
Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease
Abstract
Decades of research indicate mitochondria from Alzheimer’s disease (AD) patients differ from those of non-AD individuals. Initial studies revealed structural differences, and subsequent studies showed functional deficits. Observations of structure and function changes prompted investigators to consider the consequences, significance, and causes of AD-related mitochondrial dysfunction. Currently, extensive research argues mitochondria may mediate, drive, or contribute to a variety of AD pathologies. The perceived significance of these mitochondrial changes continues to grow, and many currently believe AD mitochondrial dysfunction represents a reasonable therapeutic target. Debate continues over the origin of AD mitochondrial changes. Some argue amyloid-β (Aβ) induces AD mitochondrial dysfunction, a view that does not challenge the amyloid cascade hypothesis and that may in fact help explain that hypothesis. Alternatively, data indicate mitochondrial dysfunction exists independent of Aβ, potentially lies upstream of Aβ deposition, and suggest a primary mitochondrial cascade hypothesis that assumes mitochondrial pathology hierarchically supersedes Aβ pathology. Mitochondria, therefore, appear at least to mediate or possibly even initiate pathologic molecular cascades in AD. This review considers studies and data that inform this area of AD research.
INTRODUCTION
Over 40 years ago, electron microscopy (EM) pictures of Alzheimer’s disease (AD) brains revealed altered mitochondrial infrastructures [1, 2]. Initial reports, though, offered limited speculation into the cause or significance of this basic observation. Later studies confirmed and extended the finding [3, 4]. In the early 1980 s, fluorodeoxyglucose positron emission tomography (FDG PET) studies showed brains from AD patients utilized less glucose than brains from control subjects [5–8], which piqued interest in a potential metabolism component for thisdisease [9–12]. Investigators subsequently attempted to explain reduced AD brain glucose utilization. Proposed hypotheses included impaired blood-brain barrier glucose transport [13], reduced tissue energy requirements due to reduced synaptic activity, a tissue-loss artifact, and lesions of energy metabolism enzymes [10].
Whether related or not to reduced FDG PET glucose utilization, around this time and shortly thereafter, studies in fact did reveal activity deficiencies in several bioenergetic flux-related enzymes. Some studies noted reductions in glycolysis enzymes [14]. Initially implicated mitochondria-localized enzymes included pyruvate dehydrogenase complex (PDHC) and α-ketoglutarate dehydrogenase complex (KGDHC) [15, 16]. Functional differences from control subjects were later demonstrated in additional Krebs cycle enzymes [17].
A single study from 1987 analyzed oxygen consumption in brain biopsy homogenates from dementia subjects [18]. Interestingly, under sub-maximal conditions oxygen consumption from the dementia patient homogenates exceeded that of the control subject homogenates. Mitochondrial uncoupling in the dementia subject homogenates potentially contributed to this perhaps unexpected finding. In contrast, PET, when used to quantify brain oxygen consumption in vivo, showed decreased oxygen consumption by AD brains [19, 20]. Consistent with the oxygen consumption PET data, Parker et al. in 1990 reported reductions in AD subject cytochrome oxidase (COX) activity [21].
Still, the origin and consequences of these seminal but descriptive observations remained unclear. An obvious consideration was whether mitochondrial or other bioenergetic lesions contributed to AD, or whether they simply represented a byproduct of the disease. Based on assumptions made by the amyloid cascade hypothesis, first proposed in the early 1990 s [22, 23], the latter possibility seemed more likely. Even when viewed from this perspective, though, accumulating data from the past two decades suggest if indeed mitochondrial-bioenergetic dysfunction represents a byproduct of more fundamental AD events, this dysfunction may yet play an important and possibly relatively upstream role in mediating AD dysfunction and degeneration. Other data, though, also argue mitochondrial dysfunction could represent an independent or perhaps even primary event in AD.
EVIDENCE OF SECONDARY MITOCHONDRIAL CASCADES IN AD
Sequential cleavages of the amyloid-β protein precursor (AβPP) produce a peptide byproduct called Aβ. A popular line of AD research argues Aβ, and perhaps specifically oligomeric conformations of a 42 amino acid-long Aβ species, initiates neurodegeneration [24, 25]. Cultured cells maintained in the presence of Aβ show reduced electron transport chain enzyme activities [26]. Aβ also impairs respiratory chain function in isolated mitochondria [27–29]. While such data indicate potential important relationships between Aβ and mitochondria might exist, they do not compellingly address the AD-relevance of an Aβ-induced mitochondrial lesion, let alone the presence of a resulting mitochondrialcascade.
A report from Cardoso et al. more directly assessed this question [30]. The authors added Aβ to the medium of human neuronal NT2 cells, which induced cell death. They similarly added Aβ to the medium of NT2 ρ0 cells, NT2 cells previously depleted of their mitochondrial DNA (mtDNA). Because they lack mtDNA, ρ0 cells do not produce key respiratory chain subunits and are respiration-incompetent. The Aβ treatment did not harm the ρ0 cells. These studies suggested mitochondria in general, and the respiratory chain specifically, may mediate Aβ toxicity. This study was perhaps the first to make a case for a possible Aβ-induced mitochondrial cascadein AD.
Subsequently, studies revealed AβPP and Aβ co-localize with mitochondria [31–37]. Prominent early observations came from models that featured an artificial expression of AβPP, and of Aβ overproduction. Because artificial expression or overproduction of a protein may affect its intracellular targeting, it is reasonable to question the physiologic relevance of these findings. However, analyses show mitochondria from AD subject autopsy brains do appear to contain Aβ [33, 35, 38, 39].
Research by Yan and colleagues sought to define how Aβ located within mitochondria might mediate mitochondrial dysfunction and result in a downstream pathologic cascade. In the first of these studies, that of Lustbader et al., Aβ bound a dehydrogenase protein the authors called the Aβ-binding alcohol dehydrogenase (ABAD) [31]. Blocking ABAD-Aβ physical interactions mitigated oxidative stress and apoptosis. In the second study, that of Du et al., Aβ bound cyclophilin D (cypD), a component of the mitochondrial transition pore. CypD knock-out reduced oxidative stress and apoptosis, and cypD knock-out preserved cognitive performance in AβPP transgenic mice [36]. The results of these studies are consistent with the possible presence of functionally important Aβ-induced, mitochondria-mediated pathologic cascades in AD.
AβPP itself contains a reported mitochondrial targeting motif [40–44]. When AβPP accesses mitochondria, it partly passes through the mitochondrial protein import apparatus, including the translocase of the outer mitochondrial membrane 40 kilodalton (TOMM40) protein pore. This passage ultimately does not complete, due to the presence of an AβPP acidic domain, and the mitochondrial import-arrested AβPP both clogs the import infrastructure and protrudes from the mitochondria and into the cytoplasm. The presence of arrested AβPP in the mitochondria (with its associated C-terminal end extended into the cytosol) reportedly reduces COX activity [40].
Altered calcium homeostasis represents yet another fundamental physiologic alteration Aβ can induce [45, 46]. In both cell and animal experimental models, Aβ appears to both enhance the ability of calcium to access the cytoplasm, and reduce the cell’s ability to lower its cytosolic calcium levels. Elevated calcium interferes with mitochondrial function, which reduces ATP production. An inhibition of oxidative phosphorylation additionally depolarizes mitochondria, which further impairs the ability of cells to buffer calcium loads. Based on this, some propose an AD “calcium hypothesis [47].” Under this scenario, Aβ-induced changes in calcium homeostasis adversely impact mitochondrial function, and through this drive other AD-associated changes in brain function. Disrupted calcium handling, therefore, could represent a mechanism via which secondary mitochondrial cascades initiated by Aβ ultimately drive this disease.
Other studies performed using transgenic mice designed to model human AD infer a meaningful role for mitochondrial damage (at least in these models). For example, Reddy et al. found that in mice expressing a mutant human AβPP transgene, an apparent compensatory upregulation of respiratory chain enzyme subunits preceded both behavioral change and Aβ plaque deposition [48].
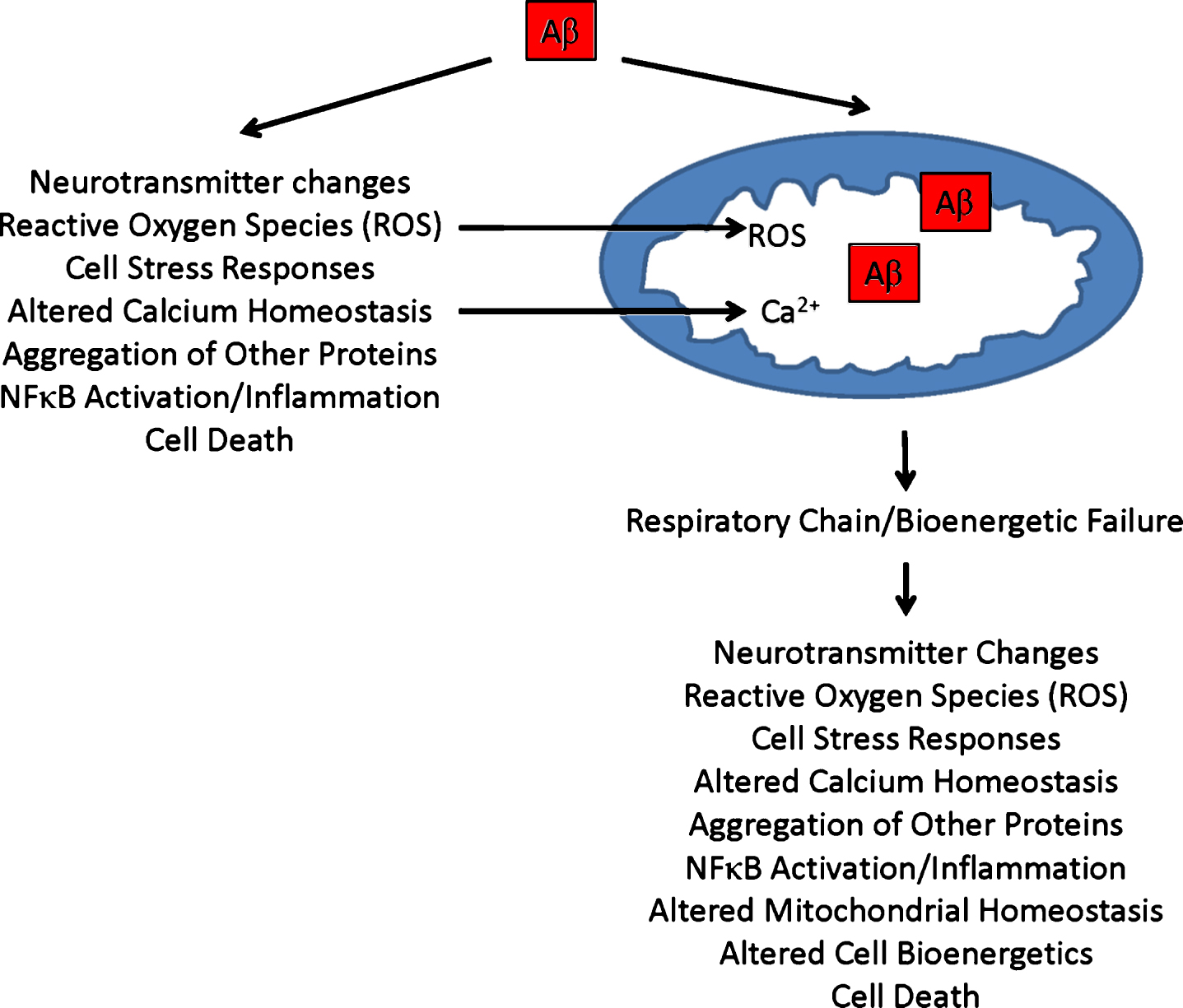
Recent reports indicate either AβPP overexpression or Aβ exposure affects various aspects of mitochondrial function [43]. In both AβPP transgenic mice and cultured cells treated with Aβ, the balance between mitochondrial fission and fusion shifts in favor of fission [49–52]. Fewer mitochondria also distribute throughout dendrites. Analyses of brains from AD patients show similar changes [3]. If Aβ causes these mitochondrial perturbations in autopsy brains, and these mitochondrial perturbations in turn contribute to neuron dysfunction or degeneration, they could represent evidence of an Aβ-induced, mitochondria-mediated cascade [43, 53]. Figure 1 illustrates potential causes and consequences of a secondary mitochondrial cascade.
Fig.1
Secondary mitochondrial cascade. Secondary mitochondrial cascades are compatible with the amyloid cascade hypothesis, and could mediate Aβ toxicity. As illustrated, Aβ can directly introduce various AD-associated functional changes and pathologies, and directly or indirectly cause mitochondrial dysfunction. Aβ-induced mitochondrial dysfunction, in turn, could further contribute to or initiate additional AD-associated functional changes and pathologies.
EVIDENCE OF A PRIMARY MITOCHONDRIAL CASCADE IN AD
Interestingly, some of the more influential discoveries of altered energy metabolism enzymes arose from studies of non-brain tissues, and were latersubsequently documented in brain parenchyma. Parker and colleagues first documented lower AD COX activity in platelets [21]. Blass, Gibson, and colleagues first showed reduced KGDHC activity in fibroblasts, and lowered transketolase activity in red blood cells [16]. Reports from the mid-1980 s noted altered patterns of glucose and oxygen consumption in fibroblasts from AD subjects [54, 55].
While biochemical defects in AD brain could represent a consequence of neurodegeneration, neurodegeneration per se should not directly cause specific biochemical defects outside the neuro-axis and in non-degenerating tissues. Other structural brain changes that could affect energy metabolism, such as synaptic loss, similarly should not directly cause non-neural biochemical defects. AβPP or Aβ could perhaps drive biochemical changes in different tissues provided their expression and production occurs in those tissues. To this point, AβPP does appear outside of the brain, but expresses as different isoforms. Aβ also exists in multiple tissues outside of the brain, including blood vessels, skin, subcutaneous tissues, intestine, and muscle [56]. To what extent peripheral processing of AβPP to Aβ occurs locally, or whether Aβ simply exports to these tissues, remains unknown although it increasingly appears that some tissues do locally generate Aβ. Two examples include muscle and platelets [57–59].
The presence of distinct, definable biochemical features outside the brains of AD subjects and within multiple tissues suggests at a biochemical level, AD is not a brain-limited process or event. At the very least, it is hard to see how a single, stochastic Aβ oligomerization-aggregation event occurring within the brain could alter either AβPP homeostasis or energy metabolism across multiple tissues. Even if altered Aβ homeostasis does contribute to non-brain bioenergetic or mitochondrial features seen in AD subjects, the question of why Aβ homeostasis itself changes across multiple tissues warrantsconsideration.
An alternative possibility is that altered energy metabolism in AD subjects exists independently of AβPP or Aβ, and that altered energy metabolism drives changes in AβPP and Aβ homeostasis. Existing data support this possibility. In cell culture experiments, Gabuzda et al. found COX inhibition shifts AβPP processing toward the amyloidogenic pathway that ultimately produces Aβ [60]. Mitochondria-generated reactive oxygen species may play a particularly important role in shifting AβPP processing to Aβ [61]. Other cell culture studies suggest interfering with cell bioenergetics shifts AβPP processing away from its non-amyloidogenic pathway, and presumably towards the amyloidogenic pathway [62, 63].
One study found transgenic mice that concurrently expressed a mutant human AβPP gene and proof reading-defective mtDNA polymerase γ (mtPOLG) showed enhanced Aβ plaque deposition [64]. Due to the presence of the mutated mtPOLG transgene, these mice acquire excess somatic mtDNA mutations and mitochondrial dysfunction [65, 66]. This finding links mitochondrial dysfunction to plaque deposition. Other studies performed using AD mouse models report similar links, although the direction of the relationship differed. Moraes and colleagues previously reported, in two studies using different approaches, that interfering with respiratory chain assembly actually reduces Aβ plaque deposition[67, 68]. In one case, the authors induced mitochondrial dysfunction through targeted knockout of the gene for COX10, which encodes a farnesyltransferase required for COX assembly. In the other, they expressed a mitochondria-targeted restriction enzyme that cleaves mtDNA. It is unclear why driving mitochondrial dysfunction through different approaches produced either increased or decreased plaque deposition. Most likely, methodologic factors are relevant. In the Moraes group studies, the mice produced less respiratory chain infrastructure, and with mtPOLG driving mitochondrial dysfunction aberrant rather than less respiratory chain infrastructure likely resulted. Other potential factors could include differential effects on cell unfolded protein responses. Regardless, studies such as these link mitochondria to plaque deposition.
In another study, Scheffler et al. used a selective breeding strategy to create AβPP transgenic mice that varied primarily in the origin of their mitochondria [69]. More specifically, the authors created mice that contained mitochondria from different mouse strains. Ultimately, the various groups of mice differed only in their mtDNA sequences. This study reflected two previous studies performed in cytoplasmic hybrid (cybrid) cell lines, in which the investigators generated human cell lines containing mtDNA from different individuals [70, 71]. Both studies created a group of cybrid lines in which the mtDNA came from individuals with AD, and a group of cybrid lines in which the mtDNA came from age-matched, control subjects. In each case, the cybrid lines that contained the mtDNA form the AD subjects produced (or at least retained) more Aβ.
The use of cybrid cell lines that contain mtDNA from AD subjects, herein referred to as “AD cybrids,” warrants additional consideration. Cybrid studies in general are consistent with the presence of a primary mitochondrial cascade in AD. AD cybrid cell lines were first created in the 1990 s to address the specific question of whether mtDNA contributes to lower platelet COX activity in AD subjects [72, 73]. First, the investigators removed the endogenous mtDNA from human neuronal cell lines, either the SH-SY5Y neuroblastoma cell line or the NT2 teratocarcinoma cell line, thereby creating SH-SY5Y and NT2 ρ0 cell lines [73–75]. Next, they isolated platelets from blood samples taken from subjects with and without AD. Briefly maintaining platelets from individual subjects with ρ0 cells in the presence of a detergent allows for the mixing of platelet and ρ0 cell cytoplasms. Selecting specifically for cells that contain a ρ0 cell nucleus and platelet mitochondria results in the isolation of a unique cybrid line. Because the nuclear component of cybrid cell lines prepared using a particular ρ0 cell line is similar if not identical, different cybrid cell lines generally contain the same nuclear DNA [76]. As for components transferred from platelets, the only component that can perpetuate over time is the mtDNA that is contained within the platelet mitochondria. As the cells divide, all other platelet-derived components degrade and dilute. Ultimately, the most fundamental difference between different cybrid cell lines prepared on the same nuclear background is their mtDNA sequence. The transfer of platelet mitochondria, and the mtDNA they contain, to ρ0 cells to create cybrid cells restores the ability of the former ρ0 cells to respire. In this case, the respiratory chains of the cybrid cells jointly consists of subunits encoded from the nuclear DNA of the original cell line nucleus and the mtDNA from the particular platelet donor [76]. Between cybrid cell lines prepared on the same nuclear background, the nuclear DNA-encoded respiratory chain subunits are presumably identical. The mtDNA-encoded respiratory chain subunits, though, differ because mtDNA sequences vary between individuals. Differences between mtDNA sequences affect respiratory chain function, so that cybrid cell lines containing mtDNA from different donors vary in terms of respiratory function. This in turn leads to differences in mitochondrial function, which mediates effects on a variety of cell characteristics.
Studies generally report that although COX activities between individual cybrid cell lines prepared from AD or control subject platelets do overlap, mean COX activity in groups of AD cybrids is lower than that of the comparative control groups [72, 73, 77–79]. In these experiments, a transfer of AβPP or Aβ is much less likely to drive the observed lower mean COX activity. Rather, cybrid data indicate mtDNA at least partly accounts for the observed reduction in AD subject platelet mitochondria COX activity. The magnitude of the difference between COX activity means varies between studies, but generally reflects a 15–40% activity reduction [80]. Methodologic differences likely contribute to this range.
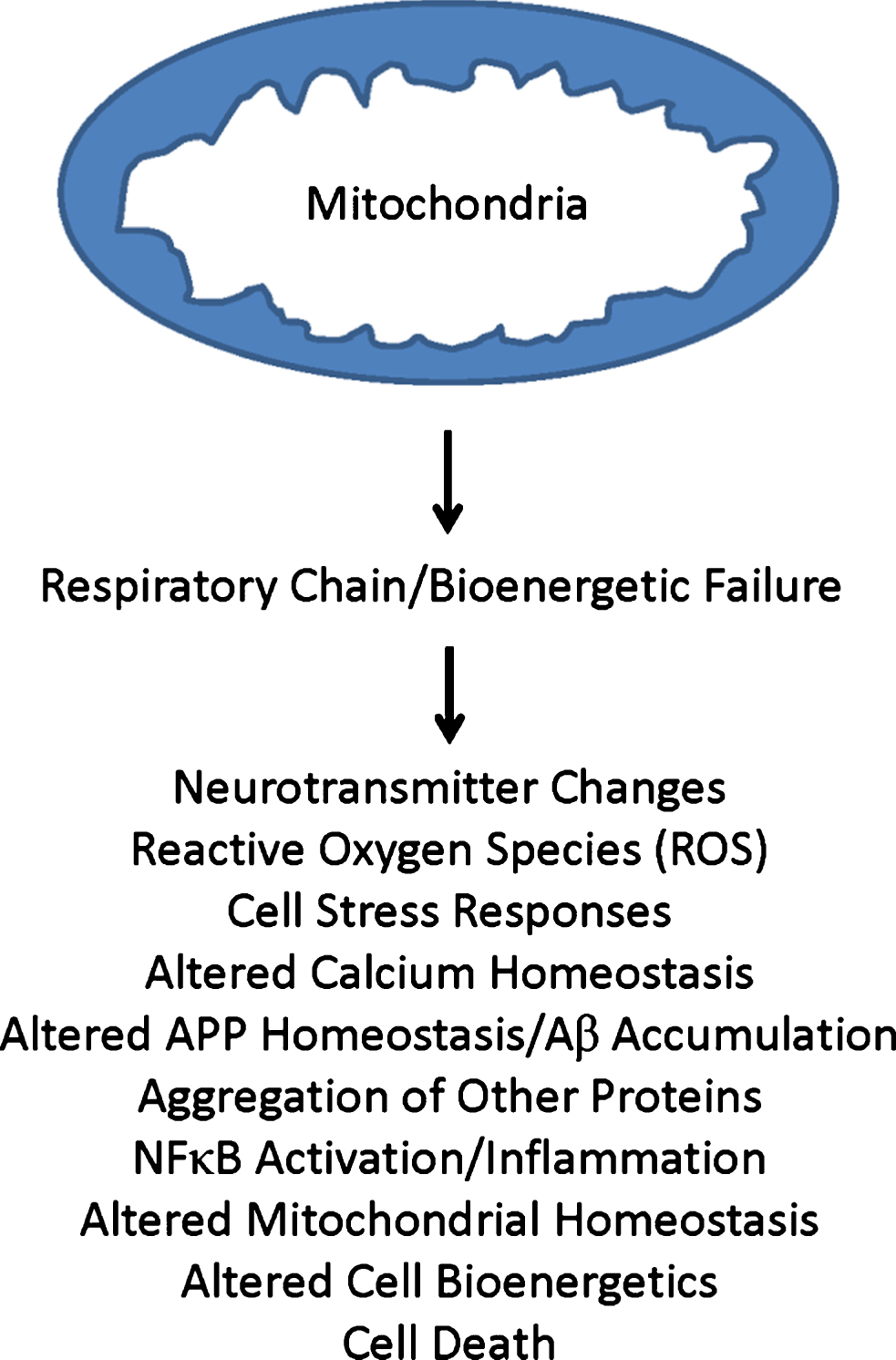
Importantly, differences between AD and control cybrid lines are not limited to COX activity. While values among AD and control lines generally overlap for measured parameters, mean values for a given group of AD cybrid lines occasionally are higher or lower than their comparative control group cybrid lines [70–73, 77–79, 81–97]. Increased parameters include markers of oxidative stress, Aβ production, stress pathway activation, NFκB activation, apoptotic signaling, and mitochondrial fission. Decreased parameters include mitochondrial membrane potential, ATP levels, oxygen consumption, glycolysis flux, calcium homeostasis, and peroxisome proliferator-activated receptor γ related complex α (PGC1α). These differences tend to recapitulate features observed in studies of brains from AD subjects. Because the only consistent difference between cybrid lines of AD and control groups is whether the mtDNA they contain originated from AD versus control subjects, and the mtDNA within the cybrid lines derived from a non-brain, non-degenerating tissue, and it is not clear how transferred AβPP or Aβ could account for the observed biochemical and molecular changes, cybrid data support the presence of a primary mitochondrial cascade in AD (Fig. 2).
Fig.2
Primary mitochondrial cascade. A primary mitochondrial cascade is incompatible with the amyloid cascade hypothesis. Under this scenario, impaired mitochondrial function and associated bioenergetic changes alter Aβ homeostasis and lead to an accumulation of Aβ. Aβ may or may not in turn contribute to the development of other AD-associated functional changes and pathologies.
The “mitochondrial cascade hypothesis” represents one formal attempt to explain how mitochondria may serve as the primary generator of AD [98–101]. This mitochondrial cascade hypothesis incorporates data from genetic, biochemical, molecular, cell biology, animal, clinical endophenotype, and epidemiologic studies. Also, as mitochondrial function generally declines during aging [102], it tries to bridge AD and aging research by providing a platform that potentially explains why advancing age represents the single greatest AD risk factor [103]. It is important to note various versions of this general postulate exist, and that some of these versions similarly identify a role for mtDNA, be it in the form of acquired, somatic mtDNA mutations or in the form of inherited variants [21, 104, 105]. The mitochondrial cascade hypothesis, with the intent of acknowledging data that support both views, proposes inherited mtDNA variants influence mitochondrial function and aging, and in the brain somatic mtDNA mutations that influence mitochondrial function accumulate with advancing age.
ASSUMPTIONS AND LIMITATIONS OF THE (PRIMARY) MITOCHONDRIAL CASCADE HYPOTHESIS
A primary mitochondrial cascade hypothesis neither depends on nor addresses the question of whether Aβ, in any of its forms, contributes to AD dysfunction or degeneration. Rather, it infers mitochondrial function or cell bioenergetic states meaningfully alter either AβPP production, intracellular AβPP targeting, processing of AβPP to Aβ monomers, the formation of Aβ oligomers or fibrils, or the removal of Aβ. Toxic or non-toxic amyloid cascades may exist, but if so, they are secondary events.
Similarly, a primary mitochondrial cascade hypothesis neither depends on nor addresses the question of whether aggregation of tau protein into tangles, or the presence of tau tangles themselves, contributes to AD dysfunction or degeneration. Instead, it infers mitochondrial function or cell bioenergetic states meaningfully alter either tau production, intracellular targeting, processing, post-translational modification, oligomer or fibril formation, or removal. Tangles or other tau protein derivatives may or may not function in a toxic fashion, but if they do, it represents a secondaryevent.
Undoubtedly, in the neurodegenerative disease field protein aggregation and mitochondrial dysfunction commonly occur. Existing literature does not resolve whether one of these pathologies consistently drives the other. It is nevertheless worth mentioning new reports that indicate on a fundamental level, bioenergetic states and mitochondria profoundly influence protein aggregation. In one study, Patel et al. showed that ATP functions as a “hydrotrope” [106, 107]. Essentially, its hydrophobic nucleotide portion associates with hydrophobic protein segments, while its hydrophilic phosphate group maintains the complex in a soluble state. At physiologic ATP concentrations, its hydrotrope properties prevent proteins that tend to self-aggregate from self-aggregating. As ATP concentrations fall to intermediate levels, this hydrotrope effect weakens and oligomers form. As ATP levels fall to low levels, fibrils then begin to assemble. In another study, Ruan et al. showed that mitochondria act as a sink for aggregation-prone proteins, and mitochondrial proteases degrade those proteins following their importation [108]. A loss of this “mitochondria as guardian in cytosol” (MAGIC; a term used by the authors to describe this phenomenon) function could in general promote protein aggregation.
When it comes to MAGIC, though, it is also conceivable that a primary protein overload may drive mitochondrial dysfunction, and through this instigate a secondary mitochondrial cascade. This could prove especially pertinent to cases of AD that occur in the presence of deterministic AβPP, presenilin 1 (PSEN1), or presenilin 2 (PSEN2) gene mutations. It is therefore possible that some of what we now consider AD arises through a primary mitochondrial cascade, while some of what we now consider AD essentially involves a secondary mitochondrial cascade.
It is still possible that mitochondrial cascades (either as primary or secondary events) are in fact not important in AD. If so, it would imply that observed differences between mitochondria from AD and non-AD brains constitute a disease-associated but non-contributing biomarker of the responsible process, or an end-stage artifact of the responsible process. It is more difficult to account for non-brain differences in mitochondria and mitochondrial function, but potential explanations are still possible. For example, a genetic parameter that influences AD risk could lead to a physiologically diffuse yet unrelated change in mitochondria or their function. APOE or perhaps TOMM40 genotypes could to some extent conceivably mediate such a phenomenon [109–112]. Similarly, over the course of many years a lifestyle characteristic could lead to a diffuse yet unrelated change tomitochondria.
Despite these caveats, extensive literature does reveal mitochondria from AD subjects (i.e., the cybrid literature) or inducing mitochondrial dysfunction recapitulates a number of AD-associated molecular events. Mitochondria also undoubtedly critically contribute to an array of cell processes, including oxidative stress, calcium homeostasis, and cell death. These factors would seem to argue anatomically widespread changes to mitochondrial function in AD subjects represent more than simply coincidental events or disease artifacts.
IMPLICATIONS FOR THE TEMPORAL AND SPATIAL POSITIONING OF AD BIOMARKERS
Investigators currently use specific cerebrospinal fluid (CSF) and neuroimaging-derived biomarkers to document transitions within the brain, and to link those transitions to AD. These include measurements of CSF Aβ and tau protein, PET-based visualization of fibrillar Aβ and tau, and magnetic resonance imaging-inferred reductions in hippocampal volume. It is possible to temporally order these biomarker changes [113, 114]. Initial shifts occur in the Aβ measurements, which manifest as decreased CSF Aβ (particularly Aβ42) or as an arbitrarily defined, supra-threshold accumulation of parenchymal Aβ plaques. Next, CSF tau levels fall, and parenchymal, fibrillar tau accumulations start to extend beyond the medial temporal lobes. After this, hippocampal volumes shrink and shortly thereafter performance on cognitive tests typically falls below expectations.
This temporal ordering is consistent with the idea of a primary amyloid cascade, but relevant studies also contribute data that are harder to incorporate. For example, Aβ plaque deposition typically precedes cognitive decline by 1–3 decades [113–118]. This suggests Aβ-induced physiologic damage lags substantially behind its physical deposition, and infers Aβ toxicity is at most subtle. In response to this, some now speculate Aβ itself is insufficient to cause the disease, but instead initiates AD by triggering a critical change in tau biology [114, 119, 120]. This change in tau biology, either by itself or by working in conjunction with Aβ (more specifically oligomers of Aβ42), ultimately destroys the hippocampus and causes cognitive decline. On a conceptual level, this possibility would not rule out the presence of a secondary mitochondrial cascade.
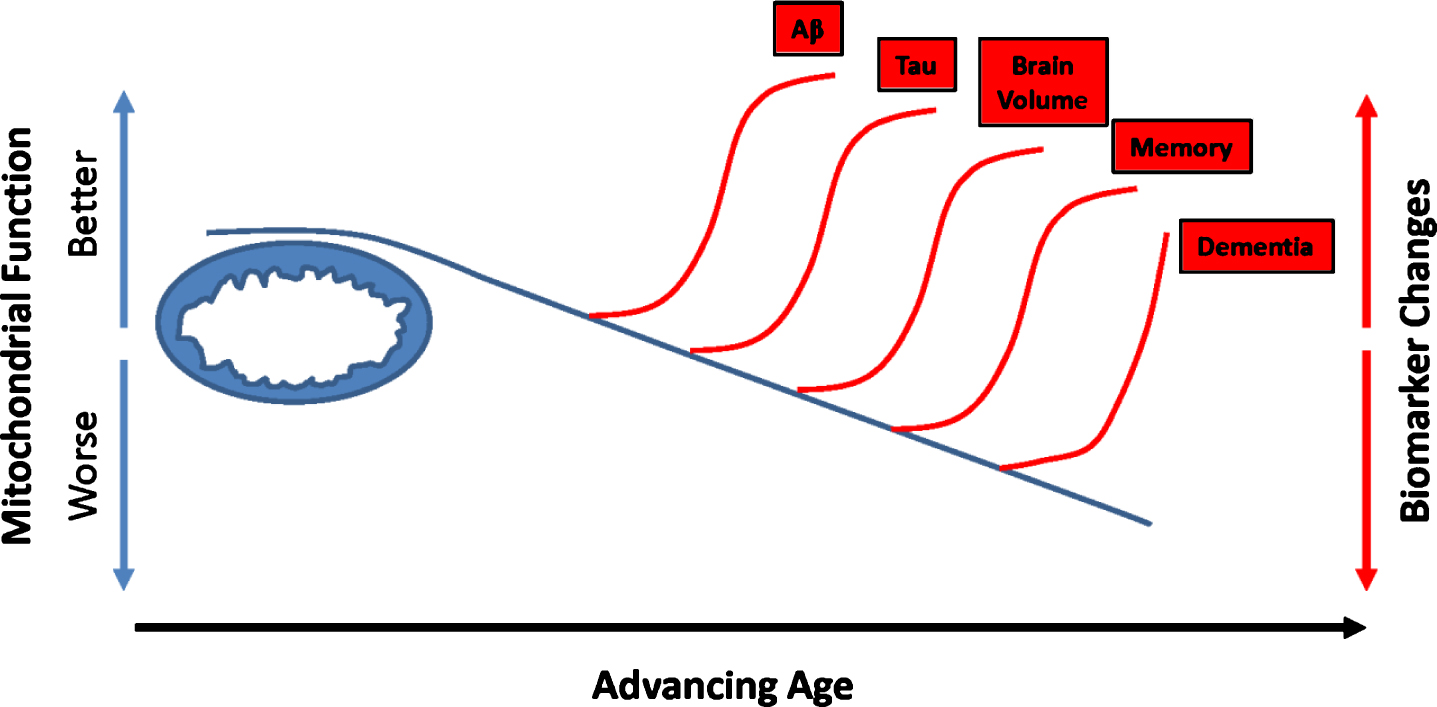
This temporal scheme emphasizes biomarker changes investigators can detect over the course of a longitudinal study, or by analyzing different cross-sectional studies. Some studies, though, suggest when it comes to predicting or at least monitoring AD risk, particular AD-relevant biomarkers potentially predate low Aβ CSF levels or Aβ plaque deposits [121]. In one relevant study, investigators analyzed diary entries from young nuns (written mostly at the beginning of their third decade) for idea density and grammatical complexity [122]. Nuns who wrote with lower idea density were more likely to develop AD in old age. In another study, individuals who reached their highest level of employment in their third or fourth decade were subsequently more likely to develop AD than individuals who continued to advance through employment hierarchies [123]. The authors speculated in this case, a relative premature peaking in one’s employment trajectory could possibly reflect a manifestation of incipient AD. Additional biomarker studies report similar findings and further suggest mitochondrial function or energy metabolism could constitute a relevant factor. Middle-aged individuals with an increased lifetime AD risk, as defined by the presence of an APOE4 allele or a maternal family history (a marker of mtDNA inheritance) of the disease, are more likely to demonstrate AD-like changes on FDG PET scans [124–126]. As a group, middle-aged people with AD-affected mothers also have reduced (relative to middle-aged individuals without an AD-affected mother) platelet mitochondria COX activity and exhibit brain regions with reduced volumes [127–132]. Middle-aged children of AD mothers who also carry an APOE4 allele perform relatively less well on memory testing [133]. Other similarly structured studies using different biomarker measurements show essentially consistent findings [134–138]. These data suggest when it comes to brain aging and AD, inherited or acquired energy metabolism parameters could predate Aβ changes. Results from at least one direct biochemical study of brain tissue are consistent with these findings from living subjects [139]. On a conceptual level, this is compatible with the presence of a primary mitochondrial cascade (Fig. 3).
Fig.3
Could mitochondrial function influence recognized AD biomarker changes? Studies that track dynamic biomarker shifts suggest changes in Aβ (lower CSF levels or plaque accumulation) precede tau changes (lower CSF tau or tangle accumulation beyond the medial temporal regions). Reductions in brain volume (hippocampal volumes) follow Aβ and tau changes. Declining memory abilities and dementia eventually occur. Additional data, though, suggest metabolism-relevant characteristics may distinguish high AD risk individual from low AD risk individuals before detectable Aβ changes occur. Mitochondrial function also changes with advancing age. It is reasonable to consider whether mitochondria define thresholds at which these biomarker changes begin to manifest.
Biomarker spatial distributions also warrant consideration. Within the brain, Aβ plaque and tau tangle deposition follow predictable anatomical patterns. Initially, plaques occur predominantly within the brain’s default mode network, a region that features high levels of aerobic glycolysis [140]. A secondary mitochondrial cascade would infer that Aβ causes aerobic glycolysis in this region, while a primary mitochondrial cascade would infer aerobic glycolysis in some way enhances Aβ deposition. Similarly, a secondary mitochondrial cascade would infer that outside the brain, brain-derived Aβ causes changes to mitochondrial function [80] and calcium homeostasis [141–143], while a primary mitochondrial cascade would infer those changes arise independent of Aβ, and perhaps drive local Aβ deposition [70, 72].
Ultimately, the cascades that drive AD biomarkers will influence how we define this disease. If a primary amyloid cascade drives AD, then persons who lack an AD clinical phenotype but have amyloid plaques may arguably have AD; a current construct refers to this state as “preclinical AD” [144]. Further, those with AD phenotypes that lack amyloid plaques and an obvious alternative clinical diagnosis arguably do not have AD; a current construct refers to this state as “suspected non-Alzheimer pathology” (SNAP) [145]. Conversely, if a primary mitochondrial cascade drives AD, then the presence of plaques would essentially function as a biomarker (rather than a cause) of changing brain bioenergetics. Those with a primary energy metabolism failure, a typical AD phenotype, but not plaques could thus potentially represent “plaque-negative” AD cases.
IMPLICATIONS FOR THERAPEUTIC DEVELOPMENT
Mitochondrial and amyloid cascades could co-exist. A secondary mitochondrial cascade may mediate damage caused by a primary amyloid cascade, or amyloid produced as part of a primary mitochondrial cascade could itself cause harm. Therapies targeting Aβ may ultimately reveal the extent to which Aβ contributes to AD. If these approaches robustly benefit AD patients, then Aβ likely plays a substantial and potentially proximal role. If Aβ toxicity plays a minimal or downstream role, such approaches will probably confer at best minimal benefits. Clinical efficacy would not rule out, though, the concomitant presence of a mitochondrialcascade. Obviously, if a primary amyloid cascade drives AD by inducing a secondary mitochondrial cascade, then eliminating Aβ would in general help to prevent the secondary mitochondrial cascade. AD investigators who believe in a secondary mitochondrial cascade are meanwhile also developing interventions that would ideally interrupt very specific parts of an Aβ-dependent mitochondrial cascade. Some efforts include deploying small molecule inhibitors of previously documented Aβ-mitochondria interactions [37, 146]. Other interventions currently in development intend to block Aβ-induced changes in mitochondrial physiology. Preventing an Aβ-induced increase in mitochondrial fission represents one such approach [147].
Addressing a primary mitochondrial cascade may require unique strategies. These strategies could focus on preventing age-related declines in mitochondrial function, for example through exercise or diet. Pharmacologic manipulations that enhance aerobic or other aspects of mitochondrial function, or overall cell bioenergetics, could prove beneficial [148]. Proposed interventions include the use of molecules that can enhance bioenergetic fluxes or increase mitochondrial mass [149].
CONCLUSIONS
AD features mitochondrial and bioenergetic alterations that could contribute to the development or progression of the disease. Upstream pathologies, including Aβ, may influence mitochondrial and bioenergetic function, and thereby initiate a secondary mitochondrial cascade. Alternatively, a primary mitochondrial cascade might represent a proximal cause of many AD cases, and directly perturb vital brain functions or introduce other pathologies that secondarily disrupt normal brain physiology and cause neurodegeneration.
Data support the possibility of both primary and secondary mitochondrial cascades in AD. The presence of a secondary mitochondrial cascade does not contradict, and is consistent with, the potential presence of a primary amyloid cascade. The presence of mitochondrial changes outside of the brain in AD, the fact that Aβ plaques deposit in brain regions defined by specific bioenergetic conditions, that mitochondrial and bioenergetic changes seem to temporally precede detectable changes in Aβ homeostasis, and the ability of mitochondrial dysfunction to affect a variety of AD-associated pathologies argue in favor of a primary mitochondrial cascade. Regardless of which view is correct, both primary and secondary mitochondrial cascades currently represent reasonable AD therapeutic targets.
ACKNOWLEDGMENTS
This author is supported by the University of Kansas Alzheimer’s Disease Center (P30AG-035982).
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/17-0585).
REFERENCES
[1] | Johnson AB , Blum NR ((1970) ) Nucleoside phosphatase activities associated with the tangles and plaques of Alzheimer’s disease: A histochemical study of natural and experimental neurofibrillary tangles. J Neuropathol Exp Neurol 29: , 463–478. |
[2] | Wisniewski H , Terry RD , Hirano A ((1970) ) Neurofibrillary pathology. J Neuropathol Exp Neurol 29: , 163–176. |
[3] | Hirai K , Aliev G , Nunomura A , Fujioka H , Russell RL , Atwood CS , Johnson AB , Kress Y , Vinters HV , Tabaton M , Shimohama S , Cash AD , Siedlak SL , Harris PL , Jones PK , Petersen RB , Perry G , Smith MA ((2001) ) Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci 21: , 3017–3023. |
[4] | Baloyannis SJ ((2006) ) Mitochondrial alterations in Alzheimer’s disease. J Alzheimers Dis 9: , 119–126. |
[5] | Ferris SH , de Leon MJ , Wolf AP , Farkas T , Christman DR , Reisberg B , Fowler JS , Macgregor R , Goldman A , George AE , Rampal S ((1980) ) Positron emission tomography in the study of aging and senile dementia. Neurobiol Aging 1: , 127–131. |
[6] | Foster NL , Chase TN , Fedio P , Patronas NJ , Brooks RA , Di Chiro G ((1983) ) Alzheimer’s disease: Focal cortical changes shown by positron emission tomography. Neurology 33: , 961–965. |
[7] | Friedland RP , Budinger TF , Ganz E , Yano Y , Mathis CA , Koss B , Ober BA , Huesman RH , Derenzo SE ((1983) ) Regional cerebral metabolic alterations in dementia of the Alzheimer type: Positron emission tomography with [18F]fluorodeoxyglucose. J Comput Assist Tomogr 7: , 590–598. |
[8] | de Leon MJ , Ferris SH , George AE , Christman DR , Fowler JS , Gentes C , Reisberg B , Gee B , Emmerich M , Yonekura Y , Brodie J , Kricheff II , Wolf AP ((1983) ) Positron emission tomographic studies of aging and Alzheimer disease. AJNR Am J Neuroradiol 4: , 568–571. |
[9] | Hoyer S ((1993) ) Brain oxidative energy and related metabolism, neuronal stress, and Alzheimer’s disease: A speculative synthesis. J Geriatr Psychiatry Neurol 6: , 3–13. |
[10] | Blass JP , Zemcov A ((1984) ) Alzheimer’s disease. A metabolic systems degeneration? Neurochem Pathol 2: , 103–114. |
[11] | Swerdlow R , Marcus DL , Landman J , Kooby D , Frey W 2nd , Freedman ML ((1994) ) Brain glucose metabolism in Alzheimer’s disease. Am J Med Sci 308: , 141–144. |
[12] | Beal MF ((1995) ) Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol 38: , 357–366. |
[13] | Marcus DL , Freedman ML ((1997) ) Decreased brain glucose metabolism in microvessels from patients with Alzheimer’s disease. Ann N Y Acad Sci 826: , 248–253. |
[14] | Meier-Ruge W , Iwangoff P , Reichlmeier K ((1984) ) Neurochemical enzyme changes in Alzheimer’s and Pick’s disease. Arch Gerontol Geriatr 3: , 161–165. |
[15] | Sorbi S , Bird ED , Blass JP ((1983) ) Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Ann Neurol 13: , 72–78. |
[16] | Gibson GE , Sheu KF , Blass JP , Baker A , Carlson KC , Harding B , Perrino P ((1988) ) Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer’s disease. Arch Neurol 45: , 836–840. |
[17] | Gibson GE , Starkov A , Blass JP , Ratan RR , Beal MF ((2010) ) Cause and consequence: Mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochim Biophys Acta 1802: , 122–134. |
[18] | Sims NR , Finegan JM , Blass JP , Bowen DM , Neary D ((1987) ) Mitochondrial function in brain tissue in primary degenerative dementia. Brain Res 436: , 30–38. |
[19] | Frackowiak RS , Pozzilli C , Legg NJ , Du Boulay GH , Marshall J , Lenzi GL , Jones T ((1981) ) Regional cerebral oxygen supply and utilization in dementia. A clinical and physiological study with oxygen-15 and positron tomography. Brain 104: , 753–778. |
[20] | Fukuyama H , Ogawa M , Yamauchi H , Yamaguchi S , Kimura J , Yonekura Y , Konishi J ((1994) ) Altered cerebral energy metabolism in Alzheimer’s disease: A PET study. J Nucl Med 35: , 1–6. |
[21] | Parker WDJr , Filley CM , Parks JK ((1990) ) Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology 40: , 1302–1303. |
[22] | Hardy J , Allsop D ((1991) ) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12: , 383–388. |
[23] | Hardy JA , Higgins GA ((1992) ) Alzheimer’s disease: The amyloid cascade hypothesis. Science 256: , 184–185. |
[24] | Hardy J , Selkoe DJ ((2002) ) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: , 353–356. |
[25] | Walsh DM , Selkoe DJ ((2007) ) A beta oligomers - a decade of discovery. J Neurochem 101: , 1172–1184. |
[26] | Pereira C , Santos MS , Oliveira C ((1998) ) Mitochondrial function impairment induced by amyloid beta-peptide on PC12 cells. Neuroreport 9: , 1749–1755. |
[27] | Canevari L , Clark JB , Bates TE ((1999) ) beta-Amyloid fragment 25-35 selectively decreases complex IV activity in isolated mitochondria. FEBS Lett 457: , 131–134. |
[28] | Casley CS , Canevari L , Land JM , Clark JB , Sharpe MA ((2002) ) Beta-amyloid inhibits integrated mitochondrial respiration and key enzyme activities. J Neurochem 80: , 91–100. |
[29] | Parks JK , Smith TS , Trimmer PA , Bennett JP Jr , Parker WD Jr ((2001) ) Neurotoxic Abeta peptides increase oxidative stress in vivo through NMDA-receptor and nitric-oxide-synthase mechanisms, and inhibit complex IV activity and induce a mitochondrial permeability transition in vitro. J Neurochem 76: , 1050–1056. |
[30] | Cardoso SM , Santos S , Swerdlow RH , Oliveira CR ((2001) ) Functional mitochondria are required for amyloid beta-mediated neurotoxicity. FASEB J 15: , 1439–1441. |
[31] | Lustbader JW , Cirilli M , Lin C , Xu HW , Takuma K , Wang N , Caspersen C , Chen X , Pollak S , Chaney M , Trinchese F , Liu S , Gunn-Moore F , Lue LF , Walker DG , Kuppusamy P , Zewier ZL , Arancio O , Stern D , Yan SS , Wu H ((2004) ) ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease. Science 304: , 448–452. |
[32] | Manczak M , Anekonda TS , Henson E , Park BS , Quinn J , Reddy PH ((2006) ) Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 15: , 1437–1449. |
[33] | Hansson Petersen CA , Alikhani N , Behbahani H , Wiehager B , Pavlov PF , Alafuzoff I , Leinonen V , Ito A , Winblad B , Glaser E , Ankarcrona M ((2008) ) The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A 105: , 13145–13150. |
[34] | Crouch PJ , Blake R , Duce JA , Ciccotosto GD , Li QX , Barnham KJ , Curtain CC , Cherny RA , Cappai R , Dyrks T , Masters CL , Trounce IA ((2005) ) Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1-42. J Neurosci 25: , 672–679. |
[35] | Caspersen C , Wang N , Yao J , Sosunov A , Chen X , Lustbader JW , Xu HW , Stern D , McKhann G , Yan SD ((2005) ) Mitochondrial Abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J 19: , 2040–2041. |
[36] | Du H , Guo L , Fang F , Chen D , Sosunov AA , McKhann GM , Yan Y , Wang C , Zhang H , Molkentin JD , Gunn-Moore FJ , Vonsattel JP , Arancio O , Chen JX , Yan SD ((2008) ) Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med 14: , 1097–1105. |
[37] | Yao J , Du H , Yan S , Fang F , Wang C , Lue LF , Guo L , Chen D , Stern DM , Gunn Moore FJ , Xi Chen J , Arancio O , Yan SS ((2011) ) Inhibition of amyloid-beta (Abeta) peptide-binding alcohol dehydrogenase-Abeta interaction reduces Abeta accumulation and improves mitochondrial function in a mouse model of Alzheimer’s disease. J Neurosci 31: , 2313–2320. |
[38] | Fernandez-Vizarra P , Fernandez AP , Castro-Blanco S , Serrano J , Bentura ML , Martinez-Murillo R , Martinez A , Rodrigo J ((2004) ) Intra- and extracellular Abeta and PHF in clinically evaluated cases of Alzheimer’s disease. Histol Histopathol 19: , 823–844. |
[39] | Beck SJ , Guo L , Phensy A , Tian J , Wang L , Tandon N , Gauba E , Lu L , Pascual JM , Kroener S , Du H ((2016) ) Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat Commun 7: , 11483. |
[40] | Anandatheerthavarada HK , Biswas G , Robin MA , Avadhani NG ((2003) ) Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol 161: , 41–54. |
[41] | Anandatheerthavarada HK , Devi L ((2007) ) Amyloid precursor protein and mitochondrial dysfunction in Alzheimer’s disease. Neuroscientist 13: , 626–638. |
[42] | Devi L , Prabhu BM , Galati DF , Avadhani NG , Anandatheerthavarada HK ((2006) ) Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci 26: , 9057–9068. |
[43] | Pagani L , Eckert A ((2011) ) Amyloid-Beta interaction with mitochondria. Int J Alzheimers Dis 2011: , 925050. |
[44] | Wang Y , Wu F , Pan H , Zheng W , Feng C , Wang Y , Deng Z , Wang L , Luo J , Chen S ((2016) ) Lost region in amyloid precursor protein (APP) through TALEN-mediated genome editing alters mitochondrial morphology. Sci Rep 6: , 22244. |
[45] | Supnet C , Bezprozvanny I ((2010) ) Neuronal calcium signaling, mitochondrial dysfunction, and Alzheimer’s disease. J Alzheimers Dis 20: (Suppl 2), S487–S498. |
[46] | Starkov AA , Beal FM ((2008) ) Portal to Alzheimer’sdisease. Nat Med 14: , 1020–1021. |
[47] | Supnet C , Bezprozvanny I ((2010) ) The dysregulation of intracellular calcium in Alzheimer disease. Cell Calcium 47: , 183–189. |
[48] | Reddy PH , McWeeney S , Park BS , Manczak M , Gutala RV , Partovi D , Jung Y , Yau V , Searles R , Mori M , Quinn J ((2004) ) Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: Up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum Mol Genet 13: , 1225–1240. |
[49] | Manczak M , Calkins MJ , Reddy PH ((2011) ) Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum Mol Genet 20: , 2495–2509. |
[50] | Wang X , Su B , Fujioka H , Zhu X ((2008) ) Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am J Pathol 173: , 470–482. |
[51] | Wang X , Su B , Lee HG , Li X , Perry G , Smith MA , Zhu X ((2009) ) Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci 29: , 9090–9103. |
[52] | Wang X , Su B , Siedlak SL , Moreira PI , Fujioka H , Wang Y , Casadesus G , Zhu X ((2008) ) Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A 105: , 19318–19323. |
[53] | Zhu X , Perry G , Smith MA , Wang X ((2013) ) Abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J Alzheimers Dis 33: (Suppl 1), S253–S262. |
[54] | Sims NR , Finegan JM , Blass JP ((1985) ) Altered glucose metabolism in fibroblasts from patients with Alzheimer’s disease. N Engl J Med 313: , 638–639. |
[55] | Sims NR , Finegan JM , Blass JP ((1987) ) Altered metabolic properties of cultured skin fibroblasts in Alzheimer’sdisease. Ann Neurol 21: , 451–457. |
[56] | Joachim CL , Mori H , Selkoe DJ ((1989) ) Amyloid beta-protein deposition in tissues other than brain in Alzheimer’s disease. Nature 341: , 226–230. |
[57] | Askanas V , Engel WK , Alvarez RB ((1992) ) Light and electron microscopic localization of beta-amyloid protein in muscle biopsies of patients with inclusion-body myositis. Am J Pathol 141: , 31–36. |
[58] | Smith CC ((1997) ) Stimulated release of the beta-amyloid protein of Alzheimer’s disease by normal human platelets. Neurosci Lett 235: , 157–159. |
[59] | Catricala S , Torti M , Ricevuti G ((2012) ) Alzheimer disease and platelets: How’s that relevant. Immun Ageing 9: , 20. |
[60] | Gabuzda D , Busciglio J , Chen LB , Matsudaira P , Yankner BA ((1994) ) Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J Biol Chem 269: , 13623–13628. |
[61] | Leuner K , Schutt T , Kurz C , Eckert SH , Schiller C , Occhipinti A , Mai S , Jendrach M , Eckert GP , Kruse SE , Palmiter RD , Brandt U , Drose S , Wittig I , Willem M , Haass C , Reichert AS , Muller WE ((2012) ) Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid Redox Signal 16: , 1421–1433. |
[62] | Webster MT , Pearce BR , Bowen DM , Francis PT ((1998) ) The effects of perturbed energy metabolism on the processing of amyloid precursor protein in PC12 cells. J Neural Transm 105: , 839–853. |
[63] | Gasparini L , Racchi M , Benussi L , Curti D , Binetti G , Bianchetti A , Trabucchi M , Govoni S ((1997) ) Effect of energy shortage and oxidative stress on amyloid precursor protein metabolism in COS cells. Neurosci Lett 231: , 113–117. |
[64] | Kukreja L , Kujoth GC , Prolla TA , Van Leuven F , Vassar R ((2014) ) Increased mtDNA mutations with aging promotes amyloid accumulation and brain atrophy in the APP/Ld transgenic mouse model of Alzheimer’s disease. Mol Neurodegener 9: , 16. |
[65] | Trifunovic A , Wredenberg A , Falkenberg M , Spelbrink JN , Rovio AT , Bruder CE , Bohlooly YM , Gidlof S , Oldfors A , Wibom R , Tornell J , Jacobs HT , Larsson NG ((2004) ) Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429: , 417–423. |
[66] | Kujoth GC , Hiona A , Pugh TD , Someya S , Panzer K , Wohlgemuth SE , Hofer T , Seo AY , Sullivan R , Jobling WA , Morrow JD , Van Remmen H , Sedivy JM , Yamasoba T , Tanokura M , Weindruch R , Leeuwenburgh C , Prolla TA ((2005) ) Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309: , 481–484. |
[67] | Fukui H , Diaz F , Garcia S , Moraes CT ((2007) ) Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 104: , 14163–14168. |
[68] | Pinto M , Pickrell AM , Fukui H , Moraes CT ((2013) ) Mitochondrial DNA damage in a mouse model of Alzheimer’s disease decreases amyloid beta plaque formation. Neurobiol Aging 34: , 2399–2407. |
[69] | Scheffler K , Krohn M , Dunkelmann T , Stenzel J , Miroux B , Ibrahim S , von Bohlen Und Halbach O , Heinze HJ , Walker LC , Gsponer JA , Pahnke J ((2012) ) Mitochondrial DNA polymorphisms specifically modify cerebral beta-amyloid proteostasis. Acta Neuropathol 124: , 199–208. |
[70] | Khan SM , Cassarino DS , Abramova NN , Keeney PM , Borland MK , Trimmer PA , Krebs CT , Bennett JC , Parks JK , Swerdlow RH , Parker WD Jr , Bennett JP Jr ((2000) ) Alzheimer’s disease cybrids replicate beta-amyloid abnormalities through cell death pathways. Ann Neurol 48: , 148–155. |
[71] | Onyango IG , Ahn JY , Tuttle JB , Bennett JP Jr , Swerdlow RH ((2010) ) Nerve growth factor attenuates oxidant-induced beta-amyloid neurotoxicity in sporadic Alzheimer’s disease cybrids. J Neurochem 114: , 1605–1618. |
[72] | Sheehan JP , Swerdlow RH , Miller SW , Davis RE , Parks JK , Parker WD , Tuttle JB ((1997) ) Calcium homeostasis and reactive oxygen species production in cells transformed by mitochondria from individuals with sporadic Alzheimer’s disease. J Neurosci 17: , 4612–4622. |
[73] | Swerdlow RH , Parks JK , Cassarino DS , Maguire DJ , Maguire RS , Bennett JP Jr , Davis RE , Parker WD Jr ((1997) ) Cybrids in Alzheimer’s disease: A cellular model of the disease? Neurology 49: , 918–925. |
[74] | Binder DR , Dunn WH Jr , Swerdlow RH ((2005) ) Molecular characterization of mtDNA depleted and repleted NT2 cell lines. Mitochondrion 5: , 255–265. |
[75] | Miller SW , Trimmer PA , Parker WD Jr , Davis RE ((1996) ) Creation and characterization of mitochondrial DNA-depleted cell lines with “neuronal-like” properties. J Neurochem 67: , 1897–1907. |
[76] | Swerdlow RH ((2007) ) Mitochondria in cybrids containing mtDNA from persons with mitochondriopathies. J Neurosci Res 85: , 3416–3428. |
[77] | Ghosh SS , Swerdlow RH , Miller SW , Sheeman B , Parker WD Jr , Davis RE ((1999) ) Use of cytoplasmic hybrid cell lines for elucidating the role of mitochondrial dysfunction in Alzheimer’s disease and Parkinson’s disease. Ann N Y Acad Sci 893: , 176–191. |
[78] | Cardoso SM , Santana I , Swerdlow RH , Oliveira CR ((2004) ) Mitochondria dysfunction of Alzheimer’s disease cybrids enhances Abeta toxicity. J Neurochem 89: , 1417–1426. |
[79] | Silva DF , Selfridge JE , Lu J , EL, Roy N , Hutfles L , Burns JM , Michaelis EK , Yan S , Cardoso SM , Swerdlow RH ((2013) ) Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines. Hum Mol Genet 22: , 3931–3946. |
[80] | Swerdlow RH ((2012) ) Mitochondria and cell bioenergetics: Increasingly recognized components and a possible etiologic cause of Alzheimer’s disease. Antioxid Redox Signal 16: , 1434–1455. |
[81] | Bijur GN , Davis RE , Jope RS ((1999) ) Rapid activation of heat shock factor-1 DNA binding by H2O2 and modulation by glutathione in human neuroblastoma and Alzheimer’s disease cybrid cells. Brain Res Mol Brain Res 71: , 69–77. |
[82] | Cassarino DS , Swerdlow RH , Parks JK , Parker WD Jr , Bennett JP Jr ((1998) ) Cyclosporin A increases resting mitochondrial membrane potential in SY5Y cells and reverses the depressed mitochondrial membrane potential of Alzheimer’s disease cybrids. Biochem Biophys Res Commun 248: , 168–173. |
[83] | Davis RE , Miller S , Herrnstadt C , Ghosh SS , Fahy E , Shinobu LA , Galasko D , Thal LJ , Beal MF , Howell N , Parker WD Jr ((1997) ) Mutations in mitochondrial cytochrome c oxidase genes segregate with late-onset Alzheimer disease. Proc Natl Acad Sci U S A 94: , 4526–4531. |
[84] | De Sarno P , Bijur GN , Lu R , Davis RE , Jope RS ((2000) ) Alterations in muscarinic receptor-coupled phosphoinositide hydrolysis and AP-1 activation in Alzheimer’s disease cybrid cells. Neurobiol Aging 21: , 31–38. |
[85] | Onyango IG , Tuttle JB , Bennett JP Jr ((2005) ) Altered intracellular signaling and reduced viability of Alzheimer’s disease neuronal cybrids is reproduced by beta-amyloid peptide acting through receptor for advanced glycation end products (RAGE). Mol Cell Neurosci 29: , 333–343. |
[86] | Onyango IG , Bennett JP Jr , Tuttle JB ((2005) ) Endogenous oxidative stress in sporadic Alzheimer’s disease neuronal cybrids reduces viability by increasing apoptosis through pro-death signaling pathways and is mimicked by oxidant exposure of control cybrids. Neurobiol Dis 19: , 312–322. |
[87] | Thiffault C , Bennett JP Jr ((2005) ) Cyclical mitochondrial deltapsiM fluctuations linked to electron transport, F0F1 ATP-synthase and mitochondrial Na+/Ca+2 exchange are reduced in Alzheimer’s disease cybrids. Mitochondrion 5: , 109–119. |
[88] | Trimmer PA , Swerdlow RH , Parks JK , Keeney P , Bennett JP Jr , Miller SW , Davis RE , Parker WD Jr ((2000) ) Abnormal mitochondrial morphology in sporadicParkinson’s and Alzheimer’s disease cybrid cell lines. Exp Neurol 162: , 37–50. |
[89] | Trimmer PA , Borland MK ((2005) ) Differentiated Alzheimer’s disease transmitochondrial cybrid cell lines exhibit reduced organelle movement. Antioxid Redox Signal 7: , 1101–1109. |
[90] | Zhang H , Liu Y , Lao M , Ma Z , Yi X ((2011) ) Puerarin protects Alzheimer’s disease neuronal cybrids from oxidant-stress induced apoptosis by inhibiting pro-death signaling pathways. Exp Gerontol 46: , 30–37. |
[91] | Silva DF , Santana I , Esteves AR , Baldeiras I , Arduino DM , Oliveira CR , Cardoso SM ((2013) ) Prodromal metabolic phenotype in MCI cybrids: Implications for Alzheimer’s disease. Curr Alzheimer Res 10: , 180–190. |
[92] | Gan X , Huang S , Wu L , Wang Y , Hu G , Li G , Zhang H , Yu H , Swerdlow RH , Chen JX , Yan SS ((2014) ) Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer’s disease cybrid cell. Biochim Biophys Acta 1842: , 220–231. |
[93] | Gan X , Wu L , Huang S , Zhong C , Shi H , Li G , Yu H , Howard Swerdlow R , Xi Chen J , Yan SS ((2014) ) Oxidative stress-mediated activation of extracellular signal-regulated kinase contributes to mild cognitive impairment-related mitochondrial dysfunction. Free Radic Biol Med 75: , 230–240. |
[94] | Yu Q , Fang D , Swerdlow RH , Yu H , Chen JX , Yan SS ((2016) ) Antioxidants rescue mitochondrial transport in differentiated Alzheimer’s disease trans-mitochondrial cybrid cells. J Alzheimers Dis 54: , 679–690. |
[95] | Costa RO , Ferreiro E , Martins I , Santana I , Cardoso SM , Oliveira CR , Pereira CM ((2012) ) Amyloid beta-induced ER stress is enhanced under mitochondrial dysfunction conditions. Neurobiol Aging 33: , 824.e825–816. |
[96] | Jeong JH , Yum KS , Chang JY , Kim M , Ahn JY , Kim S , Lapchak PA , Han MK ((2015) ) Dose-specific effect of simvastatin on hypoxia-induced HIF-1alpha and BACE expression in Alzheimer’s disease cybrid cells. BMC Neuro 15: , 127. |
[97] | Trimmer PA , Keeney PM , Borland MK , Simon FA , Almeida J , Swerdlow RH , Parks JP , Parker WD Jr , Bennett JP Jr ((2004) ) Mitochondrial abnormalities in cybrid cell models of sporadic Alzheimer’s disease worsen with passage in culture. Neurobiol Dis 15: , 29–39. |
[98] | Swerdlow RH , Khan SM ((2004) ) A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med Hypotheses 63: , 8–20. |
[99] | Swerdlow RH , Khan SM ((2009) ) The Alzheimer’s disease mitochondrial cascade hypothesis: An update. Exp Neurol 218: , 308–315. |
[100] | Swerdlow RH , Burns JM , Khan SM ((2010) ) The Alzheimer’s disease mitochondrial cascade hypothesis. J Alzheimers Dis 20: (Suppl 2), S265–S279. |
[101] | Swerdlow RH , Burns JM , Khan SM ((2014) ) The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim Biophys Acta 1842: , 1219–1231. |
[102] | Swerdlow RH , Koppel S , Weidling I , Hayley C , Ji Y , Wilkins HM ((2017) ) Mitochondria, cybrids, aging, and Alzheimer’s disease. Prog Mol Biol Transl Sci 146: , 259–302. |
[103] | Swerdlow RH ((2007) ) Is aging part of Alzheimer’s disease, or is Alzheimer’s disease part of aging? Neurobiol Aging 28: , 1465–1480. |
[104] | Parker WD ((1990) ) Sporadic neurologic disease and the electron transport chain: A hyothesis. In Proceedings of the 1989 Scientific Meeting of the American Society for Neurological Investigation: New Developments in Neuromuscular Disease, Pascuzzi RM , ed. Indiana University Printing Services, Bloomington, Indiana. |
[105] | Wallace DC ((1992) ) Mitochondrial genetics: A paradigm for aging and degenerative diseases? Science 256: , 628–632. |
[106] | Patel A , Malinovska L , Saha S , Wang J , Alberti S , Krishnan Y , Hyman AA ((2017) ) ATP as a biological hydrotrope. Science 356: , 753–756. |
[107] | Rice AM , Rosen MK ((2017) ) ATP controls the crowd. Science 356: , 701–702. |
[108] | Ruan L , Zhou C , Jin E , Kucharavy A , Zhang Y , Wen Z , Florens L , Li R ((2017) ) Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 543: , 443–446. |
[109] | Chen HK , Ji ZS , Dodson SE , Miranda RD , Rosenblum CI , Reynolds IJ , Freedman SB , Weisgraber KH , Huang Y , Mahley RW ((2011) ) Apolipoprotein E4 domain interaction mediates detrimental effects on mitochondria and is a potential therapeutic target for Alzheimer disease. J Biol Chem 286: , 5215–5221. |
[110] | Chang S , ran Ma T , Miranda RD , Balestra ME , Mahley RW , Huang Y ((2005) ) Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci U S A 102: , 18694–18699. |
[111] | Wilkins HM , Koppel SJ , Bothwell R , Mahnken J , Burns JM , Swerdlow RH ((2017) ) Platelet cytochrome oxidase and citrate synthase activities in APOE epsilon4 carrier and non-carrier Alzheimer’s disease patients. Redox Biol 12: , 828–832. |
[112] | Roses AD ((2010) ) An inherited variable poly-T repeat genotype in TOMM40 in Alzheimer disease. Arch Neurol 67: , 536–541. |
[113] | Jack CR Jr , Knopman DS , Jagust WJ , Shaw LM , Aisen PS , Weiner MW , Petersen RC , Trojanowski JQ ((2010) ) Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 9: , 119–128. |
[114] | Jack CR Jr , Knopman DS , Jagust WJ , Petersen RC , Weiner MW , Aisen PS , Shaw LM , Vemuri P , Wiste HJ , Weigand SD , Lesnick TG , Pankratz VS , Donohue MC , Trojanowski JQ ((2013) ) Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Lancet Neurol 12: , 207–216. |
[115] | Jack CR Jr , Wiste HJ , Lesnick TG , Weigand SD , Knopman DS , Vemuri P , Pankratz VS , Senjem ML , Gunter JL , Mielke MM , Lowe VJ , Boeve BF , Petersen RC ((2013) ) Brain beta-amyloid load approaches a plateau. Neurology 80: , 890–896. |
[116] | Reiman EM , Quiroz YT , Fleisher AS , Chen K , Velez-Pardo C , Jimenez-Del-Rio M , Fagan AM , Shah AR , Alvarez S , Arbelaez A , Giraldo M , Acosta-Baena N , Sperling RA , Dickerson B , Stern CE , Tirado V , Munoz C , Reiman RA , Huentelman MJ , Alexander GE , Langbaum JB , Kosik KS , Tariot PN , Lopera F ((2012) ) Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: A case-control study. Lancet Neurol 11: , 1048–1056. |
[117] | Jansen WJ , Ossenkoppele R , Knol DL , Tijms BM , Scheltens P , Verhey FR , Visser PJ , Aalten P , Aarsland D , Alcolea D , Alexander M , Almdahl IS , Arnold SE , Baldeiras I , Barthel H , van Berckel BN , Bibeau K , Blennow K , Brooks DJ , van Buchem MA , Camus V , Cavedo E , Chen K , Chetelat G , Cohen AD , Drzezga A , Engelborghs S , Fagan AM , Fladby T , Fleisher AS , van der Flier WM , Ford L , Forster S , Fortea J , Foskett N , Frederiksen KS , Freund-Levi Y , Frisoni GB , Froelich L , Gabryelewicz T , Gill KD , Gkatzima O , Gomez-Tortosa E , Gordon MF , Grimmer T , Hampel H , Hausner L , Hellwig S , Herukka SK , Hildebrandt H , Ishihara L , Ivanoiu A , Jagust WJ , Johannsen P , Kandimalla R , Kapaki E , Klimkowicz-Mrowiec A , Klunk WE , Kohler S , Koglin N , Kornhuber J , Kramberger MG , Van Laere K , Landau SM , Lee DY , de Leon M , Lisetti V , Lleo A , Madsen K , Maier W , Marcusson J , Mattsson N , de Mendonca A , Meulenbroek O , Meyer PT , Mintun MA , Mok V , Molinuevo JL , Mollergard HM , Morris JC , Mroczko B , Van der Mussele S , Na DL , Newberg A , Nordberg A , Nordlund A , Novak GP , Paraskevas GP , Parnetti L , Perera G , Peters O , Popp J , Prabhakar S , Rabinovici GD , Ramakers IH , Rami L , Resende de Oliveira C , Rinne JO , Rodrigue KM , Rodriguez-Rodriguez E , Roe CM , Rot U , Rowe CC , Ruther E , Sabri O , Sanchez-Juan P , Santana I , Sarazin M , Schroder J , Schutte C , Seo SW , Soetewey F , Soininen H , Spiru L , Struyfs H , Teunissen CE , Tsolaki M , Vandenberghe R , Verbeek MM , Villemagne VL , Vos SJ , van Waalwijk van Doorn LJ , Waldemar G , Wallin A , Wallin AK , Wiltfang J , Wolk DA , Zboch M , Zetterberg H ((2015) ) Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA 313: , 1924–1938. |
[118] | Villemagne VL , Dore V , Bourgeat P , Burnham SC , Laws S , Salvado O , Masters CL , Rowe CC ((2017) ) Abeta-amyloid and tau imaging in dementia. Semin Nucl Med 47: , 75–88. |
[119] | Bloom GS ((2014) ) Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol 71: , 505–508. |
[120] | Musiek ES , Holtzman DM ((2015) ) Three dimensions of the amyloid hypothesis: Time, space and ‘wingmen’. Nat Neurosci 18: , 800–806. |
[121] | Chen G , Shu H , Chen G , Ward BD , Antuono PG , Zhang Z , Li SJ ((2016) ) Staging Alzheimer’s disease risk by sequencing brain function and structure, cerebrospinal fluid, and cognition biomarkers. J Alzheimers Dis 54: , 983–993. |
[122] | Snowdon DA , Kemper SJ , Mortimer JA , Greiner LH , Wekstein DR , Markesbery WR ((1996) ) Linguistic ability in early life and cognitive function and Alzheimer’s disease in late life. Findings from the Nun Study. JAMA 275: , 528–532. |
[123] | Smyth KA , Fritsch T , Cook TB , McClendon MJ , Santillan CE , Friedland RP ((2004) ) Worker functions and traits associated with occupations and the development of AD. Neurology 63: , 498–503. |
[124] | Reiman EM , Caselli RJ , Yun LS , Chen K , Bandy D , Minoshima S , Thibodeau SN , Osborne D ((1996) ) Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med 334: , 752–758. |
[125] | Small GW , Mazziotta JC , Collins MT , Baxter LR , Phelps ME , Mandelkern MA , Kaplan A , La Rue A , Adamson CF , Chang L , et al. ((1995) ) Apolipoprotein E type 4 allele and cerebral glucose metabolism in relatives at risk for familial Alzheimer disease. JAMA 273: , 942–947. |
[126] | Mosconi L , Brys M , Switalski R , Mistur R , Glodzik L , Pirraglia E , Tsui W , De Santi S , de Leon MJ ((2007) ) Maternal family history of Alzheimer’s disease predisposes to reduced brain glucose metabolism. Proc Natl Acad Sci U S A 104: , 19067–19072. |
[127] | Mosconi L , de Leon M , Murray J , E L , Lu J , Javier E , McHugh P , Swerdlow RH ((2011) ) Reduced mitochondria cytochrome oxidase activity in adult children of mothers with Alzheimer’s disease. J Alzheimers Dis 27: , 483–490. |
[128] | Honea RA , Swerdlow RH , Vidoni E , Burns JM ((2011) ) Progressive regional atrophy in normaladults with a maternal history of Alzheimer disease. Neurology 76: , 822–829. |
[129] | Honea RA , Swerdlow RH , Vidoni ED , Goodwin J , Burns JM ((2010) ) Reduced gray matter volume in normal adults with a maternal family history of Alzheimer disease. Neurology 74: , 113–120. |
[130] | Berti V , Mosconi L , Glodzik L , Li Y , Murray J , De Santi S , Pupi A , Tsui W , De Leon MJ ((2011) ) Structural brain changes in normal individuals with a maternal history of Alzheimer’s. Neurobiol Aging 32: , 2325.e2317–2326. |
[131] | Andrawis JP , Hwang KS , Green AE , Kotlerman J , Elashoff D , Morra JH , Cummings JL , Toga AW , Thompson PM , Apostolova LG ((2012) ) Effects of ApoE4 and maternal history of dementia on hippocampal atrophy. Neurobiol Aging 33: , 856–866. |
[132] | Reiter K , Alpert KI , Cobia DJ , Kwasny MJ , Morris JC , Csernansky JC , Wang L ((2012) ) Cognitively normal individuals with AD parents may be at risk for developing aging-related cortical thinning patterns characteristic of AD. Neuroimage 61: , 525–532. |
[133] | Debette S , Wolf PA , Beiser A , Au R , Himali JJ , Pikula A , Auerbach S , Decarli C , Seshadri S ((2009) ) Association of parental dementia with cognitive and brain MRI measures in middle-aged adults. Neurology 73: , 2071–2078. |
[134] | Okonkwo OC , Xu G , Oh JM , Dowling NM , Carlsson CM , Gallagher CL , Birdsill AC , Palotti M , Wharton W , Hermann BP , Larue A , Bendlin BB , Rowley HA , Asthana S , Sager MA , Johnson SC ((2014) ) Cerebral blood flow is diminished in asymptomatic middle-aged adults with maternal history of Alzheimer’s disease. Cereb Cortex 24: , 978–988. |
[135] | Mosconi L , Glodzik L , Mistur R , McHugh P , Rich KE , Javier E , Williams S , Pirraglia E , De Santi S , Mehta PD , Zinkowski R , Blennow K , Pratico D , de Leon MJ ((2010) ) Oxidative stress and amyloid-beta pathology in normal individuals with a maternal history of Alzheimer’s. Biol Psychiatry 68: , 913–921. |
[136] | Mosconi L , Rinne JO , Tsui WH , Berti V , Li Y , Wang H , Murray J , Scheinin N , Nagren K , Williams S , Glodzik L , De Santi S , Vallabhajosula S , de Leon MJ ((2010) ) Increased fibrillar amyloid-beta burden in normal individuals with a family history of late-onset Alzheimer’s. Proc Natl Acad Sci U S A 107: , 5949–5954. |
[137] | Honea RA , Vidoni ED , Swerdlow RH , Burns JM ((2012) ) Maternal family history is associated with Alzheimer’s disease biomarkers. J Alzheimers Dis 31: , 659–668. |
[138] | Liu Z , Chen HH , Li TL , Xu L , Du HQ ((2013) ) A cross-sectional study on cerebrospinal fluid biomarker levels in cognitively normal elderly subjects with or without a family history of Alzheimer’s disease. CNS Neurosci Ther 19: , 38–42. |
[139] | Valla J , Yaari R , Wolf AB , Kusne Y , Beach TG , Roher AE , Corneveaux JJ , Huentelman MJ , Caselli RJ , Reiman EM ((2010) ) Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE epsilon4 allele, the major late-onset Alzheimer’s susceptibility gene. J Alzheimers Dis 22: , 307–313. |
[140] | Vlassenko AG , Vaishnavi SN , Couture L , Sacco D , Shannon BJ , Mach RH , Morris JC , Raichle ME , Mintun MA ((2010) ) Spatial correlation between brain aerobicglycolysis and amyloid-beta (Abeta) deposition. Proc Natl Acad Sci U S A 107: , 17763–17767. |
[141] | Peterson C , Gibson GE , Blass JP ((1985) ) Altered calcium uptake in cultured skin fibroblasts from patients with Alzheimer’s disease. N Engl J Med 312: , 1063–1065. |
[142] | Khachaturian ZS ((1989) ) Calcium, membranes, aging, and Alzheimer’s disease. Introduction and overview. Ann N Y Acad Sci 568: , 1–4. |
[143] | LaFerla FM ((2002) ) Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat Rev Neurosci 3: , 862–872. |
[144] | Sperling RA , Aisen PS , Beckett LA , Bennett DA , Craft S , Fagan AM , Iwatsubo T , Jack CR Jr , Kaye J , Montine TJ , Park DC , Reiman EM , Rowe CC , Siemers E , Stern Y , Yaffe K , Carrillo MC , Thies B , Morrison-Bogorad M , Wagster MV , Phelps CH ((2011) ) Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 280–292. |
[145] | Knopman DS , Jack CR Jr , Wiste HJ , Weigand SD , Vemuri P , Lowe VJ , Kantarci K , Gunter JL , Senjem ML , Mielke MM , Roberts RO , Boeve BF , Petersen RC ((2013) ) Brain injury biomarkers are not dependent on beta-amyloid in normal elderly. Ann Neurol 73: , 472–480. |
[146] | Rao VK , Carlson EA , Yan SS ((2014) ) Mitochondrial permeability transition pore is a potential drug target for neurodegeneration. Biochim Biophys Acta 1842: , 1267–1272. |
[147] | Reddy PH ((2014) ) Inhibitors of mitochondrial fission as a therapeutic strategy for diseases with oxidative stress and mitochondrial dysfunction. J Alzheimers Dis 40: , 245–256. |
[148] | Swerdlow RH ((2014) ) Bioenergetic medicine. Br J Pharmacol 171: , 1854–1869. |
[149] | Swerdlow RH ((2016) ) Bioenergetics and metabolism: A bench to bedside perspective. J Neurochem 139: (Suppl 2), 126–135. |

