
Phospho-Tau Accumulation and Structural Alterations of the Golgi Apparatus of Cortical Pyramidal Neurons in the P301S Tauopathy Mouse Model
Abstract
The Golgi apparatus (GA) is a highly dynamic organelle involved in the processing and sorting of cellular proteins. In Alzheimer’s disease (AD), it has been shown to decrease in size and become fragmented in neocortical and hippocampal neuronal subpopulations. This fragmentation and decrease in size of the GA in AD has been related to the accumulation of hyperphosphorylated tau. However, the involvement of other pathological factors associated with the course of the disease, such as the extracellular accumulation of amyloid-β (Aβ) aggregates, cannot be ruled out, since both pathologies are present in AD patients. Here we use the P301S tauopathy mouse model to examine possible alterations of the GA in neurons that overexpress human tau (P301S mutated gene) in neocortical and hippocampal neurons, using double immunofluorescence techniques and confocal microscopy. Quantitative analysis revealed that neurofibrillary tangle (NFT)-bearing neurons had important morphological alterations and reductions in the surface area and volume of the GA compared with NFT-free neurons. Since in this mouse model there are no Aβ aggregates typical of AD, the present findings support the idea that the progressive accumulation of phospho-tau is associated with structural alterations of the GA, and that these changes may occur in the absence of Aβ pathology.
INTRODUCTION
The Golgi apparatus (GA) plays a critical role in processing and targeting cellular proteins, and its alterations give rise to neuronal dysfunctions and a variety of human diseases [1, 2]. The GA adapts to different physiological and pathological conditions by plastic changes in shape and protein content [3–6]. In Alzheimer’s disease (AD), the GA of certain populations of neurons becomes fragmented and there is experimental evidence for the link between GA fragmentation and the two main pathological hallmarks of the disease, namely the accumulation of amyloid-β (Aβ) peptides and the presence of neurofibrillary tangles (NFT) made of hyperphosphorylated microtubule-associated protein tau [7–16]. In pioneering studies in the cerebral cortex from AD patients, it was reported that the GA of certain neuronal populations of neurons becomes fragmented and decreases in size [17, 18]. However, there is some controversy regarding the possible link between fragmentation of the GA in cortical neurons and the intracellular content of NFT. The first studies on the possible alterations of GA in the cerebral cortex from AD patients were controversial. For example, Salehi et al. [17] reported that there was fragmentation and a decrease in size of the GA and that these alterations were unrelated to the intracellular content of NFT, whereas Stieber et al. [18] reported that these alterations were preferentially associated with neurons devoid of NTF. However, recent studies in the neocortex and hippocampus of AD patients using different experimental approaches to those employed by Stieber et al. [18] and Salehi et al. [17] have shown that the progressive accumulation of hyperphosphorylated tau is associated with the reduction and the morphometric alterations of GA [19]. Nevertheless, there are factors that might affect the structural characteristics of the GA, such as the age of the patients, tissue fixation procedure, and postmortem period prior to fixation [19]. In addition to NFTs, AD patient tissue is characterized by other pathologic hallmarks, namely, the accumulation of extracellular fibrillar Aβ in amyloid plaques and elevated levels of soluble Aβ oligomers in the brain. This Aβ pathology occurs to variable degrees in different patients and may also affect the structure of the GA. All of these considerations might help account for the different results reported by different laboratories. Here we have studied the possible relationship between alterations of the GA with NFT in neocortical and hippocampal neurons of the P301S tauopathy mouse model. This mouse model is characterized by the absence of Aβ plaques and carries a P301S human microtubule-associated protein tau (MAPT) gene mutation. This P301S (PS19) mouse overexpresses this gene at a level fivefold higher than the expression of the endogenous tau mouse protein, triggering — from 6 months of age onwards— the formation of NFT (similar to those observed in AD) in relatively few neurons, labeled with PHF1 as well as with Gallyas staining in both cortical and hippocampal regions [20]. In addition, between 3 and 6 months of age, P301 mice undergo a progressive accumulation of insoluble and highly phosphorylated tau species that can be detected by AT8 (phospho-Ser202 and phosho-Thr205), PHF1 (phospho-Ser396 and phospho-Ser404), and AT270 (phospho-Thr181) phosphorylation-dependent antibodies, like abnormal tau in purified AD paired helical filaments [20, 21].
The present results indicate that the overexpression of this mutated human tau gene does not seem to induce general alterations in the GA of pyramidal neurons from neocortex and hippocampus. However, the presence of hyperphosphorylated tau and NFT seems to be deleterious for the structural integrity of GA as alterations in morphology and important reductions in the size were found in the GA of NFT-bearing neurons.
MATERIALS AND METHODS
In the present study, we have used transgenic mice P301S for the human tau gene and wild-type control littermates. The P301S mouse model, obtained from Jackson laboratory (B6;C3-Tg(Prnp-MAPT*P301S)PS19Vle/J), carries a mutant (P301S) human MAPT gene encoding T34-tau isoform (1N4R) driven by the mouse prion-protein promoter (Prnp) on a B6C3H/F1 genetic background. A group was analyzed at the age of 2 weeks, and a second group was analyzed at 36 weeks when these animals show learning and motor deficits [20]. A total of 5 mice aged 2 weeks (n = 2 for WT, n = 3 for P301S) and 12 mice aged 36 weeks (n = 5 for WT, n = 7 for P301S) were analyzed. All mice were housed at the “Centro de Biología Molecular Severo Ochoa” animal facility. Mice were housed four per cage with food and water available ad libitum and maintained in a temperature-controlled environment on a 12/12 h light-dark cycle with light onset at 07 : 00 h. Animal housing and maintenance protocols followed the guidelines of Council of Europe Convention ETS123, recently revised as indicated in Directive 86/609/EEC. Animal experiments were performed under protocols (P15/P16/P18/P22) approved by the Institutional Animal Care and Utilization Committee (Comité de Ética de Experimentación Animal del CBM, CEEA-CBM, Madrid, Spain). Animals were sacrificed by a lethal intraperitoneal injection of sodium pentobarbital (200 mg/kg b.w.) and were then perfused intracardially with a saline solution followed by 4% paraformaldehyde in PB. The brain of each animal was removed and post-fixed by immersion in the same fixative for 24 h at 4°C. After rinsing in PB, brains were cut in the sagital plane using a vibratome (Leica VT2100S). Serial parasagital sections (50 μm thick) were cryoprotected in 30% sucrose solution in PB and stored in ethyleneglicol/glycerol at –20°C until they were used.
For immunofluorescence experiments, free-floating serial sections were first rinsed in PB and then preincubated for 1 h in PB with 0.25% Triton-X100 and 3% normal serum of the species in which the secondary antibodies were raised (Vector Laboratories, Burlingame, CA, USA). The sections were then incubated for 48 h at 4°C in the same stock solution containing the following primary antibodies in the combinations indicated: rabbit anti-MG160 (Abcam, 1 : 100), rabbit anti-Grasp65 (Abcam, 1 : 500), mouse anti human tau (Abcam, T13, 1 : 5000), rabbit anti-NeuN (Millipore, 1 : 2000) and mouse anti-phospho-PHF-tau pSer202+ Thr205 antibody (AT8, 1 : 2000, Pierce Endogen). After rinsing in PB, the sections were first incubated for 2 h at room temperature in biotinylated goat anti-rabbit antibody (1 : 200) to amplify the GA immunoreactivity signal. Sections were then rinsed in PB and incubated for 2 h at room temperature in Alexa 594-conjugated goat anti-mouse and streptavidin-coupled Alexa 488 (1 : 1000; Molecular Probes, Eugene, OR, USA). Sections were also stained with a nuclear stain — DAPI (4,6-diamidino-2-phenylindole; Sigma, St. Louis, MO, USA). After rinsing in PB, the sections were mounted in antifade mounting medium (ProlongGold, Invitrogen) and studied by conventional fluorescence and confocal microscopy (Zeiss, 710). Confocal image stacks, from the somatosensory neocortex and CA1 hippocampal region of control and P301 S mice, were recorded at 0.35 μm intervals through separate channels with a 63x oil-immersion lens (NA, 1.40, refraction index, 1.45). ZEN 2012 software (Zeiss) was used to construct composite images from each optical series by combining the images recorded through the different channels, and the same software was used to obtain Z projection images (image resolution: 1024×1024 pixels; pixel size: 0.11 μm). Adobe Photoshop (CS4) software was used to compose figures. Fiji software (3D object counter tool) was used to analyze the volume and surface area of the elements immunostained for the different GA markers in image stacks. Based on methods used in previous studies (see [19] for a detailed description), we cropped 3D substacks from the original confocal stacks taken from AT8-MG160 or AT8-Grasp65 double-stained sections counterstained with DAPI, trying to limit the analysis to the complete GA corresponding to the somata of single neurons. In P301S animals, these cells included pyramidal neurons with (AT8+) or without (AT8 –) hyperphosphorylated tau, which were selected such that they were similar in size and from neighboring zones with similar Z depths within the stacks, in order to minimize possible sources of bias due to neuronal population heterogeneity or antibody penetration within the tissue. We measured the volume and surface area of GA elements and calculated the percentages of variation of these parameters between AT8+ and AT8- in P301 mice, and between AT8- neurons in P301 and wild-type animals. To determine differences between values obtained in wild-type and mutant P301 mice in the different age groups (2 and 36 weeks), Kruskal-Wallis one-way analysis of variance was performed followed by a Bonferroni-corrected Mann-Whitney U-test for pairwise comparisons, using SPSS software (version 22). The Wilcoxon pair test was used to compare mean values obtained in AT8+ and AT8- neurons from mutant P301 mice, whereas a Mann-Whitney U-test was used to check differences between AT8+ neurons from mutant mice and neurons from wild-type mice.
RESULTS
In the present work, we have studied the morphological features of the Golgi apparatus (GA) of pyramidal neurons from the somatosensory cortex and CA1 hippocampal region, in relation to the accumulation of hyperphosphorylated tau, in P301S transgenic and control animals aged 2 and 36 weeks. In line with previous descriptions of this mouse model [20], in the hippocampus and neocortex of 2-week-old P301S mice, we found no phospho-tau positive (AT8+) neurons despite the fact that all neurons in these regions overexpress MAPT P301S mutated tau gene, as revealed by immunostaining using anti-human tau (T13) antibodies (Fig. 1). We have studied sections counterstained with DAPI and immunostained with antibodies that recognize MG160, a sialoglycoprotein that is mainly localized in the medial cisternae of the GA, or GRASP65, a peripheral protein localized primarily in the cis-Golgi both in combination with AT8 antibodies recognizing phospho Ser202 and phospho Thr205 sites in microtubule-associated tau protein [22] (Figs. 2 and 3). In both wild-type and P301S animals at 2 weeks of age, the GA of hippocampal and neocortical pyramidal neurons (as revealed by Grasp65 and MG160 immunostaining) consisted of a network of twisted cisternae with a ribbon-like appearance distributed throughout the cell body and frequently extending partially to the apical dendrite (Fig. 2A–D). This observation was in line with previous descriptions in other mammalian species, including humans [6, 19]. Using confocal microscopy and 3D quantitative techniques, we found no differences between wild-type and P301S animals at 2 weeks of age in terms of the surface area and volume of GA elements of neocortical and CA1 hippocampal neurons (as revealed both by Grasp65 and MG160 immunostaining) (Fig. 4). In line with previous reports [20], in 36-week-old P301S animals, we found a subpopulation of phospho-tau accumulating, AT8+ neurons throughout the neocortex and hippocampal formation. AT8+ neurons were found in CA hippocampal fields; dentate gyrus and subiculum; insular cortex; primary and secondary motor cortex; primary and secondary visual cortex; retrosplenial granular cortex; and different areas of somatosensory cortex (barrel field, hindlimb, shoulder, trunk, and upper lip regions). We focused on the CA1 hippocampal region and the hindlimb region of the somatosensory cortex in which AT8+ neurons in 36-week-old P301S mice were found to be more abundant (Fig. 2E, G, I). Most of the AT8+ neurons in these regions showed either a pre-tangle pattern or an intermediate pattern (pattern IIa according to [23]) of NFTs, whereas the presence of AT8+ neurons with an advanced (IIb) pattern of NFT was scarce.
Fig.1
Expression of human tau in hippocampal CA1 neurons from P301S and wild-type mice. Trios of confocal stack projection images taken from T13/NeuN double immunostained- and DAPI counterstained-sections from the CA of P301S (A-C, G-I) and wild-type (D-F, J-L) mice aged 2 (A-F) or 36 weeks (G-L). Images show at both ages the expression of human tau (T13 immunoreactivity, green) in CA1 neurons, as revealed by NeuN immuniostaining (red), in P301 mice in contrast to wild-type mice. Scale bar shown in L indicates 22 μm.
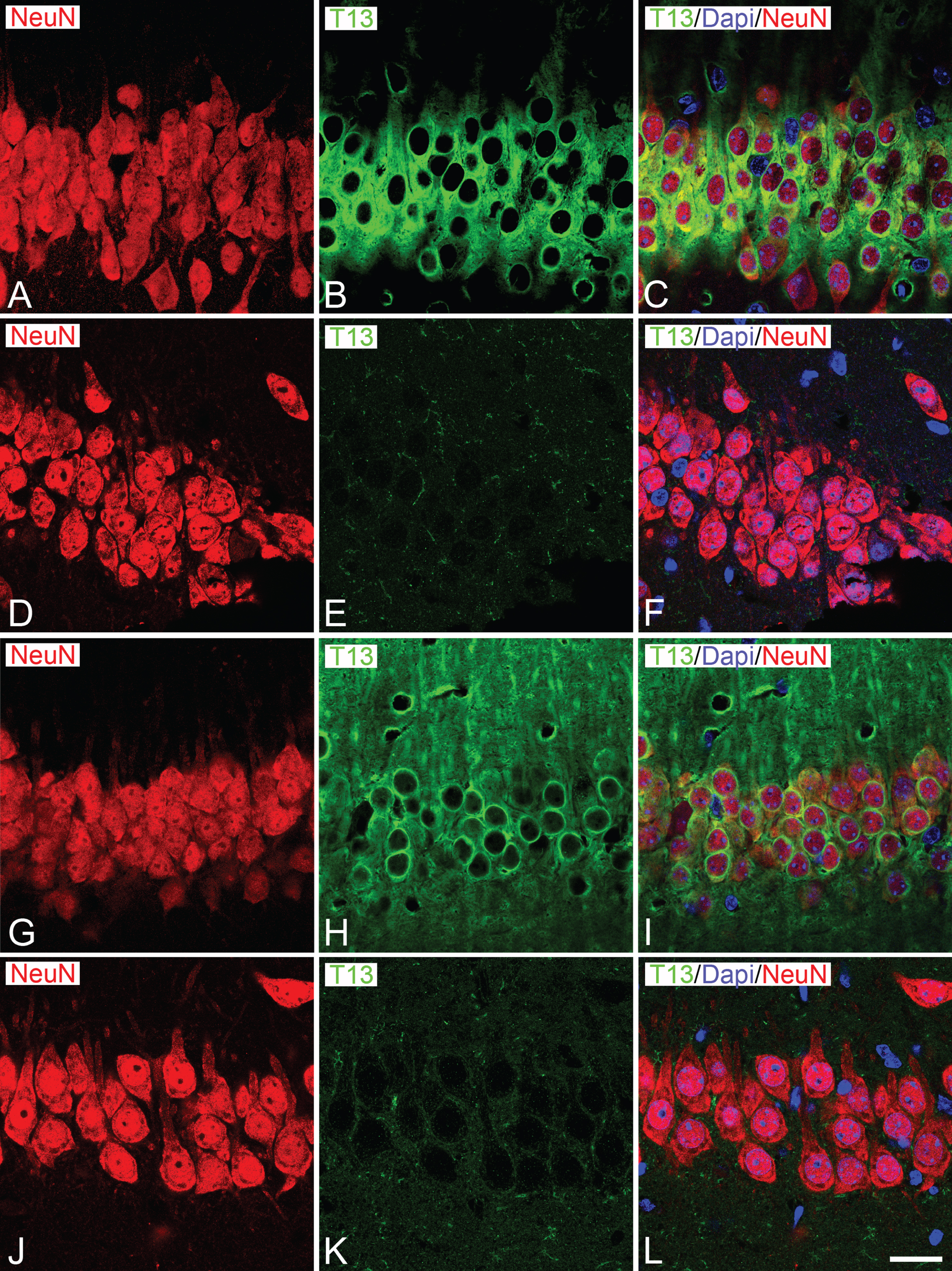
Fig.2
Distribution of Grasp65 in the GA of neurons from P301S and wild-type mice. A–D) Confocal stack projection images taken from Grasp 65/AT8 double-immunostained sections from the hindlimb region of the somatosensory neocortex (A, B) and the CA1 hippocampal region (C, D) of wild-type (A, C) and P301 (B, D) animals aged 2 weeks. Note the absence of AT8+ neurons in both regions at this age and the similar appearance of the Grasp65-ir GA elements between wild-type and P301S animals. E−J show pairs of conventional fluorescence images taken from a Grasp 65/AT8 double-immunostained parasagital section from the brain of 36-week-old P301 mice showing the distribution of AT8+ neurons in the somatosensory cortex (G, H) and the CA1 hippocampal region (I, J). Squared zones in E and F are shown at higher magnification in G−J. Scale bar shown in J indicates 7 μm in A−D, 1035 μm in E−F, 210 μm in G−H and 100 μm in I−J.
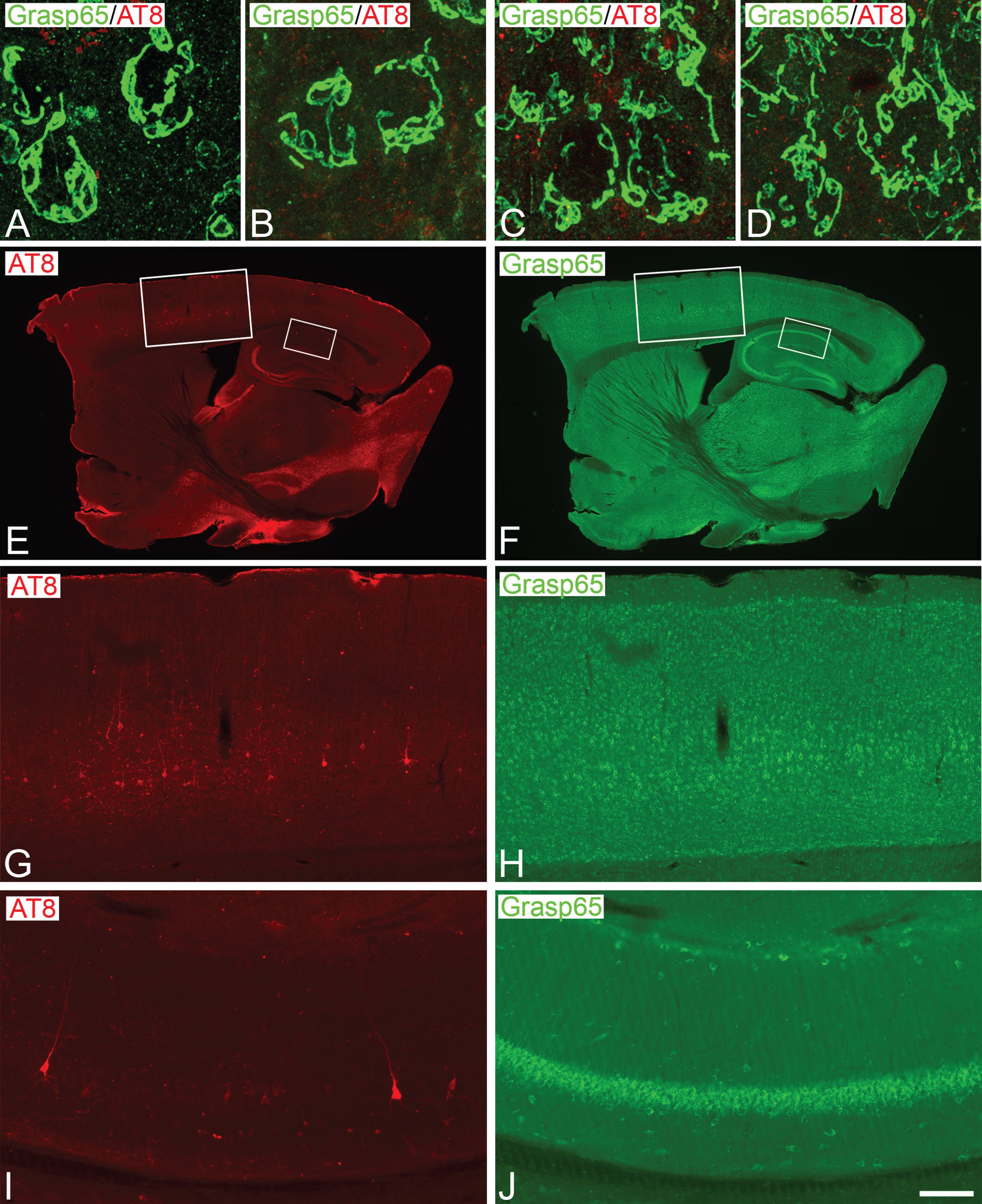
Fig.3
A–C, D–E) Trios of confocal stack projection images taken from MG160/AT8 or Grasp 65/AT8 double immunostained- and DAPI counterstained-sections from the hindlimb somatosensory cortex of P301S mice aged 36 weeks. Images show the distribution of MG160-ir (A–C) and Grasp65-ir (D–F) GA elements of AT8–and AT8+ neurons. G–J) Higher-magnification confocal projection images showing the alterations of the GA in AT8+ neurons (H, J) as compared with AT8- neurons (G, I) with each of the GA markers used. Insets show inmunofluorescence for AT8 in red. Scale bar in J indicates 21 μm in A–F and 3.6 μm in G–J. K) Bar histograms showing the percentages of AT8- and AT8+ neurons with an altered Golgi apparatus as identified by MG160 or Grasp65 immunostaining (n = 1099).
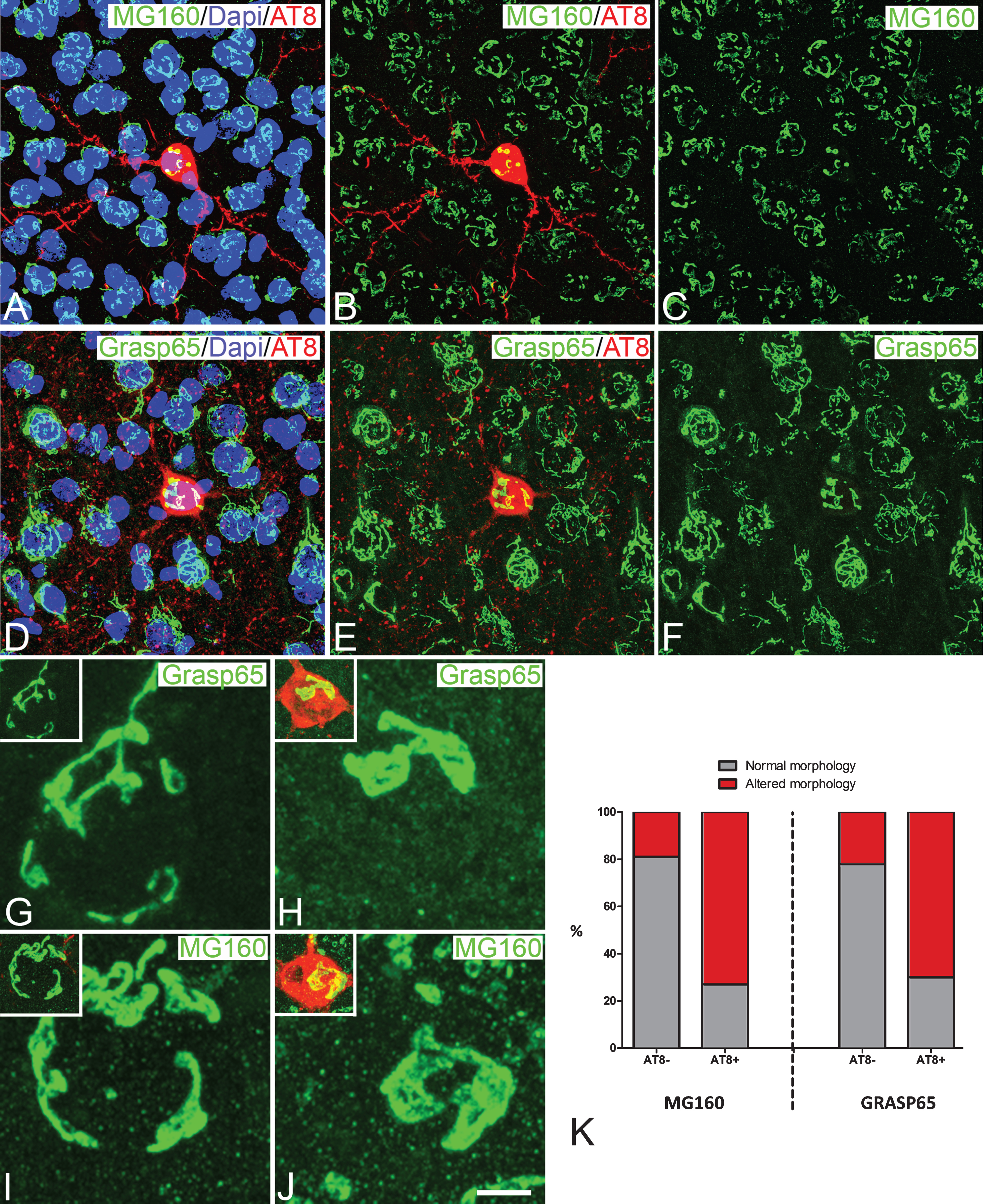
Fig.4
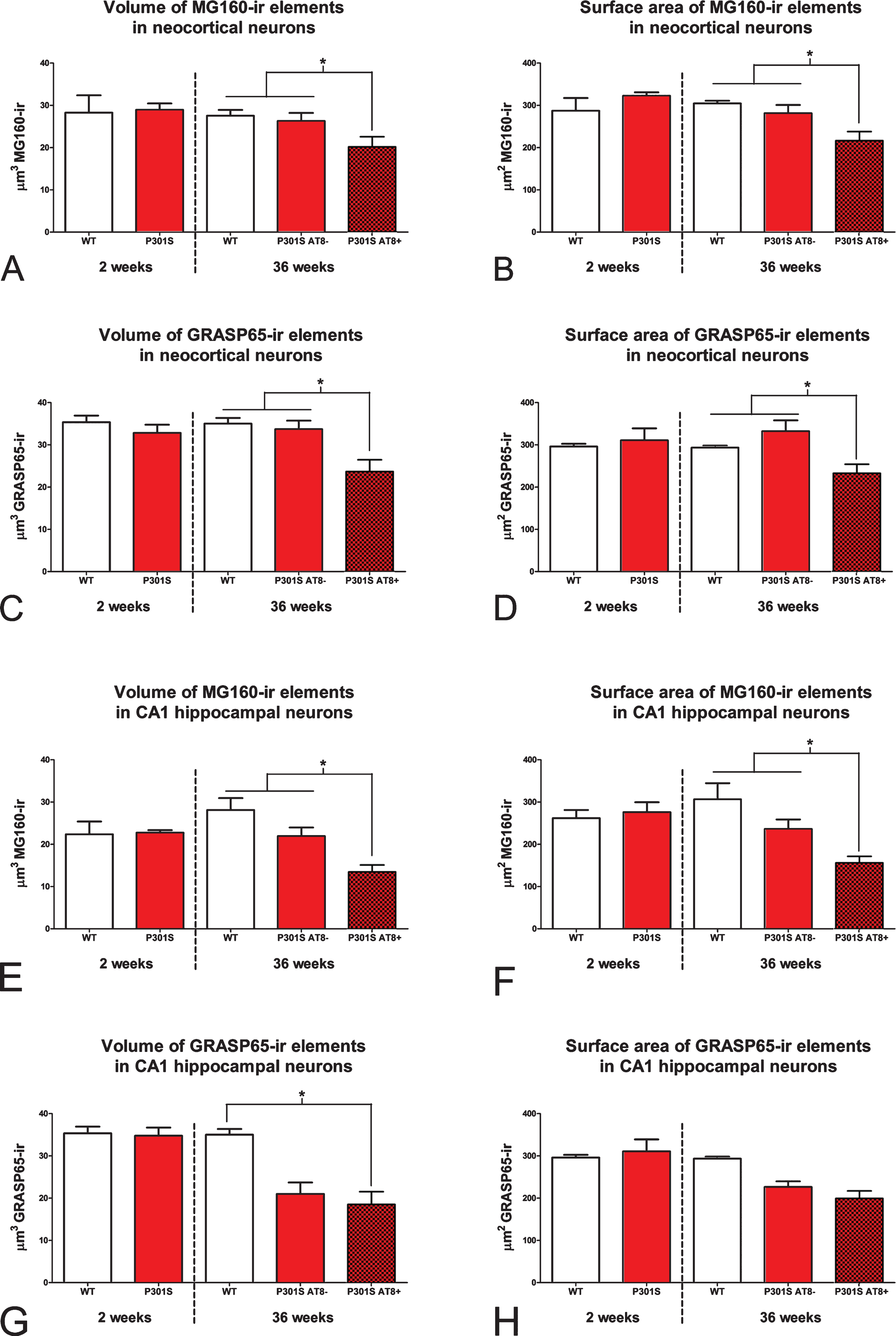
Histograms showing volume (A, C, E, and G) and surface area (B, D, F, and H) values (mean±SE) of MG160-ir (A, B, E, and F) and Grasp65-ir (C, D, G, and H) GA elements, in neurons from the neocortex (top) and hippocampus (bottom) of wild-type (white bars) and P301 transgenic (red bars) mice. Note that at 2 weeks of age (left side of each histogram), the surface area and volume of GA of neocortical and hippocampal neurons are similar in wild-type and P301S animals. In contrast, at 36 weeks of age (right side of each histogram), note the reduction of surface area and volume values in P301S AT8+ neurons (dark red) as compared to AT8–neurons (light red) both from P301S and wild-type mice (Kruskal Wallis and Wilcoxon tests, *p≤0.05). AT8–neurons (somatosensory cortex, n = 298 for Grasp65 and n = 269 for MG160; CA1, n = 76 for Grasp65 and n = 188 for MG160) and AT8+ neurons (somatosensory cortex, n = 82 for Grasp65 and n = 85 for MG160; CA1, n = 26 for Grasp65 and n = 75 for MG160).
No apparent general changes in the overall distribution of MG160 or Grasp65 (Fig. 2F, H, J) immunostaining was found between wild-type and P301S 36-week-old mice. However, the analysis of confocal image stacks at higher magnification revealed that some hippocampal and neocortical neurons displayed an altered morphology of the GA elements, as revealed by both markers, consisting of an apparent retraction and a loss of tubular structures of the GA that often showed a lobular appearance and was polarized to an extreme of the cell body (Fig. 3). We also found that the percent of neurons with an altered GA morphology was much higher in neurons with intracellular hyperphosphorylated tau aggregates (AT8+) than in adjacent AT8- neurons (Fig. 3K) (MG160 : 73.2% of AT8+ neurons versus 19.1% of AT8- neurons; Grasp 65 : 69.5% of AT8+ neurons versus 22.1% of AT8- neurons).
To quantify the possible retraction of the GA and to study whether this retraction might be related to the intracellular content of hyperphosphorylated tau aggregates, we selected — in confocal stacks from CA1 and neocortex— AT8+ and adjacent AT8- pyramidal neurons located at the same z position range within the stacks. In stacks taken from 36-week-old animals, we measured the volume and surface area of Grasp65-ir or MG160-ir GA elements and compared between the mean values in AT8+ and AT8- neurons, as well as comparing them with values obtained from age-matched wild-type animals (AT8-).
In the two regions analyzed, no significant differences were found between wild-type neurons and AT8- neurons from P301S mice in terms of the surface area and volume of the GA elements, as revealed by both markers (Fig. 4). However, in both regions, pyramidal neurons with intracellular hyperphosphorylated tau aggregates (AT8+) showed a reduction in the surface area and volume of the GA elements as compared with AT8- neurons from P301S or wild-type animals (Fig. 4). However, this reduction did not reach statistical significance in the case of the GA surface area of CA1 neurons immunostained with Grasp65 (Fig. 4G, H) probably due to the relatively low number of AT8+ NFT-bearing neurons found in this tissue (Fig. 2I, J). Finally, we calculated the mean percentage of GA decrease in AT8+ neurons as compared with AT8- neurons in 36-week-old P301S mice, taking together data from the CA1 and neocortical stacks. The mean percentage reduction in surface area and volume of GA elements was higher when analyzed with MG160 immunostaining (26.4% and 28.3%, respectively) than when estimated using Grasp65 immunostaining (14.8% and 13.2%, respectively).
DISCUSSION
The main results of the present study demonstrate that although the accumulation of tau protein of human origin in P301S mouse model is not sufficient to induce structural alterations of the GA, the hyperphosphorylation of this mutated tau and its aggregation forming NFT-like structures induce profound alterations in the GA structure as evaluated by MG160 and Grasp65 GA markers.
We have shown that at 2 weeks of age, tau was not hyperphosphorylated, as revealed by AT8 antibodies that detect phosphorylations in Ser202/Thr205. At this age, when P301S mice already overexpress MAPT P301S mutated tau gene, as revealed by T13 immunostaining (see Fig. 1) and according to previous studies [20], we found no differences between P301S mutant and wild-type animals in the general morphology of the GA of neocortical and hippocampal neurons, or in the surface area and volume of GA as evaluated with quantitative techniques. Similarly, we found no differences in the same parameters between wild-type neurons and AT8- neurons from P301 36-week-old mice, that is, an age when neurons would hypothetically accumulate more mutated tau. Therefore, we can conclude that in this model the overexpression of P301S mutated tau alone is not enough to induce GA alterations in cortical neurons. Although it is possible that tau in P301S animals at these ages might be hyperphosphorylated at residues other than Ser202/Thr205, our results demonstrate that these putative tau hyperphosphorylations (which are not detectable by the AT8 antibodies used in the present study) would not be sufficient to induce morphological alterations of neuronal GA.
Previous in vitro studies showed on one hand that the overexpression of either wild-type human Tau4R or its mutant forms like P301L in transfected rat cultured hippocampal neurons, induced GA fragmentation [16], and on the other hand, that this overexpression leads to tau hyperphosphorylation [24, 25]. However, the direct relationship between tau hyperphosphorylation and GA alteration in cortical neurons was not addressed. In the present study, we found that the accumulation of phospho-tau in P301S is associated with structural alterations of the GA since the percent of neurons with an altered morphology of the GA was much higher in AT8+ neurons than in adjacent AT8- neurons. In the hyperphosphorylated tau aggregate-bearing neurons, these alterations included a change from the typical ribbon-like appearance with multiple tubular structures to a lobulated appearance and a change from a homogeneous to a polarized distribution in the body. However, it should be noted that, in our material, we did not find evidence for GA fragmentation in AT8+ neurons, which contrasts with the fragmentation reported in CP-13+ spinal cord motor neurons (in which tau phosphorylated at serine 202 is labeled), in a study using 10–12-month-old JNPL3 mutant mice, which overexpress P301L mutated tau gene [16]. Several factors might have contributed the differences between the two studies regarding the fragmentation of the GA, namely differences in 1) the age of the animals studied [19]; 2) the type of tau mutation and the consequent type of hyperphosphorylation and aggregation; 3) the antibodies used to detect phospho-tau; and 4) the neuronal population studied. The present observations also contrast with the fragmentation of the GA described in AT8+ cortical pyramidal cells from AD patients [19], indicating that GA alterations in P301 mouse only partially reproduce those occurring during the development of the disease.
In previous studies from our laboratory using the same methodology as that used in the present work, we showed important reductions in the surface area and volume of the GA elements in AT8+ neurons in neocortical and hippocampal neurons from AD patients [19]. We suggested that the progressive accumulation of phospho-tau was associated with structural alterations of the GA. However, we could not rule out the possible involvement of different factors present in AD patients which could be deleterious for the GA integrity, such as amyloid oligomers [13–15, 26] which are bi-directionally related to tau pathology [27–29], aging, and postmortem delay period before tissue processing [19], as well as other unknown factors associated with the progression of the disease. The present results obtained from a tauopathy mouse model — in which the unique pathologic factor is the presence of human mutant tau and the development of NFT-like hyperphosphorylated tau aggregates, similar to those seen in human tissue [20]— further support the idea that the accumulation of phospho-tau is associated with structural alterations of the GA [19]. Although both processes seem to appear concomitantly, the causal relationship between them has not yet been resolved and warrants further research [8, 13, 16, 30].
The size of the GA has been correlated with cell activity levels, and the integrity of the GA is required for the proper control of the processing, sorting and transport of many important proteins that are essential for neuronal physiology [14, 15, 17, 31–34], including amyloid- β protein precursor and its processing enzymes [7, 9, 11, 12, 14]. Therefore, the retraction of the GA in cortical neurons is likely to induce functional impairment in the normal activity of those neurons affected by the hyperphosphorylated tau, and such functional impairment is likely to participate in the pathophysiology of AD.
ACKNOWLEDGMENTS
This work was supported by grants from the following entities: SAF 2015-66603-P and BFU-2016-77885-P from the Ministerio de Economía y Competitividad; Centro de Investigación en Red sobre Enfermedades Neurodegenerativas (CIBERNED, CB06/05/0066, Spain); and a grant from the Alzheimer’s Association (ZEN-15-321663). J. M-R was supported by a predoctoral fellowship from La Caixa foundation. This project received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement No. 720270.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/17-0332r1).
REFERENCES
[1] | Gonatas NK , Stieber A , Gonatas JO ((2006) ) Fragmentation of the Golgi apparatus in neurodegenerative diseases and cell death. J Neurol Sci 246: , 21–30. |
[2] | Hu Z , Zeng L , Huang Z , Zhang J , Li T ((2007) ) The study of Golgi apparatus in Alzheimer’s disease. Neurochem Res 32: , 1265–1277. |
[3] | Levine TP , Misteli T , Rabouille C , Warren G ((1995) ) Mitotic disassembly and reassembly of the Golgi apparatus. Cold Spring Harb Symp Quant Biol 60: , 549–557. |
[4] | Glick BS ((2002) ) Can the Golgi form de novo? Nat Rev Mol Cell Biol 3: , 615–619. |
[5] | Fan J , Hu Z , Zeng L , Lu W , Tang X , Zhang J , Li T ((2008) ) Golgi apparatus and neurodegenerative diseases. Int J Dev Neurosci 26: , 523–534. |
[6] | Anton-Fernandez A , Leon-Espinosa G , DeFelipe J , Munoz A ((2015) ) Changes in the Golgi apparatus of neocortical and hippocampal neurons in the hibernating hamster. Front Neuroanat 9: , 157. |
[7] | Greenfield JP , Tsai J , Gouras GK , Hai B , Thinakaran G , Checler F , Sisodia SS , Greengard P , Xu H ((1999) ) Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc Natl Acad Sci U S A 96: , 742–747. |
[8] | Sutterlin C , Hsu P , Mallabiabarrena A , Malhotra V ((2002) ) Fragmentation and dispersal of the pericentriolar Golgi complex is required for entry into mitosis in mammalian cells. Cell 109: , 359–369. |
[9] | Huse JT , Liu K , Pijak DS , Carlin D , Lee VM , Doms RW ((2002) ) Beta-secretase processing in the trans-Golgi network preferentially generates truncated amyloid species that accumulate in Alzheimer’s disease brain. J Biol Chem 277: , 16278–16284. |
[10] | Farah CA , Perreault S , Liazoghli D , Desjardins M , Anton A , Lauzon M , Paiement J , Leclerc N ((2006) ) Tau interacts with Golgi membranes and mediates their association with microtubules. Cell Motil Cytoskeleton 63: , 710–724. |
[11] | Burgos PV , Mardones GA , Rojas AL , daSilva LL , Prabhu Y , Hurley JH , Bonifacino JS ((2010) ) Sorting of the Alzheimer’s disease amyloid precursor protein mediated by the AP-4 complex. Dev Cell 18: , 425–436. |
[12] | Choy RW , Cheng Z , Schekman R ((2012) ) Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid beta (Abeta) production in the trans-Golgi network. Proc Natl Acad Sci U S A 109: , E2077–2082. |
[13] | Jiang Q , Wang L , Guan Y , Xu H , Niu Y , Han L , Wei YP , Lin L , Chu J , Wang Q , Yang Y , Pei L , Wang JZ , Tian Q ((2014) ) Golgin-84-associated Golgi fragmentation triggers tau hyperphosphorylation by activation of cyclin-dependent kinase-5 and extracellular signal-regulated kinase. Neurobiol Aging 35: , 1352–1363. |
[14] | Joshi G , Bekier ME 2nd , Wang Y ((2015) ) Golgi fragmentation in Alzheimer’s disease. Front Neurosci 9: , 340. |
[15] | Joshi G , Wang Y ((2015) ) Golgi defects enhance APP amyloidogenic processing in Alzheimer’s disease. Bioessays 37: , 240–247. |
[16] | Liazoghli D , Perreault S , Micheva KD , Desjardins M , Leclerc N ((2005) ) Fragmentation of the Golgi apparatus induced by the overexpression of wild-type and mutant human tau forms in neurons. Am J Pathol 166: , 1499–1514. |
[17] | Salehi A , Ravid R , Gonatas NK , Swaab DF ((1995) ) Decreased activity of hippocampal neurons in Alzheimer’s disease is not related to the presence of neurofibrillary tangles. J Neuropathol Exp Neurol 54: , 704–709. |
[18] | Stieber A , Mourelatos Z , Gonatas NK ((1996) ) In Alzheimer’s disease the Golgi apparatus of a population of neurons without neurofibrillary tangles is fragmented and atrophic. Am J Pathol 148: , 415–426. |
[19] | Anton-Fernandez A , Aparicio-Torres G , Tapia S , DeFelipe J , Munoz A ((2017) ) Morphometric alterations of Golgi apparatus in Alzheimer’s disease are related to tau hyperphosphorylation. Neurobiol Dis 97: , 11–23. |
[20] | Yoshiyama Y , Higuchi M , Zhang B , Huang SM , Iwata N , Saido TC , Maeda J , Suhara T , Trojanowski JQ , Lee VM ((2007) ) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53: , 337–351. |
[21] | Fernandez-Nogales M , Santos-Galindo M , Merchan-Rubira J , Hoozemans JJM , Rabano A , Ferrer I , Avila J , Hernandez F , Lucas JJ ((2017) ) Tau-positive nuclear indentations in P301S tauopathy mice. Brain Pathol 27: , 314–322. |
[22] | Goedert M , Jakes R , Vanmechelen E ((1995) ) Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett 189: , 167–169. |
[23] | Merino-Serrais P , Benavides-Piccione R , Blazquez-Llorca L , Kastanauskaite A , Rabano A , Avila J , DeFelipe J ((2013) ) The influence of phospho-tau on dendritic spines of cortical pyramidal neurons in patients with Alzheimer’s disease. Brain 136: , 1913–1928. |
[24] | Bertrand J , Plouffe V , Senechal P , Leclerc N ((2010) ) The pattern of human tau phosphorylation is the result of priming and feedback events in primary hippocampal neurons. Neuroscience 168: , 323–334. |
[25] | Bertrand J , Senechal P , Zummo-Soucy M , Plouffe V , Leclerc N ((2010) ) The formation of tau pathological phospho-epitopes in the axon is prevented by the dephosphorylation of selective sites in primary hippocampal neurons over-expressing human tau. J Neurochem 114: , 1353–1367. |
[26] | Joshi G , Chi Y , Huang Z , Wang Y ((2014) ) Abeta-induced Golgi fragmentation in Alzheimer’s disease enhances Abeta production. Proc Natl Acad Sci U S A 111: , E1230–1239. |
[27] | Oddo S , Billings L , Kesslak JP , Cribbs DH , LaFerla FM ((2004) ) Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron 43: , 321–332. |
[28] | Dai B , Li D , Xi W , Luo F , Zhang X , Zou M , Cao M , Hu J , Wang W , Wei G , Zhang Y , Liu C ((2015) ) Tunable assembly of amyloid-forming peptides into nanosheets as a retrovirus carrier. Proc Natl Acad Sci U S A 112: , 2996–3001. |
[29] | Dai CL , Tung YC , Liu F , Gong CX , Iqbal K ((2017) ) Tau passive immunization inhibits not only tau but also Abeta pathology. Alzheimers Res Ther 9: , 1. |
[30] | Lin WL , Lewis J , Yen SH , Hutton M , Dickson DW ((2003) ) Ultrastructural neuronal pathology in transgenic mice expressing mutant (P301L) human tau. J Neurocytol 32: , 1091–1105. |
[31] | Lucassen PJ , Ravid R , Gonatas NK , Swaab DF ((1993) ) Activation of the human supraoptic and paraventricular nucleus neurons with aging and in Alzheimer’s disease as judged from increasing size of the Golgi apparatus. Brain Res 632: , 105–113. |
[32] | Dries DR , Yu G ((2008) ) Assembly, maturation, and trafficking of the gamma-secretase complex in Alzheimer’s disease. Curr Alzheimer Res 5: , 132–146. |
[33] | Wang Y , Wei JH , Bisel B , Tang D , Seemann J ((2008) ) Golgi cisternal unstacking stimulates COPI vesicle budding and protein transport. PLoS One 3: , e1647. |
[34] | Xiang Y , Zhang X , Nix DB , Katoh T , Aoki K , Tiemeyer M , Wang Y ((2013) ) Regulation of protein glycosylation and sorting by the Golgi matrix proteins GRASP55/65. Nat Commun 4: , 1659. |


