
Palmitate Increases β-site AβPP-Cleavage Enzyme 1 Activity and Amyloid-β Genesis by Evoking Endoplasmic Reticulum Stress and Subsequent C/EBP Homologous Protein Activation
Abstract
Epidemiological studies implicate diets rich in saturated free fatty acids (sFFA) as a potential risk factor for developing Alzheimer’s disease (AD). In particular, high plasma levels of the sFFA palmitic acid (palmitate) were shown to inversely correlate with cognitive function. However, the cellular mechanisms by which sFFA may increase the risk for AD are not well known. Endoplasmic reticulum (ER) stress has emerged as one of the signaling pathways initiating and fostering the neurodegenerative changes in AD by increasing the aspartyl protease β-site AβPP cleaving enzyme 1 (BACE1) and amyloid-β (Aβ) genesis. In this study, we determined the extent to which palmitate increases BACE1 and Aβ levels in vitro and in vivo as well as the potential role of ER stress as cellular mechanism underlying palmitate effects. We demonstrate, in palmitate-treated SH-SY5Y neuroblastoma cells and in the hippocampi of palmitate-enriched diet-fed mice, that palmitate evokes the activation of the C/EBP Homologous Protein (CHOP), a transcription factor that is specifically responsive to ER stress. Induction of CHOP expression is associated with increased BACE1 mRNA, protein and activity levels, and subsequent enhanced amyloidogenic processing of amyloid-β protein precursor (AβPP) that culminates in a substantial increase in Aβ genesis. We further show that CHOP is an indispensable molecular mediator of palmitate-induced upregulation in BACE1 activity and Aβ genesis. Indeed, we show that Chop–/– mice and CHOP knocked-down SH-SY5Y neuroblastoma cells do not exhibit the same commensurate degree of palmitate-induced increase in BACE1 expression levels and Aβ genesis.
INTRODUCTION
Alzheimer’s disease (AD), the most common form of dementia in the elderly, is characterized by progressive neurodegeneration resulting in cognitive dysfunction and memory impairment. The histopathological hallmarks of AD include the extracellular accumulation of aggregated amyloid-β (Aβ) peptide as neuritic senile plaques and the intracellular accumulation of aggregated hyperphosphorylated protein tau as neurofibrillary tangles. Aβ is a constitutively synthesized peptide generated by the sequential two-step proteolytic cleavage of the transmembrane protein amyloid-β protein precursor (AβPP). In the first step, AβPP is cleaved by the membrane-bound aspartyl protease β-site AβPP cleaving enzyme 1 (BACE1) to generate carboxy terminal fragment β (CTFβ, or C99 fragment), which in the second step is subsequently cleaved by the γ-secretase complex to generate Aβ peptide [1]. The general consensus points to the accumulation and aggregation of the Aβ peptide as a central instigating factor that triggers a cascade of detrimental pathophysiological events that culminate in the neurodegenerative changes that characterize the onset of AD. The levels of BACE1 protein as well as its enzymatic activity are significantly augmented in the AD brain [2–5]. The etiology of AD is multifactorial with both genetic and non-genetic factors likely contributing to the disease progression. Epidemiological studies have implicated a diet rich in saturated free fatty acids (sFFA) as a significant risk factor for developing AD [6–10]. Furthermore, the degree of saturated fat or saturated fatty acids in the diet determines and dictates the degree of risk for developing AD [11, 12]. Diets rich in saturated fat also precipitate cognitive dysfunction in a multitude of murine models [13, 14]. Palmitic acid (palmitate) is the most abundant long-chain sFFA in the brain [15] and the diet [16]. Moreover, higher sFFA levels in the plasma, as observed in obesity and diabetes, inversely correlate with cognitive function [17, 18]. Additionally, sFFA in circulation such as palmitate, emanating either from the diet or from de novo lipogenesis in the liver, cross the blood-brain barrier [19] and add to the burden of saturated fatty acid pool in the brain [20, 21]. Recent studies have implicated endoplasmic reticulum (ER) stress as one of the culpable factors in initiating and fostering the neurodegenerative changes in AD [22]. A multitude of other studies have demonstrated that sFFA, such as palmitate, evoke ER stress [23–26]. Sustained ER stress culminates in the increased expression of the transcription factor C/EBP Homologous Protein (CHOP, also called growth arrest and DNA damage induced gene-153, GADD153 or DDIT3). Our earlier studies have implicated the role of CHOP in the regulation of BACE1 expression [27]. However, the extent to which palmitate modulates ER stress and subsequent CHOP expression to impinge on BACE1 expression and subsequent Aβ genesis is not well-known. In this study, we determined the impact of palmitate-enriched diet and exogenous palmitate treatment on BACE1 expression and Aβ genesis in the mouse brains and in neuroblastoma cells respectively and delineated the underlying molecular mechanisms involving ER stress and CHOP.
MATERIALS AND METHODS
Materials
Human SH-SY5Y neuroblastoma cells stably expressing the AβPP Swedish KM670/671NL double mutation (SH-SY5Y-βSwe) were cultured in DMEM:Ham’s F12 with Glutamax (1:1; v/v), 10% fetal bovine serum, and 1% antibiotic/antimycotic mix. Cells were maintained at 37°C in a saturated humidity atmosphere containing 95% air and 5% CO2. All cell culture reagents, with the exception of fetal bovine serum (Atlanta Biologicals, Lawrenceville, GA) and antibiotic/antimycotic mix (Sigma Aldrich, St. Louis, MO) were purchased from Invitrogen (Carlsbad, CA). Palmitic acid, stearic acid, palmitoleic acid, oleic acid, Tunicamycin, and 4-phenylbutyric acid were purchased from Sigma Aldrich (St. Louis, MO). The expression plasmid for overexpressing full length native CHOP (CHOP 6: mCHOP-WT-9E10-pcDNA1) was a gift from Dr. David Ron (Addgene plasmid # 21913). The expression plasmid for overexpressing the leucine zipper domain deleted CHOP mutant (CHOP LZ–) (CHOP 5: mCHOP10 [dLZ] pSRa) was a gift from Dr. David Ron (Addgene plasmid # 21912). The human and mouse CHOP double-stranded siRNA (Silencer® Select Pre-Designed & Validated siRNA) and their respective scrambled non-silencing control siRNA were purchased from Thermo Fisher Scientific (Waltham, MA). The list of siRNA and their respective targets are enumerated in Table 1. CHOP shRNA (set of 5 different shRNA) encoded in pLKO.1 lentiviral vector were purchased from Open Biosystems (GE Dharmacon, Lafayette, CO) and their respective target sequences are enumerated in Table 2.
Table 1
List of siRNA and their target sequences used for RNA interference
| Species | Gene | mRNA | RNA | RefSeq | siRNA |
| ID | target | interference | location | ||
| Human | 1649 | DDIT3 (CHOP) | siRNA | NM_001195053 | 817 |
| Human | 1649 | DDIT3 (CHOP) | siRNA | NM_001195054 | 764 |
| Human | 1649 | DDIT3 (CHOP) | siRNA | NM_001195055 | 741 |
| Human | 1649 | DDIT3 (CHOP) | siRNA | NM_001195056 | 927 |
| Human | 1649 | DDIT3 (CHOP) | siRNA | NM_001195057 | 646 |
| Human | 1649 | DDIT3 (CHOP) | siRNA | NM_004083 | 660 |
| Mouse | 12607 | Cebpz (Chop) | siRNA | NM_001024806.1 | 272 |
| Mouse | 12607 | Cebpz (Chop) | siRNA | NM_001024806.2 | 292 |
| Mouse | 13198 | Ddit3 (Chop) | siRNA | NM_001290183 | 185 |
| Mouse | 13198 | Ddit3 (Chop) | siRNA | NM_007837 | 233 |
| Mouse | 13198 | Ddit3 (Chop) | siRNA | NM_001290183 | 437 |
| Mouse | 13198 | Ddit3 (Chop) | siRNA | NM_007837 | 485 |
Table 2
List of shRNA target sequences used for RNA interference
| Species | Gene ID | mRNA target | RNA interference | Sequence |
| Human | 1649 | DDIT3 (CHOP) | shRNA | ATTGAGGGTCACATCATTGGC |
| Human | 1649 | DDIT3 (CHOP) | shRNA | TTCTTCCTCTTCATTTCCAGG |
| Human | 1649 | DDIT3 (CHOP) | shRNA | TTGGTGCAGATTCACCATTCG |
| Human | 1649 | DDIT3 (CHOP) | shRNA | TTCCAGGAGGTGAAACATAGG |
| Human | 1649 | DDIT3 (CHOP) | shRNA | TTTCCTTTCATTCTCCTGTTC |
Cell culture and treatments
SH-SY5Y-APPSwe cells were transfected with the designated vectors as previously described [28–31]. Transfected SH-SY5Y-APPSwe cells were treated with different concentrations of BSA (bovine serum albumin)-conjugated palmitate as shown previously [28]. Briefly, palmitate stock solution of 250 mM was prepared in 100% ethanol. BSA (5 mM) stock solution was prepared in MilliQ water (18 MΩ). Both, the palmitate and BSA stock solution were sterile filtered using a 0.2 μm filter. The requisite amounts of palmitate and BSA were added to sterile serum-free medium to yield the designated terminal palmitic acid concentrations with the ratio of palmitate and BSA being 6:1. The respective media were incubated for 1.5 h to conjugate the palmitic acid to the BSA. The cells were treated with the designated concentration of palmitic acid conjugated to BSA for 24 h. The BSA concentration (81.33 μM) corresponding to the highest palmitate concentration (500 μM of palmitate) was used as the experimental control. Using the highest concentration of BSA extricated any residual background effects that could be attributed to the biological effects of BSA in cells treated with different concentrations of BSA-conjugatedpalmitate
Mouse experiments
Mice harboring a homozygous targeting deletion mutation to the Ddit3 gene (Chop–/– mice) were procured from The Jackson Laboratory [B6.129S(Cg)-Ddit3 tm2.1Dron/J, Stock #005530] (Bar Harbor, ME). The corresponding C57BL/6J control mice were also procured from The Jackson Laboratory (Stock #000664). The mice were housed in individually ventilated cages at an ambient room temperature (23–25°C) and ambient relative humidity ranging between 50–70%. The mice were maintained on 12:12 h light:dark cycle and allowed access to food and water ad libitum. The Chop–/– mice and their wild-type C57BL/6J mice (all males, nine months of age) counterparts were each segregated into two groups, one fed a palmitate-enriched diet (TD 110616, Harlan Teklad, 2.2% w/w palmitic acid) and the other fed the corresponding control diet (TD 85172, Harlan Teklad, 0.8% w/w palmitic acid) for three months, to generate four different experimental cohorts of nine-month-old male mice (n = 15). The diets were isocaloric in relation to each other and the respective composition of the diets is shown in Table 3. Food-intake was monitored for the span of 24 h, once every two weeks. Body weights were measured every two weeks. No significant changes in body weight (Table 4) and food intake were observed among the different cohorts of mice. Necropsy was performed at twelve (12) months of age. All animal procedures were carried out in accordance with the U.S. Public Health Service Policy on the Humane Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at the University of North Dakota (Protocol 1506-3c).
Table 3
Composition of the control chow diet and palmitate-enriched diet
| Control chow diet | palmitate-enriched diet | |
| NIH07 open formula rodent | NIH07 open formula rodent | |
| diet – original –0.8 % | diet –palmitate enriched – | |
| palmitic acid | 2.2 % palmitic acid | |
| Protein | 23.60 % w/w | 23.60 % w/w |
| Carbohydrates | 65.80 % w/w | 65.80 % w/w |
| Total Fat | 5.60 % w/w | 5.60 % w/w |
| Total Energy | 4.08 Kcal/gram | 4.08 Kcal/gram |
| Myristic acid (14:0) | 0.10 % w/w | 0.10 % w/w |
| Palmitic acid (16:0) | 0.80 % w/w | 2.20 % w/w |
| Stearic acid (18:0) | 0.20 % w/w | 0.20 % w/w |
| Palmitoleic acid (16:1) | Trace | trace |
| Oleic acid (18:1) | 1.20 % w/w | 1.20 % w/w |
| Gadoleic acid (20:1) | Trace | trace |
| Linoleic acid (18:2 n6) | 2.20 % w/w | 0.80 % w/w |
| Linolenic acid (18:3 n3) | 0.20 % w/w | 0.20 % w/w |
| Arachadonic acid (20:4 n6) | Trace | trace |
| EPA (20:5 n3) | 0.10 % w/w | 0.10 % w/w |
| DHA (22:6 n3) | 0.30 % w/w | 0.30 % w/w |
Table 4
Body weights of mice on control chow and palmitate-enriched diet
| C57BL/6J | C57BL/6J | Chop–/– | Chop–/– | |
| Chow diet | PA diet | Chow diet | PA diet | |
| Initial weight (g) (9 months) | ||||
| (Mean±S.D, n = 15) | 28.82±2.16 | 29.91±3.15 | 27.94±1.58 | 30.87±3.56 |
| Final weight (g) (12 months) | ||||
| (Mean±S.D, n = 15) | 29.25±3.06 | 30.16±1.89 | 28.76±2.93 | 32.25±2.46 |
| Weight gain/loss (%) | 1.49 | 0.83 | 2.93 | 4.47 |
Western blot analysis
Whole cell, cytosolic, and nuclear homogenates from cells as well as the mouse cortices and hippocampi were prepared as previously described [27, 29, 32, 33]. Proteins (10–40 μg) were resolved on SDS-PAGE gels followed by transfer to a polyvinylidene difluoride membrane (BioRad, Hercules, CA) and incubation with the monoclonal antibodies listed in Table 5. The origin, source, the dilutions of the respective antibodies used for this study is compiled in Table 4. β-actin was used as a gel loading control for whole cell and cytosolic homogenates, whereas Histone H3 was used as a gel loading control for nuclear homogenates. The blots were developed with enhanced chemiluminescence (ClarityTM Western ECL blotting substrate, Bio-Rad, Hercules, CA) and imaged using a LiCOR Odyssey Fc imagingsystem.
Table 5
List of monoclonal and polyclonal antibodies used in the study
| Antibody | Dilution | Amount | Host | Manufacturer | Catalog # |
| ATF3 | 1:500 | 10 μg | rabbit | Sigma Aldrich | HPA001562 |
| ATF4 | 1:1000 | 5 μg | rabbit | Cell Signaling Technology | 11815 (D4B8) |
| ATF6 | 1:1000 | 5 μg | rabbit | Active Motif | 40962 |
| β-Actin | 1:2500 | 2 μg | mouse | Santa Cruz BioTechnology | sc-47778 (C4) |
| BACE1 | 1:1000 | 5 μg | rabbit | EMD Millipore | AB5832 |
| CHOP | 1:500 | 10 μg | rabbit | Cell Signaling Technology | 5554 (D46F1) |
| CTFα/ CTFβ | 1:400 | 12.5 μg | rabbit | BioLegend | 825001 |
| Histone H3 | 1:1000 | 5 μg | rabbit | Santa Cruz BioTechnology | sc-8654 (C16) |
| p-Ser724 IRE1α | 1:200 | 25 μg | rabbit | Abcam | ab48187 |
| IRE1α | 1:500 | 10 μg | rabbit | Cell Signaling Technology | 3294 (14C10) |
| p-Thr980 PERK | 1:500 | 10 μg | rabbit | Cell Signaling Technology | 3179 (16F8) |
| PERK | 1:500 | 10 μg | rabbit | Cell Signaling Technology | 3192 (C33E10) |
| sAβPPα | 1:500 | 10 μg | rabbit | BioLegend | 813501 |
| sAβPPβ | 1:500 | 10 μg | rabbit | BioLegend | 813401 |
Lactate Dehydrogenase (LDH) assay for cytotoxicity
The levels of LDH were measured as a surrogate measure of cytotoxic cell death in response to palmitate treatment using the “CytoTox 96® Non-Radioactive Cytotoxicity Assay” from Promega (Catalog # G1780) (Madison, WI) following manufacturer’s instructions. Data was normalized to the intensity of the absorbance in BSA-treated cells (control) (set as unit value) and expressed as a fold-change of the control±SD (six wells per one experiment from three separate experiments, n = 3).
ER stress transcription factor activation profiling plate array
The transcriptional activity of the ER-stress-associated transcription factors was determined using the “ER Stress (UPR) TF Activation Profiling Plate Array” from Signosis Inc. (Santa Clara, CA, Catalog # FA-1006) using manufacturer’s instructions and as described previously [28]. Briefly, nuclear lysate fractions were prepared, from treated SH-SY5Y-APPSwe cells or the hippocampi of mice, using a sequential cellular fraction approach. The nuclear lysate containing the equivalent of 15 μg of total protein content was used for each assay. The respective transcription factor (TF) was complexed with the respective DNA probe to generate the TF/DNA complex using the nuclear lysate and components of kit provided. The TF/DNA probe complex was separated from the free DNA probe by passing it through the isolation column and subsequently eluted from the column using the kit components following the manufacturer’s protocol. The purified TF/DNA probe complex was hybridized to a specific secondary biotin-labeled probe in a 96-well plate. The biotin and HRP-conjugated streptavidin chemistry was used to determine the luminescence signal as a surrogate measure of the transcriptional activity.
Enzyme-linked immunosorbent assay (ELISA)
Aβ1 - 42 levels in neuroblastoma cells were quantified in the conditioned media (secreted) and cellular homogenates (intracellular) using an ELISA immunoassay kit (Invitrogen, Carlsbad, CA) following the manufacturer’s protocol and as described earlier [27, 34]. Intracellular Aβ1 - 42 levels in the cellular homogenates were normalized to total protein content in the samples (pg/mg protein). Treatments were performed in quadruplet (n = 4, four biological replicates with three technical replicates within each biological replicate). The secreted Aβ1 - 42 levels measured in the culture medium are expressed in pg/mL of media. Aβ1 - 42 levels in the mouse cortex and hippocampus were quantified using the same aforementioned ELISA immunoassay kit (Invitrogen, Carlsbad, CA). The mouse cortex and hippocampal tissue was subjected to a sequential extraction procedure [35] to generate the following sub-cellular fractions containing the respective Aβ1 - 42 species - TBS-soluble Aβ1 - 42 (water soluble Aβ1 - 42), 2% SDS soluble Aβ1 - 42 (detergent soluble Aβ1 - 42), and 70% formic acid soluble Aβ1 - 42 (TBS insoluble Aβ1 - 42, detergent insoluble Aβ1 - 42) that reflects the total Aβ1 - 42 pool. Aβ1 - 42 levels in the mouse cortex and hippocampus were normalized to total protein content in the samples (pg/mg protein).
Quantitative real time RT-PCR analysis
Total RNA was isolated and extracted from treated cells using the 5 prime “PerfectPure RNA tissue kit” (5 Prime, Inc., Gaithersburg, MD) following manufacturer’s instructions and as described previously [36]. cDNA was obtained by reverse transcribing 1 μg of extracted RNA using an iScript cDNA synthesis kit” (BioRad, Hercules, CA). cDNA was obtained by reverse transcribing 1 μg of extracted RNA using an iScript cDNA synthesis kit” (BioRad, Hercules, CA). The quantitative Real-time RT-PCR was performed using TaqMan chemistry using “Assays-on-Demand” probes (ABI, Foster City, CA) for human BACE1 (BACE1 gene) (Hs01121195_m1) and mouse Bace1 (Bace1 gene) (Mm00478664_m1), The amplification was performed using the “StepOnePlus” PCR System (ABI, Foster City, CA). The expression of specific transcripts amplified was normalized to the expression of 18s rRNA. The data were quantified and expressed as fold-change compared to the control by using the ΔΔCTmethod.
FRET-based BACE1 activity assay
BACE1 activity in cellular and tissue homogenates was determined using a FRET-based kit from Sigma-Aldrich (St. Louis, MO, Catalog #CS0010) following manufacturer’s protocol. The raw data expressing the BACE1 activity in terms of percentage of substrate cleaved in respective samples was further normalized and expressed as fold-change compared to control.
2.10Statistical analysis
The significance of differences among the samples was assessed by non-parametric Kruskal-Wallis One Way Analysis of Variance followed by Dunn’s post-hoc test. Statistical analysis was performed with GraphPad Prism 6. Quantitative data for all the assays are presented as mean values±S.D (mean values±standard deviation) with unit value assigned to control and the magnitude of differences among the samples being expressed relative to the unit value of control as fold-change. Quantitative data for ELISA analysis are presented as mean values±S.D with absolute concentrations of Aβ1 - 42 reported.
RESULTS
Palmitate induces ER stress in the mouse hippocampus and the SH-SY5Y-APPSwe human neuroblastoma cells
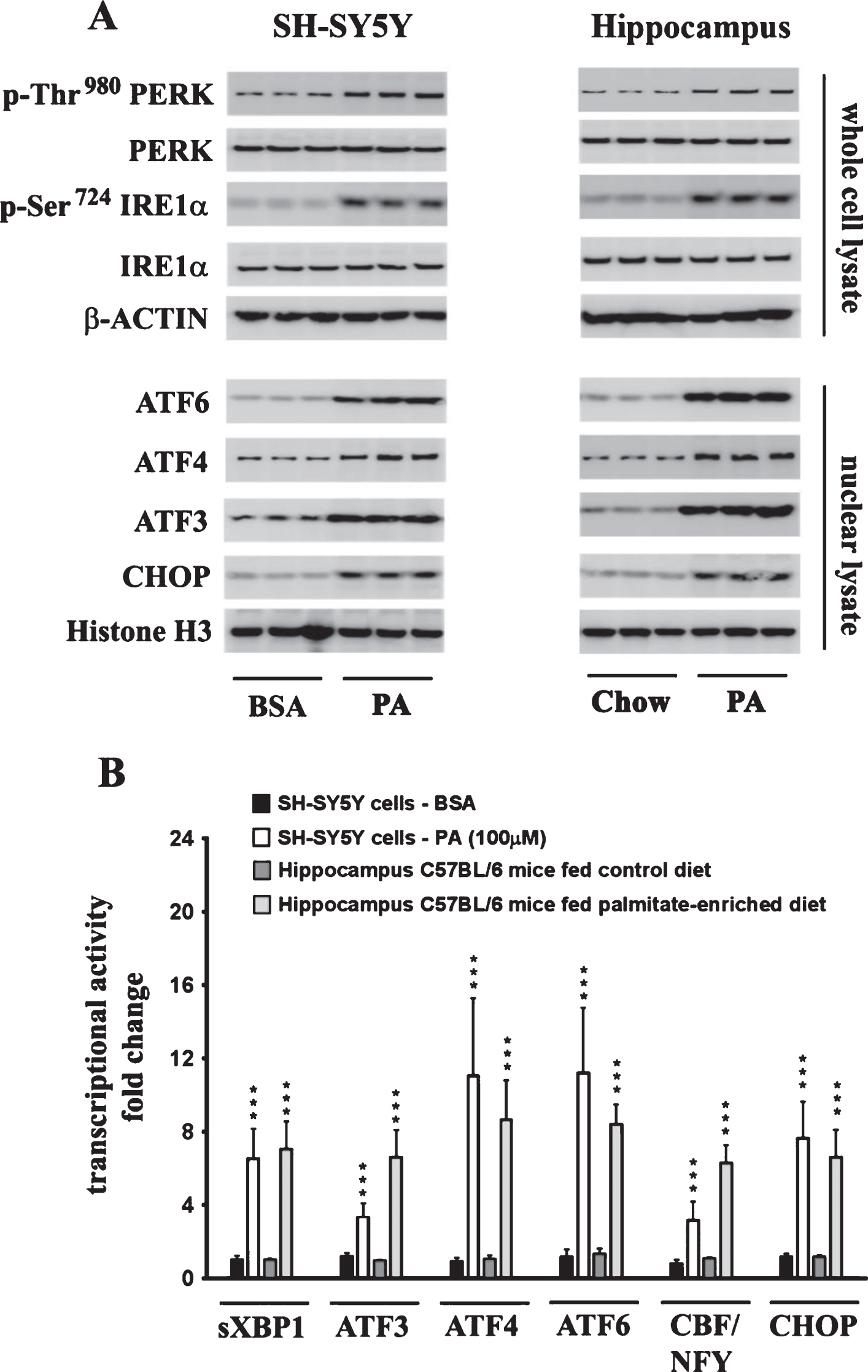
The effects of palmitate treatment on ER stress markers were determined in cultured SH-SY5Y-APPSwe human neuroblastoma cells that stably express the Swedish KM670/671NL double mutant AβPP. The brain levels of the two most abundant saturated free fatty acids, palmitate (16:0) and stearate (18:0), range from sim;60 μmoles/L (60 μM) to sim;75 μmoles/L (75 μM) and sim;50 μmoles/L (50 μM) to ∼60 μmoles/L (60 μM), respectively [37–40]. FFA in circulation are bound to serum albumin that solubilizes the hydrophobic free fatty acids and governs their bioavailability [41]. Therefore, we treated the SH-SY5Y-APPSwe cells with increasing concentrations of BSA-conjugated palmitate and stearate, as well as the mono-unsaturated fatty acids (MUFA), palmitoleate (16:1) and oleate (18:1), with concentrations ranging from 25–500 μM for 24 h. Our data demonstrates that palmitate and stearate at 100 μM and beyond, but not palmitoleate and oleate at 100 μM, evoke ER stress in SH-SY5Y-APPSwe cells as assessed by the pronounced increase in the expression of ER resident chaperones, GRP78 and GRP94, as well as ER-stress associated transcription factor CHOP (Fig. 1A-D). More importantly this concentration of palmitate and stearate (100 μM) did not elicit cell death unlike higher concentrations (250–500 μM) in cultured SH-SY5Y-APPSwe cells (Fig. 1E). Consequently, palmitate conjugated to BSA (molar ratio 6:1) at a terminal concentration of 100 μM was used to treat SH-SY5Y-APPSwe cells. Palmitate treatment of SY5Y-APPSwe cells activated the three signaling cascades of ER stress pathway as determined by a pronounced increase in the phosphorylation status of IRE1α and PERK and a significant increase in the nuclear transcloaction of ATF6 (Fig. 2A). There was a pronounced increase in nuclear translocation of other ER stress-associated transcription factors - CHOP, ATF3, and ATF4 (Fig. 2A). C57BL/6J mice fed a palmitate-enriched diet (2.2% w/w palmitic acid) for three months also exhibited profound ER stress in the hippocampus as assessed by determining the levels of the same aforementioned ER stress markers (Fig. 2A). Furthermore, the transcriptional activity of the aforementioned ER stress-associated transcription factors was also significantly increased in the treated SY5Y-APPSwe cells (Fig. 2B) as well as in the hippocampi of C57BL/6J mice fed a palmitate-enriched diet (Fig. 2B). These data are consistent with our previously published work demonstrating that palmitate evokes ER stress in neuronal cells and the mouse brain [28].
Fig.1
Dose response effects of the saturated fatty acids, palmitate and stearate, and their respective MUFA, palmitoleate and oleate, on the expression of ER stress markers and cell death in human neuroblastoma SH-SY5Y cells. A-D) Representative western blots show that treatment with exogenous palmitate (A) and stearate (B) at a concentration >100 μM for 24 h, while treatment with exogenous palmitoleate (C) and oleate (D) only at a concentration of >375 μM for 24 h, significantly increases the expression of ER stress markers - GRP78, GRP94, and CHOP in whole cell lysates from SH-SY5Y-APPSwe cells. E) Cell death assessed by the release of LDH in the conditioned medium shows that treatment with exogenous palmitate and stearate at a concentration >250 μM for 24 h, while treatment with exogenous palmitoleate and oleate only at a concentration of 500 μM for 24 h, evoked significant cell death in SH-SY5Y-APPSwe cells. Data is expressed as Mean±S.D and includes determination made in four (n = 4) separate cell culture experiments. ***p < 0.001 versus BSA-treated cells.
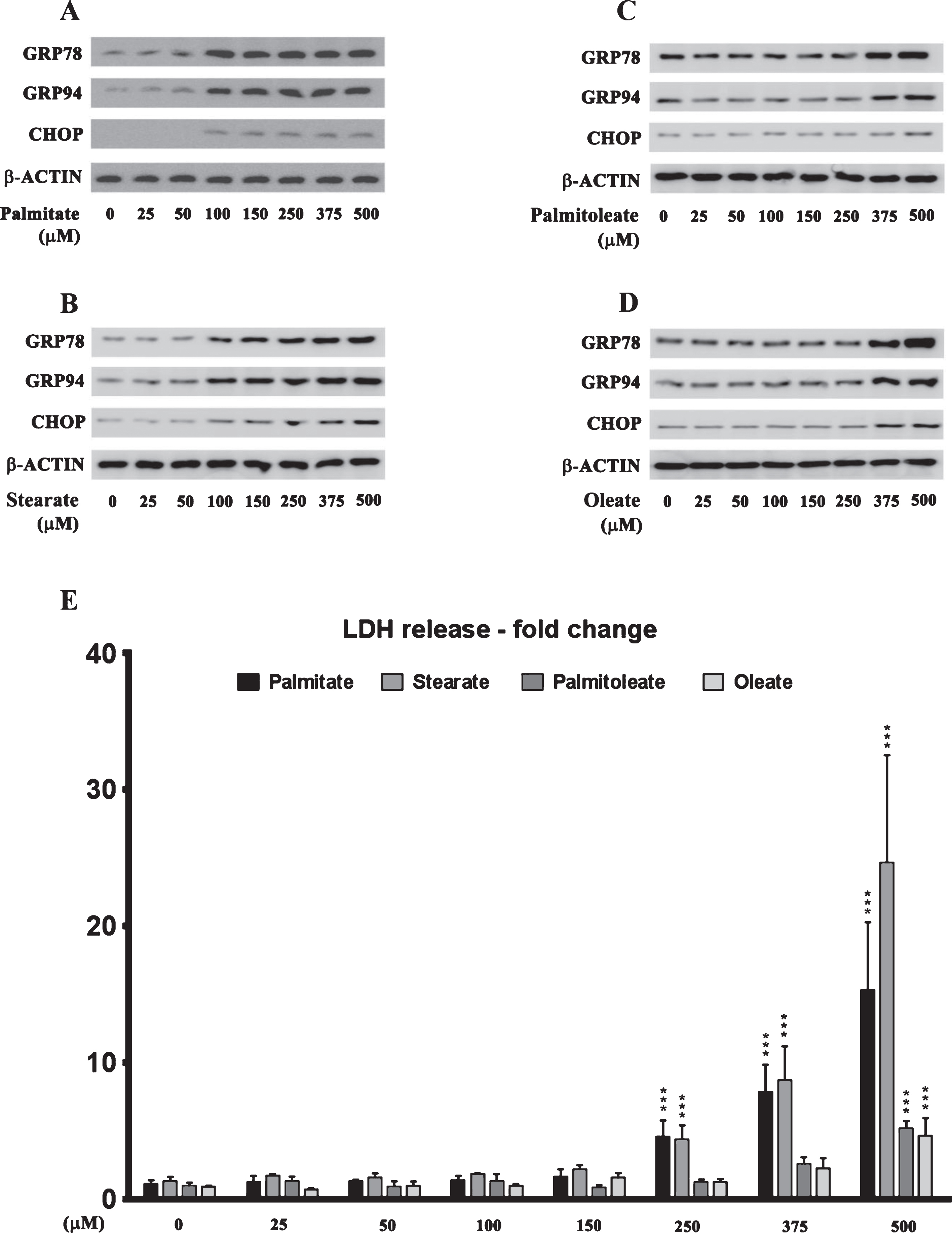
Fig.2
Exogenous palmitate treatment and a palmitate-enriched diet evoke ER stress in human neuroblastoma SH-SY5Y cells and the mouse hippocampus, respectively. A) Representative western blots show that palmitate treatment (100 μM for 24 h) of SH-SY5Y-APPSwe cells and feeding C57BL/6J wild-type mice a palmitate-enriched diet for three months, results in the activation of the three arms of ER stress signaling - IRE1α, PERK, and ATF6 pathway as assessed by an increase in the phosphorylation of IRE1α and PERK as well as the augmentation in the nuclear translocation of ATF3, ATF4, ATF6, and CHOP. B) Palmitate treatment (100 μM for 24 h) of SH-SY5Y-APPSwe cells and feeding C57BL/6J wild-type mice a palmitate-enriched diet for three months, also increases the transcriptional activities of the six transcription factors measured in the hippocampus. Data is expressed as Mean±S.D and includes determination made in three (n = 3) separate cell culture experiments and six (n = 6) different animals from each group. ***p < 0.001 versus BSA-treated control cells or C57BL/6J wild-type mice fed a control chow diet. PA, palmitic acid.
Palmitate induces BACE1 expression and the ensuing Aβ accumulation by evoking ER stress
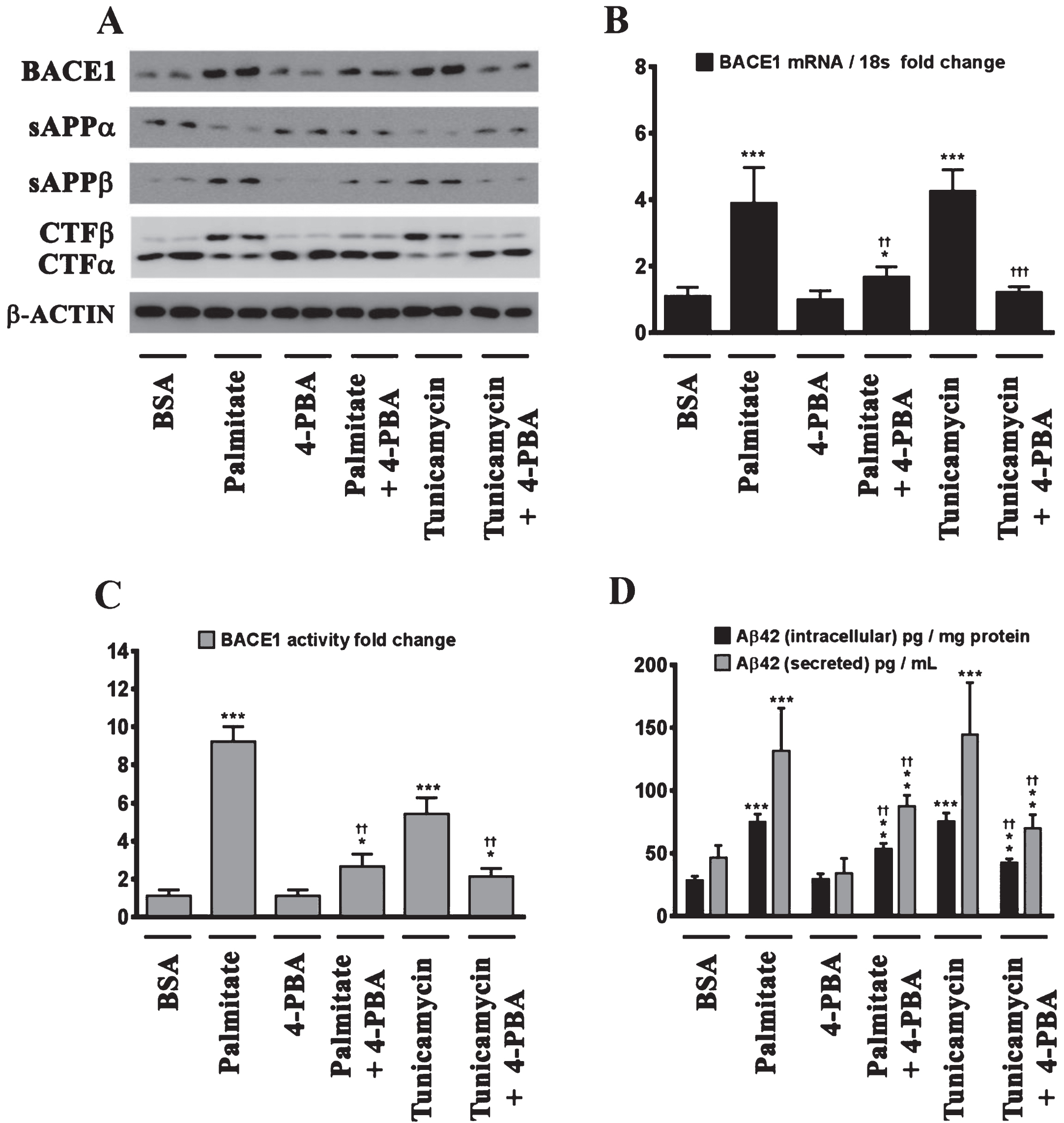
We next determined the effects of palmitate and stearate, as well as their monounsaturated counterparts, palmitoleate and oleate, respectively, on BACE1 expression levels, BACE1 activity, AβPP processing, as well as intracellular and secreted Aβ1 - 42 levels in SH-SY5Y-APPSwe cells. Both, palmitate and stearate, but not palmitoleate and oleate, significantly increased BACE1 protein levels (Fig. 3A), BACE1 mRNA expression, BACE1 enzymatic activity (Fig. 3C), and intracellular as well as secreted Aβ1 - 42 levels (Fig. 3D) in SH-SY5Y-APPSwe cells. Furthermore, this increase in Aβ1 - 42 genesis by palmitate and stearate was ascribed to increased BACE1 activity and the subsequent enhancement of amyloidogenic processing of AβPP by BACE1 as evidenced by an increase in the levels of CTFβ (C99) and sAβPPβ levels accompanied by a concomitant decrease in CTFα (C83) and sAβPPα (Fig. 3A). This aforementioned effect of palmitate and stearate was recapitulated by Tunicamycin (2 μM) (Fig. 3A-D), a well characterized inhibitor of N-linked glycosylation of nascent proteins that induces ER stress. Further evidence unequivocally implicating ER stress in the palmitate-induced effects on BACE1 expression, BACE1 activity, AβPP processing, and Aβ genesis emanated from the finding that the molecular chaperone 4-phenylbutyric acid (4-PBA), known to alleviate ER stress, significantly attenuated the palmitate induced increase in BACE1 protein levels (Fig. 4A), BACE1 mRNA expression (Fig. 4B) and BACE1 enzymaticactivity (Fig. 4C), amyloidogenic processing of AβPP (Fig. 4A), and augmentation of Aβ1 - 42 genesis(Fig. 4D).
Fig.3
BACE1 expression and subsequent Aβ genesis is induced by the sFFA, palmitate and stearate, but not by their MUFA counterparts, palmitoleate and oleate. A) Representative western blots show that exogenous palmitate and stearate treatment (100 μM for 24 h), but not palmitoleate and oleate treatment (100 μM for 24 h), significantly increases BACE1 protein levels accompanied by an increase in the amyloidogenic processing of AβPP as evidenced by an increase in sAβPPβ and CTFβ levels concomitant with a decrease in sAβPPα and CTFα levels in the whole cell homogenates from SH-SY5Y-APPSwe cells. B, C) Exogenous palmitate and stearate treatment, but not palmitoleate and oleate treatment, significantly increases BACE1 mRNA expression (B) and BACE1 activity (C) in SH-SY5Y-APPSwe cells. D) ELISA immunoassays show that exogenous palmitate and stearate treatment, but not palmitoleate and oleate treatment, significantly increases the levels of the intracellular Aβ1 - 42 species in the whole cell lysates and secreted Aβ1 - 42 species in the conditioned media, from SH-SY5Y-APPSwe cells. The ER stress inducer, Tunicamycin, also increased the following - BACE1 protein levels (A), BACE1 mRNA expression (B) and BACE1 activity (C), and the ensuing levels of intracellular as well as secreted Aβ1 - 42 species (D) in SH-SY5Y-APPSwe cells. Data is expressed as Mean±S.D and includes determination made in four (n = 4) separate cell culture experiments. ***p < 0.001 versus BSA-treated cells.
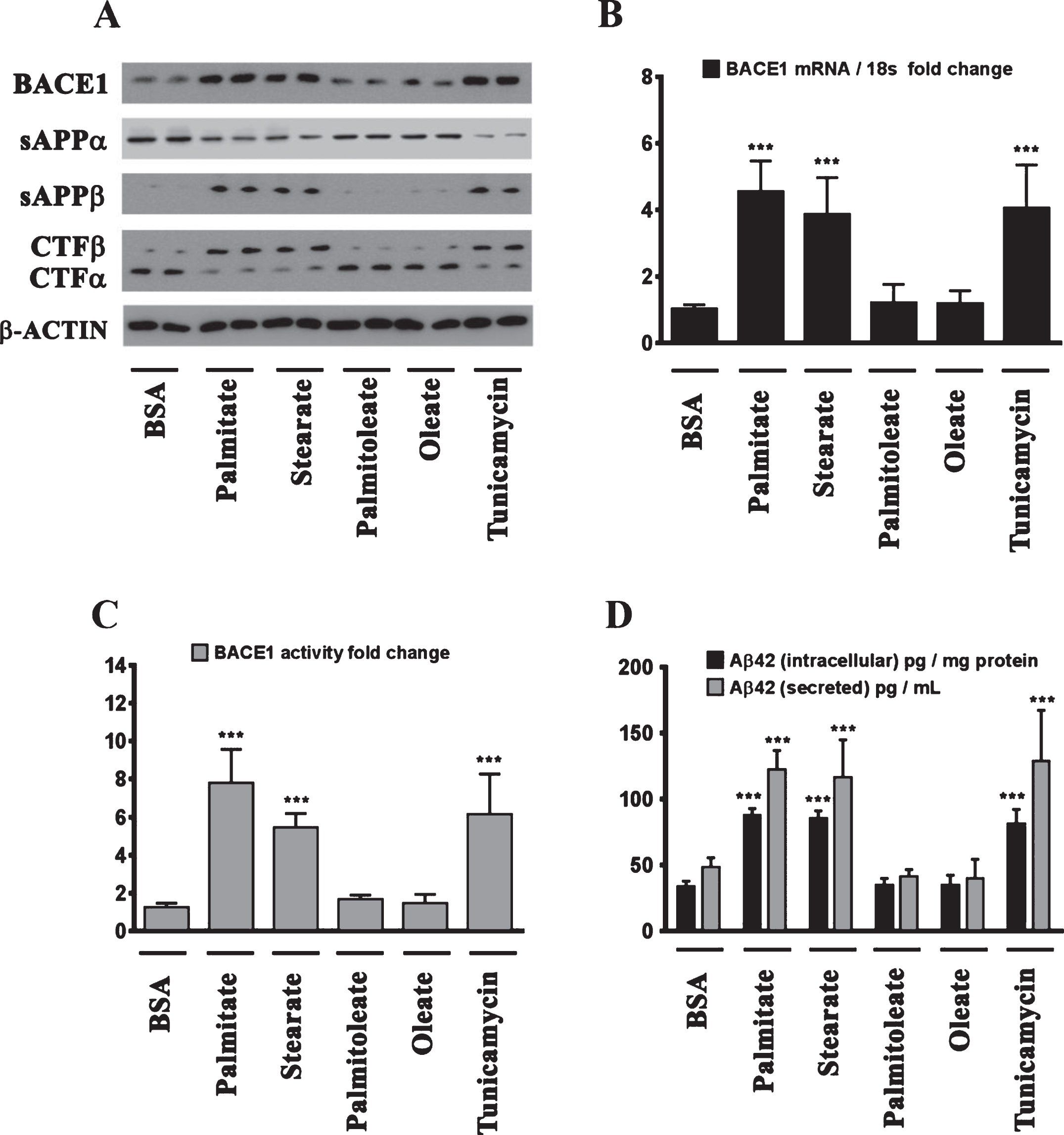
Fig.4
Palmitate induces BACE1 expression and subsequent Aβ genesis by inducing ER stress. A) Representative western blots show that pretreatment (for 2 h) of the human neuroblastoma cells with the molecular chaperone 4-PBA significantly precludes the palmitate-induced increase in BACE1 protein levels accompanied by a decrease in the amyloidogenic processing of AβPP as evidenced by a decrease in the palmitate-induced increase in sAβPPβ and CTFβ levels concomitant with an increase in the palmitate-induced decrease in sAβPPα and CTFα levels in the whole cell homogenates from SH-SY5Y-APPSwe cells. B, C) Pretreatment with 4-PBA attenuates the palmitate-induced increase in BACE1 mRNA expression (B) and BACE1 activity (C) in SH-SY5Y-APPSwe cells. D) ELISA immunoassays show that pretreatment with 4-PBA significantly attenuates the exogenous palmitate treatment-induced increase in the levels of the intracellular Aβ1 - 42 species in the whole cell lysates and secreted Aβ1 - 42 species in the conditioned media, from SH-SY5Y-APPSwe cells. Pretreatment (for 2 h) with the molecular chaperone 4-PBA also significantly precludes the Tunicamycin-induced increase in the following - BACE1 protein levels (A), BACE1 mRNA expression (B) and BACE1 activity (C), and intracellular as well as secreted Aβ1 - 42 species (D), in SH-SY5Y-APPSwe cells. Data is expressed as Mean±S.D and includes determination made in four (n = 4) separate cell culture experiments. *p < 0.05, **p < 0.01, ***p < 0.001 versus BSA-treated cells; ††p < 0.01, †††p < 0.001, versus palmitate-treated cells or Tunicamycin-treated cells.
ER-stress-induced CHOP expression is necessary for the palmitate-induced increase in BACE1 expression and ensuing Aβ genesis
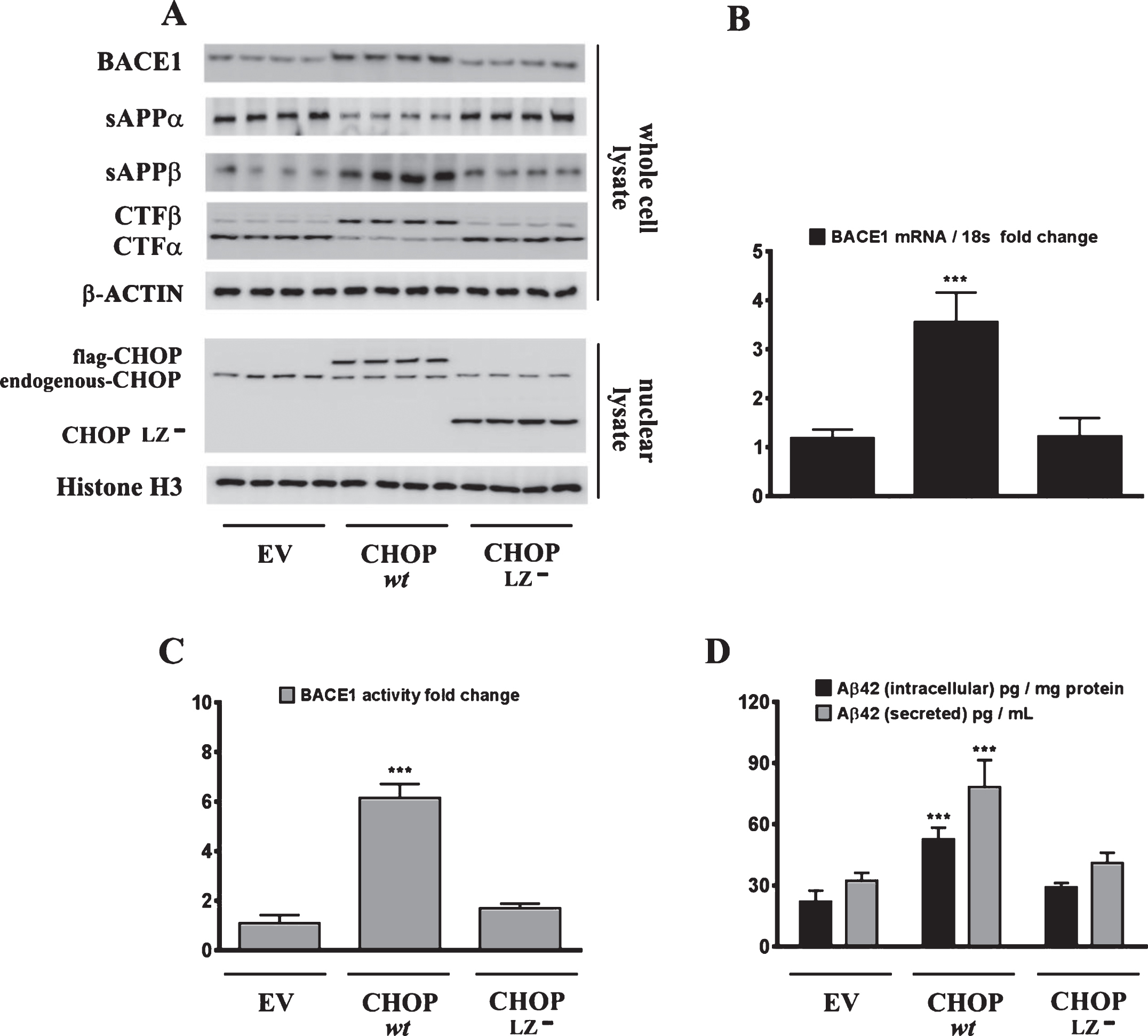
Sustained ER stress culminates in the increased expression of CHOP through the IRE1α-XBP1 pathway, ATF6 pathway, and the PERK-eIF2α-ATF4 signaling arm of ER stress signaling [22]. Studies from our laboratory have shown that the transcription factor CHOP plays an integral role in BACE1 expression and Aβ genesis [27]. We therefore determined the role of CHOP in palmitate-induced ER stress-mediated augmentation in BACE1 expression and Aβ genesis. To this end, we knocked-down CHOP expression in SH-SY5Y-APPSwe cells using a RNA interference approach followed by the challenge of palmitate treatment. Palmitate treatment failed to evoke an increase in BACE1 protein levels (Fig. 5A), BACE1 mRNA expression (Fig. 5B) and BACE1 enzymatic activity (Fig. 5C), augmentation in amyloidogenic processing of AβPP (Fig. 5D), and an increase in Aβ1 - 42 levels (Fig. 5D) to the same degree in CHOP-silenced SH-SY5Y-APPSwe cells compared to palmitate treated nativeSH-SY5Y-APPSwe cells. Knocking-down of CHOP in SH-SY5Y-APPSwe cells significantly attenuated the palmitate-induced increase in BACE1 protein levels, BACE1 activity, amyloidogenic processing of AβPP, and Aβ1 - 42 levels (Fig. 5A-D). We also generated a cohort of Chop–/– mice along with their C57BL/6J wild-type littermates (nine months of age) and fed them a control diet or a palmitate-enriched diet for three months. Feeding wild-type C57BL/6J mice a palmitate-enriched diet resulted in a pronounced increase in BACE1 protein levels (Fig. 6A), BACE1 mRNA expression (Fig. 6B) and BACE1 enzymatic activity (Fig. 6C), augmentation in amyloidogenic processing of AβPP (Fig. 6A), and an increase in total Aβ1 - 42 levels (70% formic acid soluble) (Fig. 6D) in the hippocampus region of the brain. However, Chop–/– mice fed a palmitate-enriched diet did not exhibit, to the same degree, the increase in BACE1 protein levels, BACE1 activity, amyloidogenic processing of AβPP, and total formic acid soluble Aβ1 - 42 levels (Fig. 6A-D). Interestingly, BACE1 protein levels, BACE1 activity, and total formic acid soluble Aβ1 - 42 levels were still higher in palmitate-treated CHOP knocked-down cells and in the hippocampi of Chop–/– mice compared to the BSA-treated native cells and C57BL/6J wild-type mice fed a normal chow respectively, suggesting that palmitate may mediate the aforementioned effects partially by other mechanisms that are yet to be delineated. To further substantiate the finding that palmitate-induced increase in CHOP expression underlies the augmentation in BACE1 expression and the ensuing Aβ genesis, we deleted the leucine zipper domain of CHOP that is obligatory and indispensable for CHOP transcriptional activity. We ectopically expressed either the transcriptionally dead-leucine zipper domain deleted mutant (CHOP LZ–) or the full length native CHOP in SH-SY5Y-APPSwe cells and determined BACE1 expression and activity, AβPP processing, and the ensuing Aβ genesis. Ectopic overexpression of the full-length native CHOP was sufficient by itself to significantly enhance BACE1 protein levels (Fig. 7A), BACE1 mRNA expression (Fig. 7B) and BACE1 activity (Fig. 7C), and Aβ1 - 42 levels (Fig. 7D) in SH-SY5Y-APPSwe cells. However, SH-SY5Y-APPSwe cells ectopically expressing the leucine zipper domain deleted CHOP mutant (CHOP LZ–) devoid of transcriptional activity did not exhibit any significant augmentation in BACE1 protein levels, BACE1 mRNA expression and activity, and Aβ1 - 42 levels (Fig. 7A-D).
Fig.5
CHOP mediates the palmitate-induced increase in BACE1 expression and subsequent Aβ genesis. A) Representative western blots show that knocking-down CHOP expression using a RNAi approach significantly attenuates the palmitate-induced increase in BACE1 protein levels accompanied by a decrease in the amyloidogenic processing of AβPP as evidenced by a decrease in the palmitate-induced increase in sAβPPβ and CTFβ levels concomitant with an increase in the palmitate-induced decrease in sAβPPα and CTFα levels in the whole cell homogenates from SH-SY5Y-APPSwe cells. B, C) Knocking-down CHOP expression attenuates the palmitate-induced increase in BACE1 mRNA expression (B) and BACE1 activity (C) in SH-SY5Y-APPSwe cells. D) ELISA immunoassays show that knocking-down CHOP expression significantly mitigates the exogenous palmitate treatment-induced increase in the levels of the intracellular Aβ1 - 42 species in the whole cell lysates and secreted Aβ1 - 42 species in the conditioned media, from SH-SY5Y-APPSwe cells. Data is expressed as Mean±S.D and includes determination made in four (n = 4) separate cell culture experiments. **p < 0.01, ***p < 0.001 versus BSA-treated GFP knock-down cells or BSA-treated scrambled siRNA transfected cells; †p < 0.05, ††p < 0.01, versus palmitate-treated GFP knock-down cells or palmitate-treated scrambled siRNA transfected cells. PA, palmitic acid.
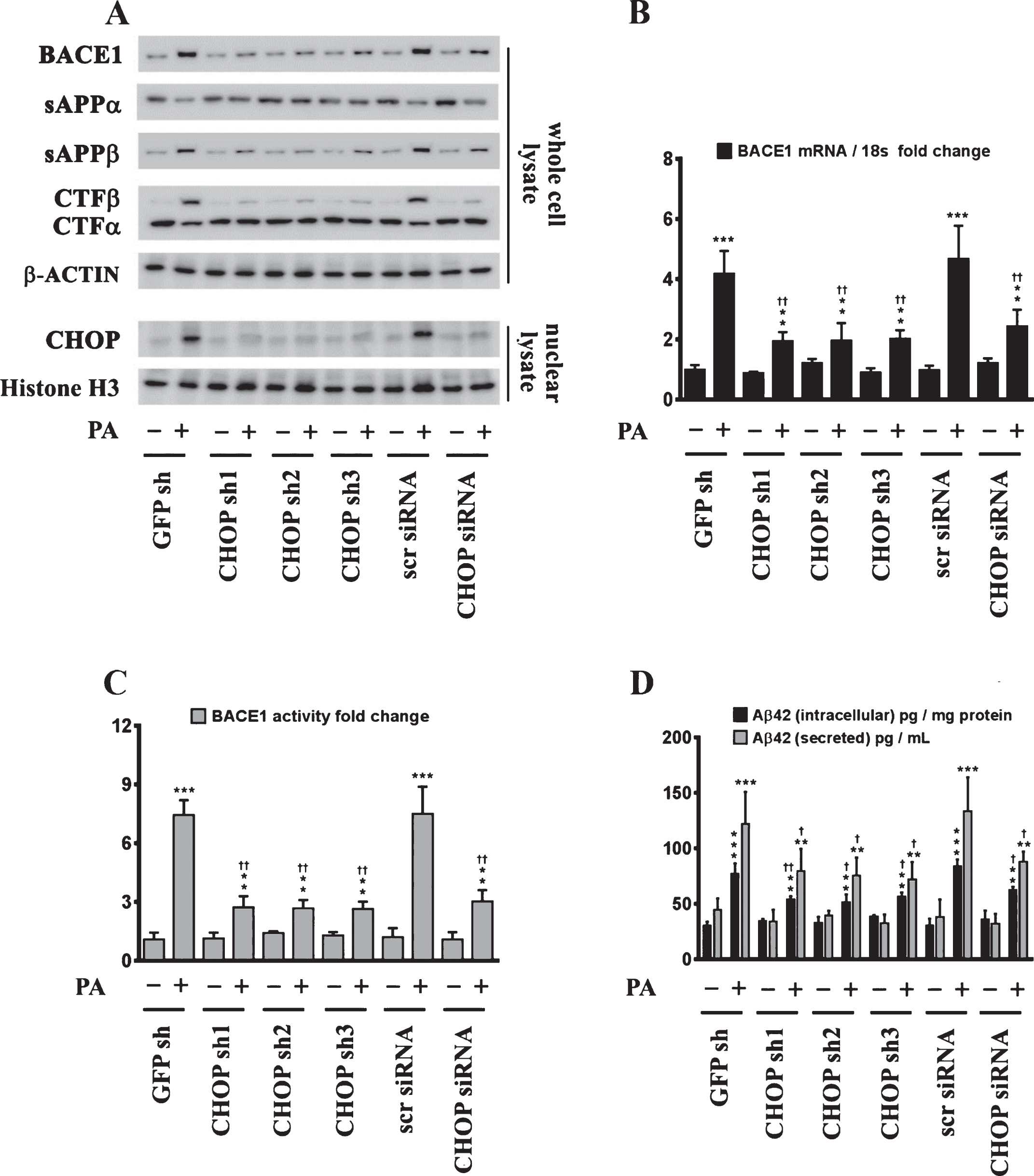
Fig.6
Chop–/– mice are significantly protected from the palmitate-enriched diet-induced increase in BACE1 expression and ensuing Aβ genesis. A) Representative western blots show that nine-month-old Chop–/– mice fed a palmitate-enriched diet for three months, do not exhibit the increase in BACE1 protein levels as well as the accompanying increase in sAβPPβ and CTFβ levels concomitant with a decrease in sAβPPα and CTFα levels, to the same degree in the hippocampal region compared to the C57BL/6J wild-type mice fed a palmitate-enriched diet. B, C) Chop–/– mice fed a palmitate-enriched diet do not exhibit the increase in BACE1 mRNA expression (B) and BACE1 activity (C), to the same degree in the hippocampal region compared to the C57BL/6J wild-type mice fed a palmitate-enriched diet. D) ELISA immunoassays show that the Chop–/– mice fed a palmitate-enriched diet have significantly lower levels of total formic acid-soluble Aβ1 - 40 and Aβ1 - 42 species in the hippocampus, compared to the C57BL/6J wild-type mice fed a palmitate-enriched diet. Data is expressed as Mean±S.D and includes determination made in six (n = 6) different animals from each group. *p < 0.05, ***p < 0.001 versus C57BL/6J wild-type mice fed a control chow diet; †p < 0.05, ††p < 0.01, versus C57BL/6J wild-type mice fed a palmitate-enriched diet; Δp < 0.05 versus Chop–/– mice fed a control chow diet. PA, palmitic acid.
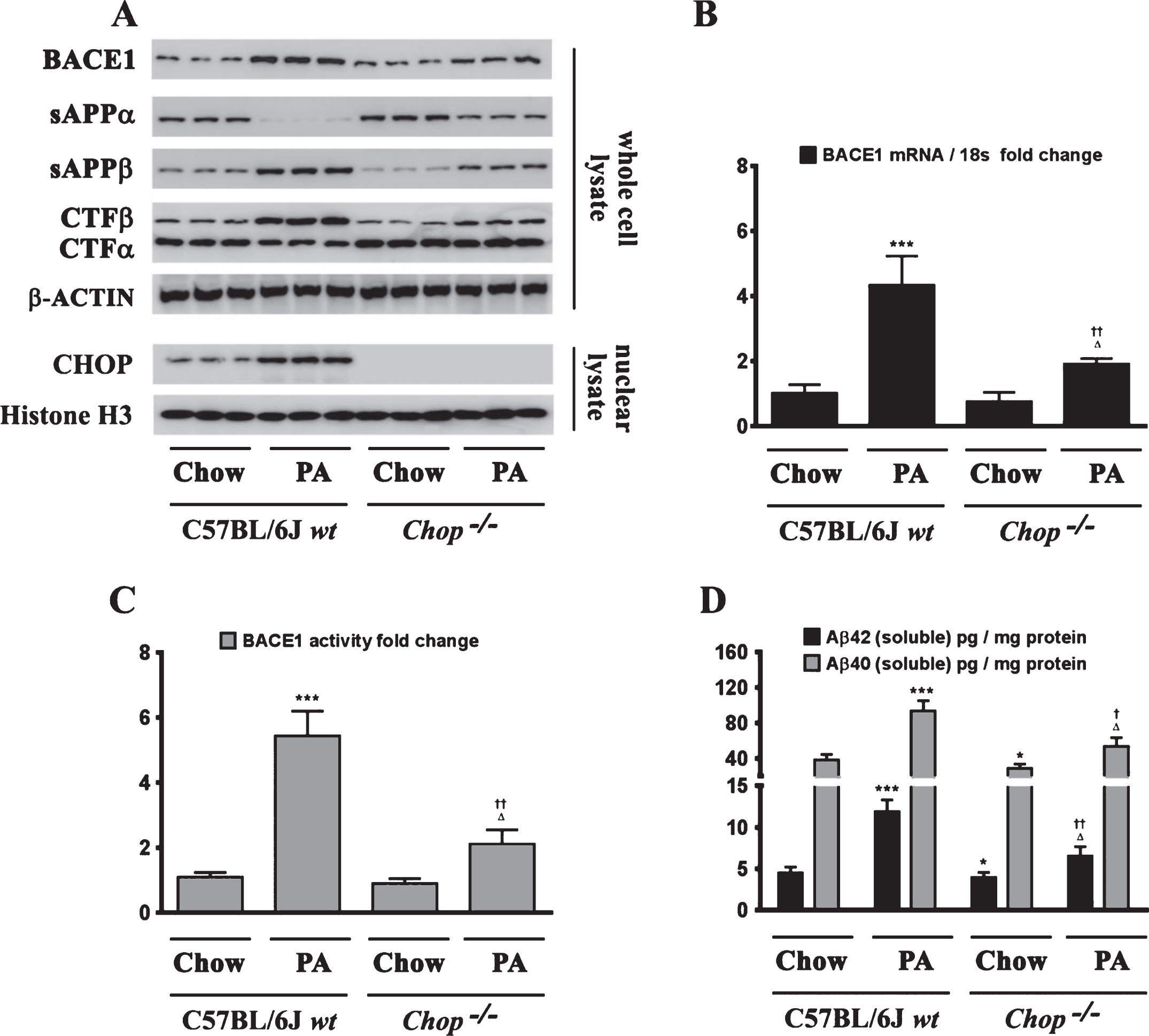
Fig.7
Ectopic overexpression of native CHOP, but not the transcriptionally dead leucine zipper domain deficient mutant CHOP (CHOP LZ–), evokes an increase in BACE1 expression and subsequent Aβ genesis. A) Representative western blots show that the leucine zipper domain of CHOP that is responsible for its transcriptional activity, is necessary to induce BACE1 protein levels as well as the accompanying increase in sAβPPβ and CTFβ levels concomitant with a decrease in sAβPPα and CTFα levels in SH-SY5Y-APPSwe cells. C, D) Ectopic overexpression of native CHOP (wt CHOP), but not the transcriptionally dead leucine zipper domain deficient mutant CHOP (CHOP LZ–), elicits an increase in BACE1 mRNA expression (B) and BACE1 activity (C) in SH-SY5Y-APPSwe cells. D) ELISA immunoassays show that the ectopic overexpression of native CHOP (wt CHOP), but not the transcriptionally dead leucine zipper domain deficient mutant CHOP (CHOP LZ–), increases the levels of the intracellular Aβ1 - 42 species in the whole cell lysates and secreted Aβ1 - 42 species in the conditioned media, from SH-SY5Y-APPSwe cells. Data is expressed as Mean±S.D and includes determination made in three (n = 3) separate cell culture experiments. ***p < 0.001 versus empty vector (EV)-transfected cells.
DISCUSSION
There is a plethora of epidemiological evidence linking the intake of a diet rich in saturated fat and the risk for developing AD [6–10]. The Rotterdam study in the Netherlands which included 5,395 participants, aged 55 years and older, concluded a positive correlation between the intake of a diet rich in saturated fat and cognitive decline with an increased risk of developing AD [7]. In the United States, The Washington Heights–Inwood Columbia Aging Project (WHICAP) and The Chicago Health and Aging Project (CHAP) that included 980 New Yorkers aged ≥65 years and 895 Chicagoans aged 65–94 respectively, also found a positive association between saturated fat intake and cognitive dysfunction leading to an increased risk for developing AD [10]. A multitude of laboratory studies have also recapitulated this detrimental aspect of a diet-enriched in saturated fat, specifically palmitate-enriched diet, on learning and memory deficits and cognitive impairment in several rodent models for assessing learning, memory, and cognitive function [13, 14]. Conditioned media from palmitate-treated astrocytes has been shown to upregulate BACE1 expression in primary cortical neurons [42] by inducing serine-palmitoyltransferase expression-mediated increase in ceramide synthesis [43] by the astrocytes that culminates in sphingomyelinase and STAT3 (Signal Transducer and Activator of Transcription 3)-induced increase in BACE1 expression levels in neurons [44, 45]. Recent studies have shown that feeding female 3xTg-AD mice a high-fat diet for nine months results in a six-fold increase in soluble Aβ levels in the cerebral cortex [46, 47]. While there is a plethora of other laboratory studies implicating high-fat diets in the positive regulation of BACE1 activity and subsequent increase in amyloidogenic processing of AβPP leading to enhanced brain deposition of Aβ in several transgenic mouse models of AD [47–52], there is a paucity of studies and data implicating a palmitate-enriched diet per se in eliciting an increase in brain Aβ levels and deposition in mice. Also, some of the aforementioned studies and other laboratory studies used a high fat–high sucrose diet [53] or a high fat–high cholesterol diet paradigm [48, 50, 54–56], that differs significantly in the macromolecular composition of the nutrients among the different studies as well as differing significantly from our diet feeding paradigm. Moreover, our study entailed and recapitulated a very important facet in energy metabolism, utilizing the respective diets that were isocaloric, unlike all the aforementioned studies that did not utilize diets that had parity in caloric content and density. To date, very few studies have determined the effects of a palmitate-enriched diet per se, and not high-fat diet, on the transcriptional regulation of BACE1, BACE1 activity, and AβPP processing in mouse models [57]. Our study emphasizes and delineates a unique mechanism that involves the induction of ER stress and CHOP expression in the hippocampi of wild-type fed a palmitate-enriched diet. The novelty of our study lies in the delineation and elucidation of a unique mechanism that involves the induction of ER stress and CHOP expression that culminates in the increase expression and activity of BACE1 in the hippocampi of wild-type mice fed a palmitate-enriched diet. Our study shows that palmitate-induced ER stress upregulates the expression and activity of BACE1 by activating the ER stress-associated transcription factor CHOP, which is pivotal in mediating the palmitate-induced increase in BACE1 expression and subsequent Aβ production.
Our study highlights a critical and indispensable role of the ER stress-associated transcription factor CHOP in mediating the effects of palmitate on inducing BACE1 expression and Aβ genesis. Changes in ER stress signaling have been reported in AD patients and various mouse models of AD [58, 59]. The phosphorylation of PERK and its downstream substrate eIF2α, as well as the expression of ATF4 and CHOP is markedly augmented in the cortex [60] and the hippocampal CA1 region [61] of the AD brain. Additionally, CHOP transcriptional activity is also enhanced in temporal cortex of AD patients [62]. Increased phosphorylation of PERK and its downstream substrate eIF2α, as well as higher levels of sXBP1, have also been reported in several mouse models of AD [58]. The induction of ER stress and phosphorylation of PERK and its downstream substrate eIF2α, as well as the expression of ATF4 and CHOP positively correlates with the Braak scores that signify the severity of AD [61]. The increased phosphorylation of PERK and eIF2α, as well as higher levels of sXBP1 that have also been reported in several mouse models of AD also positively correlate with tau tangle load in rTg4510 and 5xFAD AD mouse models [58]. Furthermore, aging, which is the single most important determinant risk factor for developing AD, is associated with increased expression of basal as well as inducible CHOP [63]. Our study clearly shows that palmitate causes ER stress in cultured neuroblastoma cells and mouse brain and that ER stress-induced CHOP activation leads to increased BACE1 expression and Aβ genesis. Obviating ER stress in neuroblastoma cells by treatment with the molecular chaperone 4-PBA, that inhibits ER stress, precludes the palmitate-induced elevation in BACE1 expression and Aβ genesis. Furthermore, we show that CHOP mediates the palmitate induced upregulation in BACE1 expression and Aβ genesis. Our study therefore suggests ER stress as a conduit for palmitate-induced deleterious effects that may serve as an instigator in the multitude of neurodegenerative processes that ensue. Interestingly, Aβ itself has been proposed to be upstream of ER stress signaling as it has been shown to induce ER stress and CHOP expression in cultured cells [64, 65]. We have shown that ER stress drives and instigates BACE1 expression and Aβ genesis, but it is also possible that Aβ may serve as a positive-feedback factor that may foster and exacerbate ER stress and consequently positively regulates BACE1 expression as well as its own production [65, 66]. However, knocking-down CHOP in SH-SY5Y cells and knocking-out Chop in C57BL/6J mice did not completely abrogate the palmitate-induced increase in BACE1 protein levels, BACE1 activity, and Aβ1 - 42 levels. This can be attributed to other signaling pathways and molecular mechanisms summoned by palmitate that could lead to an increase in BACE1 expression and activity leading to an increase in Aβ genesis. Studies using primary astrocytic and primary neuronal cultures have shown that exogenous palmitate-treated conditioned medium from astrocytes increases BACE1 expression in cultured neurons [42] via increased STAT3 transcriptional activity [45]. Exogenous palmitate treatment of astrocytes results in increased ceramide production and secretion by astrocytes [43] that causes an increase in the levels pro-inflammatory cytokines, such as TNFα (Tumor Necrosis Factor alpha) and IL1β (Interleukin 1 beta), that culminates in an increase in STAT3-mediated BACE1 expression. In rodents, peripheral administration of saturated fatty acids, such as stearate and palmitate, has been shown to result in their accumulation in the glial cells [67, 68] and evoke the expression of pro-inflammatory cytokines by astrocytes [69, 70] and microglia [71, 72] leading to a deleterious neuroinflammatory cascade [73]. Saturated fatty acids, such as palmitate, are known to activate Toll-like Receptor 2 (TLR2) and TLR4 signaling pathway-mediated increase in pro-inflammatory cytokine expression in monocytes and macrophages [74]. Recent studies have implicated deranged TLR2 and TLR4 signaling in the pathogenic mechanisms involved in AD [75–78]. Activated TLR2 and TLR4 signaling results in a profound increase in the expression of pro-inflammatory cytokines, such as TNFα, IL1β, IL6, and IL18, in the astrocytes and microglia [79], while causing apoptotic death in neurons [80]. The etio-pathological role of the aforementioned pro-inflammatory cytokines in AD is well characterized [81, 82] and the expression of BACE1 and Aβ genesis is regulated by TNFα [83–85], IL1β [84–87], IL6 [81, 85], and IL18 [82]. However, there are no published studies yet, addressing the effects of palmitate on the TLR2 and TLR4 signaling pathways in the glial cells and neurons and the extent to which these pathways contribute to palmitate-induced upregulation in pro-inflammatory cytokine expression and consequent augmentation in BACE1 expression as well as Aβ genesis. Our current studies are catered toward and honing in on these signaling pathways activated by palmitate that could lead to increased pro-inflammatory cytokine-dependent augmentation in BACE1 expression and Aβ genesis. Contemporary studies have shown that palmitate-induced pro-inflammatory cytokine expression is mediated by NF-κB (Nuclear factor of kappa-light-polypeptide gene enhancer of activated B cells) activation in 3T3-L1 adipocytes [88], endothelial cells [89], and macrophages [90]. NF-κB signaling and transcriptional activity is directly involved in the transcriptional control of BACE1 and Aβ production [85, 91–95]. Furthermore, saturated fatty acids, such as palmitate, have been shown to induce NF-κB activation via TLR signaling-dependent and-independent mechanisms [70, 90, 96–99]. Current studies in our laboratory are underway to examine the involvement of palmitate-induced NF-κB activation in the transcriptional upregulation of BACE1 and the subsequent enhancement of Aβ production. Emerging evidence has implicated the presence of the preassembled multi-protein complex inflammasome-mediated inflammatory cascade in the brain [100]. Inflammasomes composed of NLRP proteins (NOD-like receptors pyrin domain containing), NLRP1 [101, 102], NLRP2 [103], and NLRP3 [104, 105] have been identified in the brain and characterized as integral entities in eliciting neuroinflammatory responses. Furthermore, NLRP1 and NLRP3-mediated inflammatory responses are involved in the AD brain [106–109]. Palmitate, and not oleate, has been shown to activate the NLRP3 inflammatory cascade in peripheral tissues and induce insulin resistance [110, 111]. Further studies are warranted to determine the status of palmitate-induced NLRP3 inflammatory cascade in the brain and subsequent downstream deleterious consequences. The NLRC4 inflammasome (NOD like receptors family CARD domain-containing protein 4), also known as the IPAF (ice protease-activating factor) inflammasome, is also activated by palmitate in primary rat astrocytes [82, 112]. Palmitate-induced increase in the mRNA and protein levels of IPAF underlies the palmitate-induced increase in IL1β production in primary rat astrocytes [82, 112]. Furthermore, exogenous palmitate-induced increase in Aβ production in rat primary neurons is contingent on the expression and activation of the astrocytic IPAF inflammasome, as silencing IPAF in rat primary astrocytes compromises the ability of the conditioned medium from the exogenous-treated rat primary astrocytes to evoke an increase in Aβ secretion by primary neurons in culture [112]. Moreover, the mRNA expression and protein levels of IPAF are significantly increased in the neocortex of the AD brains [112]. It is also possible that the palmitate enriched diet-induced peripheral insulin resistance, fluxes in glucose disposal and hyperglycemia, and lipid dyshomeostasis can be a contributing factor and exacerbate the ER stress and other pathophysiological mechanisms relevant to AD in the hippocampus. We have therefore expanded our studies and are examining the effects of feeding our cohorts of mice a palmitate-enriched diet for six months and nine months. This feeding paradigm will allow us to determine the involvement of palmitate-induced peripheral insulin resistance and lipid dyshomeostasis as a contributing factor to the ER stress in the hippocampus.
Our current study delineates and elucidates the role of ER stress-induced CHOP expression in mediating the palmitate-induced upregulation in BACE1 expression and Aβ genesis. Our previous studies have shown that silencing CHOP expression in SH-SY5Y cells attenuates the 27-hydroxycholesterol-induced increase in BACE1 expression [27]. The present study expanded and delved further into the role of CHOP in the exogenous palmitate-mediated upregulation of BACE1 expression, BACE1 activity, and Aβ production in SH-SY5Y cells. More importantly, the present study determined the role of CHOP in the palmitate-enriched diet-induced upregulation in BACE1 expression, BACE1 activity, and Aβ genesis in the hippocampi of mice. Further studies are warranted to determine the role of ER stress-induced CHOP activation in palmitate-induced cognitive dysfunction as well as learning and memory deficits.
ACKNOWLEDGMENTS
This work was supported by a Grant from the NIH (R01AG0145264) to Dr. Othman Ghribi. We sincerely thank the effort and dedication of the personnel of the Animal Research Facility at the University of North Dakota Center for Biomedical Research.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-1130r2).
REFERENCES
[1] | Haass C , Selkoe DJ ((1993) ) Cellular processing of beta-amyloid precursor protein and the genesis of amyloid beta-peptide. Cell 75: , 1039–1042. |
[2] | Fukumoto H , Cheung BS , Hyman BT , Irizarry MC ((2002) ) Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol 59: , 1381–1389. |
[3] | Holsinger RM , McLean CA , Beyreuther K , Masters CL , Evin G ((2002) ) Increased expression of the amyloid precursor beta-secretase in Alzheimer’s disease. Ann Neurol 51: , 783–786. |
[4] | Li R , Lindholm K , Yang LB , Yue X , Citron M , Yan R , Beach T , Sue L , Sabbagh M , Cai H , Wong P , Price D , Shen Y ((2004) ) Amyloid beta peptide load is correlated with increased beta-secretase activity in sporadic Alzheimer’s disease patients. Proc Natl Acad Sci U S A 101: , 3632–3637. |
[5] | Yang LB , Lindholm K , Yan R , Citron M , Xia W , Yang XL , Beach T , Sue L , Wong P , Price D , Li R , Shen Y ((2003) ) Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med 9: , 3–4. |
[6] | Kalmijn S ((2000) ) Fatty acid intake and the risk of dementia and cognitive decline: A review of clinical and epidemiological studies. J Nutr Health Aging 4: , 202–207. |
[7] | Kalmijn S , Launer LJ , Ott A , Witteman JC , Hofman A , Breteler MM ((1997) ) Dietary fat intake and the risk of incident dementia in the Rotterdam Study. Ann Neurol 42: , 776–782. |
[8] | Morris MC , Evans DA , Bienias JL , Tangney CC , Bennett DA , Aggarwal N , Schneider J , Wilson RS ((2003) ) Dietary fats and the risk of incident Alzheimer disease. Arch Neurol 60: , 194–200. |
[9] | Parrott MD , Greenwood CE ((2007) ) Dietary influences on cognitive function with aging: From high-fat diets to healthful eating. Ann N Y Acad Sci 1114: , 389–397. |
[10] | Barnard ND , Bunner AE , Agarwal U ((2014) ) Saturated and trans fats and dementia: A systematic review. Neurobiol Aging 35: (Suppl 2), S65–S73. |
[11] | Grant WB ((1999) ) Dietary links to Alzheimer’s disease: 1999 update. J Alzheimers Dis 1: , 197–201. |
[12] | Solfrizzi V , D’Introno A , Colacicco AM , Capurso C , Del Parigi A , Capurso S , Gadaleta A , Capurso A , Panza F ((2005) ) Dietary fatty acids intake: Possible role in cognitive decline and dementia. Exp Gerontol 40: , 257–270. |
[13] | Greenwood CE , Winocur G ((1990) ) Learning and memory impairment in rats fed a high saturated fat diet. Behav Neural Biol 53: , 74–87. |
[14] | Winocur G , Greenwood CE ((1999) ) The effects of high fat diets and environmental influences on cognitive performance in rats. Behav Brain Res 101: , 153–161. |
[15] | Ulmann L , Mimouni V , Roux S , Porsolt R , Poisson JP ((2001) ) Brain and hippocampus fatty acid composition in phospholipid classes of aged-relative cognitive deficit rats. Prostaglandins Leukot Essent Fatty Acids 64: , 189–195. |
[16] | Lovejoy JC , Smith SR , Champagne CM , Most MM , Lefevre M , DeLany JP , Denkins YM , Rood JC , Veldhuis J , Bray GA ((2002) ) Effects of diets enriched in saturated (palmitic), monounsaturated (oleic), or trans (elaidic) fatty acids on insulin sensitivity and substrate oxidation in healthy adults. Diabetes Care 25: , 1283–1288. |
[17] | Greenwood CE , Winocur G ((2005) ) High-fat diets, insulin resistance and declining cognitive function. Neurobiol Aging 26: (Suppl 1), 42–45. |
[18] | Craft S , Dagogo-Jack SE , Wiethop BV , Murphy C , Nevins RT , Fleischman S , Rice V , Newcomer JW , Cryer PE ((1993) ) Effects of hyperglycemia on memory and hormone levels in dementia of the Alzheimer type: A longitudinal study. Behav Neurosci 107: , 926–940. |
[19] | Dhopeshwarkar GA , Mead JF ((1973) ) Uptake and transport of fatty acids into the brain and the role of the blood-brain barrier system. Adv Lipid Res 11: , 109–142. |
[20] | Hamilton JA , Brunaldi K ((2007) ) A model for fatty acid transport into the brain. J Mol Neurosci 33: , 12–17. |
[21] | Rapoport SI ((2001) ) In vivo fatty acid incorporation into brain phosholipids in relation to plasma availability, signal transduction and membrane remodeling. J Mol Neurosci 16: , 243–261; discussion 279-284. |
[22] | Hotamisligil GS ((2010) ) Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140: , 900–917. |
[23] | Karaskov E , Scott C , Zhang L , Teodoro T , Ravazzola M , Volchuk A ((2006) ) Chronic palmitate but not oleate exposure induces endoplasmic reticulum stress, which may contribute to INS-1 pancreatic beta-cell apoptosis. Endocrinology 147: , 3398–3407. |
[24] | Kharroubi I , Ladriere L , Cardozo AK , Dogusan Z , Cnop M , Eizirik DL ((2004) ) Free fatty acids and cytokines induce pancreatic beta-cell apoptosis by different mechanisms: Role of nuclear factor-kappaB and endoplasmic reticulum stress. Endocrinology 145: , 5087–5096. |
[25] | Wang D , Wei Y , Pagliassotti MJ ((2006) ) Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology 147: , 943–951. |
[26] | Wei Y , Wang D , Topczewski F , Pagliassotti MJ ((2006) ) Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab 291: , E275–E281. |
[27] | Marwarha G , Raza S , Prasanthi JR , Ghribi O ((2013) ) Gadd153 and NF-kappaB crosstalk regulates 27-hydroxycholesterol-induced increase in BACE1 and beta-amyloid production in human neuroblastoma SH-SY5Y cells. PLoS One 8: , e70773. |
[28] | Marwarha G , Claycombe K , Schommer J , Collins D , Ghribi O ((2016) ) Palmitate-induced Endoplasmic Reticulum stress and subsequent C/EBPalpha Homologous Protein activation attenuates leptin and Insulin-like growth factor 1 expression in the brain. Cell Signal 28: , 1789–1805. |
[29] | Marwarha G , Dasari B , Ghribi O ((2012) ) Endoplasmic reticulum stress-induced CHOP activation mediates the down-regulation of leptin in human neuroblastoma SH-SY5Y cells treated with the oxysterol 27-hydroxycholesterol. Cell Signal 24: , 484–492. |
[30] | Marwarha G , Rhen T , Schommer T , Ghribi O ((2011) ) The oxysterol 27-hydroxycholesterol regulates alpha-synuclein and tyrosine hydroxylase expression levels in human neuroblastoma cells through modulation of liver X receptors and estrogen receptors–relevance to Parkinson’s disease. J Neurochem 119: , 1119–1136. |
[31] | Marwarha G , Berry DC , Croniger CM , Noy N ((2014) ) The retinol esterifying enzyme LRAT supports cell signaling by retinol-binding protein and its receptor STRA6. FASEB J 28: , 26–34. |
[32] | Marwarha G , Prasanthi JR , Schommer J , Dasari B , Ghribi O ((2011) ) Molecular interplay between leptin, insulin-like growth factor-1, and beta-amyloid in organotypic slices from rabbit hippocampus. Mol Neurodegener 6: , 41. |
[33] | Marwarha G , Raza S , Meiers C , Ghribi O ((2014) ) Leptin attenuates BACE1 expression and amyloid-beta genesis via the activation of SIRT1 signaling pathway. Biochim Biophys Acta 1842: , 1587–1595. |
[34] | Marwarha G , Dasari B , Prasanthi JR , Schommer J , Ghribi O ((2010) ) Leptin reduces the accumulation of Abeta and phosphorylated tau induced by 27-hydroxycholesterol in rabbit organotypic slices. J Alzheimers Dis 19: , 1007–1019. |
[35] | Rostagno A , Ghiso J ((2009) ) Isolation and biochemical characterization of amyloid plaques and paired helical filaments. Curr Protoc Cell Biol Chapter 3: , Unit 3.33 3.33.1-33. |
[36] | Marwarha G , Dasari B , Prabhakara JP , Schommer J , Ghribi O ((2010) ) beta-Amyloid regulates leptin expression and tau phosphorylation through the mTORC1 signaling pathway. J Neurochem 115: , 373–384. |
[37] | Bazan NG Jr ((1970) ) Effects of ischemia and electroconvulsive shock on free fatty acid pool in the brain. Biochim Biophys Acta 218: , 1–10. |
[38] | Rehncrona S , Westerberg E , Akesson B , Siesjo BK ((1982) ) Brain cortical fatty acids and phospholipids during and following complete and severe incomplete ischemia. J Neurochem 38: , 84–93. |
[39] | Yoshida S , Harik SI , Busto R , Santiso M , Martinez E , Ginsberg MD ((1984) ) Free fatty acids and energy metabolites in ischemic cerebral cortex with noradrenaline depletion. J Neurochem 42: , 711–717. |
[40] | Yoshida S , Inoh S , Asano T , Sano K , Shimasaki H , Ueta N ((1983) ) Brain free fatty acids, edema, and mortality in gerbils subjected to transient, bilateral ischemia, and effect of barbiturate anesthesia. J Neurochem 40: , 1278–1286. |
[41] | Spector AA ((1975) ) Fatty acid binding to plasma albumin. J Lipid Res 16: , 165–179. |
[42] | Patil S , Sheng L , Masserang A , Chan C ((2006) ) Palmitic acid-treated astrocytes induce BACE1 upregulation and accumulation of C-terminal fragment of APP in primary cortical neurons. Neurosci Lett 406: , 55–59. |
[43] | Patil S , Melrose J , Chan C ((2007) ) Involvement of astroglial ceramide in palmitic acid-induced Alzheimer-like changes in primary neurons. Eur J Neurosci 26: , 2131–2141. |
[44] | Liu L , Martin R , Chan C ((2013) ) Palmitate-activated astrocytes via serine palmitoyltransferase increase BACE1 in primary neurons by sphingomyelinases. Neurobiol Aging 34: , 540–550. |
[45] | Liu L , Martin R , Kohler G , Chan C ((2013) ) Palmitate induces transcriptional regulation of BACE1 and presenilin by STAT3 in neurons mediated by astrocytes. Exp Neurol 248: , 482–490. |
[46] | Vandal M , White PJ , Tremblay C , St-Amour I , Chevrier G , Emond V , Lefrancois D , Virgili J , Planel E , Giguere Y , Marette A , Calon F ((2014) ) Insulin reverses the high-fat diet-induced increase in brain Abeta and improves memory in an animal model of Alzheimer disease. Diabetes 63: , 4291–4301. |
[47] | Julien C , Tremblay C , Phivilay A , Berthiaume L , Emond V , Julien P , Calon F ((2010) ) High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol Aging 31: , 1516–1531. |
[48] | Refolo LM , Malester B , LaFrancois J , Bryant-Thomas T , Wang R , Tint GS , Sambamurti K , Duff K , Pappolla MA ((2000) ) Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol Dis 7: , 321–331. |
[49] | Ho L , Qin W , Pompl PN , Xiang Z , Wang J , Zhao Z , Peng Y , Cambareri G , Rocher A , Mobbs CV , Hof PR , Pasinetti GM ((2004) ) Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J 18: , 902–904. |
[50] | Fitz NF , Cronican A , Pham T , Fogg A , Fauq AH , Chapman R , Lefterov I , Koldamova R ((2010) ) Liver X receptor agonist treatment ameliorates amyloid pathology and memory deficits caused by high-fat diet in APP23 mice. J Neurosci 30: , 6862–6872. |
[51] | Maesako M , Uemura K , Kubota M , Kuzuya A , Sasaki K , Asada M , Watanabe K , Hayashida N , Ihara M , Ito H , Shimohama S , Kihara T , Kinoshita A ((2012) ) Environmental enrichment ameliorated high-fat diet-induced Abeta deposition and memory deficit in APP transgenic mice. Neurobiol Aging 33: , 1011 e1011–1023. |
[52] | Maesako M , Uemura M , Tashiro Y , Sasaki K , Watanabe K , Noda Y , Ueda K , Asada-Utsugi M , Kubota M , Okawa K , Ihara M , Shimohama S , Uemura K , Kinoshita A ((2015) ) High fat diet enhances beta-site cleavage of amyloid precursor protein (APP) via promoting beta-site APP cleaving enzyme 1/adaptor protein 2/clathrin complex formation. PLoS One 10: , e0131199. |
[53] | Cao D , Lu H , Lewis TL , Li L ((2007) ) Intake of sucrose-sweetened water induces insulin resistance and exacerbates memory deficits and amyloidosis in a transgenic mouse model of Alzheimer disease. J Biol Chem 282: , 36275–36282. |
[54] | Thirumangalakudi L , Prakasam A , Zhang R , Bimonte-Nelson H , Sambamurti K , Kindy MS , Bhat NR ((2008) ) High cholesterol-induced neuroinflammation and amyloid precursor protein processing correlate with loss of working memory in mice. J Neurochem 106: , 475–485. |
[55] | Wang R , Li JJ , Diao S , Kwak YD , Liu L , Zhi L , Bueler H , Bhat NR , Williams RW , Park EA , Liao FF ((2013) ) Metabolic stress modulates Alzheimer’s beta-secretase gene transcription via SIRT1-PPARgamma-PGC-1 in neurons. Cell Metab 17: , 685–694. |
[56] | Levin-Allerhand JA , Lominska CE , Smith JD ((2002) ) Increased amyloid- levels in APPSWE transgenic mice treated chronically with a physiological high-fat high-cholesterol diet. J Nutr Health Aging 6: , 315–319. |
[57] | Geekiyanage H , Upadhye A , Chan C ((2013) ) Inhibition of serine palmitoyltransferase reduces Abeta and tau hyperphosphorylation in a murine model: A safe therapeutic strategy for Alzheimer’s disease. Neurobiol Aging 34: , 2037–2051. |
[58] | Endres K , Reinhardt S ((2013) ) ER-stress in Alzheimer’s disease: Turning the scale? Am J Neurodegener Dis 2: , 247–265. |
[59] | Marwarha G , Ghribi O ((2012) ) Leptin signaling and Alzheimer’s disease. Am J Neurodegener Dis 1: , 245–265. |
[60] | Yoon SO , Park DJ , Ryu JC , Ozer HG , Tep C , Shin YJ , Lim TH , Pastorino L , Kunwar AJ , Walton JC , Nagahara AH , Lu KP , Nelson RJ , Tuszynski MH , Huang K ((2012) ) JNK3 perpetuates metabolic stress induced by Abeta peptides. Neuron 75: , 824–837. |
[61] | Hoozemans JJ , van Haastert ES , Nijholt DA , Rozemuller AJ , Eikelenboom P , Scheper W ((2009) ) The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J Pathol 174: , 1241–1251. |
[62] | Lee JH , Won SM , Suh J , Son SJ , Moon GJ , Park UJ , Gwag BJ ((2010) ) Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Exp Mol Med 42: , 386–394. |
[63] | Kirkland JL , Tchkonia T , Pirtskhalava T , Han J , Karagiannides I ((2002) ) Adipogenesis and aging: Does aging make fat go MAD? Exp Gerontol 37: , 757–767. |
[64] | Fonseca AC , Ferreiro E , Oliveira CR , Cardoso SM , Pereira CF ((2013) ) Activation of the endoplasmic reticulum stress response by the amyloid-beta 1-40 peptide in brain endothelial cells. Biochim Biophys Acta 1832: , 2191–2203. |
[65] | Marwarha G , Ghribi O ((2012) ) Cellular model of Alzheimer’s disease–relevance to therapeutic testing. Exp Neurol 233: , 733–739. |
[66] | Marwarha G , Ghribi O ((2015) ) Does the oxysterol 27-hydroxycholesterol underlie Alzheimer’s disease-Parkinson’s disease overlap? Exp Gerontol 68: , 13–18. |
[67] | Morand O , Baumann N , Bourre JM ((1979) ) In vivo incorporation of exogenous [1-14C]stearic acid into neurons and astrocytes. Neurosci Lett 13: , 177–181. |
[68] | Mitchell RW , Hatch GM ((2011) ) Fatty acid transport into the brain: Of fatty acid fables and liid tails. Prostaglandins Leukot Essent Fatty Acids 85: , 293–302. |
[69] | Gupta S , Knight AG , Gupta S , Keller JN , Bruce-Keller AJ ((2012) ) Saturated long-chain fatty acids activate inflammatory signaling in astrocytes. J Neurochem 120: , 1060–1071. |
[70] | Milanski M , Degasperi G , Coope A , Morari J , Denis R , Cintra DE , Tsukumo DM , Anhe G , Amaral ME , Takahashi HK , Curi R , Oliveira HC , Carvalheira JB , Bordin S , Saad MJ , Velloso LA ((2009) ) Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: Implications for the pathogenesis of obesity. J Neurosci 29: , 359–370. |
[71] | Tracy LM , Bergqvist F , Ivanova EV , Jacobsen KT , Iverfeldt K ((2013) ) Exposure to the saturated free fatty acid palmitate alters BV-2 microglia inflammatory response. J Mol Neurosci 51: , 805–812. |
[72] | Duffy CM , Yuan C , Wisdorf LE , Billington CJ , Kotz CM , Nixon JP , Butterick TA ((2015) ) Role of orexin A signaling in dietary palmitic acid-activated microglial cells. Neurosci Lett 606: , 140–144. |
[73] | Cai D ((2013) ) Neuroinflammation and neurodegeneration in overnutrition-induced diseases. Trends Endocrinol Metab 24: , 40–47. |
[74] | Huang S , Rutkowsky JM , Snodgrass RG , Ono-Moore KD , Schneider DA , Newman JW , Adams SH , Hwang DH ((2012) ) Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J Lipid Res 53: , 2002–2013. |
[75] | Landreth GE , Reed-Geaghan EG ((2009) ) Toll-like receptors in Alzheimer’s disease. Curr Top Microbiol Immunol 336: , 137–153. |
[76] | Walter S , Letiembre M , Liu Y , Heine H , Penke B , Hao W , Bode B , Manietta N , Walter J , Schulz-Schuffer W , Fassbender K ((2007) ) Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol Biochem 20: , 947–956. |
[77] | Gambuzza ME , Sofo V , Salmeri FM , Soraci L , Marino S , Bramanti P ((2014) ) Toll-like receptors in Alzheimer’s disease: A therapeutic perspective. CNS Neurol Disord Drug Targets 13: , 1542–1558. |
[78] | Huang NQ , Jin H , Zhou SY , Shi JS , Jin F ((2017) ) TLR4 is a link between diabetes and Alzheimer’s disease. Behav Brain Res 316: , 234–244. |
[79] | Trudler D , Farfara D , Frenkel D ((2010) ) Toll-like receptors expression and signaling in glia cells in neuro-amyloidogenic diseases: Towards future therapeutic application. Mediators Inflamm 2010,: , pii: 497987. |
[80] | Tang SC , Arumugam TV , Xu X , Cheng A , Mughal MR , Jo DG , Lathia JD , Siler DA , Chigurupati S , Ouyang X , Magnus T , Camandola S , Mattson MP ((2007) ) Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proc Natl Acad Sci U S A 104: , 13798–13803. |
[81] | Wang WY , Tan MS , Yu JT , Tan L ((2015) ) Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med 3: , 136. |
[82] | Liu L , Chan C ((2014) ) The role of inflammasome in Alzheimer’s disease. Ageing Res Rev 15: , 6–15. |
[83] | Yamamoto M , Kiyota T , Horiba M , Buescher JL , Walsh SM , Gendelman HE , Ikezu T ((2007) ) Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol 170: , 680–692. |
[84] | Blasko I , Veerhuis R , Stampfer-Kountchev M , Saurwein-Teissl M , Eikelenboom P , Grubeck-Loebenstein B ((2000) ) Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of Abeta1-40 and Abeta1-42 by human astrocytes. Neurobiol Dis 7: , 682–689. |
[85] | Cole SL , Vassar R ((2007) ) The basic biology of BACE1: A key therapeutic target for Alzheimer’s disease. Curr Genomics 8: , 509–530. |
[86] | Shaftel SS , Griffin WS , O’Banion MK ((2008) ) The role of interleukin-1 in neuroinflammation and Alzheimer disease: An evolving perspective. J Neuroinflammation 5: , 7. |
[87] | Griffin WS , Sheng JG , Royston MC , Gentleman SM , McKenzie JE , Graham DI , Roberts GW , Mrak RE ((1998) ) Glial-neuronal interactions in Alzheimer’s disease: The potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol 8: , 65–72. |
[88] | Ajuwon KM , Spurlock ME ((2005) ) Palmitate activates the NF-kappaB transcription factor and induces IL-6 and TNFalpha expression in 3T3-L1 adipocytes. J Nutr 135: , 1841–1846. |
[89] | Maloney E , Sweet IR , Hockenbery DM , Pham M , Rizzo NO , Tateya S , Handa P , Schwartz MW , Kim F ((2009) ) Activation of NF-kappaB by palmitate in endothelial cells: A key role for NADPH oxidase-derived superoxide in response to TLR4 activation. Arterioscler Thromb Vasc Biol 29: , 1370–1375. |
[90] | Suganami T , Tanimoto-Koyama K , Nishida J , Itoh M , Yuan X , Mizuarai S , Kotani H , Yamaoka S , Miyake K , Aoe S , Kamei Y , Ogawa Y ((2007) ) Role of the Toll-like receptor 4/NF-kappaB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler Thromb Vasc Biol 27: , 84–91. |
[91] | Bourne KZ , Ferrari DC , Lange-Dohna C , Rossner S , Wood TG , Perez-Polo JR ((2007) ) Differential regulation of BACE1 promoter activity by nuclear factor-kappaB in neurons and glia upon exposure to beta-amyloid peptides. J Neurosci Res 85: , 1194–1204. |
[92] | Buggia-Prevot V , Sevalle J , Rossner S , Checler F ((2008) ) NFkappaB-dependent control of BACE1 promoter transactivation by Abeta42. J Biol Chem 283: , 10037–10047. |
[93] | Chami L , Buggia-Prevot V , Duplan E , Delprete D , Chami M , Peyron JF , Checler F ((2012) ) Nuclear factor-kappaB regulates betaAPP and beta- and gamma-secretases differently at physiological and supraphysiological Abeta concentrations. J Biol Chem 287: , 24573–24584. |
[94] | Chen CH , Zhou W , Liu S , Deng Y , Cai F , Tone M , Tone Y , Tong Y , Song W ((2012) ) Increased NF-kappaB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int J Neuropsychopharmacol 15: , 77–90. |
[95] | Chen XF , Zhang YW , Xu H , Bu G ((2013) ) Transcriptional regulation and its misregulation in Alzheimer’s disease. Mol Brain 6: , 44. |
[96] | Tsukumo DM , Carvalho-Filho MA , Carvalheira JB , Prada PO , Hirabara SM , Schenka AA , Araujo EP , Vassallo J , Curi R , Velloso LA , Saad MJ ((2007) ) Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 56: , 1986–1998. |
[97] | Lee JY , Ye J , Gao Z , Youn HS , Lee WH , Zhao L , Sizemore N , Hwang DH ((2003) ) Reciprocal modulation of Toll-like receptor-4 signaling pathways involving MyD88 and phosphatidylinositol 3-kinase/AKT by saturated and polyunsaturated fatty acids. J Biol Chem 278: , 37041–37051. |
[98] | Lee JY , Sohn KH , Rhee SH , Hwang D ((2001) ) Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem 276: , 16683–16689. |
[99] | Weatherill AR , Lee JY , Zhao L , Lemay DG , Youn HS , Hwang DH ((2005) ) Saturated and polyunsaturated fatty acids reciprocally modulate dendritic cell functions mediated through TLR4. J Immunol 174: , 5390–5397. |
[100] | Guo H , Callaway JB , Ting JP ((2015) ) Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat Med 21: , 677–687. |
[101] | de Rivero Vaccari JP , Lotocki G , Alonso OF , Bramlett HM , Dietrich WD , Keane RW ((2009) ) Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J Cereb Blood Flow Metab 29: , 1251–1261. |
[102] | Brickler T , Gresham K , Meza A , Coutermarsh-Ott S , Williams TM , Rothschild DE , Allen IC , Theus MH ((2016) ) Nonessential role for the NLRP1 inflammasome complex in a murine model of traumatic brain injury. Mediators Inflam 2016: , 6373506. |
[103] | Minkiewicz J , de Rivero Vaccari JP , Keane RW ((2013) ) Human astrocytes express a novel NLRP2 inflammasome. Glia 61: , 1113–1121. |
[104] | Liu HD , Li W , Chen ZR , Hu YC , Zhang DD , Shen W , Zhou ML , Zhu L , Hang CH ((2013) ) Expression of the NLRP3 inflammasome in cerebral cortex after traumatic brain injury in a rat model. Neurochem Res 38: , 2072–2083. |
[105] | Jha S , Srivastava SY , Brickey WJ , Iocca H , Toews A , Morrison JP , Chen VS , Gris D , Matsushima GK , Ting JP ((2010) ) The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J Neurosci 30: , 15811–15820. |
[106] | Halle A , Hornung V , Petzold GC , Stewart CR , Monks BG , Reinheckel T , Fitzgerald KA , Latz E , Moore KJ , Golenbock DT ((2008) ) The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9: , 857–865. |
[107] | Tan MS , Yu JT , Jiang T , Zhu XC , Tan L ((2013) ) The NLRP3 inflammasome in Alzheimer’s disease. Mol Neurobiol 48: , 875–882. |
[108] | Singhal G , Jaehne EJ , Corrigan F , Toben C , Baune BT ((2014) ) Inflammasomes in neuroinflammation and changes in brain function: A focused review. Front Neurosci 8: , 315. |
[109] | Saresella M , La Rosa F , Piancone F , Zoppis M , Marventano I , Calabrese E , Rainone V , Nemni R , Mancuso R , Clerici M ((2016) ) The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol Neurodegener 11: , 23. |
[110] | Wen H , Gris D , Lei Y , Jha S , Zhang L , Huang MT , Brickey WJ , Ting JP ((2011) ) Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 12: , 408–415. |
[111] | Stienstra R , Tack CJ , Kanneganti TD , Joosten LA , Netea MG ((2012) ) The inflammasome puts obesity in the danger zone. Cell Metab 15: , 10–18. |
[112] | Liu L , Chan C ((2014) ) IPAF inflammasome is involved in interleukin-1beta production from astrocytes, induced by palmitate; implications for Alzheimer’s disease. Neurobiol Aging 35: , 309–321. |


