
Neurofibrillary Tangles of Aβx-40 in Alzheimer’s Disease Brains
Abstract
The two pathognomonic lesions in the brain of AD patients are senile plaques and intraneuronal neurofibrillary tangles (NFT). Previous studies have demonstrated that amyloid-β (Aβ) is a component of both senile plaques and NFTs, and have showed that intracellular accumulation of Aβ is toxic for cells and precedes the appearance of extracellular amyloid deposits. Here we report that there are numerous intraneuronal NFT and extraneuronal NFT immunoreactive for Aβx-40 in which there is no co-localization with tau staining suggesting the existence of two different neurodegenerating populations associated with the intracellular accumulation of either tau protein or Aβx-40 in AD.
Alzheimer’s disease (AD) is a neurodegenerative disease characterized by the progressive and irreversible destruction of neurons in the cerebral cortex. There are two pathognomonic lesions in the brain of AD patients: senile plaques, composed mainly of extracellular aggregates of amyloid-β peptides (Aβ) [1], and intraneuronal neurofibrillary tangles (NFT), consisting of paired helical filaments (PHF) [2] composed primarily of hyperphosphorylated tau protein [3, 4]. The relationship between these processes is uncertain and still not completely understood. The existence of intraneuronal Aβ has been known for many years. Masters and collaborators published in 1985 that Aβ, initially termed amyloid A4, is deposited first intraneuronally as NFT and subsequently in the extracellular space, associated with senile plaques and blood vessels [5]. Later on, the immunolabeling of most intraneuronal NFT (iNFT) and extraneuronal NFT (eNFT) with anti-Aβ antibodies was replicated in several laboratories [6–8]. However, other studies failed to find Aβ immunolabeling associated with iNFT [9–11] or with both iNFT and eNFT [12].
Later on, immunostaining with specific antibodies against the C-terminal fragment of either Aβ40 or Aβ42 (Aβx-40 or Aβx- 42, respectively) enabled the visualization of Aβ42 associated with iNFT where it was seen to collocalize with tau, whereas Aβ40 did not associate at all or to a far lesser degree [13–15]. Additionally, it was reported that numerous eNFT appeared stained for Aβx-40, which was interpreted as a secondary deposition over remnants of iNFT exposed in brain tissue once the cells died; however, evidence of colocalization of Aβx-40 with tau protein in these eNFT was lacking [16].
Studies in cell cultures have shown that both Aβ42 and Aβ40 can be intracellularly produced in neurons (but apparently not in other types of cells) [17, 18], mainly in the trans-Golgi network, although production of Aβ42 is observed as well in the endoplasmic reticulum [19]. Numerous studies have shown that the intraneuronal accumulation of Aβ may be toxic and precedes its extracellular deposition both in AD brains and in transgenic mice models of the disease [14, 20–25]. Interestingly, intraneuronal accumulation of either Aβ40 or Aβ42 has been described in brains of Down syndrome patients aged less than three years [20, 26, 27]. Furthermore, it has been found that intracellular Aβ deposition precedes the appearance of immunoreactive tau iNFT [20, 28] which subsequently were found associated with Aβ42 in neurites and synapses [15]. These findings led to a modification of the amyloid cascade hypothesis in which the first step of the cascade is the intraneuronal accumulation of Aβ [29].
In this work, we report the finding of numerous Aβx-40 positive iNFT and eNFT in the entorhinal, hippocampal and, to a lesser extent, the parietal cortex of AD brains. The Aβx-40 staining did not co-localize with tau staining in the same NFTs suggesting that there may be two different neurodegenerating pathways in AD brains leading to the intraneuronal accumulation of either tau or Aβx-40.
Paraffin embedded sections from the entorhinal, hippocampus, and parietal cortex of patients (both genders, age range 36-97) diagnosed histopathologically with AD Braak & Braak (B&B; [30]) stages II-III, IV, and VI (N = 3 from each stage), as well as from two Down syndrome (DS) patients with AD (B&B stage VI; N = 2) and from 4 healthy control brains, were immunohistochemically analyzed (for demographic characteristic, see Supplementary Table 1). The tissues were obtained from the Neurological Tissue Bank of the Clinic Hospital at the University of Barcelona in accordance with the Helsinki Declaration of 1975.
Brain sections were deparaffinized incubated for 3 minutes in 90% formic acid for antigen retrieval (except for the AT8 antibody in which a microwave was used) and quenched for endogenous peroxidase with 3.3% hydrogen peroxide and 30% methanol. The antibodies used were: SAR-022 (anti-Aβx-40 polyclonal antibody; Araclon Biotech, Zaragoza) at 1 : 500; SAR-031 (anti-Aβx- 42 polyclonal antibody; Araclon Biotech, Zaragoza) at 1 : 2000; 6E10 monoclonal antibody (anti-N-terminal region of Aβ; Covance, London) at 1 : 2000; 4G8 monoclonal antibody (anti-central region of Aβ; Covance, London) at 1 : 1000; and AT8 (anti-Phospho-PHF-tau pSer202+Thr205; ThermoFisher Scientific, Waltham) at 1 : 200. The anti Aβx-40 and anti Aβx- 42 antibodies were purified by antigen-affinity chromatography using the same Aβ fragment inoculated to the rabbits (Aβ33–40 and Aβ35–42, respectively). As described elsewhere, they have practically no cross-reactivity (less than 1%) between their target Aβ species nor with Aβ1–38 (<2%), Aβ1–43 (<1%) [31].
After overnight incubation with each primary antibody and subsequent washing, the sections were incubated for 45 min in either goat anti-rabbit at 1 : 200 (Sigma, St. Louis) or goat anti-mouse at 1 : 100 (Sigma, St. Louis) for poly- or monoclonal primary antibodies, respectively; followed by 45 min incubation in avidin-biotin (Vectastain ABC kit, Vector lab, Burlingame) and then developed with diaminobencidine as chromogen. Selected sections were double immunofluorescence stained with anti-Aβx-40 and AT8 antibodies followed by rodamine-goat anti-rabbit at 1 : 100 (Sigma, St. Louis) or fluorescein-goat anti-mouse at 1 : 100 (Sigma, St. Louis). Specificity of the anti-Aβx-40 labeling was assessed by overnight preadsorption of the primary antibody with Aβ1 -40 peptide (10e-5 M).
The section labeled with the anti-Aβx–40 antibody presented (in addition to positively stained diffuse and senile plaques) numerous Aβx–40 positive NFTs. These Aβ-NFTs were intensely stained and appeared very similar to the classic tau-NFTs. The Aβ-NFTs were localized in the entorhinal cortex, mainly in the cell islands of layer II, but also in deeper cortical layers (Fig. 1A); in the hippocampus, including dentate gyrus, fields CA1-CA3, and in the subiculum (Fig. 1B) of all the patients analyzed (Braak &Braak stages II-III, IV, and VI) and, to a lesser extent, dispersed within the parietal cortex of the more advanced cases of AD. No Aβ-NFTs were observed in the preparations from the healthy controlbrains.
Fig.1
A-F) Aβx-40 immunostaining of AD brain sections (B&B stage VI). A) Clusters of Aβx-40 positive NFT are seen in layer II and scattered in deeper layers of entorhinal cortex together with slightly stained senile plaques (arrow heads). B) In the hippocampal formation, Aβx-40 positive NFT appeared mainly in the CA1-CA3 fields and in the dentate gyrus. C) Aβx-40 positive iNFT in the hippocampus (arrow heads). D) Aβx-40 eNFT and iNFT (arrow heads) in the entorhinal cortex. E, F) The sections reacted with 6E10 (E) or 4G8 (F) showing numerous senile plaques but no NFT; asterisks are in the same blood vessel for reference.
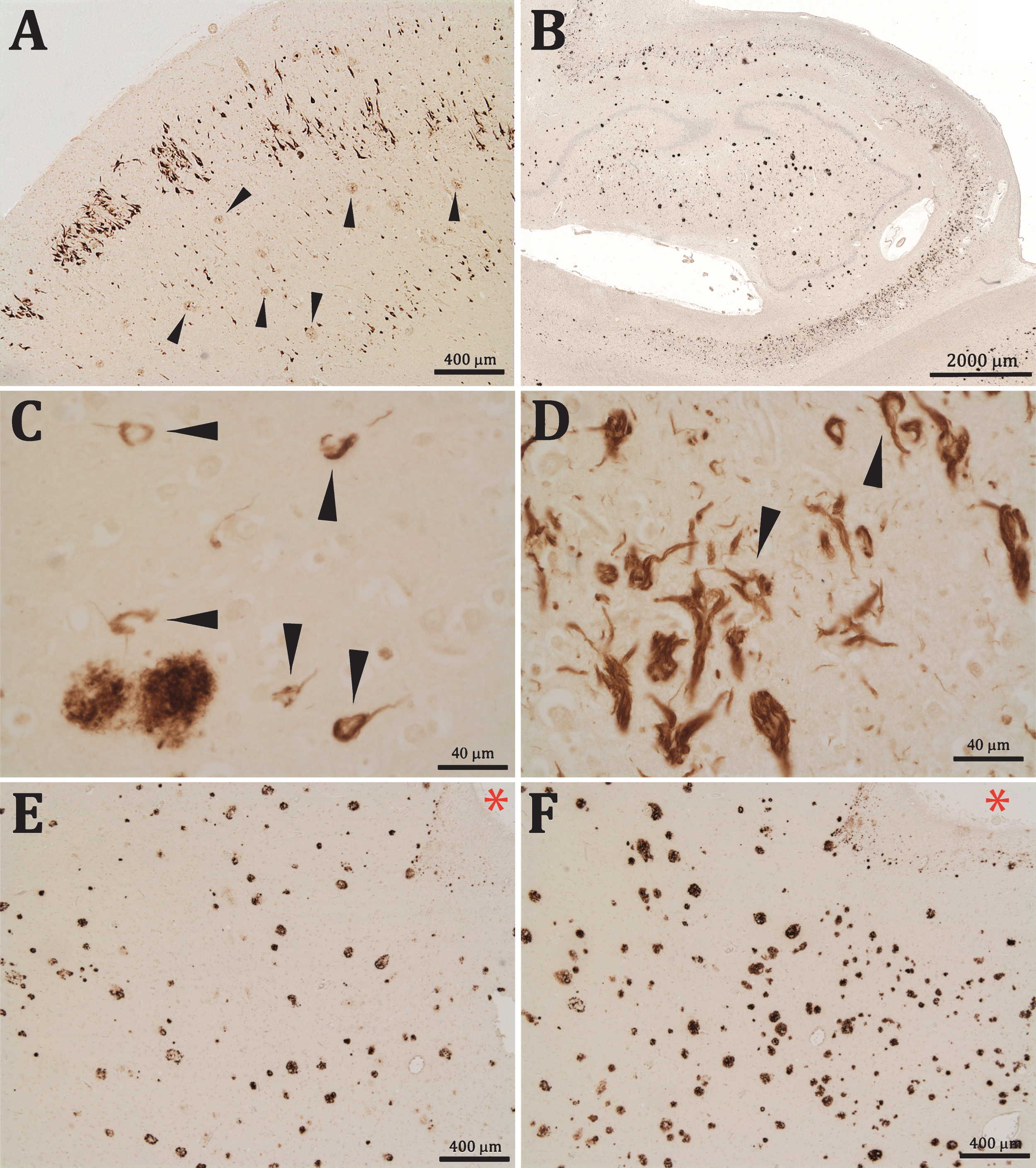
Aβx-40 positive labeling was seen in the cytoplasm of apparently functional neurons as iNFTs (Fig. 1C, D), but labeling of eNFT was more frequently seen, particularly in advanced AD stages (Fig. 1D). Many of these eNFTs maintained the shape of neurons, although they were normally larger than their unlabeled counterparts, which suggested that these profiles could represent an additional Aβx-40 deposition once the parental neurons have lost their plasmatic membrane, leaving the NFTs exposed in the neuropil.
In contrast to the intense labelling obtained with the anti-Aβx-40 antibody, we did not see NFT labelling with any of the anti-Aβ monoclonal antibodies that target the N-terminal or central parts of the Aβ peptides, which indicated that Aβ-NFTs were composed of Aβ C-terminal fragments (Fig. 1E, F). Very occasionally we found similar iNFTs and eNFTs marked with the anti-Aβ42 antibody (not shown).
Aβ-NFTs were also found in the brain of a DS patient who had been diagnosed with severe dementia of Alzheimer type. Interestingly, these Aβx-40 positive profiles were almost completely lacking in the other non-demented DS patient, in spite of both having been classified as B&B stage VI and both presented extensive Aβ42 burden (Fig. 2A-D).
Fig.2
Contiguous entorhinal sections from two Down syndrome patients, one diagnosed with severe dementia of the Alzheimer type (A-B) and other non-demented (C-D); asterisks are in the same blood vessels for reference. Sections were reacted for Aβx-40 (A and C) and Aβx- 42 (B and D). Note that whereas both brains presented numerous Aβx- 42 positive profiles, Aβx-40 positive profiles were almost completely lacking in the non-demented patient. Preadsorption of the first antibody with the immunogenic peptide resulted in a complete absence of the Aβx-40 labeling in F with regard to the contiguous positively stained section in E; asterisks are in the same blood vessels for reference. Sections A, E, and F were slightly stained with cresyl violet.
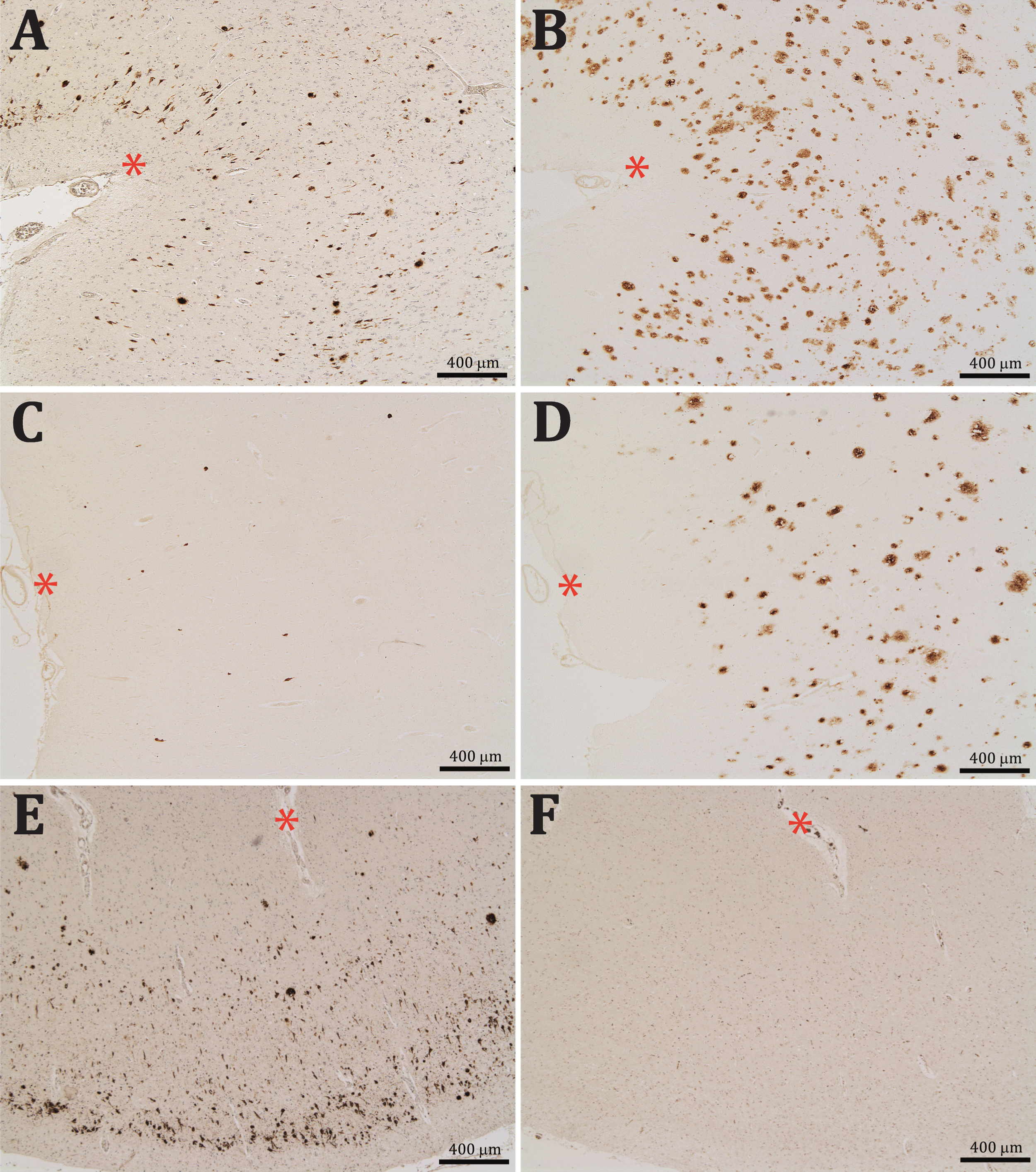
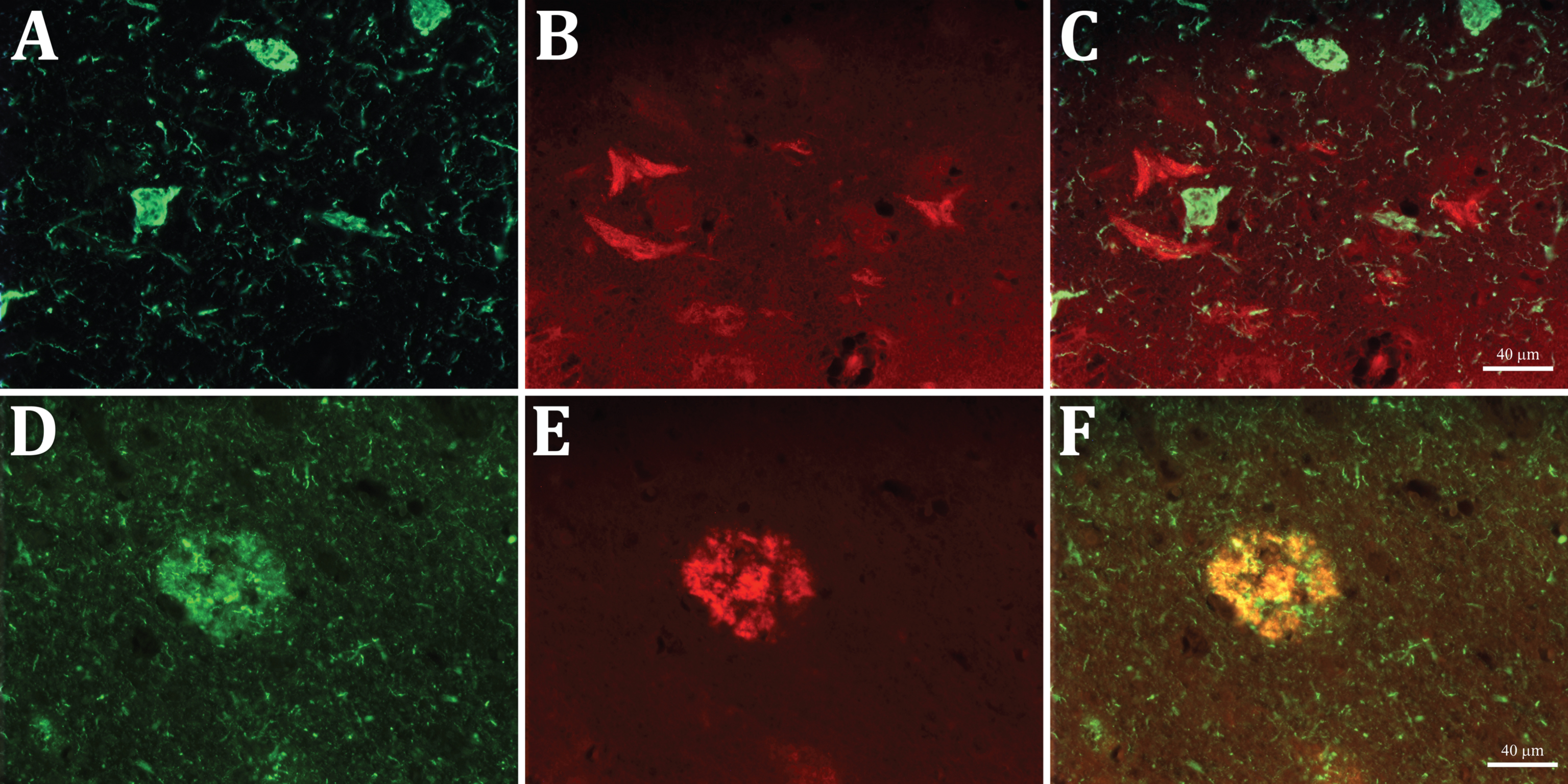
The preadsorption of the anti-Aβx-40 antibody with Aβ1 -40 led to the complete disappearance of staining in both the senile plaques and Aβ-NFTs, demonstrating that the labeling is due to the presence of the Aβ40 C-terminal fragments specifically recognized by our Aβx-40 antibody (Fig. 2E, F). Furthermore, double immunofluorescent staining showed that the epitopes for the anti-Aβ40 and AT8 antibodies do not colocalize on the same neurons, whereas they positively colocalize on the diffuse and senile plaques (Fig. 3A-F). These findings allowed us to discard the possibility of that the labelling of Aβ-NFTs was due to a cross-reactivity of our antibody with the tau-NFTs. Thus, two populations of degenerating neurons could be detected and differentiated, one filled with tau and the other with C-terminal fragments of Aβ40.
Fig.3
Double immunofluorescent staining with AT8 anti-tau (green, A and D) and anti-Aβx-40 (red, B and E) antibodies in entorhinal cortex sections. Note that the two labels do not co-localize in the NFT profiles nor in the short threads in A and D (merged in C), whereas both co-localize in the senile plaques (merged in F).
An exaggeratedly antagonistic vision of the two hallmark lesions in AD (extracellular senile plaques mainly containing Aβ and intraneuronal NFTs composed of hyperphosphorylated tau protein) has crystallized into the idea that Aβ triggers neurodegeneration from the outside of cells and a subsequent tau accumulation damages them from inside. This overly simplistic vision disregards pioneers’ results demonstrating that Aβ is a component of both senile plaques and NFTs [5, 8, 9, 32] and also more recent reports showing that intracellular accumulation of Aβ is toxic for cells and precedes the appearance of extracellular amyloid deposits [14, 20, 22–27, 29]. The presence of Aβx-40 immunoreactivity in the eNFTs as described in the present work, has been known for a long time [13, 16], yet contradictory results have also been reported [14, 20]. These discrepancies suggest that not all anti-Aβ40 specific antibodies are able to recognize the Aβ40 C-terminal fragments present in the NFT, although they could also be due to differences in the processing of brain sections[8, 33, 34].
Since the majority of the eNFTs found in those studies were Aβ40 positive, whereas, in contrast, very few iNFTs appeared labelled with anti-Aβ40 antibodies, it was assumed that eNFTs resulted from a secondary deposition of extracellular Aβ40 once the neuron had died and lost its nucleus and its plasmatic membrane. Visualization of tau positive eNFTs was not present in these studies because they were predominantly carried out with the Alz-50 antibody, which targets a tau-epitope known to be lost in the eNFTs [10, 32]. However, in the present work we have used the monoclonal AT8 anti-tau antibody, which has allowed us to label both tau positive iNFT and eNFT, in accordance with previous reports [35]. Interestingly, the Aβx-40 staining does not co-localize with the tau staining in the same NFT, while it does co-localize in the diffuse and senile plaques. Thus, two populations of degenerating neurons can be differentiated in a brain with AD, suggesting the existence of at least two different neurodegenerating pathways leading to the intraneuronal accumulation of either tau or Aβx-40.
Nevertheless, further experiments analyzing more brains, a larger panel of antibodies targeting not only Aβ40 and phosphorylated tau but also native tau, the ultrastructural characterization of Aβx-40 NFTs and their timeline throughout the disease process, will be necessary to test this hypothesis.
In spite of its limitations, the present work comments on the role of Aβ40 in AD which is also underlined by our finding of numerous Aβx-40 positive profiles in the brain of a Down syndrome patient with severe dementia while these profiles were almost completely lacking in the brain of a non-demented Down syndrome patient. Although there are controversial reports concerning which Aβ species accumulates intracellularly in DS patients, our result is congruent with previous studies showing an increase of Aβ40 immunoreactive profiles in the brains of Down syndrome patients from 30 years old onwards, when neuropathology rapidly accumulates until it reaches levels sufficient for a diagnosis of AD in most of these patients (for a recent review, see [36]). Currently, it is generally accepted that Aβ42 may be of particular relevance in triggering AD because it is more hydrophobic and prone to aggregate in vitro than Aβ40 [37]. However, it should be considered that the aggregating behavior of the various Aβ species could be different in vivo than in vitro [38] and, in this line, it is also relevant to underline some studies pointing to the relevance of Aβ40 in AD[39, 40].
In conclusion, the present work underlines the possible role for Aβ40 in AD physiopathology as our results have shown the existence of two different neurodegenerating populations alternativelyassociated with the intracellular accumulation of either tau protein or Aβx-40.
ACKNOWLEDGMENTS
This work has been financed by Araclon Biotech Ltd.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/17-0163r1).
Appendices
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-170163.
REFERENCES
[1] | Glenner GG , Wong CW , Quaranta V , Eanes ED ((1984) ) The amyloid deposits in Alzheimer’s disease: Their nature and pathogenesis. Appl Pathol 2: , 357–369. |
[2] | Mann DM ((1985) ) The neuropathology of Alzheimer’s disease: A review with pathogenetic, aetiological and therapeutic considerations. Mech Ageing Dev 31: , 213–255. |
[3] | Kosik KS , Joachim CL , Selkoe DJ ((1986) ) Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci U S A 83: , 4044–4048. |
[4] | Ballatore C , Lee VM , Trojanowski JQ ((2007) ) Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci 8: , 663–672. |
[5] | Masters CL , Multhaup G , Simms G , Pottgiesser J , Martins RN , Beyreuther K ((1985) ) Neuronal origin of a cerebral amyloid: Neurofibrillary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J 4: , 2757–2763. |
[6] | Spillantini MG , Goedert M , Jakes R , Klug A ((1990) ) Different configurational states of beta-amyloid and their distributions relative to plaques and tangles in Alzheimer disease. Proc Natl Acad Sci U S A 87: , 3947–3951. |
[7] | Spillantini MG , Goedert M , Jakes R , Klug A ((1990) ) Topographical relationship between beta-amyloid and tau protein epitopes in tangle-bearing cells in Alzheimer disease. Proc Natl Acad Sci U S A 87: , 3952–3956. |
[8] | Perry G , Cras P , Siedlak SL , Tabaton M , Kawai M ((1992) ) Beta protein immunoreactivity is found in the majority of neurofibrillary tangles of Alzheimer’s disease. Am J Pathol 140: , 283–290. |
[9] | Hyman BT , Van Hoesen GW , Beyreuther K , Masters CL ((1989) ) A4 amyloid protein immunoreactivity is present in Alzheimer’s disease neurofibrillary tangles. Neurosci Lett 101: , 352–355. |
[10] | Tabaton M , Cammarata S , Mancardi G , Manetto V , Autilio-Gambetti L , Perry G , Gambetti P ((1991) ) Ultrastructural localization of beta-amyloid, tau, and ubiquitin epitopes in extracellular neurofibrillary tangles. Proc Natl Acad SciU S A 88: , 2098–2102. |
[11] | Yamaguchi H , Nakazato Y , Shoji M , Okamoto K , Ihara Y , Morimatsu M , Hirai S ((1991) ) Secondary deposition of beta amyloid within extracellular neurofibrillary tangles in Alzheimer-type dementia. Am J Pathol 138: , 699–705. |
[12] | Ikeda K , Haga C , Oyanagi S , Iritani S , Kosaka K ((1992) ) Ultrastructural and immunohistochemical study of degenerate neurite-bearing ghost tangles. J Neurol 239: , 191–194. |
[13] | Schwab C , McGeer PL ((2000) ) Abeta42-carboxy-terminal-like immunoreactivity is associated with intracellular neurofibrillary tangles and pick bodies. Exp Neurol 161: , 527–534. |
[14] | Takahashi RH , Milner TA , Li F , Nam EE , Edgar MA , Yamaguchi H , Beal MF , Xu H , Greengard P , Gouras GK ((2002) ) Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol 161: , 1869–1879. |
[15] | Takahashi RH , Capetillo-Zarate E , Lin MT , Milner TA , Gouras GK ((2010) ) Co-occurrence of Alzheimer’s disease beta-amyloid and tau pathologies at synapses. Neurobiol Aging 31: , 1145–1152. |
[16] | Schwab C , Akiyama H , McGeer EG , McGeer PL ((1998) ) Extracellular neurofibrillary tangles are immunopositive for the 40 carboxy-terminal sequence of beta-amyloid protein. J Neuropathol Exp Neurol 57: , 1131–1137. |
[17] | Wertkin AM , Turner RS , Pleasure SJ , Golde TE , Younkin SG , Trojanowski JQ , Lee VM ((1993) ) Human neurons derived from a teratocarcinoma cell line express solely the 695-amino acid amyloid precursor protein and produce intracellular beta-amyloid or A4 peptides. Proc Natl Acad Sci U S A 90: , 9513–9517. |
[18] | Hartmann T , Bieger SC , Bruhl B , Tienari PJ , Ida N , Allsop D , Roberts GW , Masters CL , Dotti CG , Unsicker K , Beyreuther K ((1997) ) Distinct sites of intracellular production for Alzheimer’s disease A beta40/42 amyloid peptides. Nat Med 3: , 1016–1020. |
[19] | Greenfield JP , Tsai J , Gouras GK , Hai B , Thinakaran G , Checler F , Sisodia SS , Greengard P , Xu H ((1999) ) Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer beta-amyloid peptides. Proc Natl Acad Sci U S A 96: , 742–747. |
[20] | Gouras GK , Tsai J , Naslund J , Vincent B , Edgar M , Checler F , Greenfield JP , Haroutunian V , Buxbaum JD , Xu H , Greengard P , Relkin NR ((2000) ) Intraneuronal Abeta42 accumulation in human brain. Am J Pathol 156: , 15–20. |
[21] | Wirths O , Multhaup G , Czech C , Blanchard V , Moussaoui S , Tremp G , Pradier L , Beyreuther K , Bayer TA ((2001) ) Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett 306: , 116–120. |
[22] | Oddo S , Caccamo A , Shepherd JD , Murphy MP , Golde TE , Kayed R , Metherate R , Mattson MP , Akbari Y , LaFerla FM ((2003) ) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 39: , 409–421. |
[23] | Oddo S , Caccamo A , Smith IF , Green KN , LaFerla FM ((2006) ) A dynamic relationship between intracellular and extracellular pools of Abeta. Am J Pathol 168: , 184–194. |
[24] | Knobloch M , Konietzko U , Krebs DC , Nitsch RM ((2007) ) Intracellular Abeta and cognitive deficits precede beta-amyloid deposition in transgenic arcAbeta mice. Neurobiol Aging 28: , 1297–1306. |
[25] | Umeda T , Ramser EM , Yamashita M , Nakajima K , Mori H , Silverman MA , Tomiyama T ((2015) ) Intracellular amyloid beta oligomers impair organelle transport and induce dendritic spine loss in primary neurons. Acta Neuropathol Commun 3: , 51. |
[26] | Gyure KA , Durham R , Stewart WF , Smialek JE , Troncoso JC ((2001) ) Intraneuronal abeta-amyloid precedes development of amyloid plaques in Down syndrome. Arch Pathol Lab Med 125: , 489–492. |
[27] | Mori C , Spooner ET , Wisniewsk KE , Wisniewski TM , Yamaguchi H , Saido TC , Tolan DR , Selkoe DJ , Lemere CA ((2002) ) Intraneuronal Abeta42 accumulation in Down syndrome brain. Amyloid 9: , 88–102. |
[28] | Fernandez-Vizarra P , Fernandez AP , Castro-Blanco S , Serrano J , Bentura ML , Martinez-Murillo R , Martinez A , Rodrigo J ((2004) ) Intra- and extracellular Abeta and PHF in clinically evaluated cases of Alzheimer’s disease. Histol Histopathol 19: , 823–844. |
[29] | Wirths O , Multhaup G , Bayer TA ((2004) ) A modified beta-amyloid hypothesis: Intraneuronal accumulation of the beta-amyloid peptide–the first step of a fatal cascade. J Neurochem 91: , 513–520. |
[30] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[31] | Perez-Grijalba V , Fandos N , Canudas J , Insua D , Casabona D , Lacosta AM , Montanes M , Pesini P , Sarasa M ((2016) ) Validation of immunoassay-based tools for the comprehensive quantification of Abeta40 and Abeta42 peptides in plasma. J Alzheimers Dis 54: , 751–762. |
[32] | Perry G , Richey PL , Siedlak SL , Smith MA , Mulvihill P , DeWitt DA , Barnett J , Greenberg BD , Kalaria RN ((1993) ) Immunocytochemical evidence that the beta-protein precursor is an integral component of neurofibrillary tangles of Alzheimer’s disease. Am J Pathol 143: , 1586–1593. |
[33] | D’Andrea MR , Nagele RG , Wang HY , Lee DH ((2002) ) Consistent immunohistochemical detection of intracellular beta-amyloid42 in pyramidal neurons of Alzheimer’s disease entorhinal cortex. Neurosci Lett 333: , 163–166. |
[34] | D’Andrea MR , Reiser PA , Polkovitch DA , Gumula NA , Branchide B , Hertzog BM , Schmidheiser D , Belkowski S , Gastard MC , Andrade-Gordon P ((2003) ) The use of formic acid to embellish amyloid plaque detection in Alzheimer’s disease tissues misguides key observations. Neurosci Lett 342: , 114–118. |
[35] | Augustinack JC , Schneider A , Mandelkow EM , Hyman BT ((2002) ) Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol 103: , 26–35. |
[36] | Head E , Lott IT , Wilcock DM , Lemere CA ((2016) ) Aging in Down syndrome and the development of Alzheimer’s disease neuropathology. Curr Alzheimer Res 13: , 18–29. |
[37] | Selkoe DJ ((2001) ) Alzheimer’s disease: Genes, proteins, and therapy. Physiol Rev 81: , 741–766. |
[38] | Shin RW , Ogino K , Kondo A , Saido TC , Trojanowski JQ , Kitamoto T , Tateishi J ((1997) ) Amyloid beta-protein (Abeta) 1-40 but not Abeta1-42 contributes to the experimental formation of Alzheimer disease amyloid fibrils in rat brain. J Neurosci 17: , 8187–8193. |
[39] | Naslund J , Schierhorn A , Hellman U , Lannfelt L , Roses AD , Tjernberg LO , Silberring J , Gandy SE , Winblad B , Greengard P , Nordstedt C , Terenius L ((1994) ) Relative abundance of Alzheimer A beta amyloid peptide variants in Alzheimer disease and normal aging. Proc Natl Acad Sci U S A 91: , 8378–8382. |
[40] | Wang J , Dickson DW , Trojanowski JQ , Lee VM ((1999) ) The levels of soluble versus insoluble brain Abeta distinguish Alzheimer’s disease from normal and pathologic aging. Exp Neurol 158: , 328–337. |


