
Dementia Pugilistica Revisited
Abstract
Extensive exposure of boxers to neurotrauma in the early 20th century led to the so-called punch drunk syndrome, which was formally recognized in the medical literature in 1928. “Punch drunk” terminology was replaced by the less derisive ‘dementia pugilistica’ in 1937. In the early case material, the diagnosis of dementia pugilistica required neurological deficits, including slurring dysarthria, ataxia, pyramidal signs, extrapyramidal signs, memory impairment, and personality changes, although the specific clinical substrate has assumed lesser importance in recent years with a shift in focus on molecular pathogenesis. The postmortem neuropathology of dementia pugilistica has also evolved substantially over the past 90 years, from suspected concussion-related hemorrhages to diverse structural and neurofibrillary changes to geographic tauopathy. Progressive neurodegenerative tauopathy is among the prevailing theories for disease pathogenesis currently, although this may be overly simplistic. Careful examination of historical cases reveals both misdiagnoses and a likelihood that dementia pugilistica at that time was caused by cumulative structural brain injury. More recent neuropathological studies indicate subclinical and possibly static tauopathy in some athletes and non-athletes. Indeed, it is unclear from the literature whether retired boxers reach the inflection point that tends toward progressive neurodegeneration in the manner of Alzheimer’s disease due to boxing. Even among historical cases with extreme levels of exposure, progressive disease was exceptional.
INTRODUCTION
Given the necessity for self-defense in essentially all animal species, it is not hard to imagine the clenched fist appearing in human evolution alongside the act of running, or that pugilism is likely as old as humankind itself. Egyptian hieroglyphics dating from 4000 BC suggest the existence of boxing-like combat as a military expediency, with thongs wrapped around hands as a precursor to the boxing glove [1]. Similar leather-wrapped fists were noted in ancient Baghdad. Boxing as a part of various games and festivals was evident in early Greek and Roman cultures [1]. The term boxing is believed to have its origin in the clenched fist, or the folding of fingers and thumb into a box-like structure, its roots from the Greek and Latin being puxos and buxus, respectively. Implements such as the cestus, a form of lethal gauntlet, were used in Ancient Rome for contests among gladiators on feasts and holidays, but were eventually banned in favor of the common sense view that fights to the death, or near death, was not necessarily advantageous for warrior training [1]. Without the prospect of someone being killed, public interest in boxing declined in the later years of the Roman Empire. Emperor Theodosius the Great banned the Olympic games and therefore its boxing component in 393 AD [1]. The subsequent 1300 years saw a relative historical silence to boxing as a spectacle with greater emphasis on warfare and weaponry, but resurfaced in England, the “cradle of pugilism,” [1] coinciding with expansion of the proletariat of the industrial revolution. Ritualistic armed contests such as fencing that developed in the middle ages drifted into sport for the aristocracy, while boxing was something of an equalizer for the new labor class, and a diversion from the squalor of urban confinement.
By the late 19th and early 20th century, boxing gloves and rules of engagement (e.g., Queensberry Rules of 1867) had been broadly implemented, allowing more of a sporting quality compared to underground bare knuckle contests, and formal promotion of “prize fighting.” Its popularity was such that quality professional boxers could occasionally subsist solely on contest proceeds, while many others could earn a worthwhile supplement to a laborer’s wage [2]. This is perhaps among the many factors that led to increased exposure and increased numbers of those exposed in recent times. At any other time in recorded history, boxing appears to have been either compulsory or provided an unenviable sum to that which could be derived from the low wage labor market or the informal street economy. The increased popularity also meant that lower tier and undertrained boxers would continue fighting while their skills were in decline, and at times offer themselves up for a small stipend in professional contests, knowing full well that severe physical punishment was the likely outcome [2]. Moreover, when not participating in promoted fights, many boxers fought in carnival booths, as often as 30 to 40 times a day [3], in an unmonitored fashion, “taking on all comers.” Others sparred with upper tier and more punishing fighters as a separate endeavor, or in addition to promoted contests and booth exhibitions [2]. The long term effects of these ancillary activities, potentially synergistic with promoted contests, was obliquely addressed by Roberts [2], but are largely unknown to this day and likely lost to history. The plight of such rank and file boxer is summed up well by Critchley [4]:
“Of special aetiological importance is that humbler side of boxing where the contestant travels in fairs in the boxing booth (or “blood-tub”), taking on all-comers at any weight. Almost as characteristic is the story of the boxer who eventually gives up the ring, having failed to make the grade, later to become what they call a “punch-bag”- that is, one of a team of sparring partners to a first-class heavyweight. A typical story is that the boxer, after a promising early career in the ring, begins to slow up; to be knocked out more often; to win fewer contests; and to be seedy for increasingly longer periods after each affray. Most characteristic of all is the admission on the part of the boxer that he finally abandoned the ring because of his wife’s increasing disapproval of his career.”
High levels of neurotrauma exposure from boxing in the early 20th century is apparent in the early case material with professional fights numbering as many as 1,000 and after a lengthy amateur career [5]. Such high numbers are not encountered today. According to one literature review, the average boxing career since the 1930 s has declined from 19 years to 5 years, and the number of fights has declined from an average of 336 to 13 [6]. The popularity of the sport at that time was such that skilled fighters would have bouts arranged and promoted almost every week, compared to once a month or less by the 1950 s [2]. Oversight of the contests also differed. Prior to World War II, there was no inclination on the part of either the referee or the competitors to stop the fight when one of the fighters was obtunded [2]. Repetitive concussive blows leveled on an incapacitated fighter “out on his feet,” until he lay prostrate on the canvass, occurred as a matter of routine [4]. The presence of a physician was variably required among the state boxing commissions, mainly for purposes of emergency care [2]. It was not until 1979 that the New York State Athletic Commission specified that an assigned physician could step into the ring and stop a bout [7]. Exceptionally long contests with numerous two minute rounds were commonplace before 1940, and little care was taken in fight promotions to match evenly skilled or evenly weighted boxers [2]. Lighter boxing gloves (6 oz.) were common [5]. Thus, severe and protracting beatings, and numerous such beatings with no mandatory exclusion times, were typical over a boxer’s professional career, notwithstanding any additive effects of one’s career as an amateur, a sparring partner, or a booth fighter. It is perhaps not surprising, in light of the dynamics of boxing participation and boxing oversight, that a neurological condition known colloquially as ‘punch drunk’ emerged in the early 20th century and has since tapered.
The focus on neurological injury from boxing exposure tends to place emphasis on poor outcome, although it should be pointed out for balance that transcendent human benefits are still commented upon in the modern era, such as self-esteem, respect within the community, physical fitness, and avoidance of a criminal lifestyle, alcohol, and illicit drugs [8]. Boxers themselves have rejected, sometimes bitterly, the notion that boxing first and foremost encourages violence [8]. Nevertheless, heated debate among boxing advocates versus those favoring a ban on the sport have been taking place for many years and are ongoing [9]. For the purposes of this review, we do not presume to provide insight into the question of whether ordered society has the moral imperative to ban individuals from a sport like boxing, or whether the benefits of boxing outweigh the risks. We are more concerned with the nature of neurological and biological processes associated with the “model experiment” [10] of boxing, and the analysis of the largely historical entity of dementia pugilistica with the benefit of hindsight.
PUNCH DRUNK
Punch drunk appeared in the medical literature for the first time in 1928, when Martland commented on a ‘pecular condition’ among prize fighters [11]. The condition, plainly visible to boxing fans, promoters, and indeed the fighters themselves in advance of Martland’s paper, had not been codified previously as a neurological disease state. It was said to affect lower skilled fighters of the “slugger” type who tended to take considerable punishment while sizing up their opponent for the elusive knockout blow, or who were otherwise limited in defensive skills but possessed the constitution to withstand substantial physical punishment. Parker would later comment that “Quick, agile, clever boxers who guard themselves well and take little punishment seem to escape. Chiefly affected are the less expert but courageous men who take considerable injury in the hope of wearing out their opponents” [12]. Critchley drew similar conclusions regarding the style of boxing, and also suggested that the condition was much less common in African American boxers [4], although he did not provide any statistical basis for this latter conclusion.
Twenty-three fighters with the putative punch drunk condition were reported to Martland from a single, well known, boxing promoter. 15 of these were described simply as punch drunk, four were said to be committed to an asylum and not otherwise characterized, three dragged an extremity (two of these were also dysarthric, one was in ‘bad shape’), and one had a parkinsonian syndrome. Martland examined five of these subjects, although he provided no description of any of the five. Instead, he described an additional subject as case 2 in his report – a 38-year-old man who became symptomatic at age 23 with a left hand tremor and unsteadiness in his legs, after 7 years of fighting. Martland frankly acknowledged the differential diagnosis of early-onset Parkinson disease (paralysis agitans) in this subject, although he eliminated epidemic encephalitis from the time course of clinical symptoms. (case 1 in Martland’s paper consisted of the autopsy findings in a 76-year-old man who succumbed after falling down a flight of stairs; this subject had no boxing history and was presented only to illustrate Martland’s original hypothesis that punctate hemorrhages were responsible for the clinical signs of the punch drunk syndrome).
In keeping with present day discussions of chronic traumatic encephalopathy, the relationship between clinical findings, which themselves were wide-ranging, and neuropathology, was speculative. Martland described flopping of the foot or leg, unsteadiness in gate, uncertainty in equilibrium (even while actively boxing), appearance of intoxication, peculiar tilting of the head, dragging of one or both legs, a staggering or propulsive gait, parkinsonian faces, tremors, vertigo, deafness, and in some cases marked mental deterioration. The pathological substrate offered by Martland appeared more in line with acute parenchymal brain injury. His depiction of the stairway-related fall is that of diffuse axonal injury, with acute gliding contusions, hemorrhages in the corpus callosum, and small hemorrhages in the basal ganglia. Martland offered the theory that “Punch drunk bares the same relation to multiple concussion hemorrhages as do many of the post-concussion neuroses and psychoses that follow blows or falls on the head.” There was no mention of neurofibrillary tangles (NFT) or Alzheimer-related changes, or otherwise a lesion that would correspond to accumulations of phosphorylated tau (p-tau) or chronic traumatic encephalopathy as it has been recently described [13]. The potential for a manufactured condition was not lost on Martland in his index report, noting:
“While the establishment of these facts is of enormous importance to the courts and to labor compensation boards in placing many cases of cranial injuries on a firm pathologic basis, it also will have its disadvantages. A very great field is opened for the so-called expert testimony, in which malingerers and those suffering from various forms of psychoses and neuroses may claim undue compensation. The correct diagnosis during life will always be extremely difficult, as the condition can only be proved byautopsy” [11].
The latter concept of autopsy-proven disease has its own element of subjectivity. Martland’s “proof” at the time consisted of concussion-related hemorrhages, which were never seriously considered as a lesion responsible for long term sequelae after the 1940 s.
Millspaugh introduced the term “dementia pugilistica” in 1937 [14] in a somewhat meandering discussion, commenting on Martland’s findings and the possibility for microscopic ring hemorrhages. Observations by Parker, including his assertion of a multitude of individual lesions [12], and by Jokl and Gutterman [15] favoring a cerebral trauma-induced dementia, were noted. Some attention to the problems of weight differences among fighters, and rules requiring the attendance of medical officers, indicated a level of concern for serious or permanent injury at the time. Dyslalia of heterogeneous origin (laryngeal trauma, facial and dentition trauma, psychic effects), othematoma, and traumatic metacarpophalangeal arthropathies, were mentioned briefly. Psychiatric disturbances, including quasi-delusional ideation with “magnification of former prowess” was commented upon, as were gait abnormalities and Parkinsonian symptoms. Overall, dementia pugilistica was meant to convey a physical-psychic syndrome that accumulated over a lengthy boxing career, but was less derisive and less resented among boxers than ‘punch drunk’.
Despite the newly introduced term encompassing ‘dementia’, Millspaugh tended to convey a static condition, in that steady neurological deterioration was less striking than the odd psychic characteristics, neurological signs, and speech difficulties. The morbid anatomy and associated mechanism were addressed with some specificity. Particular vulnerability of the “corpora striata, corona radiata, and the basal ganglions” was suggested. Mechanistic theory included “Traumatic punctate cerebral hemorrhages, hydrostatic disequilibrium of the spinal fluid, cerebral edema, concussion injury to the cortical cells, cerebral vasomotor imbalance, reparative gliosis or degenerative lesions of cerebral parenchyma, variation in the weight and therefore inertia between gray and white matter and tension transmission by nerves, blood vessels and musculature,” which tend to suggest cumulative damage from acute injury mechanisms, as opposed to a progressive Alzheimer-like neurodegeneration.
THE NATURE OF DISEASE PROGRESSION
Modern experimental constructs implicating protein templating as an underlying mechanism [16] require a priori the conclusion that progressive neurodegeneration is caused by repetitive neurotrauma. A close examination of the earliest case material in dementia pugilistica, i.e., those with the most extensive exposure to repetitive neurotrauma, nevertheless indicate heterogeneity with regard to progressive disease, and that a trauma-induced, progressive proteinopathy model is overly simplistic. Martland stated unambiguously that “Many cases remain mild in nature and do not progress beyond that point” [11]. Parker similarly noted that “a pugilist may be only mildly affected, and may continue to fight to the end of his career, or he may be so disabled that he ultimately has to quit boxing and yet gets no worse in after life” [12]. On the other hand, a “progressive neurological syndrome may appear, putting an end to all fighting, and leading finally to mental or physical helplessness” was also suggested in some cases. Critchley also raised the issue of a “groggy state,” [4] or a form of preclinical dementia pugilistica, in which the boxer’s skills may have subtly declined, rendering him increasingly vulnerable to concussive blows to the head, from which recovery is prolonged and more likely incomplete. In this respect, an exponential, rather than linear, increase in traumatic brain damage during this era late in a boxing career may be hypothesized, emphasizing the importance of retirement once a critical point is reached (i.e., once the boxer “softens up”) [17, 18], particularly if cumulative structural injury underlies clinical progression.
Three case studies by Parker indicated injuries traceable to specific fights, and a static or improved course over time [12]. In Parker’s case 1, the boxer “attributed all his difficulties to his last and futile appearance in the ring. The unbroken chain of cause and effect is very evident in this case, and it is to be noted that in nearly two years the condition had not become worse nor was there much improvement beyond a certain point.” He went on to say that “No specific nervous syndrome appeared, such as Parkinson’s disease, but rather a medley of scattered and incomplete lesions of the brain.” Parker’s case 2 showed a number of neurological signs and deficient memory but that did not deteriorate over the 6 years prior to the report and, if anything, improved. Parker’s case 3 showed progressive symptoms “up to his final, ignominious failure in the ring, and then, for eleven years thereafter, his condition remained much the same as it was when he ceased fighting,” despite an earlier clinical diagnosis of lateral sclerosis.
Critchley on the other hand suggested the term chronic traumatic encephalopathy in 1949 [19] and then “chronic progressive traumatic encephalopathy of boxers” in 1957 [4], suggesting that lack of insight into the neurological deficits was evidence of an insidious onset, often culminating in a fatuous or euphoric dementia with emotional lability. Speech and thought became progressively slower in his experience, with mood swings, truculence, and uninhibited violent behavior. To support his views, Critchley detailed 10 cases of punchdrunkenness, although the individual case histories fit into the neurological and psychiatric spectrum of other described cases. He nevertheless favored a progressive process once initiated:
“Of great interest, pathological as well as practical, is the fact that this traumatic encephalopathy is a progressive condition. Once established it not only does not permit of reversibility, but it ordinarily advances steadily. This is the case even though the boxer has retired from the ring and repeated cranial traumata are at an end” [4].
Mawdsley and Ferguson described a series of 10 subjects with air encephalography, documenting abnormalities in the septum pellucidum and dilated lateral ventricles [20]. The case histories likewise showed a mixture of neurological signs with gait and speech abnormalities, pyramidal and extra-pyramidal signs, memory loss, and brain atrophy. Slow progression of dysarthria, disorder of movement, and memory deficits, were noted, and it was stated that 8 out of 10 subjects had a significant disability. Many were “unemployed for many years,” while the authors also commented that “Cerebrovascular lesions in ex-boxers of middle age could possibly aggravate pre-existing traumaticdamage.”
Without insight into prevalence of the presumed head-trauma induced condition, discussions of disease progression prior to the late 1960 s may have been premature. Indeed, misinterpretation of single cases, or failure to exclude coincidental neurological illness, could mean the difference between concluding progressive disease versus a stationary condition. To address this issue, the Royal College of Physicians of London set up a committee ‘to report on the medical aspects of boxing’, and appointed Dr. A. H. Roberts to carry out a large scale cross-sectional study, and the only study of its kind to date on long term effects of neurotrauma in humans [2]. This was accomplished by examining a random sample 250 boxers out of 16,781 total boxers registered by the British Boxing Board of Control between 1929 and 1955. Of those 250, 16 died, 9 emigrated, and one refused to cooperate. 224 were thus studied by a series of four neuropsychological tests, patient interview, clinical examination, and electroencephalography. The results of the studies were published in a book, ‘Brain Damage in Boxers’ in 1969. The remarkable yield of cooperation was only possible through the diligent and time-consuming house-to-house inquiries by social workers, many times with the help of boxers themselves, in order to facilitate dialogue. When one considers the diligence of the effort as well as the era of numerous boxers with extensive neurotrauma exposure, it is not surprising that this now 50-year-old study has never been replicated.
Roberts found that of the 224 boxers studied, 37 (17%) “had evidence of lesions of their central nervous systems, similar to those reported and attributed to boxing by others, and not typical of the clinical entities encountered in routine neurological practice.” Of these, 13 (6%) were more severely disabled and appeared to present two clinical syndromes: “one predominantly extra-pyramidal and apparently more clearly progressive”, and the other predominantly cerebellar. (Extra-pyramidal signs attributable to boxing have since been said to be exceedingly rare [5, 21].) The more severe nature of the thirteen cases was evident in their presentation to neurological clinics. According to Roberts, these 6% were of the kind labeled ‘punch drunk’ in boxing circles. The same could not be said of the other 24 cases, as these individuals “were on the whole only marginally or in no way disabled.” Interestingly, all of the 13 cases with more severe clinical signs recognized as punch drunk boxed prior to World War II. Roberts commented on the issue of disease progression at some length despite the cross sectional design, as the idea of a neurodegenerative disease provoked by repetitive head trauma was already being raised. He noted that the “first evidence of the condition may follow one or a series of particularly hard fights, and that it may then regress if the boxer stops fighting.” This same phenomenon appears in a number of other described cases [3], rather than after a period of latency. Roberts goes on to state:
“When there is an adequate independent account, or the individual has sufficient insight, most of these boxers appear to have developed the symptoms of the condition during their last years boxing or after a series of particularly hard fights. It seems that some degree of progression may occur which cannot be accounted for simply on the basis of normal ageing, and occurs more commonly in the extra-pyramidal lesions, but apart from a few isolated exceptions reported previously and in one case in the present series (case 2), this progression is not in general characteristic of that seen in the systems degenerations or in the commoner presenile dementias” [2].
The appearance of traumatic encephalopathy from midbrain injury after a single bout supports Roberts’ assertions [17, 22, 23]. Roberts also identified asymmetrical motor signs, including tremor and hyperreflexia, with an overall tendency to favor the boxer’s left side [2]. Given the predominance of right-handed boxers, it has been asserted that such asymmetry is due to structural injury per se to the upper brainstem, from mechanical trauma per se. Case 2 in the selected case summaries provided by Roberts, on the other hand, suggests a progressive neurological disease. Roberts commented that the condition dated from the subject’s active boxing career (as opposed to following a period of “latency”), and expressed some difficulty in excluding a co-existing neurodegenerative disease, especially given the rarity of this type of clinical progression in the series. In the final analysis, Roberts’ described uncertainty on issue of progression versus the aging process superimposed on static deficits, a possibility endorsed by Jordan [24]:
“How far it can be assumed that a progressive degenerative process involving the neuron is mirrored in the widespread neurofibrillary changes found in some ex-boxers’ brains remains unanswered. It seems certain that diffuse cellular and axonal depletion has resulted from boxing to account for the clinical syndrome related to occupational exposure found in the present study. It must therefore be assumed that cerebral trauma of a minor degree may result in permanent structural damage that is cumulative. There was good evidence in some cases that the condition had progressed for some time after retirement from boxing, particularly in those with evidence of extra-pyramidal lesions, but the information available from the study proved inadequate to settle finally the question of progression unrelated to the changes associated with ageing” [2].
In short, cases with progressive disease were the exception in Roberts’ series, and still lacked Alzheimer-like progression in the rare patients with extrapyramidal signs. At one point, Roberts commented that “There is a good deal of evidence in the present study to suggest that in most cases the condition remains stationary when the individual has stopped boxing, and indeed there are excellent independent accounts for a few of undoubted improvement after their retirement.” The contrast between Roberts’ case synthesis and classical neurodegenerative diseases is noteworthy, as the latter never remain stationary let alone improve. Only two subjects in Roberts’ series were indisputably demented judging by their inability to live unaided in the community [2]. The protein cascades currently under discussion seem, therefore, to over-run the empirical observation that a progressive neurodegenerative disease, in a manner of Alzheimer’s disease (AD), is exceptional at best, even in these early 20th century boxers whose exposure levels were extreme. It finally should be noted that none of the Roberts’ cases were accompanied by pathological confirmation. The difficulty in obtaining such cases at autopsy, then and now, speaks to the lack of a cohesive neurodegenerative disease entity, and keeps viable the possibility or probability that some cases in this largest series of boxers consisted of idiopathic progressive synucleinopathies or other sporadic neurodegenerative diseases, cerebrovascular disease, and/or an undiagnosed infectious or inflammatory processes.
NEUROPATHOLOGY DESCRIBED IN BOXERS
Historically, neurodegenerative diseases were uncovered by the appearance of an unusual and inexorably progressive neurological decline, followed by neuropathological examination at autopsy. The index case of AD, for example, occurred in a middle aged woman with relentless neurologic deterioration and autopsy demonstration of hallmark lesions [25]. In contrast, Martland’s original description [11], Millspaugh’s article introducing the term “dementia pugilistica,” [14] Roberts’ case series [2], and many other early reports describing encephalopathy in boxers [3], were unaccompanied by autopsy neuropathology.
It is further emblematic of the complexity of the topic that the first report of boxers’ encephalopathy at autopsy was not a case of dementia pugilistica or punch drunk syndrome, at all. In 1954, Brandenburg and Hallervorden described the autopsy findings in a 51-year-old man who had boxed from age 18 to 29, ultimately holding the title of amateur German middle-weight champion for 6 years [26]. At age 39, personality changes appeared, which deteriorated into insomnia, memory loss, dysphasia, parkinsonism, and frank dementia. The most remarkable finding at autopsy was extensive AD pathology, including an abundance of senile plaques of various morphological types, severe cerebral amyloid angiopathy, and numerous NFT. Noteworthy as well were numerous so-called “condensation plaques” which correspond to today’s “cotton wool” plaque. The findings thus indicate rather unambiguously the presence of early-onset AD. The abundance of cotton wool plaques also suggests familial disease associated with presenilin-1 mutation [27]. Unfortunately, no family history was available or otherwise provided in this case.
The case reported by Grahmann and Ule in 1957 [28] is often considered alongside the case report by Brandenburg and Hallervorden as among the first cases suggesting a “link” between neurofibrillary change and boxing. In this case, the findings in a 46-year-old man who boxed between the ages of 15 and 25 were described. He expired after suffering hemorrhagic venous infarction from a cerebral venous sinus thrombosis, and after a documented, progressive neurological decline starting at age 36. The case history is similar to the earlier report by Brandenburg and Hallervorden in its description of progressive neurological deterioration, which clearly suggests neurodegenerative disease. It differs, however, in the absence of senile plaques and cerebral amyloid angiopathy, and with neurofibrillary changes noted largely in the brainstem and medial temporal lobe. Cerebral atrophy was present, along with cavum septum pellucidum, as was loss of Purkinje cells and granular neurons of the cerebellum. Since limited pathological illustrations were made available, it is unclear how carefully sporadic systems degenerations were excluded. From the standpoint of Roberts’ epidemiological survey, this case would have represented an outlier, both in terms of the long symptom-free interval between boxing and onset of symptoms, and the inexorable disease progression. The possibility of coincidental neurodegenerative disease unrelated to boxing remains a consideration. To his credit, Roberts recognized the possibility of coincidental neurological disease in the above two cases, noting:
“it would appear that the kind of progression indistinguishable from Alzheimer’s disease in two of the fourteen neuropathological studies of boxers reported so far must be extremely uncommon. So much so, that the fortuitous occurrence in Boxers of Alzheimer’s disease in these two cases cannot be entirely ignored” [2].
Neubuerger et al. reported two cases in 1959 [29]. His first case presented at age 46 initially for a coronary complaint, and four years later with a complaint of headaches. Neurological examination revealed a head and neck tremor, increased tone in the right arm, defective upward gaze, a mildly abnormal EEG, and abnormalities on neurocognitive testing. Neuropathology was limited to a brain biopsy, performed for unstated reasons, which showed gliosis. His second patient presented at age 53 with neurological decline including dementia and ataxia, eventually expiring secondary to pulmonary fibrosis and cor pulmonale. He had boxed between the ages of 18 and 24. Autopsy examination showed significant frontal cortical atrophy but no neurofibrillary degeneration or senile plaques. This case thus lacks all pathological features attributed to boxing, while frontotemporal dementia would be a consideration if not likely.
Courville (1962) published what he considered a verified case of dementia pugilistica in a 49-year-old man with alcoholism and diabetes mellitus, and a four-year boxing history [30]. Autopsy, however, showed nonspecific thickening of the leptomeninges and nonspecific changes on microscopy. Spillane (1962) reported autopsy findings in a 45-year-old man with approximately 300 professional fights, among a total of five cases with heterogeneous clinical presentations [31]. Neurological signs included slurred speech beginning about the time he retired from boxing at age 32. Later neurological signs included dragging of the left lower extremity and left arm posturing, ataxia, and continued dysarthria. Autopsy was performed after he suffered an acute myocardial infarct. Areas of “softening” in multiple brain regions were noted, suggestive of cerebrovascular disease. Spillane made no mention of neurofibrillary degeneration and ultimately concluded that “Taken together, these cases illustrate the difficulties of aetiological diagnosis and emphasize the extent of our ignorance of this subject.”
Case 10 by Mawdsley and Ferguson (1963) depicted the autopsy findings in a 51-year-old ex-boxer who retired at age 35 with 300 professional fights [20]. He was said to be “punchy” with slurred speech, an expressionless face, memory loss, apathy, and brisk reflexes, expiring secondary to squamous cell carcinoma of the floor of mouth. Autopsy showed gross atrophy, cavum septum pellucidum, and septal fenestration. No microscopic examination was provided. Payne (1968) reported neuropathological findings in six professional boxers with extensive boxing histories in addition to significant vascular, psychiatric, and alcohol abuse co-morbidities [32]. In particular, he noted septal abnormalities, miniscars in the cerebral cortex, and foci of white matter degeneration. Senile plaques and early neurofibrillary changes were noted in two cases, which Payne concluded were “nonspecific degenerative phenomena.” Burger and Minarovjech (1966) reported nonspecific neuropathology in a 44-year-old former boxer who expired from intracerebral hemorrhage [33]. Betti and Ottino (1969) reported brain biopsy findings in a 38-year-old former boxer with an extensive boxing exposure as well as head trauma from a stairway-related fall [34]. Asymmetrical tremor, abnormal gait, and dysarthria were noted clinically. Brain biopsy, performed for unclear reasons, showed nonspecific loss of neurons.
The landmark study of chronic neuropathology in former boxers was published by Corsellis et al. in 1973 [35] and established the neuropathology of dementia pugilistica until about 1990. This was a retrospective analysis of 15 cases examined in the Department of Neuropathology at the Runwell Hospital Institute of Psychiatry, near London, UK. Since the limiting factor for case acquisition in the Corsellis series was examination at the Institute of Psychiatry, selection bias precludes any discussion of prevalence. The spectrum of pathology is nonetheless noteworthy and perhaps not fully appreciated in modern discussions of long-term effects of mild neurotrauma. Subjects in the Corsellis series boxed between 1900 and 1940, with ages at death ranging from 57 to 91. As noted above, boxers in this era had extensive neurotrauma exposure, vastly exceeding levels of exposure encountered today. Many of the subjects participated in hundreds of promoted fights. Some fought in booths. 7 of the 15 endorsed a history of heavy alcohol use, whereas heightened sensitivity to the effects of alcohol was reported in 6 cases. In only four cases was alcohol use specifically denied. The duration of neurological signs potentially attributed to boxing was often unclear, but ranged from 8 years to 41 years in cases providing such data. The precise onset of symptoms relative to boxing was often unclear.
Neuropathological examination emphasized neurofibrillary degeneration on von Braunmuhl silver impregnation, especially in the substantia nigra and medial temporal lobe. These were often present in large numbers in the face of limited plaque pathology, thus separating the neurofibrillary degeneration of dementia pugilistica from that of typical AD. Substantia nigra neuron loss, cerebellar scarring (with extensive measurements and neuronal counts) particularly involving the cerebellar tonsils, and septal abnormalities with an enlarged cavum and septal fenestrations were also considered among the cardinal manifestations of boxing-relating neuropathology. There was significant heterogeneity in the sample, however. Although not specifically highlighted as boxing-specific, atrophic or flattened fornices were present in several cases. Shrunken and atrophic mammillary bodies were also noted in about half of the cases. Hippocampal atrophy was present in some cases, and hippocampal sclerosis per se was depicted in case 1. Cerebrovascular disease with infarcts was present in several cases. Given the important role of cerebrovascular disease in producing focal neurological signs as well as cognitive deficits [36], and the relative lack of effective anti-hypertensive therapy in the early 20th century, the role of co-morbid cerebrovascular disease among classic cases of boxers’ encephalopathy may be underappreciated. Structural traumatic brain injury was also evident, with remote contusions and hemosiderin staining of the olfactory bulbs in some cases. A cavernous malformation was present in one case. Tabes dorsalis was present in still another case. Three cases showed no specific pathology, and one was reported to lack a boxing history on further analysis [37]. All told, the Corsellis series is a complex and heterogeneous sample with numerous co-morbidities and structural injuries involving a multitude of brain regions. Despite the heterogeneity of neuropathology in the Corsellis et al. sample, however, the major impact seems to have been the renewal of focus on neurodegenerative pathology and in particular neurofibrillarydegeneration.
As an indicator of the rarity of dementia pugilistica, studies in the 1980 s and early 1990 s often looked to the Corsellis series for application of lately available immunohistochemistry, and offered examination of only a small number of new cases. For example, Roberts [38] in 1988 examined 8 of 15 cases from the Corsellis series with anti-sera to p-tau with no additional cases. Roberts et al. [39] in 1990 examined 14 of 15 Corsellis et al. cases with anti-sera to amyloid-β (Aβ) (in addition to 5 amateur boxer cases that lacked features of classical dementia pugilistica). The 1988 study confirmed the linkage of p-tau to neurofibrillary degeneration in dementia pugilistica, with cases resembling AD, while the 1990 study highlighted variably extensive Aβ-positive diffuse plaques not previously noted with silver impregnation techniques, raising the issue of diffuse plaque pathology as a component of repetitive trauma-induced changes. Tokuda et al. [40] re-examined 7 cases from the Corsellis et al. series, and added one new case, noting extensive morphologically diverse plaque pathology with significant overlap with AD. This suggested to Tokuda et al. a role of trauma in AD pathogenesis, but on balance also suggests co-morbid aging and/or AD pathology in the original Corsellis case series. Allsop et al. [41] re-examined 6 cases from the original Corsellis series and 2 new cases, also noting more abundant Aβ pathology than previously appreciated from silver impregnation, in addition to Aβ immunoreactive NFT with advancedpathology.
Hof et al. [42] extended the specificity of neurofibrillary degeneration in dementia pugilistica by examining three boxers with extensive boxing exposure. In this study, findings consistent with Corsellis et al. were noted, including abundant neurofibrillary pathology, cerebellar cortical pathology, substantia nigra pathology, and septal pathology. However, the authors noted larger numbers of NFT in superficial cortical laminae, and thus provided an additional point of distinction between trauma-related NFT and aging/Alzheimer-related NFT. Geddes et al. [43] examined the brain of a 23-year-old boxer who suffered an acute boxing-related death. P-tau immunohistochemistry, even in this young subject, showed abundant cortical p-tau, predominantly in frontal and temporal lobes. The immunoreactivity was patchy and showed a predilection for perivascular areas. No clinical signs were ascribed to the p-tau pathology although it was noted that the decedent was “somewhat forgetful” during life.
EVOLUTION OF DEMENTIA PUGILISTICA OVER TIME
The evolution of punch drunk/dementia pugilistica since 1928 is noteworthy, particularly alongside AD in which basic understanding of the clinical substrate, cognitive trajectory, and the pathological hallmarks has remained relatively stable since 1906. As noted above, dementia pugilistica was identified initially because of purely neurological signs, such as dysarthria, pyramidal and extra-pyramidal signs, and ataxia. Concussion-related hemorrhages were the initially-offered pathologic substrate [11]. Alzheimer-like pathology was then erroneously linked to dementia pugilistica as noted above [26]. NFT as a component of dementia pugilistica was reaffirmed by Corsellis et al. [35], who then added septal abnormalities, cerebellar sclerosis, and damage to the substantia nigra to the list of changes. Patchy tauopathy with predilection for superficial cortical laminae and perivascular areas of the cortex and Aβ-positive diffuse plaques appeared in the 1990 s [42, 43], while the specificity of nigral and cerebellar pathology seems to have been abandoned. Additional patterns of tauopathy have been ascribed to chronic effects of trauma in more recent studies [44], and further refined to a single required criterion with several supportive criteria according to a consensus recommendation forged in 2015 [13]. It is further stated that as many as 20% of cases with presumably sport-related p-tau deposits may be missed by routine dementia sampling and immunostaining protocols [13].
It is of note that among the few dementia pugilistica cases reported in the recent literature, either no clinical signs were apparent, or clinical signs were attributable to other major diseases [43, 45–48]. Thus, as classical dementia pugilistica has receded over the years, dementia pugilistica diagnosed purely by p-tau immunohistochemistry, either in the absence of neurological signs or in the context of other neurological diseases, have taken their place. This leaves open the question of clinical significance of patchy p-tau immunoreactivity, identified by highly sensitive means.
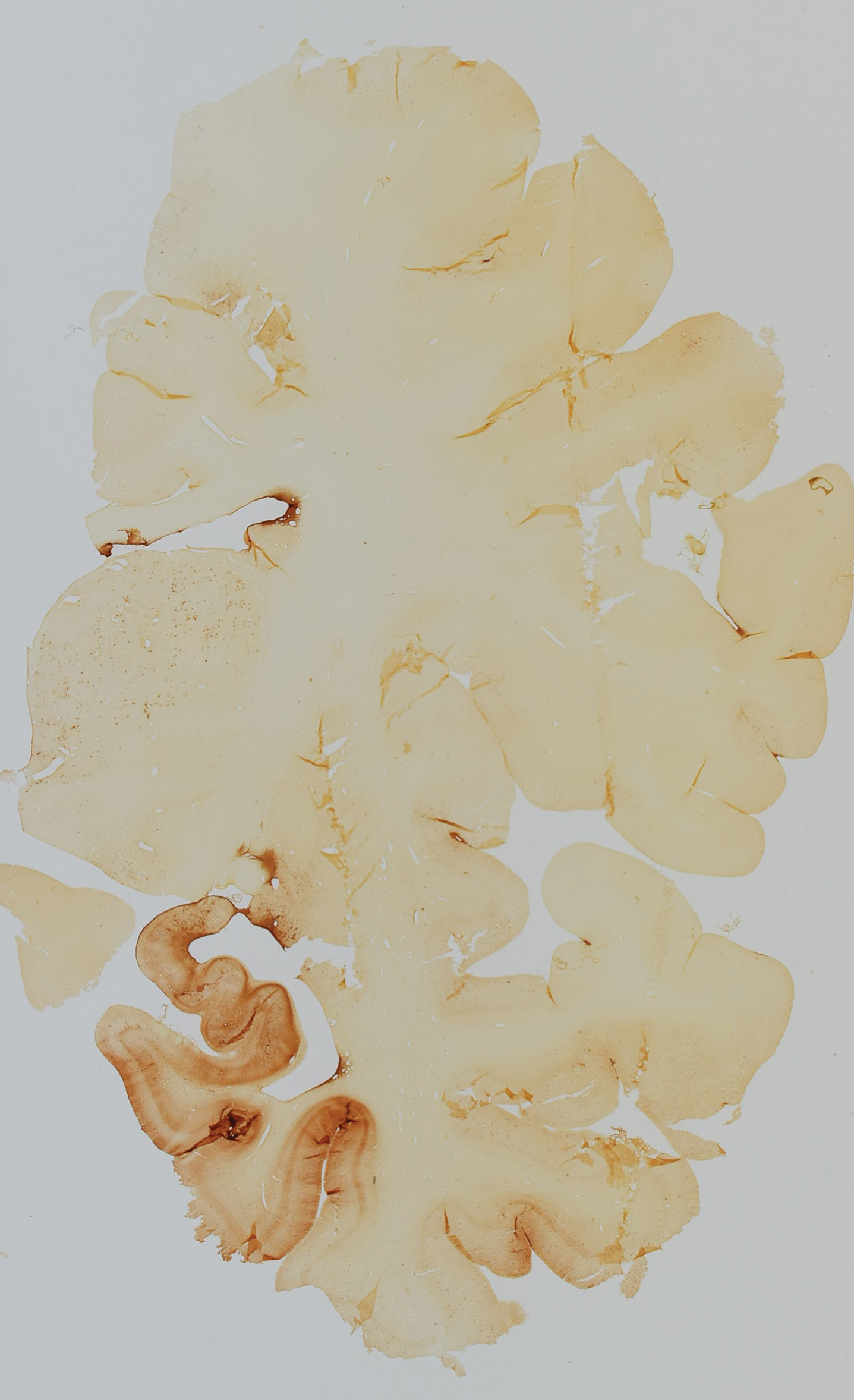
Figure 1 depicts a case in point. This section was obtained from the NIH Neurobiobank at the University of Maryland. In particular, it was obtained as a control case for a brain bank dedicated to neurodevelopmental disorders. The decedent was a 51-year-old man who died of suddenly of pneumonia complicated by cardiovascular disease, prior to which he had no health complaints whatsoever. He was neurologically and psychologically healthy. He was on no medications. He did not drink alcohol or use illicit drugs. It was gleaned from the death investigation, however, that the decedent had competed as an professional boxer during his youth. He fought in 14 professional contexts and lost 7 of them, two by knock out. At autopsy, his brain was grossly and microscopically normal. Because of his boxing history, and only because of his boxing history, extensive immunohistochemical stains for p-tau (AT8) were performed, and indeed there were accumulations of p-tau in neurons and glia, at times in patchy foci at the depths of cortical sulci, meeting modern criteria for chronic traumatic encephalopathy. Given his limited boxing exposure, it seems likely that a similar subclinical stationary tauopathy may be encountered in other asymptomatic athletes with similar or less exposure, and for that matter in non-athletes. This would not be particularly surprising given the ubiquity of more stereotyped p-tau accumulation with age and in the absence of clinical disease [49]. One could speculate about the decedent’s neurological or cognitive trajectory had he lived, but the circularity of such speculation, the lack of insight into the kinetics of such changes over time, and the poor correlation between uncomplicated p-tau reactivity (i.e., p-tau reactivity without neuronal loss) and neurological signs, are problematic.
Fig.1
Whole mount immunohistochemical stain for p-tau (AT8) in a 51-year-old retired professional boxer.
THE ROLE OF TAU PATHOLOGY IN DEMENTIA PUGILISTICA
As stated by Roberts:
“It has never been doubted, since it is implicit in the contest, that personal injury occurs in boxing, or that, rarely, accidents happen, as they do in other sports, which result in damage to cerebral functions and even death. Until recently it had not been suggested that the evidently transient incapacity usually sustained might result in permanent, slight but cumulative damage to delicate neural structures.”
The heavy neurotrauma exposure of the early 20th century boxing thus brought into medical consciousness the intuitively obvious conclusion that brain damage could occur in some participants, and that structural brain injury might be cumulative with time and exposure. A progressive Alzheimer-like neurodegenerative cascade from boxing-induced neurofibrillary degeneration, however, was hypothetical then as it is today. The modern concept of p-tau as a driver of disease likewise sets aside cumulative structural brain injury and instead opens up hypothesis-confirming lines of inquiry, perhaps prematurely, such as 1) tau biomarkers, such as in cerebrospinal fluid or on PET imaging studies [50]; 2) the role of low-n assembly intermediates of p-tau in experimental trauma versus control [51]; 3) the role of any of a number inflammatory mediators in tau phosphorylation [52]; 4) similarities and differences in tau isoform profiles in repetitive trauma versus conventional tauopathy [46]; and 5) templating behavior and prion-like conformers and strains in experimental constructs, among others [53]. The question of whether tau pathobiology is a primary or inherently pathogenic process in the first place seems reasonable to ask, given that p-tau is manifestly downstream in a multitude of conditions [54–57], and that the overwhelming majority of heavily exposed boxers are asymptomatic, with most of the rest having stationary neurological deficits.
The amyloid cascade hypothesis may provide some guidance as one attempts to understand the relationship between proteinopathy and clinical disease [58–60]. In terms of a hypothesis for disease etiology or an otherwise rate-limiting factor for disease pathogenesis [61], the role of Aβ in AD is considerably more robust scientifically than p-tau in dementia pugilistica. Pathogenic mutations in amyloid-β protein precursor (AβPP) within and near the Aβ coding region lead to familial autosomal dominant AD with near 100% penetrance. In Down syndrome patients, who possess an extra copy and overproduce AβPP, AD pathology as well as cognitive deterioration from baseline, appear consistently and early compared to sporadic AD and aging. Familial early-onset AD is associated with mutations not only in AβPP, but also in presenilin 1 and 2—components of the γ-secretase complex, required for processing of the AβPP into Aβ. Thus in AD, not only is there strict adherence to central dogma of molecular biology and genotype-phenotype relationships, there is also genetic adherence to genotype-enzyme-substrate phenotype relationships. In addition, animal models in AD are well characterized and consistently recapitulate hallmarks of AD pathology [62]. Therapeutic constructs that reduce Aβ have been shown to reduce behavioral and neurocognitive deficits in AD models [63]. These data have further been extended to clinical trials in humans, in which immunotherapy has been shown to reduce Aβ burden [64]. Yet in the case of AD, despite its robust molecular-genetic underpinnings and detailed extent to which it has been investigated in humans, all major therapeutic trials targeting Aβ have either shown no benefit or have performed worse than placebo [60]. The point here is that lesion- or protein-driven neurodegenerative disease constructs insufficiently address the complexity of neurodegenerative disease in humans, even in the case of AD, a much more homogeneous condition compared to dementia pugilistica, studied in exhaustive detail over three decades with all available technological advancements. By that analogy, the concept of p-tau as a driver of disease or target for therapy in dementia pugilistica seems premature.
CONCLUSIONS
Dementia pugilistica has evolved remarkably over time. Concussion-related hemorrhages, AD pathology, diverse neuropathology encompassing structural brain lesions, neurofibrillary pathology and diffuse Aβ plaques, and, most recently, geographic tauopathy have all been proposed as pathological substrates. The classic clinical presentation includes slurring dysarthia, pyramidal and extrapyramidal signs, ataxia, memory deficits, and personality changes, although the diagnosis is occasionally offered in more recent literature in asymptomatic young athletes and in retired athletes with other major diseases. Assuming that dementia pugilistica is exposure-related, the prevalence of dementia pugilistica or punch drunk syndrome appears to have peaked at 6%, and it makes sense in light of the markedly decreased exposure since World War II that classical dementia pugilistica has reduced to a rarity, notwithstanding subclinical and stationary p-tau deposits in asymptomatic athletes. On the other hand, since cumulative structural brain injury may have been the driver of disease for dementia pugilistica when it was most prevalent, and since single bouts have been shown, on rare occasions, to produce permanent deficits, boxing can never be said to be devoid of risk for permanent brain injury or even death.
Of particular concern is not simply the existence of structural brain injury, including cumulative structural brain injury, which obviously has occurred in some boxers, but the specific concept of repetitive trauma-induced neurodegenerative disease in the manner of AD, propagated by a proteinopathy. Clinicopathological data in boxers with extreme neurotrauma exposure leaves considerable doubt that such a process exists today with any frequency, while those anecdotal instances with evidence of progression tend to be confounded either by major co-morbidities, failure to exclude coincidental neurodegeneration disease, or outdated pathomorphological analyses that are not amenable to comparison with current concepts. Also striking is the paucity of new cases of dementia pugilistica in the literature, and in those few cases that have appeared, the lack of the neurological syndrome (dysarthria, ataxia, asymmetric hyperreflexia, etc.) that allowed identification of punch drunk syndrome or dementia pugilistica in the first place. At present, if one is rigorous with the human data, it remains unclear how, and indeed whether, traumatic brain injury from boxing crosses the threshold from structural damage to a progressive neurodegenerative cascade. On the other hand, the finding of p-tau in atypical patterns and distributions, even in asymptomatic athletes, indicates that more research is needed.
DISCLOSURE STATEMENT
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/17-0669).
REFERENCES
[1] | Arnold P ((1985) ) The Origins of Boxing. In History of Boxing, Deans International Publishing, London, pp. 6–33. |
[2] | Roberts A ((1969) ) Brain damage in boxers: A study of prevalance of traumatic encephalopathy among ex-professional boxers, Pitman Medical Scientific Publishing Co., London. |
[3] | Unterharnscheidt F , Taylor-Unterharnscheidt J ((2003) ) Chronic clinical findings. In Boxing: Medical Aspects. Elsevier Academic Press, London, pp. 322. |
[4] | Critchley M ((1957) ) Medical aspects of boxing, particularly from a neurological-standpoint. Br Med J 1: , 357–362. |
[5] | McCrory P , Zazryn T , Cameron P ((2007) ) The evidence for chronic traumatic encephalopathy in boxing. Sport Med 37: , 467–476. |
[6] | Clausen H ((2005) ) The risk of chronic traumatic brain injury in professional boxing: Change in exposure variables over the past century. Br J Sports Med 39: , 661–664. |
[7] | Battalia JE , Aschenbrener CA , Bennett DR ((1983) ) Brain injury in boxing. Council on scientific affairs. J Am Med Assoc 249: , 254–257. |
[8] | Wacquant L ((1995) ) The pugilistic point of view: How boxers think and feel about their trade. Theory Soc 24: , 489–535. |
[9] | Unterharnscheidt F , Taylor-Unterharnscheidt J ((2003) ) Pros and cons of boxing. Statements by physicians, officials, writers, moral theologians, politicians, etc. In Boxing: Medical Aspects. Elsevier Academic Press, London, pp. 636–736. |
[10] | Winterstein CE ((1937) ) Head injuries attributable to boxers. Lancet 230: , 719–722. |
[11] | Martland HS ((1928) ) Punch drunk. J Am Med Assoc 91: , 1103–1107. |
[12] | Parker HL ((1934) ) Traumatic encephalopathy (“punch drunk”) of professional pugilists. J Neurol Psychopathol 15: , 20–28. |
[13] | McKee AC , Cairns NJ , Dickson DW , Folkerth RD , Dirk Keene C , Litvan I , Perl DP , Stein TD , Vonsattel JP , Stewart W , Tripodis Y , Crary JF , Bieniek KF , Dams-O’Connor K , Alvarez VE , Gordon WA ((2016) ) The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 131: , 75–86. |
[14] | Millspaugh JA ((1937) ) Dementia pugilistica. U S Nav Med Bull 35: , 297–303. |
[15] | Jokl E , Guttman E ((1933) ) Neurologisch-psychiatrishe Untersuchungen an Boxern. Munch Med Wochenschr 80: , 560–562. |
[16] | Prusiner SB ((2012) ) A unifying role for prions in neurodegenerative diseases. Science 336: , 1511–1513. |
[17] | Critchley M ((1937) ) Nervous disorders in boxers. Med Annu 318–320. |
[18] | Carroll E ((1936) ) Punch Drunk. Am J Med Sci 191: , 706–712. |
[19] | Critchley M ((1949) ) e. Punch-drunk syndromes: The chronic traumatic encephalopathy of boxers, Maloine, Paris. |
[20] | Mawdsley C , Ferguson FR ((1963) ) Neurological disease in boxers. Lancet 2: , 795–801. |
[21] | Unterharnscheidt F ((1995) ) A neurologist’s reflections on boxing. II. Acute and chronic clinical findings secondary to central nervous system damage. Rev Neurol 23: , 833–846. |
[22] | McAlpine D , Page F ((1949) ) Midbrain syndrome in a boxer. Proc R Soc Med 42: , 792–793. |
[23] | Kremer M , Russell W , Ge S ((1947) ) A midbrain syndrome following head injury. J Neurol Neurosurg Psychiatry 10: , 49–60. |
[24] | Jordan BD ((2000) ) Chronic traumatic brain injury associated with boxing. Semin Neurol 20: , 179–185. |
[25] | Maurer K , Volk S , Gerbaldo H ((1997) ) Auguste D and Alzheimer’s disease. Lancet 349: , 1546–1549. |
[26] | Brandenburg W , Hallervorden J ((1954) ) Dementia pugilistica mit anatomischem Befund. Virchows Arch 325: , S680–S709. |
[27] | Brooks WS , Kwok JBJ , Kril JJ , Broe GA , Blumbergs PC , Tannenberg AE , Lamont PJ , Hedges P , Schofield PR ((2003) ) Alzheimer’s disease with spastic paraparesis and “cotton wool” plaques: Two pedigrees with PS-1 exon 9 deletions. Brain 126: , 783–791. |
[28] | Grahmann H , Ule G ((1957) ) Beitrag zur Kenntnis der chronischen cerebralen Krankheitsbilder bei Boxern. Psychiatr Neurol (Basel) 134: , 261–283. |
[29] | Neubuerger KT , Sinton DW , Denst J ((1959) ) Cerebral atrophy associated with boxing. AMA Arch Neurol Psychiatry 81: , 403–408. |
[30] | Courville C ((1962) ) Punch drunk: Its pathogenesis and pathology on the basis of a verified case. Bull Los Angel Neuro Soc 27: , 160–168. |
[31] | Spillane J ((1962) ) Five boxers. Br Med J 2: , 1205–1210. |
[32] | Payne E ((1968) ) Brains of boxers. Neurochirugia 11: , 173–188. |
[33] | Burger H , Minarovjech V ((1966) ) Sind die Regeln der AIBA fur den Gesundheitsschutz der Boxer ausreichend? Med Sport (Berl) 6: , 12–15. |
[34] | Betti CO , Ottino CA ((1969) ) Pugilistic encephalopathy. Acta Neurol Latinoam 15: , 47–51. |
[35] | Corsellis JA , Bruton CJ , Freeman-Browne D ((1973) ) The aftermath of boxing. Psychol Med 3: , 270–303. |
[36] | Ighodaro ET , Abner EL , Fardo DW , Lin A-L , Katsumata Y , Schmitt FA , Kryscio RJ , Jicha GA , Neltner JH , Monsell SE , Kukull WA , Moser DK , Appiah F , Bachstetter AD , Van Eldik LJ , Alzheimer’s Disease Neuroimaging Initiative. (ADNI), Nelson PT ((2017) ) Risk factors and global cognitive status related to brain arteriolosclerosis in elderly individuals. J Cereb Blood Flow Metab 37: , 201–216. |
[37] | Unterharnscheidt F , Taylor-Unterharnscheidt J ((2003) ) Chronic pathomorphological findings. In Boxing: Medical Aspects. Elsevier Academic Press, London, pp. 503. |
[38] | Roberts GW ((1988) ) Immunohistochemistry of neurofibrillary tangles in dementia pugilistica and Alzheimer’s disease: Evidence for a common genesis. Lancet 332: , 1456–1458. |
[39] | Roberts GW , Allsop D , Bruton C ((1990) ) The occult aftermath of boxing. J Neurol Neurosurg Psychiatry 53: , 373–378. |
[40] | Tokuda T , Ikeda S , Yanagisawa N , Ihara Y , Glenner GG ((1991) ) Re-examination of ex-boxers’ brains using immunohistochemistry with antibodies to amyloid β-protein and tau protein. Acta Neuropathol 82: , 280–285. |
[41] | Allsop D , Haga S , Bruton C , Ishii T , Roberts GW ((1990) ) Neurofibrillary tangles in some cases of dementia pugilistica share antigens with amyloid beta-protein of Alzheimer’s disease. Am J Pathol 136: , 255–260. |
[42] | Hof PR , Bouras C , Buee L , Delacourte A , Perl DP , Morrison JH ((1992) ) Differential distribution of neurofibrillary tangles in thecerebral cortex of dementia pugilistica and Alzheimer’s disease cases. Acta Neuropathol 85: , 23–30. |
[43] | Geddes JF , Vowles GH , Robinson SF , Sutcliffe JC ((1996) ) Neurofibrillary tangles, but not Alzheimer-type pathology, in a young boxer. Neuropathol Appl Neurobiol 22: , 12–16. |
[44] | McKee AC , Stern RA , Nowinski CJ , Stein TD , Alvarez VE , Daneshvar DH , Lee H-S , Wojtowicz SM , Hall G , Baugh CM , Riley DO , Kubilus CA , Cormier KA , Jacobs MA , Martin BR , Abraham CR , Ikezu T , Reichard RR , Wolozin BL , Budson AE , Goldstein LE , Kowall NW , Cantu RC ((2013) ) The spectrum of disease in chronic traumatic encephalopathy. Brain 136: , 43–64. |
[45] | Cabot RC , Scully RE , Mark EJ , McNeely WF , Ebeling SH , Drachman DA , Newell KL ((1999) ) Case 12-1999. N Engl J Med 340: , 1269–1277. |
[46] | Schmidt ML , Zhukareva V , Newell KL , Lee VM , Trojanowski JQ ((2001) ) Tau isoform profile and phosphorylation state in dementiapugilistica recapitulate Alzheimer’s disease. ActaNeuropathol 101: , 518–524. |
[47] | Areza-Fegyveres R , Rosemberg S , Castro RMRPS , Porto CS , Bahia VS , Caramelli P , Nitrini R ((2007) ) Dementia pugilistica with clinical features of Alzheimer’s disease. Arq Neuropsiquiatr 65: , 830–833. |
[48] | Nowak LA , Smith GG , Reyes PF ((2009) ) Dementia in a retired world boxing champion: Case report and literature review. Clin Neuropathol 28: , 275–280. |
[49] | Braak H , Thal DR , Ghebremedhin E , Del Tredici K ((2011) ) Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J Neuropathol Exp Neurol 70: , 960–969. |
[50] | Gandy S , Ikonomovic MD , Mitsis E , Elder G , Ahlers ST , Barth J , Stone JR , DeKosky ST ((2014) ) Chronic traumatic encephalopathy: Clinical-biomarker correlations and current concepts in pathogenesis. Mol Neurodegener 9: , 37. |
[51] | Ojo JO , Mouzon B , Algamal M , Leary P , Lynch C , Abdullah L , Evans J , Mullan M , Bachmeier C , Stewart W , Crawford F ((2016) ) Chronic repetitive mild traumatic brain injury results in reduced cerebral blood flow, axonal injury, gliosis, and increased T-tau and tau oligomers. J Neuropathol Exp Neurol 75: , 636–655. |
[52] | Collins-Praino LE , Corrigan F ((2017) ) Does neuroinflammation drive the relationship between tau hyperphosphorylation and dementia development following traumatic brain injury? Brain Behav Immun 60: , 369–382. |
[53] | Woerman AL , Aoyagi A , Patel S , Kazmi SA , Lobach I , Grinberg LT , McKee AC , Seeley WW , Olson SH , Prusiner SB ((2016) ) Tau prions from Alzheimer’s disease and chronic traumatic encephalopathy patients propagate in cultured cells. Proc Natl Acad Sci U S A 113: , E8187–E8196. |
[54] | Arendt T , Stieler JT , Holzer M ((2016) ) Tau and tauopathies. Brain Res Bull 126: , 238–292. |
[55] | Castellani RJ , Nunomura A , Lee H , Perry G , Smith MA ((2008) ) Phosphorylated tau: Toxic, protective, or none of the above. J Alzheimers Dis 14: , 377–383. |
[56] | Trojanowski JQ , Lee VMY ((2005) ) Pathological tau: A loss of normal function or a gain in toxicity? Nat Neurosci 8: , 1136–1137. |
[57] | Santacruz K , Lewis J , Spires T , Paulson J , Kotilinek L , Ingelsson M , Guimaraes A , DeTure M , Ramsden M , McGowan E , Forster C , Yue M , Orne J , Janus C , Mariash A , Kuskowski M , Hyman B , Hutton M , Ashe KH ((2005) ) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309: , 476–481. |
[58] | Hardy JA , Higgins GA ((1992) ) Alzheimer’s disease: The amyloid cascade hypothesis. Science 256: , 184–185. |
[59] | Castellani RJ , Lee H-G , Zhu X , Perry G , Smith MA ((2008) ) Alzheimer disease pathology as a host response. J Neuropathol Exp Neurol 67: , 523–531. |
[60] | Castellani RJ , Perry G ((2014) ) The complexities of the pathology-pathogenesis relationship in Alzheimer disease. Biochem Pharmacol 88: , 671–676. |
[61] | Selkoe DJ , Hardy J ((2016) ) The amyloid hypothesis of Alzheimer’sdisease at 25 years. EMBO Mol Med 8: , 595–608. |
[62] | Götz J , Ittner LM ((2008) ) Animal models of Alzheimer’s disease and frontotemporal dementia. Nat Rev 9: , 532–544. |
[63] | Schenk D , Barbour R , Dunn W , Gordon G , Grajeda H , Guido T , Hu K , Huang J , Johnson-Wood K , Khan K , Kholodenko D , Lee M , Liao Z , Lieberburg I , Motter R , Mutter L , Soriano F , Shopp G , Vasquez N , Vandevert C , Walker S , Wogulis M , Yednock T , Games D , Seubert P ((1999) ) Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400: , 173–177. |
[64] | Holmes C , Boche D , Wilkinson D , Yadegarfar G , Hopkins V , Bayer A , Jones RW , Bullock R , Love S , Neal JW , Zotova E , Nicoll JA ((2008) ) Long-term effects of Aβ42 immunisation in Alzheimer’s disease: Follow-up of a randomised, placebo-controlled phase I trial. Lancet 372: , 216–223. |


