
Acceleration of Amyloidosis by Inflammation in the Amyloid-Beta Marmoset Monkey Model of Alzheimer’s Disease
Abstract
Background: The immune system is increasingly mentioned as a potential target for Alzheimer’s disease (AD) treatment.
Objective: In the present pilot study, the effect of (neuro)inflammation on amyloidopathy was investigated in the marmoset monkey, which has potential as an AD animal model due to its natural cerebral amyloidosis similar to humans.
Methods: Six adult/aged marmosets (Callithrix jacchus) were intracranial injected with amyloid-beta (Aβ) fibrils at three cortical locations in the right hemisphere. Additionally, in half of the monkeys, lipopolysaccharide (LPS) was co-injected with the Aβ fibrils and injected in the other hemisphere without Aβ fibrils. The other three monkeys received phosphate buffered saline instead of LPS, as a control for the inflammatory state. The effect of inflammation on amyloidopathy was also investigated in an additional monkey that suffered from chronic inflammatory wasting syndrome. Mirror histology sections were analyzed to assess amyloidopathy and immune reaction, and peripheral blood for AD biomarker expression.
Results: All LPS-injected monkeys showed an early AD immune blood cell expression profile on CD95 and CD45RA. Two out of three monkeys injected with Aβ and LPS and the additional monkey, suffering from chronic inflammation, developed plaques. None of the controls, injected with Aβ only, developed any plaques.
Conclusion: This study shows the importance of immune modulation on the susceptibility for amyloidosis, a hallmark of AD, which offers new perspectives for disease modifying approaches in AD.
INTRODUCTION
Alzheimer’s disease (AD) is a severe age-related chronic neurodegenerative disorder with increasing prevalence for which still no cure exists. To identify targets for effective drug therapies, it is paramount to have insight into the pathogenic mechanisms. Although the exact cause of AD remains unknown, a pro-inflammatory state is increasingly associated with senile plaque formation, mainly consisting of amyloid-β (Aβ) aggregates, that characterizes AD pathogenesis [1].
This amyloidopathy is an early AD hallmark and constructs the basis of the amyloid cascade hypothesis as first stated by Hardy et al. [2]. Amyloidopathy is the result of proteolytic cleavage of the Aβ protein precursor by consecutive β- and γ-secretases [3]. Mainly the Aβ42 monomers, but also the Aβ43 monomers, are prone to form soluble oligomers and insoluble Aβ fibrils that form the basis of senile plaques [4–6].
The early intracellular accumulation of Aβ oligomers and extracellular Aβ fibrils elicits a detrimental reactive microgliosis via a tyrosine signaling response [7, 8]. During the AD associated inflammatory reaction, the ramified/inactive microglia cells become activated and secrete high levels of pro-inflammatory cytokines including IL-1β. These cytokines enhance Aβ protein precursor synthesis leading to an increase in amyloidopathy and neuronal damage [9–12]. Indeed, chronic inflammation has been acknowledged as an important part of the AD pathogenesis and a clear linkage is present with early AD amyloidopathy [1, 13–16].
However, the relation between amyloidopathy and the immune system is bi-directional and the causality is difficult to establish. Chronic inflammation increases the susceptibility for AD and Aβ accumulation in the brain can be initiated by peritoneal lipopolysaccharide (LPS) injection in mice [11, 12, 17]. Additionally, expression of CD95 (apoptosis receptor) and CD45RA (maturity marker) on CD4+ T-cell reflecting peripheral immune activation, which is predictive for AD, indicating immune interference [18, 19].
The involvement of an inflammatory mechanism in the pathogenesis of AD suggests that a pro-inflammatory cytokine profile combined with diffuse amyloid depositions may initiate a self-propagating process leading to plaque progression and thus the risk of developing AD [20]. Therefore, we have investigated the transmissibility of amyloidosis and the contribution of an inflammatory condition, induced by intracranial LPS injection, in plaque progression in the common marmoset (Callithrix jacchus). We used peripheral blood marker analysis (CD45RA downregulation and CD95 upregulation) to show alterations due to the rapid amyloidopathy and neuroinflammation to demonstrate the translatability of this model [18, 19]. Furthermore, brain tissue from a confirmed AD patient was used as positive control and a marmoset that had succumbed from wasting syndrome, a clinical condition associated with inflammatory bowel disease (chronic colitis) [21, 22], was used to investigate the positive natural amyloidosis as a result of chronic systemicinflammation.
The marmoset monkey is suggested as an AD animal model as amyloidosis is transmissible to this species [23, 24] and these monkeys can develop amyloidosis naturally at old age [24–26]. Their relative short life span (15 years) compared to other monkey species [27], the high resemblance of the immune system and aging phenotype to humans [28, 29] and the possibility of complex cognitive testing, makes the common marmoset an appealing model of human aging [27, 29] and specifically an useful (sporadic) AD animal model [26, 30, 31].
It was hypothesized that inflammation accelerates the amyloidosis. This will provide evidence for a causal relationship between a pro-inflammatory cytokine profile and protein aggregation in the presence of fibrillar Aβ and offer the opportunity to gain more insight into the etiology and to find new therapeutic targets.
MATERIALS AND METHODS
Animals
Four middle aged (5–8 y) and two aged (13-14 y)common marmosets (Callithrix jacchus) of both sexes (325–460 g) were purchased from the purpose-bred colony of the Biomedical Primate Research Centre (BPRC) in the Netherlands. In Table 1, an overview of all monkeys is given. Prior to inclusion in the study, the monkeys underwent elaborate physical examination by the institute’s veterinarian, as only healthy monkeys were included. All monkeys were experimental naïve. The monkeys were pair-housed under conventional conditions in spacious cages with a varying cage environment and were under intensive veterinary care throughout the study. The monkeys function as their own control in regard to the effect of fibrillar amyloid on amyloidogenesis, by injecting the fibrillar amyloid solely in the right hemisphere at three locations (frontal, sensorimotor, and parietal cortices), whereas both hemispheres were injected with phosphate buffered saline (PBS) or LPS to investigate the additive effect of inflammation on amyloidopathy. No randomization of monkeys was performed, but both groups were composed with monkeys of a similar age, i.e., in both groups an aged monkey of >10 years of age was included. All monkeys were observed for clinical symptoms and the body weight was measured every week. The additional monkey with wasting syndrome was 6 years and one month at the time of sacrifice.
The monkey facility was under controlled conditions of humidity (>60%), temperature (22–26°C) and lighting (12 h light/dark cycles from 7 am to 7 pm). The animals were daily fed with standard monkey-chow (Special Diet Services, Witham, Essex, UK) in combination with fruits and vegetables and ad libitum water supply. According to the Dutch law on animal experimentation, the Institute’s Ethics Committee reviewed and approved the study protocol and experimental procedure.
Aβ fibrils
Both human derived and artificial synthetic Aβ fibrils were used to determine the optimal composition of the substrate to induce amyloidosis. Aβ43 artificial fibrils were chosen to be able to distinguish the developing plaques, containing mainly Aβ42, from the inoculated Aβ43 fibrils post-mortem and due to its amyloidogenic properties [6]. The synthetic Aβ protein 1–43 was purchased from Bachem AG, Switzerland (Bachem H-1586).
The human Aβ was derived from brain material of a deceased patient with autopsy confirmed AD that was donated to the Dutch brain institute. The Aβ was isolated prior to the study, as described by Shankar et al. (section 3.2) [32]. In short, 3.7 g of human brain was cut in small pieces prior to homogenization. Sequentially, Tris-buffered saline (TBS), TBS-Triton-X-100 (TX), and 88% formic acid was used to extract Aβ from the tissue. After formic acid extraction, the sample was neutralized by addition of NaOH that restored the protein to the fibrillar stage as was conformed by a Congo red staining that demonstrates the intact beta-sheet composition of the protein. Furthermore, we have run a SDS PAGE gel showing a band containing fibrillar Aβ. After sonication, aliquots were made and stored at – 80°C.
Stereotactic intracranial injections
Under anesthesia with Alfaxan® (0.7 mL; 10 mg/mL), an incision was made in the skull and six small holes were drilled for stereotactic intracranial injections at three cortical brain areas (prefrontal: Bregma +10 mm, motor: Bregma +2 mm, and parietal cortices: Bregma – 6 mm) all 3 mm lateral from the sutura sagitalis with a ventral distance of 1.5 mm in both hemispheres. The left hemisphere was injected with LPS alone (amyloidosis control), whereas LPS combined with Aβ fibrils was injected in the right hemisphere. The LPS control animals were injected with PBS as vehicle alone (left hemisphere) or combined with Aβ fibrils (right hemisphere). The prefrontal cortex was injected with 200 pg human Aβ fibrils, whereas the motor and parietal cortices were injected with 600 pg and 200 pg artificial Aβ43, respectively.
Behavioral observations
Throughout the entire study the monkeys were observed thrice weekly (Monday, Wednesday, and Friday) by an animal caretaker and scored for clinical signs using a scoring list with ten variables (apathy, inadequate grooming, immobility, instability, huddle, stereotypic behavior, stypic behavior, facial abnormalities, attention, and fecal abnormalities). The scoring rate varied from 0 (normal) to 4 (severely aberrant) in order to detect alterations in behavior and welfare.
Blood analysis
For flow cytometry, peripheral blood monocytes were isolated from the EDTA buffered blood. The cells were incubated with antibodies against CD3 (SP34), CD45RA (5H9), CD95 (DX2) (All from BD Biosciences, San Diego, CA), CD4 (MT310; Dako, Glostrup, Denmark), and CD8 (LT8; Serotec, Düsseldorf, Germany). Finally, the samples were fixed in 2% paraformaldehyde and stored at 4°C o/n. FACS LSRII (BD biosciences) was used for the analysis using FlowJo software (Tree Star, Ashland, OR).
CD3, CD4, and CD8 antibodies were used for the gating strategy to isolate the CD4 population to investigate the expression of CD45RA and CD95, as these cell membrane proteins have AD predictive value in humans [18, 19].
Magnetic resonance imaging (MRI)
The animals were sacrificed, with pentobarbital after sedation with Alfaxan®, 5 months after the intracranial injections. The brains were removed, weighed, and sequentially immersed in 4% formaldehyde for 24 h, 2% formaldehyde for 60 h, and 0.05% sodium azide in PBS, during which MRI images were taken. Sodium azide was added to prevent microbial growth.
MRI was performed to investigate its additive value for detecting amyloid plaques in vivo, as metal particles are located in the plaques and are visible on a T2 weighted image as described by Jack Jr, et al. [33]. Furthermore, the MRI images were used to look for traces of the injections [33]. T1-IR (T1 weighed inversion recovery-longitudinal relaxation, fat = bright, fluid = dark), T2-RARE (T2 weighed rapid acquisition with relaxation enhancement, transversal relaxation, fat = intermediate-bright, fluid = bright) images were taken. ImageJ software (NIH, Bethesda, MD) was used to visualize and analyze the stacks for plaques or injection sites.
Histology (general procedure)
For plaque detection, histology was performed as described by Uchihara [34]. A strip of subsequent brain sections was used for a variety of stains to gain a good multifaceted view on the plaques. Mirror sections were analyzed with a Campbell-Switzer silver staining [35] and immunohistochemistry staining (Aβ, Aβ42, Aβ43, Iba1, and GFAP; Bachem, Switzerland) for amyloidopathy and the immune and glia reaction. With the Campbell-Switzer staining, immature/diffuse plaques can be distinguished from mature/dense-cored/congophilic amyloid plaques, which was confirmed by a variety of Aβ antibodies that were used to stain the subsequent brain sections of the strip of sections. The compositionof the plaques was determined by the specific use of Aβ42 and Aβ43 anti-bodies. To investigate the involvement of glial cells in the amyloidopathy, microglia (Iba1) and astrocyte (GFAP) stains were performed. Hematoxyline-eosine staining was also part of the staining sequence, as this stain is useful to examine the brain morphology and offers an additional method to visualize plaques [35].
Paraffin sections
Prior to paraffin embedment, the cerebrum was sectioned in five equal pieces cut through the frontal plane at +10, +5, 0, and – 5 mm from Bregma on the anterior posterior axis. The paraffin blocks were cooled to – 10°C to ease sectioning. With the Microm HM340 E microtome, strips of nine 4 μm sections were made after which 400 μm was discarded. Eighteen strips of each brain were made for analysis of which most were close to the prefrontal and sensorimotor cortex injection sites as the parietal cortices did not show traces of an injection spot during necropsy nor with the MRI. The strips were stored at 4°C until they were separately transferred to microscope slides by the use of a water bath at 37°C (Menzel-Gläser polysine slides, Thermo Fisher scientific, Waltham MA). The slides were dried o/n at 40°C after which they were stored at roomtemperature.
Campbell-Switzer staining
The paraffin sections were first deparaffinized by sequentially placement in xylene, 100% ethanol, 96% ethanol, 70% ethanol, and distilled water. Afterwards, the slides were transferred to the silver attachment substance, consisting of 1% silver nitrate, 1% potassium carbonate, and pyridine for 40 min. The sections were then sequentially placed in 1% citric acid and 4.99 pH acetate buffer to stop the attachment of the silver particles. Subsequently, the chemical transformation took place in order to visualize the metallic silver by placing the slides in the physical developer fluid (2-3 min), a mixture of sodium carbonate, ammonium nitrate, silver nitrate, tungstosilicic acid, and formaldehyde diluted in distilled water. The development was stopped and fixated by sequentially placing the slides in 4.99 pH acetate buffer, distilled water, and 0.5% sodium thiosulfate. Afterwards, the slides were dehydrated and mountedwith malinol.
Immunohistochemistry
The EnVision™ staining kit was used for the immunohistochemical analyses (G|2 Double-stain System, Rabbit/Mouse, DAB+/Permanent RED code K5361; Agilent technologies, Dako DK). The optimal antigen retrieval method and concentration was determined per antibody. After deparaffinization, the slides were washed in PBS/0.05% tween prior to antigen retrieval in the microwave (2 min 780W, 10 min 150W, and 10 min 80W) or in 70% formic acid. Subsequently, the slides were placed in cuvettes, which are part of a capillary gap staining method (Thermo Fisher Scientific, Inc. (NYSE:TMO)). Endogenous peroxidase activity was blocked by the EnVision™ dual endogenous enzyme block. After washing, a second blocking step was used consisting of PBS, 0.1% BSA, 1% NHS and 0.2% Triton X-100. Then the primary antibody (diluted in 0.1% BSA/PBS) was added for 10 min, followed by a washing step and the addition of EnVision™ polymer/HRP (secondary antibody) for 10 min. A washing step followed after which DAB+ was added to start the enzymatic reaction and visualization of the elements.
A counter stain with hematoxiline-eosine was applied for orientation purposes. After a dehydration sequence, the slides were mounted with malinol.
Statistics
The number of monkeys used was six to determine the relation between LPS and Aβ and to confirm that plaque progression take place. Each monkey acted as its own control. The statistical exact power calculation is based on simple t-tests. The formula used is: N = 2(Zα/2 + Zβ)2 * (SD/ES)2. The calculation is based on an α of 0.05 and a power of 80%.
Between-group comparisons were performed using independent t-tests, or a Mann-Whitney test in case normality was not met. Within animal comparisons were performed using a paired t-test, or a Wilcoxon-signed rank test in case normality was not met. A significance level of p < 0.05 was considered significant (Prism 6.0e for Mac OS X; GraphPad Software, San Diego, CA, USA).
RESULTS
Clinical signs
The surgery and blood withdrawals were noticeable in minor fluctuations in clinical observational score (the sum of the behavioral variables analyzed three times weekly) and on body weight with a temporary reduction of 5%. No further effects of either PBS or LPS treatment combined with Aβ intracranial injections were reflected in clinical score or body weight fluctuations (p > 0.5; not shown).
Blood biomarkers for AD
A significantly higher CD45RA expression on CD3+CD4+ cells was found in the PBS treated monkeys (m = 63.13, SE = 3.18) compared to the LPS treated monkeys (m = 40.13, SE = 6.12); t (4) = 3.34, p = 0.029. A trend was visible toward higher CD95 expression on CD3+CD4+ cells between the PBS treated monkeys (m = 29.33, SE = 4.38) and LPS treated monkeys (m = 50.07, SE = 7.03); t (4) = 2.50, p = 0.067 (Fig. 1).
Plaque identification
Histology was performed to analyze natural amyloidosis in the wasting syndrome monkey, and the amyloidopathy of the experimental monkeys as a result of the experimental treatment. In order to confirm the injections in the pilot study, the brains were analyzed with MRI and during necropsy for traces of the injections. No amyloid plaques were detected on the T1 (IR-RARE) nor the T2 (TurboRARE) weighed images, whereas some injection sites were visible (Fig. 2). As none of the parietal injections at – 6 mm from Bregma on the anterior-posterior axis left traces, amyloidopathy analyses were focused on the first (+10 mm from Bregma) and second injection sites (+2 mm from Bregma).
The Campbell-Switzer staining was the most sensitive method for senile plaque visualization, compared to Aβ immunohistochemistry and hematoxyline-eosine stains (Fig. 3). Immunostaining for Aβ (6F/3D) detected only 25% of the Campbell-Switzer positive plaques (Fig. 3A, B). This finding made us decide to use the presence of plaques on Campbell-Switzer stains and/or the presence of traces of an injection site on the first and/or eight section of the section strip (Fig. 3C, D) as a criterion for further immunohistochemical analyses.
The wasting syndrome monkey and two Aβ+LPS-injected monkeys (m06015 and m9856) demonstrated senile plaques, resembling those in the brain of the human AD patient, on the Campbell-Switzer stains (Fig. 4). The origin of wasting syndrome is believed to be chronic colitis [21, 22] and is associated with chronic systemic inflammation. Only a few plaques were present in the wasting syndrome monkey (mi031452) and monkey m06015, whereas monkey m9856 showed severe amyloidopathy throughout the brain (Figs. 3–5). Since two out of three LPS+Aβ monkeys and none of the PBS+Aβ group demonstrated plaques, a trend of an effect of LPS on amyloidopathy was recognized.
Amyloidogenesis after Aβ injection
The plaque load in the right hemisphere of monkey m9856 was significantly higher than the plaque load in the left hemisphere analyzed with Wilcoxon-signed-rank test (Z = 26, p = 0.0001; Fig. 5). Sections adjacent to the frontal (human Aβ) and middle (artificial Aβ) injection sites demonstrated a dense plaque load (Fig. 5). A trend is present when the sections surrounding the middle injection site, containing LPS and artificial Aβ43 fibrils (600 pg) (+5.5/–0.5 mm from Bregma on the anterior-posterior axis), and the sections surrounding the frontal injection site, containing LPS and human Aβ fibrils (200 pg) from a postmortem brain sample (+11/+6.5 mm from Bregma on the anterior-posterior axis), were taken together, the middle injection was associated to a higher plaque load than the frontal injection analyzed with Wilcoxon-signed-rank test (Z = 53.5,p = 0.06).
Plaque composition
23% of the Campbell-Switzer positive plaques in the right hemisphere of monkey m9856 were immunohistochemical Aβ positive. From the Aβ positive plaques, 61% was Aβ42 positive and 20.4% was Aβ43 positive. The Aβ42 and Aβ43 mainly co-localized in plaques, although more plaques were Aβ42 positive than Aβ43 positive (Fig. 6A–C). Aβ43 proteins were only located in the dense core of the senile plaques whereas Aβ42 proteins were also part of the outer ring of the dense-core plaque (Fig. 6D, E).
Reactive microglia
Reactive microglia were detected in proximity of high senile plaque densities in the brain of monkey m9856 (Fig. 7). It was notable that these senile plaque densities were comprised of immature/diffuse senile plaques (Fig. 7D, E).
DISCUSSION
This pilot study confirmed (inducible) amyloidosis in the common marmoset as described by Baker et al. and Ridley et al. [23, 24]. We hypothesize that the pre-term formation of amyloid plaques was due to the induction inflammation. This hypothesis was tested in marmosets receiving intracranial injection of Aβ fibrils with or without the Toll-like receptor 4 (TLR4) ligand LPS. The postmortem brain analysis showed that two LPS-injected monkeys developed senile plaques in addition to diffuse plaques that were found in the monkey with the wasting syndrome at an unusually young age of 6 years (mi031452). We have examined the brain pathology of the marmoset suffering from the wasting syndrome, as this syndrome is a systemic inflammatory disorder that is associated with severe inflammatory bowel disease. This would give an indication whether inflammation should be applied locally or that a systemic inflammation would be sufficient to trigger the amyloidosis. For this pilot study, we have chosen for the optimal approach by inducing a pro-inflammatory stage intracranial at the same location of the Aβ injection. The presence of plaques in the adult monkey with wasting syndrome and the LPS-injected monkeys fits the hypothesis stating that (chronic) inflammation increases the susceptibility for and occurrence of amyloidopathy. This study demonstrated that an accelerated AD pathology as amyloidopathy was found after 5 months, when Aβ fibrils were injected together with LPS, instead of a period of more then 3.5 years in other marmoset studies when only Aβ was injected [23, 24, 31].
Although clear amyloidosis was found in the monkeys with an inflammatory state, no alterations were identified on behavioral symptoms. Considering that the AD amyloidopathy in humans precedes its clinical symptoms and cognitive decline, it could have been too early for clinical symptoms to arise in these monkeys [36, 37].
The immune reaction as a result of the injections of LPS combined with Aβ fibrils found in this study, is in accordance with the literature [7, 8, 11, 14, 38, 39]. Not only the presence of amyloidogenesis in the LPS group, but also the accumulation of glial cells surrounding the plaques (Figs. 3C–D; 8) demonstrated the linked effect of Aβ fibrils and LPS. Additionally, the CD4+ T-cells in the peripheral blood of the LPS-treated monkeys demonstrated an expression profile— higher expression of CD95 and lower expression of CD45RA— similar to the AD predictive profile found in humans [18, 19].
The induced neuroinflammatory state in the marmoset monkey accelerated amyloidopathy as none of the monkeys injected with PBS combined with Aβ fibrils showed any plaques.
Albeit we found plaques after LPS combined with Aβ, only the older LPS injected monkey (m9856) demonstrated severe amyloidopathy throughout the brain, with the emphasis around the LPS injection sites (Fig. 5). However, even in the presence of clear senile plaques in the brain of this older monkey, we were not able to identify plaques with the MRI. This could be explained by the resolution of the MRI scan (7.0 Tesla instead of 9.4 Tesla) and the finding that senile plaques in the marmoset monkey are smaller (<50 μm) than the size at which plaques were detectable (>50 μm) in other studies [33]. The plaque load was predominantly present in the Aβ+LPS-injected hemisphere. It is described in literature that natural amyloid deposition is distributed symmetrical over the hemispheres [40], which makes us conclude that the lateralization in plaque load found in monkey m9856 was due to the Aβ injection in the right hemisphere, which supports the hypothesis that amyloid injection leads to amyloidogenesis. Furthermore, the inoculation of 600 pg of Aβ43 fibrils was the best way to trigger the amyloidopathy, as its injection site was associated with the highest plaque load and amyloidogenesis is additionally proved by the presence of Aβ42 proteins in addition to the injected Aβ43 proteins.
The use of synthetic Aβ43 fibrils, partly due to the proposed high amyloidogenicity by Saito et al. [6], instead of Aβ42 or Aβ40 synthetic fibrils could explain the deviation in results compared to the study by Ridley et al. [24] where they did not identify plaques after artificial fibril inoculation [6, 24]. The composition of the plaques was in accordance with the study of Parvathy et al. [5], as most of the Aβ43 proteins co-localized with Aβ42 proteins (Fig. 6A, B) and the Aβ43 proteins mostly resided in the cores of the plaques, whereas the Aβ42 proteins were also present in the outer circle (Fig. 6D, E) [5].
Increased susceptibility of monkey m9856 for amyloidopathy owing to an aged immune system could explain the discrepancy in senile plaque presentation with the other monkeys. Findings in literature describe the effect of aging on the immune system varying from subtle changes in microglia morphology to a more pro-inflammatory cytokine expression profile [41, 42], also called inflammaging, to comprehend the disturbance of the inflammatory and anti-inflammatory equilibrium of the immune system leading to an increase in susceptibility for neurodegenerative diseases [12, 41, 43]. In addition to the higher vulnerability, it is likely that natural occurring amyloidopathy could have given the old monkey of 14 years of age a head start before the others [24–26]. The life expectancy of marmoset monkeys varies from 10 years in the wild to 15–20 years in captivity.
A dual role of the immune system in the study cannot be excluded. The immune system has been described as a double-edged sword [44], as it canexert a neuroprotective or a neurotoxic effect, dependent on its stimulus [38] and age of the immune system [41, 43]. Neuroprotective immune activation could have cleared the injected Aβ fibrils, as described in literature [45, 46]. However, the described malignant effects of specifically LPS and Aβ fibril inoculation on microglia activation and amyloidosis [11, 38], makes a neuroprotectiveeffect of the immune system in our study questionable.
This is in conjunction with the finding that downregulation of the inflammation seems to have a positive effect on the prevention of AD. Rheumatoid arthritis patients that are treated with non-steroidal anti-inflammatory drugs (NSAIDs) demonstrated a lower susceptibility for AD [47]. However, many clinical studies thereafter were not able to replicate the proposed association between non-steroidal anti-inflammatory drugs use and AD risk [48, 49]. This can be the result of the timing of non-steroidal anti-inflammatory drugs treatment as Côté et al. [50] demonstrated that there is indeed a beneficial effect of non-steroidal anti-inflammatory drugs treatment in lowering the susceptibility of AD once the treatment is administered prior to any disease symptoms. This means that once the vicious circle has started it would be difficult to stop the process. Anti-inflammatory treatment can therefore be used as a neuroprotective treatment for patients at risk. The present study strongly indicates the high translational power of the common marmoset in this approach towards the pathogenic aspects of AD, as this model not only shows a natural spontaneous plaque progression at age, but also the transmissibility of amyloidopathy and the influence of the immune system on the AD pathology was conspicuous. The model can generate essential knowledge in the mechanism of amyloidopathy itself but also in AD therapies, as this study show the importance of immune modulation on the susceptibility for amyloidosis, a hallmarkof AD.
ACKNOWLEDGMENTS
This study was supported by the EU transnational access to the research infrastructure PRIMOCID-205 of EUPRIM-Net under the EU contract 262443 of the 7th Framework Program.
We want to thank the research group of Dr. Louise van der Weerd from the LUMC, Leiden, for the MRI support, Ralph Hamelink from the Netherlands Institute for Neuroscience, Amsterdam, for the advises and coordination of the stereo-tactical intracranial injections of Aβ and LPS, and the Animal Science Department of the BPRC, and animal caretakers specifically, for all the animal experimental support.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-0673r1).
REFERENCES
[1] | Akiyama H , Barger S , Barnum S , Bradt B , Bauer J , Cole GM , Cooper NR , Eikelenboom P , Emmerling M , Fiebich BL , Finch CE , Frautschy S , Griffin WS , Hampel H , Hull M , Landreth G , Lue L , Mrak R , Mackenzie IR , McGeer PL , O’Banion MK , Pachter J , Pasinetti G , Plata-Salaman C , Rogers J , Rydel R , Shen Y , Streit W , Strohmeyer R , Tooyoma I , Van Muiswinkel FL , Veerhuis R , Walker D , Webster S , Wegrzyniak B , Wenk G , Wyss-Coray T ((2000) ) Inflammation and Alzheimer’s disease. Neurobiol Aging 21: , 383–421. |
[2] | Hardy J , Higgins G ((1992) ) Alzheimer’s disease: The amyloid cascade hypothesis. Science 256: , 184–185. |
[3] | Haass C , Selkoe DJ ((2007) ) Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 8: , 101–112. |
[4] | Ahmed M , Davis J , Aucoin D , Sato T , Ahuja S , Aimoto S , Elliott JI , Van Nostrand WE , Smith SO ((2010) ) Structural conversion of neurotoxic amyloid- 1-42 oligomers to fibrils. Nat Struct Mol Biol 17: , 561–567. |
[5] | Parvathy S , Davies P , Haroutunian V , Purohit DP , Davis KL , Mohs RC , Park H , Moran TM , Chan JY , Buxbaum JD ((2001) ) Correlation between Aβx-40–, Aβx-42–, and Aβx-43–containing amyloid plaques and cognitive decline. Arch Neurol 58: , 2025–2032. |
[6] | Saito T , Suemoto T , Brouwers N , Sleegers K , Funamoto S , Mihira N , Matsuba Y , Yamada K , Nilsson P , Takano J , Nishimura M , Iwata N , Van Broeckhoven C , Ihara Y , Saido TC ((2011) ) Potent amyloidogenicity and pathogenicity of Aβ43. Nat Neurosci 14: , 1023–1032. |
[7] | Sondag CM , Dhawan G , Combs CK ((2009) ) Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J Neuroinflammation 6: , 1. |
[8] | Dhawan G , Floden A , Combs C ((2012) ) Amyloid-β oligomers stimulate microglia through a tyrosine kinase dependent mechanism. Neurobiol Aging 33: , 2247–2261. |
[9] | Cuello AC , Ferretti MT , Leon WC , Iulita MF , Melis T , Ducatenzeiler A , Bruno MA , Canneva F ((2010) ) Early-stage inflammation and experimental therapy in transgenic models of the Alzheimer-like amyloid pathology. Neurodegener Dis 7: , 96–98. |
[10] | Doens D , Fernández PL ((2014) ) Microglia receptors and their implications in the response to amyloid β for Alzheimer’s disease pathogenesis. J Neuroinflammation 11: , 48. |
[11] | Lee JW , Lee YK , Yuk DY , Choi DY , Ban SB , Oh KW , Hong JT ((2008) ) Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation 5: , 37. |
[12] | Eikelenboom P , Bate C , Van Gool WA , Hoozemans JJ , Rozemuller JM , Veerhuis R , Williams A ((2002) ) Neuroinflammation in Alzheimer’s disease and prion disease. Glia 40: , 232–239. |
[13] | Hanzel CE , Pichet-Binette A , Pimentel LS , Iulita MF , Allard S , Ducatenzeiler A , Do Carmo S , Cuello AC ((2014) ) Neuronal driven pre-plaque inflammation in a transgenic rat model of Alzheimer’s disease. Neurobiol Aging 35: , 2249–2262. |
[14] | Gao HM , Hong JS ((2008) ) Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol 29: , 357–365. |
[15] | Van Exel E , de Craen AJ , Remarque EJ , Gussekloo J , Houx P , Bootsma-van der Wiel A , Frölich M , Macfarlane PW , Blauw GJ , Westendorp RG ((2003) ) Interaction of atherosclerosis and inflammation in elderly subjects with poor cognitive function. Neurology 61: , 1695–1701. |
[16] | Wyss-Coray T , Rogers J ((2012) ) Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med 2: , 1–23. |
[17] | Remarque E , Bollen EL , Weverling-Rijnsburger AW , Laterveer JC , Blauw GJ , Westendorp RG ((2001) ) Patients with Alzheimer’s disease display a pro-inflammatory phenotype. Exp Gerontol 36: , 171–177. |
[18] | Tan J , Town T , Abdullah L , Wu Y , Placzek A , Small B , Kroeger J , Crawford F , Richards D , Mullan M ((2002) ) CD45 isoform alteration in CD4+ T cells as a potential diagnostic marker of Alzheimer’s disease. J Neuroimmunol 132: , 164–172. |
[19] | Lombardi V , Garcia M ((1999) ) Characterization of cytokine production, screening of lymphocyte subset patterns and in vitro apoptosis in healthy and Alzheimer’s disease (AD) individuals. J Neuroimmunol 97: , 163–171. |
[20] | Griffin W , Sheng JG , Roberts GW , Mrak RE ((1995) ) Interleukin-1 expression in different plaque types in Alzheimer’s disease: Significance in plaque evolution. J Neuropathol Exp Neurol 54: , 276–281. |
[21] | Logan AC , Khan KNM ((1996) ) Case report: Clinical pathologic changes in two marmosets with wasting syndrome. Toxicol Pathol 24: , 707–709. |
[22] | Nakashima E , Okano Y , Niimi K , Takahashi E ((2013) ) Detection of calprotectin and apoptotic activity in the colon of marmosets with chronic diarrhea. J Vet Med Sci 75: , 1633–1636. |
[23] | Baker HF , Ridley RM , Duchen LW , Crow TJ , Bruton CJ ((1993) ) Evidence for the experimental transmission of cerebral beta-amyloidosis to primates. Int J Exp Pathol 74: , 441–454. |
[24] | Ridley RM , Baker HF , Windle CP , Cummings RM ((2006) ) Very long term studies of the seeding of beta-amyloidosis in primates. J Neural Transm (Vienna) 113: , 1243–1251. |
[25] | Geula C , Nagykery N , Wu CK ((2002) ) Amyloid-β deposits in the cerebral cortex of the aged common marmoset (Callithrix jacchus): Incidence and chemical composition. Acta Neuropathol 103: , 48–58. |
[26] | Mansfield K ((2003) ) Marmoset models commonly used in biomedical research. Comp Med 53: , 383–392. |
[27] | Tardif SD , Mansfield KG , Ratnam R , Ross CN , Ziegler TE ((2011) ) The marmoset as a model of aging and age-related diseases. ILAR J 52: , 54–65. |
[28] | Carrion R Jr , Patterson JL ((2012) ) An animal model that reflects human disease: The common marmoset (Callithrix jacchus). Curr Opin Virol 2: , 357–362. |
[29] | Ross CN , Davis K , Dobek G , Tardif SD ((2012) ) Agingphenotypes of common marmosets (Callithrix jacchus). J Aging Res 2012: , 567143. |
[30] | Finch CE , Austad SN ((2012) ) Primate aging in the mammalian scheme: The puzzle of extreme variation in brain aging. Age 34: , 1075–1091. |
[31] | MacLean CJ , Baker HF , Ridley RM , Mori H ((2000) ) Naturally occurring and experimentally induced beta-amyloid deposits in the brains of marmosets (Callithrix jacchus). J Neural Transm 107: , 799–814. |
[32] | Shankar GM , Welzel AT , McDonald JM , Selkoe DJ , Walsh DM ((2011) ) Isolation of Low-n amyloid β-protein oligomers from cultured cells, CSF, and brain. Meth Mol Biol 670: , 33–44. |
[33] | Jack CR Jr , Garwood M , Wengenack TM , Borowski B , Curran GL , Lin J , Adriany G , Gröhn OH , Grimm R , Poduslo JF ((2004) ) In vivo visualization of Alzheimer’s amyloid plaques by MRI in transgenic mice without a contrast agent. Magn Reson Med 52: , 1263–1271. |
[34] | Uchihara T ((2007) ) Silver diagnosis in neuropathology: Principles, practice and revised interpretation. Acta Neuropathol 113: , 483–499. |
[35] | Castellani R , Alexiev BA , Phillips D , Perry G , Smith MA ((2007) ) Microscopic investigations in neurodegenerative diseases. In Modern Research and Educational Topics in Microscopy, Mendez-Vilas A , Diaz J , eds. Formatex, pp. 171–182. |
[36] | Jack CR Jr , Knopman DS , Jagust WJ , Shaw LM , Aisen PS , Weiner MW , Petersen RC , Trojanowski JQ ((2010) ) Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol 9: , 119–128. |
[37] | Villemagne VL , Burnham S , Bourgeat P , Brown B , Ellis KA , Salvado O , Szoeke C , Macaulay SL , Martins R , Maruff P , Ames D , Rowe CC , Masters CL , Australian Imaging Biomarkers Lifestyle (AIBL) Research Group ((2013) ) Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol 12: , 357–367. |
[38] | Butovsky O , Talpalar AE , Ben-Yaakov K , Schwartz M ((2005) ) Activation of microglia by aggregated beta-amyloid or lipopolysaccharide impairs MHC-II expression and renders them cytotoxic whereas IFN-gamma and IL-4 render them protective. Mol Cell Neurosci 29: , 381–393. |
[39] | Veerhuis R , Van Breemen MJ , Hoozemans JM , Morbin M , Ouladhadj J , Tagliavini F , Eikelenboom P ((2003) ) Amyloid beta plaque-associated proteins C1q and SAP enhance the Abeta1- 42 peptide-induced cytokine secretion by adult human microglia in vitro. Acta Neuropathol 105: , 135–144. |
[40] | Raji C , Becker J , Tsopelas N ((2008) ) Characterizingregional correlation, laterality and symmetry of amyloid deposition in mild cognitive impairment and Alzheimer’s disease with Pittsburgh Compound B. J Neurosci Meth 172: , 277–282. |
[41] | Baron R , Babcock AA , Nemirovsky A , Finsen B , Monsonego A ((2014) ) Accelerated microglial pathology is associated with Aβ plaques in mouse models of Alzheimer’s disease. Aging Cell 13: , 584–595. |
[42] | Streit WJ ((2006) ) Microglial senescence: Does the brain’s immune system have an expiration date? Trends Neurosci 29: , 506–510. |
[43] | Blasko I , Stampfer-Kountchev M , Robatscher P , Veerhuis R , Eikelenboom P , Grubeck-Loebenstein B ((2004) ) How chronic inflammation can affect the brain and support the development of Alzheimer’s disease in old age: The role of microglia and astrocytes. Aging Cell 3: , 169–176. |
[44] | Wyss-Coray T , Mucke L ((2002) ) Inflammation in neurodegenerative disease–a double-edged sword. Neuron 35: , 419–432. |
[45] | Solito E , Sastre M ((2012) ) Microglia function in Alzheimer’s disease. Front Pharmacol 3: , 14. |
[46] | Schwartz M , Kipnis J ((2004) ) A common vaccine for fighting neurodegenerative disorders: Recharging immunity for homeostasis. Trends Pharmacol Sci 25: , 407–412. |
[47] | McGeer PL , McGeer EG ((1996) ) Anti-inflammatory drugs in the fight against Alzheimer’s disease. Ann N Y Acad Sci 777: , 213–220. |
[48] | In ’t Veld BA , Launer LJ , Hoes AW , Ott A , Hofman A , Breteler MM , Stricker BH ((1998) ) NSAIDs and incident Alzheimer’s disease. The Rotterdam Study. Neurobiol Aging 19: , 607–611. |
[49] | Launer L ((2003) ) Nonsteroidal anti-inflammatory drug use and the risk for Alzheimer’s disease. Drugs 63: , 731–739. |
[50] | Côté S , Carmichael PH , Verreault R , Lindsay J , Lefebvre J , Laurin D ((2012) ) Nonsteroidal anti-inflammatory drug use and the risk of cognitive impairment and Alzheimer’s disease. Alzheimers Dement 8: , 219–226. |
Figures and Tables
Fig.1
Blood biomarkers for AD. CD95 and CD45RA expression on CD3+CD4+ cells expressed in percentage of the CD3+CD4+ cells. Scores were compared between the LPS and PBS groups. *Significant differences, p < 0.05 (Mann-Whitney).
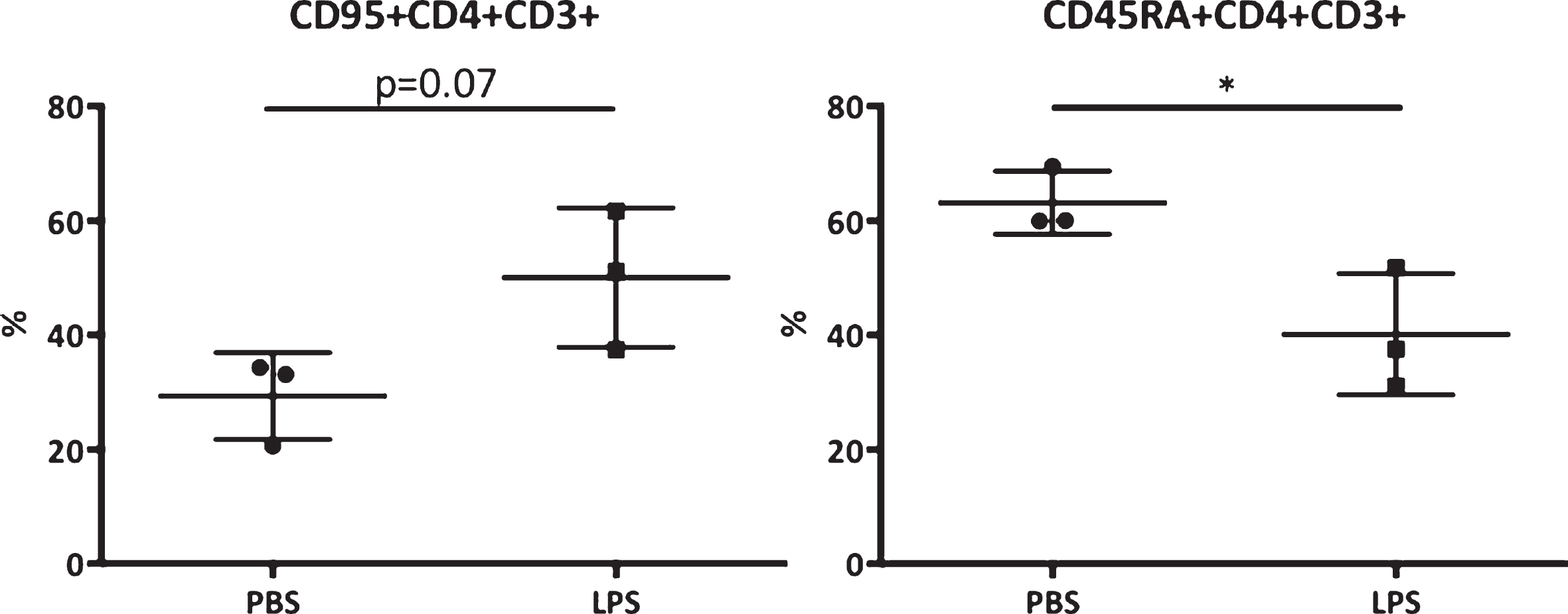
Fig.2
Postmortem MRI of a transcranial section of a marmoset brain. MRI is taken at +4 mm from Bregma on the anterior-posterior axis from an Aβ+LPS treated monkey. A) IR-RARE T1 weighed image, and B) TurboRARE T2 weighed image. The red arrow indicates the location of injection. CC, corpus callosum; R, right; L, left.
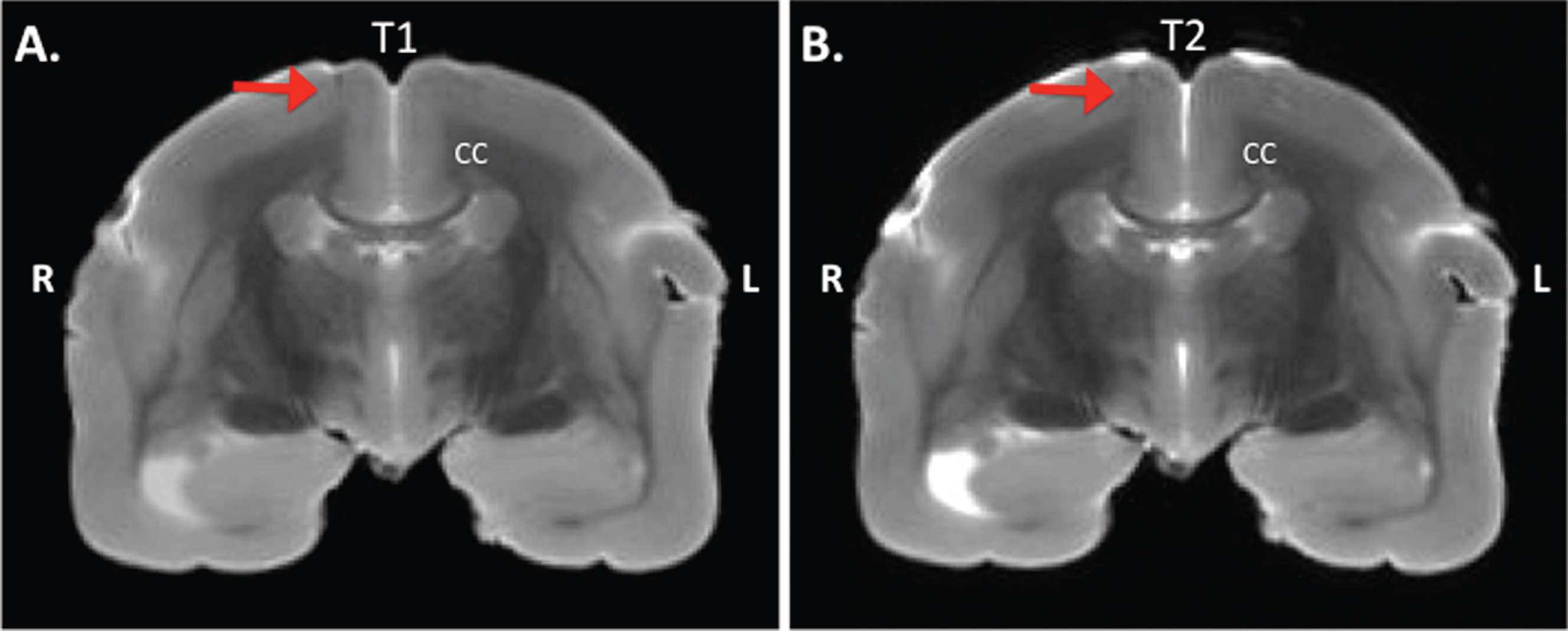
Fig.3
The amyloidopathy of monkey m9856 visualized on a staining strip. Eight plaques could be counted on Campbell-Switzer (CS) stained brain tissue (A) of monkey m9856, although only four plaques were detectable on the mirror section with an Aβ (6F/3D) immunohistochemical (IHC) staining (B). A strip of mirror sections was stained with different stainings (CS, Aβ, Iba1, GFAP, HE, Aβ42, and Aβ43) to characterize the plaques and investigate the glial response (C) and D) Not all strips could cover the complete diameter of all plaques (see the first and eight section of ‘D’). CS, Campbell-Switzer; IHC, immunohistochemistry; Aβ, amyloid-beta; HE, hematoxyline-eosine; Iba1, microglia marker; GFAP, glial fibrillar acidic protein (a marker for astrocytes).
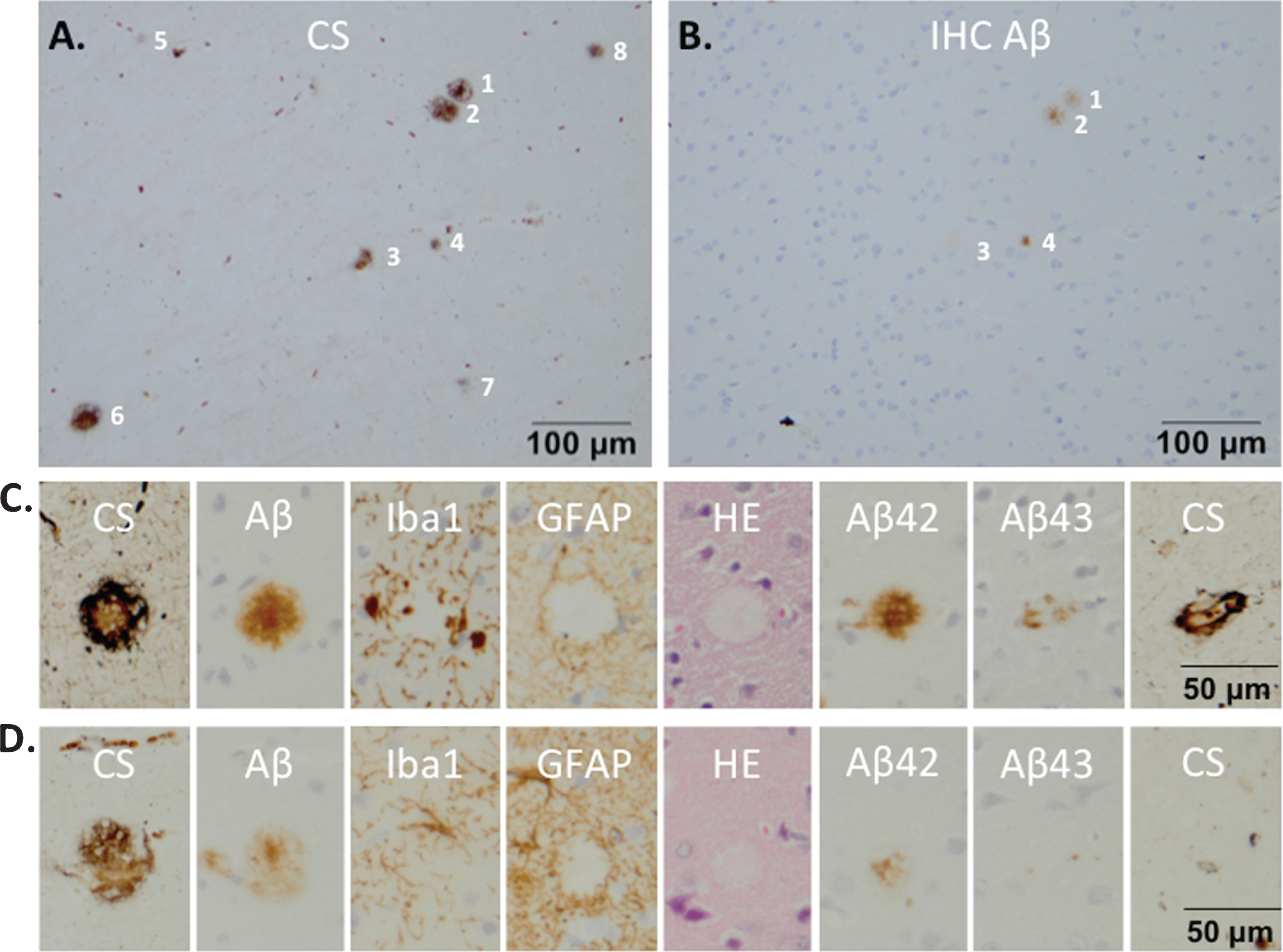
Fig.4
Senile plaques visualized with Campbell-Switzer staining. Senile plaque formation was present in brain material of an AD patient (A) and also natural (early) amyloidosis was present in the common marmoset (B) that died at an age of six due to wasting syndrome (mi031452). Experimental monkeys m06015 and m9856 that were injected with the Aβ combined with LPS also demonstrated amyloidopathy (C, D). The plaques in monkey m06015 were solely found in the right hemisphere. Diffuse plaques are indicated with a black arrow and dense-core plaques are indicated with a white arrow.
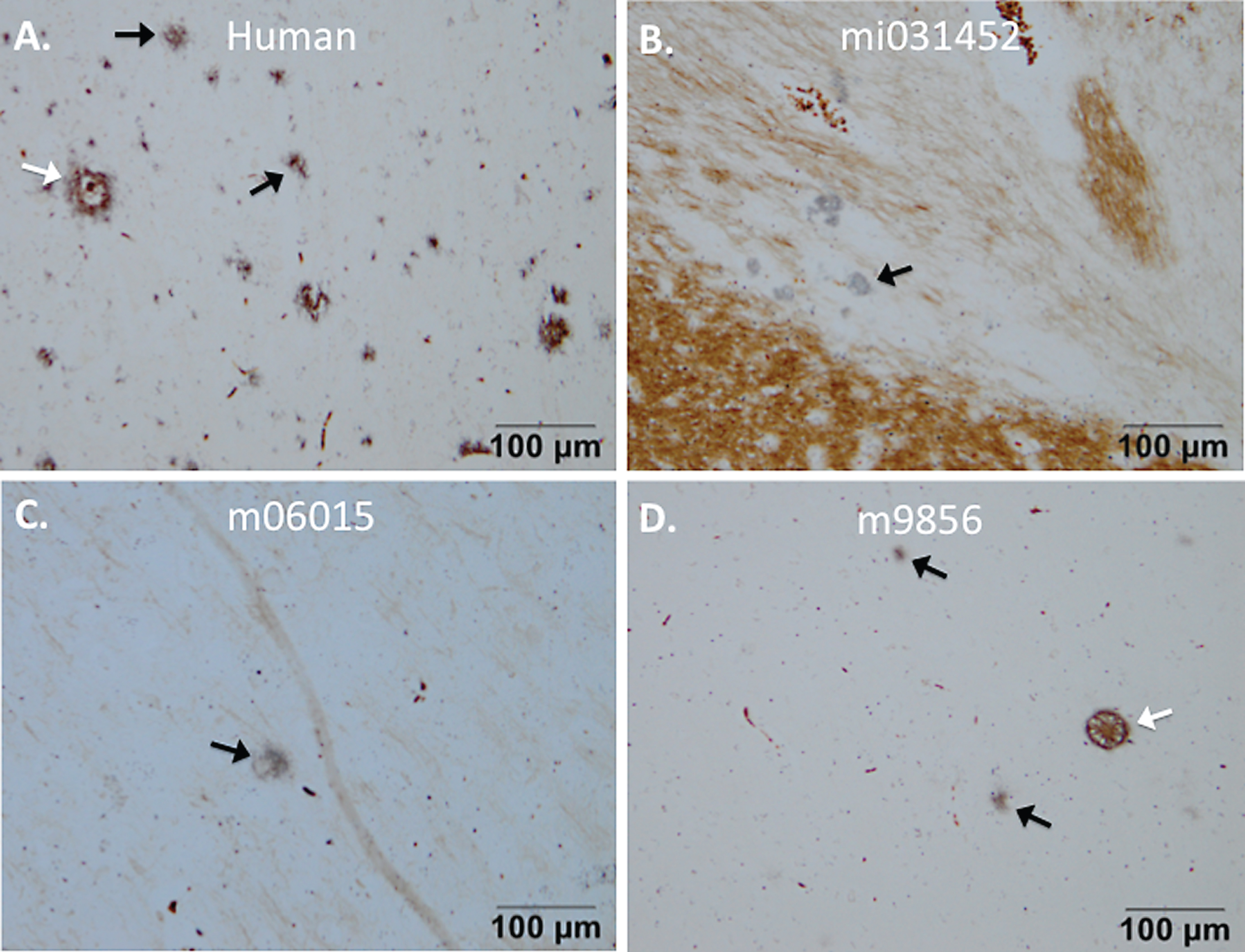
Fig.5
Plaque load and distribution throughout brain of monkey m9856. The distribution of the plaques is visualized on six transcranial sections and indicated in yellow in the right hemisphere and in red in the left hemisphere. The absolute plaque load of the right and left hemisphere on the sections analyzed are displayed on the left and the right side, respectively. The right demonstrated significantly more severe amyloidopathy than the left hemisphere (p < 0.001; Wilcoxon-signed-rank test). B, Bregma; FC, frontal cortex; CG, cingulate gyrus; AA, anterior amygdala; TG, temporal gyrus; PC, posterior cortex; CA3 & CA1, regions of hippocampus; VLA, ventrolateral anteriorextrastriate area.
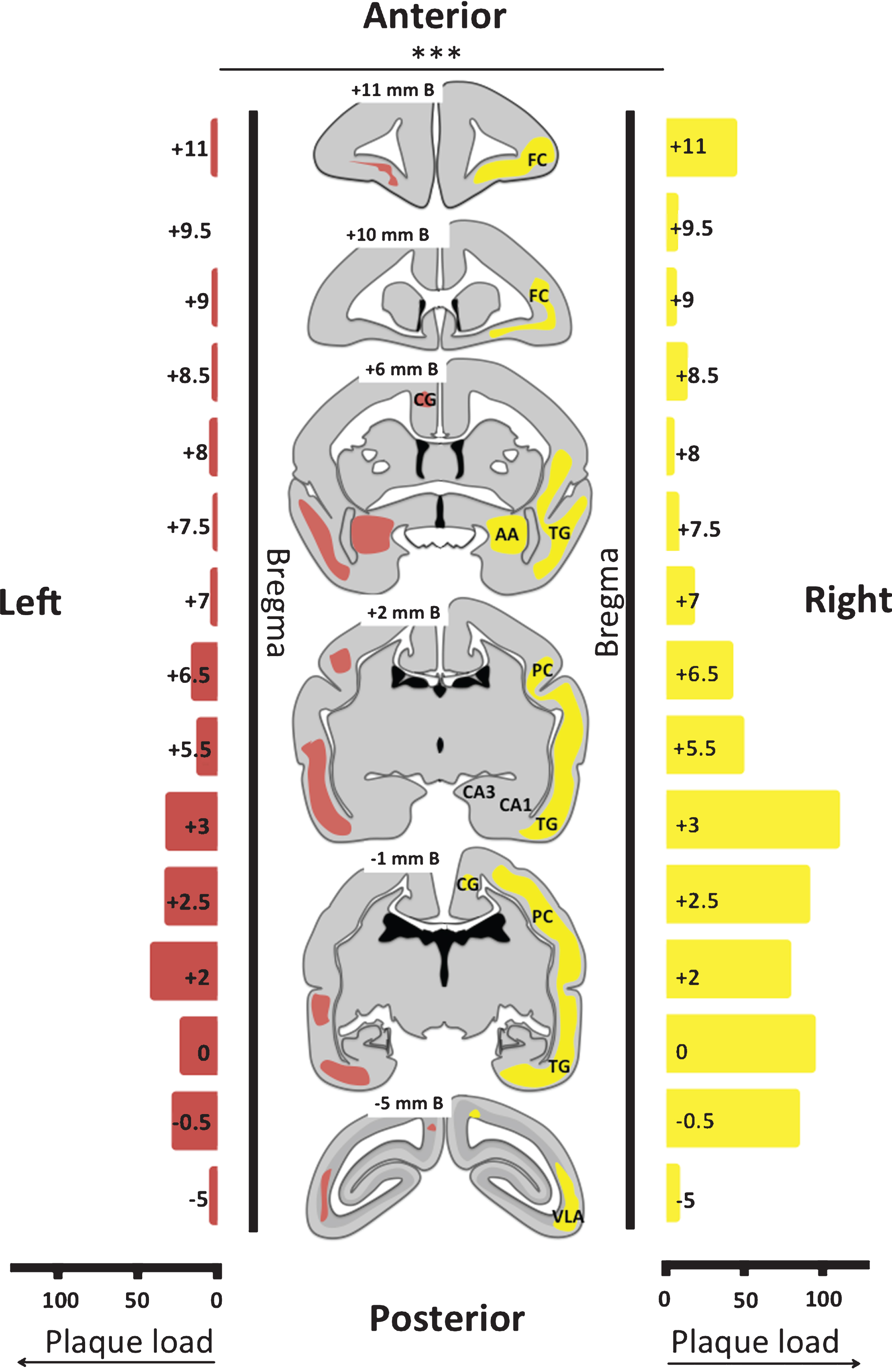
Fig.6
Composition of the plaques on brain sections from monkey m9856. Co-localization of Aβ42 (A) and Aβ43 (B) in plaques 1 and 2 are shown, which were also visible with the Campbell-Switzer staining (C). Plaque 3 was only Aβ42 positive and Campbell-Switzer positive. The vessels indicated by the asterisk (*) and plus (+) symbols, were used for navigation. Aβ42 mainly stained the center of the plaque, but also the outer circle, whereas Aβ43 only stains the center of the Campbell-Switzer positive plaque (D, E). CS, Campbell-Switzer staining; Aβ, amyloid-beta.
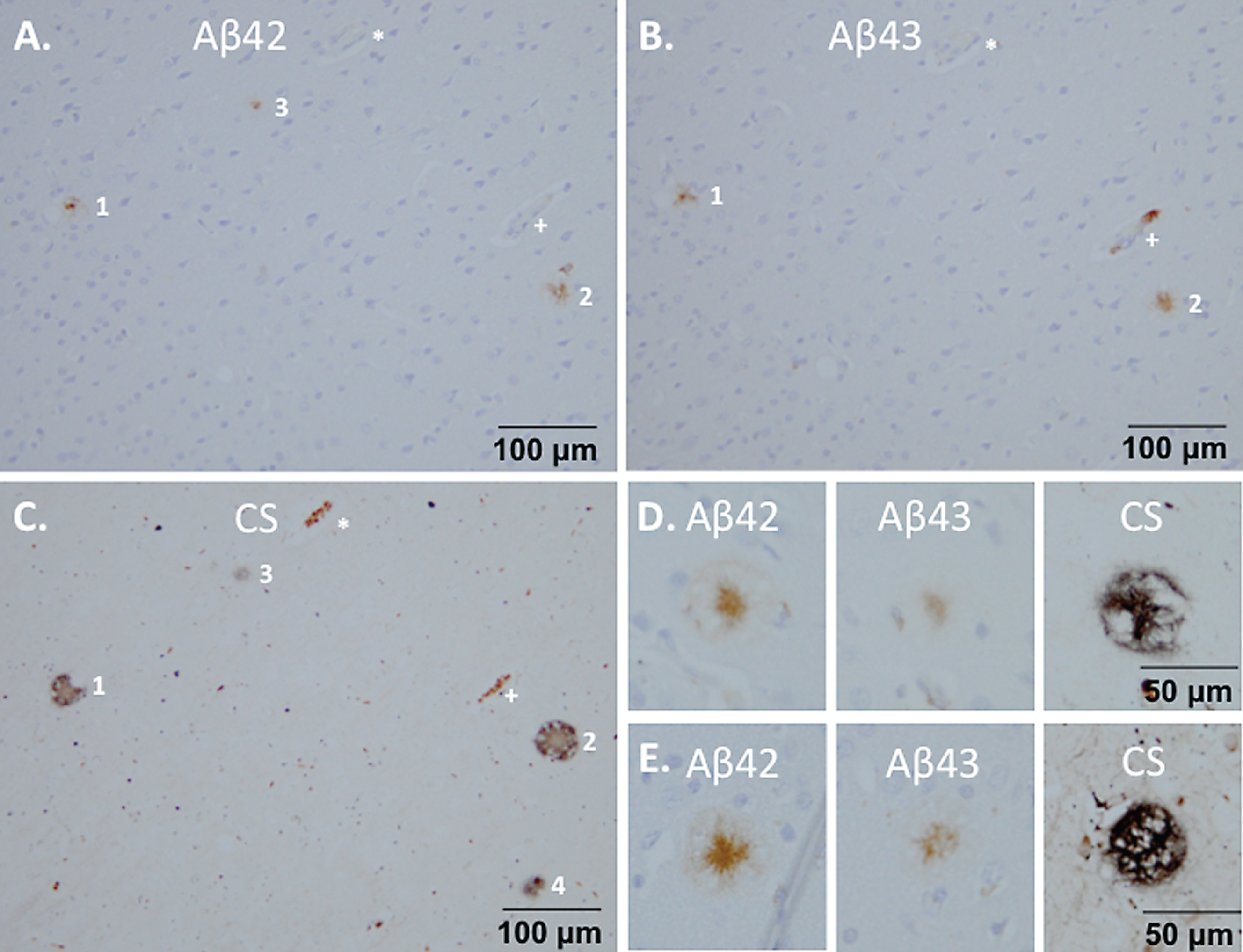
Fig.7
Reactive microglia surrounding senile plaque densities in monkey m9856. Regions with microglia accumulation were visible (A) and these cells also reflected a reactive morphology (B) compared to the ramified microglia in the other regions (C). Diffuse senile plaque formation co-localized with the reactive microglia as visible in a mirror section (D, E). The vessel indicated by the asterisk (*) symbol, was used for navigation between A, D, and E.
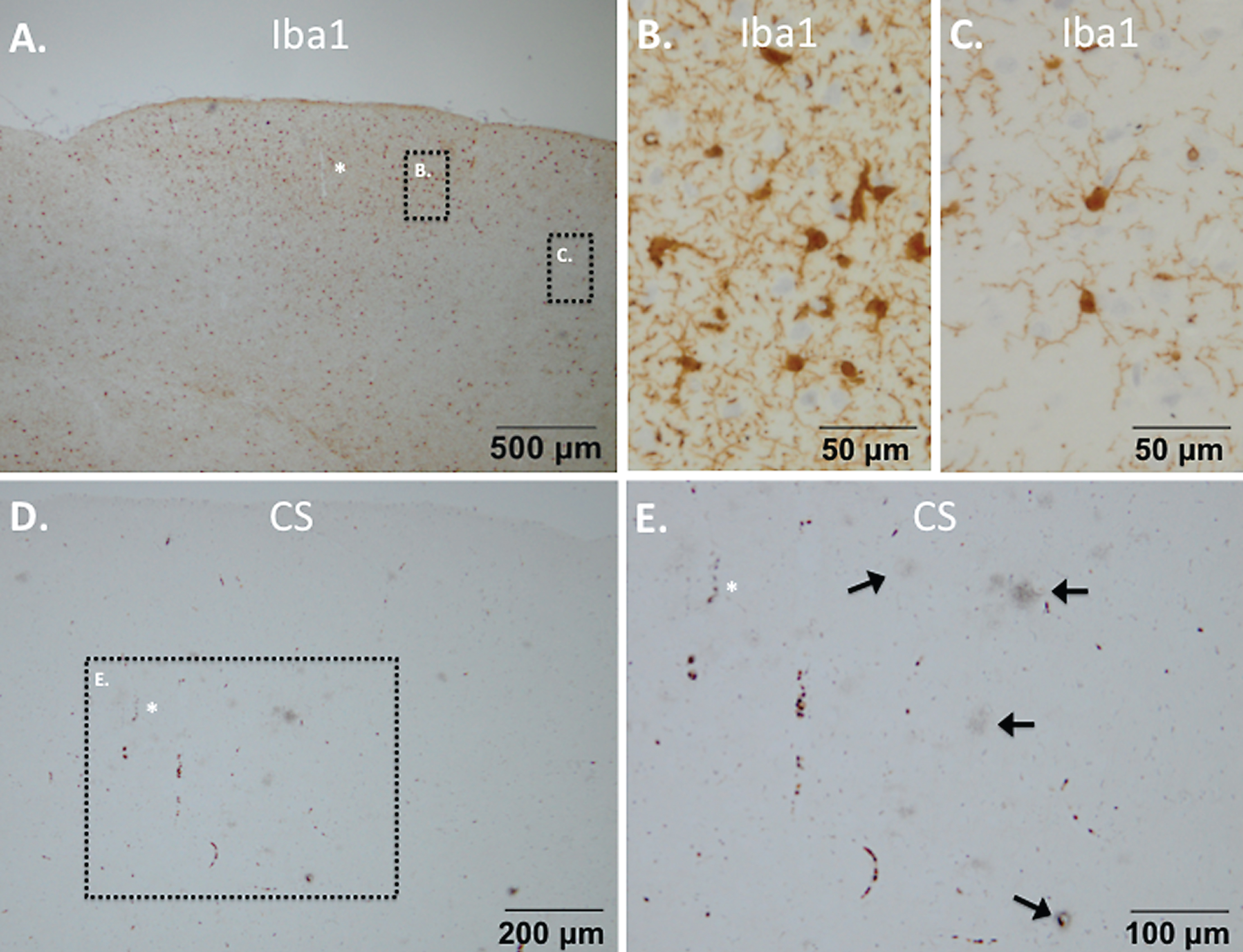
Table 1
Overview of the characteristics of each marmoset monkey
| Animal | Gender | Age (y) | Group | Body weight (g) | Aβ plaques | Aβ plaques |
| Right* | Left** | |||||
| m9856 | male | 15 | Aβ+LPS | 341 | ++ | + |
| m06015 | male | 8 | Aβ+LPS | 325 | x | – |
| m08058 | female | 6 | Aβ+LPS | 457 | – | – |
| m0011 | female | 14 | Aβ+PBS | 373 | – | – |
| m06038 | female | 8 | Aβ+PBS | 462 | – | – |
| m07068 | man | 7 | Aβ+PBS | 330 | – | – |
| mi031452 | female | 6 | MWS | 355 | x | x |
MWS, marmoset wasting syndrome; –, no plaques; x, diffuse plaques; +, ++, dense core plaques; *Right hemisphere (Aβ+LPS or PBS); **Left hemisphere (Aβ alone).


