
Oxidative Stress, Amyloid-β Peptide, and Altered Key Molecular Pathways in the Pathogenesis and Progression of Alzheimer’s Disease
Abstract
Oxidative stress is implicated in the pathogenesis and progression of Alzheimer’s disease (AD) and its earlier stage, amnestic mild cognitive impairment (aMCI). One source of oxidative stress in AD and aMCI brains is that associated with amyloid-β peptide, Aβ1-42 oligomers. Our laboratory first showed in AD elevated oxidative stress occurred in brain regions rich in Aβ1-42, but not in Aβ1-42-poor regions, and was among the first to demonstrate Aβ peptides led to lipid peroxidation (indexed by HNE) in AD and aMCI brains. Oxidatively modified proteins have decreased function and contribute to damaged key biochemical and metabolic pathways in which these proteins normally play a role. Identification of oxidatively modified brain proteins by the methods of redox proteomics was pioneered in the Butterfield laboratory. Four recurring altered pathways secondary to oxidative damage in brain from persons with AD, aMCI, or Down syndrome with AD are interrelated and contribute to neuronal death. This “Quadrilateral of Neuronal Death” includes altered: glucose metabolism, mTOR activation, proteostasis network, and protein phosphorylation. Some of these pathways are altered even in brains of persons with preclinical AD. We opine that targeting these pathways pharmacologically and with lifestyle changes potentially may provide strategies to slow or perhaps one day, prevent, progression or development of this devastating dementing disorder. This invited review outlines both in vitro and in vivo studies from the Butterfield laboratory related to Aβ1-42 and AD and discusses the importance and implications of some of the major achievements of the Butterfield laboratory in AD research.
The editors of The Journal of Alzheimer's Disease (JAD) invited Professor Butterfield to contribute a review article focused on the importance and implications of his laboratory’s research over the years. According to the editors of JAD, this invitation was based on an assessment that our laboratory was one of the journal’s top authors and a pioneer in the concept of oxidative stress as a major contributor to the pathogenesis and progression of Alzheimer’s disease (AD). Consequently, what follows is largely a compilation of some of our studies related to amyloid-β peptide (Aβ) and its oxidative and neurotoxic properties and our studies related to oxidative and nitrosative stress in brains of subjects with AD, amnestic mild cognitive impairment (aMCI), and preclinical AD (PCAD). Because of this focus per the instructions of the JAD editors, most of the work discussed emanated from our laboratory. This focus is not meant to suggest that other laboratories have not contributed to the themes of this article, but as noted, our laboratory is recognized by JAD to be a major player in areas of the focus of this paper. Moreover, alternative hypotheses of Aβ-mediated oxidative stress have been proposed, but a complete review of the amyloid hypothesis is beyond the scope of this review. Following a brief introduction to AD, the content of this review paper is organized by in vitro studies, then in vivo studies, and concluding with a discussion of the implications and importance of the research from the Butterfield laboratory to the understanding of the pathogenesis and progression of AD.
BACKGROUND OF ALZHEIMER’S DISEASE
Alzheimer’s disease (AD) is an age-associated, neurodegenerative disorder characterized clinically by progressive cognitive decline and pathologically by the presence of senile plaques (composed mostly of Aβ surrounded by dystrophic neurites), by the presence of neurofibrillary tangles (NFTs, composed of hyperphosphorylated tau protein), and by loss of synapses. Currently, it is estimated that AD affects 5.5 million Americans and it is the 6th leading cause of death in the United States, but it is projected that the number of Americans living with AD will increase to 16 million by 2050. The cost of care of this number of patients and lost productivity of patients and caregivers is estimated to be between 1–2 trillion US $ annually [1]. In our view, this is a public health crisis unless means are determined to slow the onset of this disorder. Given that the average time between diagnosis and death is 8–10 years, delaying the onset of AD by 5 years instantly cuts the number of patients in half. In order to find a means of achieving this goal, basic research into the underlying pathophysiology of this disease is necessary. Our laboratory has contributed to this effort by discovery of oxidative stress associated with Aβ peptide.
AMYLOID-β PEPTIDE AND OXIDATIVE STRESS IN VITRO
Aβ-mediated free radical generation in solution
Early studies from our laboratory utilized electron paramagnetic resonance (EPR) to study the properties of Aβ in solution [2–9]. At the time, no hypothesis had been proposed to integrate the aggregation and neurotoxic properties of synthetic Aβ, shown by Aβ1-40 and exacerbated in Aβ25-35, with the observed biochemistry in AD brain. However, spin trapping studies utilizing highly purified N-tert-butyl-α-phenylnitrone (PBN) showed that both Aβ1-40 and Aβ25-35 independently induced the conversion of PBN from the non-paramagnetic nitrone to the stable, paramagnetic nitroxide in phosphate buffered saline (PBS), which could only happen by reaction with a free radical. Consistent with its accelerated aggregation and neurotoxic properties, Aβ25-35 resulted in a characteristic EPR signal within minutes and reached maximum intensity within 1 h, while Aβ1-40 required 3 h to produce a similar PBN signal which reached maximum intensity at 6 h [2]. Similar to Aβ1-40, Aβ1-42 was also shown to yield an EPR signal in this system [10]. These results persisted in the presence of metal chelators in all solutions employed indicating that Aβ-induced free radical formation was independent of redox metals; moreover, the reverse sequence of Aβ1-40, Aβ40-1, did not lead to a signal, even though any potential contaminants of neurotoxic Aβ1-40 that would produce a free radical would be present in the latter, non-neurotoxic peptide [11, 12]. Molecular oxygen was a requirement for radical formation since all signals disappeared if nitrogen-saturated solutions were employed. These results led us to propose our hypothesis that Aβ oligomer-induced free radical generation plays a causative role in the pathogenesis of AD and that methionine 35, as the only redox active amino acid of this peptide, plays a critical role in its neurotoxic chemistry [2].
Oxidative stress
Protein oxidation and lipid peroxidation are consequences of oxidative stress due to imbalance that occurs at a molecular/cellular level when free radical production exceeds antioxidant scavenging capacity [13, 14]. Protein oxidation is indexed by protein carbonyls [13] and 3-nitrotyrosine [15], while measures of lipid peroxidation include protein-bound 4-hydroxy-2-trans-nonenal (HNE) [16].
Reactive oxygen species
Production of intracellular reactive oxygen species (ROS) can be detected by the conversion of 2,3-dichlorofluorescin to 2,3-dichlorofluorescein (DCF) and subsequent increase in fluorescence. Aβ1-40 increases intracellular ROS in hippocampal neurons [11, 17, 18] and in cortical synaptosomes [19]. More recent research has shown that DCF has some issues related to potential artifacts [20], but since the reverse peptide did not lead to a signal, the conclusions reached using DCF likely are sound.
Protein oxidation
Aβ25-35, Aβ1-40, and Aβ1-42 were shown to induce significant increases in protein carbonyls in cortical synaptosomes [21–23], cultured hippocampal neurons [9, 10, 17, 18, 24, 25], primary neuronal cultures [12, 19, 26–30], and cultured astrocytes [31, 32].
Increased protein resident 3-nitrotyrosine was detected in primary neuronal culture [30, 33, 34] and cortical synaptosomes [21] and mitochondria [22] treated with Aβ1-42. Primary neuronal cultures from APP/PS1 human double mutant knock-in mice showed an increased susceptibility to exogenous Aβ1-42 mediated oxidative and nitrosative stress [35].
Antioxidants were tested in vitro and showed protection against Aβ1-42-induced ROS and protein oxidation. DCF fluorescence in hippocampal culture was modulated by pretreatment with vitamin E [36, 37]. Vitamin E [28, 36–38], propyl gallate [17], γ-glutamyl cysteine ethyl ester (GCEE) [29], and tricyclodecan-9-xanthogenate (D609) [39] were shown to prevent Aβ1-42 mediated protein carbonyl formation, while low molecular weight thiol amides including N-acetyl cysteine amide (AD4), N-acetyl glycine cysteine amide (AD7), and N-acetyl-Cys-Gly-Pro-Cys-amide (CB4) [30], acetyl-L-carnitine [33], and quercetin [34] were shown to prevent both protein carbonyl and 3NT formation. In vivo administration of D609 [21] and ferulic acid ethyl ester (FAEE) [23] were both independently shown to abrogate Aβ1-42-mediated increase in protein carbonyls and 3NT in subsequently isolated synaptosomes. In vivo administration of D609 also prevented Aβ1-42-induced protein carbonyl and 3NT formation in mitochondria [22].
Lipid peroxidation
In what we believe was the first demonstration that Aβ peptides could lead to lipid peroxidation, EPR studies utilizing nitroxyl stearate spin labels inserted into the lipid bilayers of rodent neocortical synaptosomes demonstrated that Aβ25-35 rapidly quenched the paramagnetic signal of 12-nitroxyl stearate (12-NS), a spin label that embeds deep within the lipid bilayer, but not the paramagnetic signal of 5-nitroxyl stearate (5-NS) whose nitroxide moiety is located near the lipid-water interface. The reverse sequence Aβ35-25 had little effect on either spin label showing that Aβ25-35 had the ability to embed in the lipid bilayer and induce lipid peroxidation through free radical generation [3]. Consistent with the notion that a free radical event in the bilayer following Aβ addition occurred, we postulated that the free radical in the 12-NS spin label was reduced by a free radical associated with Aβ, resulting in loss of the EPR signal. Pretreatment with chain-breaking antioxidant vitamin E prevented this Aβ25-35-induced decreased 12-NS signal intensity [40], further supporting this notion. Aβ25-35 was further shown to display hydrogen peroxide-like reactivity and was also shown to decrease the W/S ratio of the protein-specific spin label, 4-maleimido-tempo (MAL-6) bound to rodent cortical synaptosomes. MAL-6 reacts covalently with the thiol of cysteine residues resulting in spin label covalently bound in deep pockets of proteins where steric hindrance limits rotation of the spin label (S-site) or near the protein surface in more solvent exposed sites with higher mobility (W-sites). A decrease in W/S ratio (based on the EPR spectrum) indicates that Aβ induces conformational changes in membrane protein structure [7]. Lipid peroxidation occurred more quickly than protein oxidation following Aβ addition to synaptic membrane preparations.
We investigated the effects of the lipid peroxidation product HNE in cortical synaptsomes and compared its chemistry to that of Aβ25-35. HNE decreased the W/S ratio of MAL-6 similar to Aβ25-35; however, pretreatment with vitamin E prevented the effect of Aβ25-35, but not HNE since this reactive alkenal is not a free radical. Additionally, pretreatment with vitamin E significantly prevented Aβ25-35-induced increased protein carbonyls in cortical synaptosomes and decreased cell viability in cultured hippocampal neurons. Taken together, these results showed that HNE significantly affects the conformation of membrane proteins and increases protein carbonyls in cortical synaptosomes while decreasing cell survival in cultured hippocampal neurons, similar to Aβ25-35, but by a different mechanism which does not involve generation of free radicals [41].
In addition to Aβ25-35 and Aβ1-40, the more neurotoxic Aβ1-42 also induced not only protein oxidation, but also lipid peroxidation as measured by protein-bound HNE in primary neuronal culture [26–28]. Increased protein-bound HNE following addition of these Aβ peptides was prevented by pretreatment with vitamin E [26–28], low molecular weight thiol amides [30], acetyl-L-carnitine [33], and quercetin [34]. Moreover, Aβ1-42 induced loss of phospholipid asymmetry [42], consistent with our findings in AD and aMCI discussed further below [43]. In vivo administration of both D609 and FAEE independently protected against this Aβ1-42-mediated loss of phospholipid asymmetry in cortical synaptosomes [42], consistent with free radical processes.
To investigate the role of the secondary structure of the peptide contributing to the oxidative stress associated with Aβ1-42, we performed the following study [25]. Reasoning that like essentially all type I proteins in membrane lipid bilayers, Aβ1-42 would adopt a helical conformation, subsequently demonstrated by others [44], and noting that helices exhibit the i + 4 rule in which every 4th amino acid interacts with each other, we hypothesized that Ile-31 of Aβ1-42 would interact with the Met-35 residue of this neurotoxic peptide. Using NMR, the carbonyl oxygen atom of Ile-31 was shown to be within a van der Waals distance from the S-atom of Met-35 [45]. Since O is more electronegative than S, the electron density of the lone pair of electrons on the S-atom of Met-35 would be drawn toward the O-atom of Ile-31 (Fig. 1). This would make one of these two electrons susceptible to a one-electron oxidation by an oxidant, creating a transient free radical on the S-atom of Met-35. The dipole moment of the alpha-helix of Aβ1-42 in the bilayer could stabilize this sulfuranyl free radical long enough to allow it to abstract a labile allylic H-atom on the acyl chain of phospholipids, the first step in the process of lipid peroxidation. The resulting SH+ is an acid with pKa of minus 5, meaning any nearby base can easily remove the H+, forming Met again. In other words, this is a catalytic process in which the Met-35 of Aβ1-42 is the catalyst. Note that the carbon-centered free radical on the acyl chain of phospholipids following allylic hydrogen atom abstraction immediately binds paramagnetic molecular oxygen (two unpaired electrons), which by virtue of its zero dipole moment, is non-polar and highly soluble in the low dielectric medium of the hydrophobic core of the lipid bilayer. Radical-radical recombination reactions are among the fastest known reactions, and in this case molecular oxygen reaction with the C-centered radical on the acyl chain of phospholipids leads to a lipid peroxyl free radical (COO·). This latter free radical abstracts another allylic hydrogen atom resulting in a lipid hydroperoxide (COOH) functionality. It is this moiety in the lipid bilayer that decomposes into HNE among other products [13, 14]. The dipole moment of helical Aβ1-42, by virtue of its longer length, is larger than that of Aβ1-40, meaning that stability of the sulfuranyl radical on the S-atom of Met-35 would be higher with Aβ1-42 than Aβ1-40, providing more time for this radical to initiate lipid peroxidation by abstraction of a nearby labile allylic H-atom from the acyl chain of phospholipids. Such a scenario could conceivably provide a contribution to the more neurotoxic properties of the former over the latter. To test our proposed mechanism of the role of the helical nature of Aβ1-42 in the lipid bilayer contributing to the production of a free radical on its Met-35 residue, we used Aβ1-42I31P, in which Ile-31 was replaced by the helix-breaking amino acid proline, and observed no oxidative stress, no lipid peroxidation, and no neurotoxicity compared to native Aβ1-42, supporting our proposed mechanism [25]. As discussed further below, we provide additional evidence that the single Met residue of Aβ1-42 is the major site of oxidative stress associated with this peptide.
Fig.1
Oligomeric Aβ1-42 in the lipid bilayer maintains an α-helical secondary structure in which the carbonyl oxygen of Ile-31 interacts with the sulfur atom of Met-35 drawing electron density away from the sulfur atom increasing vulnerability to oxidants in the lipid bilayer. Such a one-electron oxidation of the sulfur atom initiates a catalytic cycle in which the resulting sulfuranyl free radical (S+) can abstract a labile allylic H-atom from the acyl chain of a phospholipid in the membrane forming a lipid radical which reacts with molecular oxygen to yield a lipid peroxyl free radical (ROO·). The lipid peroxyl free radical propagates the reaction by abstracting an additional hydrogen atom to yield a lipid peroxide (ROOH) which can decompose to form HNE as a lipid peroxidation byproduct that can subsequently lead to protein oxidation. Alternatively, the sulfuranyl free radical can abstract a H-atom from a membrane protein directly initiating protein oxidation. Mutation of Ile-31 to Pro breaks this α-helical interaction and subsequent Aβ1-42 initiated free radical lipid and protein oxidation indicating the importance of this interaction in the Met-35-mediated free radical chemistry of Aβ1-42.
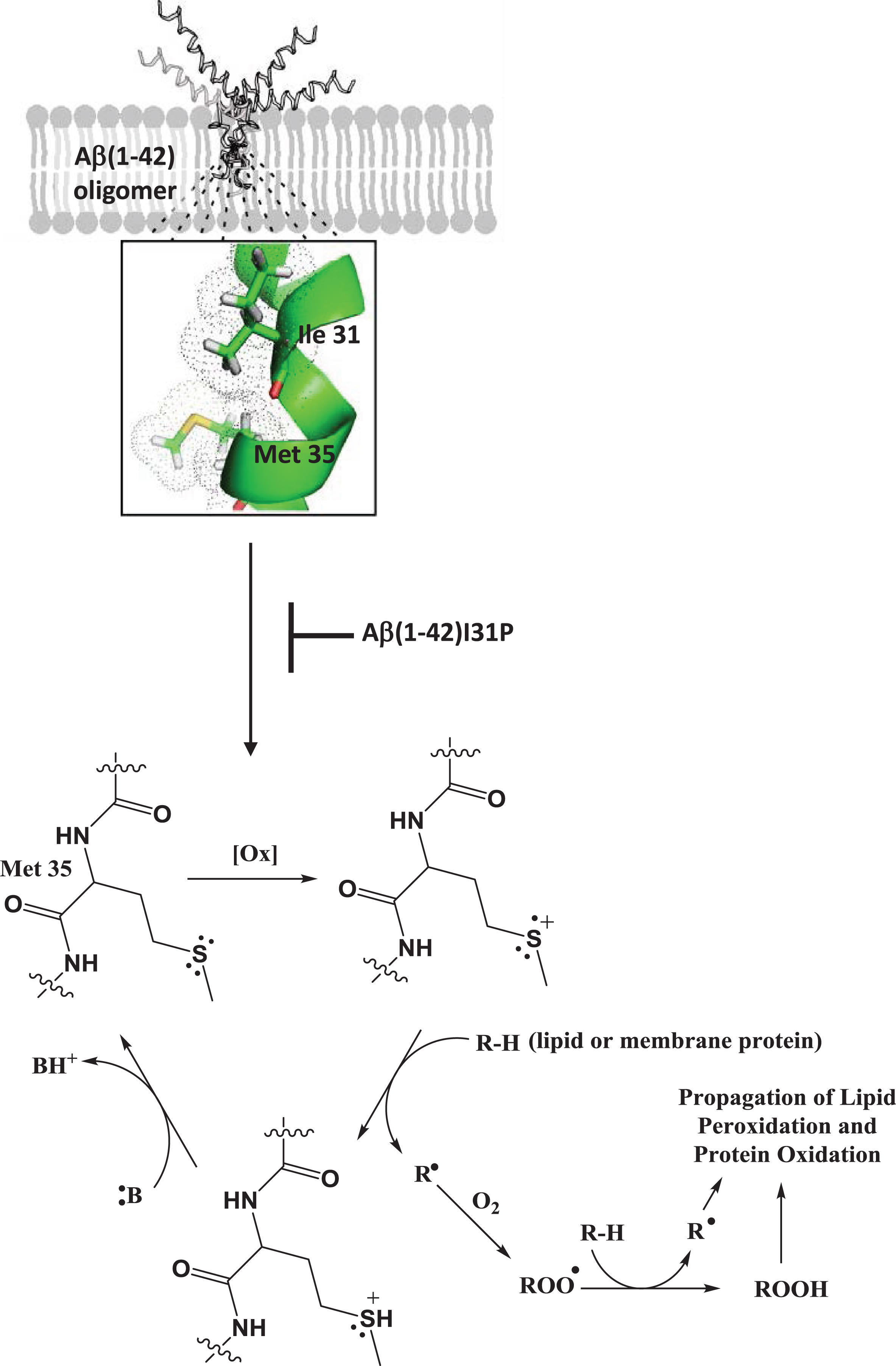
Aβ-mediated oxidative stress is modulated by apolipoprotein E
Cortical synaptosomes of apolipoprotein E (apoE) knock-out mice are more vulnerable to Aβ1-40-induced protein oxidation and lipid peroxidation as indexed by protein carbonyls and protein-bound HNE [46, 47]. ApoE isoforms modulate Aβ1-42 protein oxidation and lipid peroxidation in targeted-replacement knock-in mice in an allele specific manner with Aβ1-42-inducing the most cumulative oxidative damage in mice expressing human apoE ɛ4 followed by apoE ɛ3, while apoE ɛ2 did not exhibit any significant change in ROS generation, lipid peroxidation, nor protein oxidation compared to wild-type (WT) mice, suggesting that apoE-associated risk of AD may be related to the relationship between the apoE isoform and Aβ [48].
Aβ-induced protein oxidation and loss of function
Both Aβ1-40 and Aβ25-35 selectively mediated a significant reduction in Na+/K+-ATPase activity prior to neuronal degeneration in cultured rat hippocampal neurons, while Na+/Ca2 + exchange activity remained unchanged and oubain-insensitive Mg2 +-ATPase activity remained unaffected. The significant decrease in Na+/K+-ATPase activity induced by Aβ25-35 occurred prior to a significant increase in intracellular Ca2 + concentration. Pretreatment with vitamin E or propylgallate significantly attenuated the Aβ25-35-induced loss in Na+/K+-ATPase activity, increased intracellular Ca2 + concentration, and neuronal death. Aβ25-35 was also shown to impair Na+/K+-ATPase and Ca2 +-ATPase activity in synaptosomes prepared from neurologically normal adult hippocampus [49].
As discussed above, Aβ1-42 induces loss of phospholipid asymmetry in cortical synaptosomes [42] and similar loss of phospholipid asymmetry in AD and aMCI brain also was demonstrated [43] as discussed further below. Phospholipid asymmetry of phosphatidylserine primarily is maintained by the activity of flippase (i.e., ATP-dependent aminophospholipid translocase; Mg2 + ATPase) [50]. Flippase activity is significantly decreased in cortical synaptosomes treated with Aβ1-42; however, this is abrogated by in vivo administration with D609 or FAEE suggesting Aβ42-induced oxidation of flippase may lead to loss of function [42].
Glutamine synthase (GS) activity is significantly impaired by Aβ1-40 in solution [51], in hippocampal culture treated with Aβ1-40 [17, 51] and in cortical synaptosomes treated with Aβ25-35 [2]. Co-incubation of exogenous GS and Aβ1-40 exacerbates the neurotoxic properties of Aβ1-40 in hippocampal culture [17] due to disruption of Aβ fibril formation and partial fragmentation of Aβ1-40 [52] providing evidence of the toxicity of small soluble oligomers. GS is significantly carbonylated by Aβ25-35 in solution and correlates with significant loss in activity [53]. Taken together, these early studies showed that Aβ-induced oxidation of GS leads to loss of function and suggest that decreased GS activity in AD brain [54, 55] could be due to the effects of Aβ in vivo. Consistent with this suggestion, our group developed a spin label assay to determine GS oxidative structural alteration (and therefore functional alterations) [55]. GS has numerous Cys residues, the primary binding site for the spin label, MAL-6 (which was discussed above). We could fit the binding of MAL-6 to GS to a second-order kinetics rate equation, the slope of which was considerably smaller in previously oxidized GS, likely due to oxidation of the – SH moieties of the Cys residues in this protein. Addition of MAL-6 to GS isolated from AD brain showed a slope similar to that of the oxidized GS, consistent with the oxidation of this protein in AD brain as we determined by redox proteomics [56].
Creatine kinase (CK) BB isoform activity is significantly decreased in cortical synaptosomes incubated with Aβ25-35 [2] and in cultured hippocampal neurons treated with Aβ25-35 or Aβ1-40 even though CK expression was significantly increased [18, 24]. Pretreatment with vitamin E protected against Aβ25-35-induced loss of CK activity suggesting loss of function was due to oxidative modification of CK [36]. These results are consistent with observation of increased carbonylation of CK and decreased CK activity in AD brain [57].
Aβ25-35-mediated decreased glutamate uptake was prevented by pretreatment with the vitamin E analogue trolox in cultured rat hippocampal astrocytes. Aβ25-35 significantly increased protein carbonyls and decreased GS activity in astrocytes [31, 32]. Aβ1-42 induced oxidation of the glial glutamate transporter, GLT-1, in cortical synaptosomes assessed by significantly elevated protein-bound HNE consistent with oxidation of GLT-1 [58] and decreased GLT-1 activity in AD brain [59]. Synaptic membrane proteins covalently modified by HNE have altered conformations and likely altered function as a result [60]. Glial glutamate transport is important for removal of glutamate from the synapse, and decreased GS activity indicates an inability to efficiently convert glutamate into glutamine. Taken together inhibition of glutamate transport and impaired GS activity could lead to excitotoxicty and neuronal death [61]. Likewise, activity of choline acetyltansferase (ChAT), the enzyme which catalyzes the rate-determining step in acetylcholine synthesis, is decreased by Aβ1-42 [62] and in AD brain [63]. Aβ1-42 led to increased levels of protein-bound HNE oxidation of ChAT in cortical synaptosomes [64].
Taken together, these studies provide evidence for the role of Aβ1-42 mediated oxidative damage that induces conformational changes in cytosolic and membrane proteins resulting in loss of function and have relevance to oxidative damage observed in aMCI and AD brain [65].
Aβ-mediated protein oxidation: A redox proteomics approach
The Butterfield laboratory pioneered the use of redox proteomics [56, 66] as described further below and applied the technique to study the consequences of protein oxidation in in vitro models of Aβ1-42-induced oxidative stress. Dihydropyrimidase-related protein 2 (DRP-2), glial fibrillary acidic protein, and β-actin were found to be specifically significantly oxidized in cortical synaptosomes treated with Aβ1-42 [67], while 14-3-3 zeta, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), pyruvate kinase, and malate dehydrogenase were found to be significantly oxidatively carbonylated in primary neuronal cultures treated with Aβ1-42 [39, 68]. Pretreatment with GCEE or D609 abrogated oxidation of these vulnerable proteins [39, 68]. Proteins identified to be oxidized by Aβ1-42 in in vitro models are consistent with proteins identified to be oxidized in either AD or aMCI brain [56, 66, 69–74; reviewed in 75–77].
Aβ-mediated oxidative stress and neurotoxicity: The role of met 35
The critical role of methionine 35 in Aβ free radical generation has been previously reviewed [28, 78–80]. This oxidation of Met-35 was prevented by co-incubation with the free radical scavenger PBN demonstrating the oxidation of Met-35 is free-radical dependent [4]. Aβ peptides with scrambled [2, 32, 49, 51] or reversed [2, 3, 10, 51] amino acid sequences exhibited no oxidative stress and neurotoxicity in vitro. Deletion of Met-35 in Aβ25-35, Aβ25-34, prevented free radical production, protein oxidation, and neurotoxicity exhibited by Aβ25-35 [9]. Substitution of Met-35 of Aβ25-35, Aβ1-40, or Aβ1-42 by noreleucine, which replaced the sulfur moiety of Met with a methylene group (-CH2-), ameliorated the oxidative stress and neurotoxic properties [9, 10]. Conversion of the C-terminal carboxylate of Aβ25-35 to an amide moiety resulted in loss of the neurotoxic and oxidative properties of the peptide. Additionally, oxidation of the methionine thiol group to a sulfoxide also abrogated these properties in both Aβ25-35 and Aβ1-42 [12]. Aβ25-35 was shown to accelerate and exacerbate the oxidative and neurotoxic effects compared to Aβ1-40 and Aβ1-42, which we proposed to be a consequence of Met-35 being the C-terminal amino acid in Aβ25-35, and therefore ability to form a sulfuranyl free radical more quickly [12, 18]. As discussed below, in vivo studies of human Aβ1-42 in worms or mice models also demonstrated the key role of Met-35 of Aβ1-42 in the oxidative stress properties of this neurotoxic peptide [10, 81].
The histidine residues at positions 6, 13, and 14 were proposed to be a Cu2 + binding domain on which reduction of Cu2 + could initiate peptide radicalization [82, 83]; however, our studies showed that substitution of these residues in Aβ1-42 by tyrosine, which binds Cu2 + with at least two orders of magnitude less affinity than His, did not ameliorate the oxidative and neurotoxic properties of the peptide [11]. These results suggest that Cu2 + reduction is not a requirement for peptide-mediated radicalization [28]. Rodent Aβ1-42 was also investigated as it possesses mutations within the Cu2 + binding domain in the N-terminus of the peptide (arginine 5, tyrosine 10, and histidine 13 to glycine, phenylalanine, and arginine, respectively). Rodent Aβ1-42 was shown to have decreased Cu2 + reduction yet significant protein oxidation and lipid peroxidation which was inhibited by pretreatment with vitamin E, similar to Aβ1-42 [26].
Based on predictions of computational studies [84, 85], additional synthetic peptide substitutions were tested in vitro. Glycine 33 was proposed to play a role in Aβ1-42 free radical propagation by methionine sulfuranyl free radical abstraction of the α-hydrogen of Gly 33 [84]. Mutation of glycine 33 to valine, Aβ1-42G33V, diminished the free radical-induced oxidative stress and neurotoxic properties of the peptide indicating the potential involvement of Gly-33, but the mutant peptide still resulted in oxidative damage and neurotoxicity compared to control [19]. Phenylalanine 20 was proposed to play a role in long distance electron transfer between a sulfuranyl radical of Met-35 and Cu2 + bound in the N-terminus of the peptide [85]. Substitution of Phe-20 by alanine, Aβ1-42F20A, was shown to significantly decrease Cu2 + reduction, but did not alter the oxidative stress and neurotoxic properties of the peptide [27].
As noted above, the role of the α-helical secondary structure of the C-terminus of Aβ1-42 was investigated. Mutation of Ile-31 to proline, Aβ1-42I31P resulted in abrogation of Aβ1-42-induced neurotoxicity and oxidative damage, indicating that helix-associated interactions between Ile-31 and Met-35 play an important role [25].
Mutation of glycine 37 to aspartate, Aβ1-42G37D, introduced a highly negative charge, causing removal of Met-35 from the lipid bilayer. Consistent with the notion that with such a modified peptide added to membranes there would be no allylic H-atoms near Met-35 for abstraction to initiate lipid peroxidation, Aβ1-42G37D abolished the oxidative and neurotoxic properties of Aβ1-42, suggesting that interaction of Met-35 within the lipid bilayer is a requirement for Aβ1-42-mediated lipid peroxidation [86]. This result is consistent with the known requirement that the free radical source needed to initiate lipid peroxidation must be in the lipid bilayer [13] because of the extremely short half-lives and reactivity of most free radicals. A free radical source external to the lipid bilayer would long since have reacted prior to its entry to the lipid bilayer and could not initiate lipid peroxidation [13, 14]. Consequently, Aβ1-42 oligomers must be inserted into the lipid bilayer of brain cells to initiate lipid peroxidation, and we propose, based on this consideration and our experimental evidence outlined above and below, that lipid peroxidation, with subsequent production of HNE, is the first damaging event to occur with Aβ1-42 in AD brain.
IN VIVO STUDIES OF AD AND MODELS THEREOF FROM THE BUTTERFIELD LABORATORY
Oxidative stress in brain in subjects with AD and aMCI
As discussed above, the three major indices of oxidative and nitrosative stress used in our laboratory are measures of protein oxidation (protein carbonyls; 3-nitrotyrosine, 3NT) and lipid peroxidation (protein-bound HNE) [13]. Using brain regions obtained at autopsy with a very short postmortem interval (2–4 h) provided by the Alzheimer Disease Center of the Sanders-Brown Center on Aging at the University of Kentucky, our laboratory showed that protein carbonyls were elevated in brain of subjects with AD in regions rich in Aβ but not elevated in Aβ-poor cerebellum [87].
As noted above, lipid peroxidation in AD brain was evidenced by elevated levels of HNE bound to the glutamate transporter, GLT-1 (also called excitatory amino acid transporter-2, EAAT2) [58]. This oxidative modification of GLT-1 likely accounts for decreased activity of this transporter in AD [59], since we showed earlier that HNE binding to synaptic membrane proteins leads to altered conformation [60], and essentially always leads to altered structure and consequent altered function. Coupled to our earlier studies showing that glutamine synthetase was more oxidized in AD brain [55, 56, 88], HNE modification of GLT-1 suggests that at least two means of removing glutamate from the synapse after neurotransmission do not work well in AD brain. The consequent increased synaptic glutamate would lead to excitotoxicity [89].
One aspect of lipid peroxidation is loss of phospholipid asymmetry in brain cell membranes. Using a method by which fluorescently labeled phosphatidylserine (PtdSer), normally found on the inner bilayer lammelae, could be detected on the outer membrane bilayer lammelae, we reported that AD brain had a profound loss of phospholipid asymmetry evidenced by a large percentage of PtdSer on the outer bilayer lammelae [43]. Such loss of phospholipid asymmetry would have profound effects on transmembrane proteins, such as ion-motive ATPases, which are known to be dysfunctional [49], and PtdSer on the outside lamellae of lipid bilayers is a marker for and possibly inducer of apoptosis [43]. Elevated 3NT in AD brain also was determined [70, 90]. Consequently, AD brain is under significantly elevated oxidative and nitrosative stress with altered function of oxidatively modified proteins.
Similar studies were performed in brain of subjects with aMCI. Elevated oxidative and nitrosative stress and loss of phospholipid asymmetry in brain in aMCI (also similarly obtained with short postmortem interval) were performed in our laboratory and others and demonstrate that long before dementia occurs in the progression of AD oxidative and nitrosative stress already are present [43, 72, 91–95].
Confirmation of oxidative stress in animal models of AD
C. elegans
When the DNA of Aβ1-42 was inserted into C. elegans using a muscle wall protein promoter, elevated oxidative stress was observed evidenced by elevated protein carbonyls [10] that correlated with the amount of soluble Aβ1-42 present. When the codon for Met in residue 35 of Aβ1-42 was replaced by a codon for Cys, no oxidative stress was observed even though the amount of Aβ1-42 deposited was similar to that in worms with non-mutated Aβ1-42, consistent with the discussion above on in vitro studies of Aβ1-42 that the single Met residue of this neurotoxic peptide is critical to its oxidative stress properties in vivo as well as in vitro [12]. This point is discussed further below. In a second study [96], C. elegans with insertion of a unique surveillance system that destroyed the message of foreign genes, i.e., DNA of Aβ1-42, were used. However, if these worms were raised at a non-permissive temperature, the surveillance system did not function and Aβ1-42 could be produced for as long as the non-permissive temperature was employed. That is, one could control the time during which Aβ1-42 was produced. Using this system, protein carbonlys were determined in these C. elegans as a function of production time of Aβ1-42. At 24 h following initiation of Aβ1-42 production, protein carbonyls were highly significantly elevated, and this correlated precisely with onset of a paralysis phenotype. That is, Aβ1-42-mediated oxidation of muscle wall proteins likely led to the observed paralysis.
Mouse models
a) Indices of oxidative and nitrosative stress in brain of APP/PS-1 human double mutant knock-in mice were significantly elevated as a function of age (and therefore of increased Aβ1-42 production), and these elevated indices correlated with the level of soluble Aβ1-42 [97–99]. Providing N-acetylcysteine (NAC), which supplies the rate-limiting substrate (Cys) for the synthesis of the endogenous antioxidant glutathione, in the drinking water for these mice and corresponding WT mice for 5 months starting at either 4 months or 7 months of age (to bracket the time of Aβ deposition beginning significantly at 9 months of age) demonstrated significantly decreased levels of protein carbonyls, protein-bound HNE, and protein-resident 3NT in brain at both 9 months and 12 months of age in the APP/PS-1 human double mutant knock-in mice [100]. These results are consistent with an Aβ1-42-associated free radical oxidative stress in brain of these knock-in mice. The reduction of each oxidative or nitrosative stress parameter following NAC treatment was greater in the 9-month old mice compared to 12-month old mice, consistent with the notion that the earlier the treatment with NAC in these mice, the more pronounced its protective effects in brain. Since NAC is FDA-approved for acetaminophen overdose, we posit that NAC should be considered as a possible adjuvant treatment for persons diagnosed with either PCAD or aMCI. Based on the results mentioned above, it is likely that NAC use for late-stage AD would be much less effective. Similar to what occurs in AD and aMCI brain, lipid asymmetry is lost in brains of these mice as a function of age [99].
b) Mice with two mutations in APP, namely the Swedish/Indiana (Swe/Ind) mutations, also called the J20 mouse, exhibited significantly elevated oxidative stress in brain at 9 months of age, when pathology was apparent [81, 101–103]. If a third mutation is introduced into APP, namely substitution of Met-631 by Leu, which corresponds to the Met-35 residue of Aβ1-42, and the mice aged to 9 months, no oxidative stress occurred in brain compared to that of WT mice, confirming the findings in the C. elegans studies and showing for the first time in vivo in a mammalian system that the single Met at residue-35 of Aβ1-42 is critical to the oxidative stress associated with this neurotoxic peptide [81]. Pathologically, in contrast to the numerous senile plaques in the J20 mouse brain, the brain of the J20 mouse with Aβ1-42M35L substitution also deposit similar amounts of Aβ1-42 and Aβ1-40 but only in punctate forms. This result conceivably could support the notion that the Met residue of Aβ1-42 is critical to plaque formation as well as oxidative stress [81].
c) Seven days after in vivo injection of Aβ1-42 into rat forebrain, elevated oxidative stress in hippocampus indexed by elevated protein carbonyls was observed [104]. Redox proteomics analysis (methods discussed below) of these samples led to the identification of oxidized hippocampal proteins, among which were 14-3-3, GAPDH, phosphoglycerate mutase 1, and pyruvate dehydrogenase [104]. Comparison with redox proteomics results in aMCI and AD brains (Table 1) shows Aβ1-42 is able to replicate some of these same oxidized proteins, particularly those related to glucose metabolism.
Table 1
Redox proteomics identification of oxidatively or nitrosatively modified brain proteins from subjects with Alzheimer’s disease or amnestic mild cognitive impairment
| Cellular Function or Pathway Dysfunction | Oxidatively or Nitrosatively Modified Brain Proteins |
| Glucose Metabolism | GRP-78; Alpha-enolase; Aldoase; TPI; GAPDH; PGM; Pyruvate Kinase; Lactate Dehydrogenase; Aconitase; Malate Dehydrogenase; Carbonic Anhydrase-2; ATP Synthase-alpha |
| Excitotoxicity | Glutamine Synthetase |
| Proteasome | HSP70; Ubiquitin C-terminal Hydrolase L-1 |
| Antioxidant Defense | MnSOD; Peroxidredoxin 6; Carbonyl Reductase |
| Lipid and Cholinergic Function | Neuropolypeptide h3; GST; MRP-1 |
| Synaptic Plasticity | DRP-2 (CRMP-2); Beta-Actin; Alpha-Tubulin; Gamma-SNAP; Syntaxin Binding Protein-1; Fascin-1 |
| Tau Hyperphosphorylation, Aβ Production; Cell Cycle | Pin 1 |
| Mitochondria | VDAC-1 |
| Cell Signaling; Regulation of Antioxidant Proteins; Apoptosis | 14-3-3-Gamma; MAPK-1; p53 |
| Protein Synthesis | Initiation Factor-alpha; Elongation Factor-Tu |
Canine model
The beagle dog deposits Aβ1-42 of identical amino acid sequence as humans as a function of age. In the absence of specific interventions, beagles show behavior reminiscent of dementia by 15 years of age [105]. To investigate the role of potential interventions on brain oxidative stress, 12-year-old beagles were raised on a high antioxidant diet and provided a program of enhanced environmental stimulation and exercise. At the end of 3 years of such interventions, i.e., 15-year-old beagles, oxidative stress in brain was reduced to the levels of 4-year-old beagles and far less than brains from 15-year-old beagles maintained on a diet of dog chow [106]. Both the high antioxidant diet and behavioral enrichment were required to lower brain oxidative stress in aged beagles; neither approach alone was sufficient to lower oxidative stress in brain of the aged dogs. These beneficial effects of diet and behavioral enrichment were inversely correlated with levels of Aβ1-42 and error rate on tests on learning and memory [106]. Redox proteomics studies identified specific brain proteins that were less oxidized following the combined high antioxidant diet and behavioral enrichment programs of the aged beagles. These proteins are found in neurochemical pathways associated with synaptic remodeling (learning and memory), glucose metabolism, Ca2 + homeostasis, and membrane structure [106]. If translatable to humans, the results of this study in an animal with Aβ1-42 of the same amino acid sequence as humans would suggest that high antioxidant diets and continued learning throughout the lifespan, coupled with exercise, may reduce risk of developing AD. That is, these lifestyle-related approaches conceivably could be beneficial to lowering risk of development and/or progression of AD.
Redox proteomics identification of oxidatively or nitrosatively modified proteins in AD and aMCI and models thereof
Human studies
The field of redox proteomics, pioneered in our laboratory (reviewed in [107, 108]), was used to identify brain proteins that are oxidatively or nitrosatively modified in AD and aMCI. The methods of redox proteomics we employed [71, 75, 76, 77, 107–114] involve two-dimensional separation of proteins (isoelectric focusing followed by SDS-polyacrylamide gel electrophoresis), transfer of the separated proteins to a nitrocellulose membrane, and formation of a 2-dimensional western blot using primary antibodies specific for protein-bound HNE, protein-resident 3NT, or the protein hydrazone formed by addition of 2,4-dinitrophenylhydrazine to protein carbonyls. Sophisticated image analysis software makes a spot-by-spot comparison of separated brain proteins on gels and on western blots from AD or aMCI subjects and aged-matched control subjects that allows identification of specific elevation of oxidative or nitrosative stress markers per unit protein level for statistically differentially oxidatively or nitrosatively modified proteins. Such identified spots are cut from the gel followed by in-gel trypsin digestion, clean up using C18 Zip tip separation of ions, and subsequent submission of the tryptic peptides for tandem mass spectrometric analyses (MS/MS). This latter method provides the amino acid sequence of these peptides. Interrogation of protein data bases leads to identification of specific brain proteins that are oxidatively or nitrosatively modified, since each protein has a unique amino acid sequence. Validation of the MS/MS-identified proteins, often using western blotting methods, confirms the identification of the damaged proteins.
As noted above, oxidatively or nitrosatively modified brain proteins generally are dysfunctional. Using the above-outlined approach for redox proteomics, brain proteins and associated decreased functions were identified (Table 1). This paper provides a brief discussion of each major class of oxidatively or nitrosatively modified brain proteins in AD and/or aMCI.
a) Glucose metabolism is significantly depressed in brains of patients diagnosed with aMCI, and dramatically decreased in patients with late-stage AD as assessed by F-18dG-positron emission tomography [115]. Several glycolytic and some TCA cycle, and mitochondrial proteins, dysfunctional as a consequence of their oxidative and/or nitrosative modification, with correspondingly less ATP production, likely contribute to the defect in glucose utilization in AD and aMCI brain. Lower levels of ATP cause loss of cell potential, which would open voltage-gated Ca2 + channels, which, in turn, would initiate numerous detrimental changes in neurons, e.g., calpain activation, endonuclease and phospholipase A2 activation, ER dysfunction, and mitochondrial swelling leading to release of cytochrome C and initiation of the intrinsic pathway for apoptosis [77].
Both enolase and GAPDH are pleiotropic enzymes, with functions other than those in glycolysis [116, 117]. In addition to in glycolysis, enolase plays roles in regulation of certain pro-survival pathways and in regulation of plasminogen, a pro-enzyme that is converted to plasmin, a protease that cleaves, among other proteins, Aβ1-42. Consequently, loss of function of enolase due to oxidative modification is damaging to neurons on several levels [116]. Similarly, GAPDH has many protective functions in addition to its role in glycolysis. For example, GAPDH interacts with the sirtuin, Sirt-1, to modulate the action of p53, thereby keeping apoptosis in check. Moreover, GAPDH plays a key role in regulating the pentose phosphate shunt (PPS) (i.e., in NADPH production) and thereby keeping glutathione in a reduced state via glutathione reductase, which uses NADPH as a source of reducing equivalents. Because GAPDH has an unusually high number of thiol-containing Cys residues, this protein is particularly vulnerable to NO modification to form nitrosothiols, which interact with the transcription factor Siah-1 to form a complex that initiates apoptosis. Consequently, when oxidatively or nitrosatively modified, GAPDH loses its ability to check p53-mediated apoptosis, causes VDAC-2 to open in mitochondria with subsequent efflux of cytochrome C and initiation of apoptosis, forms high molecular weight aggregates that clog neurons, and plays as yet not well understood roles in Aβ1-42 aggregation and promotion of NFT formation [117]. Consistent with this discussion, the mitochondrial protein VDAC-1 was identified by redox proteomics as a nitrosatively modified protein in AD brain [90].
b) Glutamate-mediated excitotoxicity is an ROS-related process by which Ca2 + enters neurons and causes cell death [118]. Following its role in neurotransmission, Glu is taken up by astrocyte membrane-resident GLT-1 (EAAT2) and converted to glutamine by GS. Redox proteomics identified GS as oxidatively modified, and the resultant loss of function, coupled with loss of function of GLT-1 because of HNE modification, makes the AD brain highly vulnerable to accumulation of higher synaptic levels of Glu and induction of excitotoxicity [56].
c) Aggregated or damaged proteins are subjected to ubiquitinylation by action of E1, E2, and E3 ubiquitin ligases, forming a polyubiquitin chain on the damaged proteins. Molecular chaperones escort these polyubiquitinylated proteins to the 26S proteasome for degradation to small peptides. However, prior to or just after entering the 19S cap of the 26S cylindrically shaped proteasome, the polyubiquitinylated chain must be removed from the damage, aggregated protein. This latter function is carried out by ubiquitin C-terminal hydrolase L-1 (UCH L-1) one ubiquitin residue at a time from the C-terminal end. This is an essential function since there is a fixed pool of ubiquitin in the brain, so recycling of ubiquitin for use on other aggregated and/or damaged proteins is necessary for proper brain function. UCH L-1 is oxidatively dysfunctional as shown by our laboratory using redox proteomics [56]. Three predictions, all consonant with known pathophysiology in AD brain, result from oxidative dysfunction of UCH L-1: first, that there will be found in AD and aMCI brain an excess ubiquitinylation of many proteins [119]; second, the large number of hydrophobic ubiquitin molecules on the damaged aggregated protein that are not removed will damage the 26S proteasome [120]; and, third, inhibition of the 26S proteasome will result in a significant accumulation of aggregated, damaged proteins in the brain of AD and aMCI subjects, a quintessential pathological aspect of AD [121].
d) Since oxidative stress is suggested by our laboratory to be a key component in the progression of AD, the oxidative modification of antioxidant defense enzymes would pose a significant challenge to neuronal survival in the face of free radical attack associated in part with Aβ1-42. Peroxiredoxin 6 scavenges peroxides and H2O2 to remove this potentially pro-oxidative molecule. Consequently, oxidative or nitrosative modification of this antioxidant enzyme, as we reported occurs in early AD inferior parietal or hippocampal brain regions [71, 122], would contribute to the oxidative stress under which the early AD brain exists and thereby be damaging to neurons.
e) Neuronal signaling is altered in AD and aMCI brain [123]. Neuropolypeptide h3, also called phosphatidylethanolamine binding protein-1, has several functions, including modulation of Raf/MEK/ERK signaling, which appears to be compromised in AD brain [124]. Brain neuropolypeptide h3 was identified by redox proteomics as nitrosatively modified in early AD [122] and covalently bound by the lipid peroxidation product HNE in aMCI [74], which conceivably could contribute to the loss of function of this key signaling pathway. Other signaling-related proteins oxidatively or nitrosatively modified in AD and/or aMCI brain and likely dysfunctional include: 14-3-3 proteins (that form bridges between different proteins involved in signaling and regulates their activity) [125]; p53 (modified by nitration, HNE, and glutathionylation in AD and aMCI brain) [126–128] and which inversely regulates antioxidant protein activity [129], influences the mitochondrial proteome [130], especially MnSOD [131]; and MAPK-1, which may be modulated by neuropolypeptide h3 [74, 122, 125].
f) Synaptic plasticity is required for learning and memory. Several proteins involved in making synaptic connections (dihydropyrimidinase-related protein 2, also called collapsin response-mediated protein 2, CRMP2), neurotransmission (gamma SNAP; syntaxin binding protein-1), and axonal structural proteins (beta-actin, alpha tubulin, fascin-1) are oxidatively or nitrosatively modified and therefore structurally altered and likely less functional in AD brain [110], may contribute to the loss of cognition in aMCI and AD.
g) Oxidative stress and neuropathology. Based on our redox proteomics studies, we opine that oxidative dysfunction of a single protein, peptidyl-prolyl cis/trans-isomerase (Pin1), may be intimately associated with, if not account for, three major histopathological characteristics of AD and aMCI brain: senile plaques, NFTs, and activation of the neuronal cell cycle [132]. Pin1 is a regulatory protein that modulates the activities of proteins to which it binds. Phosphorylated Ser or Thr on the N-terminal side of a Pro is the binding sequence of the protein recognized by the WW domain of Pin1. As a hinge protein, the active site of Pin1 rolls up to change the Pro residue of the protein to which Pin1 is bound from the trans to cis conformation or vice versa. Such a change makes an enormous change in local structure of the proteins to which Pin1 binds, thereby regulating the activity of the latter.
Two key AD-related proteins to which Pin1 binds are APP and tau [133]. If Thr 668 of APP is phosphorylated, Pin1 will bind to this protein from which Aβ42 emanates. If the Pro residue on the N-terminal side of phosphorylated Thr 668 is in the trans conformation, the non-amyloidogenic pathway is engaged and the brain is protected; in contrast, if the Pro is in the cis conformation, amyloidogenic processing of APP is engaged, leading to neurotoxic Aβ1-42. That is, if the Pro adjacent to phosphorylated Thr 668 is in the cis conformation, oxidatively dysfunctional Pin1 is unable to switch the Pro of APP into a neuroprotective trans conformation, leading to excess Aβ1-42 and subsequent senile plaques. Similarly, hyperphosphorylation of tau leads to dissociation of this microtubule-stabilizing protein, forming NFTs, and the destabilized microtubules lead to loss to transport of mitochondria to the presynaptic terminus, starving the synaptic region of ATP. As a consequence, cell potentials change, opening Ca2 + channels, permitting Ca2 + enters the cells and lead to neurotoxic events as noted above. Pin1 binds to, and modulates, tau as well as to some of the primary tau kinases (such as GSK-3β) and phospho-tau phosphatases (such as PP2A) [132], contributing to the phosphorylation state of tau. We found using redox proteomics that Pin1 was oxidatively dysfunctional in both aMCI and AD brain [72, 134].
Neurons are postmitotic cells. We opine that due to the significant oxidative stress under which AD and aMCI brains exist and subsequent neurotoxicity, neurons attempt to undergo mitosis and enter the cell cycle. However, they become trapped in this cycle, and ultimately undergo apoptosis. This hypothesis was confirmed by others for AD [135] and by our laboratory for aMCI [136, 137]. Pin1 is critical to the function of the cell cycle [137], so oxidative dysfunction of Pin1 potentially contributes to the death of neurons that enter the cell cycle.
Likely explaining the loss of activity of Pin1 in AD and aMCI brain reported by us [72, 134], in collaboration with Prof. K. Ping Lu, we recently reported that the Cys-113 active site of Pin1 was highly oxidized [138]. Taken together, oxidative dysfunction of Pin1 conceivably can contribute to, if not account for, three major neuropathological alterations in AD and aMCI brain, and as such, may be a promising therapeutic target for these disorders.
h) Proteomics analyses of brains of subjects with mutations in the gene PS-1, form of inherited AD led to identification of neuronal-specific gamma-enolase, actin, UCH L-1, and dimethylarginine dimethylamino hydrolase (DMDAH), suggesting these proteins are critical to the pathogenesis of this dementing disorder [139]. These results demonstrate that inherited and sporadic AD have similarly oxidatively modified proteins. Several of these proteins were discussed above. DMDAH is associated with NO function, and therefore conceivably could contribute to the excess 3NT found in AD and aMCI brain [70, 92, 122].
In vivo studies of animal models of AD
a) C. elegans. Redox proteomics investigation of C. elegans expressing human Aβ1-42 identified oxidatively modified proteins (indexed by protein carbonyls) that occur in pathways related to fatty acid oxidation, protein synthesis, the citric acid cycle, proteasome function, PPS, and glutathione removal of HNE from cells via glutathione-S-transferase [140]. Each of these pathways are dysfunctional in AD brain. For example, glutathione-S-transferase has excess HNE covalently bound to it [141], and like other HNE-bound proteins would be expected to be dysfunctional. This phenomenon would have an effect on the higher levels of HNE-bound proteins in both human brain and C. elegans that express human Aβ1-42, both of which are known as described above. Another example of an oxidized protein found in C. elegans that express human Aβ1-42 is transketolase, a key component of the PPS, in which the major reducing agent NADPH is produced. Consequently, lower levels of NADPH would have the effect of reduction of oxidized glutathione via glutathione reductase, which is known in AD to have altered activity [142], and decreased biosynthesis in reactions that require reducing equivalents supplied by NADPH. Each of the other pathways mentioned above, known to be dysfunctional in AD brain, strengthen the notion that Aβ1-42 contributes significantly to the oxidative modification of key proteins and resulting dysfunction of important cellular pathways that impact neuronal survival in AD [140].
b) Mouse models.
1) APP/PS-1 human double mutant knock-in mice. Brains from these mice were collected as a function of age from 1–15 months of age, and redox proteomics to identify brain proteins with excess protein carbonyls was performed and compared to that of WT mice of the same background and age in order to control for the known effect of increasing protein carbonyls in brain with aging [97]. Brain proteins that were highly significantly oxidatively modified at each age group compared to the respective control brain proteins are indicated in Table 2. Examination of this table indicates that a considerable number of oxidatively or nitrosatively modified brain proteins in this mouse model of AD are the same as the human disease, suggesting that human Aβ1-42-associated oxidative stress is critical to the molecular changes observed in both cases. As noted above, NAC treatment led to decreased oxidative stress in the brain of these KI mice [100]. Proteomics analyses showed that glutamate dehydrogenase was elevated in 9-month-old, NAC-treated mice, while alpha-enolase and pyruvate kinase were both elevated at 12 months following NAC treatment [103]. Each of these enzymes are involved in ATP production via the TCA cycle (from the resulting alpha-ketoglutarate formed) or glycolysis, consistent with the neuroprotective effect of NAC to increase glucose utilization that is lost in these mice (and AD) with age.
Table 2
Proteomic identification of brain proteins with excessive levels of protein carbonyls as a function of age in APP/PS-1 knock-in mice. From [97]
| Age of the mice | Oxidatively modified proteins Identified |
| 1 month | 14-3-3 zeta/delta/gamma |
| 6 months | Alpha-enolase; Pyruvate Dehydrogenase E1 |
| 9 months | Alpha-enolase; 14-3-3 zeta/delta/gamma; Pin 1 |
| 12 months | Alpha-enolase; 14-3-3 zeta/delta/gamma; Pin 1; Beta-synuclein; ATP synthase-alpha |
| 15 months | Alpha-enolase; 14-3-3 zeta/delta/gamma; Pin 1; Beta-actin; ATP synthase-alpha |
2) The brain from the J20 mouse, with Swe/Ind human double mutant APP, was subjected to redox proteomics [101]. In the brain of the J20 mouse, we found Pin1 and phosphatidylethanolamine-binding protein-1 (also called neuropolypeptide h3 as noted above) were nitrosatively modified indexed by excess 3NT. The significance and importance of Pin1 were discussed above as was the case for neuropolypeptide h3.
c) Canine model. Redox proteomics results of the beagle model were discussed above.
Other in vivo studies in AD and aMCI
1) HO-1/BVR-A
The heme oxygenase-1 (HO-1)/bilverdin reductase-A system, which produces bilirubin, an antioxidant at low levels [143], is highly nitrosatively modified and dysfunctional in AD and aMCI brain [144–146]. Since HO-1 degrades pro-oxidant heme, dysfunction of HO-1 can contribute to oxidative stress. Similarly, since BVR-A converts biliverdin to bilirubin, oxidative dysfunction of BVR-A could lead to oxidative stress. Moreover, since BVR-A is a pleiotropic enzyme, acting as both a reductase for bilverdin and as a kinase for the insulin signaling enzyme, insulin receptor substrate-1 (IRS-1), depending on the type and location of phosphorylation, nitrosative modification of BVR-A could affect insulin signaling in AD and aMCI brain [147]. Together with our collaborator, Prof. Marzia Perluigi and her laboratory, we recently showed that this notion is indeed the case [147, 148].
2) Heat shock proteins (HSPs). Analysis of brains from subjects with aMCI demonstrated differential levels and oxidative modification of key HSPs, including HSP 27, 32, 60, 70, and 90 [149], consistent with decreased ability to refold aggregated, damaged proteins or transport such proteins for degradation through the proteostasis network that occurs early in the progression of AD.
3) The mammalian target of rampamycin (mTOR), if activated, leads to inhibition of autophagy and induction of insulin resistance [150, 151]. Activation of mTOR can occur by activation of PI3K/Akt pathway by Aβ1-42 [152]. Once activated, mTOR leads to accumulation of cellular detritus and damaged organelles due to inhibition of autophagy that can lead to neuronal death. Moreover, activated mTOR is a kinase, one substrate of which is the protein p70S6K, which then becomes a kinase, one substrate of which is IRS-1 (Ser-307) [151]. Phosphorylation of Ser-307 of IRS-1 is a biochemical marker of insulin resistance. We examined brains from subjects with AD and aMCI to demonstrate for the first time that insulin resistance is prominent in aMCI, well before dementia occurs [150]. This may have relevance to the increased risk of developing AD by those with type 2 diabetes mellitus [153]. Moreover, we showed that autophagy was inhibited in brain even at the PCAD stage of the disease [150]. Our results are consistent with the notion that inhibitors of mTOR may be a promising therapeutic approach to slow progression of AD.
4) Potential biomarkers. It is now widely accepted that neuropathology of AD occurs in brain approximately two decades prior to onset of clinical cognitive impairments [154]. Consequently, identifying biomarkers much earlier than the onset of symptoms would be great benefit to early interventions to slow or inhibit progression of the stages of AD. In a study in collaboration with our colleagues at the Universities of Valencia and Rome La Sapienza, redox proteomics for cerebrospinal (CSF) proteins modified by protein carbonyls from aMCI and AD patients were conducted [155]. Several important results and conclusions of this study were reached: a) Oxidized proteins in CSF from aMCI were largely conserved in CSF from AD patients, suggesting that oxidative stress in CSF occurs many years before onset of dementia; b) Proteins involved in inflammation, inhibition of proteases, vitamin D binding, fatty acid, and Aβ1-42 transport, among others, were oxidized in both aMCI and AD CSF, and therefore likely dysfunctional. c) Loss of function of these proteins have many pathological and potential therapeutic implications. For example, dysfunction of vitamin D-binding protein may relate to the widely-accepted notion that low vitamin D is a major risk factor for developing AD [156, 157]. Conceivably, excess oxidative modification of ApoE in both aMCI and AD is consistent with the oxidation of fatty acids of lipoproteins, which can increase levels of pro-inflammatory cytokines. Prostaglandin-H2 D-isomerase (PGHD2) was oxidized in CSF from patients with both conditions compared to controls. This protein converts prostaglandin H2 to prostaglandin D2, the latter involved in sleep regulation. It is noteworthy that lack of sufficient sleep is emerging as a significant risk factor for developing AD [158], so oxidation of PGHD2 could contribute to this risk factor. Moreover, PGHD2 may be a chaperone for Aβ1-42, suggesting oxidation of this protein could lead to more deposition of this neurotoxic peptide. We suggest that oxidation of the proteins identified in CSF in aMCI and AD could be critically important in the progression of AD and their redox proteomics identification may contribute to the panel of biomarkers that can diagnose onset of AD before clinical symptoms appear.
Similarly, we investigated mitochondria from peripheral lymphocytes in patients with AD and aMCI to demonstrate elevated oxidative stress in both cases, showing that lymphocyte-resident mitochondrial oxidative stress occurs early in the progression of AD [159, 160]. Moreover, proteomics analysis identified similar proteins as found in brain as having differential levels in both aMCI and AD, showing, like oxidative stress, that altered levels of these proteins occur early in the progression of AD. Especially elevated were ATP synthase and antioxidant proteins, possibly reflecting attempts by mitochondria in lymphocytes to combat oxidative stress present. These findings, though by themselves likely could not serve as definitive biomarkers for AD and aMCI, suggest that analysis of oxidative stress and protein alterations in CSF and peripheral lymphocyte mitochondria conceivably could be part of a panel of biomarkers that could be used to predict those who were on the path to aMCI and AD.
5) Phosphoproteomics. In addition to redox proteomics, we examined another posttranslational modification, phosphorylation, by phosphoproteomics in brains from subjects with three stages of AD: PCAD, aMCI, late-stage AD [161, 162]. Readers are directed to the primary papers for more details, but comparison across all three stages of AD demonstrated that altered phosphorylation (and therefore altered function) of two key proteins, the calcium-related protein, regucalcin, and gelsolin, a synaptic membrane remodeling-related protein required for learning and memory, were present in all stages of this disorder. These proteins are consistent with the findings in AD of calcium dyshomeostasis [163] and altered learning and memory [123], and we hypothesize that these two proteins could be therapeutically targeted in early stages of AD.
Down Syndrome (DS) and AD
DS results from trisomy of chromosome 21 entirely or in part. Both genetic alterations and environmental factors work together to determine the severity of the genetic phenotypes. Genetic instability as a consequence of the trisomy of chromosome 21 and the gene-dosage hypothesis contribute to the phenotypes presented and their severity. While there is intellectual disability from birth to about 40 years of age, for most DS patients there is a conversion between the ages of 40–50 to dementia associated with fully expressed AD neuropathology [164]. Some of the studies from the Butterfield laboratory performed in collaboration with Profs. Elizabeth Head (also at the University of Kentucky) and Marzia Perluigi (University of Rome, La Sapienza) are outlined below:
a) Oxidative stress-related genes are coded on Chr. 21, among which are Cu,Zn-superoxide dismutase (which produces pro-oxidant H2O2), Bach 1 (a transcriptional repressor of HO-1), and APP (from which Aβ1-42 is formed). Oxidative stress occurs extremely early in DS [165]: we reported that amniotic fluid of mothers carrying DS fetuses show increased oxidative stress, and redox proteomics identified oxidatively modified proteins in amniotic fluid that are consistent with the spectrum of phenotypes associated with DS [166]. Additional redox proteomics studies of brain proteins modified by protein carbonyls or protein-bound HNE from persons with DS, DS with AD, and the corresponding age-matched controls were conducted [150, 164, 167–170]. The results identified oxidatively modified and likely dysfunctional brain proteins in pathways related to glucose metabolism, the proteostasis network, mTOR activation, and neurotransmission in DS and in the transition from DS to DS with AD. That is, such proteins were quite similar, if not identical, to redox proteomics-identified brain proteins in aMCI and AD. This is the case also with polyubiquitinylated brain proteins in AD and DS with AD involving oxidized proteins associated with the ubiquitin proteasome system [119, 171].
Consequently, based on completed and ongoing oxidative stress and redox proteomics studies of brains from persons with DS at different ages both information and insights to both the molecular processes of DS and AD have emerged.
b) Other oxidative stress studies in brains from individuals with DS or DS with AD include identification of Bach1 as overexpressed and activation of p53. Bach1 represses transcription of HO-1, meaning that antioxidant bilirubin is not produced sufficiently [172]. Activation of p53 in brain with DS and DS with AD persons led to enhanced apoptosis, conceivably contributing to the cognitive dysfunction and dementia observed, respectively [173].
Implications and importance of AD-related research in the butterfield laboratory
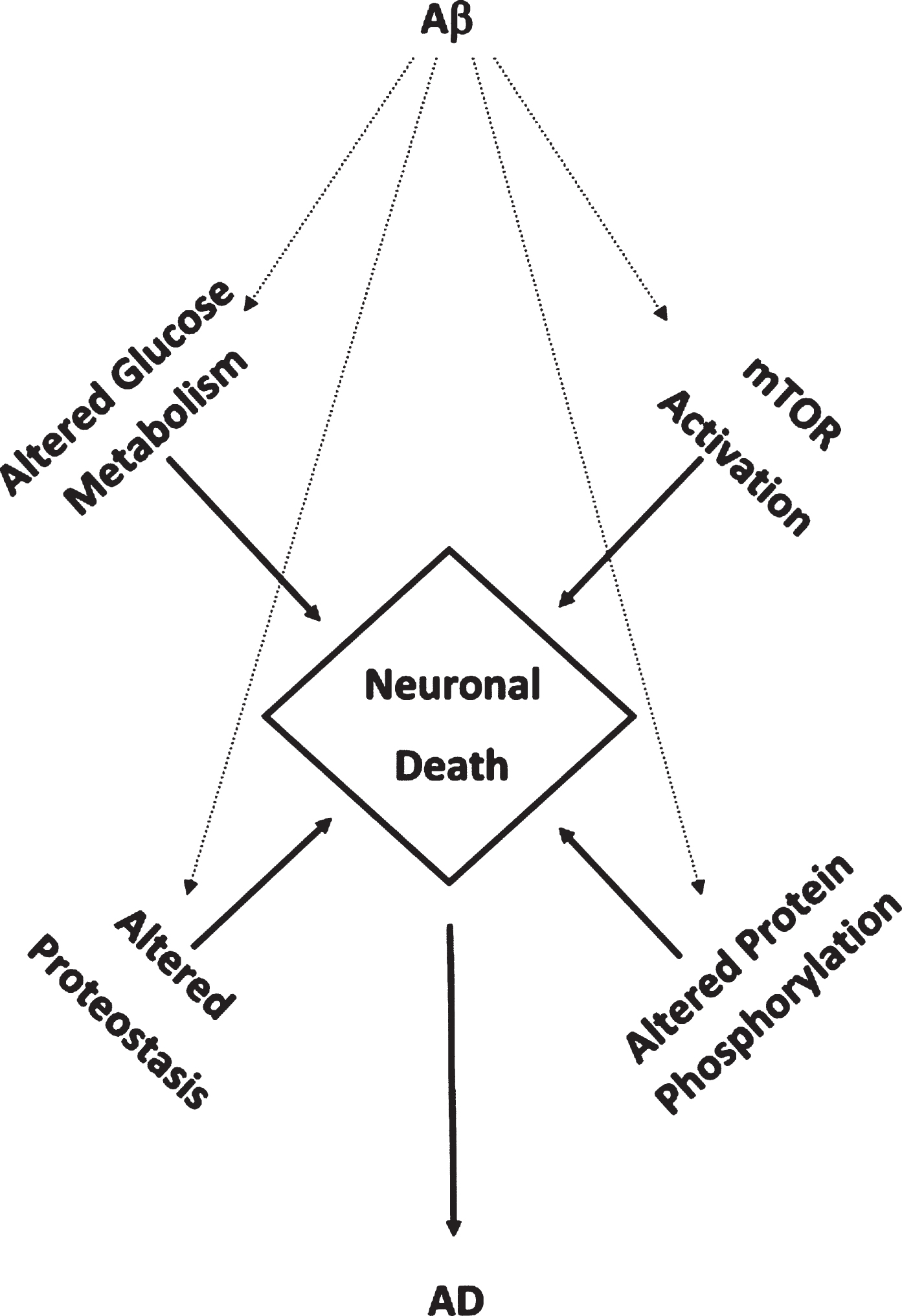
Studies employing measures of oxidative stress and redox proteomics identify four recurring altered pathways or processes secondary to oxidative damage to constituent brain proteins in these pathways or processes from persons with aMCI, AD, or DS with AD that are interrelated and contribute to neuronal death. These pathways and processes altered in these AD-related conditions named above form a “Quadrilateral of Neuronal Death” (Fig. 2) and include: glucose metabolism, mTOR activation, proteostasis network (ubiquitin proteasome system; autophagy; ER-resident unfolded protein response), and protein phosphorylation [114, 161, 162]. Some of these pathways and processes are altered even in brains of persons with PCAD. Consequently, our laboratory has contributed significantly to the notion that oxidative stress now is considered an integral part of the pathophysiology and progression of AD. In aMCI, AD, and DS with AD, Aβ1-42-associated oxidative stress or indirect oxidative stress secondary to Aβ1-42 (e.g., mitochondrial alterations) are implicated [71, 75–77, 107–114, 174, 175]. We believe that targeting these pathways pharmacologically and with lifestyle modifications potentially may provide a multi-pronged strategy to slow or perhaps one day, prevent, progression or development of this devastating dementing disorder.
Fig.2
Aβ1-42-mediates accumulated oxidative damage resulting in decreased glucose metabolism, mTOR activation, altered protein homeostasis, and altered protein phosphorylation leading in neuronal death in aMCI and AD brain.
ACKNOWLEDGMENTS
The authors thank the past and current members of the Butterfield research group for their dedication and tireless efforts to better understand oxidative stress and its sequelae in AD throughout its progression. In addition, the authors are grateful to the members and resources of the University of Kentucky Sanders-Brown Center on Aging, who provided thoroughly characterized autopsy specimens for our studies and sound advice. We are grateful to all our collaborators, and we acknowledge support from grants from the National Institutes of Health for most of the studies from our laboratory cited here.
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/17-0543r1).
REFERENCES
[1] | Alzheimer’s Association ((2017) ) Alzheimer’s disease facts and figures. Alzheimers Dement 13: , 325–373. |
[2] | Hensley K , Carney J , Mattson M , Aksenova M , Harris M , Wu JF , Floyd R , Butterfield DA ((1994) ) A new model for β-amyloid aggregation and neurotoxicity based on free radical generating capacity of the peptide: Insights into Alzheimer disease. Proc Natl Acad Sci U S A 91: , 3270–3274. |
[3] | Butterfield DA , Hensley K , Harris M , Mattson M , Carney J ((1994) ) β-amyloid peptide free radical fragments initiate synaptosomal lipoperoxidation in a sequence-specific fashion: Implications to Alzheimer’s disease. Biochem Biophys Res Commun 200: , 710–715. |
[4] | Hensley K , Aksenova M , Carney JM , Harris M , Butterfield DA ((1995) ) Amyloid β-peptide spin trapping I: Peptide enzyme toxicity is related to free radical spin trap reactivity. Neuroreport 6: , 489–492. |
[5] | Hensley K , Aksenova M , Carney JM , Harris M , Butterfield DA ((1995) ) Amyloid β-peptide spin trapping II: Evidence for decomposition of the PBN spin adduct. Neuroreport 6: , 493–496. |
[6] | Hensley K , Butterfield DA , Hall N , Cole P , Subramaniam R , Mark R , Mattson MP , Markesbery WR , Harris ME , Aksenov M , Aksenova M , Wu JF , Carney JM ((1996) ) Reactive oxygen species as causal agents in the neurotoxicity of the Alzheimer’s disease-associated amyloid beta peptide. Ann N Y Acad Sci 786: , 120–134. |
[7] | Butterfield DA , Martin L , Carney JM , Hensley K ((1996) ) Aβ(25-35) peptide displays H2O2-like reactivity towards aqueous Fe2+, nitroxide spin probes, and synaptosomal membrane proteins. Life Sci 58: , 217–228. |
[8] | Butterfield DA , Subramaniam R , Cole PS , Yatin S , Hensley K , Hall NC , Carney JM ((1997) ) Electron paramagnetic resonance studies of amyloid β-peptide- and ischemia/-reperfusion-associated oxygen free radicals and the membrane damage they cause: Relevance to Alzheimer’s disease and stroke. In Oxygen Radicals and the Disease Process, Thomas CE, Kalyanaraman B, eds. Harwood Academic Publishers, Reading, Great Britain, pp. 41–64. |
[9] | Varadarajan S , Yatin S , Kanski J , Jahanshaki F , Butterfield DA ((1999) ) Methionine residue 35 is important in amyloid β-peptide-associated free radical oxidative stress. Brain Res Bull 50: , 133–141. |
[10] | Yatin SM , Varadarajan S , Link CD , Butterfield DA ((1999) ) In-vitro and in-vivo oxidative stress associated with Alzheimer’s amyloid β-peptide (1-42). Neurobiol Aging 20: , 325–330. |
[11] | Varadarajan S , Yatin S , Aksenova M , Butterfield DA ((2000) ) Review: Alzheimer’s amyloid β-peptide-associated free radical oxidative stress and neurotoxicity. J Struct Biol 130: , 184–208. |
[12] | Varadarajan S , Kanski J , Aksenova M , Lauderback C , Butterfield DA ((2001) ) Different mechanisms of oxidative stress and neurotoxicity for Alzheimer’s Aβ(1-42) and Aβ(25-35). J Am Chem Soc 123: , 5625–5631. |
[13] | Butterfield DA , Stadtman ER ((1997) ) Protein oxidation processes in aging brain. Adv Cell Aging Gerontol 2: , 161–191. |
[14] | Halliwell B ((2007) ) Biochemistry of oxidative stress. Biochem Soc Trans 35: , 1147–1150. |
[15] | Beckman JS , Chen J , Ischiropoulos H , Crow JP ((1994) ) Oxidative chemistry of peroxynitrite. Meth Enzymol 233: , 229–240. |
[16] | Esterbauer H , Schaur RJ , Zollner H ((1991) ) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 11: , 81–128. |
[17] | Harris ME , Hensley K , Butterfield DA , Leedle RA , Carney JM ((1995) ) Direct evidence of oxidative injury produced by the Alzheimer’s amyloid beta peptide (1-40) in cultured hippocampal neurons. Exp Neurol 131: , 193–202. |
[18] | Yatin SM , Aksenova M , Aksenov M , Markesbery WR , Aulick T , Butterfield DA ((1999) ) Temporal relations between amyloid β-peptide-induced free radical oxidative stress and neuronal toxicity and neuronal defensive responses. J Mol Neurosci 11: , 183–197. |
[19] | Kanski J , Varadarajan S , Aksenova M , Butterfield DA ((2001) ) Role of glycine-33 and methionine-35 in Alzheimer’s amyloid β-peptide 1-42-associated oxidative stress and neurotoxicity. Biochim Biophys Acta 1586: , 190–198. |
[20] | Kalyanaraman B , Darley-Usmar V , Davies KJA , Dennery PA , Forman HJ , Grisham MB , Mann GE , Moore K , Roberts II LJ , Ischiropoulos H ((2012) ) Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free Radic Biol Med 52: , 1–6. |
[21] | Perluigi M , Joshi G , Sultana R , Calabrese V , De Marco C , Coccia R , Butterfield DA ((2006) ) In Vivo protection by the xanthate tricyclodecan-9-YL-xanthogenate against amyloid β-peptide (1-42)-induced oxidative stress: Implications for Alzheimer’s disease. Neuroscience 138: , 1161–1170. |
[22] | Ansari MA , Joshi G , Huang Q , Opii WO , Mohmmad Abdul H , Sultana R , Butterfield DA ((2006) ) In Vivo administration of D609 leads to protection of subsequently isolated gerbil brain mitochondria subjected to In Vitro oxidative stress induced by amyloid beta-peptide and other oxidative stressors: Relevance to Alzheimer’s disease and other oxidative stress-related neurodegenerative disorders. Free Radic Biol Med 41: , 1694–1703. |
[23] | Perluigi M , Joshi G , Sultana R , Calabrese V , De Marco C , Coccia R , Cini C , Butterfield DA ((2006) ) In Vivo protective effects of ferulic acid ethyl ester against amyloid β-peptide (1-42)-induced oxidative stress. J Neurosci Res 84: , 418–426. |
[24] | Aksenov MY , Aksenova MV , Markesbery WR , Butterfield DA ((1998) ) Amyloid β-peptide (1-40) – mediated oxidative stress in cultured hippocampal neurons: Protein carbonyl formation, CK BB expression and the level of Cu, Zn and Mn SOD mRNA. J Mol Neurosci 10: , 181–192. |
[25] | Kanski J , Aksenova M , Schoneich C , Butterfield DA ((2002) ) Substitution of isoleucine-31 by helical-breaking proline abolishes oxidative stress and neurotoxic properties of Alzheimer’s amyloid β-peptide (1-42). Free Radic Biol Med 32: , 1205–1211. |
[26] | Boyd-Kimball D , Sultana R , Mohmmad-Abdul H , Butterfield DA ((2004) ) Rodent Aβ(1-42) exhibits oxidative stress properties similar to those of human Aβ(1-42): Implications for proposed mechanisms of toxicity. J Alzheimers Dis 6: , 515–525. |
[27] | Boyd-Kimball D , Mohmmad-Abdul H , Reed T , Sultana R , Butterfield DA ((2004) ) Role of phenylalanine 20 in Alzheimer’s amyloid β-peptide (1-42)-induced oxidative stress and neurotoxicity. Chem Res Toxicol 17: , 1743–1749. |
[28] | Butterfield DA , Boyd-Kimball D ((2005) ) The critical role of methionine 35 in Alzheimer’s amyloid β-peptide (1-42)-induced oxidative stress and neurotoxicity. Biochim Biophys Acta 1703: , 149–156. |
[29] | Boyd-Kimball D , Sultana R , Mohammad-Abdul H , Butterfield DA ((2005) ) gamma-glutamylcysteine ethyl ester induced upregulation of glutathione protects neurons against Aβ(1-42)-mediated oxidative stress and neurotoxicity: Implications for Alzheimer’s disease. J Neurosci Res 79: , 700–706. |
[30] | Barton O , Sultana R , Butterfield DA , Atlas D ((2006) ) Low molecular weight thiol amides attenuate MAPK activity and protect primary neurons from Aβ(1-42) toxicity. Brain Res 1069: , 198–206. |
[31] | Harris ME , Carney JM , Cole P , Hensley K , Howard BJ , Martin L , Bummer P , Wang Y , Pedigo N , Butterfield DA ((1995) ) β-amyloid peptide-derived, oxygen-dependent free radicals inhibit glutamate uptake in cultured astrocytes: Implications to Alzheimer’s disease. Neuroreport 6: , 1875–1879. |
[32] | Harris ME , Wang Y , Pedigo NW Jr , Hensley K , Butterfield DA , Carney JM ((1996) ) Aβ(25-35) inhibits Na+-dependent glutamate uptake in rat hippocampal astrocyte cultures. J Neurochem 67: , 277–286. |
[33] | Mohmmad-Abdul H , Calabrese V , Calvani M , Butterfield DA ((2006) ) Acetyl-L-carnitine-induced up-regulation of heat shock proteins protects cortical neurons against Aβ (1-42)-mediated oxidative stress and neurotoxicity: Implications for Alzheimer’s disease. J Neurosci Res 84: , 398–408. |
[34] | Ansari MA , Mohmmad Abdul H , Joshi G , Opii WO , Butterfield DA ((2009) ) Protective effect of quercetin in primary neurons against Aβ(1-42): Relevance to Alzheimer’s disease. J Nutr Biochem 20: , 269–275. |
[35] | Mohmmad-Abdul H , Sultana R , Keller JN , St. Clair DK , Markesbery WR , Butterfield DA ((2006) ) Mutations in amyloid precursor protein and presenilin-1 genes increase the basal oxidative stress in murine neuronal cells and lead to increased sensitivity to oxidative stress mediated by amyloid β-peptide (1-42), H2O2 and kainic acid: Implications for Alzheimer’s disease. J Neurochem 96: , 1322–1335. |
[36] | Yatin SM , Aksenov M , Butterfield DA ((1999) ) The antioxidant vitamin e modulates amyloid β-peptide-induced creatine kinase activity inhibition and increased protein oxidation: Implications for the free radical hypothesis of Alzheimer’s disease. Neurochem Res 24: , 427–435. |
[37] | Yatin SM , Varadarajan S , Butterfield DA ((2000) ) Vitamin E prevents Alzheimer’s amyloid β-peptide (1-42)-induced protein oxidation and reactive oxygen species formation. J Alzheimers Dis 2: , 123–131. |
[38] | Yatin SM , Yatin M , Aulick T , Ain KB , Butterfield DA ((1999) ) Alzheimer’s amyloid β-peptide generated free radicals increase rat embryonic neuronal polyamine uptake and ODC activity: Protective effect of vitamin E. Neurosci Lett 263: , 17–20. |
[39] | Sultana R , Newman SF , Mohmmad-Abdul H , Cain J , Pierce WM , Klein JB , Merchant M , Butterfield DA ((2006) ) Protective effect of D609 against Aβ(1-42)-induced oxidative modification of neuronal proteins: Redox proteomics study. J Neurosci Res 84: , 409–417. |
[40] | Koppal T , Subramaniam R , Drake J , Prasad MR , Butterfield DA ((1998) ) Vitamin E protects against amyloid peptide (25-35)-induced changes in neocortical synaptosomal membrane lipid structure and composition. Brain Res 786: , 270–273. |
[41] | Subramaniam R , Koppal T , Green M , Yatin S , Jordan B , Butterfield DA ((1998) ) The free radical antioxidant vitamin e protects cortical synaptosomal membrane proteins from amyloid β-peptide (25-35) toxicity but not from hydroxynonenal toxicity: Relevance to the free radical hypothesis of Alzheimer’s disease. Neurochem Res 23: , 1403–1410. |
[42] | Mohmmad-Abdul H , Butterfield DA ((2005) ) Protection against amyloid beta-peptide (1-42)-induced loss of phospholipid asymmetry in synaptosomal membranes by tricyclodecan-9-xanthogenate (D609) and ferulic acid ethyl ester: Implications for Alzheimer’s disease. Biochim Biophys Acta 1741: , 140–148. |
[43] | Bader Lange ML , Cenini G , Piroddi M , Mohmmad Abdul H , Sultana R , Galli F , Memo M , Butterfield DA ((2008) ) Loss of phospholipid asymmetry and elevated brain apoptotic protein levels in subjects with amnestic mild cognitive impairment and Alzheimer’s disease. Neurobiol Dis 29: , 456–464. |
[44] | Pogocki D , Schoneich C ((2002) ) Redox properties of Met35 in neurotoxic β-amyloid peptide. A molecular modeling study. Chem Res Toxicol 15: , 408–418. |
[45] | Curtain CC , Ali F , Volitakis I , Cherny RA , Norton RS , Beyreuther K , Barrow CJ , Masters CL , Bush AI , Barnham KJ ((2001) ) Alzheimer’s disease amyloid-beta binds copper and zinc to generate an allosterically ordered membrane-penetrating structure containing superoxide dismutase-like subunits. J Biol Chem 276: , 20466–20473. |
[46] | Keller JN , Lauderback CM , Butterfield DA , Kindy MS , Markesbery WR ((2000) ) Amyloid β-peptide effects on synaptosomes from apolipoprotein E-deficient mice. J Neurochem 74: , 1579–1586. |
[47] | Lauderback CM , Hackett JM , Keller JN , Varadarajan S , Sweda L , Kindy M , Markesbery WR , Butterfield DA ((2001) ) Vulnerability of synaptosomes from Apoe knock-out mice to structural and oxidative modifications induced by Aβ(1-40): Implications for Alzheimer’s disease. Biochemistry 40: , 2548–2554. |
[48] | Lauderback CM , Kanski J , Hackett JM , Maeda N , Kindy MS , Butterfield DA ((2002) ) Apolipoprotein E modulates Alzheimer’s Aβ(1-42)-induced oxidative damage to synaptosomes in an allele-specific manner. Brain Res 924: , 90–97. |
[49] | Mark RJ , Hensley K , Butterfield DA , Mattson MP ((1995) ) Amyloid β-peptide impairs ion-motive ATPase activities: Evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J Neurosci 15: , 6239–6249. |
[50] | Auland ME , Roufogalis BD , Devaux PF , Zachowski A ((1994) ) Reconstitution of ATP-dependent aminophospholipid translocation in proteoliposomes. Proc Natl Acad Sci U S A 91: , 10938–10942. |
[51] | Aksenov MY , Aksenova MV , Harris ME , Hensley K , Butterfield DA , Carney JM ((1995) ) Enhancement of β-amyloid peptide Aβ(1–40)-mediated neurotoxicity by glutamine synthetase. J Neurochem 65: , 1899–1902. |
[52] | Aksenov MY , Aksenova MV , Butterfield DA , Hensley K , Vigo-Pelfrey C , Carney JM ((1996) ) Glutamine synthetase-induced enhancement of β-amyloid peptide Aβ(1–40) neurotoxicity accompanied by abrogation of fibril formation and Aβ fragmentation. J Neurochem 66: , 2050–2056. |
[53] | Aksenov MY , Aksenova MV , Carney JM , Butterfield DA ((1997) ) Oxidative modification of glutamine synthetase by amyloid beta peptide. Free Radic Res 27: , 267–281. |
[54] | Smith CD , Carney JM , Starke-Reed PE , Oliver CN , Stadtman ER , Floyd RA , Markesbery WR ((1991) ) Excess brain protein oxidation and enzyme dysfunction in normal aging and Alzheimer’s disease. Proc Natl Acad Sci U S A 88: , 10540–10543. |
[55] | Butterfield DA , Hensley K , Cole P , Subramaniam R , Aksenov M , Aksenova M , Bummer PM , Haley BE , Carney JM ((1997) ) Oxidatively-induced structural alteration of glutamine synthetase assessed by analysis of spin labeled incorporation kinetics: Relevance to Alzheimer’s disease. J Neurochem 68: , 2451–2457. |
[56] | Castegna A , Aksenov M , Aksenova M , Thongboonkerd V , Klein JB , Pierce WM , Booze R , Markesbery WR , Butterfield DA ((2002) ) Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: Creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med 33: , 562–571. |
[57] | Aksenov M , Aksenova M , Butterfield DA , Markesbery WR ((2000) ) Oxidative modification of creatine kinase BB in Alzheimer’s disease brain. J Neurochem 74: , 2520–2527. |
[58] | Lauderback CM , Hackett JM , Huang FF , Keller JN , Szweda LI , Markesbery WR , Butterfield DA ((2001) ) The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: Role of A β1-42 J Neurochem 78: , 413–416. |
[59] | Masliah E , Alford M , DeTeresa R , Mallory M , Hansen L ((1996) ) Deficient glutamate transport is associated with neurodegeneration in Alzheimer’s disease. Ann Neurol 40: , 759–766. |
[60] | Subramaniam R , Roediger F , Jordan B , Mattson MP , Keller JN , Waeg G , Butterfield DA ((1997) ) The lipid peroxidation product, 4-hydroxy-2-trans-nonenal, alters the conformation of cortial synaptosomal membrane proteins. J Neurochem 69: , 1161–1169. |
[61] | Lauderback CM , Harris-White ME , Wang Y , Pedigo MW Jr , Carney JM , Butterfield DA ((1999) ) Amyloid β-peptide inhibits Na+-dependent glutamate uptake. Life Sci 65: , 1977–1981. |
[62] | Pedersen WA , Kloczewiak MA , Blusztajn JK ((1996) ) Amyloid beta-protein reduces acetylcholine synthesis in a cell line derived from cholinergic neurons of the basal forebrain. Proc Natl Acad Sci U S A 93: , 8068–8071. |
[63] | Bailey JA , Lahiri DK ((2012) ) Chromatographic separation of reaction products from the choline acetyltransferase and carnitine acetyltransferase assay: Differential ChAT and CrAT activity in brain extracts from Alzheimer’s disease versus controls. J Neurochem 122: , 672–680. |
[64] | Butterfield DA , Lauderback CM ((2002) ) Lipid peroxidation and protein oxidation in Alzheimer’s disease bra, otential causes and consequences involving amyloid β-peptide-associated free radical oxidative stress. Free Radic Biol Med 32: , 1050–1060 |
[65] | Butterfield DA , Reed T , Newman SF , Sultana R ((2007) ) Roles of amyloid β-peptide associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic Biol Med 43: , 658–677. |
[66] | Castegna A , Aksenov M , Aksenova M , Thongboonkerd V , Klein JB , Pierce WM , Booze R , Markesbery WR , Butterfield DA ((2002) ) Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part II: Dihydropyrimidinase-related protein 2, α-enolase, and heat shock cognate. J Neurochem 82: , 1524–1532. |
[67] | Boyd-Kimball D , Castegna A , Sultana R , Poon HF , Petroze R , Lynn BC , Klein JB , Butterfield DA ((2005) ) Proteomic identification of proteins oxidized by Aβ(1-42) in synaptosomes: Implications for Alzheimer’s disease. Brain Res 1044: , 206–215. |
[68] | Boyd-Kimball D , Sultana R , Poon HF , Mohammad-Abdul H , Lynn BC , Klein JB , Butterfield DA ((2005) ) γ-glutamylcysteine ethyl ester protection of proteins from Aβ(1-42)-mediated oxidative stress in neuronal cell culture: A proteomics approach. J Neurosci Res 79: , 707–713. |
[69] | Aksenov MY , Aksenova MV , Butterfield DA , Geddes JW , Markesbery WR ((2001) ) Protein oxidation in the Alzheimer’s disease brain: Analysis ofrotein carbonyls by immunocytochemistry and two-dimensional western blotting. Neuroscience 103: , 373–383. |
[70] | Castegna A , Thongboonkerd V , Klein JB , Lynn B , Markesbery WR , Butterfield DA ((2003) ) Proteomic identification of nitrated proteins in Alzheimer’s disease brain. J Neurochem 85: , 1394–1401. |
[71] | Sultana R , Boyd-Kimball D , Poon HF , Cai J , Pierce WM , Klein JB , Merchant M , Markesbery WR , Butterfield DA ((2006) ) Redox proteomics identification of oxidized proteins in Alzheimer’s disease hippocampus and cerebellum: An approach to understand pathological and biochemical alterations in AD. Neurobiol Aging 27: , 1564–1576. |
[72] | Butterfield DA , Poon HF , St. Clair D , Keller JN , Pierce WM , Klein JB , Markesbery WR ((2006) ) Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: Insights into the development of Alzheimer’s disease. Neurobiol Dis 22: , 223–232. |
[73] | Sultana R , Reed T , Perluigi M , Coccia R , Pierce WM , Butterfield DA ((2007) ) Proteomic identification of nitrated brain proteins in amnestic mild cognitive impairment: A regional study. J Cell Molec Med 11: , 839–851. |
[74] | Reed T , Perluigi M , Sultana R , Pierce WM , Turner DM , Coccia R , Markesbery WR , Butterfield DA ((2008) ) Redox proteomic identification of 4-hydroxy-2-nonenal-modified proteins in amnestic mild cognitive impairment: Insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer’s disease. Neurobiol Dis 30: , 107–120. |
[75] | Sultana R , Perluigi M , Butterfield DA ((2009) ) Oxidatively modified proteins in Alzheimer’s disease, mild cognitive impairment and animal models of AD: Role of Abeta in pathogenesis. Acta Neuropath 118: , 131–150. |
[76] | Swomley AM , Forster S , Keeney J , Triplett J , Zhang Z , Sultana R , Butterfield DA ((2014) ) Abeta, oxidative stress in Alzheimer disease: Evidence based on proteomics studies. Biochim Biophys Acta 1842: , 1248–1257. |
[77] | Butterfield DA ((2014) ) The 2013 SFRBM Discovery Award: Selected discoveries from the Butterfield laboratory of oxidative stress and its sequela in brain in cognitive disorders exemplified by Alzheimer disease and chemotherapy induced cognitive impairment. Free Radic Biol Med 74: , 157–174. |
[78] | Butterfield DA , Kanski J ((2002) ) Review: Methionine residue 35 is critical for the oxidative stress and neurotoxic properties of Alzheimer’s amyloid β-peptide 1-42. Peptides 23: , 1299–1309. |
[79] | Butterfield DA , Bush AI ((2004) ) Alzheimer’s amyloid β-peptide (1-42): Involvement of methionine residue 35 in the oxidative stress and neurotoxicity properties of this peptide. Neurobiol Aging 25: , 563–568. |
[80] | Butterfield DA , Sultana R ((2011) ) Methionine-35 of Aβ(1–42): Importance for oxidative stress in Alzheimer disease. J Amino Acids 2011: , 1–10. |
[81] | Butterfield DA , Galvan V , Bader Lange M , Tang H , Sowell RA , Spilman P , Fombonne J , Gorostiza O , Zhang J , Sultana R , Bredesen DE ((2010) ) In vivo oxidative stress in brain of Alzheimer disease transgenic mice: Requirement for methionine 35 in amyloid β-peptide of APP. Free Radic Biol Med 48: , 136–144. |
[82] | Huang X , Atwood CS , Hartshorn MA , Multhaup G , Goldstein LE , Scarpa RC , Cuajungco MP , Gray DN , Lim J , Moir RD , Tanzi RE , Bush AI ((1999) ) The Aβ peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 38: , 769–7616. |
[83] | Huang X , Atwood CS , Cuajungco MP , Hartshorn MA , Tyndall J , Hanson GR , Stokes KC , Loepold M , Multhaup G , Goldstein LE , Scarpa RC , Saunders AJ , Lim J , Moir RD , Glabe C , Bowden EF , Masters CL , Fairlie DP , Tanzi RE , Bush AI ((1999) ) Cu(II) potentiation of Alzheimer Aβ neurotoxicity: Correlation with cell-free hydrogen peroxide production and metal reduction. J Biol Chem 274: , 37111–37116. |
[84] | Rauk A , Armstrong DA ((2000) ) Influence of β-sheet structure on the susceptibility of proteins to backbone oxidative damage preference for αC-centered radical formation at glycine residues of antiparallel β-sheets. J Am Chem Soc 122: , 4185–4192. |
[85] | Pogocki D ((2004) ) Mutation of the Phe(20) residue in Alzheimer’s amyloid β-peptide might decrease its toxicity due to disruption of the Met(35)-cupric site electron transfer pathway. Chem Res Toxicol 17: , 325–329. |
[86] | Kanski J , Aksenova M , Butterfield DA ((2002) ) The hydrophobic environment of Met35 of Alzheimer’s Aβ(1-42) is important for the neurotoxic and oxidative properties of the peptide. Neurotox Res 4: , 219–223. |
[87] | Hensley K , Hall N , Subramaniam R , Cole P , Harris M , Aksenov M , Aksenova M , Gabbita SP , Wu JF , Carney JM , Lovell M , Markesbery WR , Butterfield DA ((1995) ) Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J Neurochem 65: , 2146–2156. |
[88] | Butterfield DA , Boyd-Kimball D , Castegna A ((2003) ) Proteomics in Alzheimer’s disease: New insights into mechanisms of neurodegeneration. J Neurochem 86: , 1313–1327. |
[89] | Butterfield DA , Pocernich CB ((2003) ) The glutamatergic system and Alzheimer’s disease: Therapeutic implications. CNS Drugs 17: , 641–652. |
[90] | Sultana R , Boyd-Kimball D , Poon HF , Cai J , Pierce WM , Klein JB , Markesbery WR , Butterfield DA ((2006) ) Identification of nitrated proteins in Alzheimer’s disease brain using a redox proteomics approach. Neurobiol Dis 22: , 76–87. |
[91] | Butterfield DA , Reed T , Perluigi M , De Marco C , Coccia R , Cini C , Sultana R ((2006) ) Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci Letts 397: , 170–173. |
[92] | Butterfield DA , Reed T , Perluigi M , De Marco C , Coccia R , Keller JN , Markesbery WR , Sultana R ((2007) ) Elevated levels of 3-nitrotyrosine in brain from subjects with amnestic mild cognitive impairment: Implications for the role of nitration in the progression of Alzheimer’s disease. Brain Res 1148: , 243–248. |
[93] | Keller JN , Schmitt FA , Scheff SW , Ding Q , Chen Q , Butterfield DA , Markesbery WR ((2005) ) Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology 64: , 1152–1156. |
[94] | Lovell MA , Markesbery WR ((2007) ) Damage to lipids, proteins, DNA and RNA in mild cognitive impairment. Arch Neurol 64: , 954–956. |
[95] | Smith MA , Zhu X , Tabaton M , Liu G , McKeel DW Jr , Cohen ML , Wang XL , Siedlak SL , Dwyer BE , Hayashi T , Nakamura M , Nunomura A , Perry G ((2010) ) Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J Alzheimers Dis 19: , 363–372. |
[96] | Drake J , Link CD , Butterfield DA ((2003) ) Oxidative stress precedes fibrillar deposition of Alzheimer’s disease amyloid β-peptide (1-42) in a transgenic Caenorhabditis elegans model. Neurobiol Aging 24: , 415–420. |
[97] | Sultana R , Robinson RAS , Mohmmad Abdul H , Di Domenico F , St. Clair DK , Markesbery WR , Cai J , Pierce WM , Butterfield DA ((2011) ) Proteomics identification of specifically carbonylated brain proteins in APPNLh/APPNLh X PS-1P264L/PS-1P264L human double mutant knock-in mice model of Alzheimer disease as a function of age. J Proteomics 74: , 2430–2440. |
[98] | Mohmmad Abdul H , Sultana R , St. Clair DK , Markesbery WR , Butterfield DA ((2008) ) Oxidative damage in brain from human mutant APP/PS-1 double knock-in mice as a function of age. Free Radic Biol Med 45: , 1420–1425. |
[99] | Lange ML , St. Clair DK , Markesbery WR , Studzinski CM , Murphy MP , Butterfield DA ((2010) ) Age-related loss of phospholipid asymmetry in APPNLh/APPNLh x PS-1P264L/PS-1P264L human double mutant knock-in mice: Relevance to Alzheimer disease. Neurobiol Dis 38: , 104–115. |
[100] | Huang Q , Aluise C , Joshi G , Sultana R , St. Clair D , Markesbery W , Butterfield DA ((2010) ) Potential in vivo amelioration by N-Acetyl-L-Cysteine of oxidative stress in brain in human double mutant APP/PS-1 knock-in mice: Towards therapeutic modulation of mild cognitive impairment (MCI). J Neurosci Res 88: , 2618–2629. |
[101] | Robinson RAS , Bader Lange M , Sultana R , Galvan V , Fombonne J , Gorostiza O , Zhang J , Warrier G , Cai J , Pierce WM , Bredesen DE , Butterfield DA ((2011) ) Differential expression and redox proteomics analyses of an Alzheimer disease transgenic mouse model: Effects of the amyloid-β peptide of amyloid precursor protein. Neuroscience 177: , 207–222. |
[102] | Sultana R , Robinson RAS , Bader Lange M , Fiorini A , Galvan V , Fombonne J , Baker A , Gorostiza O , Zhang J , Cai T , Pierce W , Bredesen D , Butterfield DA ((2012) ) Do proteomics analyses provide insights into reduced oxidative stress in brain of an Alzheimer disease transgenic mouse model with an M631L amyloid precursor protein substitution and thereby the importance of amyloid-beta-resident methionine 35 in Alzheimer disease pathogenesis? Antioxidant Redox Signal 17: , 1507–1514. |
[103] | Robinson RAS , Joshi G , Huang Q , Sultana R , Baker AS , Cai J , Pierce WM , St. Clair DK , Markesbery WR , Butterfield DA ((2011) ) Proteomics analysis of brain proteins in APP/PS-1 human double mutant knock-in mice with increasing amyloid β-peptide deposition: Insights into the effects of in vivo treatment with N-acetylcysteine as a potential therapeutic intervention in mild cognitive impairment and Alzheimer disease. Proteomics 11: , 4243–4256. |
[104] | Boyd-Kimball D , Sultana R , Poon HF , Lynn BC , Casamenti F , Pepeu G , Klein JB , Butterfield DA ((2005) ) Proteomic identification of proteins specifically oxidized by intracerebral injection of Aβ(1-42) into rat brain: Imlications for Alzheimer’s disease. Neuroscience 132: , 313–324 |
[105] | Milgram NW , Head E , Zicker SC , Ikeda-Douglas C , Murphey H , Muggenberg MA , Siwak CT , Tapp RD , Lowry SR , Cotman CW ((2004) ) Long-term treatment with antioxidants and a program of behavioral enrichment reduces age-dependent impairment in discrimination and reversal learning in beagle dogs. Exp Gerontol 39: , 753–765. |
[106] | Opii WO , Joshi G , Head E , Milgram NW , Muggenburg BA , Klein JB , Pierce WM , Cotman CW , Butterfield DA ((2008) ) Proteomic identification of brain proteins in the canine model of human aging following a long-term treatment with antioxidants and program of behavioral enrichment: Relevance to Alzheimer’s disease. Neurobiol Aging 29: , 51–70. |
[107] | Butterfield DA , Perluigi M , Reed T , Muharib T , Hughes C , Robinson R , Sultana R ((2012) ) Redox proteomics in selected neurodegenerative disorders. From its infancy to future applications. Antioxidant Redox Signal 17: , 1610–1655. |
[108] | Butterfield DA , Gu L , Di Domenico F , Robinson RAS ((2014) ) Mass spectrometry and redox proteomics: Applications in disease. Mass Spectrom Rev 33: , 277–301. |
[109] | Sultana R , Butterfield DA ((2011) ) Identification of the oxidative stress proteome in the brain. Free Radic Biol Med 50: , 487–494. |
[110] | Sultana R , Perluigi M , Butterfield DA ((2006) ) Redox proteomics identification of oxidatively modified proteins in Alzheimer’s disease brain and in vivo and in vitro models of AD centered around Aβ(1-42). J Chromatogr B Anaylt Technol Biomed Life Sci 833: , 3–11. |
[111] | Butterfield DA , Sultana R ((2007) ) Redox proteomics identification of oxidatively modified brain proteins in Alzheimer’s disease and mild cognitive impairment: Insights into the progression of this dementing disorder. J Alzheimers Dis 12: , 61–72. |
[112] | Perluigi M , Sultana R , Cenini G , Di Domenico F , Memo M , Pierce WM , Coccia R , Butterfield DA ((2009) ) Redox proteomics identification of 4-hydroxynonenal-modified brain proteins in Alzheimer’s disease: Role of lipid peroxidation in Alzheimer’s disease pathogenesis. Proteomics–Clin Appl 3: , 682–693. |
[113] | Sultana R , Perluigi M , Butterfield DA ((2013) ) Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain. Free Radic Biol Med 62: , 157–169. |
[114] | Di Domenico F , Barone E , Perluigi M , Butterfield DA ((2017) ) The triangle of death in Alzheimer disease brain: The aberrant cross talk among energy metabolism, mTOR signaling androtein homeostasis revealed by redox proteomics. Antioxidant Redox Signal 26: , 364–387 |
[115] | Salkovic-Petrisic M , Hoyer S ((2007) ) Central insulin resistance as a trigger for sporadic Alzheimer-like pathology: An experimental approach. J Neural Transm Suppl 72: , 217–233. |
[116] | Butterfield DA , Lange ML ((2009) ) Multifunctional roles of enolase in Alzheimer disease brain: Beyond altered glucose metabolism. J Neurochem 111: , 915–933 |
[117] | Butterfield DA , Hardas SS , Bader Lange ML ((2010) ) Oxidatively modified glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and Alzheimer disease: Many pathways to neurodegeneration. J Alzheimers Dis 20: , 369–393. |
[118] | Olney JW ((1990) ) Excitotoxicity: An overview. Can Dis Wkly Rep Supp 12E: , 47–57. |
[119] | Tramutola A , Di Domenico F , Barone E , Perluigi M , Butterfield DA ((2016) ) It’s all about u(biquitin): Role of the altered ubiquitin proteasome system and UCHL1 in Alzheimer disease. Oxidative Med Cell Long 2016: , 2756068. |
[120] | Keller JN , Hannik B , Markesbery WR ((2000) ) Impaired proteasome function in Alzheimer’s disease. J Neurochem 75: , 436–439. |
[121] | Markesbery WR ((1997) ) Neuropathological criteria for the diagnosis of Alzheimer’s disease. Neurobiol Aging 18: , S13–S19. |
[122] | Reed TT , Pierce WM Jr , Turner DM , Markesbery WR , Butterfield DA ((2009) ) Proteomic identification of nitrated brain proteins in early Alzheimer’s disease inferior parietal lobule. J Cell Mol Med 8B: , 2019–2029. |
[123] | Borlikova GG , Trejo M , Mably AJ , McDonald JM , Sala Frigero C , Regan CM , Murphy KJ , Masliah E , Walsh DM ((2013) ) Alzheimer brain-derived amyloid β-protein impairs synaptic remodeling and memory consolidation. Neurobiol Aging 34: , 1315–1327. |
[124] | Keller ET , Fu Z , Brennan M ((2000) ) The role of Raf kinase inhibitor protein (RKIP) in health and disease. Biochem Pharmacol 68: , 1049–1053. |
[125] | Di Domenico F , Owen JB , Sultana R , Sowell RA , Perluigi M , Cini C , Cai J , Pierce WM , Butterfield DA ((2010) ) The wheat germ agglutinin-fractionated proteome of subjects with Alzheimer’s disease and mild cognitive impairment hippocampus and inferior parietal lobule: Implications for disease pathogenesis and progression. J Neurosci Res 88: , 3566–3577. |
[126] | Di Domenico F , Cenini G , Sultana R , Perlugi M , Uberti M , Memo M , Butterfield DA ((2009) ) Glutathionylation of the pro-apoptotic protein p53 in Alzheimer’s disease brain: Imlications for AD pathogenesis. Neurochem Res 34: , 727–733 |
[127] | Cenini C , Sultana R , Memo M , Butterfield DA ((2008) ) Elevated levels of p53 and its oxidative modification by HNE in brain from subjects with mild cognitive impairment and Alzheimer’s disease. J Cell Mol Med 12: , 987–994. |
[128] | Cenini G , Sultana R , Memo M , Butterfield DA ((2008) ) Effects of oxidative and nitrosative stress in brain on p53 proapoptotic protein in amnestic mild cognitive impairment and Alzheimer disease. Free Radic Biol Med 45: , 81–85. |
[129] | Barone E , Cenini G , Sultana R , Di Domenico F , Fiorini A , Noel T , Mancuso C , St. Clair DK , Butterfield DA ((2012) ) Lack of p53 decreases basal oxidative stress levels in brain through up-regulation of thioredoxin-1, biliverdin reductase-A, manganese superoxide dismutase, and nuclear factor-kappa B. Antioxidant Redox Signal 16: , 1407–1420. |
[130] | Fiorini A , Sultana R , Barone E , Cenini G , Perluigi M , Mancuso M , Cai J , Klein JB , St. Clair D , Butterfield DA ((2012) ) Lack of p53 affects the expression of several brain mitochondrial proteins: Insights from proteomics into important pathways regulated by p53. PLoS One 7: , e49846. |
[131] | Barone E , Cenini G , Di Domenico F , Noel T , Wang C , Perluigi M , St. Clair DK , Butterfield DA ((2015) ) Basal brain oxidative and nitrosative stress levels are finely regulated by the interplay between MnSOD and p53. J Neurosci Res 93: , 1728–1739. |
[132] | Butterfield DA , Mohmmad Abdul H , Opii W , Newman SF , Joshi G , Ansari MA , Sultana R ((2006) ) Pin 1 in Alzheimer’s disease. J Neurochem 98: , 1697–1706. |
[133] | Balastik M , Lim J , Pastorino L , Lu KP ((2007) ) Pin1 in Alzheimer disease: Multiple substrates, one regulatory mechanism? Biochim Biophys Acta 1772: , 422–429. |
[134] | Sultana R , Boyd-Kimball D , Poon HF , Cai J , Pierce WM , Klein JB , Markesbery WR , Zhou XZ , Lu KP , Butterfield DA ((2006) ) Oxidative modification and down-regulation of Pin 1 in Alzheimer’s disease hippocampus: A redox proteomics analysis. Neurobiol Aging 27: , 918–925. |
[135] | Lee HG , Casadesus G , Zhu X , Castellani RJ , McShea A , Perry G , Petersen RM , Bajc V , Smith MA ((2009) ) Cell cycle re-entry mediated neurodegeneration and its treatment role in the pathogenesis of Alzheimer’s disease. Neurochem Int 54: , 84–88. |
[136] | Sultana R , Butterfield DA ((2007) ) Regional expression of key cell cycle proteins in brain from subjects with amnestic mild cognitive impairment. Neurochem Res 32: , 655–662. |
[137] | Keeney JT , Swomley AM , Harris JL , Fiorini A , Mitov MI , Perluigi M , Sultana R , Butterfield DA ((2012) ) Cell cycle proteins in brain in mild cognitive impairment: Insights into progression to Alzheimer’s disease. Neurotox Res 22: , 220–230. |
[138] | Chen C-H , Li W , Sultana R , You M-H , Kondo A , Shahpasand K , Kim BM , Luo M , Nechama M , Lin Y-M , Yao Y , Lee TH , Zhou XZ , Swomley AM , Butterfield DA , Zhang Y , Lu KP ((2015) ) Pin 1 cysteine-113 oxidative inhibits its catalytic activity and cellular function in Alzheimer’s disease. Neurobiol Dis 76: , 13–23. |
[139] | Butterfield DA , Gnjec A , Poon HF , Castegna A , Pierce WM , Klein JB , Martins RN ((2006) ) Redox proteomics identification of oxidatively modified brain proteins in inherited Alzheimer’s disease: An initial assessment. J Alzheimers Dis 10: , 391–397. |
[140] | Boyd-Kimball D , Poon HF , Lynn BC , Cai J , Pierce WM Jr , Klein JB , Ferguson J , Link CD , Butterfield DA ((2006) ) Proteomic identification of proteins specifically oxidized inexpressing human Aβ1-42: Implications for Alzheimer’s disease. Neurobiol Aging 27: , 1239–1249. |
[141] | Sultana R , Butterfield DA ((2004) ) Oxidatively modified GST and MRP1 in Alzheimer’s disease brain: Imlications for accumulation of reactive lipid peroxidation products. Neurochem Res 29: , 2215–2220 |
[142] | Aksenov MY , Markesbery WR ((2001) ) Changes in thiol content and expression of glutathione redox system genes in the hippocampus and cerebellum in Alzheimer’s disease. Neurosci Lett 302: , 141–145. |
[143] | Mancuso C , Santangelo R , Calabrese V ((2013) ) The heme oxygenase/biliverdin reductase system: A potential drug target in Alzheimer disease. J Biol Regul Homeost Agent 27: (Suppl), 75–87. |
[144] | Barone E , Di Domenico F , Sultana R , Coccia R , Mancuso C , Perluigi M , Butterfield DA ((2012) ) Heme oxygenase-1 posttranslational modifications in the brain of subjects with Alzheimer disease and mild cognitive impairment. Free Radic Biol Med 52: , 2292–2301. |
[145] | Barone E , Di Domenico F , Cenini G , Sultana R , Coccia R , Preziosi P , Perluigi M , Mancuso C , Butterfield DA ((2011) ) Oxidative and nitrosative modifications of biliverdin reductase in the brain of subjects with Alzheimer disease and mild cognitive impairment. J Alzheimers Dis 25: , 623–633. |
[146] | Barone E , Di Domenico F , Mancuso C , Butterfield ((2014) ) The Janus face of the heme oxygenase/biliverdin reductase system in Alzheimer disease: It’s time for reconciliation. Neurobiol Dis 62: , 144–159. |
[147] | Barone E , Di Domenico F , Cassano T , Arena A , Tramutola A , Coccia R , Butterfield DA , Perluigi M ((2016) ) Impairment of biliverdin reductase-a promotes brain insulin resistance in Alzheimer disease: A new paradigm. Free Radic Biol Med 91: , 127–142. |
[148] | Barone R , Butterfield DA ((2015) ) Insulin resistance in Alzheimer disease: Is heme oxygenase-1 Achille’s heel? Neurobiol Dis 84: , 69–77. |
[149] | Di Domenico F , Sultana R , Tiu GF , Scheff NN , Perluigi M , Cini C , Butterfield DA ((2010) ) Protein levels of heat shock proteins 27, 32, 60, 70, 90 and thioredoxin-1 in amnestic mild cognitive impairment: An investigation on the role of cellular stress response in the progression of Alzheimer’s disease. Brain Res 1333: , 72–81. |
[150] | Tramutola A , Triplett J , Di Domenico F , Niedowicz DM , Murphy MP , Coccia R , Perluigi M , Butterfield DA ((2015) ) Alteration of mTOR signaling occurs early in the progression of Alzheimer disease: Analysis of brain from subjects with preclinical AD, amnestic mild cognitive impairment and late-stage AD. J Neurochem 133: , 739–749. |
[151] | Perluigi M , Di Domenico F , Butterfield DA ((2015) ) mTOR signaling in aging and neurodegeneration: At the crossroads between metabolism function and impairment of autophagy. Neurobiol Dis 84: , 39–49. |
[152] | O’Neill C , Kiely AP , Coarley MF , Manning S , Long-Smith CM ((2012) ) Insulin and IGF-1 signaling: Longevity, protein, homeostasis, and Alzheimer’s disease. Biochem Soc Trans 40: , 721–727. |
[153] | Butterfield DA , Di Domenico F , Barone E ((2014) ) Elevated risk of type 2 diabetes for development of Alzheimer disease: A key role for oxidative stress in brain. Biochim Biophys Acta 1842: , 1693–1706. |
[154] | Landau SM , Frosch MP ((2014) ) Tracking the earliest pathological changes in Alzheimer disease. Neurology 82: , 878–883. |
[155] | Di Domenico F , Pupo G , Giraldo E , Lloret A , Badia MC , Schinina ME , Giorgi A , Butterfield DA , Vina J , Perluigi M ((2016) ) Autoantibodies profile in matching CSF and serum from AD and aMCI patients: Potential pathogenic role and link to oxidative damage. Curr Alzheimers Res 13: , 112–122. |
[156] | Keeney JT , Forster S , Sultana R , Brewer LD , Latimer CS , Cai J , Klein JB , Porter NN , Butterfield DA ((2013) ) Dietary vitamin D deficiency in rats from middle- to old-age leads to elevated tyrosine nitration and proteomics changes in levels of key proteins in brain: Imlications for low vitamin D-dependent age-related cognitive decline. Free Radic Biol Med 65: , 324–334. |
[157] | Keeney JT , Butterfield DA ((2015) ) Vitamin D deficiency and Alzheimer disease: Common links. Neurobiol Dis 84: , 84–98. |
[158] | Musiek ES , Holtzman DM ((2016) ) Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science 354: , 1004–1008. |
[159] | Sultana R , Mecocci P , Mangialasche F , Cecchetti R , Butterfield DA ((2011) ) Increased protein and lipid oxidative damage in mitochondria isolated from lymphocytes from patients with Alzheimer disease: Insights into the role of oxidative stress in Alzheimer’s disease and initial investigations into a potential biomarker for this dementing disorder. J Alzheimers Dis 24: , 77–84. |
[160] | Sultana R , Baglioni M , Cecchetti R , Cai J , Klein JB , Bastiani P , Ruggiero C , Mecocci P , Butterfield DA ((2013) ) Lymphocyte mitochondria: Toward identification of peripheral biomarkers in progression of Alzheimer disease. Free Radic Biol Med 65: , 595–606. |
[161] | Di Domenico F , Sultana R , Barone E , Perluigi M , Cini C , Mancuso C , Cai J , Pierce WM , Butterfield DA ((2011) ) Quantitative proteomics analysis of phosphorylated proteins in the hippocampus of Alzheimer’s disease subjects. J Proteomics 74: , 1091–1103. |
[162] | Triplett JC , Swomley AM , Cai J , Klein JB , Butterfield DA ((2015) ) Quantitative phosphoproteomic analyses of the inferior parietal lobule from three different pathological stages of Alzheimer’s disease. J Alzheimers Dis 49: , 45–62. |
[163] | Bezprozvanny I , Mattson MP ((2008) ) Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci 31: , 454–463. |
[164] | Butterfield DA , Di Domenico F , Swomley AM , Head E , Perluigi M ((2014) ) Redox proteomics analysis to decipher the neurobiology of Alzheimer-like neurodegeneration: Overlaps in Down dyndrome and Alzheimer disease brain. Biochem J 463: , 177–189. |
[165] | Perluigi M , Butterfield DA ((2012) ) Oxidative stress and Down syndrome: A route toward Alzheimer-like dementia. Curr Gerontol Geriatr Res 2012: , 724904. |
[166] | Perluigi M , Di Domenico F , Fiorini A , Cocciolo A , Giorgi A , Foppoli C , Butterfield DA , Giorlandino M , Giorlandino C , Schinina ME , Coccia R ((2011) ) Oxidative stress occurs early in Down syndrome pregnancy: A redox proteomics analysis of amniotic fluid. Proteomics—Clin App 5: , 167–178. |
[167] | Cenini G , Dowling A , Beckett T , Barone E , Mancuso C , Murphy MP , Levine H , Lott I , Schmitt F , Butterfield DA , Head E ((2012) ) Association between frontal cortex oxidative damage and beta-amyloid as a function of age in Down syndrome. Biochim Biophys Acta 1822: , 130–138. |
[168] | Cenini G , Fiorini A , Sultana R , Perluigi M , Cai J , Klein JB , Head E , Butterfield ((2014) ) An investigation of the molecular mechanisms engaged before and after the development of Alzheimer disease neuropathology in Down syndrome: A proteomics approach. Free Radic Biol Med 76: , 89–95. |
[169] | Barone E , Head E , Butterfield DA , Perluigi M ((2017) ) HNE-modified proteins in Down syndrome: Involvement in development of Alzheimer disease neuropathology. Free Radic Biol Med 111: , 262–269. |
[170] | Di Domenico F , Tramutola A , Butterfield DA ((2017) ) Role of 4-hydroxy-2-nonenal (HNE) in the pathogenesis of Alzheimer disease and other selected age-related neurodegenerative disorders. Free Radic Biol Med 111: , 253–261. |
[171] | Tramutola A , Di Domenico F , Barone E , Giorgi A , di Francesco L , Schinina ME , Coccia R , Head E , Butterfield DA , Perluigi M ((2017) ) Ubiquityinylation profile in Down syndrome brain prior to and after development of Alzheimer neuropathology. Antioxidant Redox Signal 26: , 280–298. |
[172] | Di Domenico F , Pupo G , Mancuso C , Barone E , Paolini F , Arena A , Blarzino C , Schmitt FA , Head E , Butterfield DA , Perluigi M ((2015) ) Bach1 overexpression in Down syndrome correlates with the alteration of the HO-1/BVR-A system: Insights for transition to Alzheimer’s disease. J Alzheimers Dis 44: , 1107–1120. |
[173] | Tramutola A , Pupo G , Di Domenico F , Barone E , Arena A , Lanzillotta C , Brokeaart C , Blarzion C , Head E , Butterfield DA , Perluigi M ((2016) ) Activation of p53 in Down syndrome and in the Ts65Dn mouse brain is associated with a pro-apoptotic phenotype. J Alzheimers Dis 52: , 359–371. |
[174] | Butterfield DA , Drake J , Pocernich C , Castegna A ((2001) ) Evidence of oxidative damage in Alzheimer’s disease brain: Central role of amyloid β-eptide. Trends Mol Med 7: , 548–554. |
[175] | Tramutola A , Lanzillotta C , Perluigi M , Butterfield DA ((2017) ) Oxidative stress, protein oxidation, and Alzheimer disease. Brain Res Bull 133: , 88–96. |


