Alzheimer’s Disease: Assessing the Role of Spirochetes, Biofilms, the Immune System, and Amyloid-β with Regard to Potential Treatment and Prevention
Abstract
Alzheimer’s disease (AD) is an infectious disease caused by spirochetes, and these spirochetes form biofilms, which attract the innate immune system. The innate immune system first responder, Toll-like receptor 2, generates both NF-κB and TNF-α which try to kill the spirochetes in the biofilm, but cannot penetrate the “slime”. NF-κB is also responsible for the generation of amyloid-β (Aβ) which itself is anti-microbial. Aβ cannot penetrate the biofilm either, and its accumulation leads to destruction of the cerebral neurocircuitry. Treatment with penicillin (as in tertiary syphilis, the comparator to AD) is outlined; a biofilm dispersing agent may need to be added to the protocol.
Where spirochetes have been found in the brains of Alzheimer’s disease (AD), it may be considered an infectious disease; this is the first and most important consideration [1, 2]. It is also a chronic disease, a biofilm-associated disease, [3] and an autoimmune disease [4]. Further, it is a debilitating disease, a socially-destructive disease, an exceedingly expensive disease, and, lastly, a deadly disease [5]. This review will focus on the biofilm portion of the disorder as well as the autoimmune response. It will also touch on some rational therapeutic concepts for this most irrational of diseases.
The infectious nature of AD was revealed when spirochetes (both dental and Lyme) were shown to be present in the brains of affected patients [1]. The dental microbes travel from the oral cavity during times of disruption of the dental plaque and subsequent bacteremia following dental procedures; i.e., any time blood is seen. The hippocampus (which is the initial site of cerebral involvement in AD) is approximately 4 cm from the posterior pharynx. Lyme borrelia travel to the brain via the blood stream during the secondary stage of that disease following the erythema migrans lesion [6]. This secondary stage is characterized by fever, myalgias, arthralgias, and other systemic symptoms. The spirochetes have an affinity for neural tissue and pass through the blood-brain barrier easily [7].
Once the spirochetes are in the brain, they attach, divide (albeit very, very slowly) [8], and multiply. When they reach a quorum, they begin to spin out a biofilm (Fig. 1) [9]. This represents approximately 150 spirochetal cells which are 0.3 microns in diameter (10 cells are necessary on a two-dimensional culture plate for a quorum to begin). Because of the exceedingly slow division, it takes approximately 2 years to accumulate sufficient organisms to make one biofilm. The biofilm is protective and is a response of the organisms to ensure their survival, inasmuch as it encases them in “slime” (Fig. 2).
Quorum sensing is one triggering mechanism for the production of biofilms; other organisms in other diseases may form biofilms when subjected to different stimuli. These stimuli include salt and water, as seen in eczema and tinea versicolor [10, 11]. Low dose antibiotics and quorum sensing are seen in psoriasis [12] and arthritis [4]. Further, elevated temperatures and exposure to alcohol and other chemicals promote biofilms [13].
At some point after attachment and formation of the biofilms, the innate immune system becomes activated and attempts to destroy them [8]. Even though the spirochetes are weakly gram negative, Toll-like receptor 2 (TLR 2) has been shown to be the first responder to the organisms incorporated in the extracellular polysaccharide slime (Fig. 3) [8]. TLR 2 itself has recently been shown to be attracted to the “curli” fibers produced by the organisms within the biofilm [14]. These fibers are the major component of the proteinaceous portion of the biomass and are not only immunogenic, but are also important in the attachment of the biofilms. Ordinarily, Toll-like receptor 4, rather than TLR 2, responds to gram-negative organisms.
TLR 2 kills primarily by means of tumor necrosis factor-α (TNF-α) generated by the myeloid differentiation pathway D88 (MyD88). TLR 2 coats the microbes (Fig. 4) and generates both nuclear factor-κB (NF-kB) and TNF-α. This is the process utilized for killing when the organisms are planktonic (free floating) and not in a biofilm. Neither TLR 2 nor TNF-α can penetrate biofilm; consequently, it has been theorized that the TNF-α destroys the surrounding neural tissue instead [8].
Almost all organisms make biofilms. As has been previously stated, these biofilms protect the microbes dwelling within from noxious agents whether chemical, immunologic, or other. The bulk of a biofilm is made up of extracellular polysaccharides. Inside and out there are curli fibers; other amyloid fibers may be within and their purpose is to serve as an infrastructure for the polysaccharides. There are also DNA and water channels, as well as the microbes themselves within the biofilm [15, 16]. None of the commonly used antibiotics penetrate biofilms; and, none of the immunologic molecules from either arm of the immune system, whether innate or adaptive, are able to penetrate either.
Ordinarily, the adaptive immune system including B cells, immunoglobulins, and T cells with their cytokines are excluded from the brain by the blood-brain barrier. That is until traumatic brain injury disrupts that barrier: at that point, B lymphocytes and IgG flood the cerebrum [17]. These immunogens kill by complement, alternate complement, killer T cells, cytokines (including TNF-α and others). The killing of brain tissue around the plaques of AD is much more rapid and much more destructive with the adaptive immune system. This is without doubt the reason that AD occurs within 3 years after a cerebrovascular accident; ordinarily, it takes 30-50 years to develop. Further, it is most probably the reason that chronic traumatic encephalopathy (CTE) is so rapidly progressive after many concussions [8]. A concussion may pictorially and practically be considered an ecchymosis, and, as such, is comparable to a hemorrhagic cerebrovascular accident. CTE is currently the scourge of the National Football League where head trauma is a frequent occurrence.
Elucidation of the role of amyloid-β (Aβ) has been challenging: Aβ is a constant in AD and, in fact, it has been thought to be pathogenic by many. It, however, has been shown recently to be antimicrobial [18] and even more recently the pathway to its formation has been made apparent [19, 20] This pathway (Fig. 5) derives from the MyD88 pathway activated by TLR 2. TNF-α, generated by TLR 2, in conjugation with TNF-α converting enzyme (TACE) becomes alpha secretase and splits amyloid precursor protein (APP) to make amyloid alpha. The NF-kB generated by the same MyD88 pathway, together with Aβ converting enzyme (BACE), activates beta and gamma secretases that cleave the APP. The APP then becomes Aβ and attacks the biofilm (Figs. 6, 7) but cannot penetrate it. Consequently, it encompasses the biofilm and its buildup destroys the neurocircuitry of the brain.
This is the very essence of autoimmunity, namely the body attacking itself; this occurs when the body’s own innate immune system produces TNF-α or Aβ and attacks the biofilm encasing the spirochetes. In the process of doing this, the surrounding tissue is destroyed instead. Such is the case with the biofilm produced by staphylococcus in eczema and streptococcus in psoriasis; these biofilms call forth the innate immune system and the whole process of tissue destruction is set in motion [4]. The consequences of AD are much more dire however, because they lead to total destruction of the mind.
Any treatment of AD must take into consideration these biofilms. The pathway toward such treatment has previously been set by the treatment of syphilis. Syphilis, in its tertiary form (general paresis of the insane), has been shown to have exactly the same pathology as AD. The same plaques, neurofibrillary tangles, Aβ, and tau protein are present in both.
Where the pathology is the same and where both diseases are caused by spirochetes, and where spirochetes are sensitive to penicillin, a reasonable approach would be to follow the same treatment schedule as syphilis [21]. With that treatment, penicillin administered at any time prior to the onset of tertiary syphilis is curative. The same can reasonably be said for AD; penicillin administered any time prior to the onset of tertiary disease would also be curative. Lyme disease is most closely aligned with syphilis with erythema migrans equivalent to the chancre. In most cases, it is one tick bite compared to one chancre, so the treatment could be reasonably the same [7]. With dental organisms, exposure is ongoing; thus, the treatment would need to be tailored to the patient’s dental health. One could imagine penicillin administered once or twice yearly (or perhaps more frequently) in certain situations (CTE?). The same could be said for the 5% of AD “pre”sufferers who have the APOE ɛ4 gene for AD. CTE mimics the genetic disease. It must be stated that any neural damage is irreversible; thus, the importance and urgency of treating early in this disease course.
Treatment for patients in the early stages of dementia would need more than penicillin; they would also need an agent to disperse the biofilm [22]. Fortunately, there are such agents, and many are already being employed in AD patients. These agents include furans (citalopram), [23] thiophenes (olanzapine), [24] piperidines (donepezil), [25] pyrroles (azoles), [26] and rifampin [27]. Donepazil, for example, may be an anticholinesterase inhibitor, but it is also a biofilm disperser, so it may be helpful for a short time, but be harmful long term. The dispersal effect would potentially create many more plaques. The same may be said for haloperidol whose use in AD is already shunned.
Specifically, for early dementia, penicillin may be administered as IV or IM injections (IM would be 1.2 mu biweekly for 3 doses), probenecid 500 mg bid (to increase the serum concentration of penicillin by decreasing excretion, citalopram 20 mg daily, and rifampin 500 mg bid. These may be adjusted with the use of other medications. None of this is codified; but, the current treatment is most likely harmful with the biofilms being dispersed without the spirochetes being killed. This would conceivably lead to many more biofilms, because all the spirochetes within the previous biofilm are capable of making new biofilms.
The other major consideration is to treat in the “latent” stage for AD with penicillin by itself. Presumably, this would be similar to the treatment of latent syphilis. Also important would be to treat prior to any dental surgery just as is being done for joint implants. Consequently, the organisms would be treated before they reached the brain in the case of dental surgery and before they did damage (made biofilms) in latent disease. Syphilis, in truth, is different because its presence is revealed by a serology. However, until a serologic test is available for AD, treatment, as has been proposed herein, seems rational. It is also relatively inexpensive, both as to medical costs and the cost of ongoing care of dementia patients.
The story of AD is then one of spirochetes that make biofilms that activate the innate immune system. The first responder is TLR 2 and TLR 2 generates NF-kB and TNF-α that not only damage tissue in an attempt to kill the biofilm-encased spirochetes, but also lead to the production of Aβ. All of the foregoing leads to dementia. Treatment with a bactericidal antibiotic with a concomitant biofilm disperser seems most reasonable; but, as has been stated previously, any neurologic damage is irreversible. It is therefore of the utmost importance to treat early in the course of this disease.
ACKNOWLEDGMENTS
All the material referred to from our institution was done with the approval of the Drexel University College of Medicine Institutional Review Board.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-0388r1).
REFERENCES
[1] | Miklossy J ((2011) ) Alzheimer’s disease - a neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J Neuroinflammation 8: , 90. |
[2] | MacDonald AB ((2006) ) Spirochetal cyst forms in neurodegenerative disorders,...hiding in plain sight. Med Hypotheses 67: , 819–832. |
[3] | Allen HB , Morales D , Jones K , Joshi S ((2016) ) Alzheimer’s disease: A novel hypothesis integrating spirochetes, biofilm, and the immune system. J Neuroinfect Dis 7: , 200. |
[4] | Allen HB , Shaver CM , Etzler CA , Joshi SG ((2015) ) Autoimmune diseases of the innate and adaptive immune system including atopic dermatitis, psoriasis, chronic arthritis, lyme disease, and Alzheimer’s disease. Immunochem Immunopathol 1: , 112. |
[5] | Tejada-Vera B ((2013) ) Mortality from Alzheimer’s disease in the United States: Data for 2000 and 2010. NCHS data brief, no 116. National Center for Health Statistics, Hyattsville, MD.. |
[6] | Allen HB , Vin H , Warner C , Joshi S ((2016) ) Lyme disease: Beyond erythema migrans. J Clin Exp Dermatol Res 7: , 330. |
[7] | de Vries HE , Kuiper J , de Boer AG , Van Berkel TJ , Breimer DD ((1997) ) The blood-brain barrier in neuroinflammatory diseases. Pharmacol Rev 49: , 143–156. |
[8] | Allen HB , Morales D , Jones K , Joshi S ((2016) ) Alzheimer’s disease: A novel hypothesis for the development and the subsequent role of beta amyloid. J Neuroinfect Dis 7: , 2. |
[9] | Rutherford ST , Bassler BL ((2012) ) Bacterial quorum sensing: Its role in virulence and possibilities for its control. Cold Spring Harb Perspect Med 2: , a012427. |
[10] | Allen HB , Vaze ND , Choi C , Hailu T , Tulbert BH , Cusack CA , Joshi SG ((2014) ) The presence and impact of biofilm-producing staphylococci in atopic dermatitis. JAMA Dermatol 150: , 260–265. |
[11] | Allen HB , Goyal K , Ogrich L , Joshi S ((2015) ) Biofilm formation by as a possible mechanism of pathogenesis in Tinea versicolor. J Clin Exp Dermatol Res 6: , 311. |
[12] | Allen HB , Neidig L , Zhang J , Shave C , Cusack C ((2015) ) The etiology of psoriasis: Its close association to streptococcus. J Am Acad Dermatol 72: , AB254. |
[13] | Knobloch JK , Bartscht K , Sabottke A , Rohde H , Feucht HH , Mack D ((2001) ) Biofilm formation by depends on functional RsbU, an activator of the sigB operon: Differential activation mechanisms due to ethanol and salt stress. J Bacteriol 183: , 2624–2633. |
[14] | Tukel C , Wilson RP , Nishimori M , Pezeshki M , Chromy BA , Baumier AG ((2009) ) Responses to amyloids of microbial and host origin are mediated through toll-like receptor 2. Cell Host Microbe 6: , 45–53. |
[15] | Flemming H-C , Wingender J ((2010) ) The biofilm matrix. Nat Rev Microbiol 8: , 623–633. |
[16] | |
[17] | Doyle KP , Quach LN , Solé M , Axtell RC , Nguyen TV , Soler-Liavena GJ , Jurado S , Han J , Steinman L , Longo FM , Schneider JA , Malenka RC , Buckwalter MS ((2015) ) B-lymphocyte mediated delayed cognitive impairment following stroke. J Neurosci 35: , 2133–2145. |
[18] | Soscia SJ , Kirby JE , Washicosky KJ , Tucker SM , Ingelsson M , Burton MA , Goldstein LE , Duong S , Tanzi RE , Moir RD ((2010) ) The Alzheimer’s disease-associated β-protein is an anti-microbial peptide. PLoS One 5: , e9505. |
[19] | O’Brien RJ , Wong PC ((2011) ) Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci 34: , 185–204. |
[20] | Chami L , Checler F ((2012) ) BACE1 is at the crossroad of a toxic vicious cycle involving cellular stress and β-amyloid production in Alzheimer’s disease. Mol Neurodegener 7: , 52. |
[21] | Allen HB , Hannaway M , Joshi S ((2015) ) Tertiary treponematosis. J Clin Exp Dermatol Res 6: , 4. |
[22] | Allen HB , Kim JY , Warner C , Joshi S ((2015) ) Penicillin: The new/old wonder drug. J Drug Metab Toxicol 6: , 4. |
[23] | Baveja JK , Willcox MDP , Hume EBH , Kumar N , Odell R , Poole-Warren LA ((2004) ) Furanones as potential anti-bacterial coatings on biomaterials. Biomaterials 25: , 5003–5012. |
[24] | Liu H , Zhao Y , Shao D , Gong T , Wu Y , Han H , Xu T , Peschel A , Han S , Qu D ((2015) ) Antibacterial and anti-biofilm activities of emerging microbes and thiazolidione derivatives against clinical staphylococcus strains. Infections 4: , e17. |
[25] | Kagan S , Jabbour A , Sianov E , Alguntar AA , Steinberg D , Srebnik M , Nir-Paz R , Weiss A , Polacheck I ((2014) ) Anti-Candida albicans biofilm effect of novel heterocyclic compounds. J Antimicrob Chemother 69: , 416–27. |
[26] | Richards JJ , Reed CS , Melendez C ((2008) ) Effects of N-pyrrole substitution on the anti- biofilm activities of oroidin derivatives against Acinetobacter baumannii. Bioorg Med Chem Lett 18: , 4325–4327. |
[27] | Zheng Z , Stewart PS ((2002) ) Penetration of rifampin through Staphylococcus epidermidis biofilms. Antimicrob Agents Chemother 46: , 900–903. |
Figures and Tables
Fig.1
Hippocampal plaques consisting of biofilms. AD brain plaques: polysaccharides of biofilms stain pink with PAS. PAS 10X. From Allen et al. [8].
![Hippocampal plaques consisting of biofilms. AD brain plaques: polysaccharides of biofilms stain pink with PAS. PAS 10X. From Allen et al. [8].](https://content.iospress.com:443/media/jad/2016/53-4/jad-53-4-jad160388/jad-53-jad160388-g001.jpg)
Fig.2
Representative biofilm (slime). “Slime” represents typical biofilm on gross examination. From Allen et al. [8].
![Representative biofilm (slime). “Slime” represents typical biofilm on gross examination. From Allen et al. [8].](https://content.iospress.com:443/media/jad/2016/53-4/jad-53-4-jad160388/jad-53-jad160388-g002.jpg)
Fig.3
TLR 2 in hippocampus of AD patient. TLR 2 (CD 282) stains brown (represents activation) 10X. From Allen et al. [8].
![TLR 2 in hippocampus of AD patient. TLR 2 (CD 282) stains brown (represents activation) 10X. From Allen et al. [8].](https://content.iospress.com:443/media/jad/2016/53-4/jad-53-4-jad160388/jad-53-jad160388-g003.jpg)
Fig.4
Candidiasis–TLR 2 coats the yeasts. Activated TLR 2 coats yeasts in the stratum corneum in candidiasis; control location of TLR 2 is in epidermal basal layer (CD 282) 40X. From Allen et al. [8].
![Candidiasis–TLR 2 coats the yeasts. Activated TLR 2 coats yeasts in the stratum corneum in candidiasis; control location of TLR 2 is in epidermal basal layer (CD 282) 40X. From Allen et al. [8].](https://content.iospress.com:443/media/jad/2016/53-4/jad-53-4-jad160388/jad-53-jad160388-g004.jpg)
Fig.5
Possible pathway for development of Aβ. Schematic for production of Aβ and Aα via MyD88 pathway. From Allen et al. [8].
![Possible pathway for development of Aβ. Schematic for production of Aβ and Aα via MyD88 pathway. From Allen et al. [8].](https://content.iospress.com:443/media/jad/2016/53-4/jad-53-4-jad160388/jad-53-jad160388-g005.jpg)
Fig.6
Aβ co-localizes with PAS (biofilm) in AD plaque. Combined PAS stain and Aβ immunostain; shows co-localization of biofilm and Aβ 40X. From Allen et al. [8].
![Aβ co-localizes with PAS (biofilm) in AD plaque. Combined PAS stain and Aβ immunostain; shows co-localization of biofilm and Aβ 40X. From Allen et al. [8].](https://content.iospress.com:443/media/jad/2016/53-4/jad-53-4-jad160388/jad-53-jad160388-g006.jpg)
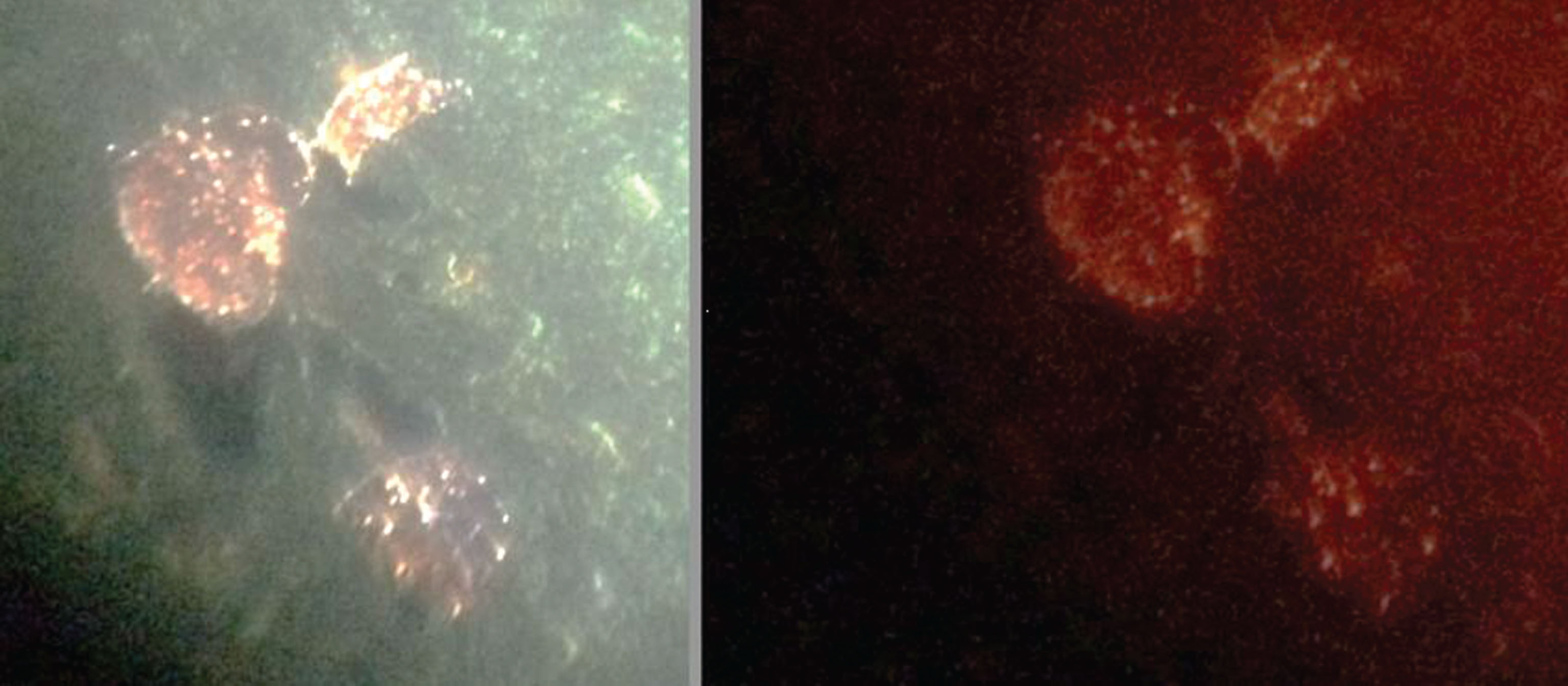
Fig.7
Same plaques in AD stained for Aβ (left) and biofilm (right). Left Congo red; right, FISH analysis of biofilm (Cy5 red label); shows Aβ and biofilm in exactly the same plaques. Dark areas show water channels, a constant finding in biofilms. Alan MacDonald, M.D., kindly provided this figure.