
CerefolinNAC Therapy of Hyperhomocysteinemia Delays Cortical and White Matter Atrophy in Alzheimer’s Disease and Cerebrovascular Disease
Abstract
We examined whether using a medical food therapy for hyperhomocysteinemia (HHcy) in patients with Alzheimer’s disease (AD) or cognitive impairment due to cerebrovascular disease (CVD) with Cerefolin®/CerefolinNAC® (CFLN: L-methylfolate, methylcobalamin, and N-acetyl-cysteine) slowed regional brain atrophy. Thirty HHcy patients with AD and related disorders (ADRD) received CFLN (HHcy+CFLN: duration [μ ± σ] = 18.6±16.1 months); a sub-sample of this group did not receive CFLN for varying periods of time (HHcy+NoCFLN: duration [μ ± σ] = 12.6±5.6 months). Thirty-seven NoHHcy patients with ADRD did not receive CFLN (NoHHcy+NoCFLN: duration [μ ± σ] = 13.3±17.7 months). No participant took supplemental B vitamins. Regional brain volumes were measured at baseline and end of study, and covariate-adjusted rates of hippocampal, cortical, and forebrain parenchymal (includes white matter) atrophy were predicted. The HHcy+CFLN group’s hippocampal and cortical atrophy adjusted rates were 4.25 and 11.2 times slower than those of the NoHHcy+NoCFLN group (p < 0.024). The HHcy+CFLN group’s forebrain parenchyma atrophy rate was significantly slower only for CVD; the rate of slowing was proportional to the degree of homocysteine lowering (p < 0.0001). CFLN was associated with significantly slowed hippocampal and cortical atrophy rates in ADRD patients with HHcy, and forebrain parenchymal atrophy rates in CVD patients with HHcy. The present results should be further validated.
INTRODUCTION
Hyperhomocysteinemia (HHcy) is a common condition worldwide with its prevalence ranging from 5.1% to 29% in persons over 65 years of age [1–3]. HHcy is associated with a wide variety of neurological disorders including stroke, Parkinson’s disease, and multiple sclerosis [4]. Compared to subjects with normal homocysteine levels, HHcy also associates with poorer cognitive performance [5], and with relatively greater regional brain atrophy [6] and ischemic lesions (i.e., white matter hyperintensities, ischemia, or infarcts) [7]. Furthermore, the odds of brain atrophy are up to 10 times higher in HHcy patients than in those with normal homocysteine levels [8], and high, compared to normal, baseline homocysteine levels predicts moderate-severe whole brain atrophy 5 years later [9].
HHcy management with B vitamin supplementation (e.g., folic acid, B12, B6) has been previously studied for its effects on cognition and brain volume. However, the results of these studies have not been consistent [7–10]. An alternative therapy of HHcy is a prescription medical food containing L-methylfolate, methylcobalamin, and N-acetyl-cysteine (CFLN: Cerefolin®/CerefolinNAC®). CFLN is specially formulated to address metabolic imbalances of HHcy and neurovascular oxidative stress, which have been associated with cognitive impairment. Several constituents of CFLN (L-methylfolate and methylcobalamin) are the reduced, immediately bioavailable forms of folate and B12, and are therefore not chemically or biologically equivalent to folic acid and B12. In addition, L-methylfolate is the form of folate that is preferentially transported into cerebrospinal fluid [11, 12]. In contrast, folic acid is not biologically active and has been shown to competitively displace L-methylfolate binding and transport to cerebrospinal fluid [12].
Our previous studies [13, 14] have shown slower cognitive decline in patients with Alzheimer’s disease and related disorders (ADRD) who were treated for HHcy with CFLN (HHcy+CFLN) compared to those without HHcy who did not receive CFLN (NoHHcy+NoCFLN), all of whom did not receive B vitamin supplementation. The slowed cognitive decline was observed in learning and memory, constructional praxis, and visual-spatial executive function, and was more significant in patients with milder baseline severity and with CFLN treatment durations of at least one year [14]. These findings are noteworthy because a significant difference between HHcy+CFLN and NoHHcy+NoCFLN groups requires at least as large of a CFLN treatment effect than HHcy+CFLN versus HHcy+NoCFLN, because of potentially greater cognitive decline found in HHcy compared to NoHHcy [15, 16]. Managing HHcy may therefore be essential for slowing cognitive decline.
The present study examined the rate of brain atrophy using the same sample [14] as was used for the previously reported cognitive outcomes. We used the same “higher bar” of comparing HHcy+CFLN to NoHHcy+NoCFLN groups. The primary between-group measures were regional brain atrophy rates of these two groups. To demonstrate a statistically significant treatment effect, CFLN must therefore overcome the more rapid atrophy due to HHcy, plus slow atrophy beyond that of patients withoutHHcy.
METHODS
Study design
Institutional review board approval was obtained for this study. Patients were recruited from a single-site community memory clinic. Inclusion criteria were that patients had: 1) a final ADRD diagnosis based on standardized evaluation with laboratory, imaging, cognitive, and functional assessment; 2) at least one prior quantitative MRI (qMRI) study; 3) previous plasma homocysteine level measured to determine whether or not they had HHcy (in this study HHcy is defined as a plasma homocysteine level ≥ 12 umol/liter); and 4) either HHcy treated with CFLN or had no HHcy. Patients who had been taking B vitamin supplementations were excluded from the study.
Study sample
67 patients met study eligibility criteria (HHcy+CFLN: n = 30; NoHHcy+NoCFLN: n = 37). All pati-ents had an ADRD diagnosis, and were treated with a cholinesterase inhibitor (ChEI) plus memantine, which was kept unchanged over the course of the study. Each patient’s baseline qMRI volumetric data were compared to the qMRI volumetric data obtained for the present study. Each patient’s “baseline” was defined as the date of their first qMRI study. Additionally, each patient was clinically followed every 3–6 months, and assessed cognitively using the MCI Screen, CERAD Drawings, Ishihara Number Naming, Trails A and B, and F-A-S verbal fluency test. Functional severity was assessed using the Functional Assessment Staging Test (FAST) procedure. There are 7 major stages and 16 sub-stages, each with a published mean duration of the untreated course of AD [17]. For both groups, blood samples were collected for homocysteine level and genetic study of single nucleotide polymorphisms. Available longitudinal data for HHcy patients had periods with and without CFLN treatment (HHcy+NoCFLN). This allowed a within-subject analysis of HHcy patients that could evaluate the effect of HHcy with and without CFLN treatment on cognitive decline. Table 1 summarizes sample characteristics.
Quantitative MRI outcomes measures
All patients had two or more qMRIs for analysis. NeuroQuant® volumetric data from hippocampus, cortex, and forebrain parenchyma were analyzed. The term, parenchyma, includes both gray matter and white matter. The forebrain parenchyma is most relevant to cerebrovascular disease because it has a large contribution from subcortical white matter.
Analyses of treatment versus control group differences
To identify covariates to adjust for group differences between HHcy+CFLN and NoHHcy+NoCFLN groups, we performed appropriate significance tests on variables belonging to demographic, diagnostic, and severity (FAST stage) categories (non-parametric χ2 tests [Stata 13.0, tab] tests for ordinal or nominal scale variables, and multivariate mean vector tests or t-tests for ratio scale variables).
Outcome measures
CFLN treatment effect on regional brain atrophy rates was expressed as a binary variable (CFLN treatment: yes/no), and as ratio scale variables (CFLN duration: years, and CFLN/Study ratio: CFLN duration normalized by study duration). Atrophy rates (slopes) were computed as volume change per year per brain region Negative volume changes per year indicate greater rates of atrophy (tissue loss).
Analytical methods
The analyzed variables were baseline and study endpoint qMRI volume per brain region, apolipoprotein E (ApoE) genotype, CFLN, ChEI and memantine use and duration, demographics, homocysteine level, ADRD diagnosis, and FAST stage severity. Exogenous, potentially confounding variables not available for analysis were B12 and folate levels, history of developmental folate supplementation, hyperlipidemia, and alcohol dependence.
Predictor variables were represented in nominal, ordinal and ratio scales (when their precision permitted). Two-way interaction effects were assessed between treatment (CFLN, memantine, ChEI), severity (FAST stage), ADRD diagnosis, and presence of 1 or more ApoE4 alleles. Each predictor variable was included as a fixed and a random effect in a multi-level model, to determine if it significantly predicted regional atrophy rates. Non-significant fixed and random effects were removed from the model.
The first model analyzed only contained patient as a random effect (unconditional means model). The second model analyzed was a growth model, which added baseline regional qMRI volume as a fixed effect to the unconditional means model. Subsequent models added various combinations of the variables analyzed to adjust for potential confounds on the treatment variables–CFLN, ChEI, and memantine. The final models estimated the confounder-adjusted, CFLN treatment effects on the intercepts and slopes of the hippocampal, cortical and forebrain parenchymal atrophy rates (predict, lfitci, Stata 13.0). When variables measuring effects of CFLN treatment or no CFLN treatment were significant in a confounder-adjusted model, t-tests were used to determine if their difference between HHcy+CFLN and NoHHcy+NoCFLN groups was significant (Stata 13.0, ttest).
RESULTS
Sample group differences
Table 1 shows that the HHcy+CFLN group had a relatively higher proportion of cognitively impaired patients with cerebrovascular disease (CVD). Significant (p < 0.05) or borderline significant (p < 0.10) t-tests (Stata 13.0, ttest) showed that HHcy+CFLN patients were older, and had smaller baseline hippocampal, cortical, and forebrain parenchymal volumes. Due to study design, HHcy+CFLN patients also had higher baseline homocysteine levels, and larger changes in homocysteine level from baseline to study endpoint (homocysteine Δ= Endpoint – Baseline Homocysteine). 87% (26/30) of the HHcY+CFLN group took the form of CFLN containing N-acetyl-cysteine for some or all of the study period.
Predicting annual rate of hippocampal atrophy
The unconditional means model for predicting hippocampal atrophy was not significant (p = 0.16), which means that hippocampal slope is not predicted by a constant. The growth model for predicting hippocampal atrophy was also not significant (p = 0.222), which means that hippocampal slope is not predicted by baseline hippocampal volume. Further analysis with the mixed effects linear model was justified.
Table 2a shows the results of the model for predicting hippocampal atrophy rate, which was highly significant (p < 0.0001). Significant or borderline significant (p < 0.10) covariate predictors were age at baseline, education, ApoE4 alleles, and duration of ChEI and memantine treatment. Significant or borderline significant predictors of reduced hippocampal atrophy rate related to CFLN treatment were: 1) greater homocysteine lowering by CFLN (homocysteine Δ); 2) longer CFLN versus NoCFLN treatment duration; and 3) milder severity (FAST stages ≤3 compared to FAST stages ≥4). CFLN treatment duration was also a significant random effect.
Figure 1a shows the predicted mean hippocampal atrophy rates and 95% confidence bands of the HHcy+CFLN, and NoHHcy+NoCFLN groups. Initially the predicted hippocampal atrophy rate was greater (more negative) for the HHcy+CFLN group than for the NoHHcy+NoCFLN group (p = 0.0000). However, the rate of hippocampal atrophy decreased over time in the HHcy+CFLN group to be 4.25 times slower than the NoHHcy+NoCFLN group (slope = 0.42 versus 0.08 mm3/y.: p < 0.024). After 2.25 years, the hippocampal volumes of the HHcy+CFLN and NoHHcy+NoCFLN groups were similar.
Predicting annual rate of cortical atrophy
Table 2b shows the results of the hierarchical model for predicting cortical atrophy rate. Overall, the model significantly predicted cortical atrophy rate (p = 0.008), and did so significantly better than a linear regression of the same model (p = 0.039). Significant or borderline significant predictors were the presence of ApoE4 alleles, CFLN (Yes/No), and duration of CFLN. The net effect of CFLN was due to the combination of these two opposing effects. Baseline homocysteine level, as a random effect, significantly improved prediction of cortical atrophy rates for individual subjects.
Figure 1b shows the predicted mean cortical atrophy rates and 95% confidence bands of the HHcy+CFLN and NoHHcy+NoCFLN groups. Initially the cortical atrophy rate of the HHcy+CFLN group was significantly greater (more negative) than that of the NoHHcy+NoCFLN group (p = 0.045). However, the predicted cortical atrophy rate of the HHcy+CFLN group decreased to be 11.2 times slower than that of the NoHHcy+NoCFLN group (slope = 4.27 versus 0.35 mm3/y.: p < 0.0001). After 2.5 years of CFLN, the predicted cortical atrophy rates of the HHcy+CFLN and NoHHcy+NoCFLN groups were similar.
Predicting annual rate of forebrain parenchymal atrophy
Table 2c shows the predicted annual rate of forebrain parenchymal atrophy after adjustment for potential confounding covariates in the model. Overall, the model was highly significant in predicting forebrain parenchymal atrophy rate of CVD patients (p < 0.00005), with significant covariate predictors of age≥65, CVD diagnosis, ChEI treatment duration, and memantine treatment (yes/no). Duration of NoCFLN, was borderline significant, and CFLN treatment duration was significant; both increased forebrain parenchymal atrophy rates.
Post-estimation testing for CVD atrophy rate differences between HHcy+CFLN and HHcy+NoCFLN groups were significant, as were CVD atrophy rate differences between HHcy+CFLN and NoHHcy+NoCFLN groups (p < 0.0065). However, there were no significant differences between HHcy+NoCFLN and NoHHcy+NoCFLN groups (p = 0.6). An interaction between homocysteine Δ and CVD diagnosis significantly reduced forebrain parenchymal atrophy rate.
The net effect of CFLN treatment in CVD patients was examined using linear combinations of CFLN versus NoCFLN treatment duration and a 5-point lowering of homocysteine for the HHcy+CFLN group. Two years of CFLN treatment slowed forebrain parenchymal atrophy rate by 8.7 mm3/y, and four years of CFLN treatment accelerated it by –6.5 mm3/y; these rates however, were not significantly different from 0 (p > 0.4). For 2 and 4 years of NoCFLN treatment, forebrain parenchymal atrophy rates accelerated by –31.4 mm3/y and –39.5 mm3/y respectively, and were highly significant (p < 0.0005). Figure 1c shows the predicted forebrain parenchymal atrophy rates and their 95% confidence bands for the CVD patients in the HHcy+CFLN and NoHHcy+NoCFLN groups.
DISCUSSION
Primary outcomes
The main finding of the present study is that CFLN treatment of HHcy in either AD or CVD patients for 2 or more years significantly delayed cortical and hippocampal atrophy compared to no CFLN treatment in no HHcy patients. For CVD patients only, CFLN treatment of HHcy significantly slowed forebrain parenchyma atrophy rates compared to no CFLN treatment of no HHcy. The timing of the slowing of hippocampal and cortical atrophy due to CFLN is consistent with studies of the effect of B-vitamin treatment of HHcy on brain atrophy [9, 18]. Those studies found that 2 or more years of B vitamin therapy slowed brain atrophy. In a separate study, a 2-year treatment period also was needed to reduce the rate of cognitive decline in mild cognitive impairment (MCI) patients with HHcy treated with B vitamins [7]. Using causal hierarchical Bayesian networks, this study also demonstrated a causal relation between homocysteine lowering and reduced cortical atrophy, cognitive and functional impairment. Our previous study using the current study patients [14] also showed that slowing of cognitive decline required at least 1 year of CFLN therapy.
Forebrain parenchymal atrophy rates were significantly reduced in the HHcy+CFLN group only for CVD patients. CVD-induced cognitive impairment is primarily associated with microvascular disease, which reduces blood flow to white matter, and has been shown to significantly increase white matter atrophy and ischemic white matter changes in HHcy patients and in cognitively normal adults, 55 years and older, with CVD [19, 20]. Cortical atrophy is also linked to microvascular disease. For example, a longitudinal study of 276 patients with CADASIL (a genetic disorder associated with severe small vessel cerebrovascular disease) identified 9 patients who developed only a single new lacunar infarct [21]. In these 9 patients, cortical areas that send or receive axons through the locus of the infarct showed atrophy. This study supports the use of measures of cortical and white matter atrophy as an indirect measure of vascular pathology, particularly in the presence of white matter ischemia, lacunar infarcts or cerebral hemorrhages.
The selective effect of CFLN in reducing forebrain parenchymal atrophy only in HHcy patients with CVD may be due to a greater proportion of white matter in the forebrain compared to hippocampus and cortex.
The selective CFLN treatment effect on forebrain parenchymal atrophy rate in HHcy patients with CVD was proportional to the degree of homocysteine lowering (homocysteine Δ). HHcy impairs nitric oxide mediated vasodilation [22], which restricts cerebral blood flow to the brain areas furthest from the heart, (i.e., white matter and subcortical nuclei). It is therefore plausible that CFLN reduces forebrain parenchymal atrophy by lowering homocysteine, which restores normal functioning of nitric oxide mediated vasodilation, and minimizes white matter atrophy and ischemia.
Structure and functional effects of CFLN treatment in HHcy
A fundamental tenet of biology is that structure is related to function, and function is related to structure. The present study shows that sustained homocysteine lowering associates with reduced rates of hippocampal, cortical, and forebrain parenchymal brain atrophy. Our previous study of the same sample showed that homocysteine lowering associates with reduced cognitive decline in ADRD patients [14]. Conjointly, these findings support the hypothesis that the homocysteine lowering effect of CFLN reduces rates of brain atrophy and of cognitive decline in ADRD patients.
The structure-function hypothesis is that CFLN reduces brain atrophy, which reduces the rate of cognitive decline. At a coarse level, HHcy lowering associates with reduced rates of brain atrophy and less cognitive decline, after adjusting for confounding factors [23]. A causal relation between HHcy lowering and reduced rates of brain atrophy and cognitive decline in AD patients has also been inferred using causal Bayesian network analysis [7]. With regard to CVD, HHcy has been associated with forebrain parenchymal atrophy, reduced white matter volume, and slowing of cognitive processing speed [19]. These findings again support the hypothesis linking structural and cognitive changes to HHcy.
At a finer level, a number of psychiatric disorders, when untreated, associate with hippocampal atrophy plus hippocampally-mediated cognitive deficits in declarative, spatial, and contextual memory performance [24]. The present study’s findings, combined with our previous study on HHcy and cognitive decline, contribute to the body of literature supporting the hypothesis of a causal relation between HHcy lowering, reduced rates of regional brain atrophy, and slowing of cognitive decline in MCI or dementia due to AD or CVD.
Homocysteine level and treatment effect
The findings on HHcy management cannot be extrapolated to treating patients with normal homocysteine levels. There are no studies of the effects of lowering normal homocysteine levels on brain atrophy. However, one retrospective study of 37,442 patients found that hypo-homocysteinemia patients had a much higher incidence of idiopathic peripheral neuropathy [25]. There is also a widely cited, randomized, double blind trial of B-vitamin treatment, in which there was no treatment effect on cognition for patients in the study who had normal baseline homocysteine levels [26].
Other possible mechanisms underlying CFLN treatment effect
Effect of N-Acetyl-Cysteine
Some of the possible mechanisms underlying the CFLN treatment effect that are not shared by B-vitamins are mediated by N-acetyl-cysteine (NAC). Since 87% of the HHcy+CFLN group took CFLN with NAC for some or all of the study, it is useful to consider its potential influence on this study’s outcomes. NAC is a precursor of the neuronal antioxidant, glutathione, which is not found in B-vitamins. Glutathione functions to regulate immune function, reduce inflammation, minimize oxidative and nitrosative stress, maintain intracellular signaling networks, reduce cytochrome c-mediated apoptosis; promote neuron survival, and regulate the methionine cycle’s epigenetic control of gene expression [30–32]. As such, glutathione depletion is implicated in the pathophysiology of various neuroimmune disorders, including depression, chronic fatigue syndrome, Parkinson’s disease, traumatic brain injury (TBI), and possibly AD. Basic research has also shown that NAC replenishes glutathione levels to protect against hypoxia, protein misfolding, oxysterol-mediated amyloid-β 1–42 production, and apoptosis [33–36]. CFLN may therefore have different treatment effects on cognitive and structural changes in AD and CVD than the B-vitamins, because of the combined effects of CFLN on HHcy and on glutathione replenishment.
Clinical studies of the effects of NAC are limited, but support the hypothesis that NAC-mediated glutathione replenishment is clinically meaningful in AD, and some other conditions. A phase II, double blind, randomized trial of 106 AD patients compared cognitive and symptom severity outcomes for placebo versus treatment consisting of folate, alpha-tocopherol, S-adenosyl methioinine, acetyl-L-carnitine, and NAC, for 3 or 6 months, followed by open-label treatment for 6 months. There was statistically significant improvement at 3 months in a constructional praxis task (Clox), and in overall dementia severity (Dementia Severity Scale), plus no decline below baseline for all subjects during six months of the open-label phase [37]. Eighty-one US soldiers actively deployed in Iraq, who had suffered mild TBI due to blast exposure in the field, were randomized to NAC or placebo and treated for 7 days. After 7 days of NAC compared to placebo, soldiers had significantly greater resolution of TBI symptoms of dizziness, headache, hearing, loss, memory loss, sleep disturbance, and cognitive impairment [38]. Major depression is another disorder whose pathophysiology includes, but is not limited to antioxidant stress. A multicenter, double blind, randomized trial of 76 major depressive disorder patients used MR spectroscopy to measure levels of glutathione, glutamate-glutamine, N-acetyl aspartate, and myoinositol in anterior cingulate cortex during trial week 12. These results support the antioxidant effect of NAC in major depressive disorder. These studies support the hypothesis that glutathione replenishment in pathophysiologies ranging from depression to TBI to AD has clinically measurable effects. They provide one explanation for why the outcomes of studies with CFLN may differ from those with B vitamins in similar patient populations.
Possible interaction effects between CFLN, memantine, and cholinesterase inhibitors
There is evidence of CFLN interaction with memantine and ChEIs, which may confer additional beneficial effects. However, given the fact that all study patients were treated with stable doses of memantine and a ChEI, the current treatment group differences cannot be accounted for by folate-memantine or folate-ChEI interactions.
At the cellular in vitro level, memantine interacts synergistically with folate to reduce mitochondrially-mediated apoptosis by 60% compared to 10% and 22% for memantine or folate alone [39]. At the animal model level, transgenic AD mice treated with memantine combined with folate memantine and folate showed better spatial memory performance, less neuronal damage, and more upregulation of genes involved in neurogenesis, neural differentiation, memory, and neurotransmission than untreated mice, as well as mice treated with either folate or memantine alone [40]. Thus, there is basic research evidence for an interaction between folate and memantine that may contribute to reduced atrophy and less cognitive decline in AD and other disorders affected by these disease mechanisms.
Interaction effects between CFLN and ChEIs are less consistently found. One randomized, double blind, placebo controlled study of vitamins B6 and B12, and folate supplementation in mild to moderately demented AD patients who had normal B12 and folate levels at baseline, and were concomitantly treated with a ChEI found no significant group differences in ADAS-Cog or functional activities after 6 months of treatment [41]. In contrast, another randomized, double blind, placebo controlled trial of folate supplementation in 57 demented AD patients concomitantly treated with a ChEI found significant group differences in the Instrumental Activities of Daily Living and Social Behavior scores of the National Institute of Clinical Excellence scale but no group differences between Mini-Mental State Examination scores [42]. The interactions between B vitamins or NAC and ChEIs await further clarification.
Study limitations
This study has several limitations. These limitations can be addressed by: 1) conducting a larger, randomized, double blind, placebo-controlled study for at least two years; 2) collecting data on potentially confounding factors not available for the present study such as alchole intake [43], and adjusting for them; and 3) examining the effect of NAC independently of the B vitamins. Potential endogenous confounding factors for which data were collected in this study were dosing and ADRD severity.
The present study used doses of L-methylfolate and B12 that were respectively 10 and 4 times larger than the folate and B12 doses used in other regional brain volumetric studies of HHcy treatment with B vitamins [6, 7, 44]. Whether such dosing difference could significantly affect regional brain atrophy rates in HHcy-treated patients is not known and should be studied. There are no studies on the effect of cognitive severity and HHcy on brain atrophy, but there are previous studies of the effects of B vitamin treatment of HHcy on cognition. In one randomized trial of mild to moderately demented AD with HHcy, folate and B12 supplementation for 2 years had no effect on specific cognitive tasks but showed a significant effect on global cognition [45]. Another study showed that HHcy treatment with B-vitamins for 2 years improved cognition more in milder severity patients [7]. The most extreme example of the impact of severity on HHcy response was reported among asymptomatic, untreated HHcy females in midlife; their late life ADRD risk assessed 35 years later was increased by 1.7-fold [46].
CFLN management of HHcy in cognitively impaired patients significantly reduced rate of hippocampal and cortical atrophy, and significantly reduced rate of forebrain parenchymal atrophy in CVD. These findings parallel the cognitive findings in the same cohort of ADRD patients with HHcy managed with CFLN and retrospectively compared to NoHHcy+NoCFLN controls. CFLN contains distinct bioactive forms of B vitamins in addition to N-acetyl-cysteine, which may contribute to the treatment effect in HHcy ADRD patients. These results warrant further investigation of CFLN in managing HHcy in a variety of cognitive disorders for at least two years.
ACKNOWLEDGMENTS
The authors thank Mr. Tushar Mangrola from Medical Care Corporation for his database support and Mr. Ali Ozgur from Department of Biology, University of California at Irvine for his assistance in preparing the manuscript. This study was sponsored by Nestlé Health Science - Pamlab, Inc. The sponsor contributed to the study design (Mrs. Barrentine and Mrs. Curole) but had no role in data collection, analysis or manuscript preparation.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-0241r1).
REFERENCES
[1] | MacFarlane AJ , Greene-Finestone LS , Shi Y ((2011) ) Vitamin B-12 and homocysteine status in a folate-replete population: Results from the Canadian Health Measures Survey. Am J Clin Nutr 94: , 1079–1087. |
[2] | Selhub J , Jacques PF , Bostom AG , Wilson PW , Rosenberg IH ((2000) ) Relationship between plasma homocysteine and vitamin status in the Framingham study population. Impact of folic acid fortification. Public Health Rev 28: , 117–145. |
[3] | Wong YY , Almeida OP , McCaul KA , Yeap BB , Hankey GJ , Flicker L ((2013) ) Homocysteine, frailty, and all-cause mortality in older men: The Health in Men Study. J Gerontol A Biol Sci Med Sci 68: , 590–598. |
[4] | Ansari R , Mahta A , Mallack E , Luo JJ ((2014) ) Hyperhomocysteinemia and neurologic disorders: A review. J Clin Neurol 10: , 281–288. |
[5] | Jochemsen HM , Kloppenborg RP , de Groot LC , Kampman E , Mali WP , van der Graaf Y , Geerlings MI , Study SMART,Group ((2013) ) Homocysteine, progression of ventricular enlargement, and cognitive decline: The Second Manifestations of ARTerial disease-Magnetic Resonance study. Alzheimers Dement 9: , 302–309. |
[6] | Gallucci M , Zanardo A , Bendini M , Di Paola F , Boldrini P , Grossi E ((2014) ) Serum folate, homocysteine, brain atrophy, and auto-CM system: The Treviso Dementia (TREDEM) study. J Alzheimers Dis 38: , 581–587. |
[7] | Douaud G , Refsum H , de Jager CA , Jacoby R , Nichols TE , Smith SM , Smith AD ((2013) ) Preventing Alzheimer’s disease-related gray matter atrophy by B-vitamin treatment. Proc Natl Acad Sci U S A 110: , 9523–9528. |
[8] | Yang LK , Wong KC , Wu MY , Liao SL , Kuo CS , Huang RF ((2007) ) Correlations between folate, B12, homocysteine levels, and radiological markers of neuropathology in elderly post-stroke patients. J Am Coll Nutr 26: , 272–278. |
[9] | Blasko I , Hinterberger M , Kemmler G , Jungwirth S , Krampla W , Leitha T , Heinz Tragl K , Fischer P ((2007) ) Conversion from mild cognitive impairment to dementia: Influence of folic acid and vitamin B12 use in the VITA cohort. J Nutr Health Aging 16: , 687–694. |
[10] | de Lau LM , Refsum H , Smith AD , Johnston C , Breteler MM ((2007) ) Plasma folate concentration and cognitive performance: Rotterdam Scan Study. Am J Clin Nutr 86: , 728–734. |
[11] | Levitt M , Nixon PF , Pincus JH , Bertino JR ((1971) ) Transport characteristics of folates in cerebrospinal fluid; a study utilizing doubly labeled 5-methyltetrahydrofolate and 5-formyltetrahydrofolate. J Clin Invest 50: , 1301–1308. |
[12] | Smith I , Hyland K , Kendall B ((1985) ) Clinical role of pteridine therapy in tetrahydrobiopterin deficiency. J Inherit Metab Dis 8: , 39–45. |
[13] | Shankle WR , Hara J , Rafii MS , Russell J ((2013) ) Impact of hyperhomocysteinemia treatment on cognitive decline due to Alzheimer’s disease and related disorders. J Aging Res Clin Pract 2: , 319–324. |
[14] | Hara J , Shankle WR , Barrentine LW , Curole MV ((2016) ) Novel therapy of hyperhomocysteinemia in mild cognitive impairment, Alzheimer’s disease, and other dementing disorders. J Nutr Health Aging 10.1007/s12603-016-0688-z |
[15] | Dufouil C , Alpérovitch A , Ducros V , Tzourio C ((2003) ) Homocysteine, white matter hyperintensities, and cognition in healthy elderly people. Ann Neurol 53: , 214–221. |
[16] | Sachdev P ((2004) ) Homocysteine, cerebrovascular disease and brain atrophy. J Neurol Sci 226: , 25–29. |
[17] | Reisberg B ((1986) ) Dementia: A systematic approach to identifying reversible causes. Geriatrics 41: , 30–46. |
[18] | de Jager CA , Oulhaj A , Jacoby R , Refsum H , Smith AD ((2012) ) Cognitive and clinical outcomes of homocysteine-lowering B-vitamin treatment in mild cognitive impairment: A randomized controlled trial. Int J Geriatr Psychiatry 27: , 592–600. |
[19] | Feng L , Isaac V , Sim S , Ng TP , Krishnan KR , Chee MW ((2013) ) Associations between elevated homocysteine, cognitive impairment, and reduced white matter volume in healthy old adults. Am J Geriatr Psychiatry 21: , 164–172. |
[20] | Kloppenborg RP , Geerlings MI , Visseren FL , Mali WP , Vermeulen M , van der Graaf Y , Nederkoorn PJ , Study SMART, Group ((2014) ) Homocysteine and progression of generalized small-vessel disease: The SMART-MR Study. Neurology 82: , 777–783. |
[21] | Duering M , Righart R , Csanadi E , Jouvent E , Hervé D , Chabriat H , Dichgans M ((2012) ) Incident subcortical infarcts induce focal thinning in connected cortical regions. Neurology 79: , 2025–2028. |
[22] | Doshi S , McDowell I , Goodfellow J , Stabler S , Boger R , Allen R , Newcombe R , Lewis M , Moat S ((2005) ) Relationship between S-adenosylmethionine, S-adenosylhomocysteine, asymmetric dimethylarginine, and endothelial function in healthy human subjects during experimental hyper- and hypohomocysteinemia. Metabolism 54: , 351–360. |
[23] | de Jager CA ((2014) ) Critical levels of brain atrophy associated with homocysteine and cognitive decline. Neurobiol Aging 35: , 35–39. |
[24] | McEwen BS ((2001) ) Plasticity of the hippocampus: Adaptation to chronic stress and allostatic load. Ann N Y Acad Sci 933: , 265–277. |
[25] | Cullen CE , Carter GT , Weiss MD , Grant PA , Saperstein DS ((2012) ) Hypohomocysteinemia: A potentially treatable cause of peripheral neuropathology? Phys Med Rehabil Clin N Am 23: , 59–65. |
[26] | Aisen PS , Schneider LS , Sano M , Diaz-Arrastia R , van Dyck CH , Weiner MF , Bottiglieri T , Jin S , Stokes KT , Thomas RG , Thal LJ , Alzheimer Disease Cooperative, study ((2008) ) High-dose B vitamin supplementation and cognitive decline in Alzheimer disease: A randomized controlled trial. JAMA 300: , 1774–1783. |
[27] | Chan A , Tchantchou F , Graves V , Rozen R , Shea TB ((2008) ) Dietary and genetic compromise in folate availability reduces acetylcholine, cognitive performance and increases aggression: Critical role of S-adenosyl methionine. J Nutr Health Aging 12: , 252–261. |
[28] | Hasselmo ME ((2006) ) The role of acetylcholine in learning and memory. Curr Opin Neurobiol 16: , 710–715. |
[29] | Okada K , Tanaka H , Temporin K , Okamoto M , Kuroda Y , Moritomo H , Murase T , Yoshikawa H ((2011) ) Akt/mammalian target of rapamycin signaling pathway regulates neurite outgrowth in cerebellar granule neurons stimulated by methylcobalamin. Neurosci Lett 495: , 201–204. |
[30] | Morris G , Anderson G , Dean O , Berk M , Galecki P , Martin-Subero M , Maes M ((2014) ) The glutathione system: A new drug target in neuroimmune disorders. Mol Neurobiol 50: , 1059–1084. |
[31] | ((2000) ) N-acetylcysteine. Altern Med Rev 5: , 467–471. |
[32] | ((2001) ) Glutathione, reduced. (GSH). Altern Med Rev 6: , 601–607. |
[33] | Jayalakshmi K , Sairam M , Singh SB , Sharma SK , Ilavazhagan G , Banerjee PK ((2005) ) Neuroprotective effect of N-acetyl cysteine on hypoxia-induced oxidative stress in primary hippocampal culture. Brain Res 1046: , 97–104. |
[34] | Otte DM , Sommersberg B , Kudin A , Guerrero C , Albayram O , Filiou MD , Frisch P , Yilmaz O , Drews E , Turck CW , Bilkei-Gorzó A , Kunz WS , Beck H , Zimmer A ((2011) ) N-acetyl cysteine treatment rescues cognitive deficits induced by mitochondrial dysfunction in G72/G30 transgenic mice. Neuropsychopharmacology 36: , 2233–2243. |
[35] | Rideau Batista Novais A , Guiramand J , Cohen-Solal C , Crouzin N , de Jesus Ferreira MC , Vignes M , Barbanel G , Cambonie G ((2013) ) N-acetyl-cysteine prevents pyramidal cell disarray and reelin-immunoreactive neuron deficiency in CA3 after prenatal immune challenge in rats. Pediatr Res 73: , 750–755. |
[36] | Unnithan AS , Choi HJ , Titler AM , Posimo JM , Leak RK ((2012) ) Rescue from a two hit, high-throughput model of neurodegeneration with N-acetyl cysteine. Neurochem Int 61: , 356–368. |
[37] | Remington R , Bechtel C , Larsen D , Samar A , Doshanjh L , Fishman P , Luo Y , Smyers K , Page R , Morrell C , Shea TB ((2015) ) A phase II randomized clinical trial of a nutritional formulation for cognition and mood in Alzheimer’s disease. J Alzheimers Dis 45: , 395–405. |
[38] | Hoffer ME , Balaban C , Slade MD , Tsao JW , Hoffer B ((2013) ) Amelioration of acute sequelae of blast induced mild traumatic brain injury by N-acetyl cysteine: A double-blind, placebo controlled study. PLoS One 8: , e54163. |
[39] | Chen TF , Tang MC , Chou CH , Chiu MJ , Huang RF ((2013) ) Dose-dependent folic acid and memantine treatments promote synergistic or additive protection against Aβ(25-35) peptide-induced apoptosis in SH-SY5Y cells mediated by mitochondria stress-associated death signals. Food Chem Toxicol 62: , 538–547. |
[40] | Chen TF , Huang RF , Lin SE , Lu JF , Tang MC , Chiu MJ ((2010) ) Folic Acid potentiates the effect of memantine on spatial learning and neuronal protection in an Alzheimer’s disease transgenic model. J Alzheimers Dis 20: , 607–615. |
[41] | Sun Y , Lu CJ , Chien KL , Chen ST , Chen RC ((2007) ) Efficacy of multivitamin supplementation containing vitamins B6 and B12 and folic acid as adjunctive treatment with a cholinesterase inhibitor in Alzheimer’s disease: A 26-week, randomized, double-blind, placebo-controlled study in Taiwanese patients. Clin Ther 29: , 2204–2214. |
[42] | Connelly PJ , Prentice NP , Cousland G , Bonham J ((2008) ) A randomised double-blind placebo-controlled trial of folic acid supplementation of cholinesterase inhibitors in Alzheimer’s disease. Int J Geriatr Psychiatry 23: , 155–160. |
[43] | Bleich S , Bandelow B , Javaheripour K , Müller A , Degner D , Wilhelm J , Havemann-Reinecke U , Sperling W , Rüther E , Kornhuber J ((2003) ) Hyperhomocysteinemia as a new risk factor for brain shrinkage in patients with alcoholism. Neurosci Lett 335: , 179–182. |
[44] | Smith AD , Smith SM , de Jager CA , Whitbread P , Johnston C , Agacinski G , Oulhaj A , Bradley KM , Jacoby R , Refsum H ((2010) ) Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: A randomized controlled trial. PLoS One 5: , e12244. |
[45] | van der Zwaluw NL , Dhonukshe-Rutten RA , van Wijngaarden JP , Brouwer-Brolsma EM , van de Rest O , In ‘t Veld PH , Enneman AW , van Dijk SC , Ham AC , Swart KM , van der Velde N , van Schoor NM , van der Cammen TJ , Uitterlinden AG , Lips P , Kessels RP , de Groot LC ((2014) ) Results of 2-year vitamin B treatment on cognitive performance: Secondary data from an RCT. Neurology 83: , 2158–2166. |
[46] | Zylberstein DE , Lissner L , Björkelund C , Mehlig K , Thelle DS , Gustafson D , Ostling S , Waern M , Guo X , Skoog I ((2011) ) Midlife homocysteine and late-life dementia in women. A prospective population study. Neurobiol Aging 32: , 380–386. |
Figures and Tables
Fig.1
Predicted rates of regional brain atrophy. Rate of regional brain volume change plotted against years after baseline qMRI study for HHcy+CFLN, and NoHHcy+NoCFLN groups for (a) hippocampus, (b) cortex, and (c) forebrain parenchyma in CVD patients. The covariate adjusted volume change rates were estimated from the final predictive models, and the mean slopes and 95% confidence bands were computed (Stata 13.0: xtmixed, postestimation predict xb, lfitci). For (c), CVD patients in the HYN+CFLN group had significantly reduced rates of forebrain parenchymal volume change compared to CVD patients in the NoHHcy+NoCFLN group.
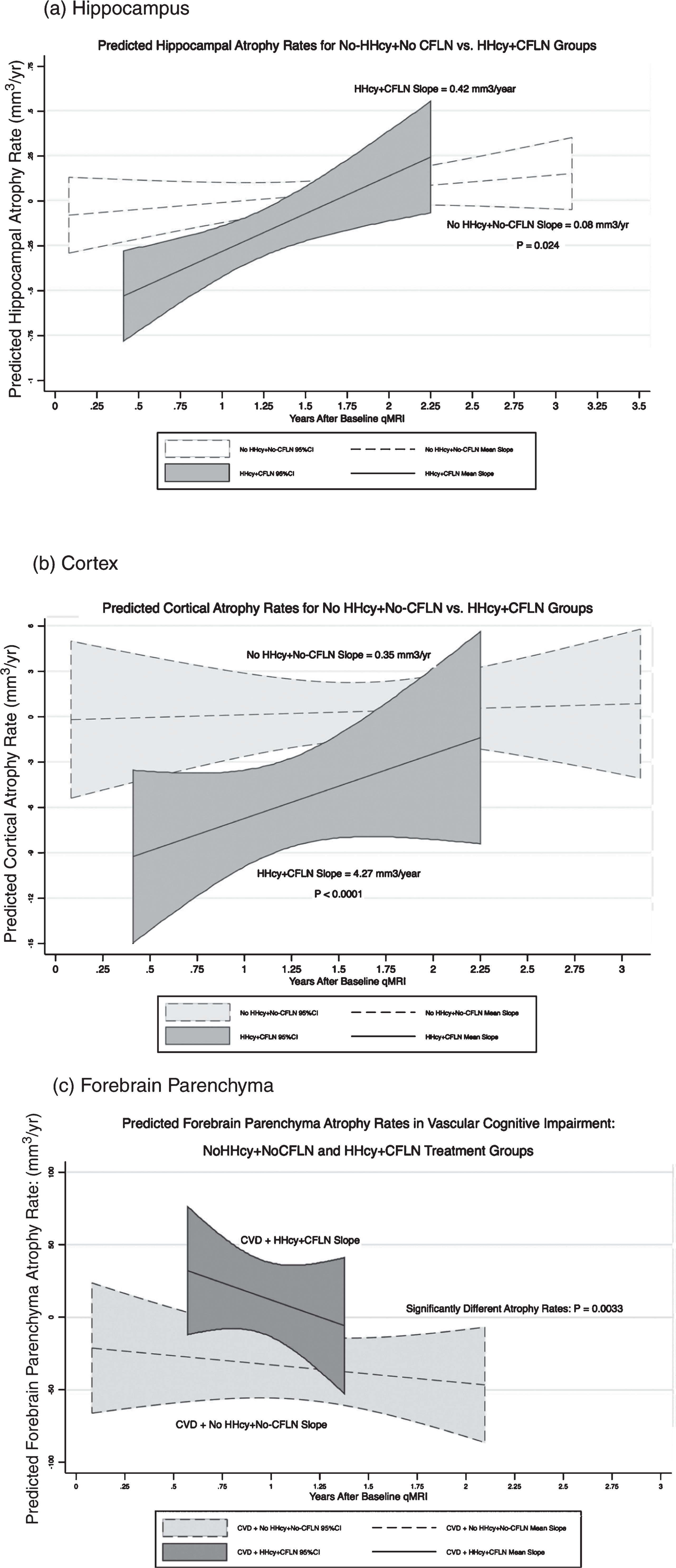
Table 1
Sample characteristics
| Sample Parameter | NoHHcy+NoCFLN | HHcy+CFLN | p |
| Total n | 37 | 30 | – |
| No. of Females: n (%)1 | 24 (65%) | 20 (63%) | 0.839 |
| ≥1 ApoE4 Alleles: n (%)1 | 16 (43%) | 16 (50%) | 0.575 |
| Age at Baseline (μ ± σ)2 | 71.6±8.7 | 76.0±10.1 | 0.064* |
| Years Education (μ ± σ)2 | 16.4±2.5 | 15.7±2.17 | – |
| Severity: n (% Demented)1 | 20 (54%) | 17 (53%) | 0.938 |
| Pure or Mixed AD: n (%)1 | 20 (54%) | 16 (50%) | 0.737 |
| Pure or Mixed CVD: n (%)1 | 6 (16%) | 12 (38%) | 0.045** |
| ADRD not due to AD or CVD: n (%)1 | 13 (35%) | 8 (25%) | 0.362 |
| Baseline MCI Screen (μ ± σ)2 | 46.2±12.9 | 43.1±13.1 | 0.318 |
| Baseline Hippocampal Vol. (μ ± σ)2 | 6.8±0.7 | 6.4±0.8 | 0.044** |
| Baseline Cortical Vol. (μ ± σ)2 | 449.7±41.7 | 428.6±49.5 | 0.068* |
| Baseline FB Parenchyma Vol. (μ ± σ)2 | 955.3±108.7 | 891.3±98.1 | 0.014** |
| Baseline Homocysteine (μ ± σ) (μ ± σ)2 | 8.95±2.66 | 11.43±4.07 | 0.005** |
| Change from Baseline Homocysteine (μ ± σ)2 | –0.56±1.89 | –2.95±3.97 | 0.003** |
| Study Duration (y) (μ ± σ)2 | 1.63±0.63 | 1.37±0.72 | 0.111 |
| CFLN Duration (y) (μ ± σ)2 | 0.0±0.0 | 1.06±1.07 | – |
| Memantine Duration (y) (μ ± σ)2 | 0.88±0.79 | 0.85±1.00 | – |
| ChEI Duration (y) (μ ± σ)2 | 1.39±2.30 | 0.85±1.27 | 0.220 |
1χ2 association using Fisher’s exact test. 2Multivariate means test: Likelihood Ratio χ2(10) = 18.75, p = 0.046. –Not Tested. **p < 0.05. *p < 0.10. HHcy, hyperhomocysteinemia; CFLN, Cerefolin®/CerefolinNAC®; AD, Alzheimer’s disease; CVD, cerebrovascular disease; MCI, mild cognitive impairment; ChEI, cholinesterase inhibitor.
Table 2
Mixed effects model predicting annual rate of atrophy
| (a) Hippocampus | |||
| Fixed Effect Predictor | Coef. | SEM | p> |z| |
| Baseline Age (y) | –0.003 | 0.001 | 0.061* |
| Male | 0.057 | 0.060 | 0.344 |
| Years of Education | 0.078 | 0.041 | 0.055* |
| 1 or 2 ApoE4 Alleles | –0.147 | 0.055 | 0.007** |
| Duration of ChEI (y) | 0.041 | 0.017 | 0.013* |
| Duration of Memantine (y) | –0.065 | 0.037 | 0.081** |
| No HHcy &NoCFLN &Demented | –0.032 | 0.069 | 0.642 |
| HHcy &CFLN &Not Demented | 0.049 | 0.140 | 0.724 |
| HHcy &CFLN &Demented | –0.263 | 0.115 | 0.022** |
| Homocysteine Δ | 0.033 | 0.011 | 0.002** |
| Duration of NoCFLN (y) | 0.060 | 0.026 | 0.019** |
| Duration of CFLN (y) | 0.130 | 0.075 | 0.083* |
| Random Effect (std. dev.) | |||
| Duration of CFLN (y) | 0.176 | 0.058 | –– |
| Residual (std. dev.) | 0.193 | 0.021 | –– |
ChEI, cholinesterase inhibitor; HHcy, hyperhomocysteinemia; CFLN, Cerefolin®/CerefolinNAC®; Coeff., fixed or random effect coefficient; SEM, standard error of the mean; Std. Dev., standard deviation; p> |z|, p-value. *p≤0.10. **p≤0.05. Log likelihood = 5.103. Wald χ2(12) = 98.37. Prob > χ2 = 0.0000. LR test versus linear regression: χ2(3) = 7.72. Prob > χ2 = 0.003.
| (b) Cortex | |||
| Fixed Effect Predictor | Coef. | SEM | p> |z| |
| Age >65 Years Old | –3.452 | 4.633 | 0.456 |
| Male | 2.715 | 3.00 | 0.365 |
| Education >12 Years | 1.554 | 4.224 | 0.713 |
| 1 or 2 ApoE4 Alleles | –7.683 | 2.790 | 0.006** |
| AD Diagnosis | –2.849 | 3.909 | 0.466 |
| ADRD not due to AD or CVD | –5.110 | 4.133 | 0.216 |
| Baseline Homocysteine Level | 0.626 | 0.438 | 0.153 |
| CFLN (Yes/No) | –10.360 | 3.793 | 0.006** |
| Duration of CFLN (y) | 2.998 | 1.743 | 0.085* |
| Duration of NoCFLN (y) | 0.764 | 1.206 | 0.526 |
| it Random Effect Predictor (std. dev.) | |||
| Baseline Homocysteine Level | 0.625 | 0.192 | –– |
| Residual Error | 8.641 | 1.446 | –– |
AD, Alzheimer’s disease; CVD, cerebrovascular disease; CFLN, Cerefolin®/CerefolinNAC®; Coeff., fixed or random effect coefficient; SEM, standard error of the mean; Std. Dev., standard deviation; p> |z|, p-value. *p≤0.10. **p≤0.05. Log likelihood = –254.0. Prob > χ2 = 0.008. Wald χ2(7) = 23.75. LR test versus linear regression: χ2(01) = 3.13. Prob > =χ2 = 0.039.
| (c) Forebrain parenchyma | |||
| Fixed Effect Predictor | Coef. | SEM | p> |z| |
| Age Category | 5.051 | 2.494 | 0.043** |
| CVD Diagnosis | –23.349 | 6.432 | 0.000** |
| ADRD not due to CVD & Homocysteine Δ | 0.689 | 0.799 | 0.388 |
| CVD Diagnosis &Homocysteine Δ | –9.444 | 1.747 | 0.000** |
| Duration of ChEI (y) | –4.190 | 1.400 | 0.003** |
| Memantine Use (Yes/No) | –15.705 | 7.162 | 0.028** |
| Duration of NoCFLN (y) | –4.042 | 2.340 | 0.084* |
| Duration of CFLN (y) | –7.584 | 2.955 | 0.010** |
| Residual (std. dev.) | 17.962 | 1.552 | –– |
Age Category: 1 = “<51”, 2 = “51-64”, 3 = “65-76”, 4 = “>76”; CVD, cerebrovascular disease; ChEI, cholinesterase inhibitor; CFLN, Cerefolin®/CerefolinNAC®; Coeff., fixed or random effect coefficient; SEM, standard error of the mean; Std. Dev., standard deviation; p> |z|, p-value. *p≤0.10. **p≤0.05. Log likelihood = –288.1. Prob > χ2 = 0.0000. Wald χ2(7) = 98.84.


