Aberrant Co-localization of Synaptic Proteins Promoted by Alzheimer’s Disease Amyloid-β Peptides: Protective Effect of Human Serum Albumin
Abstract
Amyloid-β (Aβ), Aβ40, Aβ42, and, recently, Aβ25-35 have been directly implicated in the pathogenesis of Alzheimer’s disease. We have studied the effects of Aβ on neuronal death, reactive oxygen species (ROS) production, and synaptic assembling in neurons in primary culture. Aβ25-35, Aβ40, and Aβ42 significantly decreased neuronal viability, although Aβ25-35 showed a higher effect. Aβ25-35 showed a more penetrating ability to reach mitochondria while Aβ40 did not enter the neuronal cytosol and Aβ42 was scarcely internalized. We did not observe a direct correlation between ROS production and cell death because both Aβ40 and Aβ42 decreased neuronal viability but Aβ40 did not change ROS production. Rather, ROS production seems to correlate with the penetrating ability of each Aβ. No significant differences were found between Aβ40 and Aβ42 regarding the extent of the deleterious effects of both peptides on neuronal viability or synaptophysin expression. However, Aβ40 elicited a clear delocalization of PSD-95 and synaptotagmin from prospective synapsis to the neuronal soma, suggesting the occurrence of a crucial effect of Aβ40 on synaptic disassembling. The formation of Aβ40- or Aβ42-serum albumin complexes avoided the effects of these peptides on neuronal viability, synaptophysin expression, and PSD-95/synaptotagmin disarrangement suggesting that sequestration of Aβ by albumin prevents deleterious effects of these peptides. We can conclude that Aβ borne by albumin can be safely transported through body fluids, a fact that may be compulsory for Aβ disposal by peripheral tissues.
INTRODUCTION
Alzheimer’s disease (AD) is characterized by the formation of plaques composed of amyloid-β peptides (Aβ) [1]. The more frequent Aβ found in these plaques are Aβ40 and Aβ42 which are formed from AβPP (amyloid-β protein precursor) by β- and γ-secretases [2]. Recently it has been found that these peptides can be detached from the plaques by a spontaneous mechanism that is not dependent on enzymatic catalysis [3]. In addition, the shorter Aβ peptide that remains toxic, Aβ25-35, has also been found in the brain of AD patients, presumably coming from Aβ40 cleavage [4]. Thus, normal aging would promote racemization of serine26 of Aβ40, which may result in the formation of truncated forms of Aβ40 including Aβ25-35 [5]. This suggests that Aβ25-35 together with Aβ40 and Aβ42 can play a role in the pathogenesis of AD. In this context, Aβ25-35 can reach mitochondria where it may cause the disarrangement of respiratory machinery [6]. This can be accomplished by the occlusion of TIM23/TOM40 mitochondrial protein permease system as caused by AβPP overproduction [7] or by the interaction of Aβ with the mitochondria permeability transition pore which may lead to the increase mitochondrial permeability and subsequent apoptosis [8]. Finally, an abnormal interaction of Aβ with Drp1 in neurons from AD patients can result in the inhibition of mitochondrial fission [9], a fact that can change mitochondrial number together with their distribution around synapsis (for a review see [10]).
One of the early events in neurodegeneration associated with AD is synaptic disarrangement, a phenomenon that presumably precedes Aβ plaque formation [11, 12]. Indeed, Aβ preferentially accumulates in synapsis [13, 14], which can result in the decrease in neuronal interconnections [15–17]. In agreement with this, it has been described that Aβ caused morphological and functional changes in synapsis [18, 19] and inhibited neuronal plasticity [20, 21].
Monomeric and oligomeric Aβ can be transported from brain interstitial compartment to the cerebrospinal fluid (CSF) [22] and from this to peripheral blood circulation [23, 24]. Most of the Aβ in CSF and blood is associated with serum albumin [25, 26], which that may prevent Aβ polymerization [27]. Moreover, it has been proposed that serum albumin plays an important role in Aβ clearance and elimination [28]. Aβ concentrations in the blood correlated with those observed in the brain, leading to the proposal of the “peripheral sink hypothesis” for disposal of the peptides [28]. According to this hypothesis, the “peripheral sink” would drive Aβ efflux from the brain. Indeed, serum albumin can play an important role in this process because Aβ is transported as Aβ-albumin complex by body fluids. In addition, Aβ disposal by the kidney may be mediated by serum albumin because megalin, i.e., a serum albumin receptor in the apical membrane of proximal tubular cells [29], has shown a protective effect promoting Aβ disposal [30–33]. Thus, Aβ would be transported by serum albumin from the brain to the kidney where its disposal is facilitated by the albumin-megalin endocytosis mechanism. It should be mentioned that neprilysin, a putative Aβ-protease, is abundant in the kidney brush border membrane [34], suggesting the occurrence of a convenient allocation of Aβ disposal machinery. In agreement with the role played by serum albumin in Aβ clearance from the brain, treatment of AD patients by repeat plasma replacements with Aβ-free human serum albumin decreased brain Aβ accumulations and ameliorated cognitive test results [35].
In a previous work, we showed that the presence of serum albumin promoted neuronal survival in the presence of Aβ25-35, which can be explained because serum albumin prevented amyloid entry into neurons [36]. In this work, we have studied the protective effect of human serum albumin on the changes caused by Aβ25-35, Aβ40, and Aβ42 in neuronal viability, reactive oxygen species (ROS) production, and synaptic protein expression and localization in neurons in primary culture.
MATERIALS AND METHODS
Neuronal cultures
Albino Wistar rats were obtained from the animal house of the University of Salamanca (Spain) and were used according to local and EU Ethical Committee guidelines. Cell cultures were carried out as previously described [37]. Briefly, fetuses at 17.5 days of gestation were delivered by rapid hysterectomy after cervical dislocation of the mother. Animals were decapitated and their brains immediately excised. After removing the meninges and blood vessels, the forebrains were placed in Earle’s balanced solution (EBS) containing 20 μg/ml DNase and 0.3% (w/v) BSA. The tissue was minced, washed, centrifuged at 500 × g for 4 min and incubated in 0.025% trypsin (type III) and 60 μg/ml DNase I for 15 min at 37°C. Trypsinization was terminated by the addition of DMEM containing 10% FCS. The tissue was then dissociated by gentle passing it eight times through a siliconized Pasteur pipette, and the supernatant cell suspension was recovered. This operation was repeated and the resulting cell suspension was centrifuged at 500 × g for 5 min. The cells were then resuspended in DMEM containing 10% FCS and plated on Petri dishes coated with 10 μg/ml of poly-L-lysine at a density of 1.0×105 cells/cm2. Neurons were maintained at 37°C and 5% CO2. One day after plating, cytosine arabinoside was added to avoid glial cell proliferation.
Cellular treatments
Aβ peptides (30 μM) were purchased from Bachem (Bubendorf, Switzerland) and were prepared in sterile deionized water. This concentration was chosen because dose-response experiments showed that 30 μM caused maximal effects with the three Aβ (see Supplementary Figure 1). Human Serum Albumin was obtained from Grifols (Barcelona, Spain). In order to prepare the HSA-Aβ complexes, Aβ peptides were gently dissolved in an HSA solution.
Neuronal viability assay
3 days in vitro neurons (3 DIV) were maintained in a serum-free medium (Hanks medium, pH = 7.4) with the different treatments for 20 h. Then, neuronal viability was determined by the MTT reduction assay [38]. Briefly, MTT (Thermo Fisher, Waltham, USA) was diluted in Hanks medium (0.5 mg/ml) and added to the cells. After 75 min of incubation (37°C, 5% CO2, in the darkness), the medium with MTT was replaced by dimethyl sulfoxide and cells were gently shaken for 10 min in the darkness. Finally, the absorbance was measured at 570 nm. Data are presented as percentages of cell viability as compared to non-treated cells.
Reactive oxygen species production
Production of ROS was measured using the fluorogenic 2’,7’-dichlorodihydrofluorescein-diacetate probe (H2DCFDA, Thermo Fisher) [39]. 3 days in vitro neurons (3 DIV) were incubated in Hanks medium containing 10 μM H2DCFDA for 20 h treatments. Fluorescence at 535 nm was measured at the beginning and at the end of the experiment. The fluorescence difference was normalized using cell viability data and was expressed as the percentage of ROS production as compared to non-treated cells.
Immunocytochemistry
Immunocytochemistry was essentially carried out as described by Tabernero et al. [40]. After the treatments, cells were fixed in 4% paraformaldehyde for 20 min. In those experiments where mitochondria were localized, neurons were incubated with 100 nM MitotrackerRed (Thermo Fisher) for 20 min before fixation. Once fixed, cells were washed with PBS and permeabilized with 0.25% Triton X-100 for 1 h or 100% methanol for 10 min at –20°C. Then, neurons were incubated overnight at 4°C with primary antibodies (1:200) against Aβ25-35 (LSBio, Seattle, USA), Aβ40 (Novus Biologicals, Littleton, USA), Aβ42 (LSBio), Glut3 (Novus Biologicals), APP (Sigma-Aldrich, Madrid, Spain), PSD-95 (Thermo Scientific), or synaptotagmin (Synaptic Systems, Goettingen, Germany). Then they are incubated for 2 h at room temperature with (1:1000) secondary antibodies anti-rabbit or anti-mouse Alexa Fluor 488, 594, or 647 (Thermo Fisher). Finally, nuclei were stained with TOPRO3 (Thermo Fisher). Images were taken using a Leica DM-IRE 2 TCS-SP2 confocal microscope with LCS Lite Software (Leica Microsystems, Wetzlar, Germany). Fluorescence and co-localization were analyzed using Image J software (NIH, Bethesda, MD,USA).
Western blot analysis
Cell proteins were extracted using a lysis buffer containing 5 mM Tris-HCl (pH 6.8), 2% SDS, 2 mM EDTA, 2 mM EGTA, 1 mM PMSF, and cocktail protease inhibitors (Calbiochem, Darmstadt, USA). Lysates were centrifuged at 14.000 × g for 15 min at 4°C. 20 μg of protein extract was analyzed in 10% precast commercial gels (NuPAGE Novex 10% Bis-Tris Midi Gel 1.0 mm). The buffer used for protein electrophoresis was NuPAGE MOPS SDS Running Buffer 20X. NuPAGE Sample Reducing Agent 10X and NuPAGE LDS Sample Buffer 4X were used to prepare the samples. Electrophoresis was run at room temperature using a constant voltage. After electrophoresis, gels were washed in transfer buffer (10% methanol, 0.1% NuPAGE Antioxidant diluted in NuPAGE Transfer Buffer 2X) for 10 min. Then, the proteins were transferred to a nitrocellulose membrane (iBlot Gel Transfer Stacks Nitrocellulose) for 10 min, applying a constant voltage. All products used for electrophoresis and subsequent electrotransfer were purchased from Invitrogen (Thermo Fisher). After blocking for non-specific binding, the membranes were incubated overnight at 4°C with mouse monoclonal antibody against synaptophysin 1:2500 (Santa Cruz Biotechnology, Dallas, USA). Mouse monoclonal antibody against α-tubulin (Sigma-Aldrich) was used to normalize and quantify protein expression. After several washes, membranes were incubated with secondary antibody against mouse immunoglobulin conjugated with peroxidase. Finally, membranes were incubated for 1 min with peroxidase substrates, which afforded a chemiluminescence reaction. The signal collected on the autoradiographic film was proportional to the amount of protein in the membrane. The bands were quantified using an image analysis program.
Statistical analysis
All results are presented as the mean±SEM of at least three independent experiments (n≥3). Data were analyzed for statistical significance with one-way ANOVA, followed by an appropriate post-hoc test. Dunnett test was used to compare all the values with the control and Tukey test to compare all the values among themselves. Values were considered significant when p < 0.05.
RESULTS
Effects of Aβ peptides on cell viability in neurons in primary culture
The three Aβ assayed promoted cellular death in neurons in primary culture although Aβ25-35 showed a more conspicuous deleterious effect (Fig. 1). The presence of serum albumin per se was able to decrease neuronal death caused by Aβ25-35 but not by Aβ40 or Aβ42 (Fig. 1). However, if the Aβ-serum albumin complexes were previously formed the effect of Aβ40 or Aβ42 on cell viability were fully avoided although Aβ25-35- albumin complex still showed a low deleterious effect(Fig. 1).
Effects of Aβ peptides on ROS production in neurons in primary culture
The presence of Aβ25-35 sharply increased ROS production in neurons in primary culture (Fig. 2). The effect of Aβ42 on ROS production was lower as compared to Aβ25-35. ROS production showed by the presence of Aβ40 was negligible (Fig. 2). In all the cases including controls the presence of serum albumin per se strongly prevented ROS production confirming the role of serum albumin as an inhibitor of ROS production [41]. Unexpectedly, Aβ40- or Aβ42-albumin complexes were unable to prevent ROS production suggesting that the changes in albumin conformation caused by binding to these amyloids are enough to avoid the role of albumin as an inhibitor of ROS production [41].
Cellular localization of Aβ peptides in neurons in primary culture
Aβ25-35 was clearly internalized by neurons (Fig. 3) and reached mitochondria as showed by its co-localization with MitotrakerRed (yellow), a specific marker of mitochondria (Figs. 3 and 4). Under these circumstances, Aβ40 mostly remained outside the cell and formed discrete aggregations subtly bound to the neuronal membrane (Fig. 5). Instead, Aβ42 was found inside the cell (Fig. 3) mostly clustered in lumpy aggregations firmly pasted to the neuronal membrane (Fig. 5). These results are in agreement with Nagele et al. [42] who found an accumulation of exogenous Aβ42 but not Aβ40 in neuroblastoma cells.
The presence of albumin per se did not change Aβ40 or Aβ42 cellular localization (see Supplementary Figure 3) but both Aβ-albumin complexes (C 40 and C 42; Fig. 5) did not enter neurons nor stick to their membranes but apparently they were mostly washed during the preparation of immunocytochemistry (Fig. 5).
Effects of Aβ peptides on synaptic generation and assembling
It has been suggested that an early event in AD is synaptic disruption, which resulted in the decrease in neuronal connections [15–17]. In agreement with this, our results show that the three Aβ assayed decreased synaptophysin expression (Fig. 6). The effect of Aβ25-35 cannot be prevented by albumin per se but it is mostly not observed in the presence of Aβ25-35-albumin complex (Fig. 6). Albumin per se decreased the effect of Aβ40 or Aβ42 on synaptophysin expression while Aβ-albumin complexes avoided their effects (Fig. 6).
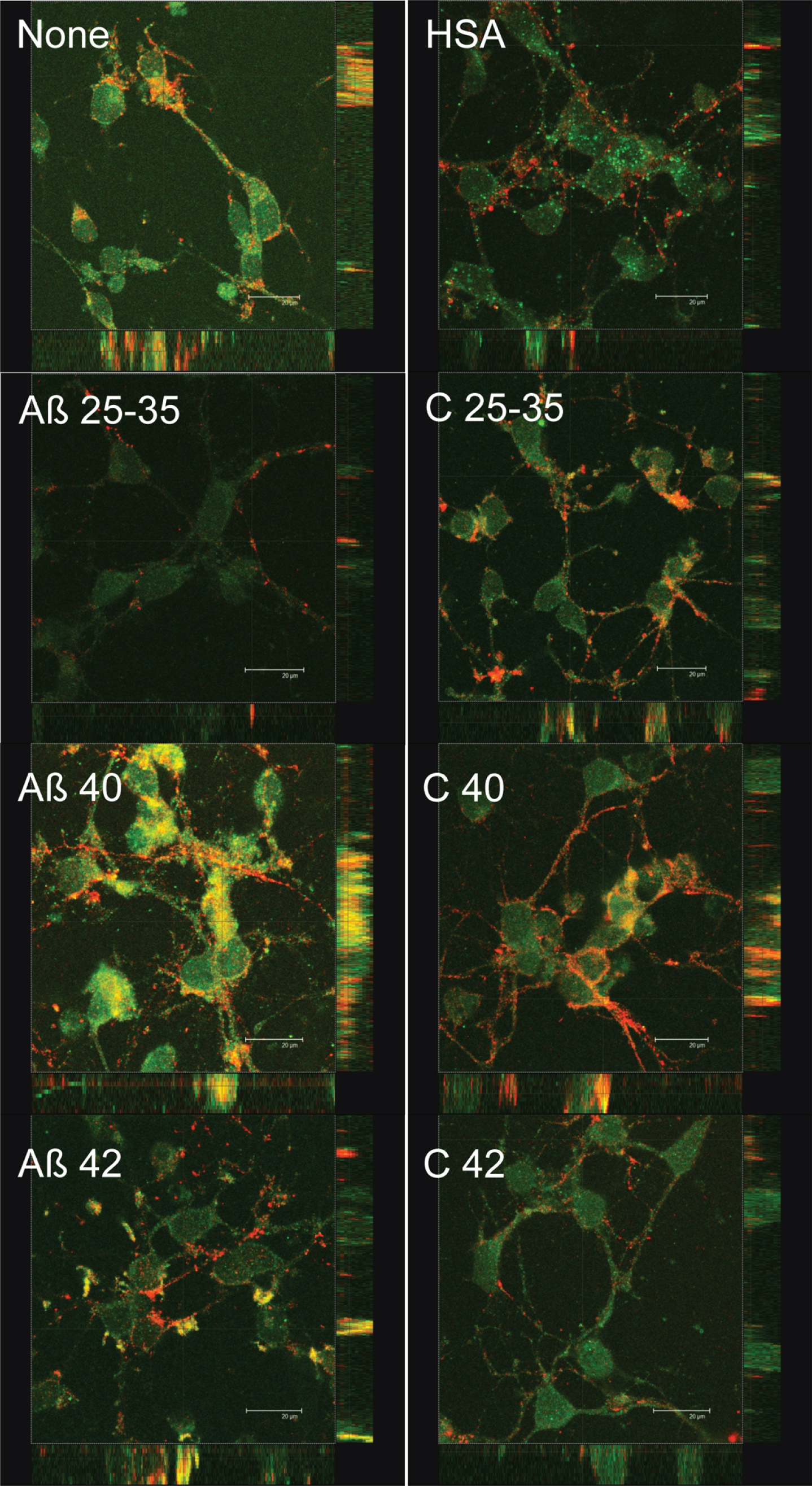
In addition, we decide to investigate the possible effect of Aβ on the localization of synaptic proteins. Since our aim was to study the effects of Aβ on synaptic assembling, we challenged neurons with Aβ before this process was completed. Indeed, preliminary experiments showed that the effect of Aβ was observed as early as after 4 days in culture. Consequently, these experiments were carried out in 4 DIV neurons. In order to know if Aβ peptides elicited some short-term effects on synaptic assembling, we studied the short-term (2 h) effects of the three amyloids on pre-synaptic (synaptotagmin) and post-synaptic (PSD-95) proteins expression (Fig. 7). Synaptotagmin fluorescence staining decreased in the presence of Aβ25-35 but not significantly in the presence of Aβ40 or Aβ42 (Fig. 7). PSD-95 slightly decreased in the presence of Aβ25-35 or Aβ40 but not in the presence of Aβ42 (Fig. 7). Co-localization between both markers measured by yellow fluorescence decreased in the presence of Aβ25-35 but not in the presence of Aβ40 or Aβ42 (Fig. 7). More intriguing were the changes in the distribution of synaptotagmin and PSD-95 elicited by the presence of Aβ (Fig. 8). Thus, in the absence of Aβ (Fig. 8, None), PSD-95 (green) was distributed by the cell soma while synaptotagmin (red) localized in neurites. Moreover, co-localization of both proteins was observed (yellow) in the so-called “synaptic points” which have been associated with prospective synapsis [43, 44]. However, the presence of Aβ40 notably enhanced PSD-95-synaptotagmin co-localization (Fig. 8; yellow) but the allocation of synaptotagmin-PSD-95 putative aggregates changed from prospective synapsis between adjacent neurons to a uniform distribution pattern over the neuronal soma (Fig. 8, Aβ40). This phenomenon was not observed with Aβ42 (Fig. 8, Aβ42) or Aβ25-35 (Fig. 8, Aβ25-35) suggesting that the effects of Aβ40 on synaptotagmin-PSD-95 distribution are exclusive. However, these effects were not observed with the Aβ40-albumin complex (Fig. 8, C 40), suggesting that the association of Aβ40 with albumin avoided the effects of this amyloid on synaptotagmin-PSD-95 disarrangement (see Supplementary Figure 4 for the scarce effect observed when serum albumin is not previously bound to Aβ).
DISCUSSION
There is increasing evidence that Aβ and others AβPP metabolites may perform as antimicrobial agents or play a physiological role regulating protein expression as transcription factors [45–48]. However, there is a consensus that Aβ accumulation is the main event in the development of neurodegeneration observed in AD. In this context, the three Aβ assayed significantly decreased neuronal viability but Aβ25-35 elicited the higher effect (Fig. 1). This may be due to a more penetrating ability performed by Aβ25-35, since under our experimental circumstances, Aβ25-35 was internalized by the neuron and it reached mitochondria (Figs. 3 and 4). This result may be relevant in the context that it has been proposed that short Aβ peptides may be released from AD plaques enhancing neurodegeneration [3]. In agreement with this, Aβ25-35 fragments have been found in the brain of AD patients [4], probably synthesized by a senile degradative process that includes racemization of serine26 in Aβ40 [5]. If so, it is reasonable to suggest that short Aβ-fractions containing Aβ25-35 amino acid sequence may be released by AD plaques contributing to neurodegeneration observed in AD. Thus, these peptides would be internalized by neurons reaching mitochondria where they interfered with the respiratory machinery [6]. It should be mentioned that mitochondrial dysfunction is an early event in the development of AD [49], a fact that has been explained by the deleterious effects of Aβ on mitochondria. Indeed, Aβ interaction with mitochondrial transition pore [8] together with the inhibition of mitochondrial fission [9] would result in mitochondrial respiratory impairment and the subsequent increase in neuronal death (Fig. 1). It should be mentioned that our results are not concurrent with the idea that there is a direct correlation between ROS production (Fig. 2) and cell viability (Fig. 1), because both Aβ40 and Aβ42 increased cell death (Fig. 1) while Aβ40 did not change ROS production (Fig. 2). Conversely, we observed that those amyloids that are able to cross the neuronal membrane (Aβ25-35 and Aβ42) increased ROS production but those remained outside (Aβ40) did not change ROS generation (Figs. 2 and 3). This is in agreement with Boldyrev et al. [50] who did not find a correlation between cell death and ROS production in dissociated granular cell neurons exposed to Aβ. Moreover, Aβ variants unable to enter neurons do not impair synaptic transmission [51].
In our hands, no significant differences were found between Aβ40 and Aβ42 regarding the effect of both peptides on neuronal viability. This is in agreement with the idea that the enhanced deleterious effect of Aβ42 as compared to Aβ40 that has been reported [52] must be due to the lower clearance rate of the former through the blood-brain barrier [53]. In this context, soluble forms of Aβ are released into the CSF [22] and transported to the blood [23, 24] by a process in which serum albumin may play a crucial role. In fact, albumin not only showed a high affinity for Aβ but also prevented Aβ polymerization [27]. In agreement with this, our results showed that Aβ-albumin complexes avoided the effects of Aβ on cell death (Fig. 1), a fact according with the idea that albumin not only helps to transport Aβ through body fluids but also protects brain structures against the deleterious effects of Aβ. In agreement with this, repeated replacements of plasma by Aβ-free serum albumin (Albutein®) ameliorated cognitive decline in AD patients [35], which is consistent with the idea that albumin plays an important role in Aβ clearance and disposal. It should be mentioned that other serum proteins such as immunoglobulins (IVIG) have also shown to perform a protective role on preventing oxidative damage associated to AD [54, 55].
It has been proposed that neurodegeneration occurring in AD started by the decrease in dendritic spines and subsequent synaptic damage resulting in the impairment of neurotransmission [2]. In this context, Aβ preferentially accumulated in synapsis [13, 14] and the decrease in synaptic function correlates with cognitive decline in AD [15–17]. In agreement with this, our results show that both Aβ40 and Aβ42 decreased synaptophysin expression (Fig. 6), suggesting that neurotransmitter storage and/or release may be compromised in these circumstances [18, 19]. Moreover, a short exposition of neurons to Aβ40 or Aβ42 did not change PSD-95 and synaptotagmin expression (Fig. 7), but Aβ40 elicited a clear delocalization of PSD-95 and synaptotagmin from cell membrane to the soma (Fig. 8). This suggests the occurrence of a short-term effect of Aβ40 on synaptic disassembling. It should be mentioned that PSD-95 and synaptotagmin co-localization in adjacent membranes between neurons is a clear indication of prospective synapsis (Fig. 8 None, yellow points) [43, 44] suggesting that under our experimental conditions, Aβ40 is disrupting synaptic structure. According to Savioz’s model, as far as dendrites are disarranged with the progress of the disease, PSD-95 decreases in dendritic spines while its presence in the soma becomes apparent [56]. In this context, our results clearly indicate that in the presence of Aβ40, PSD-95, and synaptotagmin abnormally co-localized in the cell soma before the disarrangement of neurites (Fig. 8). This suggests that abnormal co-localization of PSD-95-synaptotagmin aggregates caused by Aβ40 precedes dendrite disarrangement. If so, the presence of Aβ40 may cause the disruption of synaptic connections by leading PSD-95 and synaptotagmin to an aberrant co-localization in cell soma instead of between adjacent neurites. In our experiments, this phenomenon takes place as early as 2 h after the exposition of the neurons to Aβ40, consistent with the idea that delocalization of PSD-95 and synaptotagmin can be the first step in the process causing neurodegeneration in AD.
Aβ40- or Aβ42-serum albumin complexes avoided the effects of these peptides on neuronal viability (Fig. 1), synaptophysin expression (Fig. 6), and PSD-95/synaptotagmin disarrangement (Fig. 8) suggesting that sequestration of Aβ by albumin prevents deleterious effects of these peptides in neurons. This may be a consequence of the conformation adopted by Aβ when it is bound to albumin that avoids toxic effects. In addition, the protective effect of albumin may be also due to the fact that Aβ-serum albumin complexes inhibit Aβ polymerization and/or their internalization into neurons. This may prevent Aβ to adopt a toxic conformation and/or to reach the undesirable target. In agreement with this, our results show that Aβ-serum albumin complexes are mostly not detected by immunochemistry (Fig. 5), a fact consistent with the idea that Aβ were washed out as Aβ-serum albumin complex before staining. Consequently, the adverse effects of Aβ were not observed when the amyloids were bound to albumin suggesting that Aβ-serum albumin complex may be formed just when amyloids are synthesized, as the first step of a process designed not only for brain Aβ-clearance but also for transporting Aβ through body fluids safely. It should be mentioned that AD patients showed low serum albumin concentrations as compared to those healthy counterparts [57], a fact that may contribute to the accumulation of Aβ in the brain and to the neurodegeneration associated with this disease.
Taken together, our results show that unlike Aβ25-35 and Aβ42, Aβ40 does not enter the neuronal cytoplasm nor increases ROS production but causes delocalization of synaptic proteins, a phenomenon that may account for synaptic disarrangement and subsequent loss of inter-neuronal connections observed in AD.
ACKNOWLEDGMENTS
This work was supported by a grant from Instituto Grifols S.A., Barcelona, Spain. M. Domínguez-Prieto was the recipient of an FPI (MINECO, Spain). We are grateful to T. del Rey for her technical assistance.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-0346r2).
Appendices
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-160346.
REFERENCES
[1] | Masters CL , Selkoe DJ ((2012) ) Biochemistry of amyloid beta-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb Perspect Med 2: , a006262. |
[2] | Haass C , Kaether C , Thinakaran G , Sisodia S ((2012) ) Trafficking and proteolytic processing of APP. Cold Spring Harb Perspect Med 2: , a006270. |
[3] | Lyons B , Friedrich MG , Raftery MJ , Truscott RJ ((2016) ) Amyloid plaque in the human brain can decompose from Abeta1-40/1-42 by spontaneous non-enzymatic processes. Anal Chem 88: , 2675–2684. |
[4] | Kaneko I , Morimoto K , Kubo T ((2001) ) Drastic neuronal loss in vivo by beta-amyloid racemized at Ser(26) residue: Conversion of non-toxic [D-Ser(26)]beta-amyloid 1-40 to toxic and proteinase-resistant fragments. Neuroscience 104: , 1003–1011. |
[5] | Kubo T , Nishimura S , Kumagae Y , Kaneko I ((2002) ) In vivo conversion of racemized beta-amyloid ([D-Ser 26]A beta 1-40) to truncated and toxic fragments ([D-Ser 26]A beta 25-35/40) and fragment presence in the brains of Alzheimer’s patients. J Neurosci Res 70: , 474–483. |
[6] | Cuevas E , Lantz SM , Tobon-Velasco JC , Newport GD , Wu Q , Virmani A , Ali SF , Santamaria A ((2011) ) On the in vivo early toxic properties of A-beta 25-35 peptide in the rat hippocampus: Involvement of the Receptor-for-Advanced Glycation-End-Products and changes in gene expression. Neurotoxicol Teratol 33: , 288–296. |
[7] | Devi L , Prabhu BM , Galati DF , Avadhani NG , Anandatheerthavarada HK ((2006) ) Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci 26: , 9057–9068. |
[8] | Du H , Yan SS ((2010) ) Mitochondrial permeability transition pore in Alzheimer’s disease: Cyclophilin D and amyloid beta. Biochim Biophys Acta 1802: , 198–204. |
[9] | Manczak M , Anekonda TS , Henson E , Park BS , Quinn J , Reddy PH ((2006) ) Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet 15: , 1437–1449. |
[10] | Burte F , Carelli V , Chinnery PF , Yu-Wai-Man P ((2015) ) Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol 11: , 11–24. |
[11] | Selkoe DJ ((2002) ) Alzheimer’s disease is a synaptic failure. Science 298: , 789–791. |
[12] | Ferreira ST , Lourenco MV , Oliveira MM , De Felice FG ((2015) ) Soluble amyloid-beta oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front Cell Neurosci 9: , 191. |
[13] | Lacor PN , Buniel MC , Chang L , Fernandez SJ , Gong Y , Viola KL , Lambert MP , Velasco PT , Bigio EH , Finch CE , Krafft GA , Klein WL ((2004) ) Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. J Neurosci 24: , 10191–10200. |
[14] | Koffie RM , Meyer-Luehmann M , Hashimoto T , Adams KW , Mielke ML , Garcia-Alloza M , Micheva KD , Smith SJ , Kim ML , Lee VM , Hyman BT , Spires-Jones TL ((2009) ) Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A 106: , 4012–4017. |
[15] | DeKosky ST , Scheff SW ((1990) ) Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann Neurol 27: , 457–464. |
[16] | Terry RD , Masliah E , Salmon DP , Butters N , DeTeresa R , Hill R , Hansen LA , Katzman R ((1991) ) Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann Neurol 30: , 572–580. |
[17] | Scheff SW , Price DA , Schmitt FA , Mufson EJ ((2006) ) Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol Aging 27: , 1372–1384. |
[18] | Coleman PD , Yao PJ ((2003) ) Synaptic slaughter in Alzheimer’s disease. Neurobiol Aging 24: , 1023–1027. |
[19] | Almeida CG , Tampellini D , Takahashi RH , Greengard P , Lin MT , Snyder EM , Gouras GK ((2005) ) Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol Dis 20: , 187–198. |
[20] | Shankar GM , Li S , Mehta TH , Garcia-Munoz A , Shepardson NE , Smith I , Brett FM , Farrell MA , Rowan MJ , Lemere CA , Regan CM , Walsh DM , Sabatini BL , Selkoe DJ ((2008) ) Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med 14: , 837–842. |
[21] | Li S , Hong S , Shepardson NE , Walsh DM , Shankar GM , Selkoe D ((2009) ) Soluble oligomers of amyloid Beta protein facilitate hippocampal long-term depression by disrupting neuronal glutamate uptake. Neuron 62: , 788–801. |
[22] | Fukumoto H , Tokuda T , Kasai T , Ishigami N , Hidaka H , Kondo M , Allsop D , Nakagawa M ((2010) ) High-molecular-weight beta-amyloid oligomers are elevated in cerebrospinal fluid of Alzheimer patients. FASEB J 24: , 2716–2726. |
[23] | Roher AE , Esh CL , Kokjohn TA , Castano EM , Van Vickle GD , Kalback WM , Patton RL , Luehrs DC , Daugs ID , Kuo YM , Emmerling MR , Soares H , Quinn JF , Kaye J , Connor DJ , Silverberg NB , Adler CH , Seward JD , Beach TG , Sabbagh MN ((2009) ) Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer’s disease. Alzheimers Dement 5: , 18–29. |
[24] | Xia W , Yang T , Shankar G , Smith IM , Shen Y , Walsh DM , Selkoe DJ ((2009) ) A specific enzyme-linked immunosorbent assay for measuring beta-amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer disease. Arch Neurol 66: , 190–199. |
[25] | Stanyon HF , Viles JH ((2012) ) Human serum albumin can regulate amyloid-beta peptide fiber growth in the brain interstitium: Implications for Alzheimer disease. J Biol Chem 287: , 28163–28168. |
[26] | Biere AL , Ostaszewski B , Stimson ER , Hyman BT , Maggio JE , Selkoe DJ ((1996) ) Amyloid beta-peptide is transported on lipoproteins and albumin in human plasma. J Biol Chem 271: , 32916–32922. |
[27] | Milojevic J , Raditsis A , Melacini G ((2009) ) Human serum albumin inhibits Abeta fibrillization through a “monomer-competitor” mechanism. Biophys J 97: , 2585–2594. |
[28] | DeMattos RB , Bales KR , Cummins DJ , Paul SM , Holtzman DM ((2002) ) Brain to plasma amyloid-beta efflux: A measure of brain amyloid burden in a mouse model of Alzheimer’s disease. Science 295: , 2264–2267. |
[29] | Brunskill N ((2001) ) Mechanisms of albumin uptake by proximal tubular cells. Am J Kidney Dis 37: , S17–S20. |
[30] | Zlokovic BV , Martel CL , Matsubara E , McComb JG , Zheng G , McCluskey RT , Frangione B , Ghiso J ((1996) ) Glycoprotein 330/megalin: robable role in receptor-mediated transport of apolipoprotein J alone and in a complex with Alzheimer disease amyloid beta at the blood-brain and blood-cerebrospinal fluid barriers. Proc Natl Acad SciU S A 93: , 4229–4234. |
[31] | Deane R , Wu Z , Sagare A , Davis J , Du Yan S , Hamm K , Xu F , Parisi M , LaRue B , Hu HW , Spijkers P , Guo H , Song X , Lenting PJ , Van Nostrand WE , Zlokovic BV ((2004) ) LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 43: , 333–344. |
[32] | Carro E , Spuch C , Trejo JL , Antequera D , Torres-Aleman I ((2005) ) Choroid plexus megalin is involved in neuroprotection by serum insulin-like growth factor I. J Neurosci 25: , 10884–10893. |
[33] | Tanzi RE , Bertram L ((2005) ) Twenty years of the Alzheimer’s disease amyloid hypothesis: A genetic perspective. Cell 120: , 545–555. |
[34] | Carson JA , Turner AJ ((2002) ) Beta-amyloid catabolism: Roles for neprilysin (NEP) and other metallopeptidases? J Neurochem 81: , 1–8. |
[35] | Boada M , Ortiz P , Anaya F , Hernandez I , Munoz J , Nunez L , Olazaran J , Roca I , Cuberas G , Tarraga L , Buendia M , Pla RP , Ferrer I , Paez A ((2009) ) Amyloid-targeted therapeutics in Alzheimer’s disease: Use of human albumin in plasma exchange as a novel approach for Abeta mobilization. Drug News Perspect 22: , 325–339. |
[36] | Vega L , Arroyo AA , Tabernero A , Medina JM ((2009) ) Albumin-blunted deleterious effect of amyloid-beta by preventing the internalization of the peptide into neurons. J Alzheimers Dis 17: , 795–805. |
[37] | Tabernero A , Bolanos JP , Medina JM ((1993) ) Lipogenesis from lactate in rat neurons and astrocytes in primary culture. Biochem J 294: (Pt 3), 635–638. |
[38] | Mosmann T ((1983) ) Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods 65: , 55–63. |
[39] | Rosenkranz AR , Schmaldienst S , Stuhlmeier KM , Chen W , Knapp W , Zlabinger GJ ((1992) ) A microplate assay for the detection of oxidative products using 2’,7’-dichlorofluorescin-diacetate. J Immunol Methods 156: , 39–45. |
[40] | Tabernero A , Lavado EM , Granda B , Velasco A , Medina JM ((2001) ) Neuronal differentiation is triggered by oleic acid synthesized and released by astrocytes. J Neurochem 79: , 606–616. |
[41] | Anraku M , Chuang VT , Maruyama T , Otagiri M ((2013) ) Redox properties of serum albumin. Biochim Biophys Acta 1830: , 5465–5472. |
[42] | Nagele RG , D’Andrea MR , Anderson WJ , Wang HY ((2002) ) Intracellular accumulation of beta-amyloid(1-42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience 110: , 199–211. |
[43] | Christopherson KS , Ullian EM , Stokes CC , Mullowney CE , Hell JW , Agah A , Lawler J , Mosher DF , Bornstein P , Barres BA ((2005) ) Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120: , 421–433. |
[44] | Polo-Hernandez E , Tello V , Arroyo AA , Dominguez-Prieto M , de Castro F , Tabernero A , Medina JM ((2014) ) Oleic acid synthesized by stearoyl-CoA desaturase (SCD-1) in the lateral periventricular zone of the developing rat brain mediates neuronal growth, migration and the arrangement of prospective synapses. Brain Res 1570: , 13–25. |
[45] | Soscia SJ , Kirby JE , Washicosky KJ , Tucker SM , Ingelsson M , Hyman B , Burton MA , Goldstein LE , Duong S , Tanzi RE , Moir RD ((2010) ) The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 5: , e9505. |
[46] | Bailey JA , Maloney B , Ge YW , Lahiri DK ((2011) ) Functional activity of the novel Alzheimer’s amyloid beta-peptide interacting domain (AbetaID) in the APP and BACE1 promoter sequences and implications in activating apoptotic genes and in amyloidogenesis. Gene 488: , 13–22. |
[47] | Maloney B , Lahiri DK ((2011) ) The Alzheimer’s amyloid beta-peptide (Abeta) binds a specific DNA Abeta-interacting domain (AbetaID) in the APP, BACE1, and APOE promoters in a sequence-specific manner: Characterizing a new regulatory motif. Gene 488: , 1–12. |
[48] | Multhaup G , Huber O , Buee L , Galas MC ((2015) ) Amyloid precursor protein (APP) metabolites APP intracellular fragment (AICD), Abeta42, and tau in nuclear roles. J Biol Chem 290: , 23515–23522. |
[49] | Hauptmann S , Scherping I , Drose S , Brandt U , Schulz KL , Jendrach M , Leuner K , Eckert A , Muller WE ((2009) ) Mitochondrial dysfunction: An early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging 30: , 1574–1586. |
[50] | Boldyrev A , Koudinov A , Berezov T , Carpenter DO ((2004) ) Amyloid-beta induced cell death is independent of free radicals. J Alzheimers Dis 6: , 633–638; discussion 673-681. |
[51] | Ripoli C , Cocco S , Li Puma DD , Piacentini R , Mastrodonato A , Scala F , Puzzo D , D’Ascenzo M , Grassi C ((2014) ) Intracellular accumulation of amyloid-beta (Abeta) protein plays a major role in Abeta-induced alterations of glutamatergic synaptic transmission and plasticity. J Neurosci 34: , 12893–12903. |
[52] | Boada M , Ramos-Fernandez E , Guivernau B , Munoz FJ , Costa M , Ortiz AM , Jorquera JI , Nunez L , Torres M , Paez A ((2016) ) Treatment of Alzheimer disease using combination therapy with plasma exchange and haemapheresis with albumin and intravenous immunoglobulin: Rationale and treatment aroach of the AMBAR (Alzheimer Management By Albumin Replacement) study. Neurologia 31: , 473–481. |
[53] | Bell RD , Sagare AP , Friedman AE , Bedi GS , Holtzman DM , Deane R , Zlokovic BV ((2007) ) Transport pathways for clearance of human Alzheimer’s amyloid beta-peptide and apolipoproteins E and J in the mouse central nervous system. J Cereb Blood Flow Metab 27: , 909–918. |
[54] | Counts SE , Ray B , Mufson EJ , Perez SE , He B , Lahiri DK ((2014) ) Intravenous immunoglobulin (IVIG) treatment exerts antioxidant and neuropreservatory effects in preclinical models of Alzheimer’s disease. J Clin Immunol 34: (Suppl 1), S80–S85. |
[55] | Lahiri DK , Ray B ((2014) ) Intravenous immunoglobulin treatment preserves and protects primary rat hippocampal neurons and primary human brain cultures against oxidative insults. Curr Alzheimer Res 11: , 645–654. |
[56] | Savioz A , Leuba G , Vallet PG ((2014) ) A framework to understand the variations of PSD-95 expression in brain aging and in Alzheimer’s disease. Ageing Res Rev 18: , 86–94. |
[57] | Kim TS , Pae CU , Yoon SJ , Jang WY , Lee NJ , Kim JJ , Lee SJ , Lee C , Paik IH , Lee CU ((2006) ) Decreased plasma antioxidants in patients with Alzheimer’s disease. Int J Geriatr Psychiatry 21: , 344–348. |
Figures and Tables
Fig.1
Effect of amyloid-beta peptides on cell viability in the absence or the presence of HSA. Neurons in primary culture (3 DIV) were incubated for 20 h in Hanks medium with three different Aβ peptides: Aβ25-35, Aβ40, or Aβ42 (30 μM) in the absence or the presence of human serum albumin (HSA; 30 μM) or as HSA-Aβ complexes (30 μM). Results are expressed as percentages compared to non-treated cells and are means±SEM (n≥6). One-way ANOVA and Tukey Test were applied in order to compare different groups. Distinct characters are used to indicate statistically different groups (p < 0.05).
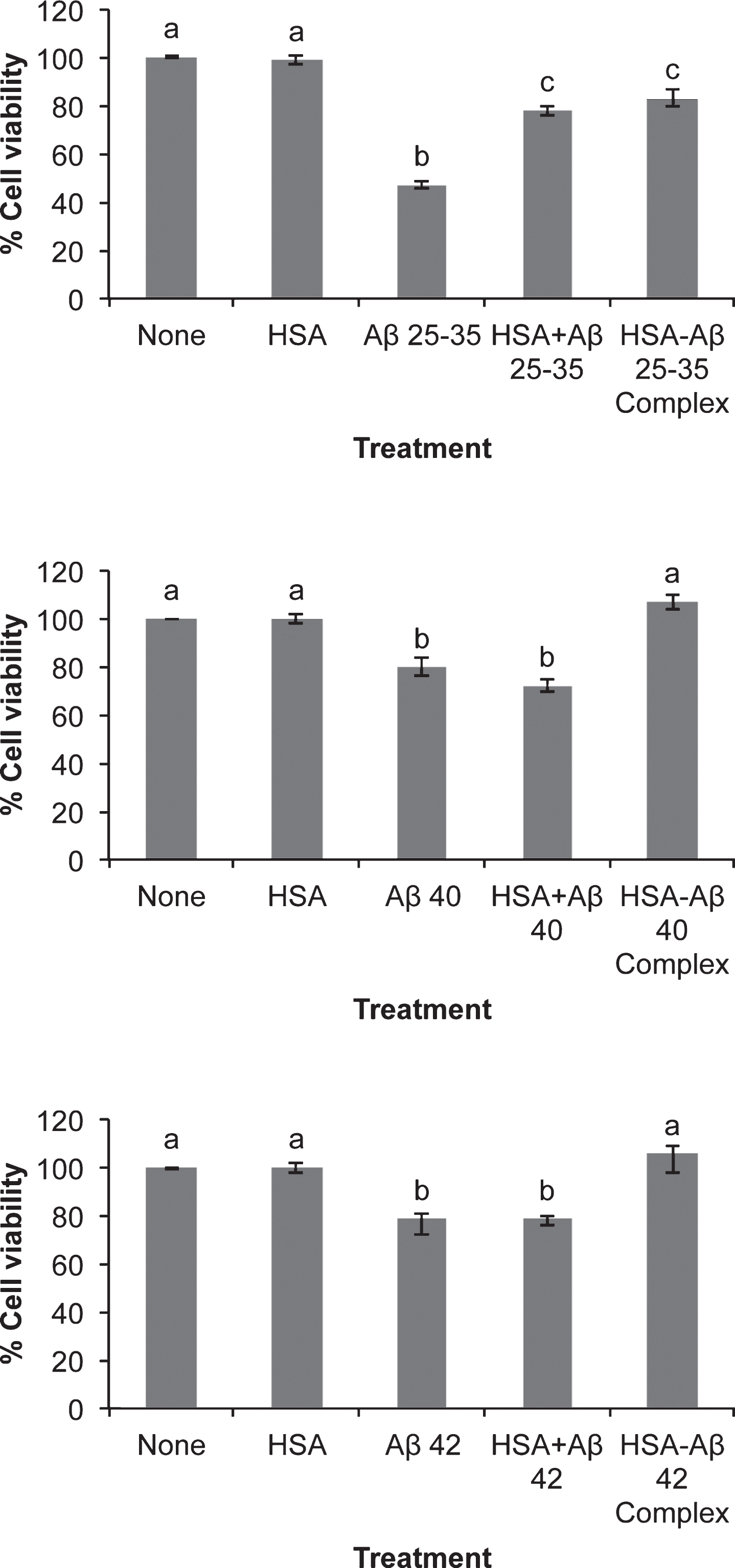
Fig.2
Effect of amyloid-beta peptides on ROS production in the absence or the presence of HSA. Neurons in primary culture (3 DIV) were incubated for 20 h in Hanks medium with three different Aβ peptides: Aβ25-35, Aβ40 or Aβ42 (30 μM) in the absence or the presence of human serum albumin (HSA; 30 μM) or as HSA-Aβ complexes (30 μM). Results were normalized using cell viability data and are expressed as percentages compared to non-treated cells. Results are means±SEM (n≥6). One-way ANOVA and Tukey Test were applied in order to compare different groups. Distinct characters are used to indicate statistically different groups (p < 0.05).
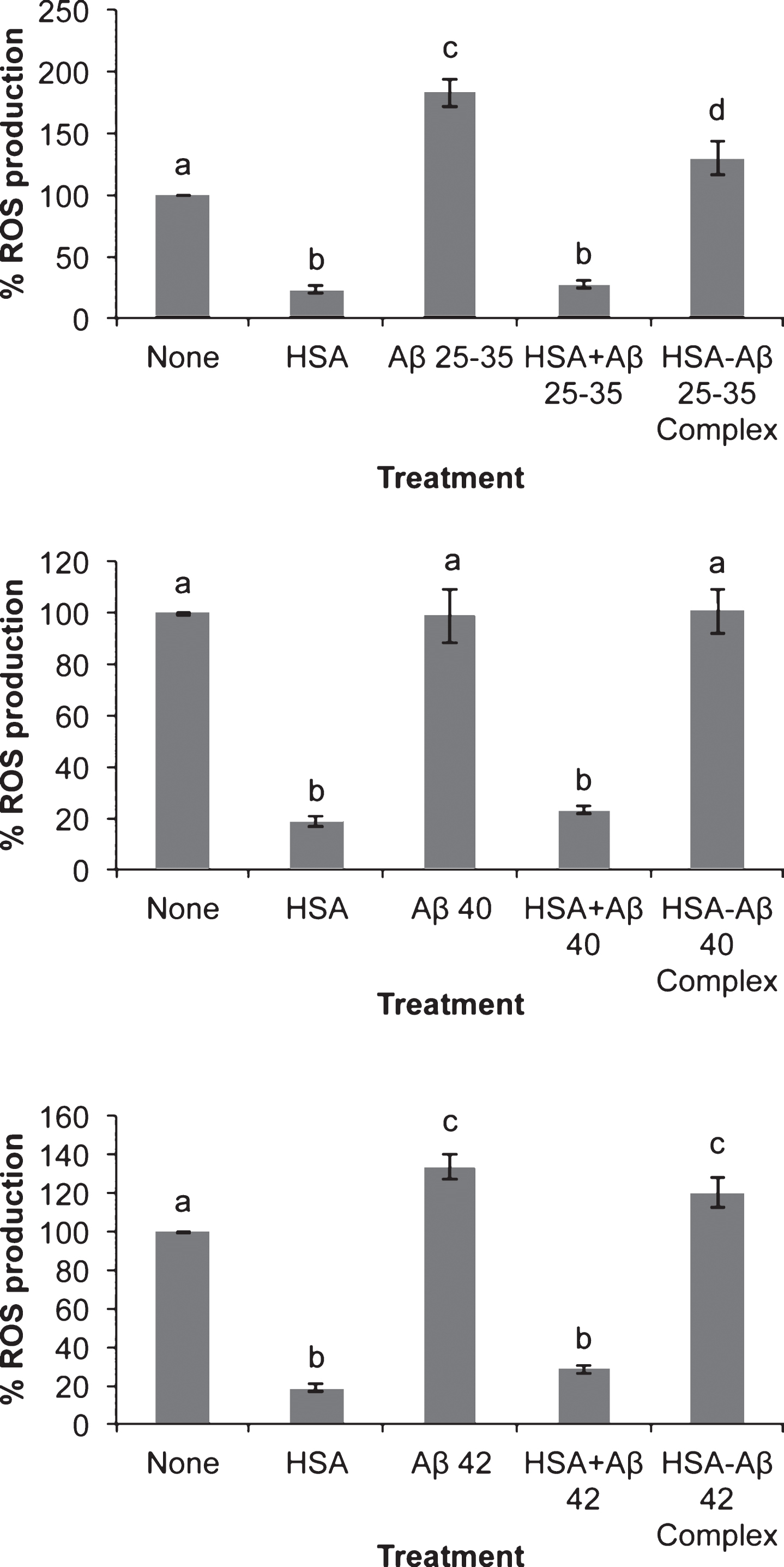
Fig.3
Cellular localization of amyloid-beta peptides in neurons in primary culture. Neurons in primary culture (3 DIV) were incubated for 5 h in Hanks medium with three different Aβ peptides: Aβ25-35, Aβ40, or Aβ42 (30 μM). After incubation, neurons were stained with MitotrakerRed, a mitochondrial marker (in red) and then were fixed. Immunocytochemistry against each Aβ was performed (in green) and nuclei were stained with TOPRO (in blue). Images were taken using confocal microscopy. Scale bar: 30 μm.
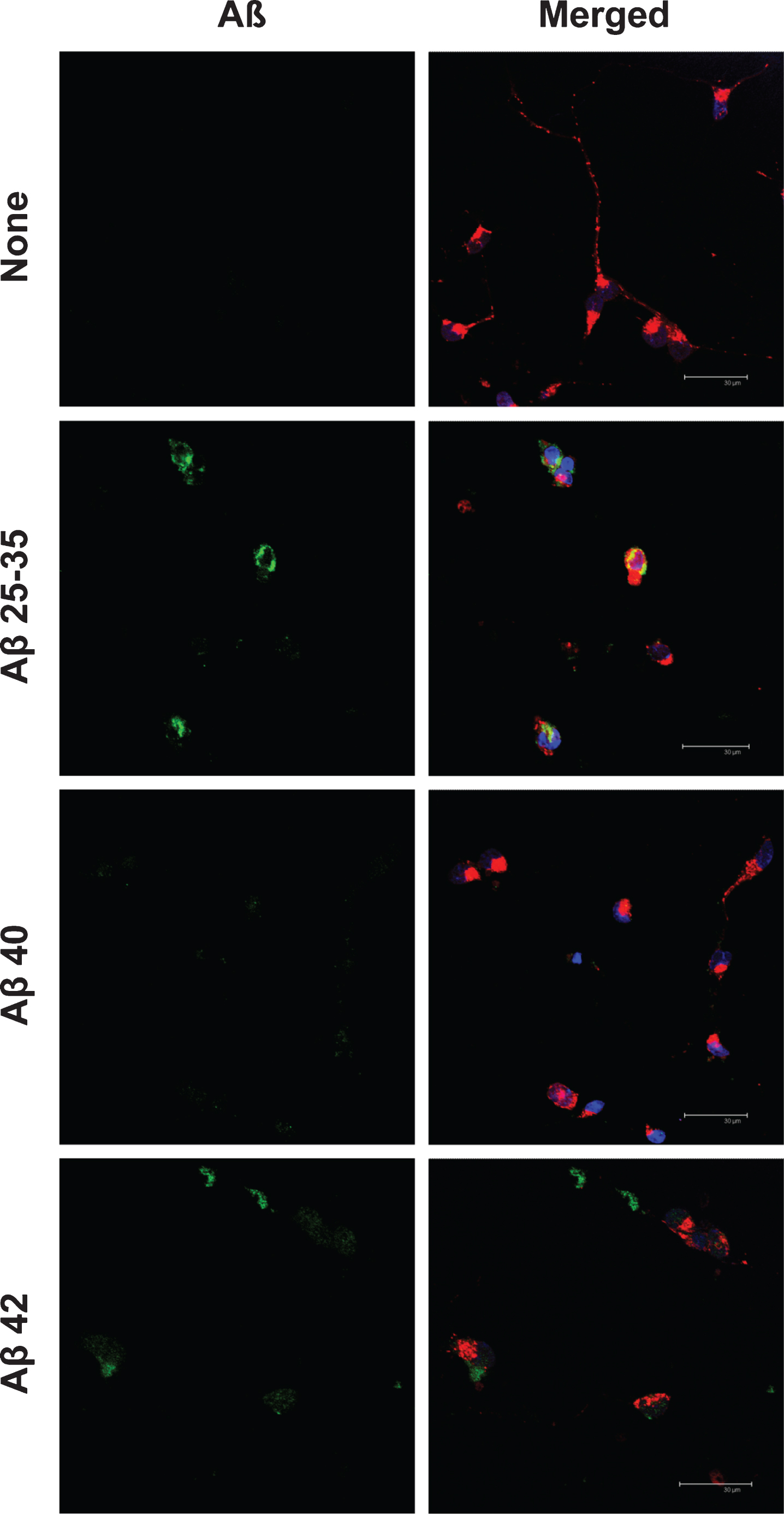
Fig.4
Subcellular localization of amyloid-beta25-35 (Aβ25-35). Neurons in primary culture (3 DIV) were incubated for 24 h in Hanks medium in the presence of Aβ25-35 (30 μM). After incubation, mitochondria were stained with MitotrackerRed (in red), and cells were fixed. Immunocytochemistry against Aβ25-35 was performed (in green) and nuclei were stained with TOPRO (in blue). Images were taken using confocal microscopy. Scale bar: 30 μm.
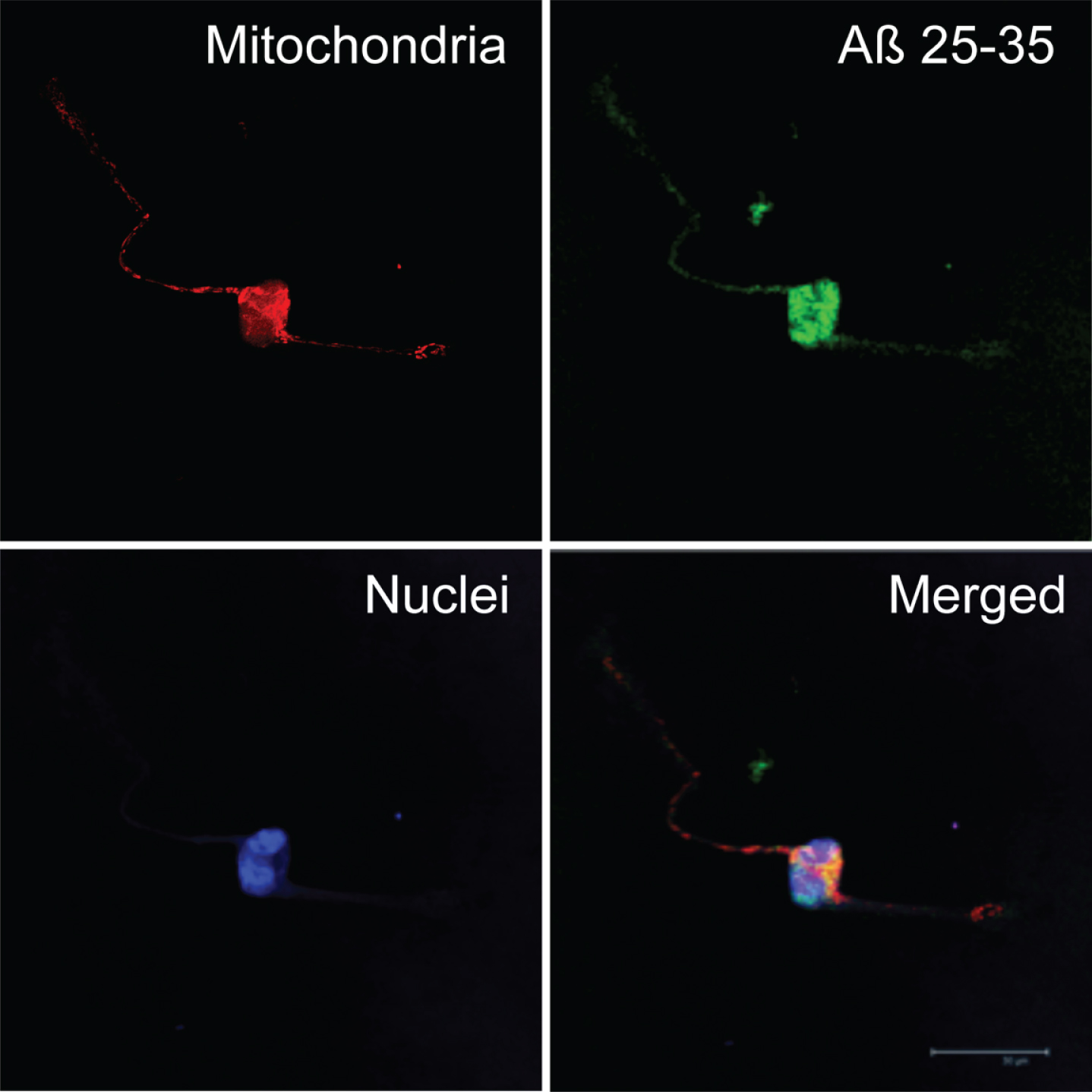
Fig.5
Effect of HSA on amyloid-beta cell entry. Neurons in primary culture (3 DIV) were incubated for 2 h in Hanks medium in the presence of 30 μM of Aβ40, Aβ42, or HSA-Aβ complexes (C40, C42). After incubation, cells were fixed and immunocytochemistry against glucose transporter 3 (in red) and against AβPP (in green) was carried out. Nuclei were stained with TOPRO (in blue). Images were taken using confocal microscopy. Scale bar: 20 μm.
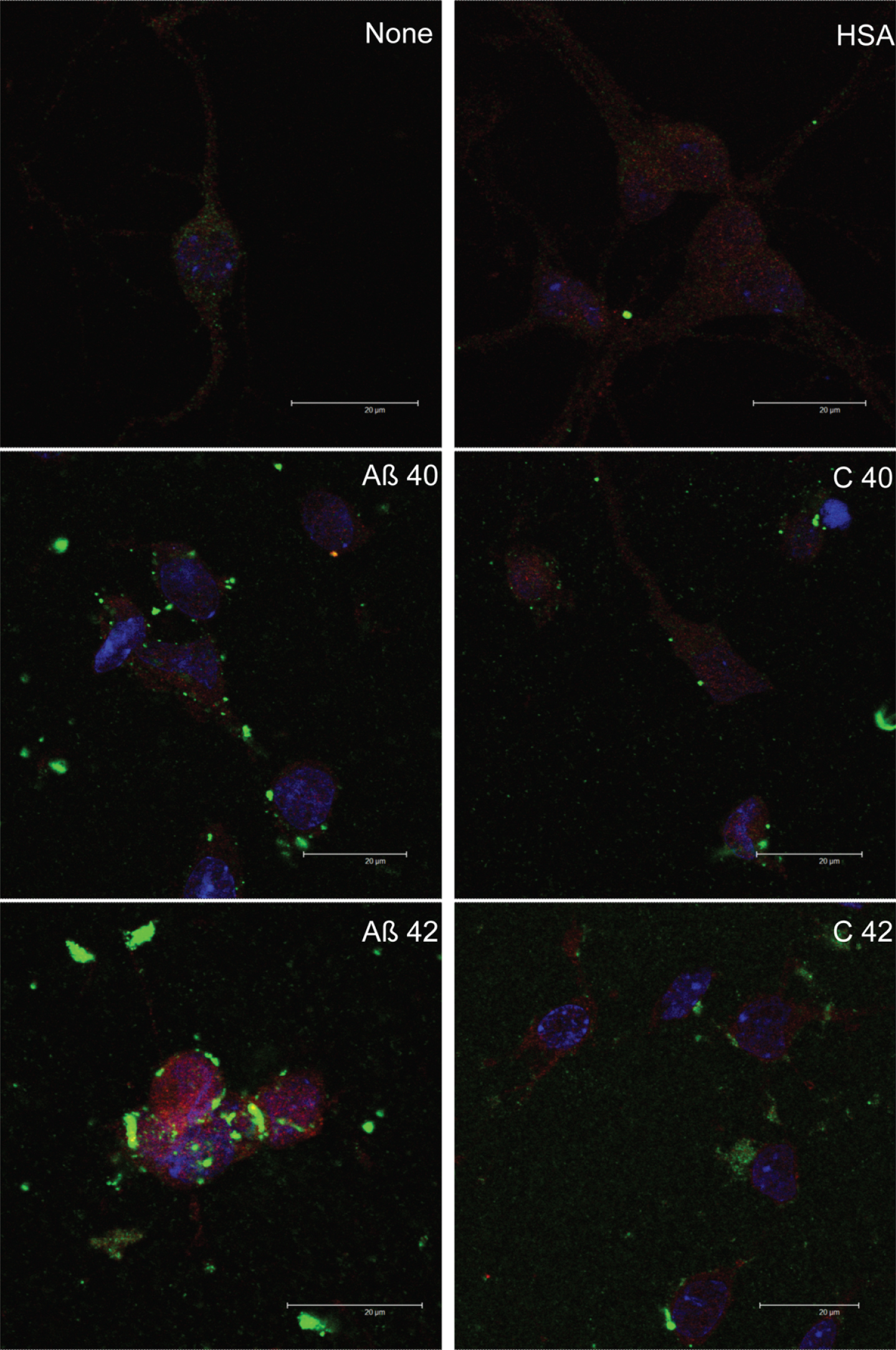
Fig.6
Effect of amyloid-beta peptides on synaptophysin expression in the absence or the presence of HSA. Neurons in primary (3 DIV) culture were incubated for 20 h in DMEM medium with three different Aβ peptides: Aβ25-35, Aβ40, or Aβ42 (30 μM) in the absence or the presence of HSA (30 μM) or as HSA-Aβ complexes (30 μM). Then proteins were obtained and analyzed by western blot. Results are expressed as percentages compared to non-treated cells and are means±SEM (n≥7). One-way ANOVA and Dunnett Test were applied in order to compare different treatments vs. control. Distinct characters are used to indicate statistically different groups as compared to the control (p < 0.05). See Supplementary Figure 2 for raw western blot.
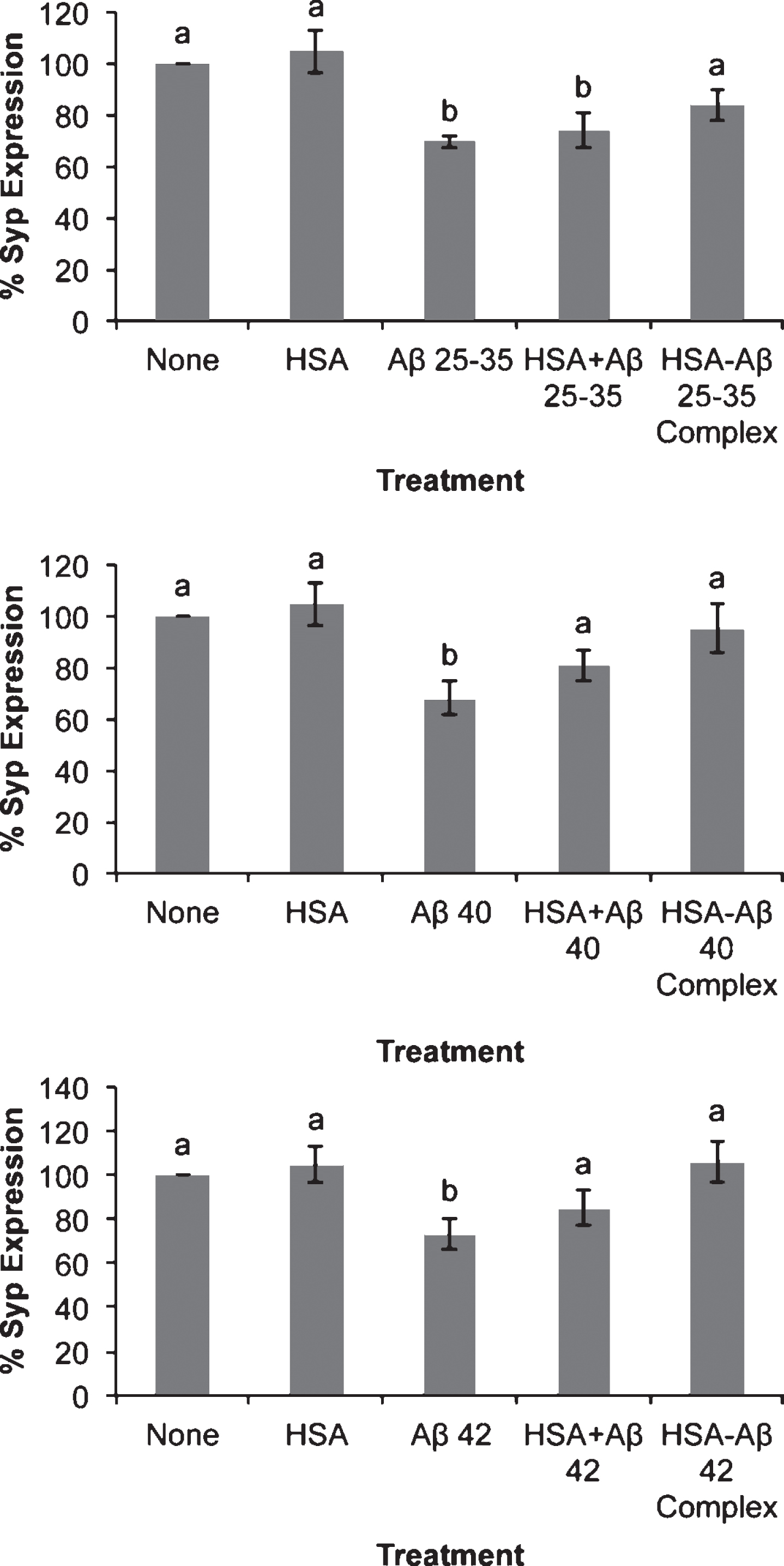
Fig.7
Quantification of PSD-95, synaptotagmin fluorescence, and co-localization. Fluorescence (Integrated density) was measured in confocal photographs as those depicted in Fig. 8 using NIH Image J Software. Co-localization fluorescence is referred to concurrence points of green (PSD-95) and red (synaptotagmin) fluorescence. Results are expressed as percentages as compared to non-treated cells and are means±SEM (n≥24). One-way ANOVA and Dunnett Test were applied in order to compare different treatments vs. control. Distinct characters are used to indicate statistically different groups as compared to the control (p < 0.05).
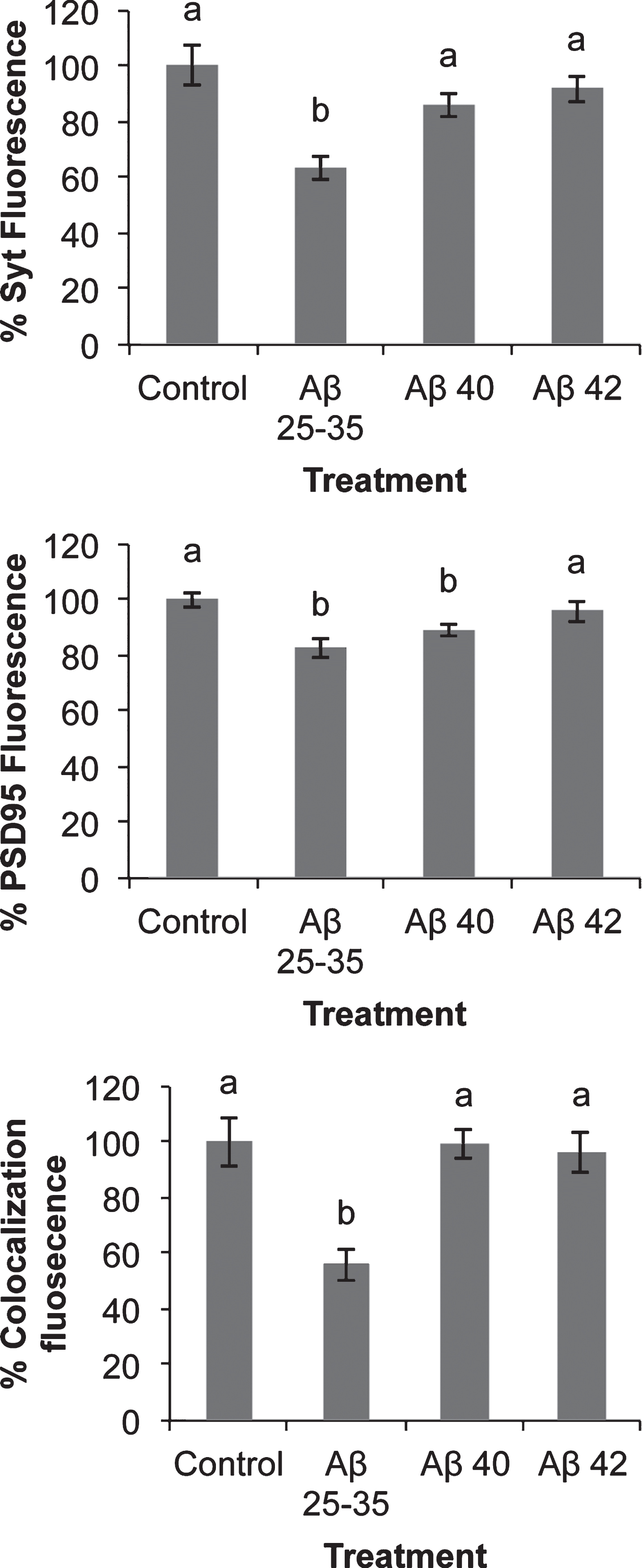
Fig.8
Effect of amyloid-beta peptides on PSD-95 and synaptotagmin localization. Neurons in primary culture (4 DIV) were incubated for 2 h in Hanks medium in the absence or the presence of 30 μM Aβ25-35, Aβ40, Aβ42, HSA, or HSA-Aβ complexes (C 25–35, C 40, C 42). After incubation, cells were fixed and immunocytochemistry against PSD-95 (in green) and against synaptotagmin (in red) was carried out. Images were taken using confocal microscopy. Orthogonal projections along the z-axis of the images are shown at the bottom and right. Scale bar: 20 μM