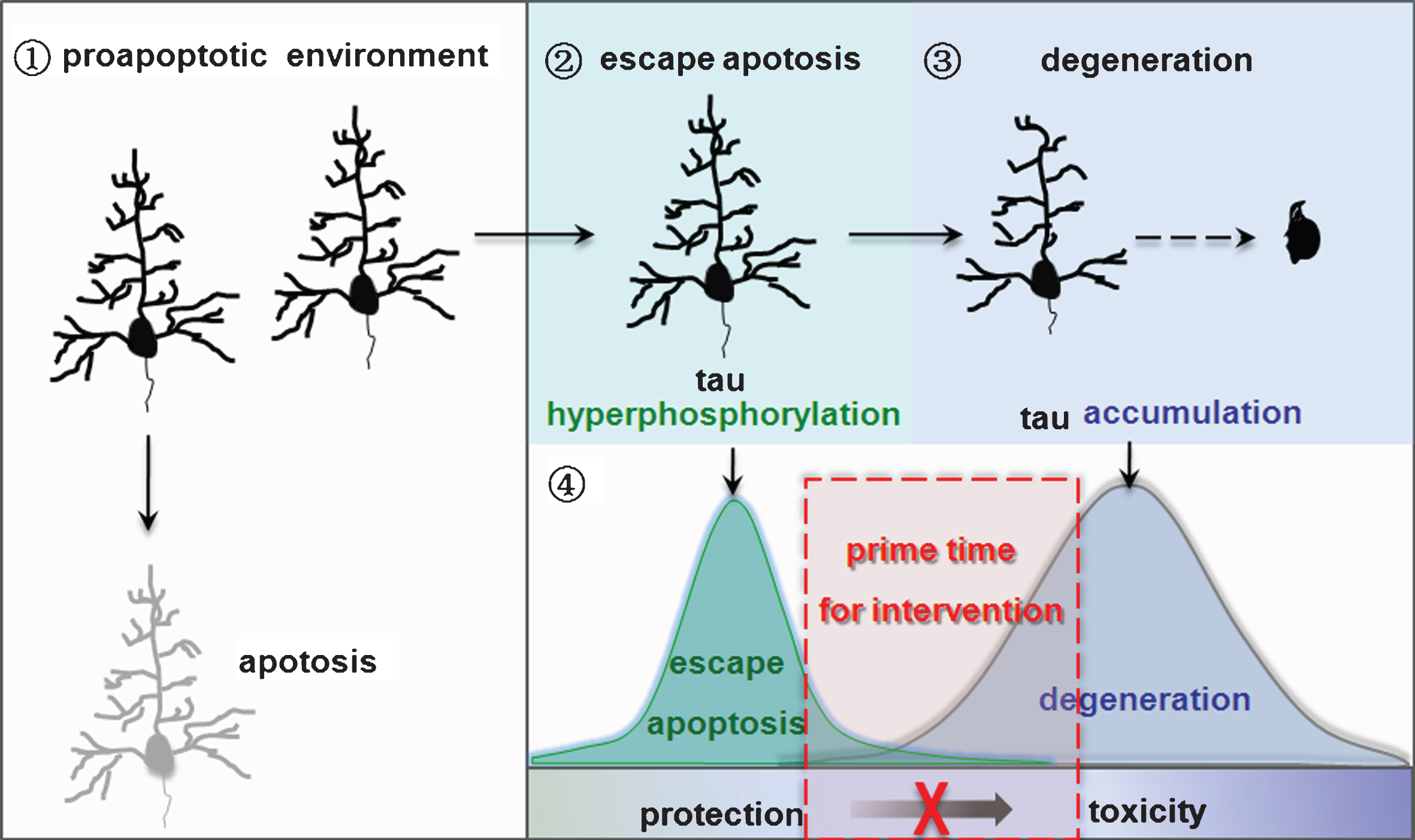
Fig.2
Nature of neurodegeneration and the implication in intervention. ➀Neurons lacking tau hyperphosphorylation enter apoptosis in a pro-apoptotic environment. ➁Tau hyperphosphorylation allows neurons to escape apoptosis. ➂Neurons that escape apoptosis go through degeneration with the increasing accumulation of hyperphosphorylated tau. ➃Targeting tau accumulation may prohibit neurodegeneration in AD and other tauopathies.
ROLE OF GSK-3β AND PP2A IN TAU AND AD PATHOLOGIES
Activation of protein kinases and/or inhibition of protein phosphatases are the direct cause of tau hyperphosphorylation. Among various protein kinases and the protein phosphatases in AD-like tau hyperphosphorylation and aggregation, GSK-3β [30] and PP2A [31–34] are among the most implicated.
GSK-3β activation mediates tau hyperphosphorylation in multiple models
GSK-3β, a Ser/Thr protein kinase abundantly expressed in neurons, plays diverse functions in physiological and neurodegenerative conditions. We demonstrated that pharmacological inhibition of phosphoinositol-3 kinase (PI3K) by wortmannin induces a transient GSK-3 activation and tau hyperphosphorylation, while simultaneous inhibition of protein kinase C (PKC) by GF-109203X and PI3K results in a sustained GSK-3 activation with tau hyperphosphorylation and impairment of spatial memory. These results indicate that inhibition of PI3K and PKC results in additive effects on GSK-3 activation and tau hyperphosphorylation [35]. The pharmacological activation of GSK-3 significantly decreases the acetylcholine level via inhibition of choline acetyl transferase rather than regulating acetylcholinesterase, while inhibition of GSK-3 by LiCl or SB216763 rescues the cholinergic functions. These data reveal the molecular mechanism that may underlie the cholinergic impairments in AD patients [21].
We also observed that AGEs induce AD-like tau hyperphosphorylation via RAGE-mediated GSK-3 activation [36]. Endogenously produced Aβ induces tau hyperphosphorylation by activating GSK-3 with downregulation of PKC, which further supports that inactivation of PKC is involved in GSK-3 activation [37]. Peroxynitrite simultaneously induces tau hyperphosphorylation, nitration, and accumulation, and activation of GSK-3β and the inhibition of proteasome activity are responsible for the peroxynitrite-induced tau hyperphosphorylation and accumulation, respectively [38]. The filtrate of H. pylori induced tau hyperphosphorylation at multiple AD-related sites in N2a cells and rat brains with activation of GSK-3β; simultaneous inhibition of GSK-3 efficiently attenuated the H. pylori-induced tau hyperphosphorylation [39].
It is to be noted that GSK-3β can robustly phosphorylate tau at basal activity under certain conditions. For instance, we observed that infusion of forskolin, an activator of protein kinase A (PKA), into rat lateral ventricle of the brain increases PKA activity by several fold and concurrently enhances tau phosphorylation at multiple AD-related sites, including Ser214, Tau-1, and PHF-1 epitopes. The forskolin-induced tau hyperphosphorylation at Ser214 was antagonized by simultaneous inhibition of PKA, but the phosphorylation at Tau-1 and PHF-1 epitopes was not affected. At the condition of PKA activation, only basal GSK-3 can induce tau hyperphosphorylation at Tau-1 and PHF-1 epitopes. These data suggest that tau becomes a more favorable substrate for GSK-3 when it is pre-phosphorylated by PKA; or in other words, phosphorylation of tau by PKA primes it for the phosphorylation by GSK-3 [40]. Additionally, Bip enhances the association of GSK-3β with tau during ER stress both in vivo and in vitro and thus promotes tau hyperphosphorylation by the kinase at basal activity [9].
GSK-3β overactivation impairs synaptic plasticity
Our studies demonstrated that overactivation of GSK-3β induces a series of damages to the brain. For instance, over-activating GSK-3β suppresses long-term potentiation (LTP) induction and presynaptic release of glutamate in rat hippocampus. GSK-3β also induces LTP-associated post-synaptic impairments, including reduced protein levels of PSD93, GluN2A, and GluN2B, thinner postsynaptic density, and broader synaptic cleft [41]. GSK-3β phosphorylates the intracellular loop-connecting domains II and III (L(II-III)) of P/Q-type calcium channel and disrupts formation of soluble N-ethylmaleimide-sensitive factor attachment protein receptor complex (SNARE). Simultaneous inhibition of GSK-3β by lithium or SB216763 or transient expression of a dominant-negative GSK-3β mutant (dnGSK-3β) attenuates the synapse impairments [42]. These data together demonstrate that over-activating GSK-3β impairs both pre- and post-synaptic structure and the functions, and it inhibits presynaptic vesicle exocytosis by phosphorylating P/Q-type calcium channel and interrupting SNARE complex formation. Whether and how tau hyperphosphorylation/accumulation contributes to the GSK-3β-induced synapse impairment deserves further verification.
GSK-3β deletion in DG excitatory neuron also impairs synaptic plasticity
The current studies on GSK-3β are mainly pan-neuron manipulated. To explore the neuron type-specific influence of GSK-3β, we recently infused AAV-CaMKII-Cre-2A-eGFP into GSK-3βlox/lox mice to selectively delete the kinase in excitatory neurons of hippocampal DG. To our surprise, we observed that GSK-3β deletion in DG excitatory neurons also induces spatial and fear memory defects but with an anti-anxiety behavior. At the molecular level, GSK-3β deletion reduces levels of several post-synaptic proteins, including NMDAR subunits (GluN1, GluN2A, and GluN2B), AMPAR subunit (GluA1), PSD93 and drebrin, and presynaptic Syn1. GSK-3β deletion also suppresses the activity-dependent neural activation and calcium/calmodulin-dependent protein kinase II (CaMKII)/CaMKIV-CREB signaling. These data demonstrate that as overactivation, GSK-3β deletion is also detrimental to the neurons, suggesting that GSK-3β in DG excitatory neurons is essential for maintaining synaptic plasticity and cognitive functions [43].
PP2A deficit in tau hyperphosphorylation and synapse impairment
PP2A is indispensable in development, and deficits of PP2A and deterioration of neuronal axons have been observed in several neurodegenerative disorders. We found that PP2A catalytic subunit (PP2Ac) is enriched in the distal axon of the hippocampal neurons. Inhibition of PP2A induces transport deficits and axonopathy [44], whereas overexpression of PP2Ac promotes formation and elongation of the functional axons, and rescues axon retardation induced by PP2A inhibition. PP2A dephosphorylates collapsin response mediator protein-2 (CRMP2) that executes axon polarization, whereas constitutive expression of phosphomimic-CRMP2 abolishes the effect of PP2A upregulation. These data reveal a mechanistic link between PP2A and axonogenesis or axonopathy [45].
Hyperhomocysteinemia is an independent risk factor of AD. We found that high plasma homocysteine induced by vena caudalis injection for 2 weeks induces tau hyperphosphorylation in rat brain hippocampus. Homocysteine inhibits PP2A shown by the increased Leu(309)-demethylation and Tyr(307)-phosphorylation [46]. Homocysteine also increases Aβ with the mechanisms involving an increased amyloid-β protein precursor (AβPP) phosphorylation [47]. These results suggest that homocysteine may be an upstream effector to induce AD-like tau hyperphosphorylation and Aβ overproduction through inactivating PP2A. Zinc also inactivates PP2A and induces tau hyperphosphorylation by Src-dependent phosphorylation of PP2A at Tyr-307 [48].
STRATEGIES TO INHIBIT TAU HYPERPHOSPHORYLATION AND ACCUMULATION
Accumulation of hyperphosphorylated tau in the cytosol [49] not only disrupts tubulin-assembling process [50, 51] but also disassembles microtubules [52], by which it plays a crucial role in AD neurodegeneration [53]. Activation of GSK-3β and/or inhibition of PP2A are direct causes for tau hyperphosphorylation and accumulation; therefore, we studied strategies for controlling the toxic turning point of tau by balancing GSK-3β and PP2A.
Strategies against GSK-3β activation
GSK-3β can phosphorylate tau at multiple AD-associated sites and activation of GSK-3β impairs synapses. Therefore, GSK-3β has been considered a promising target of AD drug development. Unfortunately, as GSK-3β possess broad critical physiological functions, direct GSK-3β deletion results in inacceptable side effects. Therefore, it is important to search for new strategies against GSK-3β overactivation. In this regard, we have tried to achieve partial inhibition of GSK-3β by activating the upstream membrane receptors, such as MT2 and EphB2 receptors.
The MT2 receptor is a principal type of G protein-coupled receptor that mainly mediates the effects of melatonin and deficit of melatonin/MT2 signaling has been shown in AD brains. We observed that upregulation of MT2 receptor activates Akt/GSK-3β/CRMP-2 signaling, and activation of MT2 receptor is necessary and sufficient to mediate the functional axonogenesis and synapse formation in neurons [54]. In Tg2576 mice, we observed that treatment with melatonin for 4 months (from 4-8 months of age) did not improve the pathology or behavioral performance of the mice. However, remarkable attenuation of tau and Aβ pathologies with memory improvement was observed when melatonin was supplied from the age of 8–12 months or 4–12 months of the mice. More importantly, the disease stage-specific alteration of GSK-3β was correlated with the alterations of the pathology and behavior, and the timely targeting of GSK-3β was critical for the efficacy of melatonin. These data suggest that melatonin treatment at the proper time can arrest AD by targeting the activated GSK-3β, which provides primary evidence for the importance and strategy in developing disease-modifying interventions of AD [55].
The EphB receptor, a bidirectional receptor, consists of an N-terminal glycosylated ligand-binding domain, a transmembrane region and an intracellular tyrosine kinase domain. The receptor preferentially binds membrane-bound ephrin-B ligands and is actively involved in nervous system. Homozygous knockout mice of EphB exhibit impaired axon guidance and vestibular function. We observed that activation of EphB2 receptor arrests tau hyperphosphorylation through its tyrosine kinase-mediated PI3K/Akt activation and the downstream GSK-3β inhibition in SK-N-SH cells, HEK293-tau cells, cultured hippocampal neurons and the hippocampus of human tau transgenic mice. These data provide membranous target for antagonizing AD-like tau pathologies [56].
The role of magnesium in AD has been debatable. We observed that level of magnesium in the streptozotocin-induced sporadic AD model rats and intraperitoneal administration of magnesium sulfate increases brain magnesium levels and thus protects learning and memory capacities. Supplement of magnesium sulfate reverses LTP impairments and synaptic plasticity, and decreases tau hyperphosphorylation by inhibiting GSK-3β and activating the upstream insulin receptor pathway [57].
Strategies for restoration of PP2A
Among various Ser/Thr protein phosphatases, PP2A has the highest specific activity in dephosphorylating tau proteins [58–60] and it is inactivated in AD brains [31, 32]. PP2A activity is repressed by the endogenous inhibitory proteins, namely inhibitor-1 of PP2A (I1PP2A) or (I2PP2A). In AD brain, I2(PP2A) is significantly increased in neocortex, and it is translocated from neuronal nuclei to the cytoplasm and cleaved into an approximately 20-kd fragment in cytosol [61]. By injecting lentivirus-shRNA targeting I2PP2A into the hippocampus and frontal cortex of 11-month-old Tg2576 mice, we demonstrated that silencing I2PP2A restores PP2A activity with attenuation of amyloidogenesis and improvement of learning and memory [62]. Downregulating I1PP2A (also termed pp32, ANP32A) also rescues synapse and cognitive impairments by chromatin remodeling in AD mouse models [63]. These data suggest that I1PP2A and I2PP2A could be promising targets for restoration of PP2A activity in AD. In astrocytes, we observed that upregulation of PP2A stimulates astrocytes migration via inhibiting p38 MAPK in Tg2576 mice [64].
Nicotinamide mononucleotide adenylyltransferase 2 (Nmnat2) is a key enzyme involved in energy metabolism and the mRNA level of Nmnat2 is reduced in the AD brains. We observed that the mRNA and protein levels of Nmnat2 were significantly decreased, while levels of p-Tyr307-PP2A (the inactive form of the phosphatase) and phosphorylated tau were increased in Tg2576 mice. In HEK293 cells with stable expression of human wild-type full-length tau, simultaneous inhibition of PP2A abolishes the Nmnat2-induced tau dephosphorylation. Overexpression of Nmnat2 activates PP2A with attenuation of tau phosphorylation, whereas downregulation of Nmnat2 inhibits PP2A with tau hyperphosphorylation. These data suggest that Nmnat2 attenuates tau phosphorylation by activating PP2A [65]. Supplementation of betaine or folate/vitamin-B12 also attenuates the hyperhomocysteinemia-induced tau hyperphosphorylation through activation activating PP2A with amelioration of memory deficits, LTP repression, and synapse impairments [66, 67].
Inhibition of asparagine endopeptidase
Asparagine endopeptidase (AEP), also named legumain, is a lysosomal cysteine protease that activates, translocate, respectively, from neuronal lysosomes and nucleus to the cytoplasm, then cleaves both AβPP and tau, mediating Aβ and tau pathology in AD [68, 69]. In AD, AEP also cleaves α-synuclein and mediates pathologies in Parkinson’s disease [70], and phosphorylation of AEP by serine/threonine-protein kinase-2 enhances its enzymatic activity and provokes AD pathogenesis [71]. An orally bioactive and brain permeable AEP inhibitor reduces tau and AβPP cleavage, ameliorates synapse loss and augments LTP, resulting in protection of memory, suggesting that this AEP inhibitor may be an effective clinical therapeutic agent for AD [72].
Regulating neurotransmitters
Neurotransmitters play a key role in neuronal signaling transmission. We found that tau accumulation induces GABA deficits, while upregulating GABA by intraperitoneal injection of midazolam, ChR2-mediated photostimulating and overexpressing vGAT, or blocking miR92a by specific antagomir or inhibitor efficiently rescues tau-induced GABAergic dysfunctions with attenuation of anxiety. These findings may lead to the development of new therapeutics for tauopathies [28]. We also observed that tau accumulation inhibits nAChRs [29] and selective dopamine receptor 4 activation mediates the hippocampal neuronal calcium response via IP3 and ryanodine receptors [73], therefore, preservation or restoration of cholinergic or regulating dopaminergic functions should be beneficial to tau pathologies.
We also tested the effects of a synthesized juxtaposition composed of an acetylcholinesterase inhibitor and a calcium channel blocker on the hyperhomocysteinemia-induced AD rat model [46, 47]. We found that administration of the juxtaposition can attenuate tau hyperphosphorylation with improvement of synaptic plasticity. These data suggest that the polytherapeutic targeting juxtaposition against cholinergic deficits and calcium imbalance may be promising for AD [74].
Blood-brain barrier is one of the major obstacles that hold back drug development in AD. To solve this problem, we constructed a novel fusion peptide by linking the active domain of brain-derived neurotrophic factor (BDNF) with an HIV-encoded transactivator of transcription (TAT) that has a strong membrane-penetrating property. By intraperitoneal injection, the eGFP-TAT could be robustly detected in different brain regions and BDNF injection significantly improved the cognitive impairments in two AD rodent models [75].
Spatial training and neural circuit stimulation
Spatial memory loss is the earliest clinical symptom in AD patients. We investigated whether spatial training in the Morris water maze could be beneficial in AD rodent models. We observed that spatial training preserves associative memory capacity with augmentation of dendrite ramification and spine generation in Tg2576 mice [76]. In Aβ1-42-injected rats, spatial training promotes short-term survival and neuron-like differentiation of newborn cells [77]. In the hyperhomocysteinemia rat model, spatial training stimulated dendrite ramification and spine generation with memory improvement in a CaMKII-dependent manner [78]. However, the paternal spatial training can only enhance an offspring’s cognitive performance and synaptic plasticity in wild-type mice but not improve memory deficit in AD transgenic mice [79].
Emotion affects learning and memory capacities, but the underlying neurobiological mechanisms remain elusive. We established models of learned helplessness (LHL) and learned hopefulness (LHF) by exposing animals to inescapable foot shocks or with anticipated avoidance trainings. By anterograde and retrograde tracing, we identified a novel excitatory monosynaptic connection between posterior basolateral amygdale (BLP) and ventral hippocampal CA1 (vCA1). We found that the LHF animals show spatial memory potentiation with upscaling BLP-vCA1 circuit, whereas the LHL show memory deficits with suppressed BLP-vCA1 connection. These data demonstrate that opposite excitatory monosynaptic scaling of BLP-vCA1 circuit controls LHF- and LHL-modulated spatial memory. It not only reveals circuit-specific mechanisms linking emotions to memory, but also suggests that stimulating the BLP-vCA1 circuit may be potential for AD therapy [80]. We also observed a long-term ameliorative effect of the antidepressant fluoxetine exposure on cognitive deficits in 3×Tg AD mice [81].
EARLY DIAGNOSIS OR PREDICTION OF AD
The long preclinical phase during the course of AD provides opportunity for intervention, but it is generally already too late for an efficient therapy when AD patients come to the clinic with complaints of memory loss. Therefore, early diagnosis of AD is critical for developing efficient interventions to postpone or prevent the disease. This requires developing new diagnostic tools to predict dementia among the high-risk populations.
Potential biomarkers in predicting AD from diabetes patients
Both type 2 diabetes mellitus (T2DM) and AD are common age-associated disorders, and the prevalence for these two diseases is increasing with population aging. Epidemiological studies show that patients with T2DM have significantly increased risk of suffering from AD. Therefore, it is important to identify who in T2DM populations will develop AD. By randomly dividing T2DM patients into two groups: T2DM with or without mild cognitive impairment (MCI), we found that aging, activation of peripheral circulating GSK-3β, expression of ApoE ɛ4, and olfactory deficit are diagnostic for MCI in T2DM patients, and combination of these biomarkers can improve diagnostic accuracy [82]. These T2DM patients with MCI also show impairment in functional magnetic resonance imaging compared with those without MCI [83]. Further follow-up studies may reveal the factors predicting which T2DM patients will most likely develop AD, thus allowing the design of non-drug interventions for these populations.
Modification of methods for detecting AD biomarkers
The enzyme linked immunosorbent assay (ELISA) has high specificity to the target molecules; however, we found that this method was not sensitive enough to measure the level of PHF-1 tau in CSF. To solve this problem, we developed an ultrasensitive bienzyme-substrate-recycle enzyme-linked immunosorbent assay for measurement of PHF-1 in CSF. We demonstrated that the assay was approximately 400 and 1,300 times more sensitive than the conventional ELISA in determining PHF-1 tau and total tau, respectively. With this method, we successfully measured total tau and PHF-1 tau in lumbar CSFs of AD and control patients. We found that elevation of PHF-1 tau in CSF is a promising diagnostic marker of AD [84]. The elevated phosphorylated neurofilament in AD patients was also detected using this method [85]. Additionally, we verified that dot-blotting was as efficient as western blotting for testing GSK-3β protein levels in peripheral blood [82].
ACKNOWLEDGMENTS
We thank our colleagues and students who have made contributions to the works presented in this review. These papers have been supported partly by grants from Natural Science Foundation of China (31730035, 91632305, 81528007, and 91632111), grants from National Key R&D Program of China (2016YFC1305800), grant from International Alzheimer Association (IIRG-09-133433), and grant from Hubei Province (201609-201908 2016CFA063).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/17-0788r1).
REFERENCES
[1] | Li HL , Wang HH , Liu SJ , Deng YQ , Zhang YJ , Tian Q , Wang XC , Chen XQ , Yang Y , Zhang JY , Wang Q , Xu H , Liao FF , Wang JZ ((2007) ) Phosphorylation of tau antagonizes apoptosis by stabilizing beta-catenin, a mechanism involved in Alzheimer’s neurodegeneration. Proc Natl Acad Sci U S A 104: , 3591–3596. |
[2] | Wang ZF , Yin J , Zhang Y , Zhu LQ , Tian Q , Wang XC , Li HL , Wang JZ ((2010) ) Overexpression of tau proteins antagonizes amyloid-beta-potentiated apoptosis through mitochondria-caspase-3 pathway in N2a cells. J Alzheimers Dis 20: , 145–157. |
[3] | Luo DJ , Feng Q , Wang ZH , Sun DS , Wang Q , Wang JZ , Liu GP ((2014) ) Knockdown of phosphotyrosyl phosphatase activator induces apoptosis via mitochondrial pathway and the attenuation by simultaneous tau hyperphosphorylation. J Neurochem 130: , 816–825. |
[4] | Duan DX , Chai GS , Ni ZF , Hu Y , Luo Y , Cheng XS , Chen NN , Wang JZ , Liu GP ((2013) ) Phosphorylation of tau by death-associated protein kinase 1 antagonizes the kinase-induced cell apoptosis. J Alzheimers Dis 37: , 795–808. |
[5] | Liu XA , Liao K , Liu R , Wang HH , Zhang Y , Zhang Q , Wang Q , Li HL , Tian Q , Wang JZ ((2010) ) Tau dephosphorylation potentiates apoptosis by mechanisms involving a failed dephosphorylation/activation of Bcl-2. J Alzheimers Dis 19: , 953–962. |
[6] | Hong XP , Peng CX , Wei W , Tian Q , Liu YH , Yao XQ , Zhang Y , Cao FY , Wang Q , Wang JZ ((2010) ) Essential role of tau phosphorylation in adult hippocampal neurogenesis. Hippocampus 20: , 1339–1349. |
[7] | Wang JZ , Liu F ((2008) ) Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog Neurobiol 85: , 148–175. |
[8] | Liu XA , Song J , Jiang Q , Wang Q , Tian Q , Wang JZ ((2012) ) Expression of the hyperphosphorylated tau attenuates ER stress-induced apoptosis with upregulation of unfolded protein response. Apoptosis 17: , 1039–1049. |
[9] | Liu ZC , Fu ZQ , Song J , Zhang JY , Wei YP , Chu J , Han L , Qu N , Wang JZ , Tian Q ((2012) ) Bip enhanced the association of GSK-3beta with tau during ER stress both in vivo and in vitro. J Alzheimers Dis 29: , 727–740. |
[10] | Li L , Xu ZP , Liu GP , Xu C , Wang ZH , Li XG , Liu EJ , Zeng J , Chai DM , Yao WL , Wang JZ ((2015) ) Expression of 1N3R-Tau isoform inhibits cell proliferation by inducing S phase arrest in N2a cells. PLoS One 10: , e0119865. |
[11] | Liu GP , Wei W , Zhou X , Zhang Y , Shi HH , Yin J , Yao XQ , Peng CX , Hu J , Wang Q , Li HL , Wang JZ ((2012) ) I(2)(PP2A) regulates p53 and Akt correlatively and leads the neurons to abort apoptosis. Neurobiol Aging 33: , 254–264. |
[12] | Grundke-Iqbal I , Iqbal K , Quinlan M , Tung YC , Zaidi MS , Wisniewski HM ((1986) ) Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem 261: , 6084–6089. |
[13] | Grundke-Iqbal I , Iqbal K , Tung YC , Quinlan M , Wisniewski HM , Binder LI ((1986) ) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 83: , 4913–4917. |
[14] | Alonso A , Zaidi T , Novak M , Grundke-Iqbal I , Iqbal K ((2001) ) Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci U S A 98: , 6923–6928. |
[15] | Luo HB , Xia YY , Shu XJ , Liu ZC , Feng Y , Liu XH , Yu G , Yin G , Xiong YS , Zeng K , Jiang J , Ye K , Wang XC , Wang JZ ((2014) ) SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination. Proc Natl Acad Sci U S A 111: , 16586–16591. |
[16] | Liu YH , Wei W , Yin J , Liu GP , Wang Q , Cao FY , Wang JZ ((2009) ) Proteasome inhibition increases tau accumulation independent of phosphorylation. Neurobiol Aging 30: , 1949–1961. |
[17] | Ren QG , Liao XM , Chen XQ , Liu GP , Wang JZ ((2007) ) Effects of tau phosphorylation on proteasome activity. FEBS Lett 581: , 1521–1528. |
[18] | Li X , Lu F , Wang JZ , Gong CX ((2006) ) Concurrent alterations of O-GlcNAcylation and phosphorylation of tau in mouse brains during fasting. Eur J Neurosci 23: , 2078–2086. |
[19] | Wang JZ , Grundke-Iqbal I , Iqbal K ((1996) ) Glycosylation of microtubule-associated protein tau: An abnormal posttranslational modification in Alzheimer’s disease. Nat Med 2: , 871–875. |
[20] | Yin Y , Gao D , Wang Y , Wang ZH , Wang X , Ye J , Wu D , Fang L , Pi G , Yang Y , Wang XC , Lu C , Ye K , Wang JZ ((2016) ) Tau accumulation induces synaptic impairment and memory deficit by calcineurin-mediated inactivation of nuclear CaMKIV/CREB signaling. Proc Natl Acad Sci U S A 113: , E3773–3781. |
[21] | Wang Y , Tian Q , Liu EJ , Zhao L , Song J , Liu XA , Ren QG , Jiang X , Zeng J , Yang YT , Wang JZ ((2017) ) Activation of GSK-3 disrupts cholinergic homoeostasis in nucleus basalis of Meynert and frontal cortex of rats. J Cell Mol Med 21: , 3515–3528. |
[22] | Wei YP , Ye JW , Wang X , Zhu LP , Hu QH , Wang Q , Ke D , Tian Q , Wang JZ ((2017) ) Tau-induced Ca2+/calmodulin-dependent protein kinase-IV activation aggravates nuclear tau hyperphosphorylation. Neurosci Bull, doi: 10.1007/s12264-017-0148-8 |
[23] | Li XC , Hu Y , Wang ZH , Luo Y , Zhang Y , Liu XP , Feng Q , Wang Q , Ye K , Liu GP , Wang JZ ((2016) ) Human wild-type full-length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci Rep 6: , 24756. |
[24] | Hu Y , Li XC , Wang ZH , Luo Y , Zhang X , Liu XP , Feng Q , Wang Q , Yue Z , Chen Z , Ye K , Wang JZ , Liu GP ((2016) ) Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 7: , 17356–17368. |
[25] | Lin L , Yang SS , Chu J , Wang L , Ning LN , Zhang T , Jiang Q , Tian Q , Wang JZ ((2014) ) Region-specific expression of tau, amyloid-beta protein precursor, and synaptic proteins at physiological condition or under endoplasmic reticulum stress in rats. J Alzheimers Dis 41: , 1149–1163. |
[26] | Jiang Q , Wang L , Guan Y , Xu H , Niu Y , Han L , Wei YP , Lin L , Chu J , Wang Q , Yang Y , Pei L , Wang JZ , Tian Q ((2014) ) Golgin-84-associated Golgi fragmentation triggers tau hyperphosphorylation by activation of cyclin-dependent kinase-5 and extracellular signal-regulated kinase. Neurobiol Aging 35: , 1352–1363. |
[27] | Wang L , Jiang Q , Chu J , Lin L , Li XG , Chai GS , Wang Q , Wang JZ , Tian Q ((2013) ) Expression of Tau40 induces activation of cultured rat microglial cells. PLoS One 8: , e76057. |
[28] | Li X , Wang Z , Tan L , Wang Y , Lu C , Chen R , Zhang S , Gao Y , Liu Y , Yin Y , Liu X , Liu E , Yang Y , Hu Y , Xu Z , Xu F , Wang J , Liu GP , Wang JZ ((2017) ) Correcting miR92a-vGAT-mediated GABAergic dysfunctions rescues human tau-induced anxiety in mice. Mol Ther 25: , 140–152. |
[29] | Yin Y , Wang Y , Gao D , Ye J , Wang X , Fang L , Wu D , Pi G , Lu C , Zhou XW , Yang Y , Wang JZ ((2016) ) Accumulation of human full-length tau induces degradation of nicotinic acetylcholine receptor alpha4 via activating calpain-2. Sci Rep 6: , 27283. |
[30] | Wang JZ , Wu Q , Smith A , Grundke-Iqbal I , Iqbal K ((1998) ) Tau is phosphorylated by GSK-3 at several sites found in Alzheimer disease and its biological activity markedly inhibited only after it is prephosphorylated by A-kinase. FEBS Lett 436: , 28–34. |
[31] | Gong CX , Singh TJ , Grundke-Iqbal I , Iqbal K ((1993) ) Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem 61: , 921–927. |
[32] | Gong CX , Shaikh S , Wang JZ , Zaidi T , Grundke-Iqbal I , Iqbal K ((1995) ) Phosphatase activity toward abnormally phosphorylated tau: Decrease in Alzheimer disease brain. J Neurochem 65: , 732–738. |
[33] | Gong CX , Lidsky T , Wegiel J , Zuck L , Grundke-Iqbal I , Iqbal K ((2000) ) Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer’s disease. J Biol Chem 275: , 5535–5544. |
[34] | Bennecib M , Gong CX , Grundke-Iqbal I , Iqbal K ((2001) ) Inhibition of PP-2A upregulates CaMKII in rat forebrain and induces hyperphosphorylation of tau at Ser 262/356. FEBS Lett 490: , 15–22. |
[35] | Liu SJ , Zhang AH , Li HL , Wang Q , Deng HM , Netzer WJ , Xu H , Wang JZ ((2003) ) Overactivation of glycogen synthase kinase-3 by inhibition of phosphoinositol-3 kinase and protein kinase C leads to hyperphosphorylation of tau and impairment of spatial memory. J Neurochem 87: , 1333–1344. |
[36] | Li XH , Lv BL , Xie JZ , Liu J , Zhou XW , Wang JZ ((2012) ) AGEs induce Alzheimer-like tau pathology and memory deficit via RAGE-mediated GSK-3 activation. Neurobiol Aging 33: , 1400–1410. |
[37] | Wang ZF , Li HL , Li XC , Zhang Q , Tian Q , Wang Q , Xu H , Wang JZ ((2006) ) Effects of endogenous beta-amyloid overproduction on tau phosphorylation in cell culture. J Neurochem 98: , 1167–1175. |
[38] | Zhang YJ , Xu YF , Liu YH , Yin J , Li HL , Wang Q , Wang JZ ((2006) ) Peroxynitrite induces Alzheimer-like tau modifications and accumulation in rat brain and its underlying mechanisms. FASEB J 20: , 1431–1442. |
[39] | Wang XL , Zeng J , Yang Y , Xiong Y , Zhang ZH , Qiu M , Yan X , Sun XY , Tuo QZ , Liu R , Wang JZ ((2015) ) Helicobacter pylori filtrate induces Alzheimer-like tau hyperphosphorylation by activating glycogen synthase kinase-3beta. J Alzheimers Dis 43: , 153–165. |
[40] | Liu SJ , Zhang JY , Li HL , Fang ZY , Wang Q , Deng HM , Gong CX , Grundke-Iqbal I , Iqbal K , Wang JZ ((2004) ) Tau becomes a more favorable substrate for GSK-3 when it is prephosphorylated by PKA in rat brain. J Biol Chem 279: , 50078–50088. |
[41] | Zhu LQ , Wang SH , Liu D , Yin YY , Tian Q , Wang XC , Wang Q , Chen JG , Wang JZ ((2007) ) Activation of glycogen synthase kinase-3 inhibits long-term potentiation with synapse-associated impairments. J Neurosci 27: , 12211–12220. |
[42] | Zhu LQ , Liu D , Hu J , Cheng J , Wang SH , Wang Q , Wang F , Chen JG , Wang JZ ((2010) ) GSK-3 beta inhibits presynaptic vesicle exocytosis by phosphorylating P/Q-type calcium channel and interrupting SNARE complex formation. J Neurosci 30: , 3624–3633. |
[43] | Liu E , Xie AJ , Zhou Q , Li M , Zhang S , Li S , Wang W , Wang X , Wang Q , Wang JZ ((2017) ) GSK-3beta deletion in dentate gyrus excitatory neuron impairs synaptic plasticity and memory. Sci Rep 7: , 5781. |
[44] | Yang Y , Yang XF , Wang YP , Tian Q , Wang XC , Li HL , Wang Q , Wang JZ ((2007) ) Inhibition of protein phosphatases induces transport deficits and axonopathy. J Neurochem 102: , 878–886. |
[45] | Zhu LQ , Zheng HY , Peng CX , Liu D , Li HL , Wang Q , Wang JZ ((2010) ) Protein phosphatase 2A facilitates axonogenesis by dephosphorylating CRMP2. J Neurosci 30: , 3839–3848. |
[46] | Zhang CE , Tian Q , Wei W , Peng JH , Liu GP , Zhou XW , Wang Q , Wang DW , Wang JZ ((2008) ) Homocysteine induces tau phosphorylation by inactivating protein phosphatase 2A in rat hippocampus. Neurobiol Aging 29: , 1654–1665. |
[47] | Zhang CE , Wei W , Liu YH , Peng JH , Tian Q , Liu GP , Zhang Y , Wang JZ ((2009) ) Hyperhomocysteinemia increases beta-amyloid by enhancing expression of gamma-secretase and phosphorylation of amyloid precursor protein in rat brain. Am J Pathol 174: , 1481–1491. |
[48] | Xiong Y , Jing XP , Zhou XW , Wang XL , Yang Y , Sun XY , Qiu M , Cao FY , Lu YM , Liu R , Wang JZ ((2013) ) Zinc induces protein phosphatase 2A inactivation and tau hyperphosphorylation through Src dependent PP2A (tyrosine 307) phosphorylation. Neurobiol Aging 34: , 745–756. |
[49] | Kopke E , Tung YC , Shaikh S , Alonso AC , Iqbal K , Grundke-Iqbal I ((1993) ) Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J Biol Chem 268: , 24374–24384. |
[50] | Alonso AC , Zaidi T , Grundke-Iqbal I , Iqbal K ((1994) ) Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A 91: , 5562–5566. |
[51] | Iqbal K , Grundke-Iqbal I , Zaidi T , Merz PA , Wen GY , Shaikh SS , Wisniewski HM , Alafuzoff I , Winblad B ((1986) ) Defective brain microtubule assembly in Alzheimer’s disease. Lancet 2: , 421–426. |
[52] | Alonso AC , Grundke-Iqbal I , Iqbal K ((1996) ) Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med 2: , 783–787. |
[53] | Hu W , Zhang X , Tung YC , Xie S , Liu F , Iqbal K ((2016) ) Hyperphosphorylation determines both the spread and the morphology of tau pathology. Alzheimers Dement 12: , 1066–1077. |
[54] | Liu D , Wei N , Man HY , Lu Y , Zhu LQ , Wang JZ ((2015) ) The MT2 receptor stimulates axonogenesis and enhances synaptic transmission by activating Akt signaling. Cell Death Differ 22: , 583–596. |
[55] | Peng CX , Hu J , Liu D , Hong XP , Wu YY , Zhu LQ , Wang JZ ((2013) ) Disease-modified glycogen synthase kinase-3beta intervention by melatonin arrests the pathology and memory deficits in an Alzheimer’s animal model. Neurobiol Aging 34: , 1555–1563. |
[56] | Jiang J , Wang ZH , Qu M , Gao D , Liu XP , Zhu LQ , Wang JZ ((2015) ) Stimulation of EphB2 attenuates tau phosphorylation through PI3K/Akt-mediated inactivation of glycogen synthase kinase-3beta. Sci Rep 5: , 11765. |
[57] | Xu ZP , Li L , Bao J , Wang ZH , Zeng J , Liu EJ , Li XG , Huang RX , Gao D , Li MZ , Zhang Y , Liu GP , Wang JZ ((2014) ) Magnesium protects cognitive functions and synaptic plasticity in streptozotocin-induced sporadic Alzheimer’s model. PLoS One 9: , e108645. |
[58] | Wang JZ , Gong CX , Zaidi T , Grundke-Iqbal I , Iqbal K ((1995) ) Dephosphorylation of Alzheimer paired helical filaments by protein phosphatase-2A and -2B. J Biol Chem 270: , 4854–4860. |
[59] | Wang JZ , Grundke-Iqbal I , Iqbal K ((1996) ) Restoration of biological activity of Alzheimer abnormally phosphorylated tau by dephosphorylation with protein phosphatase-2A, -2B and -1. Brain Res Mol Brain Res 38: , 200–208. |
[60] | Liu F , Grundke-Iqbal I , Iqbal K , Gong CX ((2005) ) Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur J Neurosci 22: , 1942–1950. |
[61] | Tanimukai H , Grundke-Iqbal I , Iqbal K ((2005) ) Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer’s disease. Am J Pathol 166: , 1761–1771. |
[62] | Liu GP , Wei W , Zhou X , Shi HR , Liu XH , Chai GS , Yao XQ , Zhang JY , Peng CX , Hu J , Li XC , Wang Q , Wang JZ ((2013) ) Silencing PP2A inhibitor by lenti-shRNA interference ameliorates neuropathologies and memory deficits in tg2576 mice. Mol Ther 21: , 2247–2257. |
[63] | Feng Q , Chai GS , Wang ZH , Hu Y , Sun DS , Li XG , Ma RH , Li YR , Ke D , Wang JZ , Liu GP ((2017) ) Knockdown of pp32 increases histone acetylation and ameliorates cognitive deficits. Front Aging Neurosci 9: , 104. |
[64] | Liu XP , Zheng HY , Qu M , Zhang Y , Cao FY , Wang Q , Ke D , Liu GP , Wang JZ ((2012) ) Upregulation of astrocytes protein phosphatase-2A stimulates astrocytes migration via inhibiting p38 MAPK in tg2576 mice. Glia 60: , 1279–1288. |
[65] | Cheng XS , Zhao KP , Jiang X , Du LL , Li XH , Ma ZW , Yao J , Luo Y , Duan DX , Wang JZ , Zhou XW ((2013) ) Nmnat2 attenuates Tau phosphorylation through activation of PP2A. J Alzheimers Dis 36: , 185–195. |
[66] | Chai GS , Jiang X , Ni ZF , Ma ZW , Xie AJ , Cheng XS , Wang Q , Wang JZ , Liu GP ((2013) ) Betaine attenuates Alzheimer-like pathological changes and memory deficits induced by homocysteine. J Neurochem 124: , 388–396. |
[67] | Wei W , Liu YH , Zhang CE , Wang Q , Wei Z , Mousseau DD , Wang JZ , Tian Q , Liu GP ((2011) ) Folate/vitamin-B12 prevents chronic hyperhomocysteinemia-induced tau hyperphosphorylation and memory deficits in aged rats. J Alzheimers Dis 27: , 639–650. |
[68] | Zhang Z , Song M , Liu X , Kang SS , Kwon IS , Duong DM , Seyfried NT , Hu WT , Liu Z , Wang JZ , Cheng L , Sun YE , Yu SP , Levey AI , Ye K ((2014) ) Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat Med 20: , 1254–1262. |
[69] | Basurto-Islas G , Grundke-Iqbal I , Tung YC , Liu F , Iqbal K ((2013) ) Activation of asparaginyl endopeptidase leads to Tau hyperphosphorylation in Alzheimer disease. J Biol Chem 288: , 17495–17507. |
[70] | Zhang Z , Kang SS , Liu X , Ahn EH , Zhang Z , He L , Iuvone PM , Duong DM , Seyfried NT , Benskey MJ , Manfredsson FP , Jin L , Sun YE , Wang JZ , Ye K ((2017) ) Asparagine endopeptidase cleaves alpha-synuclein and mediates pathologic activities in Parkinson’s disease. Nat Struct Mol Biol 24: , 632–642. |
[71] | Wang ZH , Liu P , Liu X , Manfredsson FP , Sandoval IM , Yu SP , Wang JZ , Ye K ((2017) ) Delta-secretase phosphorylation by SRPK2 enhances its enzymatic activity, provoking pathogenesis in Alzheimer’s disease. Mol Cell 67: , 812–825. |
[72] | Zhang Z , Obianyo O , Dall E , Du Y , Fu H , Liu X , Kang SS , Song M , Yu SP , Cabrele C , Schubert M , Li X , Wang JZ , Brandstetter H , Ye K ((2017) ) Inhibition of delta-secretase improves cognitive functions in mouse models of Alzheimer’s disease. Nat Commun 8: , 14740. |
[73] | Wang YL , Wang JG , Guo FL , Gao XH , Zhao DD , Zhang L , Wang JZ , Lu CB ((2017) ) Selective dopamine receptor 4 activation mediates the hippocampal neuronal calcium response via IP3 and ryanodine receptors. Brain Res 1670: , 1–5. |
[74] | Xia Y , Liu R , Chen R , Tian Q , Zeng K , Hu J , Liu X , Wang Q , Wang P , Wang XC , Wang JZ ((2014) ) Novel multipotent AChEI-CCB attenuates hyperhomocysteinemia-induced memory deficits and neuropathologies in rats. J Alzheimers Dis 42: , 1029–1039. |
[75] | Wu Y , Luo X , Liu X , Liu D , Wang X , Guo Z , Zhu L , Tian Q , Yang X , Wang JZ ((2015) ) Intraperitoneal administration of a novel TAT-BDNF peptide ameliorates cognitive impairments via modulating multiple pathways in two Alzheimer’s rodent models. Sci Rep 5: , 15032. |
[76] | Jiang X , Chai GS , Wang ZH , Hu Y , Li XG , Ma ZW , Wang Q , Wang JZ , Liu GP ((2015) ) Spatial training preserves associative memory capacity with augmentation of dendrite ramification and spine generation in Tg2576 mice. Sci Rep 5: , 9488. |
[77] | Zeng J , Jiang X , Hu XF , Ma RH , Chai GS , Sun DS , Xu ZP , Li L , Bao J , Feng Q , Hu Y , Chu J , Chai DM , Hong XY , Wang JZ , Liu GP ((2016) ) Spatial training promotes short-term survival and neuron-like differentiation of newborn cells in Abeta1-42-injected rats. Neurobiol Aging 45: , 64–75. |
[78] | Jiang X , Chai GS , Wang ZH , Hu Y , Li XG , Ma ZW , Wang Q , Wang JZ , Liu GP ((2015) ) CaMKII-dependent dendrite ramification and spine generation promote spatial training-induced memory improvement in a rat model of sporadic Alzheimer’s disease. Neurobiol Aging 36: , 867–876. |
[79] | Zhang S , Li X , Wang Z , Liu Y , Gao Y , Tan L , Liu E , Zhou Q , Xu C , Wang X , Liu G , Chen H , Wang JZ ((2017) ) Paternal spatial training enhances offspring’s cognitive performance and synaptic plasticity in wild-type but not improve memory deficit in Alzheimer’s mice. Sci Rep 7: , 1521. |
[80] | Yang Y , Wang ZH , Jin S , Gao D , Liu N , Chen SP , Zhang S , Liu Q , Liu E , Wang X , Liang X , Wei P , Li X , Li Y , Yue C , Li HL , Wang YL , Wang Q , Ke D , Xie Q , Xu F , Wang L , Wang JZ ((2016) ) Opposite monosynaptic scaling of BLP-vCA1 inputs governs hopefulness- and helplessness-modulated spatial learning and memory. Nat Commun 7: , 11935. |
[81] | Jin L , Gao LF , Sun DS , Wu H , Wang Q , Ke D , Lei H , Wang JZ , Liu GP ((2017) ) Long-term ameliorative effects of the antidepressant fluoxetine exposure on cognitive deficits in 3 x TgAD mice. Mol Neurobiol 54: , 4160–4171. |
[82] | Xu ZP , Yang SL , Zhao S , Zheng CH , Li HH , Zhang Y , Huang RX , Li MZ , Gao Y , Zhang SJ , Zhan PY , Zhang LF , Deng L , Wei S , Liu YC , Ye JW , Ren HJ , Li N , Kong CX , Wang X , Fang L , Zhou QZ , Jiang HW , Li JR , Wang Q , Ke D , Liu GP , Wang JZ ((2016) ) Biomarkers for early diagnostic of mild cognitive impairment in Type-2 diabetes patients: A multicentre, retrospective, nested case-control study. E Bio Medicine 5: , 105–113. |
[83] | Yang SQ , Xu ZP , Xiong Y , Zhan YF , Guo LY , Zhang S , Jiang RF , Yao YH , Qin YY , Wang JZ , Liu Y , Zhu WZ ((2016) ) Altered intranetwork and internetwork functional connectivity in Type 2 diabetes mellitus with and without cognitive impairment. Sci Rep 6: , 32980. |
[84] | Hu YY , He SS , Wang X , Duan QH , Grundke-Iqbal I , Iqbal K , Wang J ((2002) ) Levels of nonphosphorylated and phosphorylated tau in cerebrospinal fluid of Alzheimer’s disease patients: An ultrasensitive bienzyme-substrate-recycle enzyme-linked immunosorbent assay. Am J Pathol 160: , 1269–1278. |
[85] | Hu YY , He SS , Wang XC , Duan QH , Khatoon S , Iqbal K , Grundke-Iqbal I , Wang JZ ((2002) ) Elevated levels of phosphorylated neurofilament proteins in cerebrospinal fluid of Alzheimer disease patients. Neurosci Lett 320: , 156–160. |


