
Rare Genetic Variant in SORL1 May Increase Penetrance of Alzheimer’s Disease in a Family with Several Generations of APOE-ɛ4 Homozygosity
Abstract
Background: The major genetic risk factor for late onset Alzheimer’s disease (AD) is the APOE-ɛ4 allele. However, APOE-ɛ4 homozygosity is not fully penetrant, suggesting co-occurrence of additional genetic variants.
Objective: To identify genetic factors that, next to APOE-ɛ4 homozygosity, contribute to the development of AD.
Methods: We identified a family with nine AD patients spanning four generations, with an inheritance pattern suggestive of autosomal dominant AD, with no variants in PSEN1, PSEN2, or APP. We collected DNA from four affected and seven unaffected family members and performed exome sequencing on DNA from three affected and one unaffected family members.
Results: All affected family members were homozygous for the APOE-ɛ4 allele. Statistical analysis revealed that AD onset in this family was significantly earlier than could be expected based on APOE genotype and gender. Next to APOE-ɛ4 homozygosity, we found that all four affected family members carried a rare variant in the VPS10 domain of the SORL1 gene, associated with AβPP processing and AD risk. Furthermore, three of four affected family members carried a rare variant in the TSHZ3 gene, also associated with AβPP processing. Affected family members presented between 61 and 74 years, with variable presence of microbleeds/cerebral amyloid angiopathy and electroencephalographic abnormalities.
Conclusion: We hypothesize that next to APOE-ɛ4 homozygosity, impaired SORL1 protein function, and possibly impaired TSHZ3 function, further disturbed Aβ processing. The convergence of these genetic factors over several generations might clarify the increased AD penetrance and the autosomal dominant-like inheritance pattern of AD as observed in this family.
INTRODUCTION
Alzheimer’s disease (AD) is a complex and heterogeneous neurodegenerative disease. AD incidence increases with age, and about one third of the population aged 85 years and older is estimated to have AD [1]. AD is typically characterized by deficits in short-term memory, language, praxis, and visuospatial and executive functioning, eventually resulting in global cognitive decline. Despite intense research during past decades, the exact causes of AD are not yet understood. The leading hypothesis of AD pathogenesis is the amyloid cascade hypothesis, which proposes that aberrant processing of the amyloid-β protein precursor (AβPP) leads to increased production of amyloid-β (Aβ) peptide in the brain cells (reviewed in [2]). In turn, Aβ peptides are misfolded and accumulate into protein aggregates, ultimately leading to the formation of neurotoxic amyloid plaques that disrupt normal cellular processes. Genetic mutations in autosomal dominant AD are detected in genes involved in Aβ processing: the amyloid precursor protein (APP), which is the source of Aβ, and the presenilins (PSEN1 and PSEN2) involved in AβPP-processing[3–5].
Twin studies estimated that ∼60–80% of late onset AD risk is heritable with the remainder being environmental (LOAD, age at onset >65 years) [6]. By far the most important susceptibility gene for late onset AD is the Apolipoprotein E (APOE) gene [7]. Next to functions related with lipid and cholesterol processing, the protein product of the APOE gene, ApoE, is suggested to be involved in the clearance of Aβ from the brain (reviewed in [2]). The APOE gene contains three common allelic variants(APOE-ɛ2, APOE-ɛ3, APOE-ɛ4), which encode the ApoE2, ApoE3, and ApoE4 protein isoforms. Optimal Aβ clearance efficiency has been suggested to explain the neuroprotective nature of the ApoE2 isoform relative to the most common ApoE3 isoform, whereas presumably, the impaired Aβ clearance by the ApoE4 isoform explains the increased AD risk for APOE-ɛ4 allele carriers [2]. In fact, more than 30% of the AD cases in the population can be attributed to theAPOE-ɛ4 allele (population attributable fraction), whereas at most 8% of AD cases can be attributed to any of the genes detected in a genome-wide association studies [8].
Carrying the APOE-ɛ4 allele predisposes for AD in a dose-dependent manner: compared to non-APOE-ɛ4 carriers, AD risk is increased 3–5 fold for heterozygous APOE-ɛ4 carriers, and 10-15-fold for homozygous APOE-ɛ4 carriers [9]. However, despite this large effect size, the penetrance of APOE-ɛ4 homozygosity is incomplete. The chance thatAPOE-ɛ4 homozygotes develop AD before the age of 85 years is 50% for males and 60% for females [10]. Some APOE-ɛ4 homozygotes reach ages over 100 years while retaining their cognitive health [11]. This suggests that next to being homozygous for the APOE-ɛ4 allele, additional genetic modifiers are necessary for the development of AD. To our knowledge, it has never been investigated whether other genetic variants co-occur with APOE-ɛ4 homozygosity in AD patients.
We identified a family with AD patients with a relatively early onset of disease, spanning at least four generations, with an inheritance pattern that suggests autosomal dominant AD. DNA was available from four affected and seven unaffected family members, from the two youngest generations. We found that all four genetically tested affected family members were homozygous for the APOE-ɛ4 allele. Therefore, this family provided the unique opportunity to investigate additional genetic variants next to APOE-ɛ4 homozygosity, which might have contributed to AD. We describe the pedigree, the phenotype of the affected family members, the outcome of whole exome sequencing, and the segregation of the genetic variants.
MATERIAL AND METHODS
Pedigree and participants
We describe a family comprising nine individuals with AD symptoms who span four generations within one pedigree (Fig. 1, Supplementary Table 1A): eight were diagnosed with AD, or were reported to have symptoms of AD (0.2, I.1, I.3, I.4, II.1, II.3, II.4, and II.6) and one individual had preclinical AD (III.1). Multiple individuals did not present AD symptoms at the time of observation (youngest individuals were merged into III.4-III.5 to avoid recognition).
Four family members with (preclinical) AD and seven of the unaffected family members (aged ≤60 years) consented to participate in this study (II.3 and II.4 by consent of their proxies). Affected family member II.1 consented to the use of his clinical data for research purposes. Detailed clinical data were not available for the affected family members of the first generations (0.2, I.1, I.3, and I.4).
Two family members with AD (II.1 and II.6) and the family member with preclinical AD (III.1) visited the Alzheimer center at the VU University medical center (VUmc) in the Netherlands and underwent extensive standardized diagnostic work-up [12]. The other affected family members (II.3 and II.4) were diagnosed elsewhere in the Netherlands. All diagnoses of AD were based on the NINCDS-ADRDA criteria as described by McKhann et al. [13]. Postmortem autopsy results of individual II.4 were reviewed by our neuropathologist. The local medical ethics committee of the VUmc approved the study. We did not obtain consent to reveal disease history of family member II.8.
DNA availability
Using common procedures, DNA was isolated from peripheral blood from three affected family members (II.3, II.6, II.4) the family member with preclinical AD (III.1), and the seven unaffected participating family members (III.2, III.3, four family members merged into III.4-5, and III.6).
APOE genotyping and imputation
For all participants with DNA available, APOE genotyping was performed after genomic DNA isolation from 7–10 mL EDTA blood, using a QIAxcel DNA Fast Analysis kit (Qiagen, Venlo, The Netherlands). For the individuals comprising generation I and two individuals from generation II, blood samples could not be collected. For individuals I.1, I.2, II.1, and II.8 APOE genotypes were determined in retrospect by estimating the posterior probability of the possible APOE genotypes. To this end, we applied Bayes theorem based on (i) the known APOE genotype combinations in generations II and III, (ii) the population frequencies of APOE genotype combinations in the Dutch population published by LASA [14] (Supplementary Table 1B), and (iii) the chances for developing dementia for all APOE-genotype combinations by age and gender published by Genin et al. [10] (Supplementary Table 1C).
Analysis of AD penetrance in this family
In a Caucasian sample comprising >17,500 cases and controls, Genin et al. evaluated the AD incidence per APOE-genotype, across age at onset and gender relative to baseline AD incidence [10]. These AD incidence distributions across age allowed us to determine the a priori chance for any individual to develop AD at a certain age given their APOE genotype and gender. For each member of our family, the age at AD onset can be seen as a p-value w.r.t. to empirical the incidence distributions extracted from the Caucasian cohort.
Persons who had not yet reached 60 years at last check-up were excluded, as the chance to develop AD before this age is very low. To account for the unknown age at AD onset for several family members, we determined a p-value by repeated sampling from a uniform distribution of p-values (truncated p-value approach [15]). We used both Fisher’s approach and Stouffer’s approach to combine p-values of all family members. Apart from their genetic dependency (which we are testing), we assume that development of AD is independent between family members.
Variant detection: Exome sequencing of four family members
DNA of three affected family members (II.3, II.6, and III.1) and the oldest unaffected family member (III.2) was exome sequenced in parallel with 400 AD patients from the Amsterdam Dementia cohort (ADC) [12] who had been diagnosed with early onset dementia. The exomes were captured by the Nimblegen human exome v3 capture kit, and 100 bp paired-end sequencing reads were generated on the Illumina HiSeq 2000 platform, according to the manufacturer’s protocol. We sequenced to at least 40x mean coverage to ensure sufficient read depth for variant calling. Reads were mapped to the human reference genome sequence (UCSC hg19) using Burrows-Wheeler alignment [16]. Duplicate read removal, local sequence realignment and base quality recalibration were performed by Picard (http://picard.sourceforge.net) and with GATK (Genome analysis tool kit) [17]. Variants were called using GATK haplotype caller, and filtered using the variant filtration tool. For each variant we set the filter to PASS if the variant complied with (I) GATK quality score ≥50, (II) quality over depth ≥1.5, (III) Strand bias ≤60, (IV) total variant read depth ≥5.0. Variants were annotated and analyzed with Cartagenia (http://www.cartagenia.com/) using a filter tree specifically designed to detect variants causative for a trait with an autosomal dominant inheritance pattern. Since we aimed to identify rare pathogenic variants, we selected variants that (i) were absent in the following databases: dbSNP138 (http://www.ncbi.nlm.nih.gov/projects/SNP, build 138), the 1,000 genome project (http://www.1000genomes.org) or the National Heart Lung Blood Institute Exome Variant Server (EVS) (http://evs.gs.washington.edu/EVS/); (ii) had a prevalence of ≤5% in the whole Amsterdam Dementia cohort; (iii) were heterozygote in the 3 affected family members; and (iv) were localized in a gene listed in the OMIM database (http://www.omim.org). The predicted functional effects of the selected sequence variants were assessed by PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://sift.jcvi.org/), Mutation Taster (http://www.mutationtaster.org), and the combined annotation dependent deletion (CADD) score [18]. Information about localization and conservation of the selected variants was assessed by Uniprot (http://www.uniprot.org/uniprot/Q92673), and Alumut visual (http://www.interactive-biosoftware.com/alamut-visual/). Detected variants were confirmed by Sanger sequencing, Also, loci were genotyped in the all family members for whom DNA was available.
RESULTS
Clinical description
Family member II.1 presented with complaints of memory decline over the previous three years at the age of 67. As a child, the family member had suffered a skull fracture. The Mini-Mental State Examination (MMSE) [19] score was 25/30, and neuropsychological assessment showed impairment of episodic memory. Routine blood analysis was normal, except for increased serum cholesterol levels. Magnetic resonance imaging (MRI) showed mild bilateral hippocampal atrophy (medial temporal lobe atrophy grade 1 [20]), mild white matter hyperintensities (WMH), (Fazekas grade 1 [21], and no microbleeds. Electroencephalogram (EEG) revealed a discordant low background rhythm of 6 to 7 Hz with increased amounts of intermitting delta activity in the frontotemporal regions. Cerebrospinal fluid (CSF) was not obtained. Based on these findings, the diagnosis was mild cognitive impairment (MCI) [22]. At the age of 69, the MMSE was 21/30. Repeated neuropsychological assessment showed progression of memory impairment and impaired executive functions. At this time, MRI showed biparietal atrophy, with no progression of the hippocampal atrophy or WMH. The clinical diagnosis was probable AD [13]. Disease progression was characterized by further deterioration in all cognitive domains, including the development of behavioral disturbances (loss of initiative and increased irritability). This family member was admitted into a nursing home, suffered from episodes of focal neurological deficits probably due to recurrent strokes, and died at the age of 76 years. This family member gave consent to his physician to use his medical data for research purposes, but DNA was not available.
Family member II.3 visited a geriatrician at a local hospital at the age of 72 because of memory complaints and fatigue for two years. This family member had diabetes mellitus type 2, hypertension, and dyslipidemia, and had been treated for depression with amitriptyline for over 20 years. MMSE was 26/30 with disorientation in time, and neuropsychological testing showed impaired recall on memory tests. Computed tomography (CT) imaging showed mild diffuse cortical atrophy and aspecific hypodensities in the brainstem and in the basal ganglia. No formal diagnosis was made at that time. A second opinion by another neurologist was obtained at the age of 74. At this point, the family member reported the occurrence of headaches, and scored 23/30 on the MMSE. Routine blood analysis and EEG were normal. MRI and CSF analysis were not performed. The family member was diagnosed with probable AD [13]. Diagnostic DNA testing revealed no variants in PSEN1, PSEN2, or APP. The family member died at the age of 78 years (cause unspecified).
Family member II.4 visited a local memory clinic at the age of 70, because of progressive memory complaints, which initiated at the age of 59 after a head trauma. The family member had suffered from bacterial meningitis at the age of 69. Neuropsychological testing showed impairments in concentration and memory, disorientation in place, and dyscalculia. No additional investigations were performed. The family member was diagnosed with probable AD [13]. The patient died at the age of 74 years most likely due to a heart attack. Postmortem examination of the brain confirmed the diagnosis of severe AD (Braak stage 6/6 for tau and Thal phase 5/5 for Aβ with extensive cerebral amyloid angiopathy (CAA) type 1 [23] (Fig. 2).
Family member II.6 was evaluated at our memory clinic at the age of 70 years because of the positive family history of dementia. At this visit, this family member reported no cognitive complaints, MMSE was 30/30 and neuropsychological testing showed no abnormalities except for some difficulties with concentration. Routine blood analysis showed no abnormalities. MR imaging displayed no hippocampal atrophy (medial temporal lobe atrophy grade 0), mild WMH (Fazekas grade 1), but a high number of 47 microbleeds, suggestive of CAA (Fig. 3). EEG showed a remarkably decreased background pattern with reactive alpha-theta activity till 8 Hz. CSF analysis showed a decreased Aβ level of 232 ng/L (reference >550 ng/L), an increased total tau level of 993 ng/L (reference ≤375 ng/L), and an increased level of tau phosphorylated at threonine-181 (ptau) of 123 (reference ≤52 ng/L)). Based on the clinical examination, the family member was diagnosed with subjective cognitive decline [24]. During the following years, the family member developed memory complaints, loss of initiative, and sleeping problems. At the age of 74 years, MMSE was 29/30, and neuropsychological testing showed disturbances in episodic memory. MRI showed no progression of WMH, but the number of microbleeds had increased to 58. Repeated EEG displayed progressive slowing with theta activity of 7 Hz next to dominant posterior rhythms of 8 Hz. The family member was diagnosed with MCI. The family member was diagnosed with probable AD by a local geriatrician at the age of 82, with a MMSE score of 21 out of 30.
Family member III.1 presented at our memory clinic at 58 years with memory complaints, and self-reported difficulties with organizing and planning. This family member was treated for diabetes mellitus, hypertension, and dyslipidemia. MMSE score was 29/30, performance on neuropsychological testing was normal, and MRI showed no abnormalities. EEG was disturbed with a normal alpha background pattern of 9 Hz, but with early intermitting left predominant temporal theta activity. CSF concentrations showed a mildly decreased Aβ level of 549 ng/L (reference >550 ng/L) and increased tau level of 435 ng/L (reference ≤375 ng/L) and ptau level of 68 (reference ≤52 ng/L). Pittsburgh compound (PiB)-PET showed increased Aβ binding in all cortical areas. F18-fluorodeoxyglucose (FDG)-PET showed a normal pattern of glucose metabolism. Based on clinical evaluations, the diagnosis was subjective cognitive decline. The abnormal AD biomarkers indicated preclinical AD, with a high likelihood of underlying AD pathophysiology [25]. Over the next four years, the family member remained clinically stable.
Generation 0 and I: Family member I.2 died of cancer at age 55. Family member 1.3, a sibling of I.2, was reported to have dementia onset at 61. Family member 1.4, also a sibling of I.2, was reported to have cognitive decline prior to death at age 73. One of their parents (generation 0) was reported to have dementia symptoms suggestive of AD at age 80, Family member I.1 (partner from I.2) died at the age of 85 years from a heart attack and was reported to have had dementia with AD characteristics. For these family members, no formal diagnosis was available at the time and no DNA was secured.
Unaffected family members in generation III: The seven unaffected family members (III.2, III.3, III.4-5,and III.6) for whom DNA was available were aged 47 to 60 years and self-reported no cognitive complaints. Their partner and/or a close relative confirmed absence of signs of cognitive impairment. No formal cognitive tests were performed in these family members.
APOE genotype distribution in family structure
All affected family members with for whom DNA was available (II.3, II.4, II.6, III.1) were homozygous for the APOE-ɛ4 allele. Of the seven unaffected family members from generation III for whom DNA was available, two were genotyped APOE-ɛ4/ɛ4, four were APOE-ɛ3/ɛ4, and one was APOE-ɛ3/ɛ3 (Supplementary Table 1A, Fig. 1).
Given the frequency of all APOE genotype combinations in the Dutch population, disease status of each family member by age, and the APOE genotypes for all individuals in generations II and III, we estimated a 65% chance that individuals I.1 and I.2 are APOE-ɛ3/ɛ4 and APOE-ɛ4/ɛ4; and we estimated a 29% chance that individuals are both APOE-ɛ3/ɛ4 (grey text in Fig. 1). Chances for other possible genotypes were negligible (Supplementary Table 1D). Likewise, we estimated a 76% chance that the APOE genotype of individual II.1, who was diagnosed with AD at 69, was APOE-ɛ4/ɛ4: and a 22% that it was APOE-ɛ3/ɛ4; chances for other possible genotypes were negligible (Supplementary Table 1E). Individual II.8 has an 87% chance of being APOE-ɛ3/ɛ4 and a 12% chance of being APOE-ɛ3/ɛ3 (Supplementary Table 1F). Together, this provides support that this family included at least three generations of APOE-ɛ4 homozygotes.
Chances of developing AD in this family:A statistical analysis
We evaluated the AD incidence in this family w.r.t. empirical age at onset distributions extracted from a Caucasian cohort, given APOE-genotype and gender [10] (Supplementary Table 1C). The age of AD onset in this family was significantly earlier than expected: p = 0.0001 and p = 0.00006 using respectively Fisher’s method and Stouffer’s method of combining p-values. P-values remain significant when we use the age at diagnosis, suggesting that the finding is robust: p = 0.0290 and p = 0.0094 using Fisher’s method and Stouffer’s method respectively (Supplementary Table 1A).
Of note, the high incidence of AD may have stimulated family members to visit our hospital for cognitive testing. This may have introduced a bias at the age at AD onset level that we cannot account for. However, further family research revealed that I.3 and I.4 (two siblings of I.2, who died of cancer at age 55) were reported to have dementia at relatively early ages at onset irrespective of family bias (61 and 73 years, respectively).
Exome sequencing outcome
To investigate whether additional genetic variants occurred next to APOE-ɛ4 homozygosity, we performed exome sequencing in two siblings with AD (II.3, II.6), in one family member with preclinical AD (III.1) and in the eldest participant without cognitive complaints hitherto (III.2), all homozygous for the APOE-ɛ4 allele. Exome sequencing revealed no mutations in the PSEN1, PSEN2, and APP genes in any of the family members.
We detected 16 variants that passed filtering, several of which were rare and predicted to have a deleterious effect on protein function (Supplementary Table 2). Two of these occurred in a gene that might be functionally associated with amyloid processing or AD: (i) the missense variant c.2021A>G, p.Asn674Ser, in exon 14 of the sortilin related receptor 1 (SORL1) gene (NM.003105.5), and (ii) c.707 C>T, p.Thr236Met, in exon 2 of the teashirt zinc finger homeobox 3 (TSHZ3) gene (NM.020856.2). The coverage of SORL1 and TSHZ3 captured with the exome kit was similar to the median read depth over the whole exome.
This SORL1 variant was present in all three affected family members and the family member with preclinical AD, and in four unaffected family members (aged <60 years). The variant was not detected in any other subject in the Amsterdam Dementia cohort. One study detected the variant in a 63-year-old healthy female (control group n = 1938, MAF <0.001) [26], and the ExAC database reports only one heterozygous carrier of this variant (MAF <0.00001). The variant locus is at a highly conserved glycosylation site in the VPS10 domain of SORL1 (Fig. 4), it has a CADD score of 23.6, and it is considered deleterious by PolyPhen and Mutation Taster (although not by SIFT).
The p.Thr236Met missense variant in TSHZ3 was detected in two of the three family members with AD, in the family member with preclinical AD and in one unaffected family member, who was also homozygous for APOE-ɛ4 but did not carry the SORL1 variant. This TSHZ3 variant is located in a highly conserved amino acid, has a CADD score of 27.0 and is predicted to be deleterious by PolyPhen, Mutation Taster, and SIFT. The variant was reported in 47 individuals in the ExAC database (MAF <0.001).
DISCUSSION
We describe a family with an inheritance pattern suggestive of autosomal dominant AD of which all affected family members tested were homozygous for the APOE-ɛ4 allele. The age at AD onset was significantly earlier than expected, based on the APOE genotypes and gender of family-members, suggesting that next to a high load of APOE-ɛ4, this family is relatively enriched with other AD-associated elements. Whole exome sequencing revealed two additional variants co-inherited with APOE-ɛ4 homozygosity that might disturb Aβ processing: a rare missense variant leading to p.Asn674Ser in the SORL1 protein and a rare missense variant leading to p.Thr236Met in the TSHZ3 protein. We speculate these SORL1 and TSHZ3 variants increased the penetrance of AD in this family. Without APOE-ɛ4 homozygosity, neither variants may reach full AD penetrance, as can be observed in the pedigree where several APOE-ɛ4 heterozygous or APOE-ɛ4 negative family members carry the SORL1 and/or TSHZ3 gene variants without having the disease. It should be noted however, that these family members are still young, and may develop AD at a later age, such that we cannot exclude the possibility that either variant confers full AD penetrance. For the same reason we cannot (yet) identify the effect of APOE-ɛ4 gene dosage, by analyzing to what extent impaired SORL1 and/or TSHZ3 modulates AD penetrance in a background of heterozygous APOE-ɛ4. Moreover, this family carried several additional rare genetic variants, some of which were predicted to have a deleterious effect on protein function. Although these variants map in genes that are currently not associated with AD, we cannot a priori rule out that they modulate AD susceptibility in this family.
TSHZ3 may modulate Aβ processing
The rare variant detected in TSHZ3 is predicted to have a damaging effect on protein function by all effect predictor algorithms, and has a relatively high CADD score of 27. Although evidence is limited, this rare variant might complicate Aβ processing since the TSHZ3 protein has been found to bind to FE65, an adaptor protein that can modulate AβPP trafficking and/or processing [27]. TSHZ3 downregulates Caspase 4, which is involved cell death induced by cytotoxic AβPP peptides [28–30]. However, more evidence is needed to link disturbed TSHZ3 protein function to AD.
Impaired SORL1 increases Aβ production
In sharp contrast with TSHZ3, evidence has accumulated that impaired SORL1 function associates with AD: common genetic polymorphisms in the SORL1 locus were associated with AD in a genome-wide association studies [31], disruptive variants were only detected in AD cases and not in controls [26] and rare pathogenic SORL1 variants were found to increase the risk for early onset AD by five-fold [32].
Functional studies suggested that the SORL1 sorting receptor has a dual function: (i) SORL1 binds AβPP and prevents it from processing into Aβ [33], and (ii) SORL1 binds newly synthesized extracellular Aβ and targets it to the lysosome for degradation [34]. To exert these functions, SORL1 has two important protein domains: an AβPP-binding complement type repeat domain, and an Aβ-binding VPS10 domain [33].
The p.Asn674Ser variant in SORL1 that we detected in this family affects a highly conserved N-glycosylation site in the VPS10 domain, which is important for proper protein folding and for protein-protein interaction [33]. Previously, variants that map in the VPS10 domain (p.Glu270Lys, p.Ala528Thr) were associated with impaired retrograde sorting of AβPP and enhanced Aβ production when expressed in cells [35], suggesting that a wild-type VPS10 domain is essential for proper Aβ processing. We speculate that the p.Asn674Ser change detected in this family may at disrupt the Aβ-binding capacity of SORL1, resulting in less efficient lysosomal degradation of Aβ [34].
Combined effect of APOE-ɛ4 homozygosity, impaired SORL1 and impaired TSHZ3
Experimental studies suggest that the proteins encoded by the APOE and SORL1 genes functionally interact [36]. By binding to the complement type repeats of the SORL1 protein, ApoE4 reduced the AβPP-binding-capacity of SORL1 [37]. Furthermore, overexpression of SORL1 increases the uptake of extracellular Aβ in an ApoE-isoform-dependent manner, most efficiently in the presence of the ɛ4 isoform [36]. Therefore, the clearance of Aβ is expected to be more dependent on SORL1 expression in APOE-ɛ4 carriers than in individuals with no APOE-ɛ4 alleles. A combination of homozygous or heterozygous ApoE4 and dysfunctional SORL1 may therefore lead to abnormal increases in extracellular Aβ loads, which may underlie the neurodegenerative processes in this family.
Effect of the genotype on phenotype
Homozygous APOE-ɛ4 carriers typically present with an amnestic phenotype, however the AD phenotype of the five affected family members for whom detailed clinical data were available, was heterogeneous. The age at onset differed between affected family members and ranged between 61 and 74 years, which fits with the relatively early age of disease onset associated with APOE-ɛ4 homozygosity [38]. Homozygous APOE-ɛ4 and disrupted SORL1 are both associated with CAA, presumably as a result of the less effective Aβ clearance [39–41]. Indeed, two family members with AD had extensive microbleeds and CAA, while two others remained free of microbleeds.
Both APOE and SORL1 are involved in cholesterol metabolism/transport, and APOE-ɛ4 carriers have been found to have increased cholesterol levels [42, 43]. Indeed, three affected family members were diagnosed with hypercholesterolemia in this family. Likewise, the EEG pattern of APOE-ɛ4 allele carrier-patients shows a greater decrease of alpha activity than non-APOE-ɛ4 carrier-patients [44, 45]. However, in this family, neither microbleeds/CAA, hypercholesterolemia or EEG patterns cosegregated with APOE-ɛ4 homozygosity and/or the SORL1variant.
Conclusion
We hypothesize that the convergence of multiple genetic factors that disturb the Aβ processing pathways over several generations results in an autosomal dominant like inheritance pattern of AD in this family. Extracellular Aβ load might be abnormally increased as a result of the concerted effects of (i) ineffective clearance of extracellular Aβ from the brain by the ApoE4 protein isoform, and (ii) impaired uptake of extracellular Aβ for lysosomal degradation due to a disturbed VPS10 domain of the SORL1 protein. It is possible that a disturbed TSHZ3 function might have further contributed to impaired AβPP regulation, but compared to ApoE4 and SORL1, the evidence for an association of disrupted TSHZ3 with AD is currently very limited. Moreover, other genetic variants that were left undetected with our analysis strategy might have further influenced disease penetrance.
Given these findings, the currently unaffected family member who is homozygous for APOE-ɛ4 and who carries the SORL1 variant may be at the highest risk to develop AD. Follow-up of this family in the future will resolve these speculations. We expect that this polygenic model, possibly involving other genetic variants, might also explain autosomal dominant inheritance patterns in other APOE-ɛ4 positive families.
ACKNOWLEDGMENTS
Research of the VUmc Alzheimer center is part of the neurodegeneration research program of the Neuroscience Campus Amsterdam. The VUmc Alzheimer Center is supported by Stichting Alzheimer Nederland and Stichting VUmc fonds.
Authors disclosures available online (http://j-alz.com/manuscript-disclosures/16-0091r1).
Appendices
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-160091.
REFERENCES
[1] | Alzheimer’s Association National Plan Milestone Workgroup, Fargo KN , Aisen P , Albert M , Au R , Corrada MM , DeKosky S , Drachman D , Fillit H , Gitlin L , Haas M , Herrup K , Kawas C , Khachaturian AS , Khachaturian ZS , Klunk W , Knopman D , Kukull WA , Lamb B , Logsdon RG , Maruff P , Mesulam M , Mobley W , Mohs R , Morgan D , Nixon RA , Paul S , Petersen R , Plassman B , Potter W , Reiman E , Reisberg B , Sano M , Schindler R , Schneider LS , Snyder PJ , Sperling RA , Yaffe K , Bain LJ , Thies WH , Carrillo MC ((2014) ) 2014 Report on the Milestones for the US National Plan to Address Alzheimer’s Disease. Alzheimers Dement 10: , S430–S452. |
[2] | Yu JT , Tan L , Hardy J ((2014) ) Apolipoprotein E in Alzheimer’s disease: An update. Annu Rev Neurosci 37: , 79–100. |
[3] | Levy-Lahad E , Wasco W , Poorkaj P , Romano DM , Oshima J , Pettingell WH , Yu CE , Jondro PD , Schmidt SD , Wang K , et al. ((1995) ) Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269: , 973–977. |
[4] | Sherrington R , Rogaev EI , Liang Y , Rogaeva EA , Levesque G , Ikeda M , Chi H , Lin C , Li G , Holman K , Tsuda T , Mar L , Foncin JF , Bruni AC , Montesi MP , Sorbi S , Rainero I , Pinessi L , Nee L , Chumakov I , Pollen D , Brookes A , Sanseau P , Polinsky RJ , Wasco W , Da Silva HA , Haines JL , Perkicak-Vance MA , Tanzi RE , Roses AD , Fraser PE , Rommens JM , St George-Hyslop PH ((1995) ) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375: , 754–760. |
[5] | Goate A , Chartier-Harlin MC , Mullan M , Brown J , Crawford F , Fidani L , Giuffra L , Haynes A , Irving N , James L , Mont R , Newton P , Rooke K , Roques P , Talbot C , Pericak-Vance M , Roses A , Williamson R , Rossor M , Owen M , Hardy J ((1991) ) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349: , 704–706. |
[6] | Gatz M , Reynolds CA , Fratiglioni L , Johansson B , Mortimer JA , Berg S , Fiske A , Pedersen NL ((2006) ) Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry 63: , 168–174. |
[7] | Qiu C , Kivipelto M , Aguero-Torres H , Winblad B , Fratiglioni L ((2004) ) Risk and protective effects of the APOE gene towards Alzheimer’s disease in the Kungsholmen project: Variation by age and sex. J Neurol Neurosurg Psychiatry 75: , 828–833. |
[8] | Van Cauwenberghe C , Van Broeckhoven C , Sleegers K ((2016) ) The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet Med 18: , 421–430. |
[9] | Corder EH , Saunders AM , Strittmatter WJ , Schmechel DE , Gaskell PC , Small GW , Roses AD , Haines JL , Pericak-Vance MA ((1993) ) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261: , 921–923. |
[10] | Genin E , Hannequin D , Wallon D , Sleegers K , Hiltunen M , Combarros O , Bullido MJ , Engelborghs S , De Deyn P , Berr C , Pasquier F , Dubois B , Tognoni G , Fievet N , Brouwers N , Bettens K , Arosio B , Coto E , Del Zompo M , Mateo I , Epelbaum J , Frank-Garcia A , Helisalmi S , Porcellini E , Pilotto A , Forti P , Ferri R , Scarpini E , Siciliano G , Solfrizzi V , Sorbi S , Spalletta G , Valdivieso F , Vepsalainen S , Alvarez V , Bosco P , Mancuso M , Panza F , Nacmias B , Bossu P , Hanon O , Piccardi P , Annoni G , Seripa D , Galimberti D , Licastro F , Soininen H , Dartigues JF , Kamboh MI , Van Broeckhoven C , Lambert JC , Amouyel P , Campion D ((2011) ) APOE and Alzheimer disease: A major gene with semi-dominant inheritance. Mol Psychiatry 16: , 903–907. |
[11] | Freudenberg-Hua Y , Freudenberg J , Vacic V , Abhyankar A , Emde AK , Ben-Avraham D , Barzilai N , Oschwald D , Christen E , Koppel J , Greenwald B , Darnell RB , Germer S , Atzmon G , Davies P ((2014) ) Disease variants in genomes of 44 centenarians. Mol Genet Genomic Med 2: , 438–450. |
[12] | van der Flier WM , Pijnenburg YA , Prins N , Lemstra AW , Bouwman FH , Teunissen CE , van Berckel BN , Stam CJ , Barkhof F , Visser PJ , van Egmond E , Scheltens P ((2014) ) Optimizing patient care and research: The Amsterdam Dementia Cohort. J Alzheimers Dis 41: , 313–327. |
[13] | McKhann G , Drachman D , Folstein M , Katzman R , Price D , Stadlan EM ((1984) ) Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34: , 939–944. |
[14] | Deeg DJ , van Tilburg T , Smit JH , de Leeuw ED ((2002) ) Attrition in the Longitudinal Aging Study Amsterdam. The effect of differential inclusion in side studies. J Clin Epidemiol 55: , 319–328. |
[15] | Tang S , Ding Y , Sibille E , Mogil J , Lariviere WR , Tseng GC ((2014) ) Imputation of truncated p-values for meta-analysis methods and its genomic application. Ann Appl Stat 8: , 2150–2174. |
[16] | Li H , Durbin R ((2009) ) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: , 1754–1760. |
[17] | McKenna A , Hanna M , Banks E , Sivachenko A , Cibulskis K , Kernytsky A , Garimella K , Altshuler D , Gabriel S , Daly M , DePristo MA ((2010) ) The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: , 1297–1303. |
[18] | Kircher M , Witten DM , Jain P , O’Roak BJ , Cooper GM , Shendure J ((2014) ) A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46: , 310–315. |
[19] | Folstein MF , Folstein SE , McHugh PR ((1975) ) “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12: , 189–198. |
[20] | Scheltens P , Launer LJ , Barkhof F , Weinstein HC , van Gool WA ((1995) ) Visual assessment of medial temporal lobe atrophy on magnetic resonance imaging: Interobserver reliability. J Neurol 242: , 557–560. |
[21] | Fazekas F , Chawluk JB , Alavi A , Hurtig HI , Zimmerman RA ((1987) ) MR signal abnormalities at 1.5 T in Alzheimer’s dementia and normal aging. AJR Am J Roentgenol 149: , 351–356. |
[22] | Albert MS , DeKosky ST , Dickson D , Dubois B , Feldman HH , Fox NC , Gamst A , Holtzman DM , Jagust WJ , Petersen RC , Snyder PJ , Carrillo MC , Thies B , Phelps CH ((2011) ) The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 270–279. |
[23] | Thal DR , Ghebremedhin E , Rub U , Yamaguchi H , Del Tredici K , Braak H ((2002) ) Two types of sporadic cerebral amyloid angiopathy. J Neuropathol Exp Neurol 61: , 282–293. |
[24] | Jessen F , Amariglio RE , van Boxtel M , Breteler M , Ceccaldi M , Chetelat G , Dubois B , Dufouil C , Ellis KA , van der Flier WM , Glodzik L , van Harten AC , de Leon MJ , McHugh P , Mielke MM , Molinuevo JL , Mosconi L , Osorio RS , Perrotin A , Petersen RC , Rabin LA , Rami L , Reisberg B , Rentz DM , Sachdev PS , de la Sayette V , Saykin AJ , Scheltens P , Shulman MB , Slavin MJ , Sperling RA , Stewart R , Uspenskaya O , Vellas B , Visser PJ , Wagner M , Subjective Cognitive Decline Initiative Working Group ((2014) ) A conceptual framework for research on subjective cognitive decline in preclinical Alzheimer’s disease. Alzheimers Dement 10: , 844–852. |
[25] | Sperling RA , Aisen PS , Beckett LA , Bennett DA , Craft S , Fagan AM , Iwatsubo T , Jack CR Jr , Kaye J , Montine TJ , Park DC , Reiman EM , Rowe CC , Siemers E , Stern Y , Yaffe K , Carrillo MC , Thies B , Morrison-Bogorad M , Wagster MV , Phelps CH ((2011) ) Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 280–292. |
[26] | Verheijen J , Van den Bossche T , van der Zee J , Engelborghs S , Sanchez-Valle R , Llado A , Graff C , Thonberg H , Pastor P , Ortega-Cubero S , Pastor MA , Benussi L , Ghidoni R , Binetti G , Clarimon J , Lleo A , Fortea J , de Mendonca A , Martins M , Grau-Rivera O , Gelpi E , Bettens K , Mateiu L , Dillen L , Cras P , De Deyn PP , Van Broeckhoven C , Sleegers K ((2016) ) A comprehensive study of the genetic impact of rare variants in SORL1 in European early-onset Alzheimer’s disease. Acta Neuropathol 132: , 213–224. |
[27] | Kajiwara Y , Akram A , Katsel P , Haroutunian V , Schmeidler J , Beecham G , Haines JL , Pericak-Vance MA , Buxbaum JD ((2009) ) FE65 binds Teashirt, inhibiting expression of the primate-specific caspase-4. PLoS One 4: , e5071. |
[28] | Hitomi J , Katayama T , Eguchi Y , Kudo T , Taniguchi M , Koyama Y , Manabe T , Yamagishi S , Bando Y , Imaizumi K , Tsujimoto Y , Tohyama M ((2004) ) Involvement of caspase-4 in endoplasmic reticulum stress-induced apoptosis and Abeta-induced cell death. J Cell Biol 165: , 347–356. |
[29] | Sollberger G , Strittmatter GE , Kistowska M , French LE , Beer HD ((2012) ) Caspase-4 is required for activation of inflammasomes. J Immunol 188: , 1992–2000. |
[30] | Li C , Wei J , Li Y , He X , Zhou Q , Yan J , Zhang J , Liu Y , Liu Y , Shu HB ((2013) ) Transmembrane Protein 214 (TMEM214) mediates endoplasmic reticulum stress-induced caspase 4 enzyme activation and apoptosis. J Biol Chem 288: , 17908–17917. |
[31] | Lambert J , Ibrahim-Verbaas C , Harold D , Naj A , Sims R , Bellenguez C , Jun G , DeStefano A , Bis J , Beecham G , Grenier-Boley B , Russo G , Thornton-Wells T , Jones N , Smith A , Chouraki V , Thomas C , Ikram M , Zelenika D , Vardarajan B , Kamatani Y , Lin C , Gerrish A , Schmidt H , Kunkle B , Dunstan M , Ruiz A , Bihoreau M , Choi S , Reitz C , Pasquier F , Hollingworth P , Ramirez A , Hanon O , Fitzpatrick A , Buxbaum J , Campion D , Crane P , Baldwin C , Becker T , Gudnason V , Cruchaga C , Craig D , Amin N , Berr C , Lopez O , De Jager P , Deramecourt V , Johnston J , Evans D , Lovestone S , Letenneur L , Moron F , Rubinsztein D , Eiriksdottir G , Sleegers K , Goate A , Fievet N , Huentelman M , Gill M , Brown K , Kamboh M , Keller L , Barberger-Gateau P , McGuinness B , Larson E , Green R , Myers A , Dufouil C , Todd S , Wallon D , Love S , Rogaeva E , Gallacher J , St George-Hyslop P , Clarimon J , Lleo A , Bayer A , Tsuang D , Yu L , Tsolaki M , Bossu P , Spalletta G , Proitsi P , Collinge J , Sorbi S , Sanchez-Garcia F , Fox N , Hardy J , Naranjo M , Bosco P , Clarke R , Brayne C , Galimberti D , Mancuso M , Matthews F , European Alzheimer’s Disease Initiative (EADI), Genetic, Environmental Risk in Alzheimer’s Disease (GERAD), Alzheimer’s Disease Genetic Consortium (ADGC), Cohorts for Heart, Aging Research in Genomic Epidemiology (CHARGE), Moebus S , Mecocci P , Del Zompo M , Maier W , Hampel H , Pilotto A , Bullido M , Panza F , Caffarra P , Nacmias B , Gilbert J , Mayhaus M , Lannfelt L , Hakonarson H , Pichler S , Carrasquillo M , Ingelsson M , Beekly D , Alvarez V , Zou F , Valladares O , Younkin S , Coto E , Hamilton-Nelson K , Gu W , Razquin C , Pastor P , Mateo I , Owen M , Faber K , Jonsson P , Combarros O , O’Donovan M , Cantwell L , Soininen H , Blacker D , Mead S , Mosley T Jr , Bennett D , Harris T , Fratiglioni L , Holmes C , de Bruijn R , Passmore P , Montine T , Bettens K , Rotter J , Brice A , Morgan K , Foroud T , Kukull W , Hannequin D , Powell J , Nalls M , Ritchie K , Lunetta K , Kauwe J , Boerwinkle E , Riemenschneider M , Boada M , Hiltunen M , Martin E , Schmidt R , Rujescu D , Wang L , Dartigues J , Mayeux R , Tzourio C , Hofman A , Nothen M , Graff C , Psaty B , Jones L , Haines J , Holmans P , Lathrop M , Pericak-Vance M , Launer L , Farrer L , van Duijn C , Van Broeckhoven C , Moskvina V , Seshadri S , Williams J , Schellenberg G , Amouyel P ((2013) ) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet 45: , 1452–1458. |
[32] | Nicolas G , Charbonnier C , Wallon D , Quenez O , Bellenguez C , Grenier-Boley B , Rousseau S , Richard AC , Rovelet-Lecrux A , Le Guennec K , Bacq D , Garnier JG , Olaso R , Boland A , Meyer V , Deleuze JF , Amouyel P , Munter HM , Bourque G , Lathrop M , Frebourg T , Redon R , Letenneur L , Dartigues JF , Genin E , Lambert JC , Hannequin D , Campion D , CNR-MAJ collaborators ((2016) ) SORL1 rare variants: A major risk factor for familial early-onset Alzheimer’s disease. Mol Psychiatry 21: , 831–836. |
[33] | Willnow TE , Andersen OM ((2013) ) Sorting receptor SORLA–a trafficking path to avoid Alzheimer disease. J Cell Sci 126: , 2751–2760. |
[34] | Caglayan S , Takagi-Niidome S , Liao F , Carlo AS , Schmidt V , Burgert T , Kitago Y , Fuchtbauer EM , Fuchtbauer A , Holtzman DM , Takagi J , Willnow TE ((2014) ) Lysosomal sorting of amyloid-beta by the SORLA receptor is impaired by a familial Alzheimer’s disease mutation. Sci Transl Med 6: , 223ra220. |
[35] | Vardarajan BN , Zhang Y , Lee JH , Cheng R , Bohm C , Ghani M , Reitz C , Reyes-Dumeyer D , Shen Y , Rogaeva E , St George-Hyslop P , Mayeux R ((2015) ) Coding mutations in SORL1 and Alzheimer disease. Ann Neurol 77: , 215–227. |
[36] | Yajima R , Tokutake T , Koyama A , Kasuga K , Tezuka T , Nishizawa M , Ikeuchi T ((2015) ) ApoE-isoform-dependent cellular uptake of amyloid-beta is mediated by lipoprotein receptor LR11/SorLA. Biochem Biophys Res Commun 456: , 482–488. |
[37] | Perneczky R , Alexopoulos P , Eisele T , Förstl H , Kurz A ((2010) ) Does the Apolipoprotein E genotype influence amyloid precursor protein sorting by sortilin-related receptor: Implications for Alzheimer’s disease? Med Hypotheses Res 6: , 19–24. |
[38] | van der Flier WM , Pijnenburg YA , Fox NC , Scheltens P ((2011) ) Early-onset versus late-onset Alzheimer’s disease: The case of the missing APOE varepsilon4 allele. Lancet Neurol 10: , 280–288. |
[39] | Rannikmae K , Samarasekera N , Martinez-Gonzalez NA , Al-Shahi Salman R , Sudlow CL ((2013) ) Genetics of cerebral amyloid angiopathy: Systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 84: , 901–908. |
[40] | Bu G ((2009) ) Apolipoprotein E and its receptors in Alzheimer’s disease: Pathways, pathogenesis and therapy. Nat Rev Neurosci 10: , 333–344. |
[41] | Schuur M , van Swieten JC , Schol-Gelok S , Ikram MA , Vernooij MW , Liu F , Isaacs A , de Boer R , de Koning I , Niessen WJ , Vrooman H , Oostra BA , van der Lugt A , Breteler MM , van Duijn CM ((2011) ) Genetic risk factors for cerebral small-vessel disease in hypertensive patients from a genetically isolated population. J Neurol Neurosurg Psychiatry 82: , 41–44. |
[42] | van Vliet P ((2012) ) Cholesterol and late-life cognitive decline. J Alzheimers Dis 30: (Suppl 2), S147–S162. |
[43] | Reitz C ((2013) ) Dyslipidemia and the risk of Alzheimer’s disease. Curr Atheroscler Rep 15: , 307. |
[44] | Ponomareva NV , Korovaitseva GI , Rogaev EI ((2008) ) EEG alterations in non-demented individuals related to apolipoprotein E genotype and to risk of Alzheimer disease. Neurobiol Aging 29: , 819–827. |
[45] | de Waal H , Stam CJ , Blankenstein MA , Pijnenburg YA , Scheltens P , van der Flier WM ((2011) ) EEG abnormalities in early and late onset Alzheimer’s disease: Understanding heterogeneity. J Neurol Neurosurg Psychiatry 82: , 67–71. |
Figures and Tables
Fig.1
Pedigree of family with four generations of AD patients. Black diamonds: Family members affected with AD; White diamonds: non-affected family members at time of death or last screening; Grey diamond: no consent to reveal disease history; “W” in diamond: family member included in whole exome sequencing; ‘n’ in a diamond, multiple family members merged and represented as one; AD [number], Alzheimer’s disease with age at diagnosis; d [number], age at death. E4/E4, APOE-ɛ4 homozygosity; SORL1+, subject is positive for the variant c.2012A>G in SORL1; TSHZ3+ subject is positive for the variant c.707C>T in TSHZ3; Grey text, DNA was not available for these family members, we estimated the chances that an individual has a given APOE genotype, based on (i) frequency of genotype combinations in the Dutch population, (ii) Mendelian inheritance patterns given the genotype distribution within the family structure and (iii) disease status. The chances for APOE genotypes do not add up to 100% when (smaller) chances for other APOE genotypes remain (Supplementary Table 1). Inferred chances of carrying of SORL1 and TSHZ3 genes are based on normal Mendelian inheritance patterns; Sex is not indicated and the order of siblings is rearranged to avoid recognition of this family and individual family members. See Supplementary Table 1 for list of genotype/phenotype data per-family member.
![Pedigree of family with four generations of AD patients. Black diamonds: Family members affected with AD; White diamonds: non-affected family members at time of death or last screening; Grey diamond: no consent to reveal disease history; “W” in diamond: family member included in whole exome sequencing; ‘n’ in a diamond, multiple family members merged and represented as one; AD [number], Alzheimer’s disease with age at diagnosis; d [number], age at death. E4/E4, APOE-ɛ4 homozygosity; SORL1+, subject is positive for the variant c.2012A>G in SORL1; TSHZ3+ subject is positive for the variant c.707C>T in TSHZ3; Grey text, DNA was not available for these family members, we estimated the chances that an individual has a given APOE genotype, based on (i) frequency of genotype combinations in the Dutch population, (ii) Mendelian inheritance patterns given the genotype distribution within the family structure and (iii) disease status. The chances for APOE genotypes do not add up to 100% when (smaller) chances for other APOE genotypes remain (Supplementary Table 1). Inferred chances of carrying of SORL1 and TSHZ3 genes are based on normal Mendelian inheritance patterns; Sex is not indicated and the order of siblings is rearranged to avoid recognition of this family and individual family members. See Supplementary Table 1 for list of genotype/phenotype data per-family member.](https://content.iospress.com:443/media/jad/2017/56-1/jad-56-1-jad160091/jad-56-jad160091-g001.jpg)
Fig.2
Immunohistochemistry in temporal cortex of subject II.4. a) Immunohistochemical staining for Aβ reveals cerebral Aβ angiopathy (black arrow), classical plaques (arrow head), and diffuse plaques (white arrow) in the temporal cortex (10x obj.); b) Immunohistochemical staining for tau (mab AT8) reveals neuropil threads, (pre)tangles (arrow), and neuritic plaques (arrow head) in the temporal cortex (10x obj.).
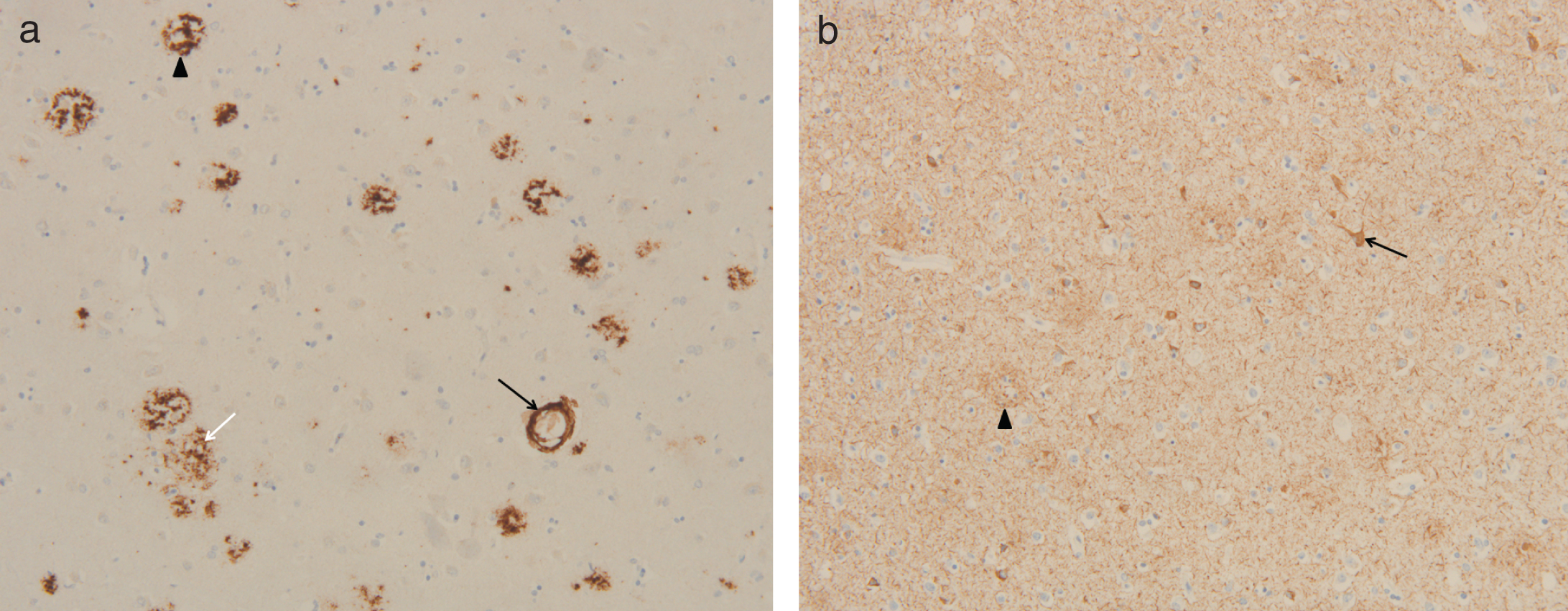
Fig.3
Baseline MR imaging of subject II.6. Cerebral MRI imaging of subject II.6 at age 70 showing several microbleeds (arrows). T2 weighted image.
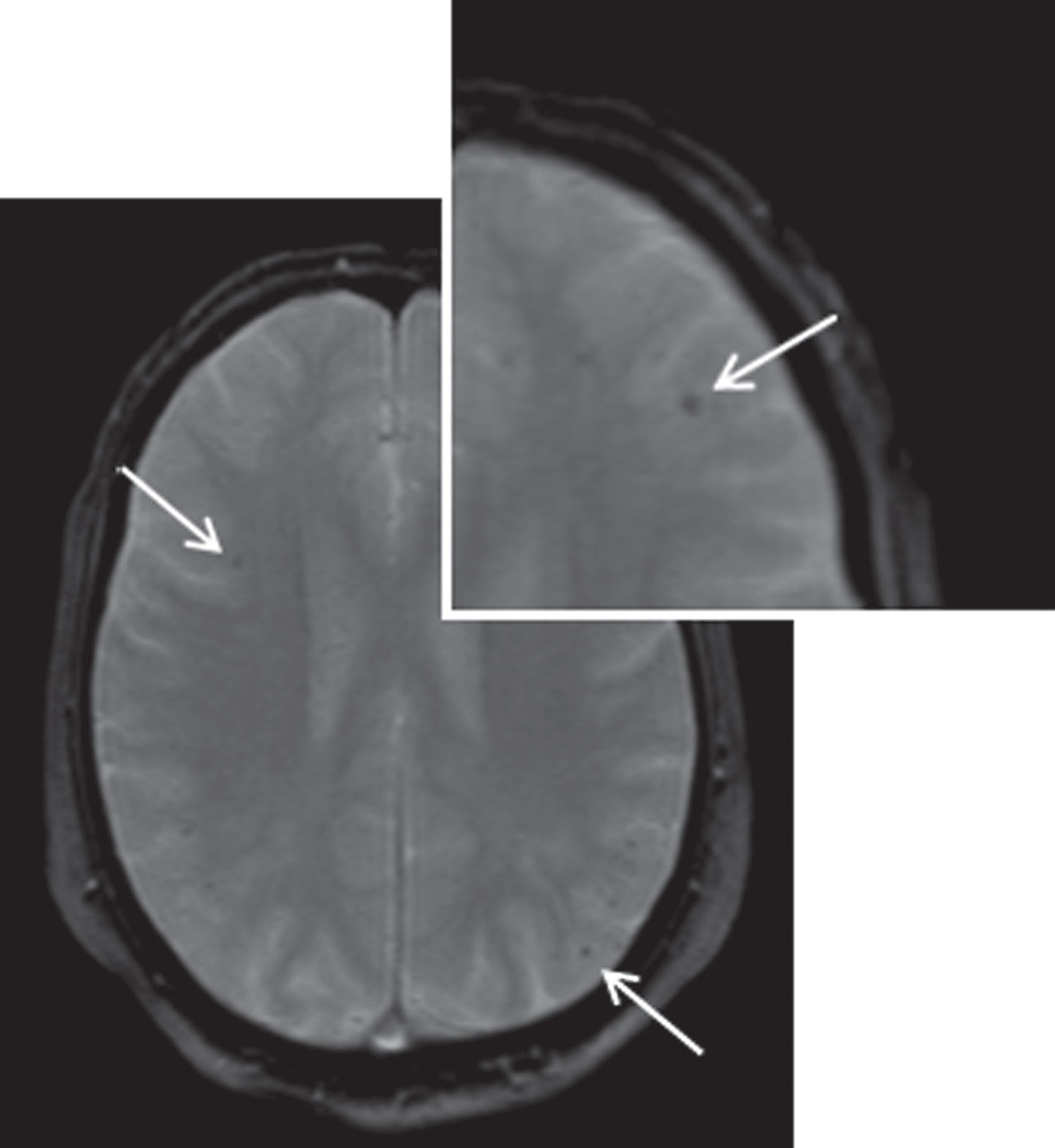
Fig.4
p.Asn674Ser in the SORL1 protein. SORL1 is located on chromosome 11q23.2-q24.2 and codes for a 250-kDa membrane protein with seven distinct domains. Blue (dark grey), extracellular domains; green (light grey), intracellular domain; arrow, the p.Asn674Ser variant we identified in this family; P, Pro-peptide; VPS10, vacuolar protein sorting domain 10; LDLR-B, LDL-receptor class B repeats; EGF, epidermal growth factor precursor type repeat; LDLR-A, LDL-receptor class A repeats; FN-III, fibronectin type-III repeats; IC, intracellular component; LDL, low density lipoprotein. The figure is based upon information from Uniprot (http://www.uniprot.org/uniprot/Q92673), transcript NM_003105.



