
A Bacterial Component to Alzheimer’s-Type Dementia Seen via a Systems Biology Approach that Links Iron Dysregulation and Inflammagen Shedding to Disease
Abstract
The progression of Alzheimer’s disease (AD) is accompanied by a great many observable changes, both molecular and physiological. These include oxidative stress, neuroinflammation, and (more proximal to cognitive decline) the death of neuronal and other cells. A systems biology approach seeks to organize these observed variables into pathways that discriminate those that are highly involved (i.e., causative) from those that are more usefully recognized as bystander effects. We review the evidence that iron dysregulation is one of the central causative pathway elements here, as this can cause each of the above effects. In addition, we review the evidence that dormant, non-growing bacteria are a crucial feature of AD, that their growth in vivo is normally limited by a lack of free iron, and that it is this iron dysregulation that is an important factor in their resuscitation. Indeed, bacterial cells can be observed by ultrastructural microscopy in the blood of AD patients. A consequence of this is that the growing cells can shed highly inflammatory components such as lipopolysaccharides (LPS). These too are known to be able to induce (apoptotic and pyroptotic) neuronal cell death. There is also evidence that these systems interact with elements of vitamin D metabolism. This integrative systems approach has strong predictive power, indicating (as has indeed been shown) that both natural and pharmaceutical iron chelators might have useful protective roles in arresting cognitive decline, and that a further assessment of the role of microbes in AD development is more than highly warranted.
INTRODUCTION
Alzheimer’s-type dementia (AD) is a neurodegenerative disorder and the most common form of dementia, already in 2013 affecting 44.4 million people globally; this number is expected to affect 75.6 million by 2030 [1]. The current cost is reckoned at $604 billion per year and this figure is expected to triple by 2050 [2]. Due to the increasing prevalence of the condition, the cost to the public health and elderly care systems to support these individuals is increasing exponentially, and posing major financial challenges [3].
Arguably, the major hurdle in understanding AD is the lack of any integrative and comprehensive knowledge about its etiology and pathogenesis (and there may be many pathways that lead to it), as the onset and risk of AD development is still mostly unexplained (and animal models are of questionable relevance) [4]. Since our genomes changed but little in the last 50 years, but the incidence of AD increased considerably [5], this increase can only to a limited extent be explained by genetic factors [6, 7], notwithstanding the signals detectable in twin and gene association studies [8, 9]. Although dementia is properly diagnosed via cognitive impairment, and true diagnoses of AD can only be done postmortem, specific lesions that characterize AD include extracellular senile plaques and intracellular neurofibrillary tangles with synaptic and neuronal loss [10–13]. In particular, the production of senile plagues, a central event in AD [14], is a result of the cleavage of the amyloid-β protein precursor (AβPP). AβPP has important developmental functions in cell differentiation and possibly in the establishment of synapses [15, 16]; however, it is also expressed by neurons in response to cell injury [17]. Neurofibrillary tangles are composed of the tau protein [18]. In healthy neurons, τau is an integral component of microtubules, which are the internal support structures that help transport nutrients, vesicles, mitochondria, and chromosomes from the cell body to the ends of the axon and backwards [19]. In AD, however, τau becomes hyperphosphorylated [18, 20]. This phosphorylation allows tau proteins to bind together and form tangled threads [21], a process that can be reversed by iron chelation [22].
Recent evidence suggests that neuroinflammation may play a major role in the pathological processes of AD progression [23–31]. Indeed, inflammation and microglial activation are known as common components of the pathogenesis of a number of neurodegenerative diseases, including AD, Parkinson’s disease, Huntington’s disease, multiple sclerosis, and amyotrophic lateral sclerosis [32]. Several neuroinflammatory mediators, including complement activators, chemokines, cytokines, and oxygen radical species, are expressed and released by microglia, astrocytes, and neurons in the AD brain. While minor signs of neuroinflammation can be found in the normal aging brain, the AD brain faces a much stronger activation of inflammatory systems, indicating that an increasing amount of (or qualitatively different) immunostimulants are present. In recent papers, we have also reviewed the comprehensive evidence that in AD the neuroinflammation is probably a systemic inflammatory condition [33, 34]. In one sense, however, the above are all manifestations or accompaniments of AD, and what we seek are the most important causative pathways. It turns out that central to all of these diseases is iron dysregulation [35, 36].
Figure 1 provides an overview of the article in the form of a ‘mind map’, while Table 1 lists some of the symptoms (some causative) accompanying the pathology of AD. This wide strategy necessarily involves a systems biology approach [37–41] as we recognize (e.g., [36, 42–47]) that this is the only reasonable strategy for approaching complex biochemical networks, each of whose components may contribute partially to the phenotype of interest.
A typical systems biology strategy (e.g., [42, 43]) has the following four elements: first we identify the actors that are most involved, and how they interact. ‘Actors’ for these purposes may be enzymes or other biochemical elements, or higher-order physiological processes (such as those in Table 1). We then adduce the order or pathway of such interactions (as in Fig. 2, below). Latterly (though we are not yet ready for this in the present problem), we seek to make quantitative these interactions, and predict their relative fluxes, contributions, and so on. We next turn to some of the main actors, starting with iron dysregulation.
IRON AND AD
Strongly and causatively related to this neuroinflammation in AD is the involvement of unliganded iron and its accompanying oxidative damage in AD etiology [48–61]. Specifically, AD is characterized by elevated brain iron levels [62–64] and the accumulation of copper and zinc in cerebral amyloid-β (Aβ) deposits (e.g., in senile plaques) [65–73].
There is evidence in the literature that the iron status of AD patients, particularly the serum ferritin (SF) levels, as measured systemically, might have clinical relevance, as this is an indication of iron dysregulation [33, 58, 72, 74, 75]. Increased iron levels are also closely linked to hematological pathology in AD, and this is indicative of systemic inflammation, which also plays an important role in the pathogenesis of the condition [54, 76, 77]. Recently, we showed that, in a randomly chosen AD population, 60% of the patients had increased SF levels, causing adverse effect on red blood cell (RBC) structure [33] as well as causing significantly thinner fibrin fiber diameters, resulting in abnormal clotting [78].
Pathology, in the presence of increased SF levels to both RBCs and fibrin formation, is indicative of a systemic inflammatory involvement of iron in AD. In the recent Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort study, increased SF levels were also measured in cerebrospinal fluid and found to be negatively associated with cognitive performance [79]. Systemically elevated SF levels therefore may have great clinical relevance in AD, as they may be useful as markers of cognitive performance.
Currently, the main therapeutic approaches in AD either attempt to prevent Aβ production (e.g., by the use of secretase inhibitors) or to clear Aβ. However, there is convincing evidence that Aβ does not spontaneously aggregate on its own, but that there is an age-dependent reaction with excess brain metal (copper, iron, and zinc), which induces the protein to precipitate into metal-enriched plaques [65]. In AD there is also a dramatic increase in brain iron content and in fact there are higher iron concentrations inside the Aβ plaques [80], suggesting that disturbances in brain iron homeostasis may contribute to AD pathogenesis [81, 82].
It is well known that excessive poorly liganded iron may cause oxidative damage [35, 83, 84], and there is ample evidence that suggests that oxidative stress and therefore aberrant redox activity is one of the earliest pathological changes in AD, and that there is a link between systemic and brain oxidative stress [50, 85].
Oxidative stress plays a significant role in the pathogenesis of AD [86–89]. Oxidative stress in AD results in increased levels of lipid peroxidation, DNA, and protein oxidation products (HNE, 8-HO-guanidine, and protein carbonyls, respectively) inside AD brains [90]. Oxidative stress participates in the development of AD by promoting Aβ deposition [91], tau protein hyperphosphorylation, and the subsequent loss of synapses and neurons. In AD, much as with the prion protein in prion diseases [35, 36, 92], Aβ can become a pro-oxidant and when complexed to iron, this can result in hydrogen peroxide formation; this process can underlie the increased oxidative stress burden [93]. The relationship between oxidative stress and AD suggests that it is an essential part of the pathological process; poorly liganded iron can participate in the Fenton reaction (Fe2 + + H2O2 ⟶ Fe3 + + . OH• + OH-), and the highly reactive hydroxyl radical OH• may be the main culprit [35]. In addition, the Haber-Weiss reaction Fe3 + + O2•- ⟶ Fe2 + + O2 reverts the Fe3 + to Fe2 + such that the ‘iron’ then becomes catalytic rather than stoichiometric [35, 94]; this is why the unliganded iron is so particularly toxic.
In a series of articles, including a number of reviews, we have shown that poorly liganded iron is key to a great variety of diseases [33, 95–97]; it also affects erythrocyte morphology and coagulation properties (touched on briefly later in this paper) [96, 98].
Ultimately, oxidative stress may be due to the combined action of mitochondrial dysfunction, increased metal levels, inflammation, and the presence of Aβ peptides [99]. However, there is a link between all the above-mentioned and the pathological presence of iron. Increased oxidative stress results in inflammation, which can be both neuroinflammation or systemic inflammation [100], and the pathologic levels of iron have been associated with both inflammation and oxidative stress in AD [23, 91]. We tend to like ideas with predictive power (such as unitary explanations for diseases with comorbidities, for which see also [101]). Thus, if iron is so important to the pathogenesis of AD, one might then suppose that its chelation (that stops the Fenton and Haber-Weiss reactions) would be expected to improve it [102, 103]. The next section looks at this.
Iron chelating improves cognition
Starting with a Lancet paper that is now a quarter of a century old [104], it has been shown that the removal of pathologic levels of free iron improves cognitive function in AD. Metal chelators such as clioquinol and desferrioxamine, and natural antioxidants such as curcumin and ginkgo extract, have had some success in altering the progression of AD symptoms [90, 105–107]. More recent and important papers, to the same effect, come from the group of Perry and colleagues [51, 54] and that of Youdim [108], while similar beneficial effects of iron chelation can be observed with Parkinson’s disease and models thereof [109–112]. We do find it slightly surprising that these indications have not been more widely picked up.
A fine control of iron regulation might play an important role in systemic iron overload [113] including AD [114], as there is a known association between diet and risk of dementia [115]. Except for pharmaceutical intervention, it is well known that a healthy diet rich in polyunsaturated fatty acids and polyphenols may have a positive effect on general health brain function [116]. In particular, the Mediterranean-type diet has a positive effect on the healthiness of AD patients [117–120], due to the presence of naturally occurring iron chelating agents found in fruit and vegetables as these agents are known scavengers as a result of their ability to chelate iron [118, 121–124]. Another route might also be calibrated phlebotomy in AD, to reduce iron stores [125].
A DORMANT MICROBIAL COMPONENT TO AD
While metals can certainly contribute significantly to the explanation of the development of AD via these Fenton-type pathways, we have recently suggested that they may do so by another and parallel means, explicitly involving the awakening of a dormant bacterial component [34, 126]. This follows from the recognition that the growth of microbes in vivo is normally limited by the availability of free iron [127–132]. Others too have noted the presence of an authentic blood microbiome even in ‘normal’ controls, based on macromolecular sequencing and other molecular approaches (e.g., [126, 133–138]), although sequencing methods cannot of themselves reflect replicative potential, of course.
In this sense, a ‘classical’, related, and well-known example is that of Helicobacter pylori and gastric ulcers. These latter had long been assumed to be due to the over-activity of the gastric H +-ATPase (which can certainly contribute). However, the pioneering (and initially ‘controversial’) work of Barry Marshall and Robin Warren showed unequivocally that they were inevitably accompanied, and the disease was essentially caused, by a hard-to-culture and little-known microaerophilic organism, subsequently codified as H. pylori. Our major thesis here (and elsewhere) is that it will turn out that a very large number of chronic, inflammatory diseases, that share many observable symptoms, will also turn out to be due to hard-to-culture organisms, many or most of which will turn out to be well known to science. The issue is that they typically lie dormant, and thus (by definition) resist culture by means that normally admit their culture.
The point of ‘dormancy’ is particularly important, as most clinical microbiologists typically consider or define microbial propagules (cells potentially capable of replication) as being ‘alive’ (i.e., so capable) or not under any conditions tested (‘dead’). However, a considerable literature (reviewed by ourselves, e.g., [126, 143–146]) and others (e.g., [147–152]) indicates that most microbes in nature are non-growing and can appear operationally ‘dead’, yet can recover culturability, by a process referred to (virtually by definition) as ‘resuscitation’. They are thus not operationally ‘dead’ and are typically and more properly referred to as ‘dormant’ (or, commonly in clinical microbiology, ‘persistent’ [150, 152–156]). One needs then to recognize that dormancy is an operational property that depends both on the cell (singular [157]) being assessed and on the means used to detect it [158]. This cannot be emphasized too strongly: the designation of a microbe as dormant implies that it is not just a property of the microbe alone but of the means by which we assess it, a phenomenon reminiscent of the “Schrödinger cat paradox” in the philosophy of quantum mechanics. One important consequence (see e.g., [126, 159–164]) of this ability of microbes to enter non-replicating physiological states is that they do not fulfill the Henle-Koch postulates regarding the microbial causality of diseases, at least in their ordinary form [165].
Particularly, the neurotoxic lipopolysaccharides (LPS) from their cell walls may be of importance (see below), since LPS molecules are highly inflammatory agents, that can even induce cell death [126]. It is of course the cell death that is the proximate cause of the loss of cognitive function. We summarize all of these pathways in Fig. 3. The especial attractions of this scheme are that (i) it provides for the necessary systems-level understanding, (ii) the elements hang together and are ‘coherent’ within the meaning of that term as used in the Philosophy of Science [126, 166], and (iii) it is rich in both predictive and explanatory power.
While recognizing the importance of various kinds of infectious agents in the pathogenesis of AD (see [34, 162, 167–198]), and that also depend for their growth on the availability of free iron, we next turn to the question of the role of prokaryotes and their inflammatory components in the pathogenesis of AD.
THE ROLE OF BACTERIA AND LPS IN AD PATHOGENESIS
Recently, immunoblotting demonstrated bands corresponding to LPS in four AD brain specimens, which were positive when screened by immunofluorescence [199]. Bacterial endotoxins may be involved in the inflammatory and pathological processes associated with AD [200]. Indeed a number of studies indicate that the LPS-induced neuroinflammation can drive Aβ formation (e.g., [201–206]).
Interestingly, it has been observed that chronic infusion of the bacterial LPS into the fourth ventricle of rats reproduces many of the inflammatory and pathological features seen in the brain of AD patients [200, 201].
Previously we have reviewed the extensive published accounts suggesting a possible link between LPS presence and the pathological process of AD [34, 126, 207–211]. It is also well known that LPS presence is at least one of the causes of inflammation [212–214], and one of the hallmarks of inflammation is a hypercoagulable state [215–221]. Previously, we have seen changes in erythrocytes (RBCs), as well as hypercoagulation in the presence of LPS, where we added LPS to whole blood of healthy individuals or to platelet poor plasma [34]. We also reported on the presence of bacteria, which will indeed point to the presence of LPS, in whole blood of AD and Parkinson’s disease patients, and also in fact inside RBCs [34]. We also discussed in detail the reasons why we might find bacteria in typically “sterile” blood, and suggested that these bacteria may be dormant (as operationally defined).
VITAMIN D, INFECTION, AND AD
While, in a sense, ‘everything is connected to everything else’, the role of the systems biologist is to highlight those metabolic networks and other processes whose variation (whether as a dependent or an independent variable – see [222]) are most pertinent to the outcomes of interest. Leaving aside the well-established roles of vitamin D in calcium and bone metabolism, it does seem to have a considerable impact on the immune system. To this end, there are some interesting clues (e.g., [223]) that link inflammation, infection, and vitamin D metabolism (and indeed elements of iron and vitamin D metabolism [224]), as well as AD [225–231]. Although the degree, and any mechanisms, of causality remain to be seen, and the inter-relations are complex and nonlinear [232, 233], there is an emerging consensus among a significant group of workers that chronic infection is intimately linked to detailed vitamin D status, and that this may provide a way in to useful therapies for a variety of chronic, inflammatory diseases (e.g., [101, 234–237]). The first issue concerns what in fact we mean by ‘vitamin D’. Specifically, vitamin D may typically refer to two distinct forms: ergocalciferol (vitamin D2) and cholecalciferol (vitamin D3), with some question as to whether D2 is indeed useful as a vitamin supplement [238, 239]. The structures and metabolic products of vitamin D2/3 (of which only the hydroxy derivatives are in fact active, and the 1α,25-dihydroxyderivative especially) are given in Fig. 4.
In particular, Mangin and colleagues [235] have suggested that that low 25(OH)D is a consequence of chronic inflammation rather than the cause, and that tissue bacteria were responsible for an inflammatory disease process which results in high 1,25(OH)2D and low 25(OH)D (see also [237]). 1,25(OH)2D activates the vitamin D receptor (VDR) [240–244], a transcription factor that serves to induce the expression of over 900 genes, including for antimicrobial peptides [101, 223, 245–251] such as cathelicidin and beta defensins which attack (presumably non-dormant) pathogens [252]. In general, the innate immune system is enhanced and the adaptive immune system is inhibited by 1,25(OH)2D [235]. The general scheme, essentially as redrawn from [235], is given in Fig. 5. Other papers have also highlighted a relationship between low 25(OH)D and AD [226, 229, 230, 253, 254] and tend to imply that vitamin D supplementation should therefore be a solution. Obviously from a systems biology point of view, this does not follow directly, and there is evidence that the opposite can in fact be true [235, 236, 255]; clearly we need to know precisely the different roles of 25(OH)D and 1,25(OH)2D, and any effects on the CYP enzymes that produce them. More particularly, however, the complex, variable quality [256], and sometimes apparently contradictory, literature [257] is arguably better explained on the basis that there are separate populations who simply respond differently to vitamin D3 supplementation [258–260]. Biomarkers (such as taurinuria; [261]) for genuine vitamin D deficiency may help disentangle this. Indeed, the contradictory nature of any kinds of phenomena in which the ‘same’ additions are made to the ‘same’ system with very different results are typically explainable on the basis of uncontrolled variation. Thus the antioxidant ascorbate is actually pro-oxidant if unliganded iron is present [35]. Another explanation of such contradictions here involves the simultaneous presence of agonist and antagonist conformers of the VDR [262–264]. Finally, and in a different vein, the apoptotic versus proliferative effects of NF-κB are determined by the frequency rather than the amplitude of the NF-κB signaling molecule [43, 265, 266]. Vitamin D has significant effects on NF-κB [267–269]. Since there are also significant oscillations in ERK [270], VDR levels are partly dependent on ERK [271], and vitamin D3 also regulates circadian genes [272], these kinds of explanations based on the timing and frequency of oscillations (rather than simple metabolite concentrations) seem well worth exploring.
At all events, the nature(s) of the intracellular pathogens (and the cells in which they reside) is probably very wide, and at least one strategy for their persistence (in terms of their ability to evade the immune systems) is the adoption of cell-wall-deficient morphologies [148] or L-forms [273]. These, as well as more conventional structures, can of course be detected microscopically.
DIRECT DETECTION OF MORPHOLOGICAL CHANGES IN THE BLOOD OF AD PATIENTS
Pathologic RBCs and hypercoagulable fibrin(ogen) in AD patients
In previous work, we showed that the erythrocytes of AD patients were of highly anomalous shape, especially when serum ferritin levels were simultaneously raised [33] and that there was likely a hypercoagulable state (ascribed to the elevated LPS [34]). Here we now also show that AD RBCs are indeed abnormal, by using RBC and antibody-based fluorescent markers for spectrin (Ab11751) (red fluorescence) and Band-3 (Ab11012) (green fluorescence). Band3 is found in three distinct protein complexes associated with the erythrocyte membrane: an ankyrin-dependent tetrameric band3 complex, a dimeric band3 complex bound to the protein 4.1-glycophorin C junctional complex, and freely diffusing dimeric band3 complexes [274, 275]. Band 3 can also bind to spectrins, the internal scaffold for erythrocyte shape, via ankyrin, suggesting that band 3 contributes to the membrane-cytoskeleton interactions that help to define erythrocyte shape and stability [276, 277]. Structural alterations to the phospholipids, as well as band 3 and spectrin, cause RBC physical shape changes, which can be detrimental to their normal functioning [97, 278]. Under normal conditions, the neutral phospholipids, phosphatidylcholine, and sphingomyelin are mostly found on the outside, and the charged phospatidylserine (PS), phosphatidylinosirol, and phosphatidylethalolamine, are found mostly on the inner membrane leaflet. However, during inflammation, the erythrocyte membrane leaflet phospholipids becomes more symmetric as PS is externalized, resulting in RBC membrane vesicle formation and ultimately microparticle shedding, with subsequent pathological shape changes of RBCs [279]. PS is normally found only on the intracellular leaflet of the plasma membrane in healthy cells, but during early eryptosis (RBC programmed cell death) [280–284], membrane asymmetry is lost and PS translocates to the external leaflet [285]. For a detailed review on the role of the RBC membrane and changes therein due to inflammation, see [286].
Figure 6A shows a typical example of confocal microscopy of a healthy RBC and Fig. 6B shows a typical scanning electron microscopy (SEM) image of a representative RBC from an age-controlled healthy individual, while Fig. 6C and D show confocal and SEM images of a representative sample from an AD individual. Figure 6A shows intense green fluorescence on the rim of the RBCs and less intense toward the inside of the RBC. There is little to no red fluorescence specifically on the rim of the RBCs indicating the presence of the spectrin. Where there is some red staining, it is more toward the inside of the RBCs and much less intense than the green band3. In the RBCs of the AD individuals (Fig. 6C), the red fluorescence is much more visible, and the red fluorescence is found not only on the inside of the RBCs but also on the rim and outside of these cells unlike the control group. This suggests a structural membrane disorder, typically associated with eryptosis, which is often enhanced by cytoplamic calcium activity and also characterized by cell membrane scrambling and cell shrinkage [287, 288]. Particularly the disarrangement of spectrin and band 3 positional changes are two important markers to determine structural damage to the membrane that will result in changes to elasticity and pliability of RBCs [286]. SEM images comparing healthy and AD RBC ultrastructure, clearly show that the RBCs from AD individuals have an eryptotic structure. Eryptosis is visible in most of the RBCs from AD patients, and also in those with Parkinson’s disease [95]. Additionally to the eryptotic structure of the RBCs, bacteria were also visible with SEM in the same AD sample (Fig. 6E, F).
As well as changes in AD RBCs, we previously found that pathologic fibrin fiber formation (associated with hypercoaguation) is also present in AD, and may therefore be used as a further and useful inflammatory indicator [34]. As seen in pathological changes in RBCs, oxidative damage, increased iron levels, and inflammation are also all reasons for the development of hypercoagulability [95–97, 289–293]. Hypercoagulability is closely associated with increased fibrin(ogen) in AD patients, while hypercoagulation has been observed in blood vessels positive for amyloid in mouse and human AD samples [294]. A changed fibrinogen structure has been implicated in the development of neuroinflammation [295, 296], and memory deficits and increased fibrinogen levels in AD are noted to be a strong indicator of cerebrovascular risk, as fibrinogen specifically binds to Aβ, thereby altering fibrin clot structure and delaying clot degradation [297]. In a previous paper, we looked at the viscoelastic and ultrastructural properties of AD plasma and whole blood by using scanning electron microscopy, thromboelastography (TEG®) and the Global Thrombosis Test (GTT®) [34]. TEG® analysis showed a hypercoagulable state in AD, while TEG® results, where LPS was added to uncitrated blood, showed the same trends as were found with the AD patients. The GTT® results (where only platelet activity is measured) were not affected by the added LPS, suggesting that LPS does not directly impact platelet function [34]. See Fig. 7 for an ultrastructural comparison of platelet poor plasma smears (treated with thrombin) from a healthy (age-controlled) individual and from an AD individual.
Although pathophysiological changes in RBCs and fibrin fiber structure are not unique to AD, they are hallmarks of systemic inflammation [96], and as noted here LPS may play a role in the biochemical pathways that may destabilize RBC and fibrin structure. As RBCs are extremely vulnerable in the presence of pro-inflammatory molecules, hydroxyl radicals, oxidative stress, and LPS, they may possibly be used as a ‘healthiness’ indicator of AD patients. Currently we have few actual markers of AD status, and we note that the latest NIH guidelines suggest that clinical medicine should focus on precision medicine [298] and that individualized medicine should in the future, form an essential part in the diagnosis and treatment of patients. We therefore suggest that RBC and fibrin morphology could be used as “health indicators”. Here we do not of course suggest that they should be used as diagnostic tools for AD per se, but rather as a healthiness indicator of the overall systemic inflammatory status of patients after diagnoses.
CONCLUSION
Modern molecular biology had become a little obsessed with a presumed need for hypotheses, and it has taken the post-genomic era to remind scientists of the virtues of scientific induction and data-driven biology [299, 300], often intertwined with a systems biology approach. A typically nice example is a hypothesis-free discovery biology paper [301] in which the authors sought to identify those pathways that were most intimately involved in the development of prion disease. Genes involved in iron metabolism were among the most highly involved [301].
In a similar vein, we have brought together the evidence underpinning a coherent and self-consistent view of the linked contributions to AD progression of iron dysregulation, the resuscitation of dormant bacteria, and the shedding of the highly inflammatory LPS that can induce both cytokines and apoptosis (see Figs. 2 and 3). As with any systems approach, it implies the need for pharmacological interventions at multiple points (e.g., [302–305]). The role of the systems pharmacologist, based on knowledge of the most important pathways proposed herein, is to develop them.
ACKNOWLEDGMENTS
We thank the Biotechnology and Biological Sciences Research Council (grant BB/L025752/1) as well as the National Research Foundation (NRF) and Medical Research Council; (MRC) of South Africa for supporting this collaboration. This is also a contribution from the Manchester Centre for Synthetic Biology of Fine and Speciality Chemicals (SYNBIOCHEM) (BBSRC grant BB/M017702/1).
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-0318r1).
REFERENCES
[1] | Vradenburg G ((2015) ) A pivotal moment in Alzheimer’s disease and dementia: How global unity of purpose and action can beat the disease by 2025. Expert Rev Neurother 15: , 73–82. |
[2] | Langa KM ((2015) ) Is the risk of Alzheimer’s disease and dementia declining? Alzheimers Res Ther 7: , 34. |
[3] | Takizawa C , Thompson PL , van Walsem A , Faure C , Maier WC ((2015) ) Epidemiological and economic burden of Alzheimer’s disease: A systematic literature review of data across Europe and the United States of America. J Alzheimers Dis 43: , 1271–1284. |
[4] | Clement C , Hill JM , Dua P , Culicchia F , Lukiw WJ ((2016) ) Analysis of RNA from Alzheimer’s disease post-mortem brain tissues. Mol Neurobiol 53: , 1322–1328. |
[5] | Rodríguez-Gómez O , Palacio-Lacambra ME , Palasí A , Ruiz-Laza A , Boada-Rovira M ((2014) ) Prevention of Alzheimer’s disease: A global challenge for next generation neuroscientists. J Alzheimers Dis 42: (Suppl 4), S515–S523. |
[6] | Morris JK , Honea RA , Vidoni ED , Swerdlow RH , Burns JM ((2014) ) Is Alzheimer’s disease a systemic disease? Biochim Biophys Acta 1842: , 1340–1349. |
[7] | Skaper SD ((2012) ) Alzheimer’s disease and amyloid: Culprit or coincidence? Int Rev Neurobiol 102: , 277–316. |
[8] | Gatz M , Reynolds CA , Fratiglioni L , Johansson B , Mortimer JA , Berg S , Fiske A , Pedersen NL ((2006) ) Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 63: , 168–174. |
[9] | Seshadri S , Fitzpatrick AL , Ikram MA , DeStefano AL , Gudnason V , Boada M , Bis JC , Smith AV , Carassquillo MM , Lambert JC , Harold D , Schrijvers EM , Ramirez-Lorca R , Debette S , Longstreth WT Jr , Janssens AC , Pankratz VS , Dartigues JF , Hollingworth P , Aspelund T , Hernandez I , Beiser A , Kuller LH , Koudstaal PJ , Dickson DW , Tzourio C , Abraham R , Antunez C , Du Y , Rotter JI , Aulchenko YS , Harris TB , Petersen RC , Berr C , Owen MJ , Lopez-Arrieta J , Varadarajan BN , Becker JT , Rivadeneira F , Nalls MA , Graff-Radford NR , Campion D , Auerbach S , Rice K , Hofman A , Jonsson PV , Schmidt H , Lathrop M , Mosley TH , Au R , Psaty BM , Uitterlinden AG , Farrer LA , Lumley T , Ruiz A , Williams J , Amouyel P , Younkin SG , Wolf PA , Launer LJ , Lopez OL , van Duijn CM , Breteler MM , Consortium C , Consortium G , Consortium E ((2010) ) Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 303: , 1832–1840. |
[10] | Reitz C ((2012) ) Alzheimer’s disease and the amyloid cascade hypothesis: A critical review. Int J Alzheimers Dis 2012: , 369808. |
[11] | Dubois B , Feldman HH , Jacova C , Cummings JL , DeKosky ST , Barberger-Gateau P , Delacourte A , Frisoni G , Fox NC , Galasko D ((2010) ) Revising the definition of Alzheimer’s disease: A new lexicon. Lancet Neurol 9: , 1118–1127. |
[12] | Kim J , Basak JM , Holtzman DM ((2009) ) The Role of Apolipoprotein E in Alzheimer’s Disease. Neuron 63: , 287–303. |
[13] | McKhann G , Drachman D , Folstein M , Katzman R , Price D , Stadlan EM ((1984) ) Clinical diagnosis of Alzheimer’s disease Report of the NINCDS-ADRDA Work Group* under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34: , 939–939. |
[14] | Joachim CL , Selkoe DJ ((1992) ) The seminal role of [beta]-amyloid in the pathogenesis of Alzheimer disease. Alzheimer Dis Assoc Disord 6: , 7–34. |
[15] | Löffler J , Huber G ((1992) ) β-amyloid precursor protein isoforms in various rat brain regions and during brain development. J Neurochem 59: , 1316–1324. |
[16] | Selkoe DJ , Podlisny MB , Joachim CL , Vickers EA , Lee G , Fritz LC , Oltersdorf T ((1988) ) Beta-amyloid precursor protein of Alzheimer disease occurs as 110-to 135-kilodalton membrane-associated proteins in neural and nonneural tissues. Proc Natl Acad Sci U S A 85: , 7341–7345. |
[17] | Baiden-Amissah K , Joashi U , Blumberg R , Mehmet H , Edwards AD , Cox PM ((1998) ) Expression of amyloid precursor protein (beta-APP) in the neonatal brain following hypoxic ischaemic injury. Neuropathol Appl Neurobiol 24: , 346–352. |
[18] | Tang Z , Ioja E , Bereczki E , Hultenby K , Li C , Guan Z , Winblad B , Pei JJ ((2015) ) mTor mediates tau localization and secretion: Implication for Alzheimer’s disease. Biochim Biophys Acta 1853: , 1646–1657. |
[19] | Goedert M , Spillantini MG ((2006) ) A century of Alzheimer’s disease. Science 314: , 777–781. |
[20] | Zhu S , Shala A , Bezginov A , Sljoka A , Audette G , Wilson DJ ((2015) ) Hyperphosphorylation of intrinsically disordered tau protein induces an amyloidogenic shift in its conformational ensemble. PLoS One 10: , e0120416. |
[21] | Braak H , Braak E , Strothjohann M ((1994) ) Abnormally phosphorylated tau protein related to the formation of neurofibrillary tangles and neuropil threads in the cerebral cortex of sheep and goat. Neurosci Lett 171: , 1–4. |
[22] | Guo C , Wang P , Zhong ML , Wang T , Huang XS , Li JY , Wang ZY ((2012) ) Deferoxamine inhibits iron induced hippocampal tau phosphorylation in the Alzheimer transgenic mouse brain. Neurochem Int 62: , 165–172. |
[23] | Ong WY , Farooqui AA ((2005) ) Iron, neuroinflammation, and Alzheimer’s disease. J Alzheimers Dis 8: , 183–200. |
[24] | Oshiro S , Morioka MS , Kikuchi M ((2011) ) Dysregulation of iron metabolism in Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Adv Pharmacol Sci 2011: , 378278. |
[25] | Heneka MT , O’Banion MK , Terwel D , Kummer MP ((2010) ) Neuroinflammatory processes in Alzheimer’s disease. J Neural Transm 117: , 919–947. |
[26] | Varley J , Brooks DJ , Edison P ((2014) ) Imaging neuroinflammation in Alzheimer’s and other dementias: Recent advances and future directions. Alzheimers Dement 11: , 1110–1120. |
[27] | Latta CH , Brothers HM , Wilcock DM ((2014) ) Neuroinflammation in Alzheimer’s disease; A source of heterogeneity and target for personalized therapy. Neuroscience 302: , 103–111. |
[28] | Dorey E , Chang N , Liu QY , Yang Z , Zhang W ((2014) ) Apolipoprotein E, amyloid-beta, and neuroinflammation in Alzheimer’s disease. Neurosci Bull 30: , 317–330. |
[29] | Filiou MD , Arefin AS , Moscato P , Graeber MB ((2014) ) ‘Neuroinflammation’ differs categorically from inflammation: Transcriptomes of Alzheimer’s disease, Parkinson’s disease, schizophrenia and inflammatory diseases compared. Neurogenetics 15: , 201–212. |
[30] | Heneka MT , Carson MJ , Khoury JE , Landreth GE , Brosseron F , Feinstein DL , Jacobs AH , Wyss-Coray T , Vitorica J , Ransohoff RM , Herrup K , Frautschy SA , Finsen B , Brown GC , Verkhratsky A , Yamanaka K , Koistinaho J , Latz E , Halle A , Petzold GC , Town T , Morgan D , Shinohara ML , Perry VH , Holmes C , Bazan NG , Brooks DJ , Hunot S , Joseph B , Deigendesch N , Garaschuk O , Boddeke E , Dinarello CA , Breitner JC , Cole GM , Golenbock DT , Kummer MP ((2015) ) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14: , 388–405. |
[31] | Steardo L Jr , Bronzuoli MR , Iacomino A , Esposito G , Steardo L , Scuderi C ((2015) ) Does neuroinflammation turn on the flame in Alzheimer’s disease? Focus on astrocytes. Front Neurosci 9: , 259. |
[32] | Qin L , Wu X , Block ML , Liu Y , Breese GR , Hong JS , Knapp DJ , Crews FT ((2007) ) Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55: , 453–462. |
[33] | Bester J , Buys AV , Lipinski B , Kell DB , Pretorius E ((2013) ) High ferritin levels have major effects on the morphology of erythrocytes in Alzheimer’s disease. Front Aging Neurosci 5: , 88. |
[34] | Bester J , Soma P , Kell DB , Pretorius E ((2015) ) Viscoelastic and ultrastructural characteristics of whole blood and plasma in Alzheimer-type dementia, and the possible role of bacterial lipopolysaccharides (LPS). Oncotarget Gerentol 6: , 35284–35303. |
[35] | Kell DB ((2009) ) Iron behaving badly: Inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC Med Genomics 2: , 2. |
[36] | Kell DB ((2010) ) Towards a unifying, systems biology understanding of large-scale cellular death and destruction caused by poorly liganded iron: Parkinson’s, Huntington’s, Alzheimer’s, prions, bactericides, chemical toxicology and others as examples. Arch Toxicol 577: , 825–889. |
[37] | Klipp E , Herwig R , Kowald A , Wierling C , Lehrach H ((2005) ) Systems biology in practice: Concepts, implementation and clinical application, Wiley/VCH, Berlin. |
[38] | Noble D ((2006) ) The music of life: Biology beyond genes, Oxford University Press, Oxford. |
[39] | Palsson BØ ((2006) ) Systems biology: Properties of reconstructed networks, Cambridge University Press, Cambridge. |
[40] | Palsson BØ ((2015) ) Systems biology: Constraint-based reconstruction and analysis, Cambridge University Press, Cambridge. |
[41] | Favrin G , Bean DM , Bilsland E , Boyer H , Fischer BE , Russell S , Crowther DC , Baylis HA , Oliver SG , Giannakou ME ((2013) ) Identification of novel modifiers of Abeta toxicity by transcriptomic analysis in the fruitfly. Sci Rep 3: , 3512. |
[42] | Kell DB ((2006) ) Metabolomics, modelling and machine learning in systems biology: Towards an understanding of the languages of cells. The 2005 Theodor Bücher lecture. FEBS J 273: , 873–894. |
[43] | Kell DB , Knowles JD ((2006) ) The role of modeling in systems biology. In System modeling in cellular biology: From concepts to nuts and bolts, Szallasi Z, Stelling J, Periwal V, eds. MIT Press, Cambridge, pp. 3–18. |
[44] | Kell DB ((2007) ) The virtual human: Towards a global systems biology of multiscale, distributed biochemical network models. IUBMB Life 59: , 689–695. |
[45] | Kell DB , Mendes P ((2008) ) The markup is the model: Reasoning about systems biology models in the Semantic Web era. J Theoret Biol 252: , 538–543. |
[46] | Herrgård MJ , Swainston N , Dobson P , Dunn WB , Arga KY , Arvas M , Blüthgen N , Borger S , Costenoble R , Heinemann M , Hucka M , Le Novère N , Li P , Liebermeister W , Mo ML , Oliveira AP , Petranovic D , Pettifer S , Simeonidis E , Smallbone K , Spasić I , Weichart D , Brent R , Broomhead DS , Westerhoff HV , Kirdar B , Penttilä M , Klipp E , Palsson BØ , Sauer U , Oliver SG , Mendes P , Nielsen J , Kell DB ((2008) ) A consensus yeast metabolic network obtained from a community approach to systems biology. Nat Biotechnol 26: , 1155–1160. |
[47] | Thiele I, Swainston N, Fleming RM, Hoppe A, Sahoo S, Aurich MK, Haraldsdottir H, Mo ML, Rolfsson O, Stobbe MD, Thorleifsson SG, Agren R, Bölling C, Bordel S, Chavali AK, Dobson P, Dunn WB, Endler L, Hala D, Hucka M, Hull D, Jameson D, Jamshidi N, Jonsson JJ, Juty N, Keating S, Nookaew I, Le Novère N, Malys N, Mazein A, Papin JA, Price ND, Selkov E Sr, Sigurdsson MI, Simeonidis E, Sonnenschein N, Smallbone K, Sorokin A, van Beek JH, Weichart D, Goryanin I, Nielsen J, Westerhoff HV, Kell DB, Mendes P, Palsson BØ ((2013) ) A community-driven global reconstruction of human metabolism. Nat Biotechnol 31: , 419–425. |
[48] | Connor JR , Snyder BS , Beard JL , Fine RE , Mufson EJ ((1992) ) Regional distribution of iron and iron-regulatory proteins in the brain in aging and Alzheimers disease. J Neuroscie Res 31: , 327–335. |
[49] | Kala SV , Hasinoff BB , Richardson JS ((1996) ) Brain samples from Alzheimer’s patients have elevated levels of loosely bound iron. Int J Neurosci 86: , 263–269. |
[50] | Casadesus G , Smith MA , Zhu X , Aliev G , Cash AD , Honda K , Petersen RB , Perry G ((2004) ) Alzheimer disease: Evidence for a central pathogenic role of iron-mediated reactive oxygen species. J Alzheimers Dis 6: , 165–169. |
[51] | Castellani RJ , Moreira PI , Liu G , Dobson J , Perry G , Smith MA , Zhu X ((2007) ) Iron: The redox-active center of oxidative stress in Alzheimer disease. Neurochem Res 32: , 1640–1645. |
[52] | Silvestri L , Camaschella C ((2008) ) A potential pathogenetic role of iron in Alzheimer’s Disease. J Cell Mol Med 12: , 1548–1550. |
[53] | Rolston RK , Perry G , Zhu X , Castellani RJ , Dwyer BE , Lee HG , Petersen RB , Smith MA ((2009) ) Iron: A pathological mediator of Alzheimer disease? Agro Food Ind Hi Tech 19: , 33–36. |
[54] | Smith MA , Zhu X , Tabaton M , Liu G , McKeel DW Jr , Cohen ML , Wang X , Siedlak SL , Dwyer BE , Hayashi T , Nakamura M , Nunomura A , Perry G ((2010) ) Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J Alzheimers Dis 19: , 363–372. |
[55] | Kupershmidt L , Amit T , Bar-Am O , Youdim MBH , Weinreb O ((2012) ) The novel multi-target iron chelating-radical scavenging compound M30 possesses beneficial effects on major hallmarks of Alzheimer’s disease. Antioxid Redox Signal 17: , 860–877. |
[56] | Castellani RJ , Moreira PI , Perry G , Zhu X ((2012) ) The role of iron as a mediator of oxidative stress in Alzheimer disease. Biofactors 38: , 133–138. |
[57] | Zecca L , Youdim MB , Riederer P , Connor JR , Crichton RR ((2004) ) Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci 5: , 863–873. |
[58] | Friedman A , Arosio P , Finazzi D , Koziorowski D , Galazka-Friedman J ((2011) ) Ferritin as an important player in neurodegeneration. Parkinsonism Relat Disord 17: , 423–430. |
[59] | Yang H , Guan H , Yang M , Liu Z , Takeuchi S , Yanagisawa D , Vincent SR , Zhao S , Tooyama I ((2015) ) Upregulation of mitochondrial ferritin by proinflammatory cytokines: Implications for a role in Alzheimer’s disease. J Alzheimers Dis 45: , 797–811. |
[60] | Wood H ((2015) ) Iron - the missing link between ApoE and Alzheimer disease? Nat Rev Neurol 11: , 369. |
[61] | Bandyopadhyay S , Rogers JT ((2014) ) Alzheimer’s disease therapeutics targeted to the control of amyloid precursor protein translation: Maintenance of brain iron homeostasis. Biochem Pharmacol 88: , 486–494. |
[62] | Collingwood JF , Mikhaylova A , Davidson M , Batich C , Streit WJ , Terry J , Dobson J ((2005) ) characterization and mapping of iron compounds in Alzheimer’s disease tissue. J Alzheimers Dis 7: , 267–272. |
[63] | Collingwood J , Dobson J ((2006) ) Mapping and characterization of iron compounds in Alzheimer’s tissue. J Alzheimers Dis 10: , 215–222. |
[64] | Collingwood JF , Chong RK , Kasama T , Cervera-Gontard L , Dunin-Borkowski RE , Perry G , Posfai M , Siedlak SL , Simpson ET , Smith MA , Dobson J ((2008) ) Three-dimensional tomographic imaging and characterization of iron compounds within Alzheimer’s plaque core material. J Alzheimers Dis 14: , 235–245. |
[65] | Bush AI ((2002) ) Metal complexing agents as therapies for Alzheimer’s disease. Neurobiol Aging 23: , 1031–1038. |
[66] | Bush AI ((2008) ) Drug development based on the metals hypothesis of Alzheimer’s disease. J Alzheimers Dis 15: , 223–240. |
[67] | Bush AI , Tanzi RE ((2008) ) Therapeutics for Alzheimer’s disease based on the metal hypothesis. Neurotherapeutics 5: , 421–432. |
[68] | Ding B , Chen KM , Ling HW , Sun F , Li X , Wan T , Chai WM , Zhang H , Zhan Y , Guan YJ ((2009) ) Correlation of iron in the hippocampus with MMSE in patients with Alzheimer’s disease. J Magn Reson Imaging 29: , 793–798. |
[69] | Gałązka-Friedman J , Bauminger ER , Szlachta K , Koziorowski D , Tomasiuk R , Jaklewicz A , Wszolek ZK , Dickson D , Kaplińska K , Friedman A ((2011) ) Iron in Alzheimer’s and control hippocampi - Mössbauer, atomic absorption and ELISA studies. Acta Physica Polonica A 119: , 81–83. |
[70] | Zhang J , Wang JH , Li K , Geng DY , Chen MR , Tang WJ , Zhao ZG , Li YH , Ma SG , Yan CG ((2010) ) Correlation between iron deposition and Alzheimer’s disease in vivo preliminary quantitative study with susceptibility-weighted imaging. Neural Regen Res 5: , 725–728. |
[71] | Raven EP , Lu PH , Tishler TA , Heydari P , Bartzokis G ((2013) ) Increased iron levels and decreased tissue integrity in hippocampus of Alzheimer’s disease detected in vivo with magnetic resonance imaging. J Alzheimers Dis 37: , 127–136. |
[72] | Quintana C , Bellefqih S , Laval JY , Guerquin-Kern JL , Wu TD , Avila J , Ferrer I , Arranz R , Patino C ((2006) ) Study of the localization of iron, ferritin, and hemosiderin in Alzheimer’s disease hippocampus by analytical microscopy at the subcellular level. J Struct Biol 153: , 42–54. |
[73] | Wang D , Li YY , Luo JH , Li YH ((2014) ) Age-related iron deposition in the basal ganglia of controls and Alzheimer disease patients quantified using susceptibility weighted imaging. Arch Gerontol Geriatr 59: , 439–449. |
[74] | Giambattistelli F , Bucossi S , Salustri C , Panetta V , Mariani S , Siotto M , Ventriglia M , Vernieri F , Dell’acqua ML , Cassetta E , Rossini PM , Squitti R ((2012) ) Effects of hemochromatosis and transferrin gene mutations on iron dyshomeostasis, liver dysfunction and on the risk of Alzheimer’s disease. Neurobiol Aging 33: , 1633–1641. |
[75] | De Sole P , Rossi C , Chiarpotto M , Ciasca G , Bocca B , Alimonti A , Bizzarro A , Rossi C , Masullo C ((2013) ) Possible relationship between Al/ferritin complex and Alzheimer’s disease. Clin Biochem 46: , 89–93. |
[76] | Barnham KJ , Bush AI ((2008) ) Metals in Alzheimer’s and Parkinson’s diseases. Curr Opin Chem Biol 12: , 222–228. |
[77] | Weinberg ED ((2010) ) The hazards of iron loading. Metallomics 2: , 732–740. |
[78] | Nielsen VG , Pretorius E , Bester J , Jacobsen WK , Boyle PK , Reinhard JP ((2015) ) Carbon monoxide and iron modulate plasmatic coagulation in Alzheimer’s disease. Curr Neurovasc Res 12: , 31–39. |
[79] | Ayton S , Faux NG ((2015) ) Ferritin levels in the cerebrospinal fluid predict Alzheimer’s disease outcomes and are regulated by APOE. Nat Commun 6: , 6760. |
[80] | Meadowcroft MD , Connor JR , Smith MB , Yang QX ((2009) ) MRI and histological analysis of beta-amyloid plaques in both human Alzheimer’s disease and APP/PS1 transgenic mice. J Magn Reson Imaging 29: , 997–1007. |
[81] | Altamura S , Muckenthaler MU ((2009) ) Iron toxicity in diseases of aging: Alzheimer’s disease, Parkinson’s disease and atherosclerosis. J Alzheimers Dis 16: , 879–895. |
[82] | Adlard PA , Bush AI ((2006) ) Metals and Alzheimer’s disease. J Alzheimers Dis 10: , 145–163. |
[83] | Jomova K , Valko M ((2011) ) Importance of iron chelation in free radical-induced oxidative stress and human disease. Curr Pharm Des 17: , 3460–3473. |
[84] | Valko M , Leibfritz D , Moncol J , Cronin MTD , Mazur M , Telser J ((2007) ) Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol 39: , 44–84. |
[85] | Cervellati C , Wood PL , Romani A , Valacchi G , Squerzanti M , Sanz JM , Ortolani B , Zuliani G ((2016) ) Oxidative challenge in Alzheimer’s disease: State of knowledge and future needs. J Investig Med 64: , 21–32. |
[86] | Smith MA , Rottkamp CA , Nunomura A , Raina AK , Perry G ((2000) ) Oxidative stress in Alzheimer’s disease. Biochim Biophys Acta 1502: , 139–144. |
[87] | Chauhan V , Chauhan A ((2006) ) Oxidative stress in Alzheimer’s disease. Pathophysiology 13: , 195–208. |
[88] | Markesbery WR ((1997) ) Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med 23: , 134–147. |
[89] | Markesbery WR , Carney JM ((1999) ) Oxidative alterations in Alzheimer’s disease. Brain Pathol 9: , 133–146. |
[90] | Smith DG , Cappai R , Barnham KJ ((2007) ) The redox chemistry of the Alzheimer’s disease amyloid beta peptide. Biochim Biophys Acta 1768: , 1976–1990. |
[91] | Jomova K , Vondrakova D , Lawson M , Valko M ((2010) ) Metals, oxidative stress and neurodegenerative disorders. Mol Cell Biochem 345: , 91–104. |
[92] | Singh N , Haldar S , Tripathi AK , Horback K , Wong J , Sharma D , Beserra A , Suda S , Anbalagan C , Dev S , Mukhopadhyay CK , Singh A ((2014) ) Brain iron homeostasis: From molecular mechanisms to clinical significance and therapeutic opportunities. Antioxid Redox Signal 20: , 1324–1363. |
[93] | Greenough MA , Camakaris J , Bush AI ((2013) ) Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem Int 62: , 540–555. |
[94] | Das TK , Wati MR , Fatima-Shad K ((2015) ) Oxidative stress gated by Fenton and Haber Weiss reactions and its association with Alzheimer’s disease. Arch Neurosci 2: , e20078. |
[95] | Pretorius E , Swanepoel AC , Buys AV , Vermeulen N , Duim W , Kell DB ((2014) ) Eryptosis as a marker of Parkinson’s disease. Aging-US 6: , 788–818. |
[96] | Kell DB , Pretorius E ((2015) ) The simultaneous occurrence of both hypercoagulability and hypofibrinolysis in blood and serum during systemic inflammation, and the roles of iron and fibrin(ogen). Integr Biol 7: , 24–52. |
[97] | Pretorius E , Kell DB ((2014) ) Diagnostic morphology: Biophysical indicators for iron-driven inflammatory diseases. Integr Biol 6: , 486–510. |
[98] | Pretorius E ((2013) ) The adaptability of red blood cells. Cardiovasc Diabetol 12: , 63. |
[99] | Chen Z , Zhong C ((2014) ) Oxidative stress in Alzheimer’s disease. Neurosci Bull 30: , 271–281. |
[100] | de la Monte SM ((2014) ) Type 3 diabetes is sporadic Alzheimer’s disease: Mini-review. Eur Neuropsychopharmacol 24: , 1954–1960. |
[101] | Proal AD , Albert PJ , Marshall TG ((2014) ) Inflammatory disease and the human microbiome. Discov Med 17: , 257–265. |
[102] | Malecki EA , Connor JR ((2002) ) The case for iron chelation and/or antioxidant therapy in Alzheimer’s disease. Drug Dev Res 56: , 526–530. |
[103] | Mandel S , Amit T , Bar-Am O , Youdim MB ((2007) ) Iron dysregulation in Alzheimer’s disease: Multimodal brain permeable iron chelating drugs, possessing neuroprotective-neurorescue and amyloid precursor protein-processing regulatory activities as therapeutic agents. Prog Neurobiol 82: , 348–360. |
[104] | Crapper McLachlan DR , Dalton AJ , Kruck TP , Bell MY , Smith WL , Kalow W , Andrews DF ((1991) ) Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 337: , 1304–1308. |
[105] | Banerjee P , Sahoo A , Anand S , Bir A , Chakrabarti S ((2015) )J Alzheimers Dis 49: , 681–693 The oral iron chelator, deferasirox, reverses the age-dependent alterations in iron and amyloid-beta homeostasis in rat bra, Imlications in the therapy of Alzheimer’s disease. |
[106] | Venigalla M , Gyengesi E , Munch G ((2015) ) Curcumin and Apigenin - novel and promising therapeutics against chronic neuroinflammation in Alzheimer’s disease. Neural Regen Res 10: , 1181–1185. |
[107] | Ghofrani S , Joghataei MT , Mohseni S , Baluchnejadmojarad T , Bagheri M , Khamse S , Roghani M ((2015) ) Naringenin improves learning and memory in an Alzheimer’s disease rat model: Insights into the underlying mechanisms. Eur J Pharmacol 764: , 195–201. |
[108] | Salkovic-Petrisic M , Knezovic A , Osmanovic-Barilar J , Smailovic U , Trkulja V , Riederer P , Amit T , Mandel S , Youdim MBH ((2015) ) Multi-target iron-chelators improve memory loss in a rat model of sporadic Alzheimer’s disease. Life Sci 136: , 108–119. |
[109] | Funke C , Schneider SA , Berg D , Kell DB ((2013) ) Genetics and iron in the systems biology of Parkinson’s disease and some related disorders. Neurochem Int 62: , 637–652. |
[110] | Finkelstein DI , Hare DJ , Billings JL , Sedjahtera A , Nurjono M , Arthofer E , George S , Culvenor JG , Bush AI , Adlard PA ((2016) ) Clioquinol improves cognitive, motor function, and microanatomy of the alpha-synuclein hA53T transgenic mice. ACS Chem Neurosci 7: , 119–129. |
[111] | Lei P , Ayton S , Appukuttan AT , Volitakis I , Adlard PA , Finkelstein DI , Bush AI ((2015) ) Clioquinol rescues Parkinsonism and dementia phenotypes of the tau knockout mouse. Neurobiol Dis 81: , 168–175. |
[112] | Billings JL , Hare DJ , Nurjono M , Volitakis I , Cherny RA , Bush AI , Adlard PA , Finkelstein DI ((2016) ) Effects of neonatal iron feeding and chronic clioquinol administration on the parkinsonian human A53T transgenic mouse. ACS Chem Neurosci 7: , 360–366. |
[113] | Toyokuni S ((2011) ) Iron as a target of chemoprevention for longevity in humans. Free Radic Res 45: , 906–917. |
[114] | Yusufov M , Weyandt LL , Piryatinsky I ((2016) ) Alzheimer’s disease and diet: A systematic review. Int J Neurosci. doi: 10.3109/00207454.2016.1155572 |
[115] | Cao L , Tan L , Wang HF , Jiang T , Zhu XC , Lu H , Tan MS , Yu JT ((2015) ) Dietary patterns and risk of dementia: A systematic review and meta-analysis of cohort studies. Mol Neurobiol. doi: 10.1007/s12035-015-9516-4 |
[116] | Gu Y , Brickman AM , Stern Y , Habeck CG , Razlighi QR , Luchsinger JA , Manly JJ , Schupf N , Mayeux R , Scarmeas N ((2015) ) Mediterranean diet and brain structure in a multiethnic elderly cohort. Neurology 85: , 1744–1751. |
[117] | Scarmeas N , Stern Y , Mayeux R , Luchsinger JA ((2006) ) Mediterranean diet, Alzheimer disease, and vascular mediation. Arch Neurol 63: , 1709–1717. |
[118] | Thaipisuttikul P , Galvin JE ((2012) ) Use of medical foods and nutritional approaches in the treatment of Alzheimer’s disease. Clin Pract (Lond) 9: , 199–209. |
[119] | Lipinski B , Pretorius E ((2013) ) The role of iron-induced fibrin in the pathogenesis of Alzheimer’s disease and the protective role of magnesium. Front Hum Neurosci 7: , 735. |
[120] | Dwyer BE , Zacharski LR , Balestra DJ , Lerner AJ , Perry G , Zhu X , Smith MA ((2010) ) Potential role of iron in a Mediterranean-style diet. Arch Neurol 67: , 1286–1287; author reply 1287-1288. |
[121] | Ayissi VB , Ebrahimi A , Schluesenner H ((2013) ) Epigenetic effects of natural polyphenols: A focus on SIRT1-mediated mechanisms. Mol Nutr Food Res 58: , 22–32. |
[122] | Feart C , Samieri C , Barberger-Gateau P ((2010) ) Mediterranean diet and cognitive function in older adults. Curr Opin Clin Nutr Metab Care 13: , 14–18. |
[123] | Gu Y , Luchsinger JA , Stern Y , Scarmeas N ((2010) ) Mediterranean diet, inflammatory and metabolic biomarkers, and risk of Alzheimer’s disease. J Alzheimers Dis 22: , 483–492. |
[124] | Hu N , Yu JT , Tan L , Wang YL , Sun L , Tan L ((2013) ) Nutrition and the risk of Alzheimer’s disease. Biomed Res Int 2013: , 524820. |
[125] | Dwyer BE , Zacharski LR , Balestra DJ , Lerner AJ , Perry G , Zhu X , Smith MA ((2009) ) Getting the iron out: Phlebotomy for Alzheimer’s disease? Med Hypotheses 72: , 504–509. |
[126] | Kell DB , Potgieter M , Pretorius E ((2015) ) Individuality, phenotypic differentiation, dormancy and ‘persistence’ in culturable bacterial systems: Commonalities in environmental, laboratory, and clinical microbiology. F1000Review 4: , 179. |
[127] | Barber MF , Elde NC ((2014) ) Nutritional immunity. Escape from bacterial iron piracy through rapid evolution of transferrin. Science 346: , 1362–1366. |
[128] | Armitage AE , Drakesmith H ((2014) ) Genetics. The battle for iron. Science 346: , 1299–1300. |
[129] | Haley KP , Skaar EP ((2012) ) A battle for iron: Host sequestration andacquisition. Microbes Infect 14: , 217–227. |
[130] | Nairz M , Haschka D , Demetz E , Weiss G ((2014) ) Iron at the interface of immunity and infection. Front Pharmacol 5: , 152. |
[131] | Nairz M , Schroll A , Sonnweber T , Weiss G ((2010) ) The struggle for iron - a metal at the host-pathogen interface. Cell Microbiol 12: , 1691–1702. |
[132] | Subashchandrabose S , Mobley HLT ((2015) ) Back to the metal age: Battle for metals at the host-pathogen interface during urinary tract infection. Metallomics 7: , 935–942. |
[133] | Nikkari S , McLaughlin IJ , Bi W , Dodge DE , Relman DA ((2001) ) Does blood of healthy subjects contain bacterial ribosomal DNA? J Clin Microbiol 39: , 1956–1959. |
[134] | Amar J , Serino M , Lange C , Chabo C , Iacovoni J , Mondot S , Lepage P , Klopp C , Mariette J , Bouchez O , Perez L , Courtney M , Marre M , Klopp P , Lantieri O , Doré J , Charles MA , Balkau B , Burcelin R , Grp DS ((2011) ) Involvement of tissue bacteria in the onset of diabetes in humans: Evidence for a concept. Diabetologia 54: , 3055–3061. |
[135] | Ribault S , Faucon A , Grave L , Nannini P , Faure IB ((2005) ) Detection of bacteria in red blood cell concentrates by the Scansystem method. J Clin Microbiol 43: , 2251–2255. |
[136] | Dinakaran V , Rathinavel A , Pushpanathan M , Sivakumar R , Gunasekaran P , Rajendhran J ((2014) ) Elevated levels of circulating DNA in cardiovascular disease patients: Metagenomic profiling of microbiome in the circulation. PLoS One 9: , e105221. |
[137] | Damgaard C , Magnussen K , Enevold C , Nilsson M , Tolker-Nielsen T , Holmstrup P , Nielsen CH ((2015) ) Viable bacteria associated with red blood cells and plasma in freshly drawn blood donations. PLoS One 10: , e0120826. |
[138] | Païssé S , Valle C , Servant F , Courtney M , Burcelin R , Amar J , Lelouvier B ((2016) ) Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion 56: , 1138–1147. |
[139] | Marshall BJ ((2001) ) One hundred years of discovery and rediscovery ofand its association with peptic ulcer disease. In Helicobacter pylori: Physiology and Genetics, Mobley HLT, Mendz GL, Hazell SL, eds. ASM Press, Washington (DC), pp. 19–24. |
[140] | Marshall BJ , Warren JR ((1984) ) Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1: , 1311–1315. |
[141] | Marshall BJ , Armstrong JA , McGechie DB , Glancy RJ ((1985) ) Attempt to fulfil Koch’s postulates for pyloric. Med J Aust 142: , 436–439. |
[142] | Marshall B ((2002) ) 20 years on. Clin Med 2: , 147–152. |
[143] | Harris CM , Kell DB ((1985) ) The estimation of microbial biomass. Biosensors 1: , 17–84. |
[144] | Kaprelyants AS , Gottschal JC , Kell DB ((1993) ) Dormancy in non-sporulating bacteria. FEMS Microbiol Rev 10: , 271–286. |
[145] | Kell DB , Young M ((2000) ) Bacterial dormancy and culturability: The role of autocrine growth factors. Curr Opin Microbiol 3: , 238–243. |
[146] | Mukamolova GV , Kaprelyants AS , Kell DB , Young M ((2003) ) Adoption of the transiently non-culturable state - a bacterial survival strategy? Adv Micr Physiol 47: , 65–129. |
[147] | Domingue GJ , Woody HB ((1997) ) Bacterial persistence and expression of disease. Clin Microbiol Rev 10: , 320–344. |
[148] | Mattman L ((2001) ) Cell wall deficient forms: Stealth pathogens, 3rd Ed., CRC Press, Boca Raton. |
[149] | Lewis K ((2007) ) Persister cells, dormancy and infectious disease. Nat Rev Microbiol 5: , 48–56. |
[150] | Lewis K ((2010) ) Persister cells. Annu Rev Microbiol 64: , 357–372. |
[151] | Shah D , Zhang Z , Khodursky A , Kaldalu N , Kurg K , Lewis K ((2006) ) Persisters: A distinct physiological state of E. coli. BMC Microbiol 6: , 53. |
[152] | Holden DW ((2015) ) Persisters unmasked. Science 347: , 30–32. |
[153] | Bigger JW ((1944) ) Treatment of staphylococcal infections with penicillin - by intermittent sterilisation. Lancet 2: , 497–500. |
[154] | Allison KR , Brynildsen MP , Collins JJ ((2011) ) Heterogeneous bacterial persisters and engineering approaches to eliminate them. Curr Opin Microbiol 14: , 593–598. |
[155] | Grant SS , Kaufmann BB , Chand NS , Haseley N , Hung DT ((2012) ) Eradication of bacterial persisters with antibiotic-generated hydroxyl radicals. Proc Natl Acad Sci U S A 109: , 12147–12152. |
[156] | Wood TK , Knabel SJ , Kwan BW ((2013) ) Bacterial persister cell formation and dormancy. Appl Environ Microbiol 79: , 7116–7121. |
[157] | Davey HM , Kell DB ((1996) ) Flow cytometry and cell sorting of heterogeneous microbial populations: The importance of single-cell analysis. Microbiol Rev 60: , 641–696. |
[158] | Kell DB , Kaprelyants AS , Weichart DH , Harwood CL , Barer MR ((1998) ) Viability and activity in readily culturable bacteria: A review and discussion of the practical issues. Antonie van Leeuwenhoek 73: , 169–187. |
[159] | Fredricks DN , Relman DA ((1996) ) Sequence-based identification of microbial pathogens - a reconsideration of Koch’s postulates. Clin Micr Rev 9: , 18–33. |
[160] | Falkow S ((1988) ) Molecular Koch’s postulates applied to microbial pathogenicity. Rev Infect Dis 10: (Suppl 2), S274–S276. |
[161] | Falkow S ((2004) ) Molecular Koch’s postulates applied to bacterial pathogenicity - a personal recollection 15 years later. Nat Rev Microbiol 2: , 67–72. |
[162] | Miklossy J ((2011) ) Alzheimer’s disease - a neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J Neuroinflammation 8: , 90. |
[163] | Segre JA ((2013) ) What does it take to satisfy Koch’s postulates two centuries later? Microbial genomics and Propionibacteria acnes. J Invest Dermatol 133: , 2141–2142. |
[164] | Byrd AL , Segre JA ((2016) ) Adapting Koch’s postulates. Science 351: , 224–226. |
[165] | Evans AS ((1976) ) Causation and disease: The Henle-Koch postulates revisited. Yale J Biol Med 49: , 175–195. |
[166] | Thagard P ((2007) ) Coherence, truth, and the development of scientific knowledge. Philosophy Sci 74: , 28–47. |
[167] | Miklossy J ((1993) ) Alzheimer’s disease–a spirochetosis? Neuroreport 4: , 841–848. |
[168] | Miklossy J ((2008) ) Chronic inflammation and amyloidogenesis in Alzheimer’s disease – role of Spirochetes. J Alzheimers Dis 13: , 381–391. |
[169] | Miklossy J ((2011) ) Emerging roles of pathogens in Alzheimer disease. Expert Rev Mol Med 13: , e30. |
[170] | Miklossy J ((2012) ) Chronic or late lyme neuroborreliosis: Analysis of evidence compared to chronic or late neurosyphilis. Open Neurol J 6: , 146–157. |
[171] | Miklossy J ((2015) ) Historic evidence to support a causal relationship between spirochetal infections and Alzheimer’s disease. Front Aging Neurosci 7: , 46. |
[172] | Itzhaki RF , Wozniak MA ((2004) ) Alzheimer’s disease, the neuroimmune axis, and viral infection. J Neuroimmunol 156: , 1–2. |
[173] | Itzhaki RF , Wozniak MA ((2008) ) Herpes simplex virus type 1 in Alzheimer’s disease: The enemy within. J Alzheimers Dis 13: , 393–405. |
[174] | Itzhaki RF , Wozniak MA ((2010) ) Alzheimer’s disease and infection: Do infectious agents contribute to progression of Alzheimer’s disease? Alzheimers Dement 6: , 93–84; author reply 85. |
[175] | Itzhaki RF , Wozniak MA ((2012) ) Could antivirals be used to treat Alzheimer’s disease? Future Microbiol 7: , 307–309. |
[176] | Itzhaki RF , Klapper P ((2014) ) Cytomegalovirus: An improbable cause of Alzheimer disease. J Infect Dis 209: , 972–973. |
[177] | Itzhaki RF , Klapper P ((2015) ) Comment on “cytomegalovirus infection and risk of Alzheimer disease in older black and white individuals,” journal of infectious diseases, 8 August 2014. J Infect Dis 211: , 2023–2024. |
[178] | Balin BJ , Gerard HC , Arking EJ , Appelt DM , Branigan PJ , Abrams JT , Whittum-Hudson JA , Hudson AP ((1998) ) Identification and localization ofin the Alzheimer’s brain. Med Microbiol Immunol 187: , 23–42. |
[179] | Balin BJ , Appelt DM ((2001) ) Role of infection in Alzheimer’s disease. J Am Osteopath Assoc 101: , S1–S6. |
[180] | Balin BJ , Little CS , Hammond CJ , Appelt DM , Whittum-Hudson JA , Gerard HC , Hudson AP ((2008) ) and the etiology of late-onset Alzheimer’s disease. J Alzheimers Dis 13: , 371–380. |
[181] | Hammond CJ , Hallock LR , Howanski RJ , Appelt DM , Little CS , Balin BJ ((2010) ) Immunohistological detection ofin the Alzheimer’s disease brain. BMC Neurosci 11: , 121. |
[182] | Allen HB , Morales D , Jones K , Joshi S ((2016) ) Alzheimer’s disease: A novel hypothesis integrating spirochetes, biofilm, and the immune system. Neuroinfect Dis 7: , 1–3. |
[183] | Olsen I , Singhrao SK ((2015) ) Can oral infection be a risk factor for Alzheimer’s disease? J Oral Microbiol 7: , 29143. |
[184] | Fong IW ((2014) ) The role of microbes in common non-infectious diseases, Springer, New York. |
[185] | Nicolson GL , Haier J ((2009) ) Role of chronic bacterial and viral infections in neurodegenerative, neurobehavioural, psychiatric, autoimmune and fatiguing illnesses: Part 1. Br J Med Pract 2: , 20–28. |
[186] | Nicolson GL , Haier J ((2010) ) Role of chronic bacterial and viral infections in neurodegenerative, neurobehavioural, psychiatric, autoimmune and fatiguing illnesses: Part 2. Br J Med Pract 3: , 301–310. |
[187] | Alonso R , Pisa D , Marina AI , Morato E , Rabáno A , Carrasco L ((2014) ) Fungal infection in patients with Alzheimer’s disease. J Alzheimers Dis 41: , 301–311. |
[188] | Bu XL , Yao XQ , Jiao SS , Zeng F , Liu YH , Xiang Y , Liang CR , Wang QH , Wang X , Cao HY , Yi X , Deng B , Liu CH , Xu J , Zhang LL , Gao CY , Xu ZQ , Zhang M , Wang L , Tan XL , Xu X , Zhou HD , Wang YJ ((2014) ) A study on the association between infectious burden and Alzheimer’s disease. Eur J Neurol 22: , 1519–1525. |
[189] | Pisa D , Alonso R , Juarranz A , Rábano A , Carrasco L ((2015) ) Direct visualization of fungal infection in brains from patients with Alzheimer’s disease. J Alzheimers Dis 43: , 613–624. |
[190] | Bhattacharjee S , Lukiw WJ ((2013) ) Alzheimer’s disease and the microbiome. Front Cell Neurosci 7: , 153. |
[191] | Noble JM , Scarmeas N , Celenti RS , Elkind MSV , Wright CB , Schupf N , Papapanou PN ((2014) ) Serum IgG antibody levels to periodontal microbiota are associated with incident Alzheimer disease. PLoS One 9: , e114959. |
[192] | Shoemark DK , Allen SJ ((2015) ) The microbiome and disease: Reviewing the links between the oral microbiome, aging, and Alzheimer’s disease. J Alzheimers Dis 43: , 725–738. |
[193] | Hill JM , Clement C , Pogue AI , Bhattacharjee S , Zhao Y , Lukiw WJ ((2014) ) Pathogenic microbes, the microbiome, and Alzheimer’s disease (AD). Front Aging Neurosci 6: , 127. |
[194] | Hill JM , Bhattacharjee S , Pogue AI , Lukiw WJ ((2014) ) The gastrointestinal tract microbiome and potential link to Alzheimer’s disease. Front Neurol 5: , 43. |
[195] | Hill JM , Lukiw WJ ((2015) ) Microbial-generated amyloids and Alzheimer’s disease (AD). Front Aging Neurosci 7: , 9. |
[196] | Bibi F , Yasir M , Sohrab SS , Azhar EI , Al-Qahtani MH , Abuzenadah AM , Kamal MA , Naseer MI ((2014) ) Link between chronic bacterial inflammation and Alzheimer disease. CNS Neurol Disord Drug Targets 13: , 1140–1147. |
[197] | Maheshwari P , Eslick GD ((2015) ) Bacterial infection and Alzheimer’s disease: A meta-analysis. J Alzheimers Dis 43: , 957–966. |
[198] | Itzhaki RF , Lathe R , Balin BJ , Ball MJ , Bearer EL , Braak H , Bullido MJ , Carter C , Clerici M , Cosby SL , Del Tredici K , Field H , Fulop T , Grassi C , Griffin WS , Haas J , Hudson AP , Kamer AR , Kell DB , Licastro F , Letenneur L , Lovheim H , Mancuso R , Miklossy J , Lagunas CO , Palamara AT , Perry G , Preston C , Pretorius E , Strandberg T , Tabet N , Taylor-Robinson SD , Whittum-Hudson JA ((2016) ) Microbes and Alzheimer’s Disease. J Alzheimers Dis 51: , 979–984. |
[199] | Poole S , Singhrao SK , Kesavalu L , Curtis MA , Crean S ((2013) ) Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J Alzheimers Dis 36: , 665–677. |
[200] | Asti A , Gioglio L ((2014) ) Can a bacterial endotoxin be a key factor in the kinetics of amyloid fibril formation? J Alzheimers Dis 39: , 169–179. |
[201] | Hauss-Wegrzyniak B , Wenk GL ((2002) ) Beta-amyloid deposition in the brains of rats chronically infused with thiorphan or lipopolysaccharide: The role of ascorbic acid in the vehicle. Neurosci Lett 322: , 75–78. |
[202] | Sheng JG , Bora SH , Xu G , Borchelt DR , Price DL , Koliatsos VE ((2003) ) Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid beta peptide in APPswe transgenic mice. Neurobiol Dis 14: , 133–145. |
[203] | Lee JW , Lee YK , Yuk DY , Choi DY , Ban SB , Oh KW , Hong JT ((2008) ) Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation 5: , 37. |
[204] | Lee YK , Yuk DY , Lee JW , Lee SY , Ha TY , Oh KW , Yun YP , Hong JT ((2009) ) (-)-Epigallocatechin-3-gallate prevents lipopolysaccharide-induced elevation of beta-amyloid generation and memory deficiency. Brain Res 1250: , 164–174. |
[205] | Spitzer P , Herrmann M , Klafki HW , Smirnov A , Lewczuk P , Kornhuber J , Wiltfang J , Maler JM ((2010) ) Phagocytosis and LPS alter the maturation state of beta-amyloid precursor protein and induce different Abeta peptide release signatures in human mononuclear phagocytes. J Neuroinflammation 7: , 59. |
[206] | Katafuchi T , Ifuku M , Mawatari S , Noda M , Miake K , Sugiyama M , Fujino T ((2012) ) Effects of plasmalogens on systemic lipopolysaccharide-induced glial activation and beta-amyloid accumulation in adult mice. Ann N Y Acad Sci 1262: , 85–92. |
[207] | Fassbender K , Walter S , Kuhl S , Landmann R , Ishii K , Bertsch T , Stalder AK , Muehlhauser F , Liu Y , Ulmer AJ , Rivest S , Lentschat A , Gulbins E , Jucker M , Staufenbiel M , Brechtel K , Walter J , Multhaup G , Penke B , Adachi Y , Hartmann T , Beyreuther K ((2004) ) The LPS receptor (CD14) links innate immunity with Alzheimer’s disease. FASEB J 18: , 203–205. |
[208] | Lee DC , Rizer J , Selenica ML , Reid P , Kraft C , Johnson A , Blair L , Gordon MN , Dickey CA , Morgan D ((2010) ) LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J Neuroinflammation 7: , 56. |
[209] | Liu Y , Walter S , Stagi M , Cherny D , Letiembre M , Schulz-Schaeffer W , Heine H , Penke B , Neumann H , Fassbender K ((2005) ) LPS receptor (CD14): A receptor for phagocytosis of Alzheimer’s amyloid peptide. Brain 128: , 1778–1789. |
[210] | Ripollés Piquer B , Nazih H , Neunlist M , Huvelin JM , Bard JM ((2004) ) Effect of LPS on basal and induced apo E secretion by 25-OH chol and 9cRA in differentiated CaCo-2. J Cell Biochem 91: , 786–795. |
[211] | Cunningham C , Wilcockson DC , Campion S , Lunnon K , Perry VH ((2005) ) Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci 25: , 9275–9284. |
[212] | Yang J , Zhao Y , Shao F ((2015) ) Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr Opin Immunol 32: , 78–83. |
[213] | Płóciennikowska A , Hromada-Judycka A , Borzęcka K , Kwiatkowska K ((2015) ) Co-operation of TLR4 and raft proteins in LPS-induced pro-inflammatory signaling. Cell Mol Life Sci 72: , 557–581. |
[214] | Jialal I , Rajamani U ((2014) ) Endotoxemia of metabolic syndrome: A pivotal mediator of meta-inflammation. Metab Syndr Relat Disord 12: , 454–456. |
[215] | Aksu G , Ozturk C , Kavakli K , Genel F , Kutukculer N ((2007) ) Hypercoagulability: Interaction between inflammation and coagulation in familial Mediterranean fever. Clin Rheumatol 26: , 366–370. |
[216] | Choi G , Schultz MJ , Levi M , van der Poll T ((2006) ) The relationship between inflammation and the coagulation system. Swiss Med Wkly 136: , 139–144. |
[217] | Cicala C , Cirino G ((1998) ) Linkage between inflammation and coagulation: An update on the molecular basis of the crosstalk. Life Sci 62: , 1817–1824. |
[218] | Levi M , van der Poll T ((2010) ) Inflammation and coagulation. Crit Care Med 38: , S26–S34. |
[219] | Petäjä J ((2011) ) Inflammation and coagulation. An overview. Thromb Res 127: (Suppl 2), S34–S37. |
[220] | Strukova S ((2006) ) Blood coagulation-dependent inflammation. Coagulation-dependent inflammation and inflammation-dependent thrombosis. Front Biosci 11: , 59–80. |
[221] | van der Poll T , de Boer JD , Levi M ((2011) ) The effect of inflammation on coagulation and vice versa. Curr Opin Infect Dis 24: , 273–278. |
[222] | Kell DB , Oliver SG ((2014) ) How drugs get into cells: Tested and testable predictions to help discriminate between transporter-mediated uptake and lipoidal bilayer diffusion. Front Pharmacol 5: , 231. |
[223] | Bartley J ((2010) ) Vitamin D: Emerging roles in infection and immunity. Expert Rev Anti Infect Ther 8: , 1359–1369. |
[224] | Zughaier SM , Alvarez JA , Sloan JH , Konrad RJ , Tangpricha V ((2014) ) The role of vitamin D in regulating the iron-hepcidin-ferroportin axis in monocytes. J Clin Transl Endocrinol 1: , 19–25. |
[225] | Annweiler C , Rolland Y , Schott AM , Blain H , Vellas B , Herrmann FR , Beauchet O ((2012) ) Higher vitamin D dietary intake is associated with lower risk of alzheimer’s disease: A 7-year follow-up. J Gerontol A Biol Sci Med Sci 67: , 1205–1211. |
[226] | Lu‘o’ng KV , Nguyen LT ((2013) ) The role of vitamin D in Alzheimer’s disease: Possible genetic and cell signaling mechanisms. Am J Alzheimers Dis Other Demen 28: , 126–136. |
[227] | Afzal S , Bojesen SE , Nordestgaard BG ((2014) ) Reduced 25-hydroxyvitamin D and risk of Alzheimer’s disease and vascular dementia. Alzheimers Dement 10: , 296–302. |
[228] | Annweiler C , Dursun E , Feron F , Gezen-Ak D , Kalueff AV , Littlejohns T , Llewellyn DJ , Millet P , Scott T , Tucker KL , Yilmazer S , Beauchet O ((2015) ) ‘Vitamin D and cognition in older adults’: Updated international recommendations. J Intern Med 277: , 45–57. |
[229] | Banerjee A , Khemka VK , Ganguly A , Roy D , Ganguly U , Chakrabarti S ((2015) ) Vitamin D and Alzheimer’s disease: Neurocognition to therapeutics. Int J Alzheimers Dis 2015: , 192747. |
[230] | Shen L , Ji HF ((2015) ) Vitamin D deficiency is associated with increased risk of Alzheimer’s disease and dementia: Evidence from meta-analysis. Nutr J 14: , 76. |
[231] | Karakis I , Pase MP , Beiser A , Booth SL , Jacques PF , Rogers G , DeCarli C , Vasan RS , Wang TJ , Himali JJ , Annweiler C , Seshadri S ((2016) ) Association of serum vitamin D with the risk of incident dementia and subclinical indices of brain aging: The Framingham Heart Study. J Alzheimers Dis 51: , 451–461. |
[232] | Cantorna MT , Yu S , Bruce D ((2008) ) The paradoxical effects of vitamin D on type 1 mediated immunity. Mol Aspects Med 29: , 369–375. |
[233] | Bordbar A , Mo ML , Nakayasu ES , Schrimpe-Rutledge AC , Kim YM , Metz TO , Jones MB , Frank BC , Smith RD , Peterson SN , Hyduke DR , Adkins JN , Palsson BØ ((2012) ) Model-driven multi-omic data analysis elucidates metabolic immunomodulators of macrophage activation. Mol Syst Biol 8: , 558. |
[234] | Proal AD , Albert PJ , Marshall TG , Blaney GP , Lindseth IA ((2013) ) Immunostimulation in the treatment for chronic fatigue syndrome/myalgic encephalomyelitis. Immunol Res 56: , 398–412. |
[235] | Mangin M , Sinha R , Fincher K ((2014) ) Inflammation and vitamin D: The infection connection. Inflamm Res 63: , 803–819. |
[236] | Proal AD , Albert PJ , Marshall TG ((2015) ), Infection and autoimmunity, Academic Press, New York. |
[237] | Waterhouse JC , Perez TH , Albert PJ ((2009) ) Reversing bacteria-induced vitamin D receptor dysfunction is key to autoimmune disease. Ann N Y Acad Sci 1173: , 757–765. |
[238] | Houghton LA , Vieth R ((2006) ) The case against ergocalciferol (vitamin D2) as a vitamin supplement. Am J Clin Nutr 84: , 694–697. |
[239] | Tripkovic L , Lambert H , Hart K , Smith CP , Bucca G , Penson S , Chope G , Hypponen E , Berry J , Vieth R , Lanham-New S ((2012) ) Comparison of vitamin D2 and vitamin D3 supplementation in raising serum 25-hydroxyvitamin D status: A systematic review and meta-analysis. Am J Clin Nutr 95: , 1357–1364. |
[240] | Norman AW ((2006) ) Minireview: Vitamin D receptor: New assignments for an already busy receptor. Endocrinology 147: , 5542–5548. |
[241] | Norman AW ((2008) ) From vitamin D to hormone D: Fundamentals of the vitamin D endocrine system essential for good health. Am J Clin Nutr 88: , 491s-499s. |
[242] | Carlberg C , Campbell MJ ((2013) ) Vitamin D receptor signaling mechanisms: Integrated actions of a well-defined transcription factor. Steroids 78: , 127–136. |
[243] | Schauber J , Dorschner RA , Coda AB , Buchau AS , Liu PT , Kiken D , Helfrich YR , Kang S , Elalieh HZ , Steinmeyer A , Zügel U , Bikle DD , Modlin RL , Gallo RL ((2007) ) Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism. J Clin Invest 117: , 803–811. |
[244] | Kongsbak M , Levring TB , Geisler C , von Essen MR ((2013) ) The vitamin d receptor and T cell function. Front Immunol 4: , 148. |
[245] | Liu PT , Stenger S , Li H , Wenzel L , Tan BH , Krutzik SR , Ochoa MT , Schauber J , Wu K , Meinken C , Kamen DL , Wagner M , Bals R , Steinmeyer A , Zugel U , Gallo RL , Eisenberg D , Hewison M , Hollis BW , Adams JS , Bloom BR , Modlin RL ((2006) ) Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 311: , 1770–1773. |
[246] | Youssef DA , Miller CW , El-Abbassi AM , Cutchins DC , Cutchins C , Grant WB , Peiris AN ((2011) ) Antimicrobial implications of vitamin D. Dermatoendocrinol 3: , 220–229. |
[247] | Fabri M , Stenger S , Shin DM , Yuk JM , Liu PT , Realegeno S , Lee HM , Krutzik SR , Schenk M , Sieling PA , Teles R , Montoya D , Iyer SS , Bruns H , Lewinsohn DM , Hollis BW , Hewison M , Adams JS , Steinmeyer A , Zugel U , Cheng G , Jo EK , Bloom BR , Modlin RL ((2011) ) Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci Transl Med 3: , 104ra102. |
[248] | Coussens AK , Martineau AR , Wilkinson RJ ((2014) ) Anti-inflammatory and antimicrobial actions of vitamin D in combating TB/HIV. Scientifica (Cairo) 2014: , 903680. |
[249] | Liu PT , Schenk M , Walker VP , Dempsey PW , Kanchanapoomi M , Wheelwright M , Vazirnia A , Zhang X , Steinmeyer A , Zugel U , Hollis BW , Cheng G , Modlin RL ((2009) ) Convergence of IL-1beta and VDR activation pathways in human TLR2/1-induced antimicrobial responses. PLoS One 4: , e5810. |
[250] | Sonawane A , Santos JC , Mishra BB , Jena P , Progida C , Sorensen OE , Gallo R , Appelberg R , Griffiths G ((2011) ) Cathelicidin is involved in the intracellular killing of mycobacteria in macrophages. Cell Microbiol 13: , 1601–1617. |
[251] | Nickel D , Busch M , Mayer D , Hagemann B , Knoll V , Stenger S ((2012) ) Hypoxia triggers the expression of human beta defensin 2 and antimicrobial activity against Mycobacterium tuberculosis in human macrophages. J Immunol 188: , 4001–4007. |
[252] | Nnoaham KE , Clarke A ((2008) ) Low serum vitamin D levels and tuberculosis: A systematic review and meta-analysis. Int J Epidemiol 37: , 113–119. |
[253] | Littlejohns TJ , Henley WE , Lang IA , Annweiler C , Beauchet O , Chaves PH , Fried L , Kestenbaum BR , Kuller LH , Langa KM , Lopez OL , Kos K , Soni M , Llewellyn DJ ((2014) ) Vitamin D and the risk of dementia and Alzheimer disease. Neurology 83: , 920–928. |
[254] | Miller JW , Harvey DJ , Beckett LA , Green R , Farias ST , Reed BR , Olichney JM , Mungas DM , DeCarli C ((2015) ) Vitamin D status and rates of cognitive decline in a multiethnic cohort of older adults. JAMA Neurol 72: , 1295–1303. |
[255] | Marshall TG ((2008) ) Vitamin D discovery outpaces FDA decision making. Bioessays 30: , 173–182. |
[256] | Nama N , Menon K , Iliriani K , Pojsupap S , Sampson M , O’Hearn K , Zhou LL , McIntyre L , Fergusson D , McNally JD ((2016) ) A systematic review of pediatric clinical trials of high dose vitamin D. Peer J 4: , e1701. |
[257] | Kearns MD , Alvarez JA , Seidel N , Tangpricha V ((2015) ) Impact of vitamin D on infectious disease. Am J Med Sci 349: , 245–262. |
[258] | Carlberg C , Seuter S , de Mello VD , Schwab U , Voutilainen S , Pulkki K , Nurmi T , Virtanen J , Tuomainen TP , Uusitupa M ((2013) ) Primary vitamin D target genes allow a categorization of possible benefits of vitamin D(3) supplementation. PLoS One 8: , e71042. |
[259] | Ryynänen J , Neme A , Tuomainen TP , Virtanen JK , Voutilainen S , Nurmi T , de Mello VD , Uusitupa M , Carlberg C ((2014) ) Changes in vitamin D target gene expression in adipose tissue monitor the vitamin D response of human individuals. Mol Nutr Food Res 58: , 2036–2045. |
[260] | Saksa N , Neme A , Ryynänen J , Uusitupa M , de Mello VD , Voutilainen S , Nurmi T , Virtanen JK , Tuomainen TP , Carlberg C ((2015) ) Dissecting high from low responders in a vitamin D3 intervention study. J Steroid Biochem Mol Biol 148: , 275–282. |
[261] | Chesney RW , Dabbagh S , Han X ((2015) ) Newer insights into the taurinuria of vitamin D deficiency: A review. Adv Exp Med Biol 803: , 651–664. |
[262] | Mizwicki MT , Norman AW ((2009) ) The vitamin D sterol-vitamin D receptor ensemble model offers unique insights into both genomic and rapid-response signaling.re. Sci Signal 2: , re4. |
[263] | Haussler MR , Jurutka PW , Mizwicki M , Norman AW ((2011) ) Vitamin D receptor (VDR)-mediated actions of 1alpha,25(OH)(2)vitamin D(3): Genomic and non-genomic mechanisms. Best Pract Res Clin Endocrinol Metab 25: , 543–559. |
[264] | Anami Y , Itoh T , Egawa D , Yoshimoto N , Yamamoto K ((2014) ) A mixed population of antagonist and agonist binding conformers in a single crystal explains partial agonism against vitamin D receptor: Active vitamin D analogues with 22R-alkyl group. J Med Chem 57: , 4351–4367. |
[265] | Nelson DE , Ihekwaba AEC , Elliott M , Gibney CA , Foreman BE , Nelson G , See V , Horton CA , Spiller DG , Edwards SW , McDowell HP , Unitt JF , Sullivan E , Grimley R , Benson N , Broomhead DS , Kell DB , White MRH ((2004) ) Oscillations in NF-kB signalling control the dynamics of gene expression. Science 306: , 704–708. |
[266] | Ashall L , Horton CA , Nelson DE , Paszek P , Ryan S , Sillitoe K , Harper CV , Spiller DG , Unitt JF , Broomhead DS , Kell DB , Rand D , Sée V , White MRH ((2009) ) Pulsatile stimulation determines timing and specificity of NFkappa-B-dependent transcription. Science 324: , 242–246. |
[267] | Szeto FL , Sun J , Kong J , Duan Y , Liao A , Madara JL , Li YC ((2007) ) Involvement of the vitamin D receptor in the regulation of NF-kappaB activity in fibroblasts. J Steroid Biochem Mol Biol 103: , 563–566. |
[268] | Wu S , Xia Y , Liu X , Sun J ((2010) ) Vitamin D receptor deletion leads to reduced level of IkappaBalpha protein through protein translation, protein-protein interaction, and post-translational modification. Int J Biochem Cell Biol 42: , 329–336. |
[269] | Chen Y , Zhang J , Ge X , Du J , Deb DK , Li YC ((2013) ) Vitamin D receptor inhibits nuclear factor kappaB activation by interacting with IkappaB kinase beta protein. J Biol Chem 288: , 19450–19458. |
[270] | Waters KM , Cummings BS , Shankaran H , Scholpa NE , Weber TJ ((2014) ) ERK oscillation-dependent gene expression patterns and deregulation by stress response. Chem Res Toxicol 27: , 1496–1503. |
[271] | Ordóñez-Morán P , Muñoz A ((2009) ) Nuclear receptors: Genomic and non-genomic effects converge. Cell Cycle 8: , 1675–1680. |
[272] | Gutierrez-Monreal MA , Cuevas-Diaz Duran R , Moreno-Cuevas JE , Scott SP ((2014) ) A role for 1alpha,25-dihydroxyvitamin d3 in the expression of circadian genes. J Biol Rhythms 29: , 384–388. |
[273] | Allan EJ , Hoischen C , Gumpert J ((2009) ) Bacterial L-forms. Adv Appl Microbiol 68: , 1–39. |
[274] | Baines AJ , Bennett PM , Carter EW , Terracciano C ((2009) ) Protein 4.1 and the control of ion channels. Blood Cells Mol Dis 42: , 211–215. |
[275] | van den Akker E , Satchwell TJ , Williamson RC , Toye AM ((2010) ) Band 3 multiprotein complexes in the red cell membrane; of mice and men. Blood Cells Mol Dis 45: , 1–8. |
[276] | Jay DG ((1996) ) Role of band 3 in homeostasis and cell shape. Cell 86: , 853–854. |
[277] | Kaestner L , Bogdanova A ((2014) ) Regulation of red cell life-span, erythropoiesis, senescence and clearance, Frontiers E-books. |
[278] | Buys AV , Van Rooy MJ , Soma P , Van Papendorp D , Lipinski B , Pretorius E ((2013) ) Changes in red blood cell membrane structure in type 2 diabetes: A scanning electron and atomic force microscopy study. Cardiovasc Diabetol 12: , 25. |
[279] | Sirachainan N , Thongsad J , Pakakasama S , Hongeng S , Chuansumrit A , Kadegasem P , Tirakanjana A , Archararit N , Sirireung S ((2012) ) Normalized coagulation markers and anticoagulation proteins in children with severe beta-thalassemia disease after stem cell transplantation. Thromb Res 129: , 765–770. |
[280] | Lang E , Qadri SM , Lang F ((2012) ) Killing me softly - Suicidal erythrocyte death. Int J Biochem Cell Biol 44: , 1236–1243. |
[281] | Lang F , Abed M , Lang E , Föller M ((2013) ) Oxidative stress and suicidal erythrocyte death. Antioxid Redox Signal 21: , 138–153. |
[282] | Lang F , Gulbins E , Lang PA , Zappulla D , Föller M ((2010) ) Ceramide in suicidal death of erythrocytes. Cell Physiol Biochem 26: , 21–28. |
[283] | Lang F , Lang E , Foller M ((2012) ) Physiology and pathophysiology of eryptosis. Transfus Med Hemother 39: , 308–314. |
[284] | Lang F , Qadri SM ((2012) ) Mechanisms and significance of eryptosis, the suicidal death of erythrocytes. Blood Purif 33: , 125–130. |
[285] | Qadri SM , Donkor DA , Bhakta V , Eltringham-Smith LJ , Dwivedi DJ , Moore JC , Pepler L , Ivetic N , Nazi I , Fox-Robichaud AE , Liaw PC , Sheffield WP ((2016) ) Phosphatidylserine externalization and procoagulant activation of erythrocytes induced by Pseudomonas aeruginosa virulence factor pyocyanin. J Cell Mol Med 20: , 710–720. |
[286] | Pretorius E , Olumuyiwa-Akeredolu OO , Mbotwe S , Bester J ((2016) ) Erythrocytes and their role as health indicator: Using structure in a patient-orientated precision medicine approach. Blood Rev. doi: 10.1016/j.blre.2016.01.001 |
[287] | Qadri SM , Mahmud H , Lang E , Gu S , Bobbala D , Zelenak C , Jilani K , Siegfried A , Foller M , Lang F ((2012) ) Enhanced suicidal erythrocyte death in mice carrying a loss-of-function mutation of the adenomatous polyposis coli gene. J Cell Mol Med 16: , 1085–1093. |
[288] | Zidova Z , Kapralova K , Koralkova P , Mojzikova R , Dolezal D , Divoky V , Horvathova M ((2014) ) DMT1-mutant erythrocytes have shortened life span, accelerated glycolysis and increased oxidative stress. Cell Physiol Biochem 34: , 2221–2231. |
[289] | Pretorius E , Bester J , Vermeulen N , Lipinski B ((2013) ) Oxidation inhibits iron-induced blood coagulation. Curr Drug Targets 14: , 13–19. |
[290] | Pretorius E , Bester J , Vermeulen N , Lipinski B , Gericke GS , Kell DB ((2014) ) Profound morphological changes in the erythrocytes and fibrin networks of patients with hemochromatosis or with hyperferritinemia, and their normalization by iron chelators and other agents. PLoS One 9: , e85271. |
[291] | Pretorius E , Lipinski B ((2012) ) Differences in morphology of fibrin clots induced with thrombin and ferric ions and its pathophysiological consequences. Heart Lung Circ 22: , 447–449. |
[292] | Pretorius E , Vermeulen N , Bester J , Lipinski B , Kell DB ((2013) ) A novel method for assessing the role of iron and its functional chelation in fibrin fibril formation: The use of scanning electron microscopy. Toxicol Mech Methods 23: , 352–359. |
[293] | Undas A , Ariëns RAS ((2011) ) Fibrin clot structure and function: A role in the pathophysiology of arterial and venous thromboembolic diseases. Arterioscler Thromb Vasc Biol 31: , e88–e99. |
[294] | Cortes-Canteli M , Paul J , Norris EH , Bronstein R , Ahn HJ , Zamolodchikov D , Bhuvanendran S , Fenz KM , Strickland S ((2010) ) Fibrinogen and beta-amyloid association alters thrombosis and fibrinolysis: A possible contributing factor to Alzheimer’s disease. Neuron 66: , 695–709. |
[295] | Cortes-Canteli M , Mattei L , Richards AT , Norris EH , Strickland S ((2015) ) Fibrin deposited in the Alzheimer’s disease brain promotes neuronal degeneration. Neurobiol Aging 36: , 608–617. |
[296] | Davalos D , Ryu JK , Merlini M , Baeten KM , Le Moan N , Petersen MA , Deerinck TJ , Smirnoff DS , Bedard C , Hakozaki H , Gonias Murray S , Ling JB , Lassmann H , Degen JL , Ellisman MH , Akassoglou K ((2012) ) Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun 3: , 1227. |
[297] | Ahn HJ , Glickman JF , Poon KL , Zamolodchikov D , Jno-Charles OC , Norris EH , Strickland S ((2014) ) A novel Abeta-fibrinogen interaction inhibitor rescues altered thrombosis and cognitive decline in Alzheimer’s disease mice. J Exp Med 211: , 1049–1062. |
[298] | Collins FS , Varmus H ((2015) ) A new initiative on precision medicine. N Engl J Med 372: , 793–795. |
[299] | Kell DB , Oliver SG ((2004) ) Here is the evidence, now what is the hypothesis? The complementary roles of inductive and hypothesis-driven science in the post-genomic era. Bioessays 26: , 99–105. |
[300] | Kell DB ((2012) ) Scientific discovery as a combinatorial optimisation problem: How best to navigate the landscape of possible experiments? Bioessays 34: , 236–244. |
[301] | Hwang D , Lee IY , Yoo H , Gehlenborg N , Cho JH , Petritis B , Baxter D , Pitstick R , Young R , Spicer D , Price ND , Hohmann JG , Dearmond SJ , Carlson GA , Hood LE ((2009) ) A systems approach to prion disease. Mol Syst Biol 5: , 252. |
[302] | Hopkins AL ((2008) ) Network pharmacology: The next paradigm in drug discovery. Nat Chem Biol 4: , 682–690. |
[303] | Kell DB ((2013) ) Finding novel pharmaceuticals in the systems biology era using multiple effective drug targets, phenotypic screening, and knowledge of transporters: Where drug discovery went wrong and how to fix it. FEBS J 280: , 5957–5980. |
[304] | Xie L , Xie L , Kinnings SL , Bourne PE ((2012) ) Novel computational approaches to polypharmacology as a means to define responses to individual drugs. Annu Rev Pharmacol Toxicol 52: , 361–379. |
[305] | Kell DB , Goodacre R ((2014) ) Metabolomics and systems pharmacology: Why and how to model the human metabolic network for drug discovery. Drug Disc Today 19: , 171–182. |
Figures and Tables
Fig.1
A mind map summarizing the content of this paper.
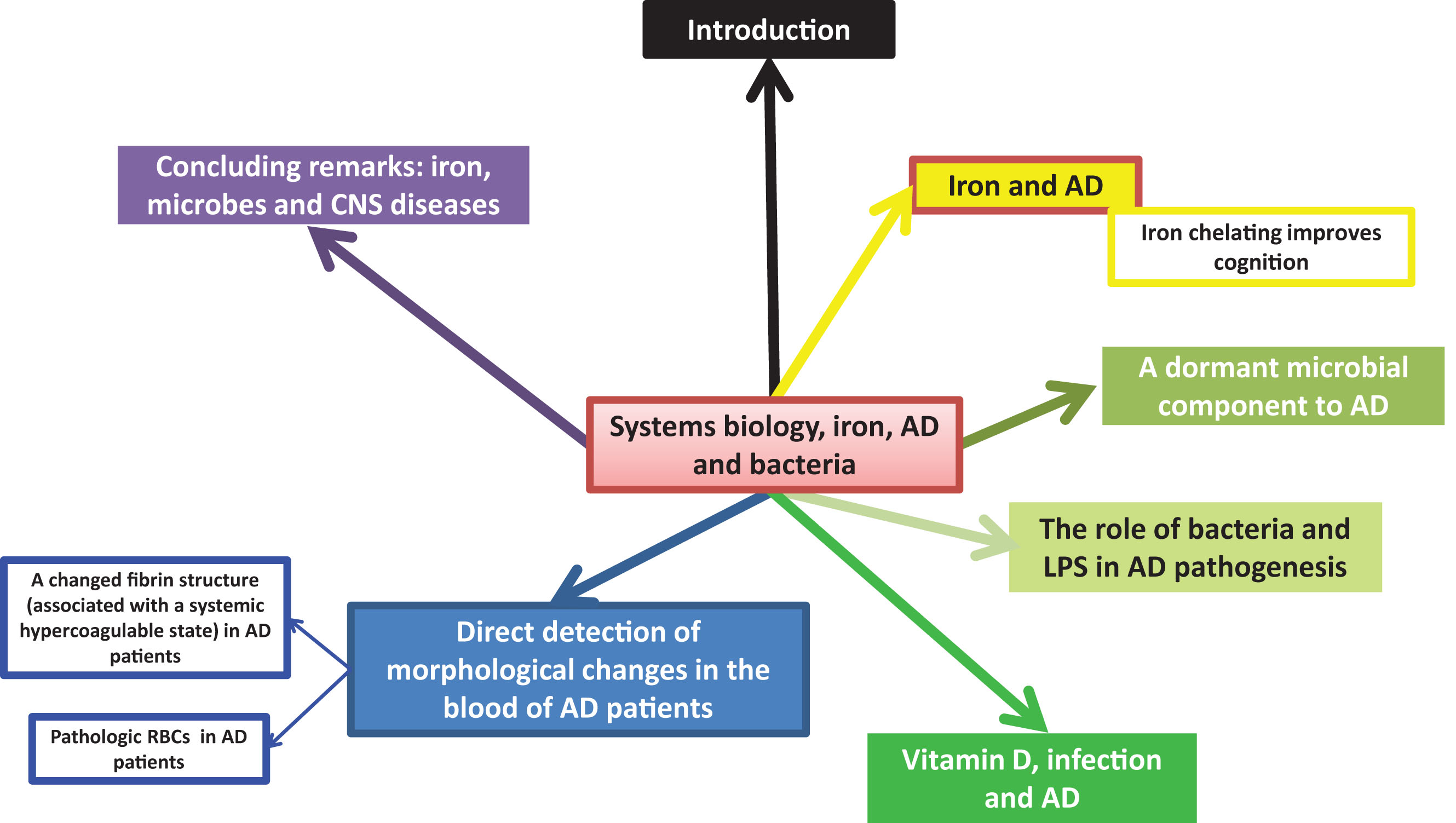
Fig.2
The order or pathway of major and potentially causative interactions in Alzheimer’s- type dementia between enzymes or biochemical elements, following a systems biology strategy.
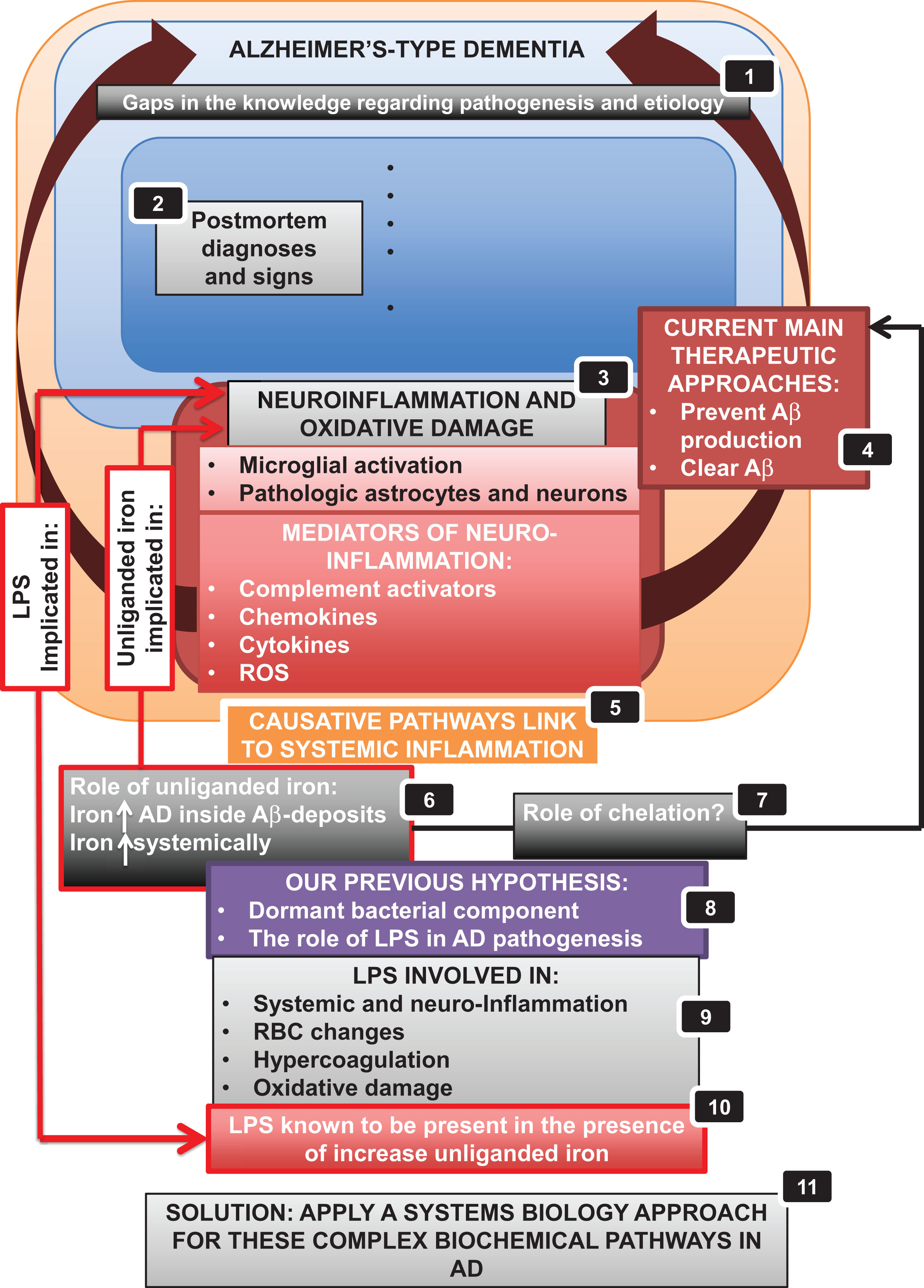
Fig.3
A generalized systems scheme for microbial/iron-driven inflammatory disease in Alzheimer’s-type dementia.
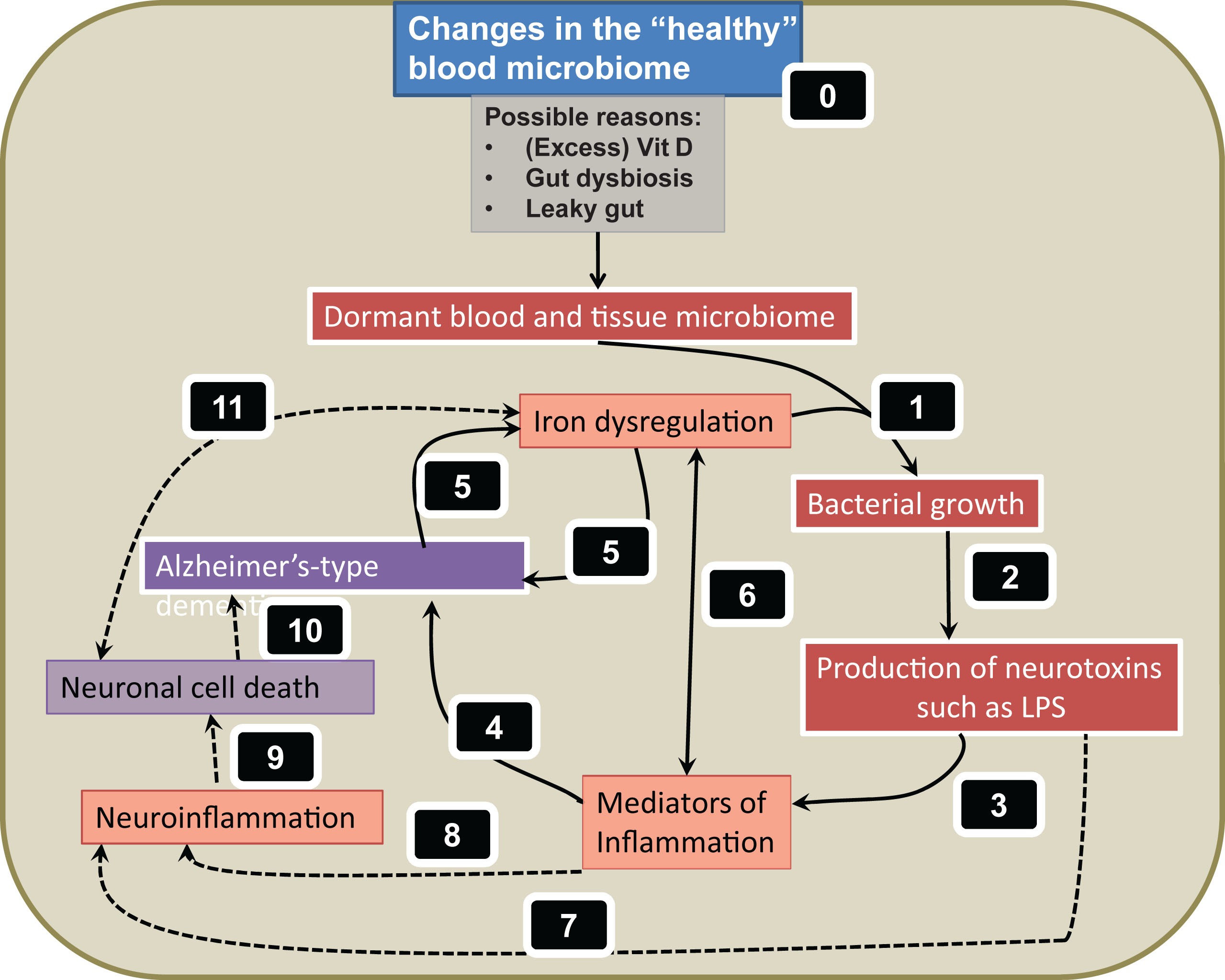
Fig.4
The structures and major metabolic products of vitamin D2/3. The dihydrohylated derivatives are by far the most active in terms of binding to the vitamin D receptor.
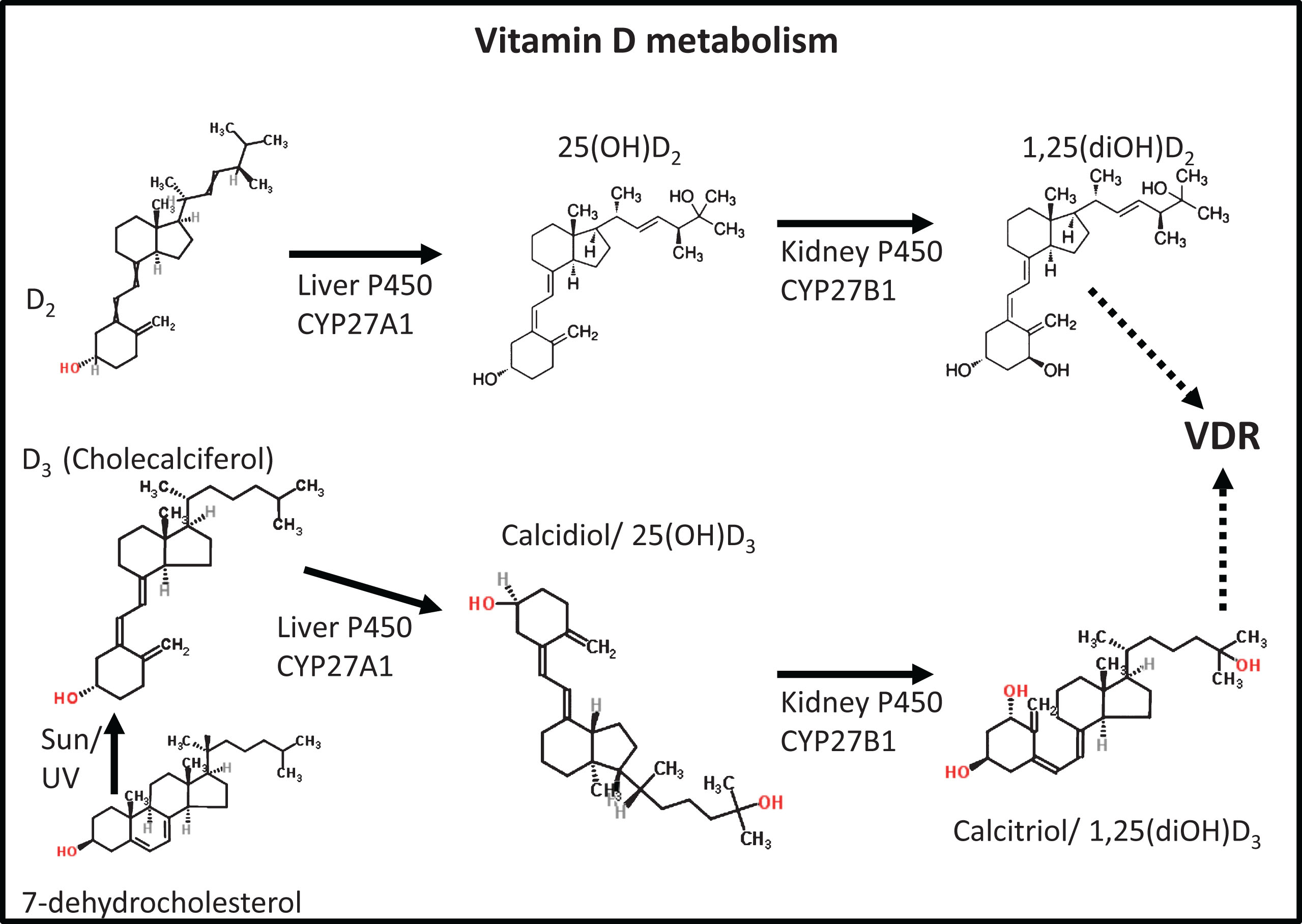
Fig.5
A general scheme of some of the roles of vitamin D and its metabolites in chronic infection: (essentially as redrawn from [235]).
![A general scheme of some of the roles of vitamin D and its metabolites in chronic infection: (essentially as redrawn from [235]).](https://content.iospress.com:443/media/jad/2016/53-4/jad-53-4-jad160318/jad-53-jad160318-g005.jpg)
Fig.6
Confocal and scanning electron microcopy (SEM) of health and Alzheimer’s-type dementia RBCs. The fluorescent markers spectrin (Ab11751) (red fluorescence) and Band-3 (Ab11012) (green fluorescence) were used in confocal microscopy. A) Confocal micrograph of a healthy RBC; B) SEM micrograph of a healthy RBC; C) Confocal micrograph of an Alzheimer’s-type dementia RBC; D) SEM micrograph of an Alzheimer’s-type dementia RBC; E) SEM micrograph showing bacteria between RBCs; and F) of bacteria and matted fibrin. Scale bar of SEM micrographs: 1μm; and for confocal: 5μm.
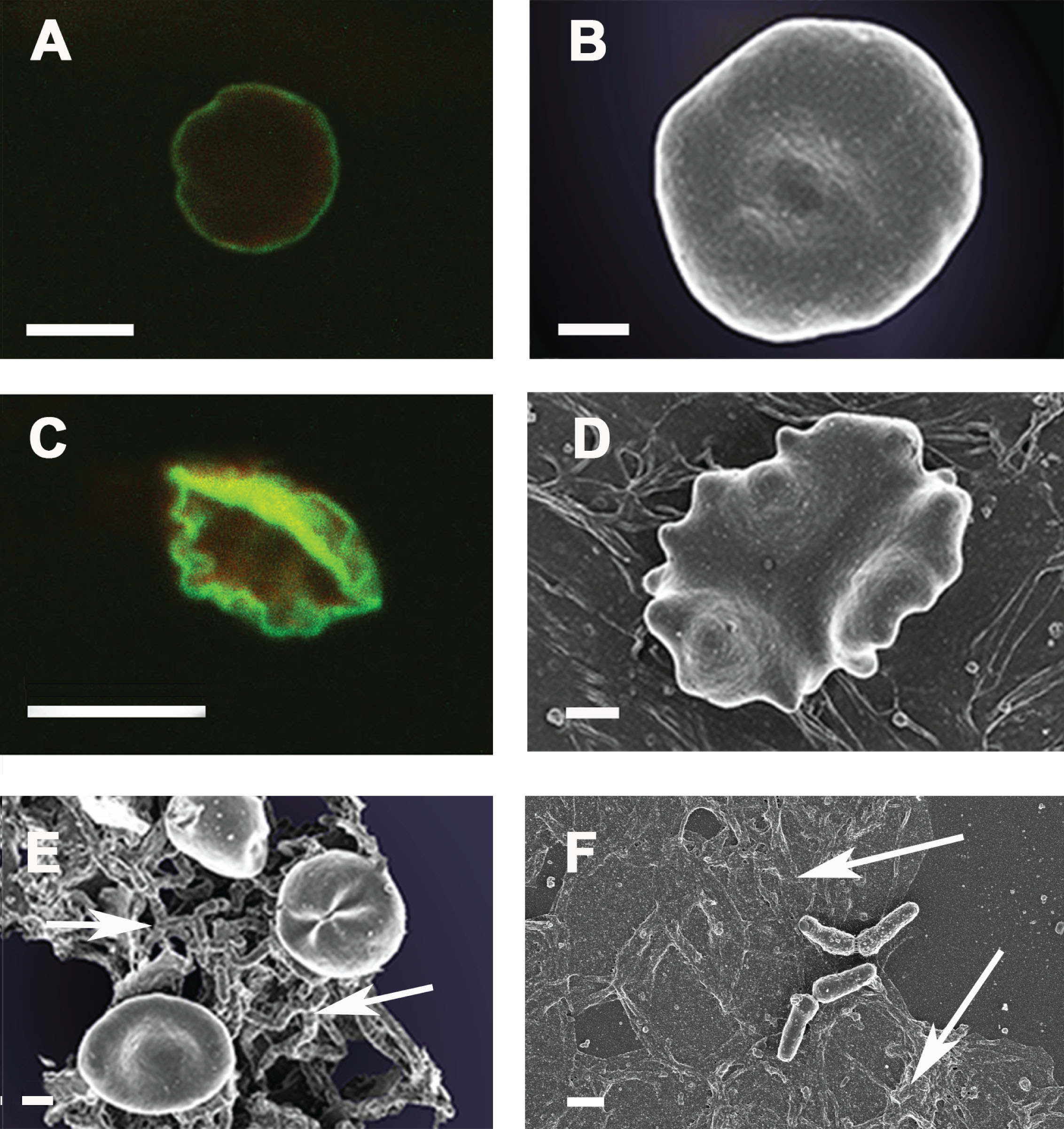
Fig.7
Platelet poor plasma of A) healthy (age-controlled) individual; and B) an Alzheimer’s-type dementia individual. Thrombin (20 U.mL–1) was added at a final concentration of 57.7 nM. Scale bar: 1μm.

Table 1
Some of the most well-known Alzheimer’s-type dementia symptoms. Some may be causative
| Most well-known (some causative) Alzheimer’s-type dementia symptoms |
| •Pathological loss of microglia, astrocytes and neurons |
| •Neurofibrillary tangles composed of hyperphosphorylated tau |
| •Cerebral amyloid-β (Aβ) or senile plaques |
| •Upregulation of complement activators, chemokines, cytokines |
| •Reactive oxygen species generation |
| •Iron dysregulation |
| •Accumulation of metals in cerebral Aβ deposits (e.g., in senile plaques) |
| •Neuroinflammation |


