
Negative 11C-PIB PET Predicts Lack of Alzheimer’s Disease Pathology in Postmortem Examination
Abstract
Our aim was to assess whether in vivo 11C-PIB negative memory-impaired subjects may nonetheless exhibit brain Alzheimer’s disease (AD) pathology. We re-evaluated the PET images and systematically characterized the postmortem neuropathology of six individuals who had undergone clinically indicated amyloid PET. The single case with negligible amyloid-β (Aβ) pathology had Lewy body disease, where concomitant AD changes are often seen. Further, the subject’s plaques were predominantly diffuse. The predictive value of a negative 11C-PIB scan appears to be good, even in memory-impaired populations. Our results suggest that considerable neuritic Aβ plaque pathology in the absence of specific/cortical 11C-PIB binding upon PET is unlikely.
INTRODUCTION
Amyloid imaging with positron emission tomography (PET) has established itself as a supplementary diagnostic tool in the differential diagnosis of Alzheimer’s disease (AD) from other dementias [1]. However, a Cochrane review stressed that it is important to clearly demonstrate its accuracy before it can be widely applied [2]. Another review pointed out that as amyloid deposition is not unique to AD, PET amyloid imaging agents cannot be said to be diagnostic of AD, although they may have utility in excluding it in suspected cases [3].
A number of case reports and studies have assessed correlates of 11C-labeled Pittsburgh Compound B (11C-PIB) and other amyloid fibril binding PET tracer uptake values in postmortem or in vivo biopsy brain tissue [4–19]. PET imaging with these agents has high sensitivity and specificity for the presence of moderate or frequent neuritic plaque densities, but none of them are sensitive enough to reliably detect the presence of sparse neuritic plaques. The amyloid tracers 11C-PIB, 18F-florbetapir, 18F-florbetapen, and 18F-flutemetamol have relatively similar fibrillar Aβ binding site affinities [3]. Robust associations between regional amyloid tracer uptake and neuritic plaque density as well as immunohistochemically measured Aβ have been reported [4–9, 18, 19].
Some studies have also provided sensitivity and specificity estimates for binary visual assessment of amyloid PET images in relation to postmortem brain tissue Aβ detection. For 18F-flutemetamol, they have been 88–91% and 88–90%. respectively [15, 16]. With 18F-florbetaben, sensitivity of 98% and specificity of 89% have been reported [18]. For 18F-florbetapir, the corresponding values for detection of moderate to frequent plaques (as opposed to none or sparse) were 92% and 100% [13]. Overall, false negatives by amyloid imaging do occur, and it still remains unclear whether notable Aβ or other AD pathology could exist in brain tissue when prior in vivo 11C-PIB PET has been negative.
Only a few single case reports [20, 21] have specifically focused on amyloid imaging negativity. One described a subject who underwent 11C-PIB PET as cognitively intact but experienced memory impairment afterwards, and autopsy performed approximately 2.5 years thereafter revealed foci of frequent neocortical diffuse amyloid plaques but only sparse neuritic plaques and neurofibrillary tangles [20]. Another report assessed a subject who had clinical diagnoses of probable dementia with Lewy bodies and possible AD, 11C-PIB PET-scanned 17 months prior to death [21]. Aβ1 - 40 levels in several cortical regions were comparable to the values of an 11C-PIB positive comparison case. The authors concluded that Aβ pathology may be associated with low or undetectable 11C-PIB uptake upon in vivo PET.
Distinct from previous end-of-life and clinically heterogeneous studies, the aim of this study was to specifically address whether individuals with a memory impairment but an 11C-PIB negative clinically indicated amyloid PET scan may exhibit AD neuropathology upon postmortem brain tissue examination.
SUBJECTS AND METHODS
Permission for the study was granted by the research sites according to local legislation. The study plan and the created privacy policy, in line with the Personal Data Act (Finland, 22.4.1999/523), were reviewed and accepted by the national Data Protection Ombudsman.
Subjects
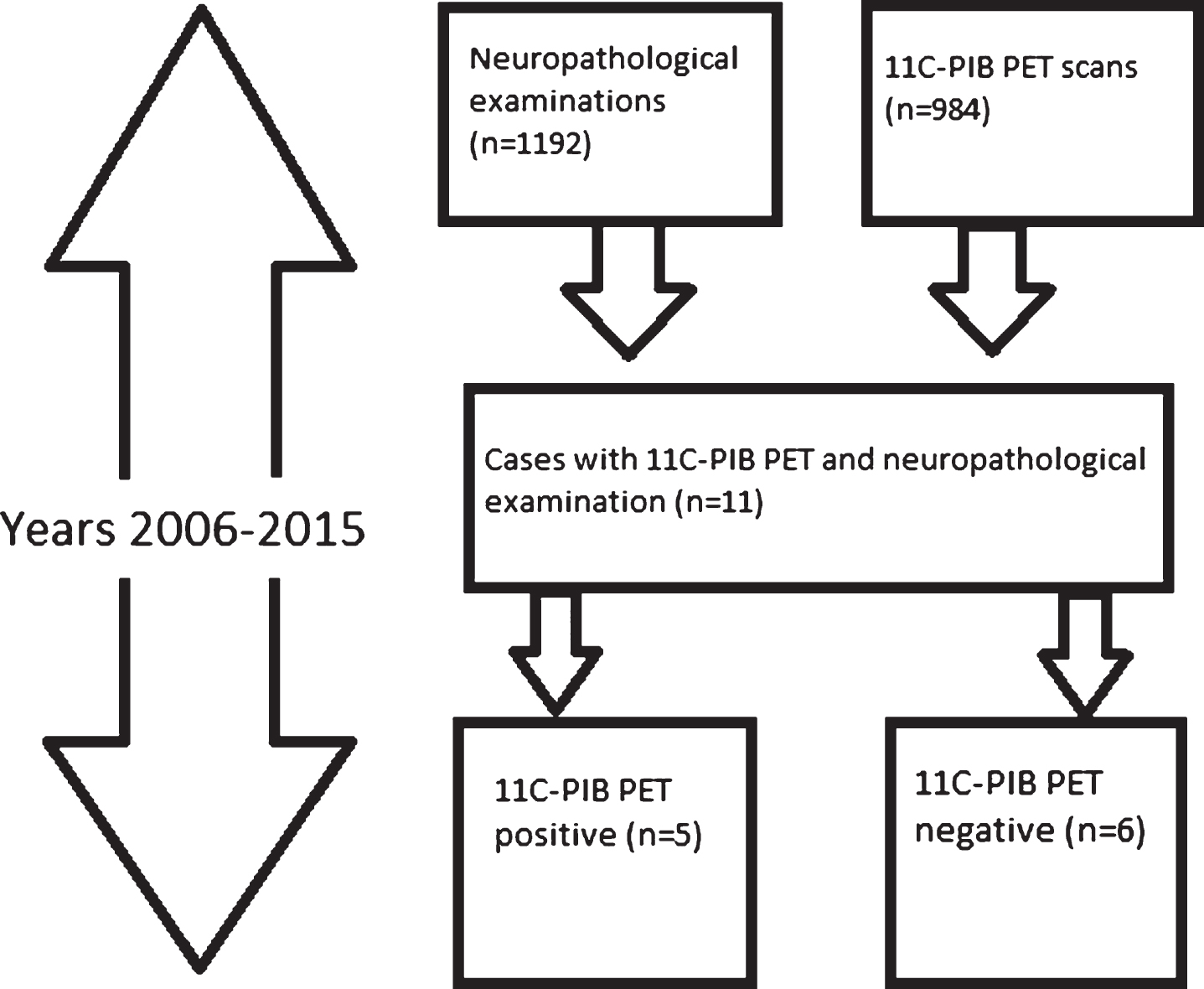
The subjects were identified by screening the list of the deceased who had undergone neuropathological examination at the Turku University Hospital during 2006–2015 and had previously been in a clinically indicated 11C-PIB PET scan at the Turku PET Centre.
Six individuals had been 11C-PIB negative according to visual assessment (see Fig. 1). The images were re-evaluated and 11C-PIB negativity was visually confirmed. The age at imaging ranged from 54 to 81 years, the interval between 11C-PIB PET and death from 15 to 67 months (Table 2). All cases had had subjective memory problems corroborated by their relatives. In addition, cognitive testing (CERAD test or detailed neuropsychological investigation) had been performed for everyone.
Fig.1
The identification of study subjects. The final sample is in the lower right corner.
Neuropathology
The neuropathological reports and slides were re-examined to determine potential AD pathology. The material included H&E and Bielschowsky silver stainings in sections from a minimum of 12 brain regions. Immunohistochemical stainings for Aβ, phospho-tau, α-synuclein, TDP-43, and p62 were performed using a Ventana Benchmark XT Immunostainer (Ventana Medical Systems, Tucson, AZ). The used antibodies, clones and manufacturers are listed in Table 1. Scoring of AD changes was carried out according to NIA-AA guidelines for neuropathological assessment of AD using the ABC principle [22] with minor modifications, except for one case (#6, Table 2) for whom only the neuropathologist’s report was available. The A stands for Aβ plaque Thal phase [23], B for neurofibrillary stage by phospho-tau staining according to Braak [24], and C for neuritic plaque scoring modified from CERAD [25]. The Neuropathological diagnostic criteria for frontotemporal lobar dementia (FTLD) and FTLD+ motor neuron disease (MND) were used as proposed by the Consortium for Frontotemporal Lobar degeneration [26]. Lewy body disease (LBD) was diagnosed according to guidelines of the DLB Consortium [27, 28].
Table 1
The antibodies, clones and manufacturers used in the immunohistochemical stainings
| Antibody | Clone | Manufacturer |
| Phospho-tau | AT-8 | Innogenetics |
| α-synuclein | KM51 | Novocastra |
| TDP-43 | pS409/410 | CosmoBio |
| p62 | 3/P62 LCK ligand | BD Biosciences |
| Amyloid-β | 6F/3D | Novocastra |
Table 2
Clinical and neuroradiological features and neuropathological findings of the subjects. Subjects #1-2 and #5-6 were female
| Subject | Indication for 11C-PIB PET imaging | Age at 11C-PIB PET | MMSE | Atrophy in MRI | Hypo-metabolism in 18F-FDG PET | Time 11C-PIB PET to death | Neuropathological findings |
| #1 | Progressive memory impairment at young age | 54 | 24/30 | No significant atrophy. | Temporoparietal symmetrical hypometabolism | 31 mo | Acute trauma related, no neurodegenerative disease. |
| #2 | AD suspicion: subjective and next-of-kin report of MCI features | 75 | 27/30 | Hippocampal atrophy II/IV. | NA | 15 mo | Acute intracerebral hemorrhage. Small vessel disease. |
| #3 | AD suspicion according to neuropsychological findings | 77 | 21/30 | Mild central atrophy. Hippocampal atrophy I/IV. | NA | 67 mo | Lewy body disease. Mild AD changes, including sparse neuritic plaques (A1B1C1). |
| #4 | Progressive memory impairment at young age | 69 | 26/30 | Widespread central, cortical and cerebellar atrophy. Hippocampal atrophy II/IV right, I/IV left. | Temporoparietal hypometabolism. No frontal hypometabolism. | 17 mo | FTLD+MND (TDP-43+) |
| #5 | Progressive memory impairment at young age | 64 | 26/30 | Prominent atrophy in anterior right temporal lobe, milder in left anterior temporal lobe. Hippocampal atrophy IV/IV. | Right dominant temporal hypometabolism. Milder general cortical hypometabolism. | 41 mo | FTLD (TDP-43+) |
| #6 | Screening for inclusion to clinical AD drug trial | 81 | 23/30 | Marked temporomedial atrophy. Hippocampal atrophy IV/IV right, III/IV left. | NA | 31 mo | Amyloid-β negative by immunohistochemistry but tau positive. |
MMSE, MRI and possible 18F-FDG PET had been conducted close to 11C-PIB PET. All six had exhibited only non-specific white matter 11C-PIB binding in amyloid PET according to visual assessment.
RESULTS
All subjects had been 11C-PIB PET imaged according to appropriate use criteria for amyloid PET [29, 30] or in the case of one subject (#6), as a part of screening for an AD drug trial. Only non-specific white matter 11C-PIB uptake was seen upon image re-evaluation. The Mini-Mental State Examination (MMSE) scores as a proxy for severity of cognitive impairment, as well as the MRI and 18F-FDG PET findings of the subjects, where available, are given in Table 2.
Histologically, two cases had an A0B0C0 and two cases had an A0B1C0 score according to NIA-AA guidelines [22], i.e., the level of AD pathology was considered insufficient to explain cognitive impairment. AD neuropathological changes of level “low” were found in only one (#3) of the six cases. This subject had neuropathological findings diagnostic for LBD, limbic type with concomitant mild AD changes (A1B1C1), meaning that mainly diffuse Aβ plaques were present in isocortical areas. AD neuropathological changes are common in LBD [27, 31]. In retrospect, the medical records of patient #3 had mentions of motor signs of LBD. These had been interpreted as a side effect of decades of antipsychotic agent use. AD had been suspected for the cognitive decline.
The principal pathologies are summarized in Table 2. They include one TDP-43 positive FTLD with mild concomitant α-synuclein pathology, and another case with TDP-43 positive FTLD + MND, and one case of small vessel disease. One patient had no neurodegenerative disease, only acute trauma related findings. This case had several years’ history of progressive memory impairment, verified in neuropsychological testing. The subject died because of a traffic accident and the trauma related changes seen in neuropathological examination are attributable to this. Another (#6) had neurodegenerative findings that were not specific of any neuropathological disease; phospho-tau positive neurofibrillary change was seen in neurons but Aβ immunohistochemistry was negative. This was the case for whom only the neuropathologist’s report was available.
DISCUSSION
To summarize our results, none of the 11C-PIB negative memory-impaired patients had such AD changes upon neuropathological examination that would have been sufficient to explain their cognitive decline. The subjects exhibited no Aβ (n = 5) or a low level of Aβ plaques (n = 1). The single case with any Aβ pathology had LBD, where concomitant AD changes are often seen [27, 31].
In a recent end-of-life PET-autopsy study with 18F-flutemetamol, 7 out of 106 subjects were false negatives by imaging, of whom 4 had unequivocally abnormal Aβ burdens in histopathology. The authors discussed the possibility of severe cortical atrophy being partly responsible for the misclassifications, although they acknowledged that equivalent atrophy was seen in many patients whose images were correctly interpreted [16]. In comparison, only one of our subjects exhibited widespread atrophy (#4). In another end-of-life study, two false negatives by imaging (in relation to having moderate or frequent Aβ plaques as opposed to none or sparse) occurred [13].
Overall, the literature on 18F labeled amyloid tracers and subsequent neuropathology is vaster than that on 11C-PIB, and strong associations between regional tracer uptake and brain Aβ have been reported [8, 18, 32]. However, the associations could be driven mostly by high amyloid tracer uptake and high levels of Aβ in brain tissue. Further, the studies have often been conducted with end-of-life subjects and not necessarily memory-impaired patients. Moreover, the reports have not discussed in further detail the amyloid PET negative subjects.
The one subject with low level AD pathology in our study could suggest that the predictive value of a negative 11C-PIB PET scan is less than perfect. However, this LBD case exhibited mainly immature/diffuse Aβ plaques that are often present even in non-demented elderly individuals [33], and very few core plaques, where 11C-PIB would typically bind. The interval between the PET scan and the autopsy was long (67 months, i.e., 5.6 years) and thus, the plaques may have developed after the amyloid imaging, which has also been suggested for the false negatives in previous studies [13, 16]. Our LBD case is also in line with Aβ being often seen in LBD [27, 31], as well as partly comparable with a previous 11C-PIB positive LBD case report, where greatest tracer retention corresponded with Aβ density in the same regions in postmortem neuropathology, but overall, neocortical neuritic plaques were sparse whereas diffuse plaques were frequent. The subject had low likelihood of AD by NIA-Reagan criteria [14].
Up to 10–30% of cognitively healthy elderly individuals exhibit positive amyloid PET [34–37] and Aβ pathology can be seen in up to 45% or more of elderly healthy subjects. On the other hand, some degree of tangles or tau pathology is universal in elderly subjects [38, 39]. It has not been established whether the former (Aβ) findings mean pending AD; this also motivated us to focus on amyloid PET negativity.
The modest sample size is an acknowledged limitation. Almost 1,000 11C-PIB PET studies had been conducted in 2006–2015 at the Turku PET Centre, but very few of these individuals had gone on to postmortem neuropathological examinations. No end-of-life amyloid imaging studies had been performed at our site during the study period, i.e., only a small proportion of our 11C-PIB PET imaged subjects have deceased to date. Also, as this was a register study, the subjects had not specifically been enrolled for neuropathology. To the best of our knowledge, ours is, however, the largest sample to date, in a study that focuses on the subsequent neuropathological findings of 11C-PIB negative cases in a clinical sample. In one earlier study, a postmortem neuropathological examination was conducted in six individuals with various clinical diagnoses and previous 11C-PIB imaging. Three of the individuals were 11C-PIB negative, and these were later neuropathologically found to have sparse diffuse amyloid plaques or a low amount of dense core plaques. The neuropathological diagnoses were sporadic Creutzfeldt-Jakob disease, Parkinson’s disease dementia, and minimal senile change (the last case being a healthy volunteer) [19]. Granted that the primary objective of that study was to correlate 11C-PIB imaging findings and amyloid plaque density across diverse clinical diagnoses and not in addressing the use of 11C-PIB imaging in the differential diagnostics of suspected AD, we find the results supportive to the conclusions of the present study. Small amounts of amyloid plaques remain undetected by 11C-PIB imaging, but AD is in this setting an unlikely cause for cognitive decline.
As a strength, we were able to characterize the subjects not only in terms of amyloid PET and thorough neuropathology, but also had information on the clinical features, MRI findings as well as 18F-FDG PET data on some of the individuals. Of note, all three patients that had undergone 18F-FDG PET exhibited cortical temporal or temporoparietal hypometabolism, together with negative 11C-PIB suggesting the presence of a non-AD etiology. Another notion pointing in amyloid PET’s added value in investigating clinically undetermined cognitive decline is the fact that some of our subjects also exhibited marked hippocampal atrophy, a cardinal MR imaging feature of AD [40].
Overall, our findings suggest that in clinical settings, when cognitive decline is evident and 11C-PIB PET is negative, efforts should be directed at investigating other less common causes of dementia. This is especially encouraged when the patient is relatively young, as the likelihood of 11C-PIB positivity increases with age [41], and when severe cortical atrophy is not present, as it might preclude correct PET image interpretation [16]. 11C-PIB PET negativity is associated with not having clinically relevant Aβ pathology, and this further validates the use of amyloid PET also in subject stratification in treatment trials targeting Aβ. For instance, in the bapineuzumab (anti amyloid antibody) PET substudy, 6.5% of APOE ɛ4 carriers and 36.1% of noncarriers had a baseline global cortical 11C-PIB average less than the prespecified threshold for amyloid positivity [42]. This could have partly contributed to the negative clinical results. An important caveat should however be remembered: it is still not possible, even with PET amyloid imaging, to reliably detect subjects with sparse neuritic plaques. PET amyloid imaging may thus still not be feasible for selecting subjects at the very earliest stages of Aβ deposition, when anti-amyloid therapy might be expected to be most useful.
ACKNOWLEDGMENTS
The staffs of the Turku PET Centre, Department of Pathology of the Turku University Hospital and of the Welfare Division of Turku City are thanked for their dedicated work and co-operation. The funding for this study was provided by Sigrid Juselius Foundation and the Finnish Governmental Research Funding for Turku University Hospital (ERVA).
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/17-0569r2).
REFERENCES
[1] | Jack CR , Albert MS , Knopman DS , Mckhann GM , Sperling RA , Carrillo MC , Thies B , Phelps CH ((2011) ) Introduction to the recommendations from the National Institute on Aging and the Alzheimer’s Association workgroup on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 257–262. |
[2] | Zhang S , Smailagic N , Hyde C , Noel-Storr AH , Takwoingi Y , McShane R , Feng J ((2014) ) (11)C-PIB-PET for the early diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database Syst Rev 7: , CD010386. |
[3] | Gelosa G , Brooks DJ ((2012) ) The prognostic value of amyloid imaging. Eur J Nucl Med Mol Imaging 39: , 1207–1219. |
[4] | Ikonomovic MD , Klunk WE , Abrahamson EE , Mathis CA , Price JC , Tsopelas ND , Lopresti BJ , Ziolko S , Bi W , Paljug WR , Debnath ML , Hope CE , Isanski BA , Hamilton RL , DeKosky ST ((2008) ) Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain 131: , 1630–1645. |
[5] | Driscoll I , Troncoso JC , Rudow G , Sojkova J , Pletnikova O , Zhou Y , Kraut MA , Ferrucci L , Mathis CA , Klunk WE , O’Brien RJ , Davatzikos C , Wong DF , Resnick SM ((2012) ) Correspondence between in vivo (11)C-PiB-PET amyloid imaging and postmortem, region-matched assessment of plaques. Acta Neuropathol 124: , 823–831. |
[6] | Rinne JO , Wong DF , Wolk DA , Leinonen V , Arnold SE , Buckley C , Smith A , McLain R , Sherwin PF , Farrar G , Kailajärvi M , Grachev ID ((2012) ) [(18)F]Flutemetamol PET imaging and cortical biopsy histopathology for fibrillar amyloid β detection in living subjects with normal pressure hydrocephalus: Pooled analysis of four studies. Acta Neuropathol 124: , 833–845. |
[7] | Leinonen V , Rinne JO , Virtanen KA , Eskola O , Rummukainen J , Huttunen J , von Und Zu Fraunberg M , Nerg O , Koivisto AM , Rinne J , Jääskeläinen JE , Buckley C , Smith A , Jones PA , Sherwin P , Farrar G , McLain R , Kailajärvi M , Heurling K , Grachev ID ((2013) ) Positron emission tomography with [18F]flutemetamol and [11C]PiB for in vivo detection of cerebral cortical amyloid in normal pressure hydrocephalus patients. Eur J Neurol 20: , 1043–1052. |
[8] | Wong DF , Moghekar AR , Rigamonti D , Brašić JR , Rousset O , Willis W , Buckley C , Smith A , Gok B , Sherwin P , Grachev ID ((2013) ) An in vivo evaluation of cerebral cortical amyloid with [18F]flutemetamol using positron emission tomography compared with parietal biopsy samples in living normal pressure hydrocephalus patients. Mol Imaging Biol 15: , 230–237. |
[9] | Leinonen V ((2008) ) Assessment of β-amyloid in a frontal cortical brain biopsy specimen and by positron emission tomography with carbon 11– labeled Pittsburgh Compound B. Arch Neurol 65: , 1304. |
[10] | Bacskai BJ , Frosch MP , Freeman SH , Raymond SB , Augustinack JC , Johnson KA , Irizarry MC , Klunk WE , Mathis CA , Dekosky ST , Greenberg SM , Hyman BT , Growdon JH ((2007) ) Molecular imaging with Pittsburgh Compound B confirmed at autopsy: A case report. Arch Neurol 64: , 431–434. |
[11] | Kadir A , Marutle A , Gonzalez D , Schöll M , Almkvist O , Mousavi M , Mustafiz T , Darreh-Shori T , Nennesmo I , Nordberg A ((2011) ) Positron emission tomography imaging and clinical progression in relation to molecular pathology in the first Pittsburgh Compound B positron emission tomography patient with Alzheimer’s disease. Brain 134: , 301–317. |
[12] | Sojkova J , Driscoll I , Iacono D , Zhou Y , Codispoti KE , Kraut MA , Ferrucci L , Pletnikova O , Mathis CA , Klunk WE , O’Brien RJ , Wong DF , Troncoso JC , Resnick SM ((2011) ) In vivo fibrillar β-amyloid detected using [11C]PiB positron emission tomography and neuropathologic assessment in older adults. Arch Neurol 68: , 232–240. |
[13] | Clark CM , Pontecorvo MJ , Beach TG , Bedell BJ , Coleman RE , Doraiswamy PM , Fleisher AS , Reiman EM , Sabbagh MN , Sadowsky CH , Schneider JA , Arora A , Carpenter AP , Flitter ML , Joshi AD , Krautkramer MJ , Lu M , Mintun MA , Skovronsky DM ((2012) ) Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-β plaques: A prospective cohort study. Lancet Neurol 11: , 669–678. |
[14] | Kantarci K , Yang C , Schneider JA , Senjem ML , Reyes DA , Lowe VJ , Barnes LL , Aggarwal NT , Bennett DA , Smith GE , Petersen RC , Jack CR , Boeve BF ((2012) ) Ante mortem amyloid imaging and β-amyloid pathology in a case with dementia with Lewy bodies. Neurobiol Aging 33: , 878–885. |
[15] | Curtis C , Gamez JE , Singh U , Sadowsky CH , Villena T , Sabbagh MN , Beach TG , Duara R , Fleisher AS , Frey KA , Walker Z , Hunjan A , Holmes C , Escovar YM , Vera CX , Agronin ME , Ross J , Bozoki A , Akinola M , Shi J , Vandenberghe R , Ikonomovic MD , Sherwin PF , Grachev ID , Farrar G , Smith APL , Buckley CJ , McLain R , Salloway S ((2015) ) Phase 3 trial of flutemetamol labeled with radioactive fluorine 18 imaging and neuritic plaque density. JAMA Neurol 72: , 287–294. |
[16] | Ikonomovic MD , Buckley CJ , Heurling K , Sherwin P , Jones PA , Zanette M , Mathis CA , Klunk WE , Chakrabarty A , Ironside J , Ismail A , Smith C , Thal DR , Beach TG , Farrar G , Smith APL ((2016) ) Post-mortem histopathology underlying β-amyloid PET imaging following flutemetamol F 18 injection. Acta Neuropathol Commun 4: , 130. |
[17] | Seo SW , Ayakta N , Grinberg LT , Villeneuve S , Lehmann M , Reed B , DeCarli C , Miller BL , Rosen HJ , Boxer AL , O’Neil JP , Jin LW , Seeley WW , Jagust WJ , Rabinovici GD ((2016) ) Regional correlations between [11C]PIB PET and post-mortem burden of amyloid-beta pathology in a diverse neuropathological cohort. Neuroimage Clin 13: , 130–137. |
[18] | Sabri O , Sabbagh MN , Seibyl J , Barthel H , Akatsu H , Ouchi Y , Senda K , Murayama S , Ishii K , Takao M , Beach TG , Rowe CC , Leverenz JB , Ghetti B , Ironside JW , Catafau AM , Stephens AW , Mueller A , Koglin N , Hoffmann A , Roth K , Reininger C , Schulz-Schaeffer WJ ((2015) ) Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer’s disease: Phase 3 study. Alzheimers Dement 11: , 964–974. |
[19] | Hatsuta H , Takao M , Ishii K , Ishiwata K , Saito Y , Kanemaru K , Arai T , Suhara T , Shimada H , Shinotoh H , Tamaoka A , Murayama S ((2015) ) Amyloid beta accumulation assessed with (1)(1)C-Pittsburgh compound B PET and postmortem neuropathology. Curr Alzheimer Res 12: , 278–286. |
[20] | Cairns NJ , Ikonomovic MD , Benzinger T , Storandt M , Fagan AM , Shah A , Schmidt RE , Perry A , Reinwald LT , Carter D , Felton A , Holtzman DM , Mintun MA , Klunk WE , Morris JC ((2009) ) Absence of Pittsburgh compound B detection of cerebral amyloid beta in a patient with clinical, cognitive, and cerebrospinal fluid markers of Alzheimer disease: A case report. Arch Neurol 66: , 1557–1562. |
[21] | Ikonomovic MD , Abrahamson EE , Price JC , Hamilton RL , Mathis CA , Paljug WR , Debnath ML , Cohen AD , Mizukami K , DeKosky ST , Lopez OL , Klunk WE ((2012) ) Early AD pathology in a [C-11]PiB-negative case: A PiB-amyloid imaging, biochemical, and immunohistochemical study. Acta Neuropathol 123: , 433–447. |
[22] | Hyman BT , Phelps CH , Beach TG , Bigio EH , Cairns NJ , Carrillo MC , Dickson DW , Duyckaerts C , Frosch MP , Masliah E , Mirra SS , Nelson PT , Schneider JA , Thal DR , Thies B , Trojanowski JQ , Vinters HV , Montine TJ ((2012) ) National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8: , 1–13. |
[23] | Thal DR , Rub U , Orantes M , Braak H ((2002) ) Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 58: , 1791–1800. |
[24] | Braak H , Alafuzoff I , Arzberger T , Kretzschmar H , Del Tredici K ((2006) ) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112: , 389–404. |
[25] | Mirra SS , Heyman A , McKeel D , Sumi SM , Crain BJ , Brownlee LM , Vogel FS , Hughes JP , van Belle G , Berg L ((1991) ) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD): Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41: , 479–479. |
[26] | Cairns NJ , Bigio EH , Mackenzie IRA , Neumann M , Lee VM-Y , Hatanpaa KJ , White CL , Schneider JA , Grinberg LT , Halliday G , Duyckaerts C , Lowe JS , Holm IE , Tolnay M , Okamoto K , Yokoo H , Murayama S , Woulfe J , Munoz DG , Dickson DW , Ince PG , Trojanowski JQ , Mann DMA , Consortium for Frontotemporal Lobar Degeneration ((2007) ) Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: Consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 114: , 5–22. |
[27] | McKeith IG ((2005) ) Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium.1455; author reply. Neurology 65: , 1455. |
[28] | McKeith IG , Boeve BF , Dickson DW , Halliday G , Taylor J-P , Weintraub D , Aarsland D , Galvin J , Attems J , Ballard CG , Bayston A , Beach TG , Blanc F , Bohnen N , Bonanni L , Bras J , Brundin P , Burn D , Chen-Plotkin A , Duda JE , El-Agnaf O , Feldman H , Ferman TJ , Ffytche D , Fujishiro H , Galasko D , Goldman JG , Gomperts SN , Graff-Radford NR , Honig LS , Iranzo A , Kantarci K , Kaufer D , Kukull W , Lee VMY , Leverenz JB , Lewis S , Lippa C , Lunde A , Masellis M , Masliah E , McLean P , Mollenhauer B , Montine TJ , Moreno E , Mori E , Murray M , O’Brien JT , Orimo S , Postuma RB , Ramaswamy S , Ross OA , Salmon DP , Singleton A , Taylor A , Thomas A , Tiraboschi P , Toledo JB , Trojanowski JQ , Tsuang D , Walker Z , Yamada M , Kosaka K ((2017) ) Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 89: , 88–100. |
[29] | Johnson KA , Minoshima S , Bohnen NI , Donohoe KJ , Foster NL , Herscovitch P , Karlawish JH , Rowe CC , Carrillo MC , Hartley DM , Hedrick S , Pappas V , Thies WH , Alzheimer’s Association Society of Nuclear Medicine and Molecular Imaging Amyloid Imaging Taskforce ((2013) ) Appropriate use criteria for amyloid PET: A report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer’s Association. Alzheimers Dement 9: , e-1–16. |
[30] | Johnson K a , Minoshima S , Bohnen NI , Donohoe KJ , Foster NL , Herscovitch P , Karlawish JH , Rowe CC , Carrillo MC , Hartley DM , Hedrick S , Pappas V , Thies WH ((2013) ) Appropriate use criteria for amyloid PET: A report of the Amyloid Imaging Task Force, the Society of Nuclear Medicine and Molecular Imaging, and the Alzheimer’s Association. J Nucl Med 54: , 476–490. |
[31] | Schneider JA , Arvanitakis Z , Bang W , Bennett DA ((2007) ) Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69: , 2197–2204. |
[32] | Leinonen V , Rinne JO , Virtanen KA , Eskola O , Rummukainen J , Huttunen J , Von und zu Fraunberg M , Nerg O , Koivisto AM , Rinne J , Jääskeläinen JE , Buckley C , Smith A , Jones PA , Sherwin P , Farrar G , Mclain R , Kailajärvi M , Heurling K , Grachev ID ((2013) ) Positron emission tomography with [18F]flutemetamol and [11C]PiB for in vivo detection of cerebral cortical amyloid in normal pressure hydrocephalus patients. Eur J Neurol 20: , 1043–1052. |
[33] | Morris JC , Storandt M , McKeel DW , Rubin EH , Price JL , Grant EA , Berg L ((1996) ) Cerebral amyloid deposition and diffuse plaques in normal aging - evidence for presymptomatic and very mild Alzheimer’s disease. Neurology 46: , 707–719. |
[34] | Villemagne VL , Pike KE , Chételat G , Ellis KA , Mulligan RS , Bourgeat P , Ackermann U , Jones G , Szoeke C , Salvado O , Martins R , O’Keefe G , Mathis CA , Klunk WE , Ames D , Masters CL , Rowe CC ((2011) ) Longitudinal assessment of Aβ and cognition in aging and Alzheimer disease. Ann Neurol 69: , 181–192. |
[35] | Aizenstein HJ , Nebes RD , Saxton JA , Price JC , Mathis CA , Tsopelas ND , Ziolko SK , James JA , Snitz BE , Houck PR , Bi W , Cohen AD , Lopresti BJ , DeKosky ST , Halligan EM , Klunk WE ((2008) ) Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol 65: , 1509. |
[36] | Scheinin NM , Aalto S , Koikkalainen J , Lötjönen J , Karrasch M , Kemppainen N , Viitanen M , Någren K , Helin S , Scheinin M , Rinne JO ((2009) ) Follow-up of [11C]PIB uptake and brain volume in patients with alzheimer disease and controls. Neurology 73: , 1186–1192. |
[37] | Rowe CC , Ng S , Ackermann U , Gong SJ , Pike K , Savage G , Cowie TF , Dickinson KL , Maruff P , Darby D , Smith C , Woodward M , Merory J , Tochon-Danguy H , O’Keefe G , Klunk WE , Mathis CA , Price JC , Masters CL , Villemagne VL ((2007) ) Imaging beta-amyloid burden in aging and dementia. Neurology 68: , 1718–1725. |
[38] | Petersen RC , Parisi JE , Dickson DW , Johnson KA , Knopman DS , Boeve BF , Jicha GA , Ivnik RJ , Smith GE , Tangalos EG , Braak H , Kokmen E ((2006) ) Neuropathologic features of amnestic mild cognitive impairment. Arch Neurol 63: , 665–672. |
[39] | Bouras C , Hof PR , Giannakopoulos P , Michel JP , Morrison JH ((1994) ) Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of elderly patients: A quantitative evaluation of a one-year autopsy population from a geriatric hospital. Cereb Cortex 4: , 138–150. |
[40] | Mattson MP ((2004) ) Pathway towards and away from Alzheimer’s disease. Nature 430: , 631–639. |
[41] | Vlassenko AG , Mintun MA , Xiong C , Sheline YI , Goate AM , Benzinger TLS , Morris JC ((2011) ) Amyloid-beta plaque growth in cognitively normal adults: Longitudinal [11C]Pittsburgh compound B data. Ann Neurol 70: , 857–861. |
[42] | Liu E , Schmidt ME , Margolin R , Sperling R , Koeppe R , Mason NS , Klunk WE , Mathis CA , Salloway S , Fox NC , Hill DL , Les AS , Collins P , Gregg KM , Di J , Lu Y , Tudor IC , Wyman BT , Booth K , Broome S , Yuen E , Grundman M , Brashear HR ((2015) ) Amyloid-β 11C-PiB-PET imaging results from 2 randomized bapineuzumab phase 3 AD trials. Neurology 85: , 692–700. |

