
Dissecting the Role of 5-Lipoxygenase in the Homocysteine-Induced Alzheimer’s Disease Pathology
Abstract
Alzheimer’s disease (AD) affects over 40 million patients around the world and poses a huge economic burden on society since no effective therapy is available yet. While the cause(s) for the most common sporadic form of the disease are still obscure, lifestyle and different environmental factors have emerged as modulators of AD susceptibility. Hyperhomocysteinemia (HHCY), a condition of high circulating levels of homocysteine, is an independent but modifiable risk factor for AD. Studies in AD mouse models have linked HHCY with memory impairment, amyloidosis, tau pathology, synaptic dysfunction, and neuroinflammation. However, the exact mechanism by which HHCY affects AD pathogenesis is unclear. The 5-lipoxygenase (5LO) is a protein upregulated in postmortem AD brains and plays a functional role in AD pathogenesis. Recently, in vitro and in vivo studies showed that HHCY effects on amyloid-β and tau pathology, synapse and memory impairments are dependent on the activation of the 5LO enzymatic pathway, since its genetic absence or pharmacological inhibition prevents them. HHCY induces 5LO gene upregulation by lowering the methylation of its promoter, which results in increased translation and transcription of its mRNA. Based on these findings, we propose that epigenetic modification of 5LO represents the missing biological link between HHCY and AD pathogenesis, and for this reason it represents a viable therapeutic target to prevent AD development in individuals bearing this risk factor.
INTRODUCTION
Alzheimer’s disease (AD) is the first cause of dementia and fifth cause of death among the elderly accounting for over 40 million patients around the world and posing a huge economic burden on the entire society. With the aging of the general population, AD incidence and its related costs are predicted to increase drastically in the next 30 years [1]. Despite extensive research effort, no effective therapy isavailable to block or slow down AD pathogenesis leaving millions of patients with no hope. AD can be classified in two main forms: familial early-onset (FAD) and sporadic late-onset AD. While the causes for the most common sporadic form of the disease are still completely obscure, in recent years a lot of attention has been focused on an individual’s lifestyle and environmental factors as functional and active players in modulating the susceptibility for sporadic AD. Epidemiological studies have reported that elevated levels of circulating homocysteine, also known as hyperhomocysteinemia (HHCY), are strongly correlated with AD development later in life. Prospective studies on AD patients have identified HHCY as a contributing risk factor to AD pathogenesis rather than a biomarker for AD susceptibility [2]. Pre-clinical studies using transgenic mouse models of AD have linked HHCY with memory impairment, amyloidosis, tau pathology, synaptic dysfunction, and neuroinflammation. However, the exact mechanism by which HHCY affects AD pathogenesis remains unclear.
The 5-lipoxygenase (5LO) is a protein enzyme widely distributed in the central nervous system (CNS), which is upregulated in postmortem brain of AD patients and plays a functional role in AD pathogenesis [3, 4]. In particular, 5LO promotes cognitive deficits and affects AD pathophysiology by promoting both amyloid and tau pathology via γ-secretase and cyclin-dependent kinase 5 (CDK-5) mechanisms, respectively [5].
Recent findings demonstrate that HHCY promotes 5LO enzymatic pathway upregulation by diminishing methylation of its promoter [6]. In vivo and in vitro studies have shown that HHCY promotes memory decline, amyloidosis, tau pathology, synaptic dysfunction, and neuroinflammation via a 5LO-dependent mechanism [7]. We hypothesize that 5LO is the key mechanism mediating HHCY-induced effects on neurodegenerative processes which are of functional importance for AD pathogenesis. This new information opens new therapeutic avenues viable for preventing and/or halting the effect of HHCY on AD development.
ALZHEIMER’S DISEASE
AD is an irreversible neurodegenerative disorder characterized by progressive accumulation of misfolded proteins in the brain and neuronal cell death that ultimately lead to memory loss and cognitive dysfunction. The principal hallmarks of AD are extracellular deposits of amyloid-β (Aβ) peptides, also known as Aβ plaques, and intracellular accumulation of hyperphosphorylated tau protein (tau neurofibrillary tangles) in specific brain areas important for memory and learning functions [8].
Aβ is formed by the cleavage of amyloid-β protein precursor (AβPP) by either α- or β-secretase enzyme followed by an additional cleavage via the γ-secretase. The α-secretase promotes non-amyloidogenic cleavage of AβPP while β-secretase promotes amyloidogenic cleavage of AβPP. Amyloidogenic cleavage of AβPP leads to Aβ formation and release in the extracellular compartment [9]. While in the healthy brain, the two pathways are in balance, in AD the amyloidogenic pathway prevails leading to Aβ accumulation as oligomers, fibrils, and finally fully developed plaques. Tau is a microtubule associated protein that normally binds to tubulin to promote microtubule stability. In the axon, tau protein is a crucial component of the cytoskeleton promoting structural stability and healthy axonal transport [10]. In AD, tau protein is hyperphosphorylated due to increased activity of specific pathogenic kinases such as CDK5 and GSK3β, or a reduction in the activity of phosphatases such as PP2A, which are involved in tau de-phosphorylation. Hyperphosphorylated tau loses affinity for the microtubules and form paired helical filaments that constitute the core of tau neurofibrillary tangles, and as result axonal structure and transport becomes severely compromised [11–13].
HOMOCYSTEINE METABOLISM
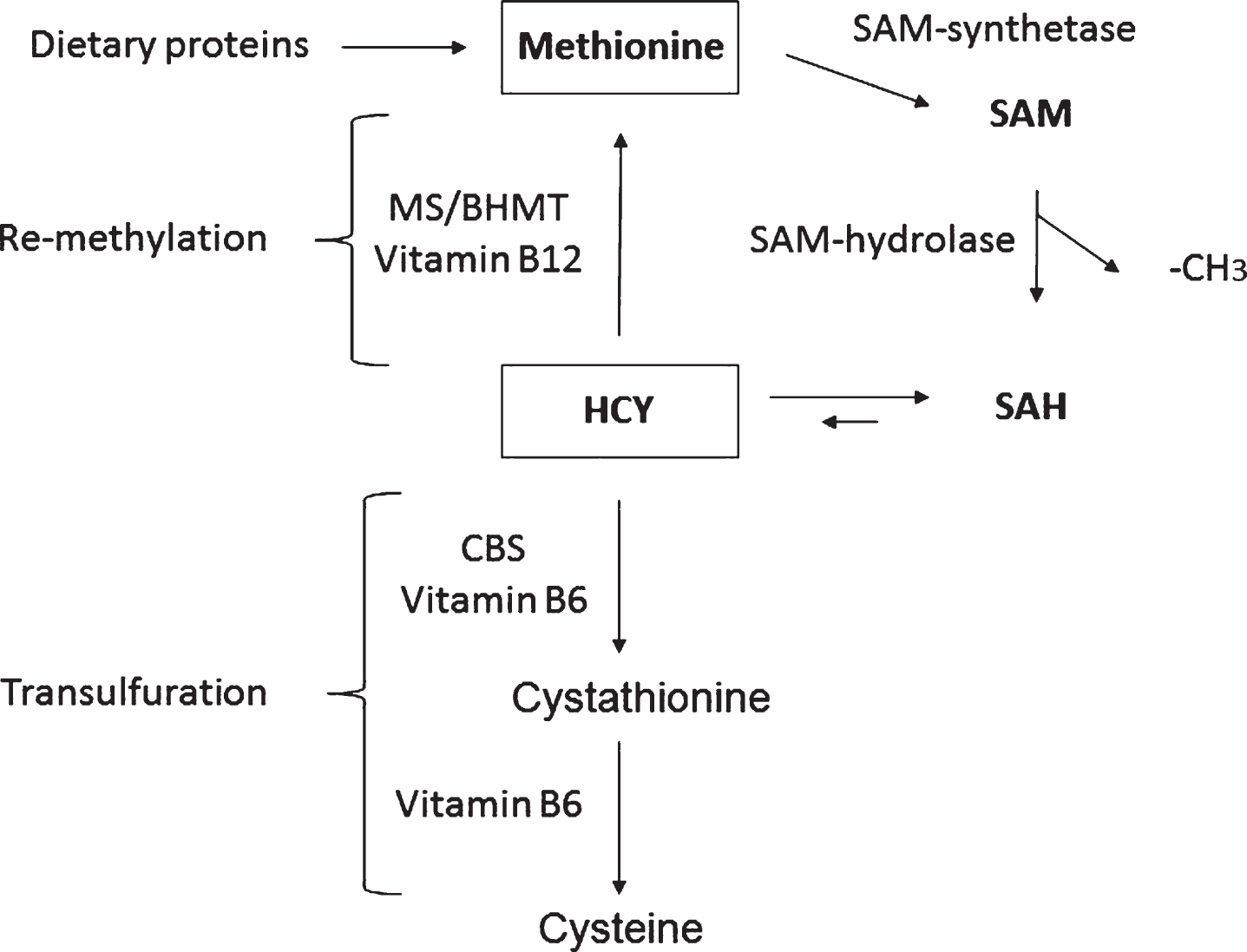
Homocysteine (HCY) is a non-essential sulfur containing amino acid produced from methionine as part of the methionine recycling pathway (Fig. 1) [14]. In this pathway, methionine is first converted to S-adenosylmethionine (SAM) by the action of SAM synthase, which condenses ATP with methionine to form SAM. SAM, in turn, is the key methyl donor in almost all biological methylation reactions. As a consequence of the methyl group donation, SAM is converted to S-adenosylhomocysteine (SAH), which in turn is acted upon by SAH hydrolase to yield HCY and adenosine (Fig. 1). It should be noticed that this reaction is entirely reversible, but it is generally driven in the forward direction in vivo due to the low concentration of adenosine and HCY inside the cell. The resulting HCY molecule has two potential metabolic fates, either re-methylation to form methionine, or transfulfuration to form cystathionine and eventually cysteine to be eliminated in the urine (Fig. 1). The re-methylation reaction is carried out by the methionine synthase (MS) and betaine-dependent Hcy methyltransferase (BHMT), enzymes requiring vitamin B12 in the form of methyl cobalamin, while the transulfuration reaction is catalyzed by cystathionine β-synthase (CBS) which uses vitamin B6 as cofactor. Because of the cyclic nature of the recycling pathway, CBS is the rate limiting enzyme in the catabolism of HCY [15]. Compared with other organs, HCY brain metabolism is peculiar since it is strictly dependent on folate and cobalaminsupplies [14].
Fig.1
The homocysteine/methionine cycle. S-adenosylmethionine (SAM), S-adenosylhomocysteine (SAH), homocysteine (HCY), methionine synthase (MS), HCY methyltransferase (BHMT), and cystathionine beta-synthase (CBS).
HYPERHOMOCYSTEINEMIA
Hyperhomocysteinemia (HHCY) is a condition characterized by high circulating levels of HCY. In humans, plasma HCY levels are usually between 5 and 15μM. Several degrees of HHCY can be classified based on its plasma concentration levels. In particular, moderate HHCY is characterized by plasma levels between 16 and 30μM, intermediate between 31 and 100μM, and severe if plasma concentration exceeds 100μM [14]. Several factors can lead to HHCY. Genetic deficiency in the enzymes involved in HCY metabolism as well as a diet scarce in folate, vitamin B12 or B6 and finally aging can all promote HHCY [15]. HHCY is a known risk factor for several diseases including cardiovascular diseases and neurodegenerative diseases, including among others AD, Parkinson’s disease, and progressive supranuclear palsy [14–16].
HYPERHOMOCYSTEINEMIA IN ALZHEIMER’S DISEASE
The first report of high HCY levels in AD dates back to Regland’s study done in the 1990s that documented elevated HCY levels in 22 primary degenerative dementia patients [2]. Since then, retrospective studies have confirmed the association between HHCY levels and AD. However, no changes were found in cerebrospinal fluid HCY levels between AD patients and healthy controls [17, 18]. To better understand whether HHCY is cause or effect of AD, several prospective studies have been performed on normal subjects, mild cognitive impairment (MCI) subjects, and AD patients. These studies have concluded that HHCY precedes AD development, correlates with cognitive decline and may predict the evolution of MCI to AD [19, 20]. Based on this evidence, HHCY seems to be the cause rather than consequence of AD pathogenesis. However, no interventional study utilizing HCY lowering therapies or folate supplementation has shown consistent success in terms of blocking AD pathogenesis [21, 22]. Although we do not have a clear answer to interpret these data, below are some general considerations that attempt to reconcile the discrepancy between observational and interventional studies. It is possible that better designed clinical trials are required to prove whether or not there is a cause-effect relationship between HHCY and AD. For instance, some of the published studies have recruited individuals with HCY levels in the normal range for the particular age of the population. Future prevention studies with HCY-reducing strategies should recruit subjects with actual HHCY and treat them for an appropriate length of time. Implementation of better and more appropriate cognitive tests, particularly the ones focusing on memory and information-processing speed, should be included in any future HHCY-lowering study on AD patients. Finally, a recent hypothesis (see below) postulates that HHCY is only a marker of altered cell methylation potential, which by affecting the expression of particular genes ultimately promotes mechanisms of importance for AD pathogenesis. Thus, folic acid supplementation, despite reducing plasma HCY levels, would not influence cell methylation levels and, actually, by facilitating methionine synthesis, could paradoxically even exacerbate hypomethylativereactions.
Numerous studies have been conducted in transgenic mouse models of AD with the main goal to investigate the mechanism(s) whereby HHCY modulates AD pathogenesis. In particular, crossing a transgenic mouse model of AD-like amyloidosis with a mouse model of CBS gene deficiency resulted in an exacerbation of Aβ pathology [23]. Several studies demonstrated that low folate diet exacerbates amyloid pathology, tau hyperphosphorylation synaptic dysfunction, and neuroinflammation in other transgenic mouse models of AD [24]. Moreover, low folate diet promoted cerebral amyloid angiopathy in the same mouse model of AD [25].
THE 5-LIPOXYGENASE ENZYMATIC PATHWAY
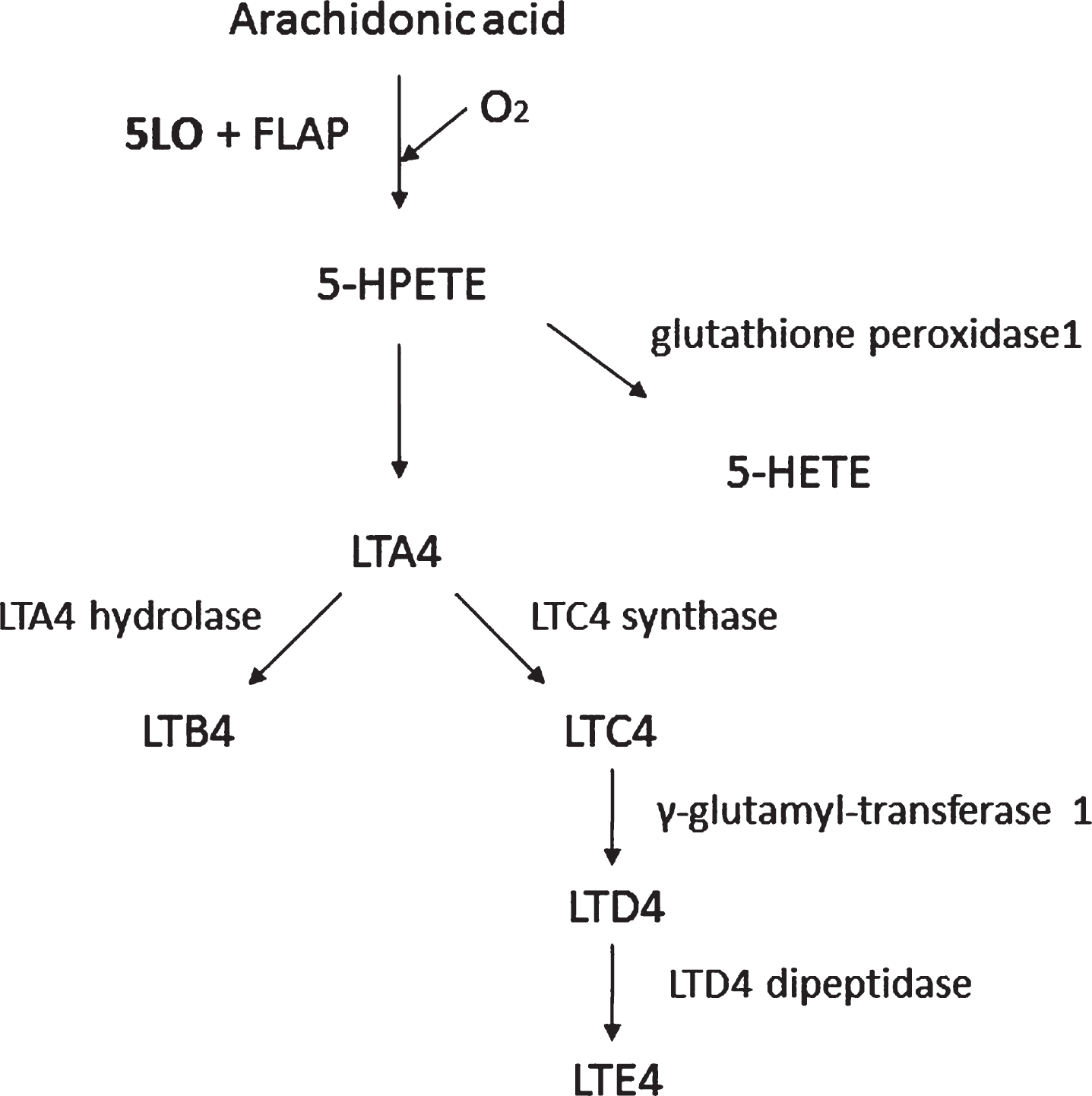
Lipoxygenases are monomeric non-heme iron-containing enzymes that catalyze the dioxygenation of free and esterified polyunsaturated fatty acids into conjugated hydroperoxides, which are involved in several autocrine, paracrine and endocrine signaling mechanisms [26]. 5LO catalyzes the oxygenation on the carbon in position 5 of the arachidonic acid to form leukotrienes with the aid of 5LO activating protein (FLAP) [27] (Fig. 2). The5-hydroperoxyeicosatetraenoic acid (5-HPETE) is the first product of 5LO action on its substrate that is later converted to 5-hydroxyeicosatetraenoic acid (5-HETE) or leukotriene A4 (LTA4). LTA4 can be metabolized either to leukotriene B4 or C4 (LTB4 or LTC4), with LTC4 further being metabolized to LTD4 and LTE4, potent bioactive inflammatory lipid mediators [28].
Fig.2
The 5LO pathway. 5LO activating protein (FLAP), 5-hydro-peroxyeicosatetraenoic acid (5-HPETE), 5-hydroxyeicosatetra-enoic acid (5-HETE), leukotriene A4 (LTA4), leukotriene B4 (LTB4), leukotriene C4 (LTC4), leukotriene D4 (LTD4), and leukotriene E4 (LTE4). The 5LO converts arachidonic acid to 5-HPETE with the aid of FLAP. 5-HPETE is then converted to 5-HETE or LTA4. Finally, LTA4 can be metabolized via a hydrolase to LTB4 or a synthase to LTC4, which via the γ-glutamyl-transferase can ultimately be transformed into LTE4 by the action of a specific dipeptidase.
The 5LO is widely expressed in the cardiovascular system, that is, aorta, coronary, and carotid arteries, as well as in macrophages and neutrophils. Interestingly, its expression levels are increased in aortas of old animals when compared with young ones [29]. This enzymatic pathway is also widely expressed in the CNS, where it localizes mainly in neuronal cells of the hippocampus and cortex, and, similar to the vasculature, its levels increase significantly with aging [30, 31]. The expression of 5LO is susceptible to hormonal regulation, since higher levels are observed in conditions of melatonin deficiency and/or hyperglucocorticoidemia [32, 33], both of which are common in elderly subjects [34]. Although in general upregulation of 5LO might serve a physiological purpose, during the aging process, it may also increase the vulnerability of the cardiovascular system and CNS to different insults/stressors [35]. Given that older subjects are at greater risk of health complications and mortality stemming from altered inflammatory and immune functions, and aging, via the upregulation of 5-LO, can be an important risk factor, the effects of stressors on this enzymatic pathway are of particular importance.
5LO AND AD PATHOGENESIS
The 5LO is upregulated in postmortem brains of AD patients, and in a small study, polymorphisms of the 5LO gene have been associated with early- and late-onset AD [36]. In vitro studies have demonstrated that 5LO overexpression promotes Aβ formation via the γ-secretase pathway. On the other hand, 5LO knockdown or inhibition in the same in vitro system reduces expression of γ-secretase steady level components lowering Aβ production [37]. 5LO is also involved in regulating tau phosphorylation. Indeed, its overexpression in vitro promotes tau hyperphosphorylation via CDK-5 pathway. At the same time, 5LO knockdown or inhibition reduces tau phosphorylation in vitro by reducing CDK5 activation [38]. These results were confirmed in different transgenic mouse models of AD [39, 40]. In particular, 5LO knockout or pharmacologic inhibition mitigates the cognitive deficit and synaptic dysfunction in both Tg2576 and 3xTg mice, models of pure amyloidosis, and both amyloid and tau pathology, respectively [37–41]. In the same animals, 5LO knockdown or pharmacologic inhibition reduced amyloid burden, tau hyperphosphorylation, synaptic pathology, and neuroinflammation [37–41]. Finally, treatment of aged 3xTg animals with zileuton, a 5LO specific inhibitor, after the AD-like phenotype is well established rescued cognitive decline, amyloidosis, tau pathology, synaptic dysfunction, and neuroinflammation [5].
IS 5LO THE MISSING LINK BETWEEN HHCY AND AD?
Despite HHCY is a well-characterized modifiable risk factor for AD, the mechanism(s) by which HHCY promotes AD pathogenesis are not clear. Several hypotheses have been proposed such as increased oxidative stress, cerebrovascular damage, excitatory damage due to NMDA overstimulation and dysregulation of DNA methylation [2]. HHCY is associated with increased levels of SAH, an inhibitor of DNA methyltransferase enzymes, and depletion of SAM, a major methyl donor in the cell [42]. These changes represent the biochemical signature of a significant reduction of the methylation status within the cell. As result, HHCY has been associated with decreased DNA methylation of several genes involved in AD pathology such as β-secretase and presenilin 1, a major component of the γ-secretase complex [43, 44].
Interestingly, 5LO expression level is also tightly regulated by DNA methylation and demethylation of its own promoter, and in this sense, it is a prerequisite for its gene expression, protein synthesis, and enzymatic activity [45]. With this knowledge, we recently embarked in a series of studies aimed at investigating whether HCY activates the 5LO enzymatic pathway by influencing the methylation status of its promoter.
To this end, we implemented brains from mice with HHCY and neuronal cells exposed to HCY to study whether methylation was the mechanisms responsible for the HHCY-dependent 5LO expression and its effect on AD pathophysiology.
HHCY induced by either low folate diet, or CBS gene deficiency resulted in 5LO protein and mRNA upregulation, which was associated with a significant increase of SAH/SAM ratio, reduced DNA methyltransferase enzymes, and hypomethylation of 5LO DNA. In vitro studies confirmed these results and demonstrated that the mechanism involved in the HCY-dependent 5LO activation and Aβ formation is 5LO DNA hypomethylation secondary to the elevated levels of SAH [6]. To prove a causative role of the elevation of SAH for this novel biological effect on 5LO we set-up experiments with neuronal cells transiently overexpressing SAH hydrolase, the only known enzyme to catalyze the breakdown of SAH and effectively reducing its intracellular levels. These studies showed that SAH overexpressing cells were protected from the HHCY-dependent reduction of all three methyltransferase enzymes and subsequent 5LO DNA hypomethylation and its protein upregulation. Additionally, we showed that the effect of HCY on the 5LO enzymatic pathway was secondary to an increase in transcription of 5LO mRNA rather than an increase in its stability as it was blocked by actinomycin-D. Similarly, blockade of de novo protein synthesis by cycloheximide prevented the HHCY-dependent upregulation of 5LO and increase in Aβ formation [6].
More recently, we have demonstrated that essential role of the 5LO pathway in the deleterious effect that HHCY has also on AD pathogenesis in vivo [7, 46]. Triple transgenic mice (3xTg), which are known to develop both Aβ plaques and tau tangles in the brain, starting at 5 months of age were chronically treated with a diet deficient in folate to induce brain HHCY, or a diet low in folate but also administered with zileuton, a 5LO specific pharmacologic inhibitor, until they were 12 months old.
At this age timepoint, animals were tested for cognitive function, brain amyloidosis, tau pathology, synaptic integrity, and neuroinflammation. Compared with controls receiving regular diet, low folate diet treated animals showed a significant increase in brain HCY levels, which was associated with 5LO upregulation (mRNA, protein, enzyme activity) secondary to a significant reduction in the methylation status of its promoter. The same animals showed marked cognitive impairments when challenged in three different paradigms assessing working and spatial memory, and learning: Y-maze, fear conditioning, and Morris water maze tests. Moreover, low folate diet-induced HHCY promoted brain amyloidosis and tau hyperphosphorylation secondary to an activation of the γ-secretase and the CDK-5 pathways, respectively. In vitro studies confirmed the effect of HCY in promoting amyloidosis and tau phosphorylation and the mechanisms involved. Moreover, the effect of HHCY on tau phosphorylation was totally independent from the effect on Aβ since blockade of γ-secretase activity was not sufficient to prevent HCY-induced tau hyperphosphorylation [46]. Finally, animals with diet-induced brain HHCY expressed lower levels of synaptic integrity marker postsynaptic density protein 95 (PSD-95), and higher levels of GFAP, an astrocytosis marker. By contrast, low folate diet animals receiving at the same time zileuron (the 5LO specific inhibitor) despite the HHCY in their brains did not manifest behavioral impairments, and were indistinguishable from the controls in terms of brain amyloidosis, tau phosphorylation, synaptic integrity, and neuroinflammatory markers [7]. In vitro studies in neuronal cell lines confirmed that 5LO pharmacologic blockade was sufficient to prevent HCY-induced Aβ accumulation [7].
Similar results were obtained utilizing a purely genetic approach in wild type animals [47]. In particular, wild type and 5LO knockout animals were chronically treated with low folate diet or regular chow for 7 months. At 12 months of age, animals were tested for cognitive function, brain amyloidosis, tau pathology, synaptic integrity and neuroinflammation. Low folate diet treated wild type animals showed increased brain HCY levels and behavioral deficits when challenged in the fear conditioning paradigm which measure short term retention memory, compared to controls. The same animals showed increased Aβ levels and tau phosphorylation in brain tissue compared to controls. Moreover, low folate diet animals displayed higher expression of astrocytosis marker GFAP, but lower levels of synaptophysin and PSD-95, two markers of synaptic integrity. By contrast, mice genetically deficient for 5LO despite the HHCY secondary to the low folate diet did not manifest any cognitive impairment, and no changes in Aβ levels, tau phosphorylation, neuroinflammation, and synaptic integrity when compared with controls receiving the regular diet [47]. Taken together, these findings further confirm and establish the novel and necessary role that the 5LO protein enzyme plays in mediating the negative effect of HHCY on the development of the entire ADphenotype.
CONCLUDING REMARKS
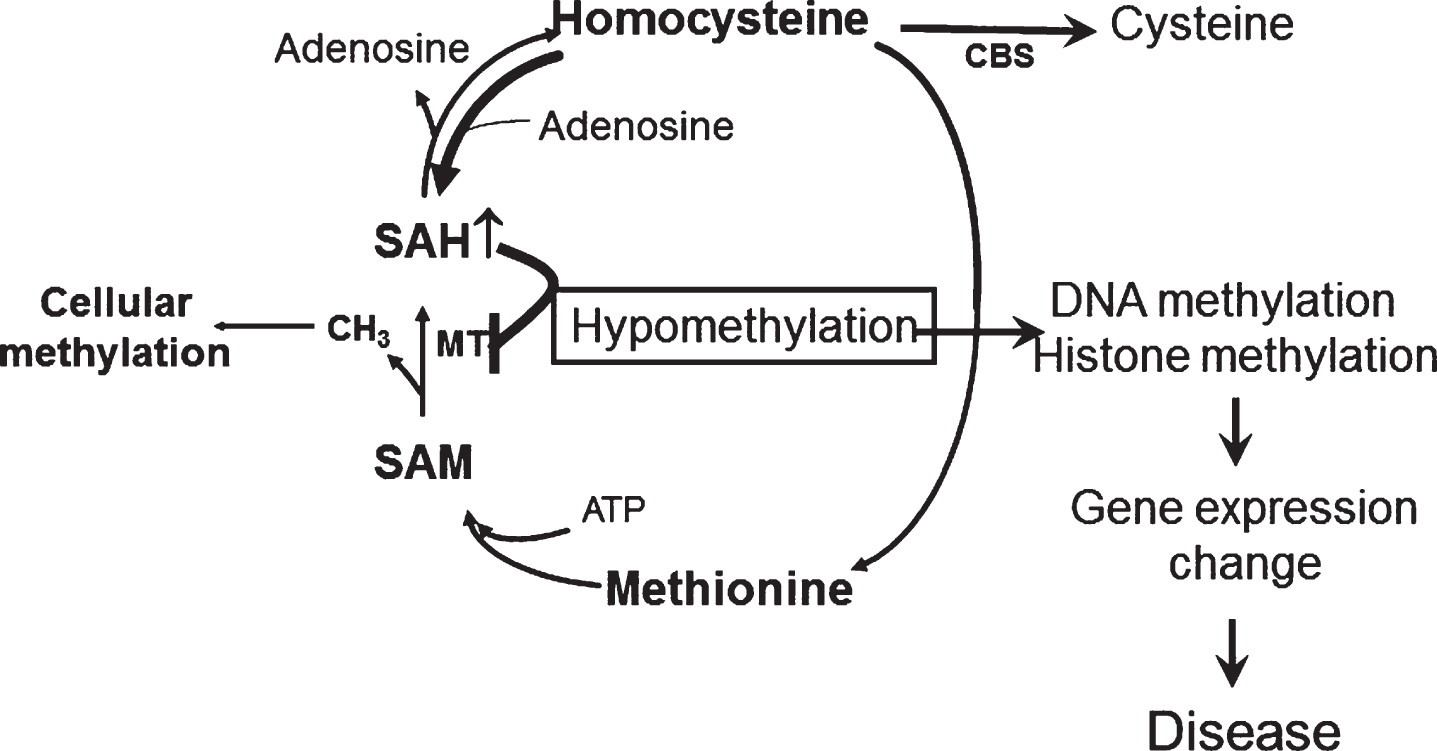
In its sporadic form, AD is a chronic neurodegenerative disease most likely resulting from a complex interplay between endogenous (i.e., genes) and exogenous (i.e., environment) factors. Although our knowledge of these risk factors is still incomplete, consistent evidence suggests that there are some modifiable ones which can directly influence its pathogenesis. Among them, epidemiological and clinical studies have revealed that HHCY double the risk for developing AD independently of several other major variables [48, 49]. However, the original hypothesis which predicted a simple and direct effect of HHCY on AD pathophysiology has been challenged by the conflicting results of the clinical trials aimed at lowering its circulating levels [22, 50]. By contrast, our body of work is consistent with a new working hypothesis in which HHCY via the increased intracellular SAH levels is a metabolic marker for a basic underlying cellular defect (hypomethylation) that by regulating specific gene expression influences the neuronal cell phenotype and ultimately causes the onset of the neurodegenerative disease (Fig. 3). Thus, we demonstrated for the first time that HHCY in the CNS upregulates the 5LO enzymatic pathway via the hypomethylation of its promoter secondary to the increased SAH levels, and that this activation then modulates the entire AD phenotype. Importantly, we have also established the necessary role for the HHCY-dependent effect on the cellular and molecular epigenetic events that drive neurodegenerative processes in vivo and in vitro.
Fig.3
The methylation hypothesis. HHCY promotes SAH increase that blocks DNA and histone methylation by inhibiting DNA methyltranferase (MT). This process results in altered expression of disease associated genes.
Importantly, our novel hypothesis could provide a biochemical explanation for the negative results of strategies using vitamins to reduce HCY levels. Thus, despite the fact that folic acid reduces plasma HCY, it does not influence SAH concentration and, in fact by facilitating methionine synthesis can indirectly even increase SAH production, further aggravating hypomethylation. In this sense, the real target in a condition of HHCY would be SAH and not HCY itself. However, at the moment there is no available therapeutic strategy which specifically targets intracellular SAH. Studies targeting SAH by gene therapy in animal models of HHCY and AD arewarranted.
The biological relevance of our new findings is underscored by the observation that this biological effect can be reproduced by using a dietary as well as a genetic approach, and, most importantly, by implementing not only transgenic mouse models but also wild type animals supporting the concept that this effect is independent from the transgene and therefore a generalized one.
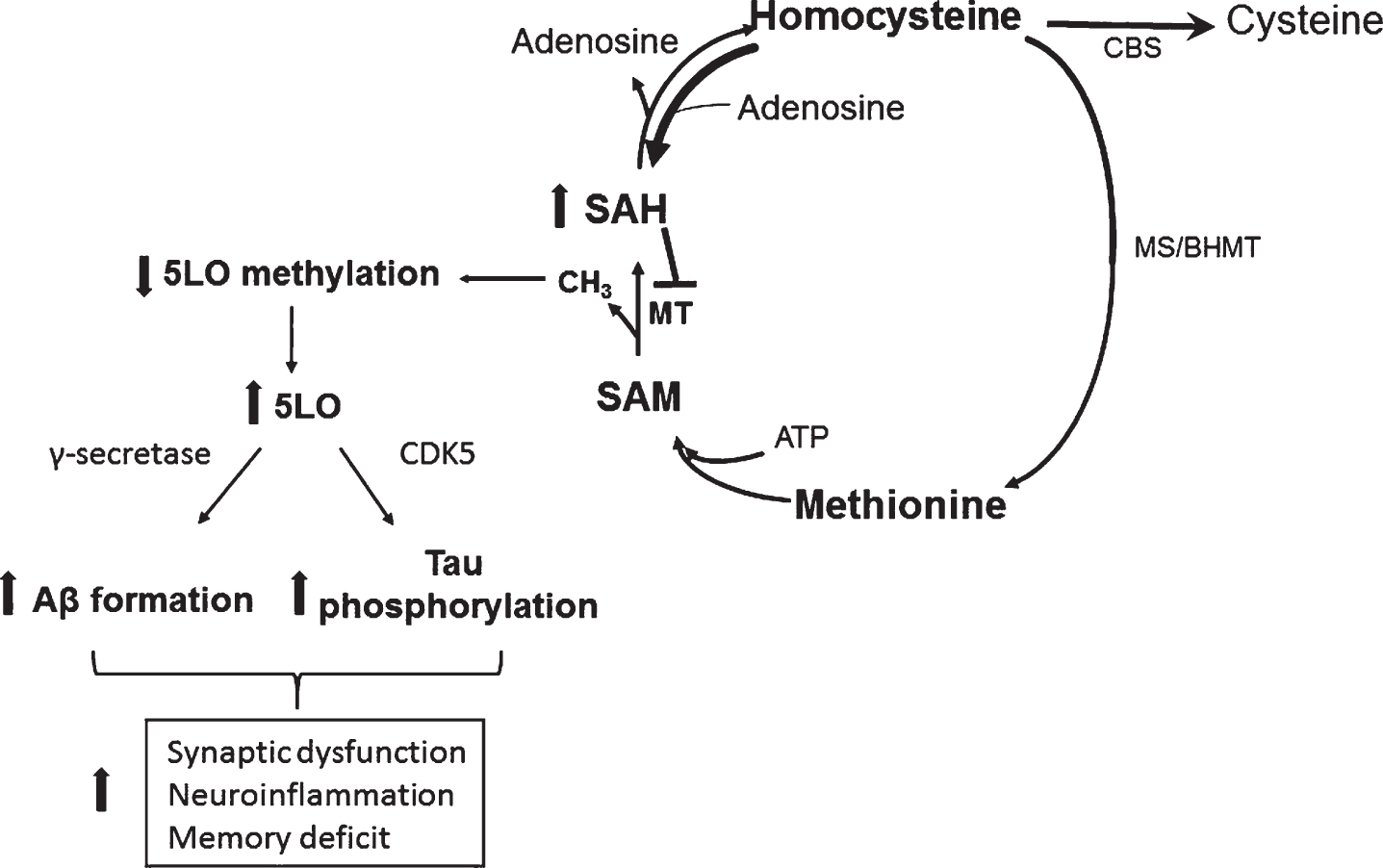
In conclusion, the significance of our work lies in the fact that it establishes a novel and strong biological link between HCY (environmental risk factor), and 5LO (genetic risk factor) with the AD pathogenesis via epigenetic mechanisms (DNA hypomethylation) (Fig. 4). Having established the central and necessary role for 5LO in the HCY-dependent AD phenotype development, and elucidated the molecular mechanisms responsible for it, our studies are now providing important and novel insights into the understanding of HCY as active player in the neurobiology of AD and important new therapeutic clues for the treatment of individuals bearing this risk factor.
Fig.4
Effect of HHCY on AD pathogenesis through 5LO. In a condition of HHCY the resulting increase in SAH levels reduce 5LO promoter methylation, which then translates in its own activation and higher 5LO gene expression. Higher 5LO levels promote amyloid-β formation and tau phosphorylation through the γ-secretase and CDK5 pathways, respectively. These events ultimately result in synaptic dysfunction and pathology, neuroinflammation and memory deficit.
ACKNOWLEDGMENTS
The work presented and or discussed in the current paper from the authors’ laboratory was supported in part by grants from the National Institute of Health (HL112966, and AG051684).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/17-0700r1).
REFERENCES
[1] | Alzheimer’s Association ((2016) ) Alzheimer’s disease facts and figures. Alzheimers Dement 12: , 459–509. |
[2] | Zhuo JM , Wang H , Praticò D ((2011) ) Is hyperhomocysteinemia an Alzheimer’s disease (AD) risk factor, an AD marker, or neither? Trends Pharmacol Sci 32: , 562–571. |
[3] | Firuzi O , Zhuo J , Chinnici CM , Wisniewski T , Praticò D ((2008) ) 5-Lipoxygenase gene disruption reduces amyloid-beta pathology in a mouse model of Alzheimer’s disease. FASEB J 22: , 1169–1178. |
[4] | Ikonomovic MD , Abrahamson EE , Uz T , Manev H , Dekosky ST ((2008) ) Increased 5-lipoxygenase immunoreactivity in the hippocampus of patients with Alzheimer’s disease. J Histochem Cytochem 56: , 1065–1073. |
[5] | Di Meco A , Lauretti E , Vagnozzi AN , Praticò D ((2014) ) Zileuton restores memory impairments and reverses amyloid and tau pathology in aged Alzheimer’s disease mice. Neurobiol Aging 35: , 2458–2464. |
[6] | Li JG , Barrero C , Gupta S , Kruger WD , Merali S , Praticò D ((2017) ) Homocysteine modulates 5-lipoxygenase expression level via DNA methylation. Aging Cell 16: , 273–280. |
[7] | Li JG , Barrero C , Merali S , Praticò D ((2017) ) Five lipoxygenase hypomethylation mediates the homocysteine effect on Alzheimer’s phenotype. Sci Rep 7: , 46002. |
[8] | Querfurth HW , LaFerla FM ((2010) ) Alzheimer’s disease. N Engl JMed 362: , 329–344. |
[9] | Iwata N , Tsubuki S , Takaki Y , Shirotani K , Lu B , Gerard NP , Hama E , Lee HJ , Saido TC ((2001) ) Metabolic regulation of brain Abeta by neprilysin. Science 292: , 1550–1552. |
[10] | Iqbal K , Alonso AeC , Chen S , Chohan MO , El-Akkad E , Gong CX , Khatoon S , Li B , Liu F , Rahman A , Tanimukai H , Grundke-Iqbal I ((2005) ) Tau pathology in Alzheimer disease and other tauopathies. Biochim Biophys Acta 1739: , 198–210. |
[11] | Lee VM , Goedert M , Trojanowski JQ ((2001) ) Neurodegenerative tauopathies. Annu Rev Neurosci 24: , 1121–1159. |
[12] | Mattsson N , Zetterberg H , Hansson O , Andreasen N , Parnetti L , Jonsson M , Herukka SK , van der Flier WM , Blankenstein MA , Ewers M , Rich K , Kaiser E , Verbeek M , Tsolaki M , Mulugeta E , Rosén E , Aarsland D , Visser PJ , Schröder J , Marcusson J , de Leon M , Hampel H , Scheltens P , Pirttilä T , Wallin A , Jönhagen ME , Minthon L , Winblad B , Blennow K ((2009) ) CSF biomarkers andincipient Alzheimer disease in patients with mild cognitiveimpairment. JAMA 302: , 385–393. |
[13] | López Salon M , Morelli L , Castaño EM , Soto EF , Pasquini JM ((2000) ) Defective ubiquitination of cerebral proteins in Alzheimer’s disease. J Neurosci Res 62: , 302–310. |
[14] | Petras M , Tatarkova Z , Kovalska M , Mokra D , Dobrota D , Lehotsky J , Drgova A ((2014) ) Hyperhomocysteinemia as a risk factor for the neuronal system disorders. J Physiol Pharmacol 65: , 15–23. |
[15] | Sharma M , Tiwari M , Tiwari RK ((2015) ) Hyperhomocysteinemia: Impact on neurodegenerative diseases. Basic Clin Pharmacol Toxicol 117: , 287–296. |
[16] | Levin J , Bötzel K , Giese A , Vogeser M , Lorenzl S ((2010) ) Elevated levels of methylmalonate and homocysteine in Parkinson’s disease, progressive supranuclear palsy and amyotrophic lateral sclerosis. Dement Geriatr Cogn Disord 29: , 553–559. |
[17] | Mizrahi EH , Jacobsen DW , Debanne SM , Traore F , Lerner AJ , Friedland RP , Petot GJ , Jacobsen DW , Debanne SM , Traore F , Lerner AJ , Friedland RP ((2003) ) Plasma total homocysteine levels, dietary vitamin B6 and folate intake in AD and healthy aging. J Nutr Health Aging 7: , 160–165. |
[18] | Serot JM , Barbé F , Arning E , Bottiglieri T , Franck P , Montagne P , Nicolas JP ((2005) ) Homocysteine and methylmalonic acidconcentrations in cerebrospinal fluid: Relation with age andAlzheimer’s disease. J Neurol Neurosurg Psychiatry 76: , 1585–1587. |
[19] | Annerbo S , Wahlund LO , Lökk J ((2006) ) The significance of thyroid-stimulating hormone and homocysteine in the development of Alzheimer’s disease in mild cognitive impairment: A 6-year follow-up study. Am J Alzheimers Dis Other Demen 21: , 182–188. |
[20] | Oulhaj A , Refsum H , Beaumont H , Williams J , King E , Jacoby R , Smith AD ((2010) ) Homocysteine as a predictor of cognitive decline in Alzheimer’s disease. Int J Geriatr Psychiatry 25: , 82–90. |
[21] | Connelly PJ , Prentice NP , Cousland G , Bonham J ((2008) ) A randomised double-blind placebo-controlled trial of folic acid supplementation of cholinesterase inhibitors in Alzheimer’s disease. Int J Geriatr Psychiatry 23: , 155–160. |
[22] | Aisen PS , Schneider LS , Sano M , Diaz-Arrastia R , van Dyck CH , Weiner MF , Bottiglieri T , Jin S , Stokes KT , Thomas RG , Thal LJ , Alzheimer Disease Cooperative, Study ((2008) ) High-dose B vitamin supplementation and cognitive decline in Alzheimer disease: A randomized controlled trial. JAMA 300: , 1774–1783. |
[23] | Pacheco-Quinto J , Rodriguez de Turco EB , DeRosa S , Howard A , Cruz-Sanchez F , Sambamurti K , Refolo L , Petanceska S , Pappolla MA ((2006) ) Hyperhomocysteinemic Alzheimer’s mouse model of amyloidosis shows increased brain amyloid beta peptide levels. Neurobiol Dis 22: , 651–656. |
[24] | Zhuo JM , Portugal GS , Kruger WD , Wang H , Gould TJ , Pratico D ((2010) ) Diet-induced hyperhomocysteinemia increases amyloid-beta formation and deposition in a mouse model of Alzheimer’s disease. Curr Alzheimer Res 7: , 140–149. |
[25] | Li JG , Praticò D ((2015) ) High levels of homocysteine results in cerebral amyloid angiopathy in mice. J Alzheimers Dis 43: , 29–35. |
[26] | Maccarrone M , Melino G , Finazzi-Agrò A ((2001) ) Lipoxygenases and their involvement in programmed cell death. Cell Death Differ 8: , 776–784. |
[27] | Rådmark O , Samuelsson B ((2010) ) Regulation of the activity of 5-lipoxygenase, a key enzyme in leukotriene biosynthesis. Biochem Biophys Res Commun 396: , 105–110. |
[28] | Bishayee K , Khuda-Bukhsh AR ((2013) ) 5-lipoxygenase antagonist therapy: A new approach towards targeted cancer chemotherapy. Acta Biochim Biophys Sin (Shanghai) 45: , 709–719. |
[29] | Zou Y , Kim DH , Jung KJ , Heo HS , Kim CH , Baik HS , Yu BP , Yokozawa T , Chung HY ((2009) ) Lysophosphatidylcholine enhances oxidative stress via the 5-lipoxygenase pathway in rat aorta during aging. Rejuvenation Res 12: , 15–24. |
[30] | Lammers CH , Schweitzer P , Facchinetti P , Arrang JM , Madamba SG , Siggins GR , Arrang JM , Madamba SG , Siggins GR , Piomelli D ((1996) ) Arachidonate 5-lipoxygenase and its activating protein: rominent hippocampal expression and role in somatostatin signaling. J Neurochem 66: , 147–152. |
[31] | Chinnici CM , Yao Y , Praticò D ((2007) ) The 5-lipoxygenase enzymatic pathway in the mouse brain: Young versus old. Neurobiol Aging 28: , 1457–1462. |
[32] | Uz T , Longone P , Manev H ((1997) ) Increased hippocampal 5-lipoxygenase mRNA content in melatonin-deficient, pinealectomized rats. J Neurochem 69: , 2220–2223. |
[33] | Uz T , Dwivedi Y , Qeli A , Peters-Golden M , Pandey G , Manev H ((2001) ) Glucocorticoid receptors are required for up-regulation of neuronal 5-lipoxygenase (5-LOX) expression by dexamethasone. FASEB J 15: , 1792–1794. |
[34] | Magri F , Locatelli M , Balza G , Molla G , Cuzzoni G , Fioravanti M , Solerte SB , Ferrari E ((1997) ) Changes in endocrine circadian rhythms as markers of physiological and pathological brain aging. Chronobiol Intern 14: , 385–396. |
[35] | Frank MG , Baratta MV , Sprunger DB , Watkins LR , Maier SF ((2007) ) Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immunity 21: , 47–59. |
[36] | Qu T , Manev R , Manev H ((2001) ) 5-Lipoxygenase (5-LOX) promoter polymorphism in patients with early-onset and late-onset Alzheimer’s disease. J Neuropsychiatry Clin Neurosci 13: , 304–305. |
[37] | Chu J , Praticò D ((2011) a) 5-lipoxygenase as an endogenous modulator of amyloid β formation in vivo. Ann Neurol 69: , 34–46. |
[38] | Chu J , Praticò D ((2013) ) 5-Lipoxygenase pharmacological blockade decreases tau phosphorylation in vivo: Involvement of the cyclin-dependent kinase-5. Neurobiol Aging 34: , 1549–1554. |
[39] | Chu J , Praticò D ((2011) b) Pharmacologic blockade of 5-lipoxygenase improves the amyloidotic phenotype of an Alzheimer’s disease transgenic mouse model involvement of γ-secretase. Am J Pathol 178: , 1762–1769. |
[40] | Giannopoulos PF , Chu J , Joshi YB , Sperow M , Li JG , Kirby LG , Praticò D ((2013) ) 5-lipoxygenase activating protein reduction ameliorates cognitive deficit, synaptic dysfunction, and neuropathology in a mouse model of Alzheimer’s disease. Biol Psychiatry 74: , 348–356. |
[41] | Giannopoulos PF , Chu J , Joshi YB , Sperow M , Li JG , Kirby LG , Pratico D ((2014) ) Gene knockout of 5-lipoxygenase rescues synaptic dysfunction and improves memory in the triple-transgenic model of Alzheimer’s disease. Mol Psychiatry 19: , 511–518. |
[42] | De La Haba G , Cantoni GL ((1959) ) The enzymatic synthesis of S-adenosyl-L-homocysteine from adenosine and homocysteine. J Biol Chem 234: , 603–608. |
[43] | Lin HC , Hsieh HM , Chen YH , Hu ML ((2009) ) S-Adenosylhomocysteine increases beta-amyloid formation in BV-2microglial cells by increased expressions of beta-amyloidprecursor protein and presenilin 1 and by hypomethylation of thesegene promoters. Neurotoxicology 30: , 622–627. |
[44] | Fuso A , Seminara L , Cavallaro RA , D’Anselmi F , Scarpa S ((2005) ) S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Mol Cell Neurosci 28: , 195–204. |
[45] | Uhl J , Klan N , Rose M , Entian KD , Werz O , Steinhilber D ((2002) ) The 5-lipoxygenase promoter is regulated by DNA methylation. J Biol Chem 277: , 4374–4379. |
[46] | Li JG , Chu J , Barrero C , Merali S , Praticò D ((2014) ) Homocysteine exacerbates β-amyloid pathology, tau pathology, and cognitive deficit in a mouse model of Alzheimer disease with plaques and tangles. Ann Neurol 75: , 851–863. |
[47] | Li JG , Barrero C , Merali S , Praticò D ((2017) ) Genetic absence of ALOX5 protects from homocysteine-induced memory impairment, tau phosphorylation and synaptic pathology. Hum Mol Genet 26: , 1855–1862. |
[48] | Shen L , Ji HF ((2015) ) Association between homocysteine, folic acid, vitamin B12 and Alzheimer’s disease: Insights from meta-analysis. J Alzheimers Dis 46: , 777–790. |
[49] | Beysoun MA , Beydoun HA , Gamaldo AA , Teel A , Zonderman AB , Wang Y ((2014) ) Epidemiologic studies of modifiable factors associated withcognition and dementia: Systemic review and meta-analysis. BMC Public Health 14: , 643. |
[50] | Clarke R , Bennet D , Parish S , Lewington S , Skeaff M , Eussen SJ , Lewerin C , Stott DJ , Armitage J , Hankey GJ , Lonn E , Spence JD , Galan P , de Groot LC , Halsey J , Dangour AD , Collins R , Grodstein F , B-Vitamin Treatment Trialists’ Collaboration ((2014) ) Effects of homocysteine lowering with B vitamins on cognitive aging: Meta-analysis of 11 trials with cognitive data on 22,000 individuals. Am J Clin Nutr 100: , 657–666. |


