
Chinese Herbal Medicine Xueshuantong Enhances Cerebral Blood Flow and Improves Neural Functions in Alzheimer’s Disease Mice
Abstract
Reduced cerebral blood flow in Alzheimer’s disease (AD) may occur in early AD, which contributes to the pathogenesis and/or pathological progression of AD. Reversing this deficit may have therapeutic potential. Certain traditional Chinese herbal medicines (e.g., Saponin and its major component Xueshuantong [XST]) increase blood flow in humans, but whether they could be effective in treating AD patients has not been tested. We found that systemic XST injection elevated cerebral blood flow in APP/PS1 transgenic mice using two-photon time-lapse imaging in the same microvessels before and after injection. Subchronic XST treatment led to improved spatial learning and memory and motor performance in the APP/PS1 mice, suggesting improved neural plasticity and functions. Two-photon time lapse imaging of the same plaques revealed a reduction in plaque size after XST treatment. In addition, western blots experiments showed that XST treatment led to reduced processing of amyloid-β protein precursor (AβPP) and enhanced clearance of amyloid-β (Aβ) without altering the total level of AβPP. We also found increased synapse density in the immediate vicinity of amyloid plaques, suggesting enhanced synaptic function. We conclude that targeting cerebral blood flow can be an effective strategy in treating AD.
INTRODUCTION
Alzheimer’s disease (AD) is one of the most devastating diseases that affect the life and health of the aging population. Decades of effort have been devoted to investigate the pathological mechanisms underlying AD in search of more effective therapeutic drugs and treatments. Current AD drugs (such as cholinergic esterase inhibitors and NMDA receptor antagonists) provide symptomatic relief without altering AD progression [1]. Past efforts have been mostly focused on amyloids, driven by the amyloid hypothesis [2, 3], with a goal to reduce amyloid plaques in the brain by reducing Aβ production or enhancing Aβ clearance. It is widely believed that elevated Aβ level leads to synapse loss and neuronal death, and thus Aβ drives AD progression [3]. However, clinical trials on drugs that target amyloid plaques have not demonstrated significant functional improvement, although these drugs are effective in reducing plaque load in AD patients [4, 5]. As an alternative to amyloid hypothesis, tau-related pathology has been proposed to play a driver role in AD pathology [6–9]. It has also been recognized that early treatment in AD might be necessary to halt or slow down its progression.
In addition to Aβ and tau, other pathological alterations associated with AD have been revealed in the past years, including reduced cerebral blood flow (CBF) and increased neuroinflammation [10–16]. It has become increasingly clear that AD is a multi-factor and multi-system disease, although the chronological sequence of and contributions from the involved systems/processes are not fully understood. What is unclear is whether the involved systems/factors act separately with their own unique contributions or if they interact to induce system-wide alterations. The latter appears to be more likely. Reduced CBF has been observed in a large percentage of AD patients and has been proposed to contribute to the pathogenesis and progression of AD [17–22]. Age-related cerebral microvascular pathologies have also been extensively observed in AD human samples, including tortuous blood vessels and capillaries, low vascular density, and microembolic brain injury. All of these alterations lead to insufficient blood supply. Cerebral hypoperfusion has been shown to occur several years before AD onset [11, 23] and may be considered an early pathological event in AD. Changes in CBF mirror the accumulation of brain amyloid, especially during the preclinical phases of AD. In addition to providing the necessary nutrition and oxygen for the brain, the cerebral vascular system also serves as a drainage system to remove waste (such as Aβ) from the brain. It has been shown that Aβ leaves the brain via this system, and thus an impaired drainage system contributes to decreased Aβ removal from the brain [11, 24]. However, there is only limited evidence that enhancing CBF is effective in ameliorating AD pathology, either in AD patients or animal models. Reduced CBF has also been reported in the APP/PS1 mice [25].
Traditional Chinese herbal medicines have been used in China for thousands of years to treat many ailments, especially chronic diseases. In traditional Chinese medical practice, brain diseases are thought to start as malfunctions or alterations in systems other than the brain itself, and malfunctions in the brain are only evident at more advanced disease stages. Thus brain diseases are usually treated as diseases of other systems, including the cardiovascular system. This school of thinking can be quite useful for AD treatment since the most effective practice in Chinese herbal medicine is to treat diseases before they become serious and to target multiple systems in an interactive manner. Numerous Chinese herbal drugs show efficacy in elevating CBF. One such example is saponin which has been widely used in improving blood circulation in humans [26]. Xueshuantong (XST) is extracted from the roots of a 3-year grown plant saponin and has been used to facilitate recoveries from strokes in the clinical settings for decades in China. XST used in the current study has five known major components and the percentage of each component is within a range. One mechanism underlying XST’s effect is the dilation of blood vessels by acting on endothelial cells [27].
In this study, we tested whether systemic injection of XST can improve neural functions and reduce AD pathology in transgenic AD mice (APP/PS1). We have utilized a combination of behavior tests, in vivo two-photon time-lapse imaging, laser Doppler blood flow measurements, immunohistochemistry and western blots. Our results revealed elevated CBF, improved cognitive and neural functions and reduced AD pathology in the XST-treated APP/PS1 mice. Therefore, improving CBF may be a viable option in treating AD.
METHODS
Animals and drug injection
APPswe/PSEN1dE9 mice with a C57BL/6 background were obtained from the Jackson Laboratory. APP/PS1 mice and wild-type littermate mice were genotyped by PCR analysis of genomic DNA from tail biopsies. Animals were maintained at 22±2 °C with a 12-h light:dark cycle (lights on at 8 AM, lights off at 8 PM) with free access to food and water. All mouse care and experimental procedures were conducted in accordance with the institutional guidelines of Peking University Shenzhen Graduate School.
XST is consisted of the total saponins from Panax notoginseng, and was provided by Guangxi Wuzhou Zhongheng Group Co., Ltd. Its major components are Notoginsenoside R1 (9.4%, PubChem CID: 441934), Ginsenoside Rg1 (38.0%, PubChem CID: 441923), Ginsenoside Re (4.8%, PubChem CID: 441921), Ginsenoside Rb1 (20.0%, PubChem CID: 9898279), and Ginsenoside Rd (1.1%, PubChem CID: 11679800). Freeze-dried powder of XST was dissolved in saline and administrated intraperitoneally (100 mg/kg), and saline injection was used as control. For experiments involving 30-day injection, XST were injected daily for 15 days in two sessions with a break of 2 days in-between; for 15-day injection, XST was injected for 15 days without any break; for measuring changes in the short-term (h) effect of XST, XST was injected only once.
Two photon time lapse imaging of blood flow and plaques
Seven-month-old male APP/PS1 mice and wild-type littermate controls were randomly assigned into four groups: WT-saline, WT-XST, APP/PS1-saline, and APP/PS1-XST. Since the effects of treatment were compared to the level prior to injection, differences in the absolute level (plaque load or CBF) do not affect the results. The age of mice was selected to maximize the opportunity of seeing drug effect, since amyloid plaques show rapid growth at this age of APP/PS1 mice [28]. Mice were injected XST or saline daily for 15 days, and the same microvessels or plaques were imaged on day 0 and day 15 on two-photon microscope. To identify the same plaques or microvessels over 15 days, images with different zooms were taken to facilitate the identification of major landmarks (such as blood vessels) in the vicinity of the plaques or microvessels. By finding these landmarks and using the relative position of the plaques/microvessels to these landmarks and to each other, we were able to find the same plaques/microvessels over 15 days.
Two-photon imaging was performed on an Olympus BX 51 microscope, using published methods [29, 30]. To measure blood flow in the brain microvessels (diameter 3–6μm), FITC-dextran (20 kD, 0.05 mL 5% (w/v)) was injected through the mouse tail vein, 1 h prior to imaging. Since FITC-dextran does not enter red blood cells (RBCs) and only labels the plasma, RBCs appear as dark objects moving inside the microvessels. A line was scanned repetitively inside a selected microvessel in a direction parallel to the vessel. Excitation was set to 800 nm. Several frame images with different zooms were collected to facilitate the identification of the same set of vessels by using landmarks in these images. The speed of the movement of the RBCs can be calculated from the stripes in the line scan images. The instantaneous velocity of RBCs is calculated as V =Δy/Δt; Flux = 1/Δt; Linear density = 1/Δy (see Fig. 1B for illustration).
Imaging of amyloid plaques was done using systemic injection of methoxy-XO4 (10 mg/mL in 10% DMSO, 45% propylene glycol and 45% saline), a fluorescent compound that crosses the blood-brain barrier and binds to amyloid plaques [31]. A set of plaques were identified using a 25×objective (Olympus, NA = 1.05). XO4 was injected again the day before the second imaging session. Plaque size was quantified using ImageJ (Version 1.42l, Wayne Rasband, National Institutes of Health, USA). Volumes of the amyloid plaques are calculated as follows: 1) a step of 2μm was used when collecting z-stack images 2) In ImageJ, the areas of each plaque at a given focus level were measured; 3) In order to estimate the volume of plaques with irregular shapes, the area at each focus level was multiplied by the interval between the sections (2μm) to yield the total volume of the entire plaque.
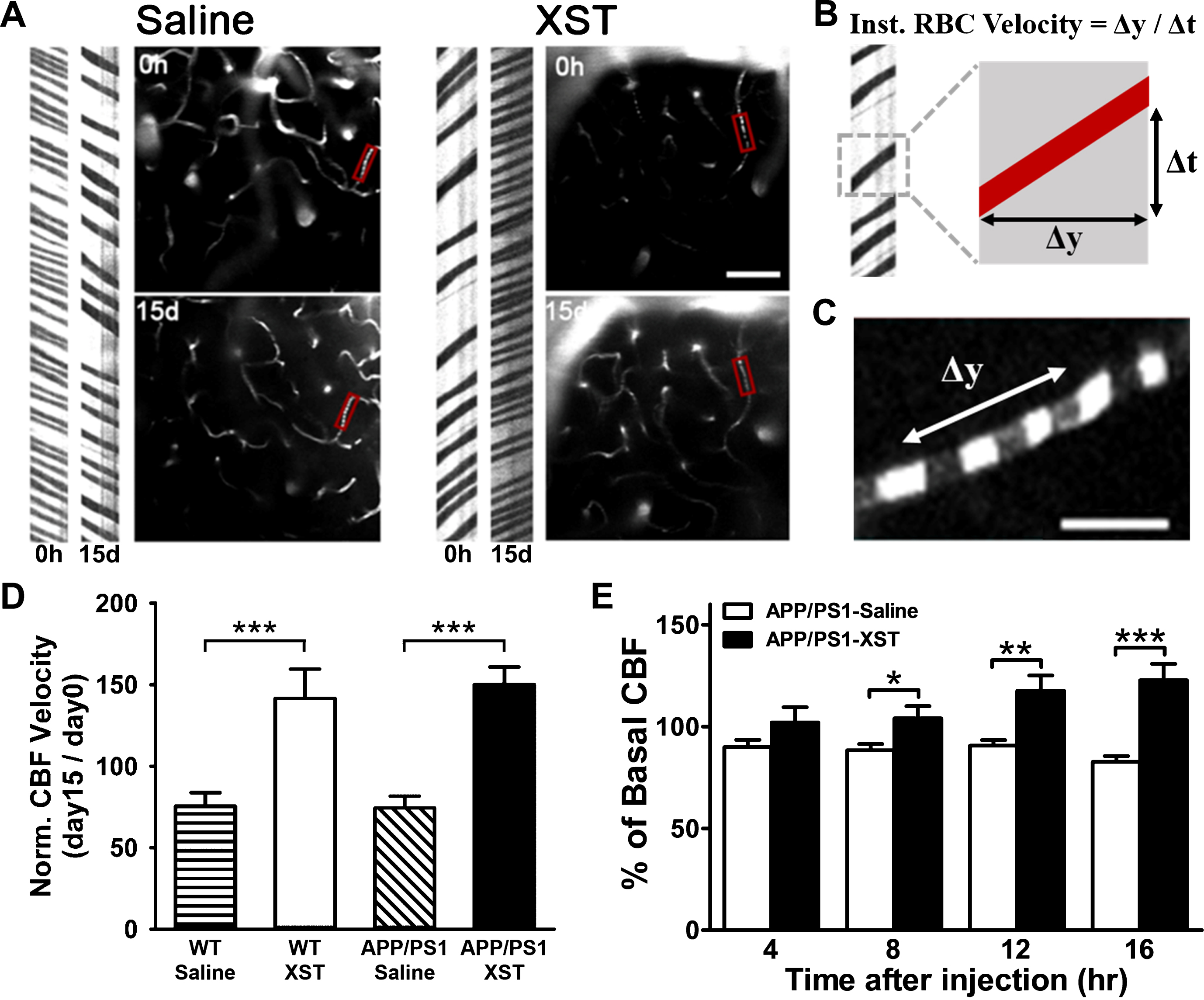
Fig.1
XST enhanced CBF in microvessels in APP/PS1 mice. A) Sample image of blood vessels (right side) and line scan image used for measuring blood flow (left side). Compared to the saline-treated APP/PS1 mice (left), 15 days of XST treatment significantly increased CBF (right) in the same microvessels. In this example, saline treatment led to reduced blood flow. Scale bar, 50μm. B) Line-scan data in (A) was used to quantify the velocity of RBCs moving through microvessels by calculating the inverse of the slope of these streaks. C) Image of RBCs (dark objects in the image) moving through microvessels. Scale bar, 10μm. D) Elevated CBF was seen in XST-treated mice, regardless of their genotypes. WT-Saline, 63 blood vessels/5 mice, 75.31±8.39% of baseline; WT-XST, 106 vessels/6 mice, 141.61±17.96%; APP/PS1-Saline, 94 vessels/7 mice, 74.24±7.38%; APP/PS1-XST, 92 vessels/6 mice, 144.99±10.86%. ***p < 0.001. E) Mean CBF measured using laser Doppler was significantly higher in XST-injected APP/PS1 mice, compared to the control group at 8 to 16 h following XST injection. n = 5 (APP/PS1-Saline), n = 5 (APP/PS1-XST). *p = 0.025; **p = 0.002; ***p < 0.001. Data represented were means±S.E.M.
Laser Doppler flow measurements
Eight-month-old female APP/PS1 mice were randomly assigned into XST-treated group or saline-treated group. We measured short-time relative changes in cerebral microvascular perfusion by laser Doppler blood flow assessment (Moor VMS-LDF1, Moor Instruments, Axminster, UK) [32]. A low red laser (785 nm) is sued to penetrate the skull and cover an area of approximately 1 mm2. During measurement, APP/PS1 mice were placed on the stereotaxic instruments (RWD Life Science Co., Ltd., China), anaesthetized with isoflurane with body temperature maintained using ThermoStar homeothermic monitoring system (RWD Life Science Co., Ltd., China). After 60 min baseline measurement, mice were treated with XST or saline. Scanning was continued for 16 h after injection every 4 h. The mean blood flow from randomly selected area was measured with the moorVMS-PC V3.1 software and expressed as normalized change (% of basal CBF).
Behavioral tests
Eight-month-old female APP/PS1 mice and littermate control mice were used in the behavioral assessments. Before drug injection, baseline locomotor ability of each mouse was assayed at a constant rotating speed of 10 rpm for 180 s. Mice were separated into different groups balanced for body weights and ability to stay on the rod (measured by latency to fall off the rod). All groups were given XST or saline injection for 30 days (Fig. 2A).
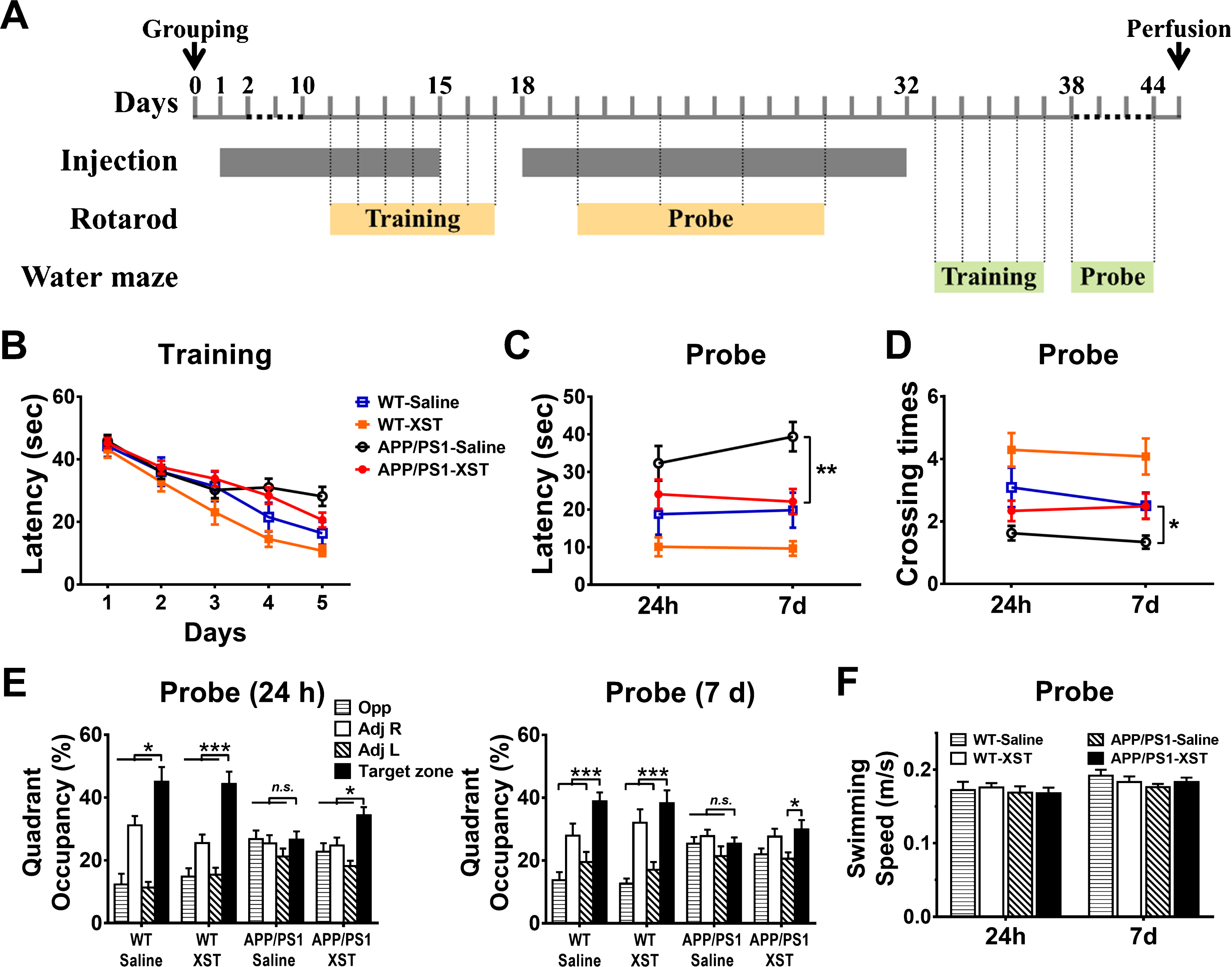
Fig.2
XST improved Morris water maze performance in the APP/PS1 mice. A) Time line of the behavioral assessments. B) Latency (time to reach the target platform) during training days. C) Latency (time to reach the original platform position) during probe trials. **p = 0.002. D) Number of crossings over the original platform position during probe trials. *p = 0.026. E) Percentage of quadrant occupancy during probe trials, 24 h (left) or 7 days (right) after training. XST-injected APP/PS1 mice showed a focus on the target zone equally well as the WT mice (*p < 0.05; ***p < 0.001 versus target zone), while there was no significant focus in the saline-injected APP/PS1 group (p > 0.05, versus target zone). F) Swimming speed during probe trials. No significant difference was found among four groups (p > 0.05). n = 12 mice (WT-Saline), n = 14 (WT-XST), n = 21 (APP/PS1-Saline), n = 27 (APP/PS1-XST).
Rotarod
Rotarod testing started 10 days after either XST or saline injection, and daily injection continued throughout the period of training and testing (Fig. 2A). A rotarod system with six individual lanes (Chengdu TME Technology Co., Ltd, China) was used. Each rod was 30 mm in diameter and 63 mm in length, and was suspended at a height of 220 mm above the foam-covered base. An electronically controlled motor was used to maintain the rod speed, and forced animals to run forward. Sensors detected the falling of a mouse in each lane automatically, with the latency registered on the computer (with a resolution of 0.1 s). Mice were trained for 7 consecutive days, and the acceleration speed was from 0 to 100 rpm in 100 s. After training was concluded, mice were tested every other two days four times. During testing, speed of the rod increased progressively from 0 to 60 rpm in 180 s. Each training or testing session consisted of 3 trials with one-minute break in between. The latency to fall off the rod was used to measure their locomotor performance and learning.
Morris water maze
Water maze training and testing were performed after 30-day XST or saline injection in the APP/PS1 mice (Fig. 2A). Morris water maze was performed in a circular pool (120 cm in diameter and 50 cm in height) with a featureless inner surface. The pool was filled to a depth of 26 cm with opaque nontoxic white dye. The tank was placed in a dimly lit, soundproof test room surrounded by distinct extra maze cues. The pool was equally divided into four quadrants. A white platform (12 cm in diameter and 25 cm high) was placed randomly in one of the quadrants and submerged 1 cm below the water surface to ensure that the platform was invisible. A pre-training day was dedicated for swim training (60 s) without the platform, and conducted in the subsequent 5 consecutive days. During training days, mice received 2 training sessions per day, with each session containing 3 trials (10 min between trials) for a total of 10 hidden sessions. The drop location was changed semi-randomly between trials with the platform location fixed throughout the training process. After a mouse located the platform, it was permitted to stay on it for 10 s. A mouse was guided to the platform and placed on it for 10 s if it cannot locate the platform within 60 s. The mouse was taken to its home cage and allowed to dry up under a dimly lamp after each trial. The time interval between each session was 30 min. Probe trials were conducted after the last training session (24 h and 7 days after). During the probe trial, the platform was removed and mice were allowed to swim for 60 s. All mice were placed at the same starting point, which was the farthest dropping location of the platform.
The cued test was conducted 24 h after the second probe trial and each mouse underwent three trials. A red flag was put on the platform to make it visible. The procedure was similar to training, except that the platform was moved to a different quadrant. Mice with visual impairment were excluded. During all trials, escape latencies, swim paths, swim speeds, and percentage of time spent in each quadrant was recorded using a video camera-based Any-maze System (Stoelting, Wood Dale, USA).
Immunohistochemistry
After 15/30-day injection with either XST or saline, mice were perfused with PBS, followed by 4% paraformaldehyde, through the heart. The whole brain was removed and fixed in 4% paraformaldehyde overnight, then transferred to 15%, 20%, and 30% sucrose solution, each for 24 h to allow sufficient dehydration. After embedding in Optical Cutting Temperature (OCT, Tissue-Cut), 20μm frozen sections were cut. Excessive embedding solution was removed with PBS. Sections were then treated with 0.3% Triton X-100 and 10% goal serum for 1 h at room temperature. The following primary antibodies were used: rat anti-amyloid fibril OC (Millipore, 1:500), rabbit anti-synaptophysin (Abcam). Secondary antibodies goat anti-rabbit IgGs, goat anti-mouse IgGs and goat anti-rat IgGs, conjugated to Alexa Fluor 488 or 546 (all 1:400, Life Technologies) were used for incubation at room temperature for 1 h, washed in PBS and cover-slipped for imaging. Thioflavine S staining was done on brain sections by treating with 0.5% thioflavine S (in 50% ethanol) for 10 min, washed in 70% ethanol twice, and followed by PBS wash once. To quantitatively measure changes in synapse density around plaques, the fluorescence intensity within a zone 10μm wider in diameter than the edge of the plaque was measured, divided by the fluorescence intensity of regions with strong and uniform synaptophysin staining.
Western blots
After laser Doppler flow measurements or 15-day injection, mice were perfused with PBS through the heart. Freshly dissected mouse brains were treated with RIPA lysis and homogenized by S10-High Speed Homogenizer (Xinzhi Biotechnology Co., Ltd., China), then centrifuged at 12,000 rpm for 10 min at 4 °C. Protein concentration was measured with a BCA Protein Assay Kit (Pierce). Clarified cell extracts were mixed with 6×SDS sample buffer. Protein samples were run on 8% or 12% SDS-PAGE using a Bio-Rad gel system and transferred onto nitrocellulose membranes. Loading controls (GAPDH) were run on the same gel. Membranes were then probed with antibodies with the appropriate dilutions, including anti-AβPP (catalog no. ab32136; Abcam; 1:20000), anti-CTFs (catalog no. A8717; Sigma-Aldrich; 1:2000), anti-BACE1 (catalog no. ab183612; Abcam; 1:1000), anti-neprilysin (catalog no. AB5458; Millipore; 1:2000), anti-insulin-degrading enzyme (catalog no. ab133561; Abcam; 1:5000), anti-presenilin 1 (catalog no. ab76083; Abcam; 1:20000). ImageJ was used for densitometric analysis.
Statistical analysis
Statistical analyses were performed using GraphPad Prism version 5.01 (GraphPad) and SPSS Statistics version 22 (IBM). For two group comparisons we used Student’s unpaired t-test. For multiple groups we used one-way or two-way ANOVA, with repeated measures. For post hoc testing we used Bonferroni multiple comparison test. A p value of less than 0.05 was considered statistically significant. In all of graphs, data were presented as mean±standard error of the mean (S.E.M.). Tissue processing and outcome measurements were conducted under treatment-blind conditions.
RESULTS
XST improves cerebral blood flow in APP/PS1 mice
Among traditional Chinese herbal drugs, XST is known to increase blood flow both in the body and brain; this may at least partially underlie its medical effect [26, 33]. To ascertain that XST elevated CBF in the APP/PS1 transgenic AD model mice, we measured CBF before and after XST injection. For these experiments, we have focused on small blood vessels (diameter 3–6μm) since these smaller vessels are shown to be altered in AD [26, 33]. Due to the heterogeneity of blood vessels (especially capillaries), comparisons between different brain regions in different mice makes it difficult to detect reliable changes. To overcome this difficulty, we utilized in vivo two photon time-lapse imaging on the same microvessels over a period of 15 days during which either XST or saline was injected. Fluorescent dyes were injected through the tail vein and labeled the plasma, while the non-labeled red blood cells appeared as dark objects moving inside the vessels (Fig. 1C). By using line scans parallel to the direction of blood flow (parallel to the blood vessels), we obtained images shown in Fig. 1A, which allowed us to calculate the rate of blood flow (see Methods; Fig. 1B). There was a significant increase in CBF velocity in the XST-injected APP/PS1 mice compared to saline-injected mice, and a significant increase in XST-injected WT mice compared to saline-injected WT mice (Fig. 1D), suggesting that XST is effective in elevating CBF velocity in both APP/PS1 and WT mice. Using laser Doppler flow measurements, we found significant increase in CBF in the APP/PS1 mice starting at 8 h after XST injection and last for at least 16 h (the end of these experiments) (Fig. 1E). The CBF velocity of saline-treated APP/PS1 mice remained unchanged for up to 16 h (Fig. 1E). These results suggest that it takes a few hours for elevation in CBF to take effect after XST injection and this elevation is persistent for at least a few hours after a single injection.
XST improves spatial learning and memory in APP/PS1 mice
The most pronounced deficit in AD patients and AD mice are their impaired ability to learn and to remember. In APP/PS1 mice, this is manifested as reduced performance in the Morris water maze which measures spatial learning and memory. During the training period, saline-treated APP/PS1 group showed longer latency in reaching the target platform, and training effects were modest before a plateau was reached where no further improvement was observed (Fig. 2B). In contrast, the other three groups of mice showed similar rates of latency reduction with training (Fig. 2B). This result suggests that XST improved spatial learning in the APP/PS1 mice. After training, we tested spatial memory of these mice 1 day (24 h) and 7 days. For escape latency (time takes to reach the original platform position), there was a significant difference between saline-treated APP/PS1 group and the other 3 groups, while the XST-treated APP/PS1 group was indistinguishable from the saline-treated WT mice at 7 days (Fig. 2C). For the number of crossings over the original platform position, we observed similar differences as in latency (Fig. 2D). These results suggest that XST improved the APP/PS1 mice’s memory function to a level comparable to the WT mice. We further analyzed quadrant occupancy by plotting the percentage of time mice spent in specific quadrants. Twenty-four hours after water maze training was terminated, WT mice and XST-injected APP/PS1 mice spent significantly more time in the target quadrant than other quadrants (Fig. 2E), indicating good retention of spatial memory. However, this retention was reduced after 7 days in XST-treated APP/PS1 mice since they only showed preference over one quadrant rather than three (Fig. 2E). Unfortunately, placing home cages in the Adj R area outside the water maze during testing may have affected the animals’ preferences (i.e., percentage time spent in Adj R was higher than in Opp or Adj L). The observed reduction in latency could be caused by improvement in swimming speed in the APP/PS1 mice by XST. However, we found no significant difference in swimming speed across all groups, as shown in Fig. 2F.
XST improves motor learning and performance in APP/PS1 mice
In addition to cognitive functions (such as memory), the general life quality of AD patients is severely reduced, including the ability to take care of one-self and cope with daily life. In AD transgenic mice in general, and in APP/PS1 mice specifically, reduction in motor function has been reported [26, 33]. Certain manipulations have also been shown to ameliorate this deficit or prevent further deterioration [26, 33]. Hence, we examined whether motor performance deficits might be ameliorated by XST treatment, using rotarod test. With training (speed from 0 to 100 rpm in 100 sec), WT mice (both saline- and XST-injected) showed substantial increase in latency to fall (time takes for a mouse to fall off the rotating rod) (Fig. 3A). While there was no significant improvement in the saline-injected APP/PS1 mice during the 7-day training period, XST-injected APP/PS1 mice showed improvement similar to that of WT mice (Fig. 3A). After termination of training, we tested mice in accelerating probe trials (speed from 0 to 60 rpm in 180 s). XST injection led to a large increase in the latency to fall in the APP/PS1 mice (26.0% compared to saline-injected; Fig. 3B). Interestingly, XST-injected WT mice showed a small but significant improvement (10.1% compared to saline-injected; Fig. 3B).
Fig.3
XST improved rotarod performance in the APP/PS1 mice. A) Latency to fall on rotarod with accelerating speed (from 0 to 100 rpm in 100 s). Over 7 training days, only saline-injected APP/PS1 mice showed no improvement. n = 31 mice (WT-Saline), n = 25 (WT-XST), n = 19 (APP/PS1-Saline), n = 24 (APP/PS1-XST). B) Latency to fall during probe trials (acceleration speed from 0 to 60 rpm in 180 s), from the same groups of mice in (A), normalized to the average value of WT-saline group. *p = 0.023; ***p < 0.001. C) Latency to fall during training on the accelerating rod (from 0 to 60 rpm in 180 s). XST-injected mice showed improvement from day 4 on, while saline-injected APP/PS1 mice did not change over the seven training days. n = 7 mice (APP/PS1-Saline), n = 10 (APP/PS1-XST). *p = 0.032; #p = 0.027. D) Latency to fall during probe trials for the same groups of mice in (C). Performance in the XST-injected APP/PS1 mice was significantly better compared to the saline-injected mice. ***p < 0.001. E) Latency to fall for every trial during the training period using data from (C). The dotted lines were drawn to illustrate the apparent 3 distinct levels of these latency values.
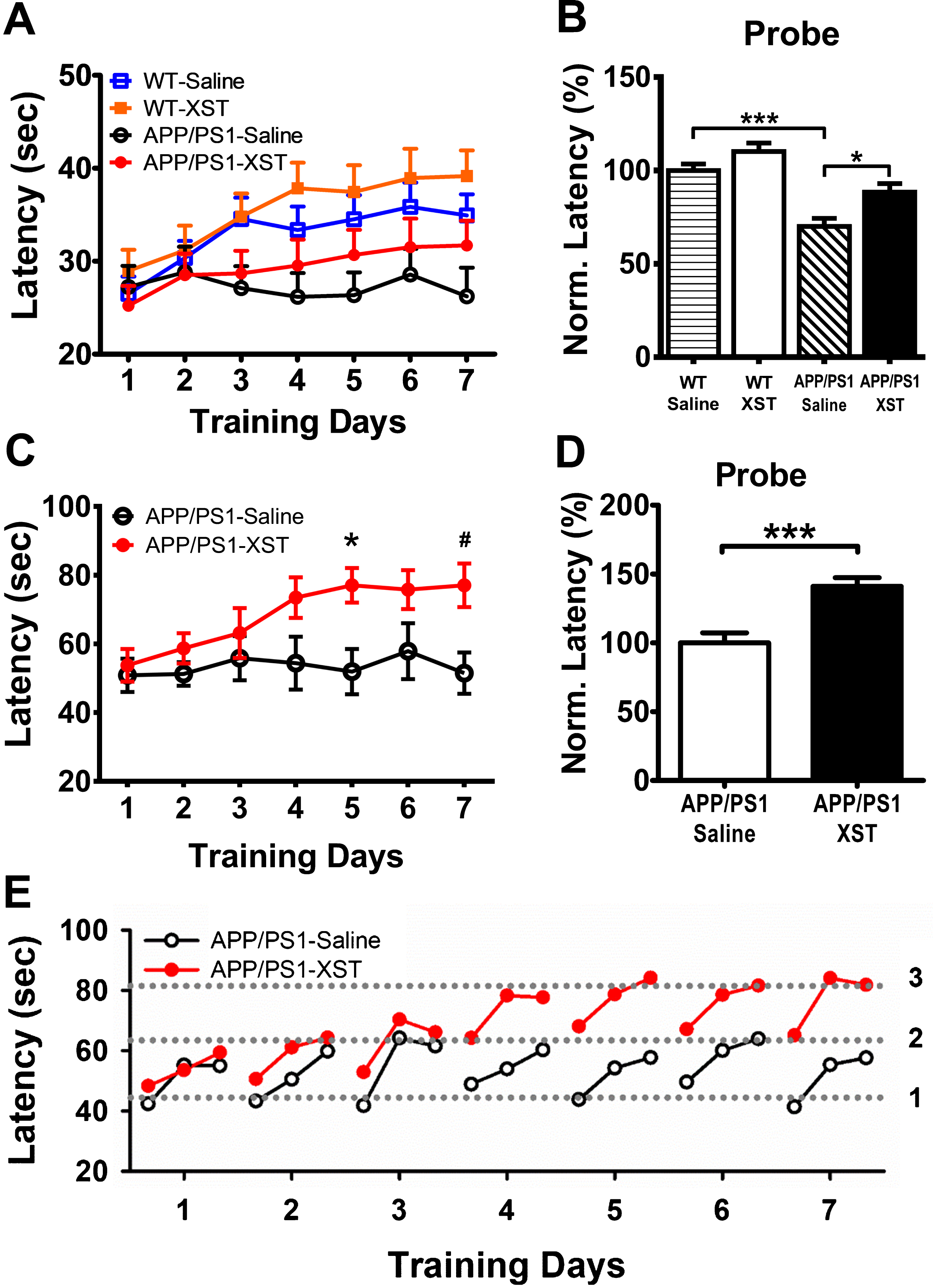
In the above experiments, the acceleration speed of rotating rod was fairly high which was evident by the low latency values, hence the benefit of XST may not be fully revealed. Thus, in a separate set of experiments, we reduced the acceleration speed during training trials (from 0 to 60 rpm in 180 s) and repeated the above experiments in APP/PS1 mice. We found larger improvement that took places sooner than at higher rotarod speed (Fig. 3C), and the benefit was also sustained throughout the probe test period (Fig. 3D). Taken together, these results indicate that XST injection significantly improves motor learning and performance in both APP/PS1 and WT mice.
The improvement in motor learning and performance can reflect improved motor function given that mice normally do not exercise regularly and vigorously. In addition, it may also reflect improved learning ability since saline-injected APP/PS1 mice did not show any consistent improvement during training. To further understand the underlying mechanism of this XST-induced improvement, we analyzed the latency values of individual trials of APP/PS1 mice during training (data from Fig. 3C). The latency values started low on the first trial of the day, progressively improved and reached the maximal level on the third (last) trial of the day (Fig. 3E), consistent with previous finding [26, 33]. For saline-injected APP/PS1 mice, the latency fluctuated between two values (level 1 and 2 in Fig. 3E) on each day and there was no steady or persistent change during the training period. For the XST-injected APP/PS1 mice, their performance was similar to the saline-injected group for the first three days (between level 1 and 2); but on the fourth day, the latency values jumped to the next level and fluctuated between level 2 and 3 (Fig. 3E). There was no further improvement for the rest of the training period, suggesting that a plateau have been reached. These results suggest that both short-term (within-day improvement) and long-term (cross-day improvement) plasticity processes occur during training, and only the long-term plasticity process is impaired in APP/PS1 mice. Furthermore, only the long-term plasticity process was improved by XST injection. To determine whether XST-induced improvement in rotarod performance was caused by a better motor function, we tested how long each mouse could stay on a rotating rod with a low constant speed (10 rpm with a maximum duration of 180 sec). No difference was found in this measurement (data not shown), and thus suggests that improved motor function is unlikely to contribute significantly to the increase in latency.
Higher synapse density near plaques in XST-treated APP/PS1 mice
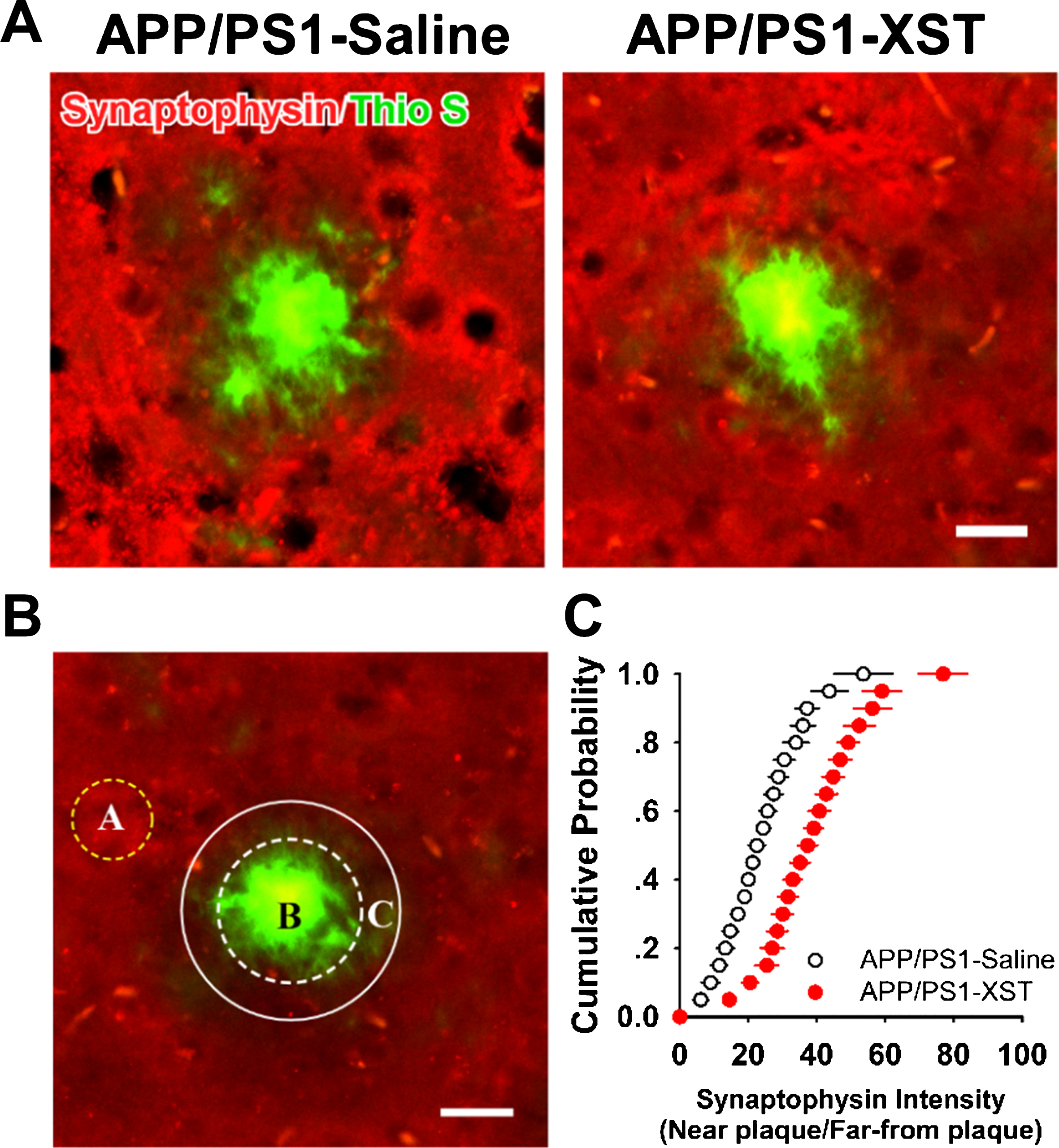
One likely mechanism underlying the improved learning and memory function in XST-treated APP/PS1 mice is an improved synaptic function or synaptic plasticity, such as higher synapse density. In APP/PS1 mice, it was widely observed that synapse/spine density is reduced near the vicinity of amyloid plaques, likely caused by an elevated Aβ level in this region [34–36]. To estimate synapse density, we stained fixed brain sections with antibodies against synaptophysin. In saline-injected APP/PS1 mice, there was a clear “dead” zone around many large plaques that were devoid of synaptophysin staining, indicating reduced synapse density or absence of synapses (Fig. 4A). However, in the brain sections from XST-injected mouse, such “dead” zones were observed far less frequently (Fig. 4A). To quantify changes in synapse density, we measured the fluorescence intensity within a zone surrounding the plaque with a diameter 10μm wider than the edge of the plaque (for only plaque with a diameter greater than 15μm) and divided this value by the fluorescence intensity within a region away from and devoid of plaques (with strong and uniform synaptophysin staining) (Fig. 4B). The fluorescence intensity was higher around the vicinity of plaques in the XST-injected than that in the saline-injected APP/PS1 mice, reflected as a rightward shift in cumulative distribution (Fig. 4C). XST treatment either led to regaining of the lost synapses, or prevented synapse loss. We cannot distinguish between these two possibilities based on data in Fig. 4.
Fig.4
Higher synapse density around amyloid plaques in the XST-treated APP/PS1 mouse brain. A) Sample images of fixed brain sections from saline-treated APP/PS1 mouse (left) and XST-treated APP/PS1 mouse brain (right). Sections were stained with thioflavin S (green) for amyloid plaques and synaptophysin (red) for presynaptic terminals. A clear zone low in the synaptophysin fluorescence intensity was seen in sections from saline-treated APP/PS1 mouse but much less so in the XST-treated ones. Scale bar, 20μm. B) A diagram showing the analysis method. A, region far away from the plaques where synaptophysin fluorescence intensity is high. B, plaque. C, region adjacent to the plaque (with a diameter 10μm wider than that of the plaque) within which synaptophysin intensity was measured. The ratio of fluorescence intensity inside region C/region A was calculated. C) Cumulative distribution of synaptophysin intensity ratio for saline-treated and XST-treated APP/PS1 mice. APP/PS1-Saline, n = 5 mice (3 sections/mice); APP/PS1-XST, n = 5 mice (3 sections/mice).
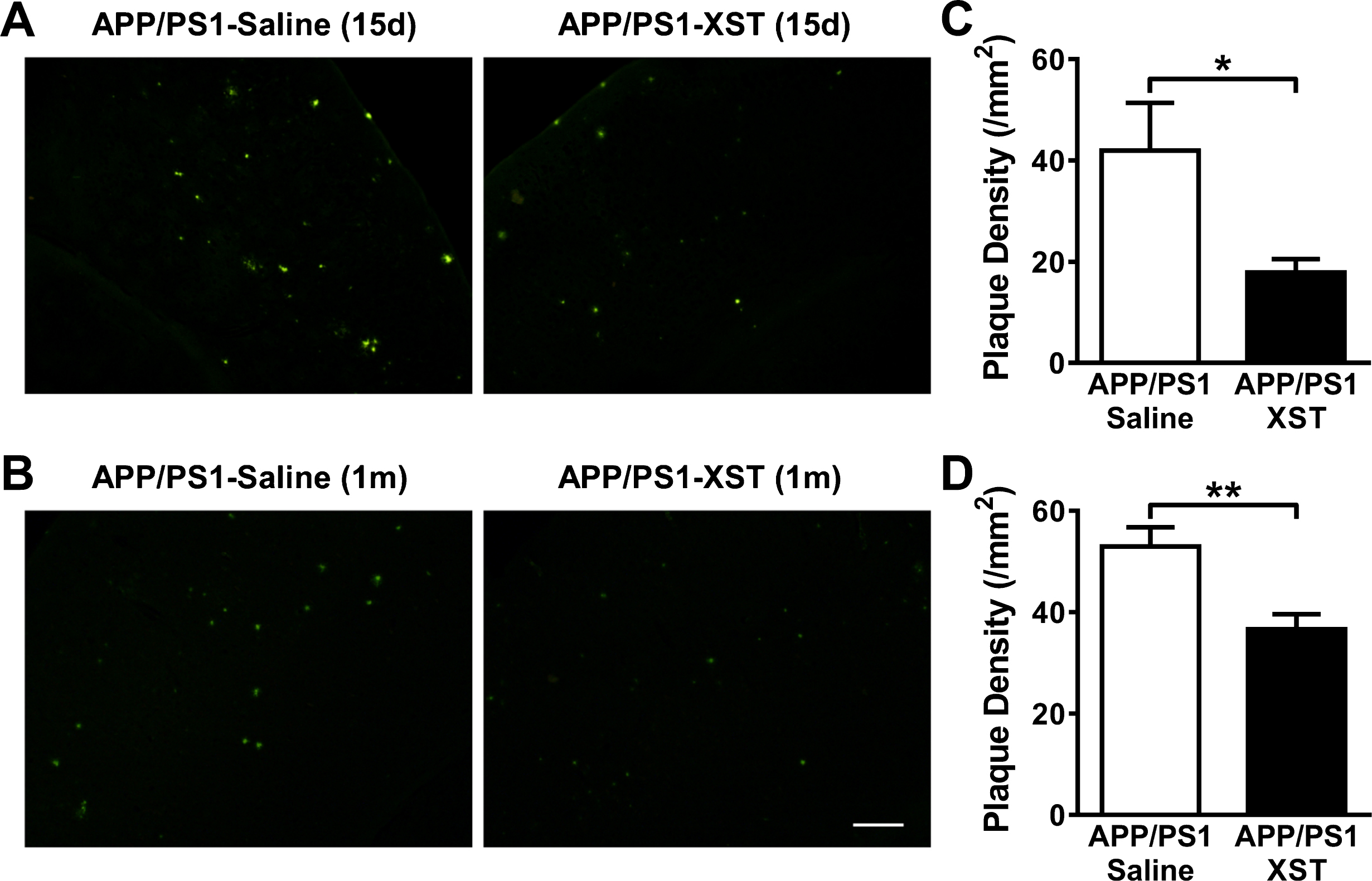
XST reduces amyloid plaque density and size in the APP/PS1 mouse brain
Amyloid plaques first appear around 6 month in the APP/PS1 mice and they become progressively larger and more numerous with disease progression [28]. Plaques were stained with thioflavin S in fixed brain sections. XST treatment in APP/PS1 mice for either 15 days (Fig. 5A, C) or 1 month (Fig. 5B, D) led to a significant reduction in plaque density in the cerebral cortex.
Fig.5
XST treatment reduced plaque density in the APP/PS1 mouse brain. A, B) Sample images of plaques in the cerebral cortex in brain sections from APP/PS1 mice treated with XST or saline for 15 days (A) or 1 month (B). Scale bar, 100μm. C) Plaque density in APP/PS1 mice treated with either XST or saline for 15 days. APP/PS1-Saline, n = 4 mice (3 sections/mice), 41.93±9.44 mm2, 100.0±22.52%; APP/PS1-XST, n = 4 mice (3 sections/mice), 17.86±2.62 mm2, 42.59±6.24%. *p = 0.049. D) Plaque density in APP/PS1 mice treated with either XST or saline for 1 month. APP/PS1-Saline, n = 13 mice (3 sections/mice), 52.94±3.78 mm2, 100.0±7.13%; APP/PS1-XST, n = 18 mice (3 sections/mice), 36.66±2.92 mm2, 69.25±5.52%. **p = 0.002.
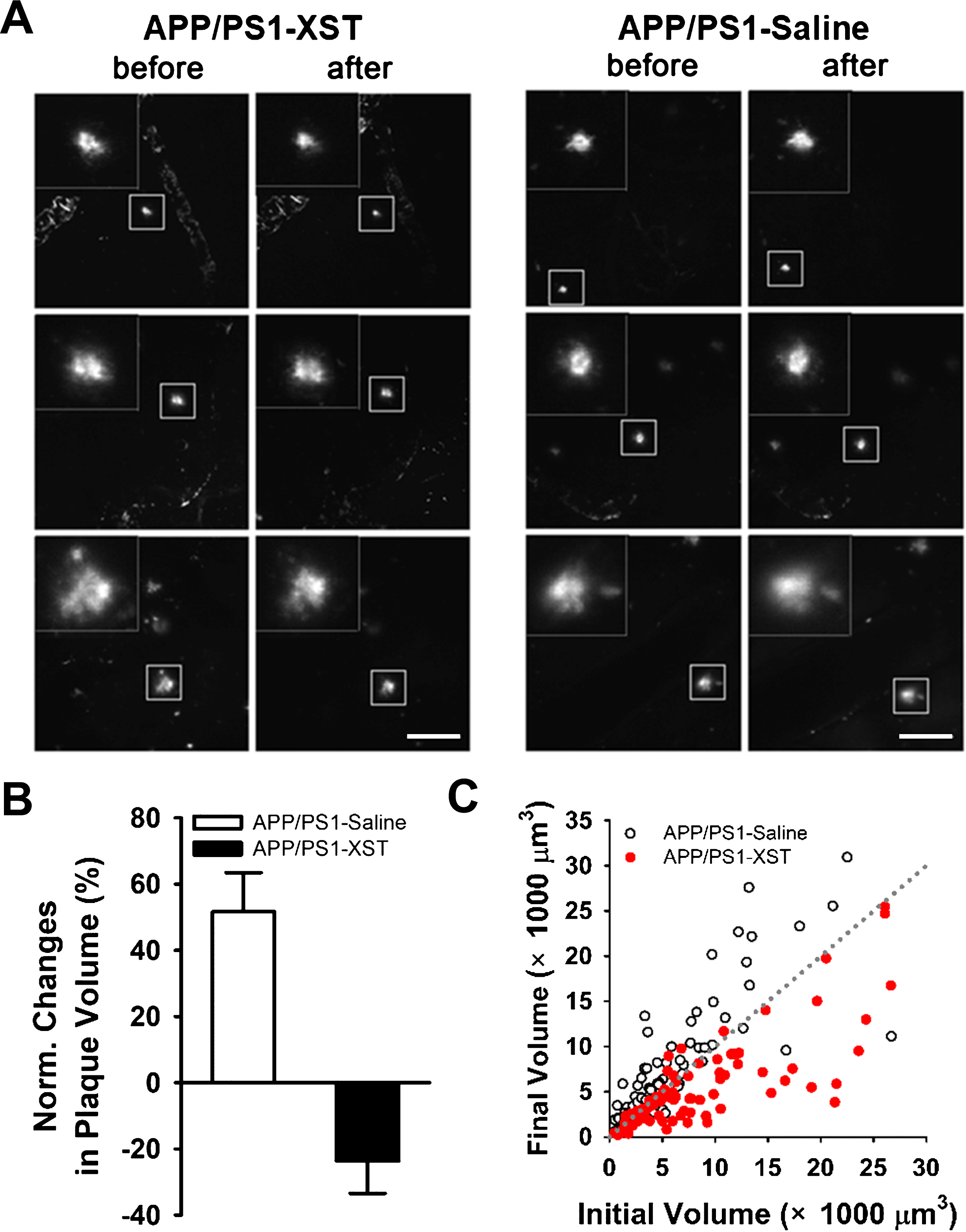
A few possible mechanisms may underlie the observed reduction in plaque density: 1) XST reduces the growth of pre-existing plaques; 2) XST causes shrinkage of the pre-existing plaques; or 3) XST reduces the rate of formation of new plaques. We cannot distinguish among the above possibilities based on the results in Fig. 5 since comparison was made among sections from different mouse groups. To overcome this problem, we monitored the size of the same set of plaques within a 15-day period using in vivo two photon time-lapse imaging. We selected an age (7-month) when plaque growth was rapid in APP/PS1 mice. In general, plaques from saline-injected APP/PS1 mice showed an increase in size during this 15-day period, while plaques became smaller in XST-injected APP/PS1 mice (Fig. 6A). There was an average of 23.7±9.7% reduction in plaque volume in the XST-treated APP/PS1 mice, compared to an average of 51.6±11.8% increase in saline-treated (Fig. 6B). As a group, the majority of plaques became smaller in the XST-treated mice while the majority of plaques became larger in the saline-treated mice (Fig. 6C). This result suggests that XST reduces the size of existing plaques within 15 days. Since the newly emerged plaques were small and often difficult to distinguish from the random non-specific background staining, we did not study newly emerged plaques. Hence, we will not address whether XST has any impact on the emergence of new plaques.
Fig.6
XST treatment induced reduction in plaque size revealed by repetitive two-photon time-lapse imaging. A) Sample images of the same plaques imaged before and after saline (right) or XST (left) injection for 15 days. Plaques in the box are shown in higher magnification in the insert. Scale bars, 100μm. B) Normalized changes in plaque volume after injection of either saline or XST. C) When the initial plaque volume (day 0, prior to injection) was plotted against the final volume (day 15, after injection), most of the values in the saline-treated group were above the dotted line (no change), while most of the values in the XST-treated group were below the line (reduction). APP/PS1-Saline, 84 plaques/5 mice; APP/PS1-XST, 84 plaques/5 mice.
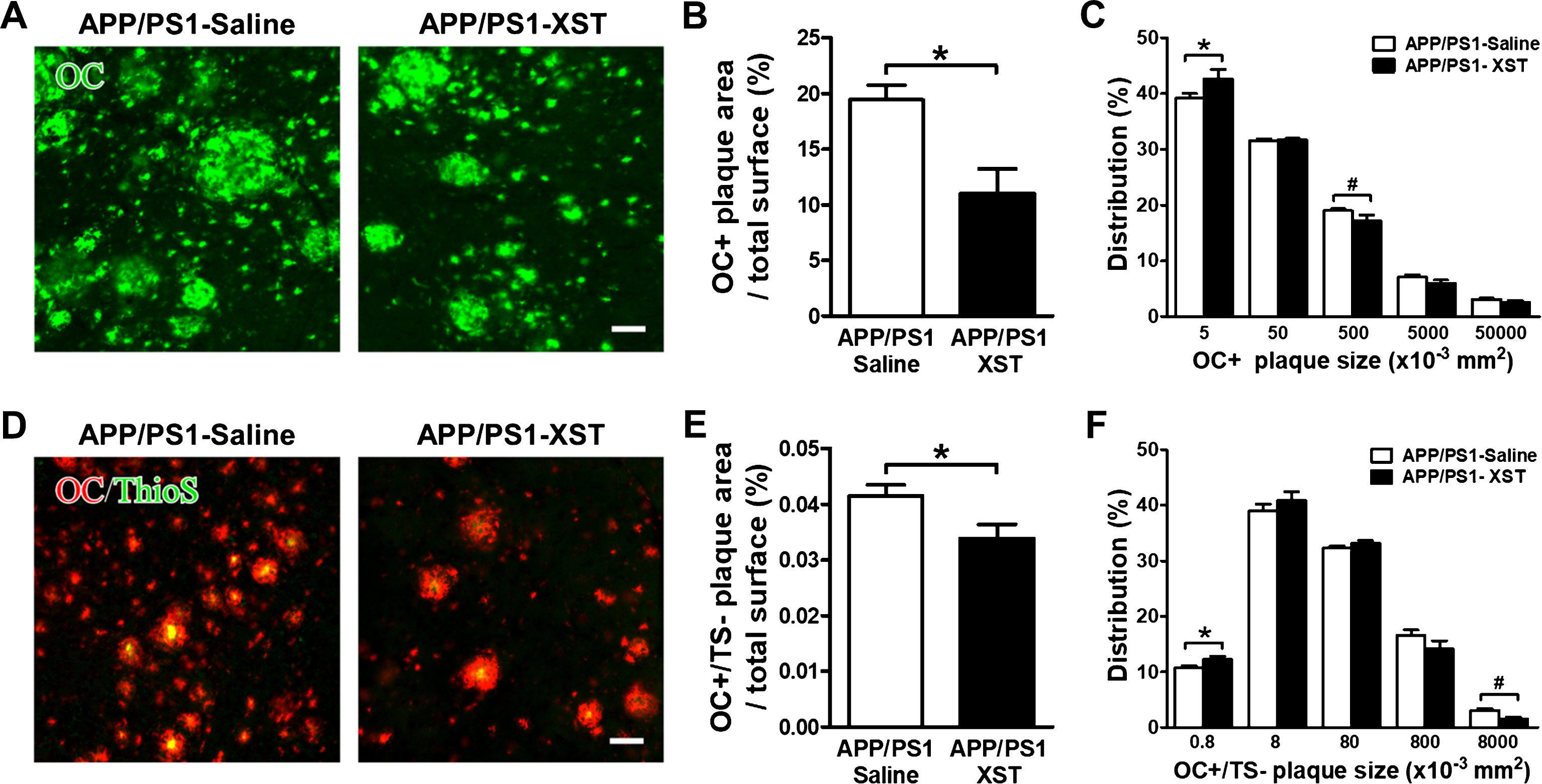
In addition to plaques with a clear dense core that is thioflavin S positive (which reflects the fibrillar form of Aβ), the soluble form of Aβ (such as the oligomer forms) are also present in AD human brain and in AD transgenic mice. These soluble forms are generally viewed as the more toxic forms of Aβ [37–39]. Kayed et al. showed that OC antibody specifically recognizes fibrils, but not random coil monomer or prefibrillar Aβ oligomers. OC also recognizes soluble fibrillar oligomers ranging from dimer to greater than 250 kDa oligomers [40]. OC antibody stained all types of amyloid deposits in human AD brain sections, and the diffuse amyloid deposits are thioflavin S negative. To understand whether this oligomer form of Aβ could be affected by XST, we stained brain sections with OC antibody [41, 42]. In comparison to thioflavin S (Fig. 7A), OC antibody stained a much larger area, some surround a thioflavin S-positive plaque core while others were only positive for OC. In general, there was a large reduction in the OC-positive area in XST-treated AD mice (Fig. 7A, B; XST-treated was 56.71±11.43% of saline-treated). Distribution of plaque size showed a decrease in larger OC-positive plaques with an increase in smaller plaques after treatment with XST (Fig. 7C). Since we have seen a significant reduction in the thioflavin S-positive plaques in the XST-treated APP/PS1 mice, the reduction observed with OC staining could be mostly reflecting reduced oligomers associated with the plaque cores. We addressed this question by double staining using OC and thioflavin S (Fig. 7D). We quantified the areas that were OC-positive but thioflavin S-negative. There was a small but significant reduction in the area (Fig. 7E; XST was 81.68±6.15% of saline). Furthermore, distribution of plaque size showed an increase in smaller OC+/TS- plaques and decrease in larger ones in the XST-treated APP/PS1 mice (Fig. 7F). Taken together, these results indicate that XST leads to a reduction in the remaining soluble, oligomer form (OC-positive) of Aβ.
Fig.7
XST treatment induced a reduction in OC-positive Aβ in the APP/PS1 mouse brain. A) Sample images of OC-positive Aβ in the APP/PS1 mouse brain sections from either saline- (left) or XST-treated (right) group. Scale bars, 50μm. B) Quantification of the areas of OC-positive Aβ over the total area showed a significant reduction in the XST-treated APP/PS1 mouse. APP/PS1-Saline, n = 5 mice (3 sections/mice); APP/PS1-XST, n = 5 mice (3 sections/mice). *p = 0.012. C) Distribution of OC+ plaques. The percentage of smaller plaques was higher while the percentage of larger plaques was lower in the APP/PS1-XST mice. *p = 0.04; #p = 0.013. D) Sample images of double-staining of OC and thioflavin S in APP/PS1 mouse brain sections. Scale bars, 50μm. E) A small but significant reduction in the areas of OC (+) but thioflavin S (–) staining over the total area in the XST-treated group. APP/PS1-Saline, n = 5 mice (3 sections/mice); APP/PS1-XST, n = 5 mice (3 sections/mice). *p = 0.048. F) Distribution of OC+/TS- plaques. There was a significant decrease in the size of smaller OC+/TS- plaques in the brain of APP/PS1-XST mice, compared to APP/PS1-saline mice. *p = 0.044; #p = 0.015.
XST alters Aβ processing and clearance in APP/PS1 mouse
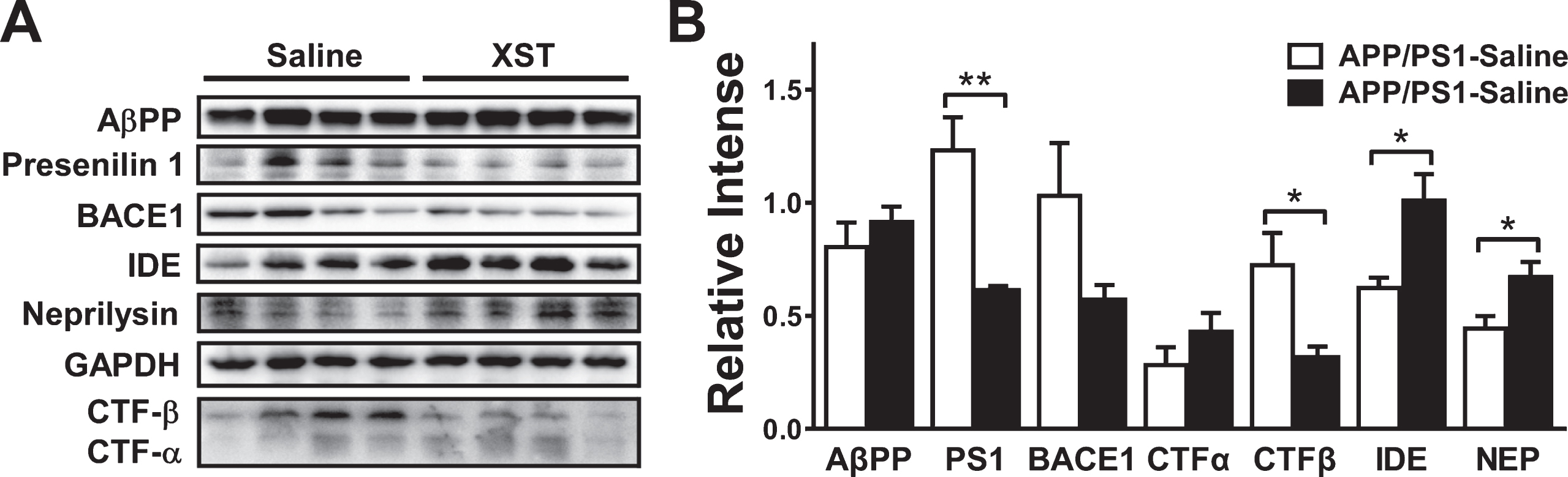
Aβ levels are determined by a balance between anabolic and catabolic activity. In the amyloidogenic pathway, processing of AβPP by β-secretase and γ-secretase leads to the production of Aβ peptides [43]. β-secretase 1 (BACE1) is considered to be a rate-limiting enzyme in Aβ pathogenic accumulation. Upregulation of CTF-β, a cleaved C-terminal fragment of AβPP by BACE1, has been shown to induce Aβ accumulation. Presenilin 1 is one of the four essential components of γ-secretase complexes. Several Aβ degrading enzymes, such as neprilysin and insulin-degrading enzyme (IDE), are also shown to affect Aβ accumulation/clearance [44]. Previous studies showed that certain ginsenosides (such as Rg1), the major components of XST, modulate Aβ-related pathology in AD model [45–50]. To investigate whether XST alters Aβ generation or enzymatic degradation, we performed western blots on AβPP, presenilin 1, BACE1, CTF-α/β, IDE and neprilysin (Fig. 8A, B). XST treatment for 15 days did not alter AβPP level in APP/PS1 mice. Although there was a trend for reduction in BACE1 level, this reduction was not statistically significant. We found a significant reduction in the levels of presenilin 1 and CTF-β; however, CTF-α level was not affected, indicating a reduced Aβ production. In addition, we found a significant increase in the levels of IDE and neprilysin, indicating an enhanced Aβ clearance. Put together, XST reduced the amyloidogenic cleavage and enhanced Aβ clearance without affecting the AβPP level.
Fig.8
Effect of XST treatment on the expression of key regulator of Aβ production and clearance in the APP/PS1 mouse brain. A) Representative western blots for the levels of various molecules examined. GAPDH was used as the control for protein loading and transfer efficiency. B) XST did not affect the protein levels of AβPP and CTF-α, but reduced the levels of presenilin 1 (**p = 0.0058) and CTF-β (*p = 0.035). The trend of reduction in BACE1 was not significant. In addition, XST increased the levels of IDE (*p = 0.022) and neprilysin (*p = 0.038). n = 4 mice per group.
Since we have found significant elevation in CBF starting at 8 h after XST injection (Fig. 1E), we were wondering whether the above changes in molecules involved in Aβ processing/clearance might occur at this time point. To do so, we repeated western blots analysis on presenilin 1, CTF-α/β, IDE and neprilysin, which showed significant alterations after XST treatment for 15 days. We did not find any significant change in any of the above molecules (Supplementary Figure 1A, B), suggesting that Aβ processing and/or clearance may take longer than 8 h to occur. Consistent with this conclusion, we did not find any significant change in the density of plagues 8 h after XST injection (Supplementary Figure 1C, D).
DISCUSSION
Our study demonstrated that systemic injection of XST (Chinese herbal medicine known to elevate CBF in humans) in transgenic AD model mice (APP/PS1) led to significant enhancement in CBF, significant improvements in brain functions (spatial learning and memory, motor learning and memory) and significant reduction in AD-associated pathology (amyloid plaque size/density), likely mediated by reduced Aβ production and enhanced Aβ clearance. We have also observed significantly higher synapse density around plaques, which may underlie these observed functional improvements. Effective AD treatment should lead to improved cognitive functions and reduced pathology. Here we showed that XST treatment can satisfy these requirements.
Improved functions after XST treatment
After XST injection, improvement in spatial learning and memory, and motor learning and performance was observed in the APP/PS1 mice, suggesting improved functions. We observed two types of plasticity processes during rotarod testing, one short-term (within-session improvement) and one long-term (cross-day improvement). Interestingly, only the long-term plasticity process was impaired in APP/PS1 mice and improved by XST treatment. We hypothesized that long-term plasticity process reflects memory formation, while short-term plasticity process is related to improvement in motor performance. This proposal is consistent with the recent findings of Hayashi-Takagi et al. [51], who showed that motor learning on rotarod resulted in enhanced Arc signaling and spine expansion in a subset of spines in the motor cortex. By expressing a photoactivatable GTPase Rac1 in spines, they showed that prolonged photo-activation led to spine shrinkage (reversed spine expansion) and loss of recently acquired motor memory. This finding of improved motor learning and performance is important giving that deficits in motor performance occur in a significant portion of AD patients and have been suggested to occur years prior to the onset of cognitive symptoms [52, 53].
Reduced AD pathology after XST treatment
Reduced synapse density and/or impaired synapse plasticity are very likely to underlie impaired neural functions in AD. Studies in human AD patients showed that reduced synapse density is more closely correlated to the severity of cognitive deficits than plaque burden [52, 53]. Impaired synaptic plasticity was shown in AD transgenic mice [54–56]. Individuals with amnestic mild cognitive impairment (MCI) and AD have significantly fewer synapses and significantly lower levels of synaptic proteins [57]. In transgenic AD mice, synapse loss and morphological changes can take place prior to amyloid deposition [54, 58]. Importantly, this loss of synapse/spine is more prominent in the vicinity of plaques, suggesting that these peri-plaque synapses are more vulnerable to degeneration [34, 35, 59]. Thus, preventing the loss of these “vulnerable synapses” should be of high priority in the treatment of AD to preserve cognitive function or to slow down its decay. Functional recovery or amelioration of AD pathology has been shown to be associated with increased synapse density or elevated expression of synaptic proteins (e.g., [60, 61]). We have observed higher density of synaptophysin staining in the vicinity of plaques. At this time, we cannot distinguish between the possibilities of whether XST prevents synapse loss or gains back the lost synapses. Nonetheless, we favor the former since it is mechanistically simpler, especially when taken into consideration that synapse loss may be accompanied by the loss of presynaptic terminals.
How can XST modulate synapse loss in AD mice? A dominant hypothesis in the field is that synapse removal is triggered by toxic Aβ. This model is supported by the observed toxicity of Aβ on neurons when they are added acutely to or over-expressed in neurons [62]. Targeting soluble Aβ oligomers has been proposed as a viable therapeutic strategy to ameliorate or prevent AD [38, 63]. OC-positive Aβ are significantly elevated in multiple brain regions of AD patients, and its level is significantly correlated to the level of cognitive decline (MMSE scores) in AD patients [37–39]. We showed a rapid reduction in the size and density of both fibrillar (plaques with cores) and soluble form of Aβ in the APP/PS1 mouse brains after XST treatment. A gradient of Aβ oligomer from the amyloid plaques were demonstrated which could explain the more severe loss of synapses in the vicinity of plagues [64]. In reducing the size of both plaques and surrounding soluble Aβ, XST can lead to higher synapse densities. The observed reduction in plaque/soluble Aβ can be at least partially medicated by enhanced CBF and this will be discussed in the next section. We have further provided evidence of reduced Aβ production based on reduced levels of presenilin 1 and CTF-β. In addition, we showed an enhanced Aβ degradation based on an elevated level of IDE and neprilysin. The total AβPP level was not altered by XST treatment. Prior studies using the major components of XST have focused on the production of Aβ, and have revealed a reduction in level of BACE1 by Ginsenoside Rg1 [45, 46] and by Ginsenoside Re [49]. In addition, Quan et al. [50] also found Ginsenoside Rg1 increased IDE level in a rat AD model. We have tested a wider range of key molecules involved in Aβ production and clearance and have demonstrated that XST treatment directly alters the metabolism of Aβ, including both production and degradation. Note that the majority of prior studies on Ginsenosides were performed on cultured cells, while our results were obtained from AD transgenic mice, and have thus extended the previous findings to animal models of AD.
Enhanced cerebral blood flow by XST and its importance in treating AD
Patients with MCI show decreased resting CBF [17, 65], suggesting that reduced CBF may occur early in AD. Activity-induced CBF response is impaired in AD patients [66] and APP/PS1 mice [67, 68], and this alteration may occur prior to neurodegenerative changes [19, 69–71]. Vascular dysfunction may also influence the amyloid pathway to reduce Aβ clearance and to increase Aβ production, which result in an elevated brain Aβ level [11]. In addition to enzymatic degradation, Aβ is cleared from the brain by transcytosis across the blood-brain barrier, uptake via microglia, and through perivascular drainage with interstitial fluid (ISF) [72]. The efficacy of the ISF drainage system in Aβ clearance may depend on the functional status of cerebral vasculature and CBF [24]. Reduced perivascular clearance of Aβ was reported in Aβ−overexpressing mice, and ISF drainage requires a functioning vascular system since impaired cerebral perfusion leads to reduced solute clearance [73]. Reduced CBF (such as during a stroke) leads to reduced ISF clearance [73, 74]. Thus, restoring CBF may be beneficial in treating AD by elevating Aβ clearance via the ISF drainage system. Future study can examine whether XST improves ISF clearance of Aβ.
One interesting question to consider is the causal relationship among the above reported changes as whether one process could serve as a trigger for the others. A clear demonstration of this relationship is rather difficult. Hence, we have thus asked a simple question: does change in one parameter occur at an earlier time point than the others? The assumption is that events occur earlier after XST injection are likely upstream of, or might even be a cause to, the events occur at later time points. For the parameters that we have examined in this study, we have decided to focus on changes in CBF, expression of a few key proteins related to Aβ-processing and plaque density. Since behavioral performance takes a long time to test, it cannot be completed in a short time frame (within a day) and hence cannot be used for this comparison. Starting at 8 h after XST injection, we have found significant elevation in CBF, but no changes in Aβ-related processing or amyloid plaques. Hence, this simple analysis suggests that elevated CBF is an early event and might drive the occurrence of the other changes associated with XST injection. Alternatively, these processes are independent of each other and may also have distinct time courses. More experiments will be required to distinguish between these two possibilities.
XST is extracted from the widely used traditional Chinese medicinal herb Panax notoginseng, and is reported to correct endothelium dysfunction in both cellular and in vivo studies [75, 76]. In clinical settings, XST has been reported to be beneficial in treating acute ischemic stroke [77]. Numerous beneficial effects of XST on the cerebrovascular system have been demonstrated, including the prevention of neuroexcitotoxicity [78], inhibiting vascular smooth muscle cell proliferation [33], inhibiting inflammation-induced monocyte adhesion and expression of endothelial adhesion molecules [79]. In addition to its strong effect in modulating/enhancing CBF, other effects (such as modulating inflammatory responses) have been reported to occur during XST treatment; these processes may contribute to the benefits we have observed. The XST used in our study has been used widely in clinical settings in China for decades, with 5 known major components. Studies using some of the major components have been conducted, and they have demonstrated efficacy on improving blood flow, reducing inflammation and oxidative stress [80–84]. Future investigation of whether the beneficial effects that we have observed here is mediated by one or a few of the major components will be interesting and informative.
An easy and effective way to elevate blood flow is exercise. It is interesting to note that exercise has been shown effective in reducing cognitive decline in AD and MCI patients, and non-demented persons. A few studies revealed that regular physical activity was associated with a significantly reduced risk of AD and dementia, and better performance in neuropsychological testing [85–88]. Six randomized controlled trials showed a decreased rate of cognitive decline and positive effects on global cognitive function in patients with AD who participated in an exercise program [89]. In addition, in the APP/PS1 mice, treadmill running improved hippocampus-based memory (contextual fear memory) and the soluble Aβ level [90], or reduce Aβ deposition and tau phosphorylation [91]. Therefore, XST may be viewed as an exercise pill.
In summary, although it is being increasingly recognized that altered cerebral vascular functions may have critical contributions to the genesis and pathological progression of AD, this topic has not caught much attention compared to amyloid pathology and neuroinflammation. It is also important to note that the altered vascular processes appear to occur in the early stages of AD. In this context, enhancing CBF may have significant therapeutic potential. This, however, has not been tested directly or systematically in AD patients or AD animal models. Our study is one step in this direction. The bigger question being raised by our study is whether enhancing certain basic body functions (such as CBF) rather than targeting an AD-specific pathology (such as Aβ) can serve as an effective AD treatment. In a broader sense, is enhancing the body/brain’s innate ability to cope with pathological conditions, be it AD, stroke or epilepsy, sufficient or potentially more effective than target-based therapy? A similar debate is on-going in cancer therapy in that whether enhancing the body’s own immune capacity is ultimately a better and safer treatment option. For AD, and neurodegenerative diseases in general, the answer awaits us. Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/17-0763r2).
ACKNOWLEDGMENTS
This work is supported by grants from Guangxi Wuzhou Zhongheng Group Co., Ltd. and Shenzhen Science and Technology Innovation Committee (JSGG20140703163838793, JCYJ20150529153646078, JCYJ20150629144658017, JCYJ20160428154156417, JCYJ20170306165021201, ZDSYS201504301539161, JCYJ20170412150845848). We thank Oliver Zhou for editorial assistance.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-170763.
REFERENCES
[1] | Cummings JL , Banks SJ , Gary RK , Kinney JW , Lombardo JM , Walsh RR , Zhong K ((2013) ) Alzheimer’s disease drug development: Translational neuroscience strategies. CNS Spectr 18: , 128–138. |
[2] | Panza F , Solfrizzi V , Imbimbo BP , Logroscino G ((2014) ) Amyloid-directed monoclonal antibodies for the treatment of Alzheimer’s disease: The point of no return?. Expert Opin Biol Ther 14: , 1465–1476. |
[3] | Selkoe DJ , Hardy J ((2016) ) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8: , 595–608. |
[4] | Spencer B , Masliah E ((2014) ) Immunotherapy for Alzheimer’s disease: Past, present and future. Front Aging Neurosci 6: , 114. |
[5] | Prins ND , Scheltens P ((2013) ) Treating Alzheimer’s disease with monoclonal antibodies: Current status and outlook for the future. Alzheimers Res Ther 5: , 56. |
[6] | Bloom GS ((2014) ) Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71: , 505–508. |
[7] | Zempel H , Mandelkow E ((2014) ) Lost after translation: Missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci 37: , 721–732. |
[8] | Frost B , Gotz J , Feany MB ((2015) ) Connecting the dots between tau dysfunction and neurodegeneration. Trends Cell Biol 25: , 46–53. |
[9] | Gruninger F ((2015) ) Invited review: Drug development for tauopathies. Neuropathol Appl Neurobiol 41: , 81–96. |
[10] | Altman R , Rutledge JC ((2010) ) The vascular contribution to Alzheimer’s disease. Clin Sci (Lond) 119: , 407–421. |
[11] | Zlokovic BV ((2011) ) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 12: , 723–738. |
[12] | Grammas P ((2011) ) Neurovascular dysfunction, inflammation and endothelial activation: Implications for the pathogenesis of Alzheimer’s disease. J Neuroinflammation 8: , 26. |
[13] | Murray IV , Proza JF , Sohrabji F , Lawler JM ((2011) ) Vascular and metabolic dysfunction in Alzheimer’s disease: A review. Exp Biol Med (Maywood) 236: , 772–782. |
[14] | Iadecola C ((2013) ) The pathobiology of vascular dementia. Neuron 80: , 844–866. |
[15] | Canobbio I , Abubaker AA , Visconte C , Torti M , Pula G ((2015) ) Role of amyloid peptides in vascular dysfunction and platelet dysregulation in Alzheimer’s disease. Front Cell Neurosci 9: , 65. |
[16] | de la Torre JC ((2002) ) Alzheimer disease as a vascular disorder: Nosological evidence. Stroke 33: , 1152–1162. |
[17] | Dai W , Lopez OL , Carmichael OT , Becker JT , Kuller LH , Gach HM ((2009) ) Mild cognitive impairment and Alzheimer disease: Patterns of altered cerebral blood flow at MR imaging. Radiology 250: , 856–866. |
[18] | Iadecola C ((2010) ) The overlap between neurodegenerative and vascular factors in the pathogenesis of dementia. Acta Neuropathol 120: , 287–296. |
[19] | Knopman DS , Roberts R ((2010) ) Vascular risk factors: Imaging and neuropathologic correlates. J Alzheimers Dis 20: , 699–709. |
[20] | Benzinger TL , Blazey T , Jack CR Jr , Koeppe RA , Su Y , Xiong C , Raichle ME , Snyder AZ , Ances BM , Bateman RJ , Cairns NJ , Fagan AM , Goate A , Marcus DS , Aisen PS , Christensen JJ , Ercole L , Hornbeck RC , Farrar AM , Aldea P , Jasielec MS , Owen CJ , Xie X , Mayeux R , Brickman A , McDade E , Klunk W , Mathis CA , Ringman J , Thompson PM , Ghetti B , Saykin AJ , Sperling RA , Johnson KA , Salloway S , Correia S , Schofield PR , Masters CL , Rowe C , Villemagne VL , Martins R , Ourselin S , Rossor MN , Fox NC , Cash DM , Weiner MW , Holtzman DM , Buckles VD , Moulder K , Morris JC ((2013) ) Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease.E. Proc Natl Acad Sci U S A 110: , 4502–4509. |
[21] | Binnewijzend MA , Kuijer JP , Benedictus MR , van der Flier WM , Wink AM , Wattjes MP , van Berckel BN , Scheltens P , Barkhof F ((2013) ) Cerebral blood flow measured with 3D pseudocontinuous arterial spin-labeling MR imaging in Alzheimer disease and mild cognitive impairment: A marker for disease severity. Radiology 267: , 221–230. |
[22] | Chui HC , Zarow C , Mack WJ , Ellis WG , Ling ZM , Jagust WJ , Dan M , Reed BR , Kramer JH , Decarli CC ((2006) ) Cognitive impact of subcortical vascular and Alzheimer’s disease pathology. Ann Neurol 60: , 677–687. |
[23] | Iadecola C ((2004) ) Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci 5: , 347–360. |
[24] | Weller RO , Massey A , Newman TA , Hutchings M , Kuo YM , Roher AE ((1998) ) Cerebral amyloid angiopathy: Amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am J Pathol 153: , 725–733. |
[25] | Zerbi V , Jansen D , Wiesmann M , Fang X , Broersen LM , Veltien A , Heerschap A , Kiliaan AJ ((2014) ) Multinutrient diets improve cerebral perfusion and neuroprotection in a murine model of Alzheimer’s disease. Neurobiol Aging 35: , 600–613. |
[26] | Gui Q , Yang Y , Ying S , Zhang M ((2013) ) Xueshuantong improves cerebral blood perfusion in elderly patients with lacunar infarction. Neural Regen Res 8: , 792–801. |
[27] | Pan C , Huo Y , An X , Singh G , Chen M , Yang Z , Pu J , Li J ((2012) ) Panax notoginseng and its components decreased hypertension via stimulation of endothelial-dependent vessel dilatation. Vascul Pharmacol 56: , 150–158. |
[28] | Garcia-Alloza M , Robbins EM , Zhang-Nunes SX , Purcell SM , Betensky RA , Raju S , Prada C , Greenberg SM , Bacskai BJ , Frosch MP ((2006) ) Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis 24: , 516–524. |
[29] | Schaffer CB , Friedman B , Nishimura N , Schroeder LF , Tsai PS , Ebner FF , Lyden PD , Kleinfeld D ((2006) ) Two-photon imaging of cortical surface microvessels reveals a robust redistribution in blood flow after vascular occlusion. PLoS Biol 4: , e22. |
[30] | Chaigneau E , Oheim M , Audinat E , Charpak S ((2003) ) Two-photon imaging of capillary blood flow in olfactory bulb glomeruli. Proc Natl Acad Sci U S A 100: , 13081–13086. |
[31] | Klunk WE , Bacskai BJ , Mathis CA , Kajdasz ST , McLellan ME , Frosch MP , Debnath ML , Holt DP , Wang Y , Hyman BT ((2002) ) Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol 61: , 797–805. |
[32] | Roehl AB , Zoremba N , Kipp M , Schiefer J , Goetzenich A , Bleilevens C , Kuehn-Velten N , Tolba R , Rossaint R , Hein M ((2012) ) The effects of levosimendan on brain metabolism during initial recovery from global transient ischaemia/hypoxia. BMC Neurol 12: , 81. |
[33] | Zhang W , Chen G , Deng CQ ((2012) ) Effects and mechanisms of total Panax notoginseng saponins on proliferation of vascular smooth muscle cells with plasma pharmacology method. J Pharm Pharmacol 64: , 139–145. |
[34] | Tsai J , Grutzendler J , Duff K , Gan WB ((2004) ) Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci 7: , 1181–1183. |
[35] | Spires TL , Meyer-Luehmann M , Stern EA , McLean PJ , Skoch J , Nguyen PT , Bacskai BJ , Hyman BT ((2005) ) Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci 25: , 7278–7287. |
[36] | Hanson JE , Meilandt WJ , Gogineni A , Reynen P , Herrington J , Weimer RM , Scearce-Levie K , Zhou Q ((2014) ) Chronic GluN2B antagonism disrupts behavior in wild-type mice without protecting against synapse loss or memory impairment in Alzheimer’s disease mouse models. J Neurosci 34: , 8277–8288. |
[37] | Hu NW , Smith IM , Walsh DM , Rowan MJ ((2008) ) Soluble amyloid-beta peptides potently disrupt hippocampal synaptic plasticity in the absence of cerebrovascular dysfunction in vivo. Brain 131: , 2414–2424. |
[38] | Hefti F , Goure WF , Jerecic J , Iverson KS , Walicke PA , Krafft GA ((2013) ) The case for soluble Abeta oligomers as a drug target in Alzheimer’s disease. Trends Pharmacol Sci 34: , 261–266. |
[39] | Viola KL , Klein WL ((2015) ) Amyloid beta oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol 129: , 183–206. |
[40] | Kayed R , Head E , Sarsoza F , Saing T , Cotman CW , Necula M , Margol L , Wu J , Breydo L , Thompson JL , Rasool S , Gurlo T , Butler P , Glabe CG ((2007) ) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener 2: , 18. |
[41] | Tomic JL , Pensalfini A , Head E , Glabe CG ((2009) ) Soluble fibrillar oligomer levels are elevated in Alzheimer’s disease brain and correlate with cognitive dysfunction. Neurobiol Dis 35: , 352–358. |
[42] | Krishnan R , Goodman JL , Mukhopadhyay S , Pacheco CD , Lemke EA , Deniz AA , Lindquist S ((2012) ) Conserved features of intermediates in amyloid assembly determine their benign or toxic states. Proc Natl Acad Sci U S A 109: , 11172–11177. |
[43] | Selkoe DJ ((2001) ) Alzheimer’s disease: Genes, proteins, and therapy. Physiol Rev 81: , 741–766. |
[44] | De Strooper B ((2010) ) Proteases and proteolysis in Alzheimer disease: A multifactorial view on the disease process. Physiol Rev 90: , 465–494. |
[45] | Wang YH , Du GH ((2009) ) Ginsenoside Rg1 inhibits beta-secretase activity in vitro and protects against Abeta-induced cytotoxicity in PC12 cells. J Asian Nat Prod Res 11: , 604–612. |
[46] | Chen LM , Lin ZY , Zhu YG , Lin N , Zhang J , Pan XD , Chen XC ((2012) ) Ginsenoside Rg1 attenuates beta-amyloid generation via suppressing PPARgamma-regulated BACE1 activity in N2a-APP695 cells. Eur J Pharmacol 675: , 15–21. |
[47] | Fang F , Chen X , Huang T , Lue LF , Luddy JS , Yan SS ((2012) ) Multi-faced neuroprotective effects of Ginsenoside Rg1 in an Alzheimer mouse model. Biochim Biophys Acta 1822: , 286–292. |
[48] | Yan X , Hu G , Yan W , Chen T , Yang F , Zhang X , Zhao G , Liu J ((2017) ) Ginsenoside Rd promotes non-amyloidogenic pathway of amyloid precursor protein processing by regulating phosphorylation of estrogen receptor alpha. Life Sci 168: , 16–23. |
[49] | Cao G , Su P , Zhang S , Guo L , Zhang H , Liang Y , Qin C , Zhang W ((2016) ) Ginsenoside Re reduces Abeta production by activating PPARgamma to inhibit BACE1 in N2a/APP695 cells. Eur J Pharmacol 793: , 101–108. |
[50] | Quan Q , Wang J , Li X , Wang Y ((2013) ) Ginsenoside Rg1 decreases Abeta(1-42) level by upregulating PPARgamma and IDE expression in the hippocampus of a rat model of Alzheimer’s disease. PLoS One 8: , e59155. |
[51] | Hayashi-Takagi A , Yagishita S , Nakamura M , Shirai F , Wu YI , Loshbaugh AL , Kuhlman B , Hahn KM , Kasai H ((2015) ) Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature 525: , 333–338. |
[52] | Terry RD , Masliah E , Salmon DP , Butters N , DeTeresa R , Hill R , Hansen LA , Katzman R ((1991) ) Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann Neurol 30: , 572–580. |
[53] | Clare R , King VG , Wirenfeldt M , Vinters HV ((2010) ) Synapse loss in dementias. J Neurosci Res 88: , 2083–2090. |
[54] | Jacobsen JS , Wu CC , Redwine JM , Comery TA , Arias R , Bowlby M , Martone R , Morrison JH , Pangalos MN , Reinhart PH , Bloom FE ((2006) ) Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 103: , 5161–5166. |
[55] | D’Amelio M , Cavallucci V , Middei S , Marchetti C , Pacioni S , Ferri A , Diamantini A , De Zio D , Carrara P , Battistini L , Moreno S , Bacci A , Ammassari-Teule M , Marie H , Cecconi F ((2011) ) Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nat Neurosci 14: , 69–76. |
[56] | Ricobaraza A , Cuadrado-Tejedor M , Marco S , Perez-Otano I , Garcia-Osta A ((2012) ) Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus 22: , 1040–1050. |
[57] | Scheff SW , Price DA , Schmitt FA , DeKosky ST , Mufson EJ ((2007) ) Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 68: , 1501–1508. |
[58] | Lanz TA , Carter DB , Merchant KM ((2003) ) Dendritic spine loss in the hippocampus of young PDAPP and Tg2576 mice and its prevention by the ApoE2 genotype. Neurobiol Dis 13: , 246–253. |
[59] | Serrano-Pozo A , Betensky RA , Frosch MP , Hyman BT ((2016) ) Plaque-associated local toxicity increases over the clinical course of Alzheimer disease. Am J Pathol 186: , 375–384. |
[60] | Kaufman AC , Salazar SV , Haas LT , Yang J , Kostylev MA , Jeng AT , Robinson SA , Gunther EC , Van Dyck CH , Nygaard HB ((2015) ) Fyn inhibition rescues established memory and synapse loss in Alzheimer mice. Ann Neurol 77: , 953–971. |
[61] | An K , Jung JH , Jeong AY , Kim HG , Jung SY , Lee K , Kim HJ , Kim SJ , Jeong TY , Son Y ((2014) ) Neuritin can normalize neural deficits of Alzheimer’s disease. Cell Death Dis 5: , e1523. |
[62] | Zhou Q ((2014) ) GluN2B-NMDA receptors in Alzheimer’s disease: Beyond synapse loss and cell death. Neural Regen Res 9: , 1878–1879. |
[63] | Walsh DM , Selkoe DJ ((2007) ) A beta oligomers - a decade of discovery. J Neurochem 101: , 1172–1184. |
[64] | Koffie RM , Meyer-Luehmann M , Hashimoto T , Adams KW , Mielke ML , Garcia-Alloza M , Micheva KD , Smith SJ , Kim ML , Lee VM , Hyman BT , Spires-Jones TL ((2009) ) Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A 106: , 4012–4017. |
[65] | Lacalle-Aurioles M , Mateos-Perez JM , Guzman-De-Villoria JA , Olazaran J , Cruz-Orduna I , Aleman-Gomez Y , Martino ME , Desco M ((2014) ) Cerebral blood flow is an earlier indicator of perfusion abnormalities than cerebral blood volume in Alzheimer’s disease. J Cereb Blood Flow Metab 34: , 654–659. |
[66] | Mentis MJ , Alexander GE , Krasuski J , Pietrini P , Furey ML , Schapiro MB , Rapoport SI ((1998) ) Increasing required neural response to expose abnormal brain function in mild versus moderate or severe Alzheimer’s disease: PET study using parametric visual stimulation. Am J Psychiatry 155: , 785–794. |
[67] | Nicolakakis N , Aboulkassim T , Ongali B , Lecrux C , Fernandes P , Rosa-Neto P , Tong XK , Hamel E ((2008) ) Complete rescue of cerebrovascular function in aged Alzheimer’s disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor gamma agonist. J Neurosci 28: , 9287–9296. |
[68] | Niwa K , Kazama K , Younkin L , Younkin SG , Carlson GA , Iadecola C ((2002) ) Cerebrovascular autoregulation is profoundly impaired in mice overexpressing amyloid precursor protein. Am J Physiol Heart Circ Physiol 283: , H315–323. |
[69] | Ruitenberg A , den Heijer T , Bakker SL , van Swieten JC , Koudstaal PJ , Hofman A , Breteler MM ((2005) ) Cerebral hypoperfusion and clinical onset of dementia: The Rotterdam Study. Ann Neurol 57: , 789–794. |
[70] | Smith CD , Andersen AH , Kryscio RJ , Schmitt FA , Kindy MS , Blonder LX , Avison MJ ((1999) ) Altered brain activation in cognitively intact individuals at high risk for Alzheimer’s disease. Neurology 53: , 1391–1396. |
[71] | Bookheimer SY , Strojwas MH , Cohen MS , Saunders AM , Pericak-Vance MA , Mazziotta JC , Small GW ((2000) ) Patterns of brain activation in people at risk for Alzheimer’s disease. N Engl J Med 343: , 450–456. |
[72] | Bakker EN , Bacskai BJ , Arbel-Ornath M , Aldea R , Bedussi B , Morris AW , Weller RO , Carare RO ((2016) ) Lymphatic clearance of the brain: Perivascular, paravascular and significance for neurodegenerative diseases. Cell Mol Neurobiol 36: , 181–194. |
[73] | Arbel-Ornath M , Hudry E , Eikermann-Haerter K , Hou S , Gregory JL , Zhao L , Betensky RA , Frosch MP , Greenberg SM , Bacskai BJ ((2013) ) Interstitial fluid drainage is impaired in ischemic stroke and Alzheimer’s disease mouse models. Acta Neuropathol 126: , 353–364. |
[74] | Garcia-Alloza M , Gregory J , Kuchibhotla KV , Fine S , Wei Y , Ayata C , Frosch MP , Greenberg SM , Bacskai BJ ((2011) ) Cerebrovascular lesions induce transient beta-amyloid deposition. Brain 134: , 3697–3707. |
[75] | Wan JB , Lee SM , Wang JD , Wang N , He CW , Wang YT , Kang JX ((2009) ) Panax notoginseng reduces atherosclerotic lesions in ApoE-deficient mice and inhibits TNF-alpha-induced endothelial adhesion molecule expression and monocyte adhesion. J Agric Food Chem 57: , 6692–6697. |
[76] | Wang N , Wan JB , Chan SW , Deng YH , Yu N , Zhang QW , Wang YT , Lee SM ((2011) ) Comparative study on saponin fractions from Panax notoginseng inhibiting inflammation-induced endothelial adhesion molecule expression and monocyte adhesion. Chin Med 6: , 37. |
[77] | Gong X , Shen JH , Guo-Ping WU ((2009) ) Effect of Xueshuantong injection on cerebral infarction. Hainan Yixueyuan Xuebao 15: , 603–604. |
[78] | Luo FC , Wang SD , Li K , Nakamura H , Yodoi J , Bai J ((2010) ) Panaxatriol saponins extracted from Panax notoginseng induces thioredoxin-1 and prevents 1-methyl-4-phenylpyridinium ion-induced neurotoxicity. J Ethnopharmacol 127: , 419–423. |
[79] | Luo FC , Wang SD , Qi L , Song JY , Lv T , Bai J ((2011) ) Protective effect of panaxatriol saponins extracted from Panax notoginseng against MPTP-induced neurotoxicity in vivo. J Ethnopharmacol 133: , 448–453. |
[80] | Sun K , Wang CS , Guo J , Horie Y , Fang SP , Wang F , Liu YY , Liu LY , Yang JY , Fan JY , Han JY ((2007) ) Protective effects of ginsenoside Rb1, ginsenoside Rg1, and notoginsenoside R1 on lipopolysaccharide-induced microcirculatory disturbance in rat mesentery. Life Sci 81: , 509–518. |
[81] | Sun XC , Ren XF , Chen L , Gao XQ , Xie JX , Chen WF ((2016) ) Glucocorticoid receptor is involved in the neuroprotective effect of ginsenoside Rg1 against inflammation-induced dopaminergic neuronal degeneration in substantia nigra. J Steroid Biochem Mol Biol 155: , 94–103. |
[82] | Ye J , Yao JP , Wang X , Zheng M , Li P , He C , Wan JB , Yao X , Su H ((2016) ) Neuroprotective effects of ginsenosides on neural progenitor cells against oxidative injury. Mol Med Rep 13: , 3083–3091. |
[83] | Zheng X , Liang Y , Kang A , Ma SJ , Xing L , Zhou YY , Dai C , Xie H , Xie L , Wang GJ , Hao HP ((2014) ) Peripheral immunomodulation with ginsenoside Rg1 ameliorates neuroinflammation-induced behavioral deficits in rats. Neuroscience 256: , 210–222. |
[84] | Zhu J , Jiang Y , Wu L , Lu T , Xu G , Liu X ((2012) ) Suppression of local inflammation contributes to the neuroprotective effect of ginsenoside Rb1 in rats with cerebral ischemia. Neuroscience 202: , 342–351. |
[85] | Hamer M , Chida Y ((2009) ) Physical activity and risk of neurodegenerative disease: A systematic review of prospective evidence. Psychol Med 39: , 3–11. |
[86] | Sattler C , Erickson KI , Toro P , Schroder J ((2011) ) Physical fitness as a protective factor for cognitive impairment in a prospective population-based study in Germany. J Alzheimers Dis 26: , 709–718. |
[87] | Buchman AS , Boyle PA , Yu L , Shah RC , Wilson RS , Bennett DA ((2012) ) Total daily physical activity and the risk of AD and cognitive decline in older adults. Neurology 78: , 1323–1329. |
[88] | Strohle A , Schmidt DK , Schultz F , Fricke N , Staden T , Hellweg R , Priller J , Rapp MA , Rieckmann N ((2015) ) Drug and exercise treatment of Alzheimer disease and mild cognitive impairment: A systematic review and meta-analysis of effects on cognition in randomized controlled trials. Am J Geriatr Psychiatry 23: , 1234–1249. |
[89] | Farina N , Rusted J , Tabet N ((2014) ) The effect of exercise interventions on cognitive outcome in Alzheimer’s disease: A systematic review. Int Psychogeriatr 26: , 9–18. |
[90] | Lin TW , Shih YH , Chen SJ , Lien CH , Chang CY , Huang TY , Chen SH , Jen CJ , Kuo YM ((2015) ) Running exercise delays neurodegeneration in amygdala and hippocampus of Alzheimer’s disease (APP/PS1) transgenic mice. Neurobiol Learn Mem 118: , 189–197. |
[91] | Liu HL , Zhao G , Zhang H , Shi LD ((2013) ) Long-term treadmill exercise inhibits the progression of Alzheimer’s disease-like neuropathology in the hippocampus of APP/PS1 transgenic mice. Behav Brain Res 256: , 261–272. |


