
Detecting At-Risk Alzheimer’s Disease Cases
Abstract
While APOE ɛ4 is the major genetic risk factor for Alzheimer’s disease (AD), amyloid dysmetabolism is an initial or early event predicting clinical disease and is an important focus for secondary intervention trials. To improve identification of cases with increased AD risk, we evaluated recruitment procedures using pathological CSF concentrations of Aβ42 (pAβ) and APOE ɛ4 as risk markers in a multi-center study in Norway. In total, 490 subjects aged 40–80 y were included after response to advertisements and media coverage or memory clinics referrals. Controls (n = 164) were classified as normal controls without first-degree relatives with dementia (NC), normal controls with first-degree relatives with dementia (NCFD), or controls scoring below norms on cognitive screening. Patients (n = 301) were classified as subjective cognitive decline or mild cognitive impairment. Subjects underwent a clinical and cognitive examination and MRI according to standardized protocols. Core biomarkers in CSF from 411 and APOE genotype from 445 subjects were obtained. Cases (both self-referrals (n = 180) and memory clinics referrals (n = 87)) had increased fractions of pAβ and APOE ɛ4 frequency compared to NC. Also, NCFD had higher APOE ɛ4 frequencies without increased fraction of pAβ compared to NC, and cases recruited from memory clinics had higher fractions of pAβ and APOE ɛ4 frequency than self-referred. This study shows that memory clinic referrals are pAβ enriched, whereas self-referred and NCFD cases more frequently are pAβ negative but at risk (APOE ɛ4 positive), suitable for primary intervention.
INTRODUCTION
Alzheimer’s disease (AD) is the major cause of memory loss and dementia and a main cause for increasing costs for health care and death. Several strategies for AD treatment are being pursued, and early diagnostics and intervention will be necessary. AD has been shown to encompass long pre-clinical and pre-dementia periods, but among those seeking help at memory clinics for early cognitive symptoms, non-AD causes (e.g. stress, worrying, and depression) are predominant [1–3]. Though memory symptoms are a major cause for concern among the elderly, few seek medical help for these symptoms [4]. Better characterization of target groups and more accurate and efficient screening procedures for early AD disease activity are crucial to recruit people with incipient AD and early stage disease for clinical trials.
A long AD pre-dementia period encompassing amyloid-β protein precursor dysmetabolism, amyloid plaque formation and inflammation has been described, initially without subjective cognitive dysfunction, then in some individuals with subjective cognitive decline (SCD) and ultimately with mild cognitive impairment (MCI) [2, 5–7]. Several definitions of AD manifestations in this period have been developed. Two pre-clinical AD stages, (1) asymptomatic amyloidosis and (2) asymptomatic amyloidosis + neurodegeneration have been suggested as precursors for SCD and MCI [3]. PET amyloid imaging and cerebrospinal fluid (CSF) amyloid-β (Aβ)42 measurements are considered as equivalent biomarkers for brain amyloid deposition, though the CSF measure of low CSF Aβ42 (pAβ) has been reported to give an earlier signal [8]. Subtle loss of grey and white matter integrity is closely coupled to cognitive impairment in the pre-dementia AD stage [9, 10]. The apolipoprotein E (APOE) ɛ4 allele is a well-established genetic risk factor for AD, and several polymorphisms associated with AD have been related to Aβ metabolism [11–16]. The exact mechanisms causing early neurodegeneration remain to be established, but most current treatment trials focus on altering amyloid metabolism at the MCI stage [17, 18].
The brain has extensive compensatory mechanisms for gradually acquired neuronal damage. Realizing that not only dementia, but also MCI appears late in the AD disease process, the prevalence of AD-like pathology among cognitively normal cases has attracted increased interest and some data has been published [19, 20].
We aimed to identify subjects suitable for primary AD intervention studies, and therefore investigated whether recruitment strategies focusing on self-referred cases with cognitive symptoms and ordinary referrals to memory clinics identified cases with pAβ and at-risk subjects without AD pathology differently.
METHODS
As part of “Dementia Disease Initiation” (DDI), a co-operation between all Norwegian health regions and university hospitals, subjects with self-reported cognitive reduction and healthy controls were recruited from January 2013 till January 2017 and examined following a standard protocol. Anonymized data were collected in a customized database. Recruitment was based on two main sources: 1) Cases were self-referred following advertisements in media, newspapers or news bulletins, or 2) recruited among referrals to local memory clinics. In addition, cognitively healthy controls were also included from spouses of patients with dementia/cognitive disorder, and from patients who completed lumbar puncture for orthopedic surgery. Participants were staged as controls, SCD or MCI using published criteria based on the comprehensive assessment program (see below) [5, 21]. The controls were further classified as having normal or abnormal cognitive screening and with or without first-degree relative with dementia. Criteria for inclusion were age between 40 and 80 and a native language of Norwegian, Swedish, or Danish. Exclusion criteria were brain trauma or disorder, including clinical stroke, dementia, severe psychiatric disease, severe somatic disease that might influence the cognitive functions, or intellectual disability or other developmental disorders.
A case report form was developed which included medical history from subject and informant, and physical and neurological examinations including cognitive examination and the 15-item Geriatric Depression Score (GDS) [22]. Educational levels were operationalized according to normative classification [23]; 0 = Primary school (7–8 y), 1 = High School (9–11 y), 2 = College (12 y), 3 = Bachelor degree (13–15 y), 4 = Master or equivalent (16–17 y), 5 = Higher university degree/PhD (18–20 y). The cognitive examination included the Mini Mental State Examination (MMSE-NR) [24], non-verbal cognitive screening (the clock drawing test) [25], verbal memory (CERAD word list) [26], visuoperceptual ability (VOSP silhouettes), psychomotor speed and divided attention (Trail making A and B and word fluency (COWAT) [27].
All subjects gave their written consent, and the Regional Committee for Medical and Health Research Ethics South-East evaluated (based on the Norwegian Health and Research Act and the Helsinki Declaration of 1964; revised 2013) and approved the study. All further study conduct was in line with these guidelines.
Cognitive staging
Symptomatic subjects with normal performance on standardized tests were classified as SCD, as defined in the framework by the working group of SCD [5]. The NIA-AA criteria for MCI were used for cases with lower performance than expected in one or more cognitive domains, but yet preserved independence in functional ability and not fulfilling the criteria of dementia, as defined in NIA-AA guidelines [21, 28]. Performance was classified as normal or abnormal according to published norms for the different tests [24–27, 29–31]. The cutoff values for SCD versus MCI (defined as normal or abnormal cognition) were results equal to or 1.5 standard deviation below normative mean on either CERAD word list (delayed recall), VOSP silhouettes, TMT-B or COWAT, or having MMSE score equal to or below 27. Cognitive functioning was also assessed by the Clinical Dementia Rating scale (CDR) [32]. Cases with dementia were excluded if CDR was >0.5 [33].
Biomarkers
Lumbar puncture was performed before noon, and CSF was collected in polypropylene tubes (Thermo Nunc) and centrifuged within 4 h at 2000 g for 10 min at room temperature. The supernatant was transferred to new tubes and frozen at –80°C prior to analysis. All CSF samples were analyzed at the Department of Interdisciplinary Laboratory Medicine and Medical Biochemistry at Akershus University Hospital, and samples from other sites were frozen before sending to this laboratory. CSF Aβ42, total tau, and phosphorylated tau were determined using ELISA (Innotest β-Amyloid (1–42), Innotest h-Tau Ag and Innotest Phospho-Tau (181P), Fujirebio, Ghent, Belgium).
APOE genotyping was performed on EDTA blood samples either at Akershus University Hospital (Gene Technology Division, Department of Interdisciplinary Laboratory Medicine and Medical Biochemistry) according to the laboratory’s routine protocol using real-time PCR combined with a TaqMan assay (Applied Biosystems, Thermo Fisher Scientific, Waltham, USA) or at the University Hospital of Trondheim according to the protocol for the Fast Start DNA Master HybProbe Kit (Roche, Basel, Switzerland) in combination with the LightMix ApoE C112R R158C kit from TiB MolBiol (Berlin, Germany) followed by LightCycler technology (Roche, Basel, Switzerland).
All subjects were referred to an MRI scan, and if available also to FDG-PET and amyloid PET.
Data analysis
The subjects with cognitive symptoms were categorized both by stage (Table 1) and by recruitment method (Table 2). The derived variable “APOE ɛ4 positive” was defined as having at least one APOE ɛ4 allele. The CSF Aβ42 measurements were dichotomized using a threshold of 708 ng/L, with values below the threshold defined as positive. This current CSF Aβ42 threshold for research use was determined based on best fit to flutemetamol PET results (based on 42 subjects for whom CSF and flutemetamol PET was available, of whom 16 subjects was flutemetamol PET positive and 26 subjects was flutemetamol PET negative), as described in [34] (sensitivity = 0.93, specificity = 0.93, AUC = 0.985; final results are due for publication in Kalheim et al., in submission).
Table 1
Demographic characteristics, cognitive test results, APOE alleles and CSF Aβ pathology of the control and symptomatic subjects classified by cognitive stage
| Control subjects | Subjects with cognitive | |||||
| symptoms | ||||||
| Total | Without | With family | Abnormal | SCD | MCI | |
| family | history | cognitive | ||||
| History | Screening | |||||
| Age at inclusion (SD) | 62.9 (9.4) | 63.2 (9.6) | 58.9 (8.9) | 65.2 (7.6) | 62.3 (8.9) | 65.4 (9.8) |
| n = 463 | n = 46 | n = 86 | n = 32 | n = 163 | n = 136 | |
| p < 0.05a | p = n.s.a | p = n.s.a | p = n.s.a | |||
| Female/Total | 253/465 | 22/46 | 53/86 | 21/32 | 93/164 | 64/137 |
| 54.4% | 47.8% | 61.6% | 65.6% | 56.7% | 46.7% | |
| p = n.s.c | p = n.s. c | p = n.s.c | p = n.s.c | |||
| Education level (IQR) | 3.0 (1.0) | 3.0 (2.0) | 3.0 (2.0) | 2.0 (2.0) | 3.0 (3.0) | 3.0 (2.0) |
| n = 461 | n = 46 | n = 85 | n = 31 | n = 163 | n = 136 | |
| p = 0.05b | p = n.s.b | p = n.s.b | p = n.s.b | |||
| MMSE (IQR) | 29.0 (2.0) | 29.0 (1.0) | 30.0 (1.0) | 28.0 (2.0) | 29.0 (1.0) | 28.0 (3.0) |
| n = 461 | n = 45 | n = 86 | n = 32 | n = 163 | n = 135 | |
| p = n.s.b | p < 0.001b | p = n.s.b | p < 0.001b | |||
| CERAD word list recall | 48.9 (14.0) | 50.1 (13.6) | 57.9 (8.7) | 48.3 (11.5) | 53.1 (10.3) | 37.5 (14.4) |
| T-score (SD) | n = 453 | n = 45 | n = 85 | n = 32 | n = 161 | n = 130 |
| p < 0.01a | p = n.s.a | p = n.s.a | p < 0.001a | |||
| VOSP silhouettes | 49.2 (11.1) | 50.6 (8.0) | 53.8 (9.2) | 43.1 (13.6) | 52.3 (9.8) | 43.6 (10.9) |
| T-score (SD) | n = 419 | n = 38 | n = 82 | n = 29 | n = 143 | n = 127 |
| p = n.s.a | p < 0.05a | p = n.s.a | p < 0.001a | |||
| TMT-B | 47.5 (10.2) | 53.7 (8.3) | 50.5 (8.9) | 44.2 (7.9) | 49.6 (8.7) | 41.3 (11.0) |
| T-score (SD) | n = 443 | n = 43 | n = 84 | n = 32 | n = 161 | n = 123 |
| p = n.s.a | p < 0.001a | p < 0.05a | p < 0.001a | |||
| COWAT | 48.9 (9.8) | 51.1 (6.8) | 50.2 (8.1) | 43.7 (10.1) | 51.5 (9.9) | 45.6 (10.1) |
| T-score (SD) | n = 452 | n = 44 | n = 84 | n = 32 | n = 160 | n = 132 |
| p = n.s.a | p < 0.01a | p = n.s.a | p < 0.001a | |||
| GDS score (IQR) | 1.0 (3.0) | 0.0 (1.0) | 0.0 (1.0) | 0.5 (1.0) | 2.0 (3.0) | 3.0 (3.0) |
| n = 448 | n = 45 | n = 81 | n = 32 | n = 156 | n = 134 | |
| p = n.s.b | p = n.s.b | p < 0.001b | p < 0.001b | |||
| APOE ɛ4 positivity | 196/430 | 9/43 | 45/82 | 12/32 | 64/147 | 66/126 |
| 45.6% | 20.9% | 54.9% | 37.5% | 43.5% | 52.4% | |
| p < 0.001c | p = n.s.c | p < 0.01c | p < 0.001c | |||
| APOE ɛ4 allele frequency | 25.7% | 10.5% | 28.7% | 20.3% | 24.8% | 31.3% |
| p < 0.01c | p = n.s.c | p < 0.01c | p < 0.001c | |||
| APOE | n = 430 | n = 43 | n = 82 | n = 32 | n = 147 | n = 126 |
| ɛ2/ɛ2 | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) | 0 (0.0%) |
| – | – | – | – | |||
| ɛ2/ɛ3 | 35 (8.1%) | 7 (16.3%) | 2 (2.4%) | 6 (18.8%) | 14 (9.5%) | 6 (4.8%) |
| p < 0.01d | p = n.s.c | p = n.s.d | p < 0.05d | |||
| ɛ3/ɛ3 | 199 (46.3%) | 27 (62.8%) | 35 (42.7%) | 14 (43.8%) | 69 (46.9%) | 54 (42.9%) |
| p < 0.05c | p = n.s.c | p = n.s.c | p < 0.05c | |||
| ɛ2/ɛ4 | 15 (3.5%) | 1 (2.3%) | 6 (7.3%) | 1 (3.1%) | 3 (2.0%) | 4 (3.2%) |
| p = n.s.d | p = n.s.d | p = n.s.d | p = n.s.d | |||
| ɛ3/ɛ4 | 156 (36.3%) | 8 (18.6%) | 37 (45.1%) | 10 (31.3%) | 52 (35.4%) | 49 (38.9%) |
| p < 0.01c | p = n.s.c | p < 0.05c | p < 0.05c | |||
| ɛ4/ɛ4 | 25 (5.8%) | 0 (0.0%) | 2 (2.4%) | 1 (3.1%) | 9 (6.1%) | 13 (10.3%) |
| p = n.s.d | p = n.s.d | p = n.s.d | p < 0.05d | |||
| CSF Aβ42 positivity | 89/393 | 2/39 | 4/62 | 6/25 | 24/145 | 53/122 |
| 22.7% | 5.1% | 6.5% | 24.0% | 16.6% | 43.4% | |
| p = n.s.d | p < 0.05d | p = n.s.c | p < 0.001c | |||
Continuous variables of assumed normal distribution (age at inclusion, CERAD word list recall T-score, VOSP silhouettes T-score, TMT-B T-score, and COWAT T-score) are summarized by mean (standard deviation) and compared with one-way ANOVA with predefined contrasts (a). Continuous variables of non-normal distribution (MMSE and GDS) and the ordinal variable (education level) are described by median (interquartile range) and compared with Mann-Whitney U tests (b). Binary variables (sex, APOE ɛ4 positivity, APOE ɛ4 allele frequency, APOE allele distribution and CSF pAβ positivity) are described with observed numbers and percentages and compared with Pearson’s Chi square tests (c) or Fisher’s exact tests when expected count is less than 5 (d).
Table 2
Demographic characteristics, distribution of cognitive stage (SCD, MCI) for symptomatic subject groups, APOE alleles and CSF Aβ pathology of the control and symptomatic subjects classified by recruitment method
| Control subjects | Subjects with cognitive symptoms | ||||||
| Without | With | Abnormal | Recruited | Self- | Memory | Self-versus | |
| family | family | cognitive | as control | referral | clinic | memory | |
| history | history | screening | subjects | referral | clinic referral | ||
| Age at inclusion (SD) | 63.2 (9.6) | 58.9 (8.9) | 65.2 (7.6) | 66.6 (6.5) | 64.4 (9.7) | 61.5 (9.1) | p < 0.05a |
| n = 46 | n = 86 | n = 32 | n = 11 | n = 179 | n = 86 | ||
| p < 0.05a | p = n.s.a | p = n.s.a | p = n.s.a | p = n.s.a | |||
| Female/Total | 22/46 | 53/86 | 21/32 | 5/11 | 97/180 | 46/87 | p = n.s.c |
| 47.8 % | 61.6 % | 65.6 % | 45.5 % | 53.9 % | 52.9 % | ||
| p = n.s.c | p = n.s.c | p = n.s.c | p = n.s.c | p = n.s.c | |||
| Education level (IQR) | 3.0 (2.0) | 3.0 (2.0) | 2.0 (2.0) | 2.0 (2.0) | 3.0 (2.0) | 2.5 (2.0) | p < 0.01b |
| n = 46 | n = 85 | n = 31 | n = 11 | n = 179 | n = 86 | ||
| p = 0.05b | p = n.s.b | p = n.s.b | p = n.s.b | p = n.s.b | |||
| MMSE (IQR) | 29.0 (1.0) | 30.0 (1.0) | 28.0 (2.0) | 27.5 (2.0) | 29.0 (2.0) | 29.0 (3.0) | p < 0.05b |
| n = 45 | n = 86 | n = 32 | n = 10 | n = 179 | n = 86 | ||
| p = n.s.b | p < 0.001b | p < 0.001b | p < 0.05b | p < 0.001b | |||
| SCD | – | – | – | 3/11 | 110/180 | 41/87 | p < 0.05c |
| 27.3 % | 61.1 % | 47.1 % | |||||
| MCI | – | – | – | 8/11 | 70/180 | 46/87 | p < 0.05c |
| 72.7 % | 38.9 % | 52.9 % | |||||
| APOE ɛ4 positive | 9/43 | 45/82 | 12/32 | 2/11 | 71/160 | 50/79 | p < 0.01c |
| 20.9 % | 54.9 % | 37.5 % | 18.2 % | 44.4 % | 63.3 % | ||
| p < 0.001c | p = n.s.c | p = n.s.d | p < 0.01c | p < 0.001c | |||
| APOE ɛ4 allele frequency | 10.5 % | 28.7 % | 20.3 % | 9.1 % | 25.0 % | 38.0 % | p < 0.01c |
| p < 0.01c | p = n.s.c | p = n.s.d | p < 0.05c | p < 0.001c | |||
| CSF Aβ42 positive | 2/39 | 4/62 | 6/25 | 1/11 | 37/155 | 29/79 | p < 0.05c |
| 5.1 % | 6.5 % | 24.0 % | 9.1 % | 23.9 % | 36.7 % | ||
| p = n.s.d | p < 0.05d | p = n.s.d | p < 0.01c | p < 0.001c | |||
The continuous variable of assumed normal distribution (age at inclusion) is summarized by mean (standard deviation) and compared with one-way ANOVA with predefined contrasts (a). The continuous variable of non-normal distribution (MMSE) and the ordinal variable (education level) are described by median (interquartile range) and compared with Mann-Whitney U tests (b). Binary variables (sex, APOE ɛ4 positivity, APOE ɛ4 allele frequency, and CSF pAβ positivity) are described with observed numbers and percentages and compared with Pearson’s Chi square tests (c) or Fisher’s exact tests when expected count is less than 5 (d).
For continuous variables with assumed normal distribution (age at inclusion, CERAD word list recall T-score, VOSP silhouettes T-score, TMT-B T-score, and COWAT T-score) means for the different groups were compared with one-way ANOVA (analysis of variance) with pre-defined contrasts. Normality was assessed by visual inspection of frequency distributions, Q-Q-plots, and box-plots. Equal variance was assumed for all except CERAD word list delayed recall, VOSP silhouettes, and COWAT T-score, given by Levene’s test. Continuous variables of non-normal distribution (MMSE and GDS) were compared with Mann-Whitney U tests, and education level was also tested with Mann-Whitney U, being an ordinal variable. The binary variables (sex, APOE ɛ4 positivity, APOE ɛ4 allele frequency, APOE allele distribution, and CSF pAβ positivity) were compared with Pearson’s Chi square test or Fisher’s exact test when expected counts were less than 5. All analyses were performed in the Statistical Package for Social Sciences version 24 (Chicago, IL, USA).
RESULTS
Of 577 cases considered, 87 withdrew before finishing the assessment program or did not fulfill the inclusion criteria (see Fig. 1). Of the 490 cases included, 465 were staged at the time of analysis. 46 were normal controls without (NC, age 63.2, SD 9.6) and 86 with first degree relative with dementia (NCFD, age 58.9, SD 8.9), 32 subjects were controls with abnormal cognitive screening (ACS, age 65.2, SD 7.6), 164 were SCD (age 62.3, SD 8.9), and 137 were MCI (age 65.4 SD, 9.8). Further characteristics are shown in Table 1.
Fig.1
Initially 577 subjects were considered for inclusion, whereof 87 did not fulfill inclusion criteria or withdrew before finishing the assessment program. 465 subjects were staged, as either normal controls (NC), normal controls with first degree relative (NCFD), controls with abnormal cognitive screening results (ACS), subjective cognitive decline (SCD) or mild cognitive impairment (MCI). At the time of analysis, APOE genotyping was available for 445 subjects, whereof 202 were APOE ɛ4 positive. Cerebrospinal fluid data was available for 411 subjects, whereof 96 subjects had pathological levels of Aβ42.
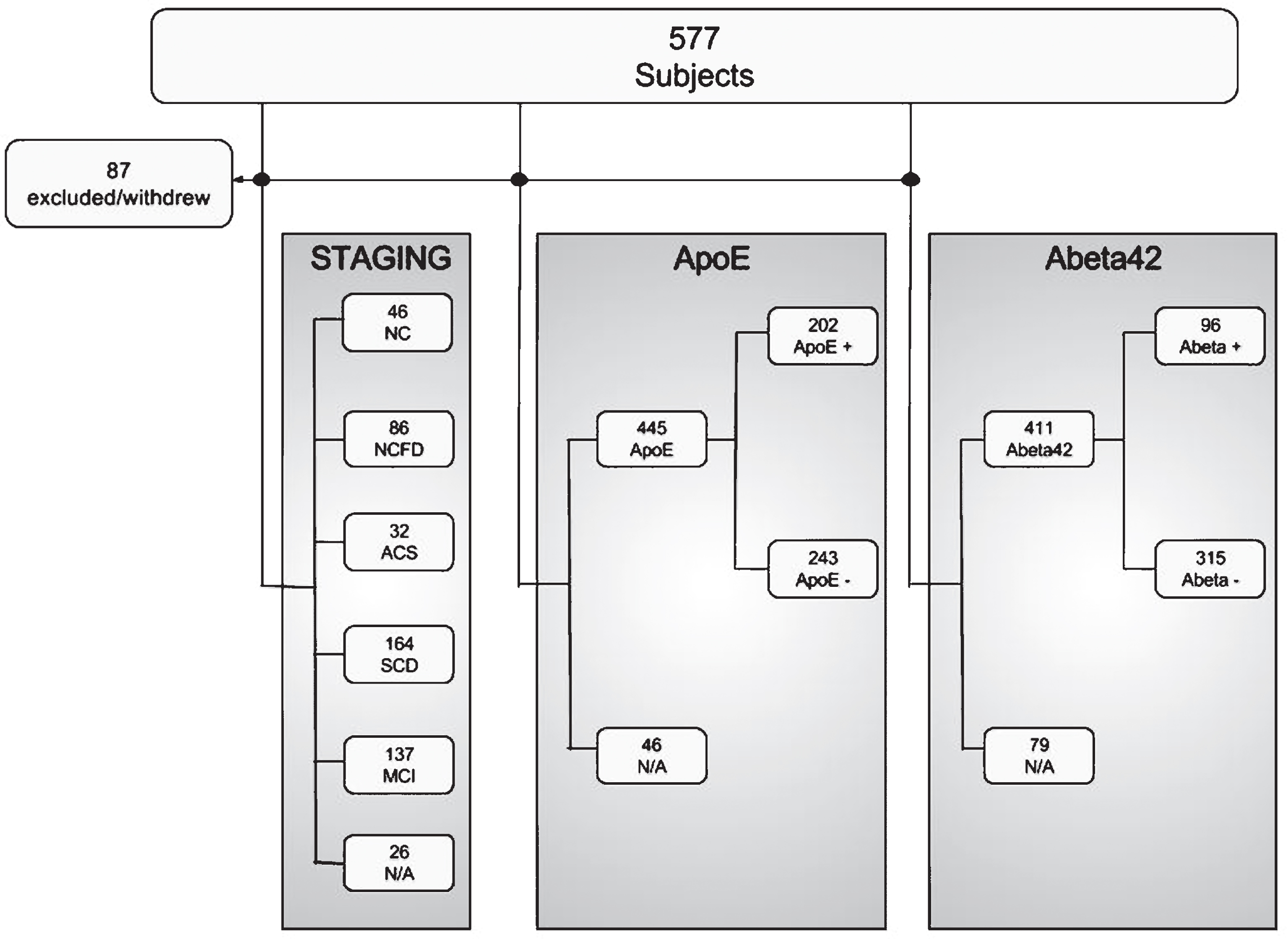
Out of 411 cases with available CSF Aβ42 values, 96 had increased fractions of pAβ (23.4%). 5.1% of the normal control, 6.5% of controls with first-degree relatives, and 16.6% of the SCD cases were pAβ, compared to 43.3% of MCI cases (p < 0.001 MCI versus NC, Table 2). Out of 445 cases with available APOE ɛ4 status, 243 were negative and 202 were positive. In the NC group, 20.9% were APOE ɛ4 allele positive (close to population normal), whereas in the NCFD, ACS, SCD, and MCI group 54.9% (p < 0.001), 37.5% (p = n.s.), 43.5% (p < 0.01), and 52.4% (p < 0.001) were APOE ɛ4 allele positive, respectively when compared to the NC group.
When the symptom subjects were stratified based on family history of dementia, there were 74 SCD subjects without and 90 SCD subjects with family history and 95 MCI subjects without and 42 MCI subjects with family history. There were no significant differences in age, gender, cognitive test results, APOE positivity, or CSF Aβ42 positivity when SCD subjects with and without family history or MCI subjects with and without family history were compared (data not shown).
In the cognitive symptom group (SCD+MCI) 23.9% of self-referred cases had pAβ, compared to 36.7% in the group referred from memory clinics, which is significantly higher fractions than the NC group (p < 0.01 and p < 0.001, respectively). Similarly, in this self-referred group 44.4% were APOE ɛ4 allele positive and 63.3% of those referred to memory clinics, which is significantly more than in the NC group (p < 0.01 and p < 0.001, respectively) (Table 2).
Comparing the two recruitment sources, there was a significantly higher percentage of CSF pAβ and APOE ɛ4 allele carriers in the group referred to memory clinic compared to self-referred subjects (p < 0.05 and p < 0.01, respectively). There were also significant differences in age (p < 0.05), education level (p < 0.01), MMSE (p < 0.05), and fractions of SCD and MCI (p < 0.05) between these two groups.
DISCUSSION
The DDI project examines incipient disease activity and AD risk factors in pre-dementia and presumptive pre-disease patient and control cohorts. Here, we compared our recruitment strategies to find optimal cohorts consisting of pre-disease at-risk cases and pre-dementia cases with signs of amyloid deposition (Table 2). Compared to NC, we found increased pAβ frequencies in both the self-referred and memory clinic-referred groups. Age did not differ significantly between these groups, and relevant patient concern connected to underlying pathology may have been a factor driving recruitment also in the self-referred group.
As expected, frequencies of pAβ differed significantly between the control groups and SCD cases compared to MCI cases. Furthermore, while self- and memory clinic-referrals frequently had pAβ, the group referred to memory clinics had clearly higher proportions than the self-referral group and are preferred as a source for secondary prevention treatment trials. Controls with first-degree relatives with dementia did not have an increased fraction of pAβ, but harbored an increased frequency of risk APOE ɛ4 alleles compared to controls without. Self-referred cases were also at-risk, enriched for APOE ɛ4 alleles, and pAβ negative. Thus, the majority among of SCD cases, self-referred cases and NCFD are suitable for primary intervention.
These differences in pAβ fractions were not mirrored in APOE ɛ4 allele frequencies. Both the self-referred group and the group referred to memory clinics had significantly higher frequencies, as had the control group with first-degree relatives with dementia indicating that all these groups are at increased risk for AD dementia. Though MCI cases had significantly lower MMSE-scores (and a higher age), APOE ɛ4 allele frequencies were not significantly different compared to SCD and first degree-relative control cases (data not shown). APOE ɛ4 allele frequency was higher in our control groups overall than expected from published Norwegian population frequencies established from healthy blood donors (22.0% compared to 19.8% allele frequency). The NC group (without first degree relatives with dementia) had a lower APOE ɛ4 allele frequency than previously published for an equivalent control group (10.5% versus 14.3%), but close to the frequency in our orthopedic-surgery control cases (11.5%), suggesting that this level may be realistic for the present population [35, 36].
To some extent, inclusion strategies and selection of cutoff levels will always cause bias. The present CSF Aβ42 cutoff gives the best fit to flutemetamol PET in (Kalheim et al., in submission). Highly standardized pre-analytical procedures and laboratory handling procedures may have contributed to numerically high values. Clinical follow-up is necessary for ultimate evaluation of CSF Aβ cutoff levels with the highest predictive power for dementia. Herein, and for other research purposes (inclusion of patients for longitudinal studies or for secondary intervention), a relatively high cutoff allowing for sensitive inclusion of patients at risk (few false negatives) may be beneficial, whereas for clinical purposes fewer false positives may be optimal.
One limitation of this study is the specific mention of first-degree relatives in the advertisements. As such, the present control group has a larger proportion of subjects with first-degree relatives with dementia. Because of this, we have split the control group and this has enabled us to specifically examine first-degree relatives.
In summary, symptomatic cases, both self- and memory clinic-referrals, harbor an increased proportion of cases with incipient AD pathology compared to the normal control group without first-degree relatives with dementia. Controls with first-degree relatives with dementia do not have an increased fraction of pAβ, but harbor an increased frequency of risk APOE ɛ4 alleles. There were significantly higher fractions of subjects with pAβ, APOE ɛ4 allele carriers and MCI subjects in the group referred from memory clinic compared to self-referred subjects.
Whereas memory clinic referrals are most enriched for cases amenable for secondary intervention studies, self-referred cases are enriched for at-risk, pAβ negative cases, putatively suitable for primary intervention.
ACKNOWLEDGMENTS
The project was funded by Norwegian Research Council, NASATS (Dementia Disease Initiation) and the JPND (APGeM) and funding from the regional health authority (Helse Sør-Øst).
We thank Claus Albretsen, Elisabeth Gundersen, Mari Thoresen Løkholm, Line Sæther, Erna Utnes, Marianne Wettergreen, Berglind Gisladottir, Marit Knapstad, Reidun Meling, and Synnøve Bremer Skarpenes for clinical examinations and essential help with the project.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/17-0231r1).
REFERENCES
[1] | Eckerstrom M , Berg AI , Nordlund A , Rolstad S , Sacuiu S , Wallin A ((2016) ) High prevalence of stress and low prevalence of Alzheimer disease CSF biomarkers in a clinical sample with subjective cognitive impairment. Dement Geriatr Cogn Disord 42: , 93–105. |
[2] | Buchhave P , Minthon L , Zetterberg H , Wallin AK , Blennow K , Hansson O ((2012) ) Cerebrospinal fluid levels of beta-amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch Gen Psychiatry 69: , 98–106. |
[3] | Sperling R , Mormino E , Johnson K ((2014) ) The evolution of preclinical Alzheimer’s disease: Implications for prevention trials. Neuron 84: , 608–622. |
[4] | Begum A , Morgan C , Chiu CC , Tylee A , Stewart R ((2012) ) Subjective memory impairment in older adults: Aetiology, salience and help seeking. Int J Geriatr Psychiatry 27: , 612–620. |
[5] | Jessen F , Amariglio RE , van Boxtel M , Breteler M , Ceccaldi M , Chetelat G , Dubois B , Dufouil C , Ellis KA , van der Flier WM , Glodzik L , van Harten AC , de Leon MJ , McHugh P , Mielke MM , Molinuevo JL , Mosconi L , Osorio RS , Perrotin A , Petersen RC , Rabin LA , Rami L , Reisberg B , Rentz DM , Sachdev PS , de la Sayette V , Saykin AJ , Scheltens P , Shulman MB , Slavin MJ , Sperling RA , Stewart R , Uspenskaya O , Vellas B , Visser PJ , Wagner M , Subjective Cognitive Decline Initiative Working Group ((2014) ) A conceptual framework for research on subjective cognitive decline in preclinical Alzheimer’s disease. Alzheimers Dement 10: , 844–852. |
[6] | Winblad B , Palmer K , Kivipelto M , Jelic V , Fratiglioni L , Wahlund LO , Nordberg A , Backman L , Albert M , Almkvist O , Arai H , Basun H , Blennow K , de Leon M , DeCarli C , Erkinjuntti T , Giacobini E , Graff C , Hardy J , Jack C , Jorm A , Ritchie K , van Duijn C , Visser P , Petersen RC ((2004) ) Mild cognitive impairment–beyond controversies, towards a consensus: Report of the International Working Group on Mild Cognitive Impairment. J Intern Med 256: , 240–246. |
[7] | Jansen WJ , Ossenkoppele R , Knol DL , Tijms BM , Scheltens P , Verhey FR , Visser PJ , Amyloid Biomarker Study G, Aalten P , Aarsland D , Alcolea D , Alexander M , Almdahl IS , Arnold SE , Baldeiras I , Barthel H , van Berckel BN , Bibeau K , Blennow K , Brooks DJ , van Buchem MA , Camus V , Cavedo E , Chen K , Chetelat G , Cohen AD , Drzezga A , Engelborghs S , Fagan AM , Fladby T , Fleisher AS , van der Flier WM , Ford L , Forster S , Fortea J , Foskett N , Frederiksen KS , Freund-Levi Y , Frisoni GB , Froelich L , Gabryelewicz T , Gill KD , Gkatzima O , Gomez-Tortosa E , Gordon MF , Grimmer T , Hampel H , Hausner L , Hellwig S , Herukka SK , Hildebrandt H , Ishihara L , Ivanoiu A , Jagust WJ , Johannsen P , Kandimalla R , Kapaki E , Klimkowicz-Mrowiec A , Klunk WE , Kohler S , Koglin N , Kornhuber J , Kramberger MG , Van Laere K , Landau SM , Lee DY , de Leon M , Lisetti V , Lleo A , Madsen K , Maier W , Marcusson J , Mattsson N , de Mendonca A , Meulenbroek O , Meyer PT , Mintun MA , Mok V , Molinuevo JL , Mollergard HM , Morris JC , Mroczko B , Van der Mussele S , Na DL , Newberg A , Nordberg A , Nordlund A , Novak GP , Paraskevas GP , Parnetti L , Perera G , Peters O , Popp J , Prabhakar S , Rabinovici GD , Ramakers IH , Rami L , Resende de Oliveira C , Rinne JO , Rodrigue KM , Rodriguez-Rodriguez E , Roe CM , Rot U , Rowe CC , Ruther E , Sabri O , Sanchez-Juan P , Santana I , Sarazin M , Schroder J , Schutte C , Seo SW , Soetewey F , Soininen H , Spiru L , Struyfs H , Teunissen CE , Tsolaki M , Vandenberghe R , Verbeek MM , Villemagne VL , Vos SJ , van Waalwijk van Doorn LJ , Waldemar G , Wallin A , Wallin AK , Wiltfang J , Wolk DA , Zboch M , Zetterberg H ((2015) ) Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA 313: , 1924–1938. |
[8] | Morris JC , Roe CM , Xiong C , Fagan AM , Goate AM , Holtzman DM , Mintun MA ((2009) ) APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol 67: , 122–131. |
[9] | Grambaite R , Reinvang I , Selnes P , Fjell AM , Walhovd KB , Stenset V , Fladby T ((2011) ) Pre-dementia memory impairment is associated with white matter tract affection. J Int Neuropsychol Soc 17: , 143–153. |
[10] | Villemagne VL , Burnham S , Bourgeat P , Brown B , Ellis KA , Salvado O , Szoeke C , Macaulay SL , Martins R , Maruff P , Ames D , Rowe CC , Masters CL , Australian Imaging Biomarkers and Lifestyle Research Group ((2013) ) Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol 12: , 357–367. |
[11] | Hardy J , Allsop D ((1991) ) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12: , 383–388. |
[12] | Villegas-Llerena C , Phillips A , Garcia-Reitboeck P , Hardy J , Pocock JM ((2016) ) Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr Opin Neurobiol 36: , 74–81. |
[13] | Jonsson T , Atwal JK , Steinberg S , Snædal J , Jonsson PV , Björnsson S , Stefansson H , Sulem P , Gudbjartsson D , Maloney J , Hoyte K , Gustafson A , Liu Y , Lu Y , Bhangale T , Graham RR , Huttenlocher J , Bjornsdottir G , Andreassen OA , Jönsson EG , Palotie A , Behrens TW , Magnusson OT , Kong A , Thorsteinsdottir U , Watts RJ , Stefansson K ((2012) ) A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488: , 96–99. |
[14] | Karch CM , Goate AM ((2015) ) Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry 77: , 43–51. |
[15] | Zhu JB , Tan CC , Tan L , Yu JT ((2017) ) State of play in Alzheimer’s disease genetics. J Alzheimers Dis 58: , 631–659. |
[16] | Naj AC , Jun G , Reitz C , Kunkle BW , Perry W , Park YS , Beecham GW , Rajbhandary RA , Hamilton-Nelson KL , Wang LS , Kauwe JS , Huentelman MJ , Myers AJ , Bird TD , Boeve BF , Baldwin CT , Jarvik GP , Crane PK , Rogaeva E , Barmada MM , Demirci FY , Cruchaga C , Kramer PL , Ertekin-Taner N , Hardy J , Graff-Radford NR , Green RC , Larson EB , St George-Hyslop PH , Buxbaum JD , Evans DA , Schneider JA , Lunetta KL , Kamboh MI , Saykin AJ , Reiman EM , De Jager PL , Bennett DA , Morris JC , Montine TJ , Goate AM , Blacker D , Tsuang DW , Hakonarson H , Kukull WA , Foroud TM , Martin ER , Haines JL , Mayeux RP , Farrer LA , Schellenberg GD , Pericak-Vance MA , Albert MS , Albin RL , Apostolova LG , Arnold SE , Barber R , Barnes LL , Beach TG , Becker JT , Beekly D , Bigio EH , Bowen JD , Boxer A , Burke JR , Cairns NJ , Cantwell LB , Cao C , Carlson CS , Carney RM , Carrasquillo MM , Carroll SL , Chui HC , Clark DG , Corneveaux J , Cribbs DH , Crocco EA , DeCarli C , DeKosky ST , Dick M , Dickson DW , Duara R , Faber KM , Fallon KB , Farlow MR , Ferris S , Frosch MP , Galasko DR , Ganguli M , Gearing M , Geschwind DH , Ghetti B , Gilbert JR , Glass JD , Growdon JH , Hamilton RL , Harrell LE , Head E , Honig LS , Hulette CM , Hyman BT , Jicha GA , Jin LW , Karydas A , Kaye JA , Kim R , Koo EH , Kowall NW , Kramer JH , LaFerla FM , Lah JJ , Leverenz JB , Levey AI , Li G , Lieberman AP , Lin CF , Lopez OL , Lyketsos CG , Mack WJ , Martiniuk F , Mash DC , Masliah E , McCormick WC , McCurry SM , McDavid AN , McKee AC , Mesulam M , Miller BL , Miller CA , Miller JW , Murrell JR , Olichney JM , Pankratz VS , Parisi JE , Paulson HL , Peskind E , Petersen RC , Pierce A , Poon WW , Potter H , Quinn JF , Raj A , Raskind M , Reisberg B , Ringman JM , Roberson ED , Rosen HJ , Rosenberg RN , Sano M , Schneider LS , Seeley WW , Smith AG , Sonnen JA , Spina S , Stern RA , Tanzi RE , Thornton-Wells TA , Trojanowski JQ , Troncoso JC , Valladares O , Van Deerlin VM , Van Eldik LJ , Vardarajan BN , Vinters HV , Vonsattel JP , Weintraub S , Welsh-Bohmer KA , Williamson J , Wishnek S , Woltjer RL , Wright CB , Younkin SG , Yu CE , Yu L ((2014) ) Effects of multiple genetic loci on age at onset in late-onset Alzheimer disease: A genome-wide association study. JAMA Neurol 71: , 1394–1404. |
[17] | Liu-Seifert H , Siemers E , Holdridge KC , Andersen SW , Lipkovich I , Carlson C , Sethuraman G , Hoog S , Hayduk R , Doody R , Aisen P ((2015) ) Delayed-start analysis: Mild Alzheimer’s disease patients in solanezumab trials, 3.5 years. Alzheimers Dement Trans Res Clin Interv 1: , 111–121. |
[18] | Sevigny J , Chiao P , Bussiere T , Weinreb PH , Williams L , Maier M , Dunstan R , Salloway S , Chen T , Ling Y , O’Gorman J , Qian F , Arastu M , Li M , Chollate S , Brennan MS , Quintero-Monzon O , Scannevin RH , Arnold HM , Engber T , Rhodes K , Ferrero J , Hang Y , Mikulskis A , Grimm J , Hock C , Nitsch RM , Sandrock A ((2016) ) The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 537: , 50–56. |
[19] | Rowe CC , Ellis KA , Rimajova M , Bourgeat P , Pike KE , Jones G , Fripp J , Tochon-Danguy H , Morandeau L , O’Keefe G , Price R , Raniga P , Robins P , Acosta O , Lenzo N , Szoeke C , Salvado O , Head R , Martins R , Masters CL , Ames D , Villemagne VL ((2010) ) Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 31: , 1275–1283. |
[20] | Randall C , Mosconi L , de Leon M , Glodzik L ((2013) ) Cerebrospinal fluid biomarkers of Alzheimer’s disease in healthy elderly. Front Biosci (Landmark Ed) 18: , 1150–1173. |
[21] | Albert MS , DeKosky ST , Dickson D , Dubois B , Feldman HH , Fox NC , Gamst A , Holtzman DM , Jagust WJ , Petersen RC , Snyder PJ , Carrillo MC , Thies B , Phelps CH ((2011) ) The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 270–279. |
[22] | Mitchell AJ , Bird V , Rizzo M , Meader N ((2010) ) Diagnostic validity and added value of the Geriatric Depression Scale for depression in primary care: A meta-analysis of GDS30 and GDS15. J Affect Disord 125: , 10–17. |
[23] | Heaton RK ((2004) ) Revised Comprehensive Norms for an Expanded Halstead-Reitan Battery: Demographically Adjusted Neuropsychological Norms for African American and Caucasian Adults: Professional Manual, Psychological Assessment Resources. |
[24] | Folstein MF , Folstein SE , McHugh PR ((1975) ) “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12: , 189–198. |
[25] | Shulman KI ((2000) ) Clock-drawing: Is it the ideal cognitive screening test? Int J Geriatr Psychiatry 15: , 548–561. |
[26] | Fillenbaum GG , van Belle G , Morris JC , Mohs RC , Mirra SS , Davis PC , Tariot PN , Silverman JM , Clark CM , Welsh-Bohmer KA , Heyman A ((2008) ) Consortium to Establish a Registry for Alzheimer’s Disease (CERAD): The first twenty years. Alzheimers Dement 4: , 96–109. |
[27] | Benton AL , Hamsher KD ((1989) ) Multilingual Aphasia Examination, AJA Associates, Iowa City. |
[28] | McKhann GM , Knopman DS , Chertkow H , Hyman BT , Jack CR Jr , Kawas CH , Klunk WE , Koroshetz WJ , Manly JJ , Mayeux R , Mohs RC , Morris JC , Rossor MN , Scheltens P , Carrillo MC , Thies B , Weintraub S , Phelps CH ((2011) ) The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 263–269. |
[29] | Warrington EK , James M ((1991) ) The Visual Object and Space Perception Battery, Thames Valley Test Company, Bury St Edmunds, England. |
[30] | Reitan RM , Wolfson D ((1985) ) The Halstead-Reitan Neuropsychological Test Battery, Neuropsychology Press, Tucson. |
[31] | Sotaniemi M , Pulliainen V , Hokkanen L , Pirttila T , Hallikainen I , Soininen H , Hanninen T ((2012) ) CERAD-neuropsychological battery in screening mild Alzheimer’s disease. Acta Neurol Scand 125: , 16–23. |
[32] | Hughes CP , Berg L , Danziger WL , Coben LA , Martin RL ((1982) ) A new clinical scale for the staging of dementia. Br J Psychiatry 140: , 566–572. |
[33] | Petersen RC ((2004) ) Mild cognitive impairment as a diagnostic entity. J Intern Med 256: , 183–194. |
[34] | Almdahl IS , Lauridsen C , Selnes P , Kalheim LF , Coello C , Gajdzik B , Moller I , Wettergreen M , Grambaite R , Bjornerud A , Brathen G , Sando SB , White LR , Fladby T ((2017) ) Cerebrospinal fluid levels of amyloid beta 1-43 mirror 1-42 in relation to imaging biomarkers of Alzheimer’s disease. Front Aging Neurosci 9: , 9. |
[35] | Kumar T , Liestol K , Maehlen J , Hiorth A , Jettestuen E , Lind H , Brorson SH ((2002) ) Allele frequencies of apolipoprotein E gene polymorphisms in the protein coding region and promoter region (-491A/T) in a healthy Norwegian population. Hum Biol 74: , 137–142. |
[36] | Sando SB , Melquist S , Cannon A , Hutton ML , Sletvold O , Saltvedt I , White LR , Lydersen S , Aasly JO ((2008) ) APOE epsilon 4 lowers age at onset and is a high risk factor for Alzheimer’s disease; a case control study from central Norway. BMC Neurol 8: , 9. |

