Evidence For and Against a Pathogenic Role of Reduced γ-Secretase Activity in Familial Alzheimer’s Disease
Abstract
The majority of mutations causing familial Alzheimer’s disease (fAD) have been found in the gene PRESENILIN1 (PSEN1) with additional mutations in the related gene PRESENILIN2 (PSEN2). The best characterized function of PRESENILIN (PSEN) proteins is in γ-secretase enzyme activity. One substrate of γ-secretase is encoded by the gene AMYLOID BETA A4 PRECURSOR PROTEIN (AβPP/APP) that is a fAD mutation locus. AβPP is the source of the amyloid-β (Aβ) peptide enriched in the brains of people with fAD or the more common, late onset, sporadic form of AD, sAD. These observations have resulted in a focus on γ-secretase activity and Aβ as we attempt to understand the molecular basis of AD pathology. In this paper we briefly review some of the history of research on γ-secretase in AD. We then discuss the main ideas regarding the role of γ-secretase and the PSEN genes in this disease. We examine the significance of the “fAD mutation reading frame preservation rule” that applies to PSEN1 and PSEN2 (and AβPP) and look at alternative roles for AβPP and Aβ in fAD. We present a case for an alternative interpretation of published data on the role of γ-secretase activity and fAD-associated mutations in AD pathology. Evidence supports a “PSEN holoprotein multimer hypothesis” where PSEN fAD mutations generate mutant PSEN holoproteins that multimerize with wild type holoprotein and dominantly interfere with an AD-critical function(s) such as autophagy or secretion of Aβ. Holoprotein multimerization may be required for the endoproteolysis that activates PSENs’ γ-secretase activity.
DISCOVERY OF THE AUTOSOMAL DOMINANT FAMILIAL ALZHEIMER’S DISEASE LOCI AβPP, PSEN1 & PSEN2
In 1906, Alois Alzheimer first identified foci (plaques) in the cerebral cortex staining in a similar manner to starch (hence “amyloid”) in the brain of the late Auguste Deter, a patient who had suffered from presenile dementia [1]. Nearly 80 years later (in 1985), the discovery that Alzheimer’s disease (AD) brains and Down syndrome brains share vascular deposits of amyloid-β (Aβ) led to the suggestion that AD may be caused by a genetic defect on Chromosome 21 [2, 3]. The subsequent discovery that Aβ is also found in neuritic plaques in AD brains [4, 5] founded the amyloid hypothesis that Aβ is central to the pathological process causing the disease. Great weight was lent to the amyloid hypothesis by the subsequent identification of a mutation causing familial AD (fAD) in the AβPP gene [6] and, after the PSEN1 and PSEN2 fAD loci were identified in 1995 [7, 8], by the realization that mutations in the PSEN genes could alter Aβ synthesis [9–12]. (Other, rarer fAD loci have since been identified, e.g., SORL1 [13, 14]). Subsequent molecular analyses including inhibitor studies have shown that the PSEN proteins form the catalytic core of the γ-secretase enzyme complexes that cleave AβPP to form Aβ (e.g., [15–17]). While the effects of fAD mutations on Aβ production have been seen to be variable (for example, see summaries in [18, 19]), a consistent characteristic is a shift in the ratio of longer versus shorter forms of Aβ and this is thought to favor the formation of purportedly toxic aggregates [19, 20]. Nevertheless, during the past decade, the amyloid hypothesis has come under increasing criticism for a number of reasons including the lack of overt neuronal loss following deposition of human Aβ in mouse brains [21] (although synaptic toxicity is apparent [22]) and the failure of therapeutic approaches based on reduction of Aβ production or aggregation [23] (although this may be due to the advanced disease stages at which treatment has been attempted [24]). However, it is not the purpose of this paper to review the vast literature that now addresses the role of Aβ in AD. (Interested readers can refer to a number of excellent reviews on that topic such as [25–27].) Instead we will focus on the effects of mutations in the PSEN genes that represent the overwhelming majority of known fAD mutations.
Before γ-secretase can cleave a type I transmembrane domain protein (having a luminal/extracellular N-terminus and a cytosolic C-terminus), the luminal/extracellular domain must be almost entirely excised [28]. The luminal/extracellular domain of AβPP can be cleaved by a number of enzyme activities (Fig. 1A, 1B) but only cleavage at AβPP’s “β-site” by a “β-secretase” allows production of the Aβ peptide. Subsequent cleavage by γ-secretase of the remaining transmembrane fragment of AβPP, C99, releases Aβ and the AβPP intracellular domain, AICD (see later and Fig. 1A).
ARE PRESENILINs THE ONLY γ-SECRETASES?
For a decade following the identification of the PSEN loci, there was uncertainty regarding whether PRESENILIN proteins were γ-secretases or the only source of γ-secretase activity. For example, in 2001, Armogida et al. [29] showed that mouse embryonic fibroblasts that apparently lacked Psen1 and Psen2 gene activity, indeed lost the ability to cleave within the transmembrane domain of an introduced Notch1 protein γ-secretase substrate. However, they could still produce 40 and 42 amino acid residue forms of Aβ (Aβ40 and Aβ42) from endogenous mouse AβPP. In 2002, Wilson et al. [30] showed that neurons harvested from mouse embryos lacking Psen1 and Psen2 activity could still produce, specifically, Aβ42 in the endoplasmic reticulum (ER)/intermediate compartment indicating the presence of an additional γ-secretase activity in the early secretory pathway. In 2003, Taniguchi et al. fractionated membranes from cells lacking PSEN1 and PSEN2 activity and identified a fraction with apparent γ-secretase activity but lacking PSEN protein [31]. Lai et al. [32] in 2006 analyzed γ-secretase-like activity in the membranes of blastocyst-derived cells and saw continued production of Aβ that could not be inhibited with the γ-secretase inhibitor L-685,458. However, the activity could be inhibited with the general aspartyl protease inhibitor pepstatin A. The production of Aβ was also observed in membranes from blastocyst-derived cells lacking PSEN proteins. Interestingly, the optimal pH for production of Aβ40 and Aβ42 by this activity was pH 6.0 compared to the measured optimum pH for Aβ production when the L-685,458 was not present of closer to pH 7.0. This suggests that the two activities may be distributed in different subcellular compartments. The PSEN-independent γ-secretase-like activity was also responsible for a greater proportion of total Aβ42 production than of total Aβ40. Other evidence that PRESENILINs might not be essential for production of Aβ included that expression of human AβPP in a yeast species apparently lacking PRESENILIN orthologs, Pichia pastoris, nevertheless permitted formation of Aβ peptides [33]. However, the PRESENILINs were, ultimately, conclusively shown to function as γ-secretases by the demonstration that transition state analogue inhibitors of γ-secretase bind to the PSENs [34], by reconstruction of the enzyme complex (consisting of the four proteins NCSTN, PSENEN, APH1 and PSEN) in the yeast Saccharomyces cerevisiae [17] and by the demonstration that recombinant PSEN alone, when expressed in and purified from bacteria and then reconstituted into liposomes, retained some ability to cleave AβPP [35].
The studies above suggest that a non-PSEN-dependent form of γ-secretase-like activity can be present in animal cells and can produce Aβ. One protein that may provide this activity is the acidic aspartyl protease Cathepsin D (CTSD). CTSD is capable of producing Aβ in assays where AβPP fragments equivalent to those produced after cleavage by β-secretase are provided [36, 37]. In human brain, BACE1 protein appears to provide most of the β-secretase activity generating Aβ [38] (although in vitro studies have indicated that CTSD may also cleave AβPP at its β-site raising the possibility that this one protease may be sufficient to produce Aβ from AβPP [39]). CTSD shows maximum activity at pH 3.8 [40] similar to BACE1’s optimum of pH 4.5 [41]. Curiously, BACE1 is nearly inactive at pH 6.0 which is the optimum for the γ-secretase-like activity described by Lai et al. above [32] who also showed that γ-secretase has a pH optimum closer to 7! This is despite the fact that a protein complex has been detected containing both PSEN1 and BACE1 (together with OPIOID RECEPTOR, DELTA-1; OPRD1) [42] indicating that the γ-secretase and β-secretase enzyme activities may (at least in some situations) be spatially co-located. However, to confirm that CTSD provides the γ-secretase-like activity in cells lacking PRESENILINs it will be necessary to analyze Aβ production in triple-mutant cells lacking all of PSEN1, PSEN2, and CTSD.
Finally, it is worth mentioning the fascinating case of another γ-secretase substrate, the NERVE GROWTH FACTOR RECEPTOR, NGFR (p75(NTR)) and its close homolog NRH1. NRH1 is found in non-mammalian vertebrates while a truncated form, NRH2 (also known as NRADD) exists in mammals [43]. The sequence similarity between NGFR and NRH1 points to a relatively recent evolutionary origin by a gene duplication event in vertebrates. However, unlike NGFR, the apparent intramembraneous cleavage of NRH1 (and NRH2) cannot be inhibited by the γ-secretase inhibitors DAPT and WPE [43]. Unpublished preliminary investigation of the zebrafish orthologue of NRH1 in Lardelli’s laboratory supports these observations. However, another report on NRH2/NRADD did see inhibition of cleavage of NRH2 with γ-secretase inhibitors L685,458; DAPT and Compound E [44] and resolution of these conflicting observations is required. Was the common ancestor of the NGFR and NRH1 proteins cleaved by γ-secretase or the “other” unidentified γ-secretase-like enzyme? What determines the susceptibility of a transmembrane protein to γ-secretase cleavage when no clear cleavage site recognition sequence or motif has been identified? [45].
CURRENT THINKING ON THE ROLE OF PRESENILINS IN AD
Less than 1% of AD cases are thought to be due to autosomal dominant mutations [46]. Nevertheless, research focused on how these mutations cause AD at the cellular and molecular level has had enormous influence on how we view the etiology of both early onset fAD and the far more common late onset sporadic AD (sAD). Over 205 different fAD mutations have been identified in the PSEN1 gene [47]. In a large French study of 130 families showing autosomal dominant early-onset AD [14], 82 families (63%) possessed missense mutations in PSEN1. A further 7 families (5%) showed missense mutations in PSEN2. Therefore, mutations in the PSEN genes accounted for over two-thirds of autosomal dominant fAD in the study. 14 missense mutations in AβPP and 11 duplications of AβPP (i.e., 19% of families) were also found. The predominant focus on Aβ as the cause of AD pathology has meant that most research on the role of PSEN1 and PSEN2 mutations in fAD has examined their effects on cleavage of AβPP and Aβ production.
It is a very striking characteristic of the fAD mutations in PSEN1, PSEN2, and AβPP that none of the hundreds of these fAD mutations cause loss of the gene (e.g., by deletion) or truncation of the coding sequences (e.g., by nonsense or frameshift mutations) [47]. Even mutations causing exon loss from transcripts due to deletion of exon DNA [48] or through effects on splicing [49–51] result in transcripts with open reading frames coding for normal C-terminal sequences. (There are some rare exceptions to this “fAD mutation reading frame preservation rule” that are discussed below.) The concentration of the AβPP fAD mutations near the α-, β-, and γ-secretase cleavage sites in the protein supports that the mutations alter these cleavage events (reviewed in [52]). However, the fAD mutations in the PSEN proteins are scattered throughout their peptide chains with a somewhat higher frequency in their numerous transmembrane domains (see Fig. 1C, D). Three basic ideas have been suggested to explain this lack of any focus to the position of the fAD mutations in PSEN1 or PSEN2:
1. Mutations in PSEN Proteins Shift the Profile of Aβ Production toward Longer Peptides
γ-Secretase cleavage of AβPP occurs after the luminal/extracellular domain of the protein is excised by α– or β-secretase activity. If AβPP is cleaved by β-secretase at its β-site (rather than at its β’-site [41, 53] or at the α-site, see Fig. 1), then Aβ peptides of around 40 amino acid residues length can subsequently be formed. Following α– or β-secretase cleavage of AβPP, the membrane-bound fragment is further cleaved by the γ-secretase enzyme complex at what is termed the ɛ-site near the cytosolic extent of AβPP’s transmembrane domain [54–56] (Fig. 1A, 1B). This “endopeptidase” cleavage by PRESENILIN releases AβPP’s cytosolic “intracellular domain” (AICD) that interacts with a number of intracellular signaling pathways and acts as a transcription factor (reviewed in [57, 58]). Subsequent “carboxypeptidase” cleavages by PRESENILIN are thought to shorten the remaining, membrane-embedded portion of AβPP until its hydrophobicity is sufficiently reduced that it can escape from the lipid bilayer as soluble Aβ [59, 60]. The position of the initial ɛ-site cleavage is variable [56] and subsequent cleavages by PRESENILIN may occur progressively from the carboxy-terminal (the “progressive proteolysis hypothesis” [61, 62]) along either of two “product lines” governed by the initial cleavage [61, 63]. An attractive aspect of this idea is that the many and widely distributed fAD mutations in the PSEN proteins can be thought of as generally reducing the efficiency of γ-secretase cleavage of AβPP resulting in the commonly cited phenomenon of increased Aβ peptide length in fAD (e.g., [9, 10]) and sAD [64] brains. This idea also aligns the apparent effects of fAD mutations in both of the PSENs with mutations in AβPP that affect its cleavage by γ-secretase, i.e., either type of mutation leads to formation of longer Aβ forms ([65] see below).
In support of this apparently unifying molecular mechanism underlying fAD are the results of cell-free γ-secretase assays published in 2012 by Ch
Another troubling aspect of the idea that promotion of Aβ42 over Aβ40 production is critical in AD is the fact that some fAD mutations in PSEN1 dramatically decrease the ability of γ-secretase to produce Aβ while the shift in the Aβ42/Aβ40 ratio need not be dramatic (e.g. [71]). It is difficult to understand why a slight isoform ratio shift should be important in promoting pathology via Aβ aggregation when, presumably, aggregation would be inhibited by a decrease in the total Aβ concentration (including, as observed in [71], either no significant change or a decrease in the concentration of Aβ42).
2. Changes in the Level of γ-Secretase Activity are Critical
The γ-secretase enzyme cleaves over 90 substrate proteins including those involved in critical forms of cell signaling such as Notch receptors and NGFR (reviewed by [72]). In 2007, Shen and Kelleher proposed the Presenilin hypothesis which states that changes in the activity of the PRESENILINs underlie fAD and, possibly, sAD [18]. Aβ is incorporated into the model by postulating that it may inhibit normal γ-secretase activity (something that has actually been observed under non-physiological conditions in cell culture [73]) and that it interferes with synaptic function and signal transduction. While acknowledging the diverse functions of the PRESENILINs including in regulation of Wnt signaling (e.g., by regulating phosphorylation of β-catenin [74]) and in Ca2 + ion homeostasis, the hypothesis is primarily γ-secretase focused.
In 2013, Shen, Kelleher, and co-workers published an analysis stated to support a dominant negative interaction between fAD mutant forms of PSEN1 and the wild type (non-mutant) form as would be found in heterozygous individuals possessing dominant fAD mutations [75]. In the study, they co-tranfected constructs expressing mutant and wild type PSEN1 in a 1:1 ratio at (purportedly) sub-saturating levels into mouse embryonic fibroblasts lacking mouse Psen genes. They saw reductions in γ-secretase activity as evidenced by reduced production of AICD from co-transfection with the C99 fragment of AβPP or of the Notch intracellular domain, NICD, from a truncated form of the Notch1 receptor. (N-terminally truncated forms of transmembrane protein such as C99 are commonly used in γ-secretase assays since these do not require prior membrane-proximal, extracellular/luminal cleavage in order to become γ-secretase substrates.) However, the reductions in AICD and NICD were not >50% as one might expect for adominant negative effect. While this may be explained by the claim that the levels of PSEN1 were not saturating, one needs to remember that, in cell transfection experiments, only a subset of the cells present are competent to absorb the expression vector DNAs [76] and the expression levels of the vectors in these individual cells can be quite high, while the expression measured over the total cell population will be lower. Also, as Thinakaran et al. showed in 1997 [77], cells appear to possess a mechanism that limits total PSEN protein to a particular level.
In 2015 the debate around the importance of changes in γ-secretase activity gained additional intensity [78] with the publication of another paper by Shen, Kelleher, and co-workers with direct relevance to the question of whether fAD mutations in the PSENs have dominant negative effects on γ-secretase activity. Xia et al. [71] had introduced fAD mutations of human PSEN1 (L435F or C410Y) into the endogenous Psen1 gene of mice and found that levels of de novo Aβ production (reflecting γ-secretase activity) were reduced by approximately half in in vitro γ-secretase assays of brain tissue from embryonic and 3 month old heterozygous L435F mice. This does not support a dominant negative action of L435F mutant Psen1 on the γ-secretase activity of wild type Psen1. The L435F or C410Y mutant alleles showed almost complete loss of AβPP cleavage when homozygous. Interestingly, both mutations cause great increases in Psen1 holoprotein levels in heterozygous mouse brains (presumably due to decreased auto-endoproteolysis of Psen1 that activates the γ-secretase activity of the protein [79]). Endogenous levels of both Aβ40 and Aβ42 were decreased in the detergent-resistant (“insoluble”) fraction from 3-month-old heterozygous L435F mouse brains but little change was seen for the soluble fraction. This suggests either that clearance of Aβ was reduced (as might be expected from, e.g., inhibited lysosome function) or even that a homeostatic mechanism exists to maintain a particular level of Aβ expression in brain. However, the Aβ42/Aβ40 ratio was significantly increased in both fractions. We note also that in the recent study by Ch
The 2015 Xia et al. paper is important, since it provides evidence of a loss of γ-secretase activity in PSEN1 fAD mutant brains. However, it does not resolve whether this or changes in the Aβ42/Aβ40 ratio are critical to the disease. Indeed, another Psen1 mutation knock-in model from the laboratory of Shen and Kelleher — the G183V mutation [80] that apparently causes Pick disease (a frontotemporal dementia) without Aβ deposition [81, 82] — also shows decreased γ-secretase activity and Aβ production. Why should the same phenomena (decreased γ-secretase activity and Aβ production) produce two different diseases? This does not support that decreased γ-secretase specifically causes fAD. However, both Pick disease and AD are tauopathies where neurons show inclusions containing MICROTUBULE-ASSOCIATED PROTEIN TAU (MAPT) [83] and aberrant splicing of PSEN1 transcripts has been observed in sporadic frontotemporal dementia [84]. Takashima et al. [85] showed that PSEN1 protein binds MAPT together with a kinase that phosphorylates the latter, GLYCOGEN SYNTHASE KINASE 3-BETA (GSK3B). They also showed that transfection of fAD mutant forms of PSEN1 into COS-7 cells could increase phosphorylation of MAPT. Saura et al. [86] showed that loss of both PSEN1 and PSEN2 from the postnatal forebrain of mice causes hyperphosphorylation of MAPT. These observations suggest that decreased γ-secretase activity may be involved with the formation of MAPT inclusions although Watanabe et al. [80] did not observe these (or changes in the phosphorylation of MAPT) in the brains of their homozygous G183V mouse model.
We should note that the G183V mutation of human PSEN1 is unusual since it affects the spice donor site of exon 6 and produces both normally spliced transcripts and transcripts with truncated open reading frames [81, 83]. The Watanabe et al.paper shows that these aberrant transcripts are not completely degraded in brain tissue although nonsense-medicated mRNA decay appears to remove them completely in other tissues. We have analyzed the putative protein products of these aberrant transcripts using assays in zebrafish embryos and have seen dominant negative effects specifically on Notch signaling but not on γ-secretase cleavage of an AβPP orthologue [87]. Zebrafish Psen1 peptides equivalent to those putatively translated from the aberrant transcripts also incorporate specifically and avidly into detergent-resistant higher molecular weight complexes and bind to both Psen1 and Psen2 proteins [87]. Therefore, it is possible that the action of truncated forms of PSEN1 protein may underlie the frontotemporal dementia pathology of the G183V mutation of human PSEN1.
A very significant advance in our understanding of γ-secretase came in 2015 with publication of the first detailed atomic structure of the enzyme complex by Bai et al. [88]. Notably, these researchers introduced ten different fAD mutations into PSEN1 and saw no consistent change in γ-secretase activity. Some mutations abolished cleavage of C99, some had little influence, while other actually increased γ-secretase activity! As Bai et al. stated, “These observations strongly suggest a disconnection between the total protease activity of γ-secretase and the development of fAD”. They also observed, “All eight mutations for which the Aβ42/Aβ40 ratio can be calculated led to increased Aβ42/Aβ40 ratios compared with wild-type γ-secretase … The generally increased ratios of Aβ42 over Aβ40 may suggest a causal relationship for development of fAD, but could also be explained by other possibilities.” We describe two such possibilities later in this paper.
3. Changes in the Non-γ-Secretase Activity(ies) of PSEN Underlie AD Pathology
The γ-secretase enzyme is a complex of four proteins and, if reduced γ-secretase activity was the cause of fAD, then we might expect fAD mutations to occur in the other γ-secretase complex components. Remarkably, these have never been found but mutations in PSEN1, NCSTN, and PSENEN have been found in another disease, inherited acne inversa [89] (also known as hidradenitis suppurativa). It is also very significant that the single known mutation in PSEN1 causing acne inversa, P242LfsX11 (Fig. 1C), does not follow the “fAD mutation reading frame preservation rule” since it causes a frameshift in exon 7 and truncation of the open reading frame. This mutation does not appear to cause fAD [89].
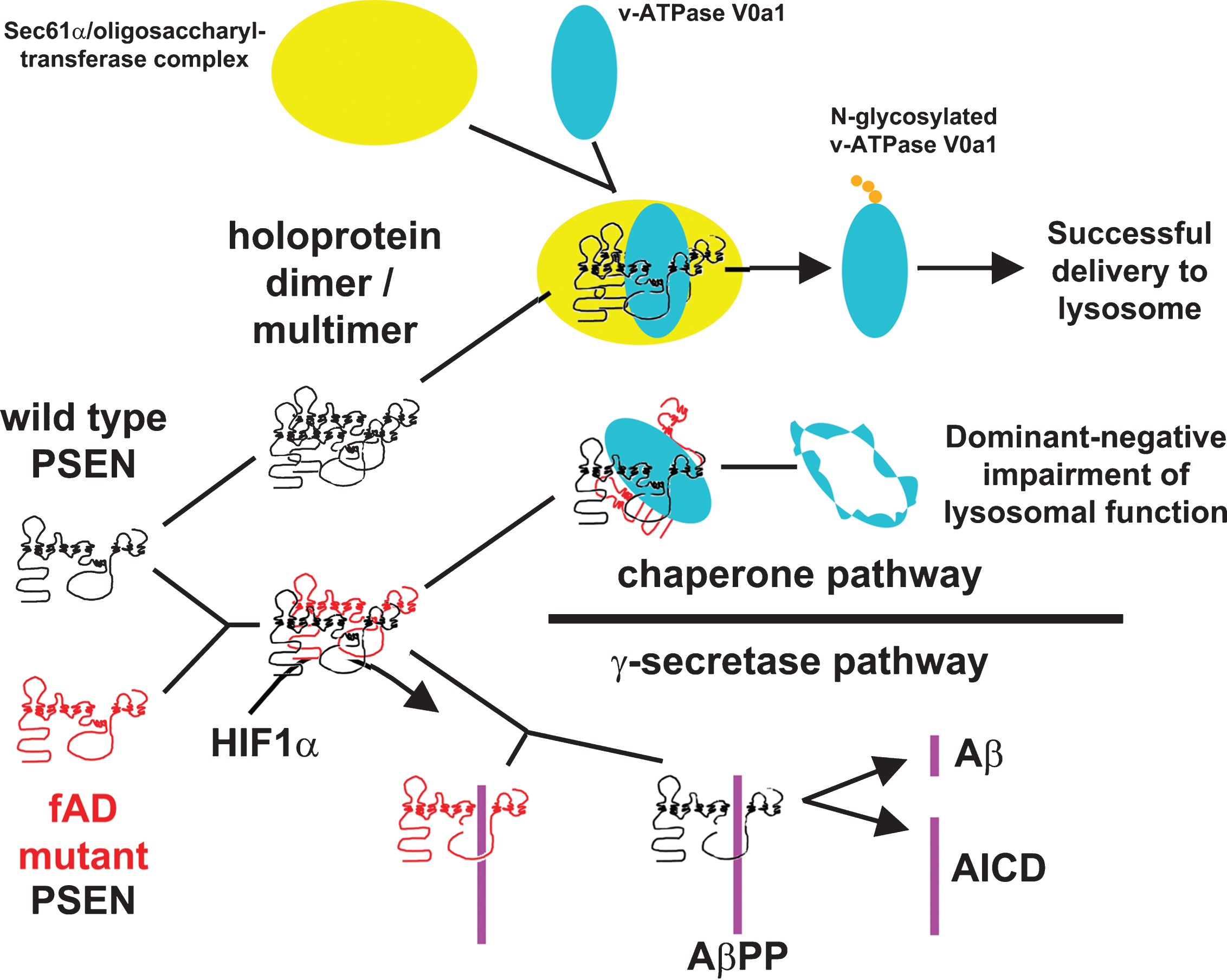
The existence of 205+ fAD mutations in PSEN1 following the “fAD mutation reading frame preservation rule” and a single PSEN1 mutation that does not preserve the open reading frame and that causes a completely different disease, strongly supports that mutations in PSEN1 can only cause fAD when they preserve the “full-length” of the protein. A parsimonious explanation for this phenomenon may have been revealed by the Heilig et al. paper [75], that showed strong association between mutant and wild type forms of the PSEN1 holoprotein (a phenomenon also noted by others [90, 91]), and by work from Nixon and co-workers demonstrating an effect of fAD PSEN1 mutations on autophagy [92]. In 2010, Lee et al. showed that heterozygous fAD mutations in PSEN1 inhibit macroautophagy by reducingN-glycosylation of the V0a1 subunit of vacuolar [H+] ATPase leading to reduced acidification of lysosomes. Subsequently, two other laboratories disputed these findings [93, 94] leading to a very “vigorous” debate at the AlzForum online site [95]. However, increased lysosomal pH due to mutations in PSEN1 has since been supported by additional work [96].
The Lee et al. paper notes three remarkable phenomena: a) It is the PSEN1 holoprotein, not its endoproteolysed form, that complexes with the proteins required for N-glycosylation of the V0a1 subunit of the vacuolar [H+] ATPase; b) loss of the γ-secretase complex component NCSTN does not affect macroautophagy; and, c) chemical inhibition of γ-secretase activity does not affect macroautophagy. These three phenomena exactly align with what we know of mutations in PSEN1 causing fAD, i.e., a) only mutations that preserve the full length of the PSEN1 holoprotein cause fAD; b) only mutations in PSEN1 and not other γ-secretase complex components cause fAD and; c) decreased γ-secretase activity does not cause fAD (since it is observed in mouse models of human PSEN1 mutations causing either fAD or FTD and so does not differentiate between them and since reduced γ-secretase activity causes acne inversa without fAD). This idea is also consistent with a single, fundamental mechanism for both fAD and sAD since disturbances of lysosomal function have been observed in human fAD brains [97] and fibroblasts [92] and disturbances of autophagy have been observed in sporadic late onset AD brains [98]. Therefore, lysosomal dysfunction provides a unifying pathological mechanism underlying AD. Such a unifying mechanism is important because, at the genetic level, there is little overlap between mutations causing fAD and the genetic risk loci for sAD identified in genome-wide association studies (GWAS). To date, only the gene SORL1 has been identified as a locus in both fAD and sAD, although other connections are apparent. For example, a search using human PSEN1 in the gene co-expression database COXPRESdb [99] shows that the most tightly co-regulated gene is the sAD risk locus PICALM (Ebrahimie, Lardelli et al., unpublished results), a locus repeatedly identified in GWAS [100–103] and that influences autophagy [104]. Also, a truncated isoform of PSEN2, “PS2V”, is upregulated in sAD brains [105]. PS2V is induced by hypoxia/oxidative stress, is associated with increased production of Aβ [106, 107], can suppress the unfolded protein response (UPR) and can boostγ-secretase activity [107, 108] through an unknown mechanism that may involve interaction with full-length PSEN protein [87].
Other activities of the PSEN proteins have also been seen to be dependent on the holoprotein rather than the endoproteolytically cleaved, γ-secretase-active form. PSEN holoproteins have been proposed to act as Ca2 + “leak channels” in the ER with fAD mutants unable to perform this function [65, 109]. In 2009, Schon and co-workers showed that the PSEN proteins are concentrated in a particular, specialized functional domain of the ER where it is closely juxtaposed to mitochondria, the “mitochondria associated membranes” (MAM, see [110]). Formation of protein disulphide bonds occurs in the MAM involving relatively large amounts of H2O2 formation (oxidative protein folding, reviewed by [111]). The MAM is comprised of detergent resistant membrane with the characteristics of a lipid raft [110, 112]. It is responsible for cholesteryl ester and phospholipid synthesis [112], controls mitochondrial activity and apoptosis via Ca2 + signaling (reviewed in [113]), is a site of autophagosome initiation [114] and is required for innate immune inflammatory responses [115]. Therefore, the MAM is a nexus for cellular activities and phenomena commonly associated with AD pathology. Indeed, Schon’s laboratory has shown that fibroblasts from both fAD and sAD individuals exhibit increased MAM formation leading to the hypothesis that it is dysfunction in communication between the ER and mitochondria that causes AD (the “MAM hypothesis”, [116]). While changes in PSEN expression and function can alter the degree of MAM formation in cells, MAM formation does not appear to be dependent on γ-secretase activity, implying that it is changes in PSEN holoprotein function that are critical.
In summary, we suggest that a parsimonious explanation for the PSEN “fAD mutation reading frame preservation rule” and the dominant nature of these mutations is a “PSEN holoprotein multimer hypothesis” whereby fAD mutations in PSEN1 or PSEN2 cause disruption of a holoprotein function (or functions) that are critical for maintenance of cellular homeostasis in an aging brain. The mutations are dominant due to inhibitory multimerization of mutant holoproteins with wild type holoprotein. The hypothesis is summarized in Fig. 2 (showing the possibility that the fAD-critical function disrupted by mutation of the holoprotein is N-glycosylation of the V0a1 subunit of vacuolar [H+] ATPase as suggested by Leeet al., [92]).
REAL AND ARTEFACTUAL EXCEPTIONS TO THE “fAD MUTATION READING FRAME PRESERVATION RULE”
Until recently no possible exceptions were known to the rule that fAD mutations preserve the open reading frames of PSEN1, PSEN2, and A βPP. However, in 2010, Jayadev et al. published a report on fAD mutations in PSEN2 that included discovery of a two nucleotide deletion causing a frameshift near the downstream end of exon 4 followed shortly thereafter by a premature termination of the open reading frame. If this mutant allele, K115Efs, produces a protein product then it would resemble the PS2V isoform mentioned previously that can both increase γ-secretase activity (and hence Aβ production) and inhibit the UPR by an unknown mechanism [107, 108] (see Fig. 1D). (The two phenomena may be mechanistically related [117].) Since PS2V is observed to be upregulated in sAD brains, the K115Efs mutation may provide another link between the pathologies of fAD and sAD [108]. However, K115Efs may be mechanistically unique among the PSEN fAD mutations since it may enhance Aβ production (or AβPP signaling) via increasing γ-secretase activity or it may promote Aβ oligomerization via suppression of the UPR (or both) rather than inhibiting Aβ breakdown and/or secretion via autophagy [118–120].
In 2014, Kadmiri et al. published two papers reporting the discovery of frameshift mutations in Moroccan fAD families [121, 122]. In both papers it is claimed that 17 sporadic cases of AD and eight families with AD were examined, so presumably the same patient cohort forms the basis for both studies. In one paper they claim to have discovered seven novel frameshift mutations in A βPP, three of which occur in families [121]. Remarkably, one particular mutant allele was discovered in three apparently separate families and, in one family, the proband possessed two separate frameshift alleles while an unaffected relative had a mutation affecting an AβPP transcript splice site! No mis-sense mutations in AβPP were reported. If we consider that the Alzheimer Disease & Frontotemporal Dementia Mutation Database lists 26 different AβPP mutations discovered for AD (10 complete gene duplications and 16 missense mutations) from tens of separate publications, the likelihood that so many novel, unprecedented fAD mutations in AβPP would be discovered in one study is infinitesimally small. In their second paper [122] Kadmiri et al. claim to have found frameshift mutations in exon 10 of PSEN1 (Fig. 1C) and in exons 5 and 9 of PSEN2 (Fig. 1D). The mutations were apparently found in three different families with no mutations found in sAD cases. However, no analysis of DNA from two or more affected individuals within a family is reported. Once again, with over 220 different mutations found collectively in PSEN1 and PSEN2 in dozens of separate studies, none of which destroy the open reading frame of the genes, the probability is extremely small that three different such mutations could be found within only eight AD families. For these reasons we consider it highly unlikely that the data in these two papers is reliable and verification of the data is essential before they can be regarded as part of the fAD mutation knowledge base. Unfortunately, the “mutations” the papers report have already been cited uncritically elsewhere in the scientific literature [123].
WHAT THEN OF AβPP, Aβ, AND THE Aβ42/Aβ40 RATIO?
The fact that mutations in the three major fAD genes all affect production of the Aβ peptide has been the most convincing evidence for the centrality of Aβ in the etiology of fAD. Therefore, if we are to discount changes in γ-secretase activity as being causative of fAD, another mechanism must be found by which mutations affecting the PSENs and AβPP can have inter related effects. In this light it may well be significant that AβPP is a substrate of chaperone-mediated autophagy [124] and that autophagy is involved in both the secretion and breakdown of Aβ [119]. AβPP has also been shown to have a very wide range of cellular functions. AβPP influences Fe ion homeostasis via its interaction with Ferroportin (SLC40A1, [125]) and AβPP affects embryo development, including vascular development, possibly via secretion of its extracellular/luminal domain [126, 127] and Aβ [128]. AβPP also influences the rate of protein synthesis [129], neurite outgrowth [130], and axonal pruning [131] as well as LPS-mediated innate immune responses [132]. Signaling via AICD regulates genes including MEMBRANE METALLOENDOPEPTIDASE (MME, also known as neprilysin) [133] and TRANSTHYRETIN, TTR [134] that encode proteins involved in degradation and clearance of Aβ from the CNS, respectively. One or more of these functions may interact with PSEN holoprotein activities. For example, alterations in PSEN gene expression affect metal homeostasis [135] and PSEN2 is also known to influence LPS-mediated innate immune responses while PSEN1 may not [136].
There are many different lengths of peptide cleaved from the membrane-proximal region of AβPP. These can also carry various forms of chemical modification that may influence their role in AD pathogenesis (reviewed by [137]). However, the most intensively studied peptides derived from AβPP are Aβ40 and Aβ42. For many years these forms of Aβ were regarded as unfortunately toxic but otherwise functionless by-products of AβPP cleavage [138]. However, numerous lines of evidence now argue against this. The sequence of Aβ is highly conserved in species as distantly related as primates (humans) and lobe-finned fishes (Coelacanth) [128] implying that mutations in Aβ are selectively disadvantageous. Aβ has neuroprotective properties [139] and may act variously as an antioxidant [140–145], in vascular development [128, 146] and function [147], as a regulator of long term potentiation [148] and even as an antimicrobial peptide of the innate immune system [149]. Aβ has also been implicated as a transcription factor. In 2011, Lahiri and colleagues claimed the existence of a DNA sequence motif binding Aβ in the promoters of the A βPP, BACE1, and APOE genes [150, 151]. In 2012, Piccini et al. [152] showed upregulation of BACE1 expression specifically by Aβ42 (but not Aβ40) applied to neuroblastoma cells apparently via the JNK signaling pathway and in 2014 Baracker et al. [153] showed that cells can absorb Aβ and transport it into nuclei where, specifically, Aβ42 (but not other isoforms) can form transcriptional complexes to repress expression of the genes LRP1 and KAI1. Aβ is upregulated by cellular stress, in particular hypoxia [154–156]. This is a selectively advantageous protective response since the molecular mechanism for upregulating Aβ under hypoxia/oxidative stress appears conserved in both zebrafish and humans, species that shared a common ancestor almost half a billion years ago [157]. The production of Aβ is very responsive to the level of expression of BACE1 [158] which itself is sensitive to hypoxia [154–156]. The “protective mutation” in AβPP (A673T) identified in studies of the Icelandic population and that reduces BACE1 cleavage of AβPP by around 40% [159] can be seen in this light. The mutation may well be reducing the cellular dysfunction that can accompany an over-vigorous protective response, much as reducing inflammatory responses can reduce cancer risk [160] or improve wound healing [161]. This is supported by the work of Weihong Song and colleagues [162] who showed that, in fact, the most common site of cleavage of AβPP by BACE1 is the β’ site that prevents Aβ formation by forming a shorter peptide (see Fig. 1). They also showed that the fAD-causing “Swedish” double mutation of AβPP inhibits this cleavage so that cleavage at the alternative β-site producing Aβ becomes more frequent. In this case the Swedish mutation may be seen as causing an upregulated stress response.
Recently, Villa et al. [163] showed that the master regulator of cellular responses to hypoxia, HYPOXIA-INDUCIBLE FACTOR 1, ALPHA SUBUNIT (HIF1α), binds directly to γ-secretase complexes to upregulate γ-secretase activity under low oxygen. This upregulation did not require increased transcription of γ-secretase complex components. Rather it appeared to be due to activation of existing “inactive γ-secretase”. It is possible that, under hypoxic stress, HIF1α promotes the endoproteolysis of a pre-existing pool of PSEN holoprotein to form active γ-secretase. Hypoxia is a form of cellular stress increasingly identified in AD [164, 165] and one implication of Villa and colleagues’ analysis is that fAD mutations might affect the effectiveness of this cellular response via alteration of an interaction (direct or indirect) between PSEN holoprotein and HIF1α. Hypoxia also upregulates autophagy [166] consistent with the role of PSEN1 holoprotein in lysosomal acidification and the concentration of PSEN proteins in the MAM. It is particularly intriguing that, in transgenic mice, expression of human fAD mutant forms of PSEN1 exclusively in neurons can cause a microvascular phenotype of abnormally looped vessels as well as “string vessels” [167]. Aβ peptides are known to be vasoactive [168] and their secretion requires autophagy [118] making them a candidate for a mechanism of communication between neurons and brain microvasculature. Since Aβ levels are so responsive to hypoxia (e.g., [169, 170]), we can speculate that the increase in brain Aβ deposition seen decades before onset of sAD [171] may be driven by the inability of aging brain vasculature to deliver sufficient oxygen. PSEN fAD mutations that promote abnormal microvascular phenotypes may thereby predispose to, and accelerate the onset of, AD. (See also a recent comprehensive review of the role of vasculature in AD by Di Marco et al., [172]).
Lastly, we should examine another of the strongest pieces of evidence used to support the pathological role of Aβ42 (and hence the pathological role of apparent changes in the endopeptidase activity of γ-secretase): The close and highly statistically significant inverse-correlation of variation in Aβ42/Aβ40 ratio with the mean age of onset of various fAD mutations in PSEN [173, 174]. As ever, we must remember that correlation is not causation and that change in the Aβ42/Aβ40 ratio may be acting as a very tight marker of the severity of another change in PSEN function that is actually causing the disease. In 1997, Cook et al. [175] used three independent methods to show that preventing the movement of AβPP from the ER/intermediate compartment to the Golgi in neurons greatly reduced Aβ40 while leaving Aβ42 production unaffected. In the same year, Hartmann et al. [176] used labeling of subcellular structures with monoclonal antibodies specifically binding either Aβ42 or Aβ40 to show the specific production of Aβ42 in the ER of neurons. As Area-Gomez et al. [110] showed, most of the γ-secretase activity in the ER is located in the MAM so, if change in PSEN function leads to increased association between the ER and mitochondria (increased MAM), we could expect the ratio of Aβ42 to Aβ40 to increase even if overall γ-secretase activity is decreased. Alternatively, Lai et al. [32] showed that the PSEN-independent, γ-secretase-like activity they detected specifically favors Aβ42 production so reduction in PSEN-dependent γ-secretase activity would again be expected to increase the ratio of Aβ42 to Aβ40. As quoted previously, Bai et al. saw increases in the Aβ42/Aβ40 ratio for all of the various fAD mutations that they introduced into human PSEN1 and that showed detectable Aβ production. However, their data support that the increase in the ratio is an intrinsic property of the mutant PSEN molecule itself since they apparently analyzed the activity of highly purified γ-secretase in an in vitro assay system [88].
CONCLUSION
If we accept that there is strong evidence implicating a primary role for PSEN holoproteins in fAD then particular questions arise. Is lysosomal acidification the relevant function of PSEN holoproteins affected in fAD, or is it changes in Ca2 + homeostasis or some other, yet unknown function? Where within cells do pools of PSEN holoprotein exist and what affects their equilibrium with γ-secretase complexes? What controls holoprotein stability? Do PSEN holoproteins function as dimers/multimers? How do fAD mutations affect holoprotein multimerization and function? With what molecules do holoproteins specifically interact and what different forms of holoprotein complex exist? Does the presence of mutant PSEN1 holoprotein increase the association of the ER with mitochondria and, if so, how does this occur? In what cell types are the function of PSEN holoproteins critical for fAD? Is the fAD-critical function of PSEN holoproteins mainly in neurons or are there critical functions in astrocytes, microglia and other neural cell types? How do changes in the rate of α-, β-, and γ-secretase cleavage of AβPP influence the fAD-critical PSEN holoprotein functions? It is clear that there is still a long way to travel to understand how mutations in the PSEN and AβPP genes cause AD but testing of this “PSEN holoprotein multimer hypothesis” may make the path a little clearer. A 2015 paper from Nixon and coworkers has shown that raised lysosomal pH due to loss of PSEN1 contributes to failure of cytosolic Ca2+ ion homeostasis. Lee et al., 2015, Cell Reports 12, 1430–1444.
ACKNOWLEDGMENTS
The authors declare no conflicts of interest, financial or otherwise, in publication of this paper. TJ is supported by a Commonwealth Scholarship. MN is supported by National Health and Medical Research Council (NHMRC) Project Grant APP1061006. SHMN is supported by NHMRC Project Grant APP1045507. GV is supported by the NHMRC (APP1045507 and APP1105698) and the Curtin University Senior Research Fellowship (CRF140196). GS is a current Sydney Medical School Foundation Fellow. GM, IM and ML are academic employees of their respective universities. ML is supported by the NHMRC (APP1045507 and APP1061006).
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/15-1186r1).
REFERENCES
[1] | Maurer K , Volk S , Gerbaldo H ((1997) ) Auguste D and Alzheimer’s disease. Lancet 349: , 1546–1549. |
[2] | Glenner GG , Wong CW ((1984) ) Alzheimer’s disease and Down’s syndrome: Sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun 122: , 1131–1135. |
[3] | Glenner GG , Wong CW ((1984) ) Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120: , 885–890. |
[4] | Wong CW , Quaranta V , Glenner GG ((1985) ) Neuritic plaques and cerebrovascular amyloid in Alzheimer disease are antigenically related. Proc Natl Acad Sci U S A 82: , 8729–8732. |
[5] | Masters CL , Simms G , Weinman NA , Multhaup G , McDonald BL , Beyreuther K ((1985) ) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A 82: , 4245–4249. |
[6] | Goate A , Chartier-Harlin MC , Mullan M , Brown J , Crawford F , Fidani L , Giuffra L , Haynes A , Irving N , James L , et al ((1991) ) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349: , 704–706. |
[7] | Sherrington R , Rogaev EI , Liang Y , Rogaeva EA , Levesque G , Ikeda M , Chi H , Lin C , Li G , Holman K , Tsuda T , Mar L , Foncin JF , Bruni AC , Montesi MP , Sorbi S , Rainero I , Pinessi L , Nee L , Chumakov I , Pollen D , Brookes A , Sanseau P , Polinsky RJ , Wasco W , Da Silva HA , Haines JL , Perkicak-Vance MA , Tanzi RE , Roses AD , Fraser PE , Rommens JM , St George-Hyslop PH ((1995) ) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375: , 754–760. |
[8] | Levy-Lahad E , Wasco W , Poorkaj P , Romano DM , Oshima J , Pettingell WH , Yu CE , Jondro PD , Schmidt SD , Wang K , et al ((1995) ) Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269: , 973–977. |
[9] | Scheuner D , Eckman C , Jensen M , Song X , Citron M , Suzuki N , Bird TD , Hardy J , Hutton M , Kukull W , Larson E , Levy-Lahad E , Viitanen M , Peskind E , Poorkaj P , Schellenberg G , Tanzi R , Wasco W , Lannfelt L , Selkoe D , Younkin S ((1996) ) Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med 2: , 864–870. |
[10] | Duff K , Eckman C , Zehr C , Yu X , Prada CM , Perez-tur J , Hutton M , Buee L , Harigaya Y , Yager D , Morgan D , Gordon MN , Holcomb L , Refolo L , Zenk B , Hardy J , Younkin S ((1996) ) Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature 383: , 710–713. |
[11] | Borchelt DR , Thinakaran G , Eckman CB , Lee MK , Davenport F , Ratovitsky T , Prada CM , Kim G , Seekins S , Yager D , Slunt HH , Wang R , Seeger M , Levey AI , Gandy SE , Copeland NG , Jenkins NA , Price DL , Younkin SG , Sisodia SS ((1996) ) Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron 17: , 1005–1013. |
[12] | Herreman A , Serneels L , Annaert W , Collen D , Schoonjans L , De Strooper B ((2000) ) Total inactivation of gamma-secretase activity in presenilin-deficient embryonic stem cells. Nat Cell Biol 2: , 461–462. |
[13] | Rovelet-Lecrux A , Legallic S , Wallon D , Flaman JM , Martinaud O , Bombois S , Rollin-Sillaire A , Michon A , Le Ber I , Pariente J , Puel M , Paquet C , Croisile B , Thomas-Anterion C , Vercelletto M , Levy R , Frebourg T , Hannequin D , Campion D , Investigators of the Gp ((2012) ) A genome-wide study reveals rare CNVs exclusive to extreme phenotypes of Alzheimer disease. Eur J Hum Genet 20: , 613–617. |
[14] | Pottier C , Hannequin D , Coutant S , Rovelet-Lecrux A , Wallon D , Rousseau S , Legallic S , Paquet C , Bombois S , Pariente J , Thomas-Anterion C , Michon A , Croisile B , Etcharry-Bouyx F , Berr C , Dartigues JF , Amouyel P , Dauchel H , Boutoleau-Bretonniere C , Thauvin C , Frebourg T , Lambert JC , Campion D , Collaborators PG ((2012) ) High frequency of potentially pathogenic SORL1 mutations in autosomal dominant early-onset Alzheimer disease. Mol Psychiatry 17: , 875–879. |
[15] | De Strooper B , Saftig P , Craessaerts K , Vanderstichele H , Guhde G , Annaert W , Von Figura K , Van Leuven F ((1998) ) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391: , 387–390. |
[16] | Wolfe MS , Xia W , Ostaszewski BL , Diehl TS , Kimberly WT , Selkoe DJ ((1999) ) Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 398: , 513–517. |
[17] | Edbauer D , Winkler E , Regula JT , Pesold B , Steiner H , Haass C ((2003) ) Reconstitution of gamma-secretase activity. Nat Cell Biol 5: , 486–488. |
[18] | Shen J , Kelleher RJ ((2007) ) The presenilin hypothesis of Alzheimer’s disease: Evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A 104: , 403–409. |
[19] | De Strooper B ((2007) ) Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep 8: , 141–146. |
[20] | Jarrett JT , Lansbury PT Jr ((1993) ) Seeding one-dimensional crystallization of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 73: , 1055–1058. |
[21] | Irizarry MC , Soriano F , McNamara M , Page KJ , Schenk D , Games D , Hyman BT ((1997) ) Abeta deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci 17: , 7053–7059. |
[22] | Mucke L , Selkoe DJ ((2012) ) Neurotoxicity of amyloid beta-protein: Synaptic and network dysfunction. Cold Spring Harb Perspect Med 2: , a006338. |
[23] | Karran E , Mercken M , De Strooper B ((2011) ) The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat Rev Drug Discov 10: , 698–712. |
[24] | ((2010) ) Why are drug trials in Alzheimer’s disease failing? Lancet 376: , 658. |
[25] | Herrup K ((2015) ) The case for rejecting the amyloid cascade hypothesis. Nat Neurosci 18: , 794–799. |
[26] | Musiek ES , Holtzman DM ((2015) ) Three dimensions of the amyloid hypothesis: Time, space and wingmen. Nat Neurosci 18: , 800–806. |
[27] | Murphy MP , LeVine H ((2010) ) Alzheimer’s disease and the amyloid-beta peptide. J Alzheimers Dis 19: , 311–323. |
[28] | Struhl G , Adachi A ((2000) ) Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol Cell 6: , 625–636. |
[29] | Armogida M , Petit A , Vincent B , Scarzello S , da Costa CA , Checler F ((2001) ) Endogenous beta-amyloid production in presenilin-deficient embryonic mouse fibroblasts. Nat Cell Biol 3: , 1030–1033. |
[30] | Wilson CA , Doms RW , Zheng H , Lee VM ((2002) ) Presenilins are not required for A beta 42 production in the early secretory pathway. Nat Neurosci 5: , 849–855. |
[31] | Taniguchi Y , Karlstrom H , Lundkvist J , Mizutani T , Otaka A , Vestling M , Bernstein A , Donoviel D , Lendahl U , Honjo T ((2002) ) Notch receptor cleavage depends on but is not directly executed by presenilins. Proc Natl Acad SciU S A 99: , 4014–4019. |
[32] | Lai MT , Crouthamel MC , DiMuzio J , Pietrak BL , Donoviel DB , Bernstein A , Gardell SJ , Li YM , Hazuda D ((2006) ) A presenilin-independent aspartyl protease prefers the gamma-42 site cleavage. J Neurochem 96: , 118–125. |
[33] | Le Brocque D , Henry A , Cappai R , Li QX , Tanner JE , Galatis D , Gray C , Holmes S , Underwood JR , Beyreuther K , Masters CL , Evin G ((1998) ) Processing of the Alzheimer’s disease amyloid precursor protein in Pichia pastoris: Immunodetection of alpha-, beta-, and gamma-secretase products. Biochemistry 37: , 14958–14965. |
[34] | Li YM , Xu M , Lai MT , Huang Q , Castro JL , DiMuzio-Mower J , Harrison T , Lellis C , Nadin A , Neduvelil JG , Register RB , Sardana MK , Shearman MS , Smith AL , Shi XP , Yin KC , Shafer JA , Gardell SJ ((2000) ) Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature 405: , 689–694. |
[35] | Ahn K , Shelton CC , Tian Y , Zhang X , Gilchrist ML , Sisodia SS , Li YM ((2010) ) Activation and intrinsic gamma-secretase activity of presenilin 1. Proc Natl Acad Sci U S A 107: , 21435–21440. |
[36] | Evin G , Cappai R , Li QX , Culvenor JG , Small DH , Beyreuther K , Masters CL ((1995) ) Candidate gamma-secretases in the generation of the carboxyl terminus of the Alzheimer’s disease beta A4 amyloid: Possible involvement of cathepsin D. Biochemistry 34: , 14185–14192. |
[37] | Sadik G , Kaji H , Takeda K , Yamagata F , Kameoka Y , Hashimoto K , Miyanaga K , Shinoda T ((1999) ) In vitro processing of amyloid precursor protein by cathepsin D. Int J Biochem Cell Biol 31: , 1327–1337. |
[38] | Ahmed RR , Holler CJ , Webb RL , Li F , Beckett TL , Murphy MP ((2010) ) BACE1 and BACE2 enzymatic activities in Alzheimer’s disease. J Neurochem 112: , 1045–1053. |
[39] | Chevallier N , Vizzavona J , Marambaud P , Baur CP , Spillantini M , Fulcrand P , Martinez J , Goedert M , Vincent JP , Checler F ((1997) ) Cathepsin D displays in vitro beta-secretase-like specificity. Brain Res 750: , 11–19. |
[40] | Turk V , Kregar I , Lebez D ((1968) ) Some properties of cathepsin E from bovine spleen. Enzymologia 34: , 89–100. |
[41] | Vassar R , Bennett BD , Babu-Khan S , Kahn S , Mendiaz EA , Denis P , Teplow DB , Ross S , Amarante P , Loeloff R , Luo Y , Fisher S , Fuller J , Edenson S , Lile J , Jarosinski MA , Biere AL , Curran E , Burgess T , Louis JC , Collins F , Treanor J , Rogers G , Citron M ((1999) ) Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286: , 735–741. |
[42] | Teng L , Zhao J , Wang F , Ma L , Pei G ((2010) ) A GPCR/secretase complex regulates beta- and gamma-secretase specificity for Abeta production and contributes to AD pathogenesis. Cell Res 20: , 138–153. |
[43] | Kanning KC , Hudson M , Amieux PS , Wiley JC , Bothwell M , Schecterson LC ((2003) ) Proteolytic processing of the p75 neurotrophin receptor and two homologs generates C-terminal fragments with signaling capability. J Neurosci 23: , 5425–5436. |
[44] | Gowrishankar K , Zeidler MG , Vincenz C ((2004) ) Release of a membrane-bound death domain by gamma-secretase processing of the p75NTR homolog NRADD. J Cell Sci 117: , 4099–4111. |
[45] | Beel AJ , Sanders CR ((2008) ) Substrate specificity of gamma-secretase and other intramembrane proteases. Cell Mol Life Sci 65: , 1311–1334. |
[46] | Campion D , Dumanchin C , Hannequin D , Dubois B , Belliard S , Puel M , Thomas-Anterion C , Michon A , Martin C , Charbonnier F , Raux G , Camuzat A , Penet C , Mesnage V , Martinez M , Clerget-Darpoux F , Brice A , Frebourg T ((1999) ) Early-onset autosomal dominant Alzheimer disease: Prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet 65: , 664–670. |
[47] | Cruts M , Theuns J , Van Broeckhoven C ((2012) ) Locus-specific mutation databases for neurodegenerative brain diseases. Hum Mutat 33: , 1340–1344. |
[48] | Smith MJ , Kwok JB , McLean CA , Kril JJ , Broe GA , Nicholson GA , Cappai R , Hallupp M , Cotton RG , Masters CL , Schofield PR , Brooks WS ((2001) ) Variable phenotype of Alzheimer’s disease with spastic paraparesis. Ann Neurol 49: , 125–129. |
[49] | De Jonghe C , Cruts M , Rogaeva EA , Tysoe C , Singleton A , Vanderstichele H , Meschino W , Dermaut B , Vanderhoeven I , Backhovens H , Vanmechelen E , Morris CM , Hardy J , Rubinsztein DC , St George-Hyslop PH , Van Broeckhoven C ((1999) ) Aberrant splicing in the presenilin-1 intron 4 mutation causes presenile Alzheimer’s disease by increased Abeta42 secretion. Hum Mol Genet 8: , 1529–1540. |
[50] | Steiner H , Romig H , Grim MG , Philipp U , Pesold B , Citron M , Baumeister R , Haass C ((1999) ) The biological and pathological function of the presenilin-1 Deltaexon 9 mutation is independent of its defect to undergo proteolytic processing. J Biol Chem 274: , 7615–7618. |
[51] | Kwok J , Hallupp M , Badenhop RF , Schofield PR , Halliday GM , Brooks WS , Dolois G , Wang R , Murayama O , Takashima A , Vickers J , Gandy SE ((2002) ) Presenilin-1 mutation (L271V) results in altered exon 8 splicing and Alzheimer’s disease with non-cored plaques and no neuritic dystrophy. Neurobiol Aging 23: , S277–S277. |
[52] | Chow VW , Mattson MP , Wong PC , Gleichmann M ((2010) ) An overview of APP processing enzymes and products. Neuromolecular Med 12: , 1–12. |
[53] | Sidera C , Liu C , Austen B ((2002) ) Pro-domain removal in ASP-2 and the cleavage of the amyloid precursor are influenced by pH. BMC Biochem 3: , 25. |
[54] | Sastre M , Steiner H , Fuchs K , Capell A , Multhaup G , Condron MM , Teplow DB , Haass C ((2001) ) Presenilin-dependent gamma-secretase processing of beta-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep 2: , 835–841. |
[55] | Pinnix I , Musunuru U , Tun H , Sridharan A , Golde T , Eckman C , Ziani-Cherif C , Onstead L , Sambamurti K ((2001) ) A novel gamma -secretase assay based on detection of the putative C-terminal fragment-gamma of amyloid beta protein precursor. J Biol Chem 276: , 481–487. |
[56] | Gu Y , Misonou H , Sato T , Dohmae N , Takio K , Ihara Y ((2001) ) Distinct intramembrane cleavage of the beta-amyloid precursor protein family resembling gamma-secretase-like cleavage of Notch. J Biol Chem 276: , 35235–35238. |
[57] | Schettini G , Govoni S , Racchi M , Rodriguez G ((2010) ) Phosphorylation of APP-CTF-AICD domains and interaction with adaptor proteins: Signal transduction and/or transcriptional role–relevance for Alzheimer pathology. J Neurochem 115: , 1299–1308. |
[58] | Pardossi-Piquard R , Checler F ((2012) ) The physiology of the beta-amyloid precursor protein intracellular domain AICD. J Neurochem 120: (Suppl 1), 109–124. |
[59] | De Strooper B ((2010) ) Proteases and proteolysis in Alzheimer disease: A multifactorial view on the disease process. Physiol Rev 90: , 465–494. |
[60] | Kakuda N , Funamoto S , Yagishita S , Takami M , Osawa S , Dohmae N , Ihara Y ((2006) ) Equimolar production of amyloid beta-protein and amyloid precursor protein intracellular domain from beta-carboxyl-terminal fragment by gamma-secretase. J Biol Chem 281: , 14776–14786. |
[61] | Takami M , Nagashima Y , Sano Y , Ishihara S , Morishima-Kawashima M , Funamoto S , Ihara Y ((2009) ) gamma-Secretase: Successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J Neurosci 29: , 13042–13052. |
[62] | Wolfe MS ((2007) ) When loss is gain: Reducedresenilin proteolytic function leads to increased Abeta42/Abeta40. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep 8: , 136–140. |
[63] | Chavez-Gutierrez L , Bammens L , Benilova I , Vandersteen A , Benurwar M , Borgers M , Lismont S , Zhou L , Van Cleynenbreugel S , Esselmann H , Wiltfang J , Serneels L , Karran E , Gijsen H , Schymkowitz J , Rousseau F , Broersen K , De Strooper B ((2012) ) The mechanism of gamma-Secretase dysfunction in familial Alzheimer disease. EMBO J 31: , 2261–2274. |
[64] | Iwatsubo T , Odaka A , Suzuki N , Mizusawa H , Nukina N , Ihara Y ((1994) ) Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: Evidence that an initially deposited species is A beta 42(43). Neuron 13: , 45–53. |
[65] | Small DH , Klaver DW , Foa L ((2010) ) Presenilins and the gamma-secretase: Still a complex problem. Mol Brain 3: , 7. |
[66] | Urano Y , Hayashi I , Isoo N , Reid PC , Shibasaki Y , Noguchi N , Tomita T , Iwatsubo T , Hamakubo T , Kodama T ((2005) ) Association of active gamma-secretase complex with lipid rafts. J Lipid Res 46: , 904–912. |
[67] | Vetrivel KS , Cheng H , Lin W , Sakurai T , Li T , Nukina N , Wong PC , Xu H , Thinakaran G ((2004) ) Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J Biol Chem 279: , 44945–44954. |
[68] | Winkler E , Kamp F , Scheuring J , Ebke A , Fukumori A , Steiner H ((2012) ) Generation of Alzheimer disease-associated amyloid beta42/43 peptide by gamma-secretase can be inhibited directly by modulation of membrane thickness. J Biol Chem 287: , 21326–21334. |
[69] | Szaruga M , Veugelen S , Benurwar M , Lismont S , Sepulveda-Falla D , Lleo A , Ryan NS , Lashley T , Fox NC , Murayama S , Gijsen H , De Strooper B , Chavez-Gutierrez L ((2015) ) Qualitative changes in human gamma-secretase underlie familial Alzheimer’s disease. J Exp Med 212: , 2003–2013. |
[70] | Mawuenyega KG , Sigurdson W , Ovod V , Munsell L , Kasten T , Morris JC , Yarasheski KE , Bateman RJ ((2010) ) Decreased clearance of CNS beta-amyloid in Alzheimer’s disease. Science 330: , 1774. |
[71] | Xia D , Watanabe H , Wu B , Lee SH , Li Y , Tsvetkov E , Bolshakov VY , Shen J , Kelleher RJ ((2015) ) Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron 85: , 967–981. |
[72] | Haapasalo A , Kovacs DM ((2011) ) The many substrates of presenilin/gamma-secretase. J Alzheimers Dis 25: , 3–28. |
[73] | Sotthibundhu A , Sykes AM , Fox B , Underwood CK , Thangnipon W , Coulson EJ ((2008) ) Beta-amyloid(1-42) induces neuronal death through the p75 neurotrophin receptor. J Neurosci 28: , 3941–3946. |
[74] | Kang DE , Soriano S , Xia X , Eberhart CG , De Strooper B , Zheng H , Koo EH ((2002) ) Presenilin couples the paired phosphorylation of beta-catenin independent of axin: Imlications for beta-catenin activation in tumorigenesis. Cell 110: , 751–762. |
[75] | Heilig EA , Gutti U , Tai T , Shen J , Kelleher RJ ((2013) ) Trans-dominant negative effects of pathogenic PSEN1 mutations on gamma-secretase activity and Abeta production. J Neurosci 33: , 11606–11617. |
[76] | Zhang M , Guller S , Huang Y ((2007) ) Method to enhance transfection efficiency of cell lines and placental fibroblasts. Placenta 28: , 779–782. |
[77] | Thinakaran G , Harris CL , Ratovitski T , Davenport F , Slunt HH , Price DL , Borchelt DR , Sisodia SS ((1997) ) Evidence that levels of presenilins (PS1 and PS2) are coordinately regulated by competition for limiting cellular factors. J Biol Chem 272: , 28415–28422. |
[78] | Mutant Presenilin Knock-in Mice Mimic Knockouts,Stir Old Debate, http://www.alzforum.org/news/research-news/mutant-presenilin-knock-mice-mimic-knockouts-stir-old-debate. |
[79] | Fukumori A , Fluhrer R , Steiner H , Haass C ((2010) ) Three-amino acid spacing of presenilin endoproteolysis suggests a general stepwise cleavage of gamma-secretase-mediated intramembrane proteolysis. J Neurosci 30: , 7853–7862. |
[80] | Watanabe H , Xia D , Kanekiyo T , Kelleher RJ , Shen J ((2012) ) Familial frontotemporal dementia-associated presenilin-1 c.548G>T mutation causes decreased mRNA expression and reduced presenilin function in knock-in mice. J Neurosci 32: , 5085–5096. |
[81] | Dermaut B , Kumar-Singh S , Engelborghs S , Theuns J , Rademakers R , Sacrens J , Pickut BA , Peeters K , van den Broeck M , Vennekens K , Claes S , Cruts M , Cras P , Martin JJ , Van Broeckhoven C , De Deyn PP ((2004) ) A novel presenilin 1 mutation associated with Pick’s disease but not beta-amyloid plaques. Ann Neurol 55: , 617–626. |
[82] | Dermaut B , Kumar-Singh S , Engelborghs S , Theuns J , Rademakers R , Peeters K , Claes S , Cruts M , Martin JJ , De Deyn P , Van Broeckboven C ((2004) ) A novel presenilin 1 mutation (Gly183Val) is associated with Pick’s disease in the absence of beta-amyloid plaques. Neurobiol Aging 25: , S61–S61. |
[83] | Dermaut B , Kumar-Singh S , Rademakers R , Theuns J , Cruts M , Van Broeckhoven C ((2005) ) Tau is central in the genetic Alzheimer-frontotemporal dementia spectrum. Trends Genet 21: , 664–672. |
[84] | Evin G , Smith MJ , Tziotis A , McLean C , Canterford L , Sharples RA , Cappai R , Weidemann A , Beyreuther K , Cotton RG , Masters CL , Culvenor JG ((2002) ) Alternative transcripts of presenilin-1 associated with frontotemporal dementia. Neuroreport 13: , 917–921. |
[85] | Takashima A , Murayama M , Murayama O , Kohno T , Honda T , Yasutake K , Nihonmatsu N , Mercken M , Yamaguchi H , Sugihara S , Wolozin B ((1998) ) Presenilin 1 associates with glycogen synthase kinase-3beta and its substrate tau. Proc Natl Acad Sci U S A 95: , 9637–9641. |
[86] | Saura CA , Choi SY , Beglopoulos V , Malkani S , Zhang D , Shankaranarayana Rao BS , Chattarji S , Kelleher RJ , Kandel ER , Duff K , Kirkwood A , Shen J ((2004) ) Loss of presenilin function causes impairments of memory and synaptic plasticity followed by age-dependent neurodegeneration. Neuron 42: , 23–36. |
[87] | Newman M , Wilson L , Verdile G , Lim A , Khan I , Moussavi Nik SH , Pursglove S , Chapman G , Martins RN , Lardelli M ((2014) ) Differential, dominant activation and inhibition of Notch signalling and APP cleavage by truncations of PSEN1 in human disease. Hum Mol Genet 23: , 602–617. |
[88] | Bai XC , Yan C , Yang G , Lu P , Ma D , Sun L , Zhou R , Scheres SH , Shi Y ((2015) ) An atomic structure of human gamma-secretase. Nature 525: , 212–217. |
[89] | Wang B , Yang W , Wen W , Sun J , Su B , Liu B , Ma D , Lv D , Wen Y , Qu T , Chen M , Sun M , Shen Y , Zhang X ((2010) ) Gamma-secretase gene mutations in familial acne inversa. Science 330: , 1065. |
[90] | Schroeter EH , Ilagan MX , Brunkan AL , Hecimovic S , Li YM , Xu M , Lewis HD , Saxena MT , De Strooper B , Coonrod A , Tomita T , Iwatsubo T , Moore CL , Goate A , Wolfe MS , Shearman M , Kopan R ((2003) ) A presenilin dimer at the core of the gamma-secretase enzyme: Insights from parallel analysis of Notch 1 and APP proteolysis. Proc Natl Acad Sci U S A 100: , 13075–13080. |
[91] | Brautigam H , Moreno CL , Steele JW , Bogush A , Dickstein DL , Kwok JB , Schofield PR , Thinakaran G , Mathews PM , Hof PR , Gandy S , Ehrlich ME ((2015) ) Physiologically generated presenilin 1 lacking exon 8 fails to rescue brain PS1-/- phenotype and forms complexes with wildtype PS1 and nicastrin. Sci Rep 5: , 17042. |
[92] | Lee JH , Yu WH , Kumar A , Lee S , Mohan PS , Peterhoff CM , Wolfe DM , Martinez-Vicente M , Massey AC , Sovak G , Uchiyama Y , Westaway D , Cuervo AM , Nixon RA ((2010) ) Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 141: , 1146–1158. |
[93] | Zhang X , Garbett K , Veeraraghavalu K , Wilburn B , Gilmore R , Mirnics K , Sisodia SS ((2012) ) A role for presenilins in autophagy revisited: Normal acidification of lysosomes in cells lacking PSEN1 and PSEN2. J Neurosci 32: , 8633–8648. |
[94] | Coen K , Flannagan RS , Baron S , Carraro-Lacroix LR , Wang D , Vermeire W , Michiels C , Munck S , Baert V , Sugita S , Wuytack F , Hiesinger PR , Grinstein S , Annaert W ((2012) ) Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J Cell Biol 198: , 23–35. |
[95] | Alzforum.com (2012) Presenilins and calcium: A lysosomal stew with acid controversy. http://www.alzforum.org/news/research-news/presenilins-and-calcium-lysosomal-stew-acid-controversy |
[96] | Wolfe DM , Lee JH , Kumar A , Lee S , Orenstein SJ , Nixon RA ((2013) ) Autophagy failure in Alzheimer’s disease and the role of defective lysosomal acidification. Eur J Neurosci 37: , 1949–1961. |
[97] | Cataldo AM , Peterhoff CM , Schmidt SD , Terio NB , Duff K , Beard M , Mathews PM , Nixon RA ((2004) ) Presenilin mutations in familial Alzheimer disease and transgenic mouse models accelerate neuronal lysosomal pathology. J Neuropathol Exp Neurol 63: , 821–830. |
[98] | Nixon RA , Wegiel J , Kumar A , Yu WH , Peterhoff C , Cataldo A , Cuervo AM ((2005) ) Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J Neuropathol Exp Neurol 64: , 113–122. |
[99] | Okamura Y , Aoki Y , Obayashi T , Tadaka S , Ito S , Narise T , Kinoshita K ((2015) ) COXPRESdb in 2015: Coexpression database for animal species by DNA-microarray and RNAseq-based expression data with multiple quality assessment systems. Nucleic Acids Res 43: , D82–D86. |
[100] | Harold D , Abraham R , Hollingworth P , Sims R , Gerrish A , Hamshere ML , Pahwa JS , Moskvina V , Dowzell K , Williams A , Jones N , Thomas C , Stretton A , Morgan AR , Lovestone S , Powell J , Proitsi P , Lupton MK , Brayne C , Rubinsztein DC , Gill M , Lawlor B , Lynch A , Morgan K , Brown KS , Passmore PA , Craig D , McGuinness B , Todd S , Holmes C , Mann D , Smith AD , Love S , Kehoe PG , Hardy J , Mead S , Fox N , Rossor M , Collinge J , Maier W , Jessen F , Schurmann B , Heun R , van den Bussche H , Heuser I , Kornhuber J , Wiltfang J , Dichgans M , Frolich L , Hampel H , Hull M , Rujescu D , Goate AM , Kauwe JS , Cruchaga C , Nowotny P , Morris JC , Mayo K , Sleegers K , Bettens K , Engelborghs S , De Deyn PP , Van Broeckhoven C , Livingston G , Bass NJ , Gurling H , McQuillin A , Gwilliam R , Deloukas P , Al-Chalabi A , Shaw CE , Tsolaki M , Singleton AB , Guerreiro R , Muhleisen TW , Nothen MM , Moebus S , Jockel KH , Klopp N , Wichmann HE , Carrasquillo MM , Pankratz VS , Younkin SG , Holmans PA , O’Donovan M , Owen MJ , Williams J ((2009) ) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 41: , 1088–1093. |
[101] | Seshadri S , Fitzpatrick AL , Ikram MA , DeStefano AL , Gudnason V , Boada M , Bis JC , Smith AV , Carassquillo MM , Lambert JC , Harold D , Schrijvers EM , Ramirez-Lorca R , Debette S , Longstreth WT Jr , Janssens AC , Pankratz VS , Dartigues JF , Hollingworth P , Aspelund T , Hernandez I , Beiser A , Kuller LH , Koudstaal PJ , Dickson DW , Tzourio C , Abraham R , Antunez C , Du Y , Rotter JI , Aulchenko YS , Harris TB , Petersen RC , Berr C , Owen MJ , Lopez-Arrieta J , Varadarajan BN , Becker JT , Rivadeneira F , Nalls MA , Graff-Radford NR , Campion D , Auerbach S , Rice K , Hofman A , Jonsson PV , Schmidt H , Lathrop M , Mosley TH , Au R , Psaty BM , Uitterlinden AG , Farrer LA , Lumley T , Ruiz A , Williams J , Amouyel P , Younkin SG , Wolf PA , Launer LJ , Lopez OL , van Duijn CM , Breteler MM , Consortium C , Consortium G , Consortium E ((2010) ) Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 303: , 1832–1840. |
[102] | Beecham GW , Hamilton K , Naj AC , Martin ER , Huentelman M , Myers AJ , Corneveaux JJ , Hardy J , Vonsattel JP , Younkin SG , Bennett DA , De Jager PL , Larson EB , Crane PK , Kamboh MI , Kofler JK , Mash DC , Duque L , Gilbert JR , Gwirtsman H , Buxbaum JD , Kramer P , Dickson DW , Farrer LA , Frosch MP , Ghetti B , Haines JL , Hyman BT , Kukull WA , Mayeux RP , Pericak-Vance MA , Schneider JA , Trojanowski JQ , Reiman EM , Alzheimer’s Disease Genetics C, Schellenberg GD , Montine TJ ((2014) ) Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet 10: , e1004606. |
[103] | Naj AC , Jun G , Reitz C , Kunkle BW , Perry W , Park YS , Beecham GW , Rajbhandary RA , Hamilton-Nelson KL , Wang LS , Kauwe JS , Huentelman MJ , Myers AJ , Bird TD , Boeve BF , Baldwin CT , Jarvik GP , Crane PK , Rogaeva E , Barmada MM , Demirci FY , Cruchaga C , Kramer PL , Ertekin-Taner N , Hardy J , Graff-Radford NR , Green RC , Larson EB , St George-Hyslop PH , Buxbaum JD , Evans DA , Schneider JA , Lunetta KL , Kamboh MI , Saykin AJ , Reiman EM , De Jager PL , Bennett DA , Morris JC , Montine TJ , Goate AM , Blacker D , Tsuang DW , Hakonarson H , Kukull WA , Foroud TM , Martin ER , Haines JL , Mayeux RP , Farrer LA , Schellenberg GD , Pericak-Vance MA , Alzheimer Disease Genetics C, Albert MS , Albin RL , Apostolova LG , Arnold SE , Barber R , Barnes LL , Beach TG , Becker JT , Beekly D , Bigio EH , Bowen JD , Boxer A , Burke JR , Cairns NJ , Cantwell LB , Cao C , Carlson CS , Carney RM , Carrasquillo MM , Carroll SL , Chui HC , Clark DG , Corneveaux J , Cribbs DH , Crocco EA , DeCarli C , DeKosky ST , Dick M , Dickson DW , Duara R , Faber KM , Fallon KB , Farlow MR , Ferris S , Frosch MP , Galasko DR , Ganguli M , Gearing M , Geschwind DH , Ghetti B , Gilbert JR , Glass JD , Growdon JH , Hamilton RL , Harrell LE , Head E , Honig LS , Hulette CM , Hyman BT , Jicha GA , Jin LW , Karydas A , Kaye JA , Kim R , Koo EH , Kowall NW , Kramer JH , LaFerla FM , Lah JJ , Leverenz JB , Levey AI , Li G , Lieberman AP , Lin CF , Lopez OL , Lyketsos CG , Mack WJ , Martiniuk F , Mash DC , Masliah E , McCormick WC , McCurry SM , McDavid AN , McKee AC , Mesulam M , Miller BL , Miller CA , Miller JW , Murrell JR , Olichney JM , Pankratz VS , Parisi JE , Paulson HL , Peskind E , Petersen RC , Pierce A , Poon WW , Potter H , Quinn JF , Raj A , Raskind M , Reisberg B , Ringman JM , Roberson ED , Rosen HJ , Rosenberg RN , Sano M , Schneider LS , Seeley WW , Smith AG , Sonnen JA , Spina S , Stern RA , Tanzi RE , Thornton-Wells TA , Trojanowski JQ , Troncoso JC , Valladares O , Van Deerlin VM , Van Eldik LJ , Vardarajan BN , Vinters HV , Vonsattel JP , Weintraub S , Welsh-Bohmer KA , Williamson J , Wishnek S , Woltjer RL , Wright CB , Younkin SG , Yu CE , Yu L ((2014) ) Effects of multiple genetic loci on age at onset in late-onset Alzheimer disease: A genome-wide association study. JAMA Neurol 71: , 1394–1404. |
[104] | Moreau K , Fleming A , Imarisio S , Lopez Ramirez A , Mercer JL , Jimenez-Sanchez M , Bento CF , Puri C , Zavodszky E , Siddiqi F , Lavau CP , Betton M , O’Kane CJ , Wechsler DS , Rubinsztein DC ((2014) ) PICALM modulates autophagy activity and tau accumulation. Nat Commun 5: , 4998. |
[105] | Sato N , Hori O , Yamaguchi A , Lambert JC , Chartier-Harlin MC , Robinson PA , Delacourte A , Schmidt AM , Furuyama T , Imaizumi K , Tohyama M , Takagi T ((1999) ) A novel presenilin-2 splice variant in human Alzheimer’s disease brain tissue. J Neurochem 72: , 2498–2505. |
[106] | Sharman MJ , Moussavi Nik SH , Chen MM , Ong D , Wijaya L , Laws SM , Taddei K , Newman M , Lardelli M , Martins RN , Verdile G ((2013) ) The guinea pig as a model for sporadic Alzheimer’s disease (AD): the impact of cholesterol intake on expression of AD-related genes. PLoS One 8: , e66235. |
[107] | Sato N , Imaizumi K , Manabe T , Taniguchi M , Hitomi J , Katayama T , Yoneda T , Morihara T , Yasuda Y , Takagi T , Kudo T , Tsuda T , Itoyama Y , Makifuchi T , Fraser PE , St George-Hyslop P , Tohyama M ((2001) ) Increased production of beta-amyloid and vulnerability to endoplasmic reticulum stress by an aberrant spliced form of presenilin 2. J Biol Chem 276: , 2108–2114. |
[108] | Moussavi Nik SH , Newman M , Wilson L , Ebrahimie E , Wells S , Musgrave I , Verdile G , Martins RN , Lardelli M ((2015) ) Alzheimer’s disease-related peptide PS2V plays ancient, conserved roles in suppression of the unfolded protein response under hypoxia and stimulation of gamma-secretase activity. Hum Mol Genet 24: , 3662–3678. |
[109] | Tu H , Nelson O , Bezprozvanny A , Wang Z , Lee SF , Hao YH , Serneels L , De Strooper B , Yu G , Bezprozvanny I ((2006) ) Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer’s disease-linked mutations. Cell 126: , 981–993. |
[110] | Area-Gomez E , de Groof AJ , Boldogh I , Bird TD , Gibson GE , Koehler CM , Yu WH , Duff KE , Yaffe MP , Pon LA , Schon EA ((2009) ) Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol 175: , 1810–1816. |
[111] | Simmen T , Lynes EM , Gesson K , Thomas G ((2010) ) Oxidative protein folding in the endoplasmic reticulum: Tight links to the mitochondria-associated membrane (MAM). Biochim Biophys Acta 1798: , 1465–1473. |
[112] | Area-Gomez E , Del Carmen Lara Castillo M , Tambini MD , Guardia-Laguarta C , de Groof AJ , Madra M , Ikenouchi J , Umeda M , Bird TD , Sturley SL , Schon EA ((2012) ) Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J 31: , 4106–4123. |
[113] | Raturi A , Simmen T ((2013) ) Where the endoplasmic reticulum and the mitochondrion tie the knot: The mitochondria-associated membrane (MAM). Biochim Biophys Acta 1833: , 213–224. |
[114] | Hamasaki M , Furuta N , Matsuda A , Nezu A , Yamamoto A , Fujita N , Oomori H , Noda T , Haraguchi T , Hiraoka Y , Amano A , Yoshimori T ((2013) ) Autophagosomes form at ER-mitochondria contact sites. Nature 495: , 389–393. |
[115] | Misawa T , Takahama M , Kozaki T , Lee H , Zou J , Saitoh T , Akira S ((2013) ) Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol 14: , 454–460. |
[116] | Schon EA , Area-Gomez E ((2013) ) Mitochondria-associated ER membranes in Alzheimer disease. Mol Cell Neurosci 55: , 26–36. |
[117] | Katayama T , Imaizumi K , Manabe T , Hitomi J , Kudo T , Tohyama M ((2004) ) Induction of neuronal death by ER stress in Alzheimer’s disease. J Chem Neuroanat 28: , 67–78. |
[118] | Nilsson P , Loganathan K , Sekiguchi M , Matsuba Y , Hui K , Tsubuki S , Tanaka M , Iwata N , Saito T , Saido TC ((2013) ) Abeta secretion and plaque formation depend on autophagy. Cell Rep 5: , 61–69. |
[119] | Nilsson P , Saido TC ((2014) ) Dual roles for autophagy: Degradation and secretion of Alzheimer’s disease Abeta peptide. Bioessays 36: , 570–578. |
[120] | Correia SC , Resende R , Moreira PI , Pereira CM ((2015) ) Alzheimer’s disease-related misfolded proteins and dysfunctional organelles on autophagy menu. DNA Cell Biol 34: , 261–273. |
[121] | El Kadmiri N , Zaid N , Hachem A , Zaid Y , Dube MP , Hamzi K , El Moutawakil B , Slassi I , Nadifi S ((2014) ) Novel mutations in the amyloid precursor protein gene within Moroccan patients with Alzheimer’s disease. J Mol Neurosci 53: , 189–195. |
[122] | El Kadmiri N , Zaid N , Zaid Y , Tadevosyan A , Hachem A , Dube MP , Hamzi K , El Moutawakil B , Slassi I , Nadifi S ((2014) ) Novel presenilin mutations within Moroccan patients with Early-Onset Alzheimer’s Disease. Neuroscience 269: , 215–222. |
[123] | Cai Y , An SS , Kim S ((2015) ) Mutations in presenilin 2 and its implications in Alzheimer’s disease and other dementia-associated disorders. Clin Interv Aging 10: , 1163–1172. |
[124] | Nixon RA ((2007) ) Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci 120: , 4081–4091. |
[125] | Duce JA , Tsatsanis A , Cater MA , James SA , Robb E , Wikhe K , Leong SL , Perez K , Johanssen T , Greenough MA , Cho HH , Galatis D , Moir RD , Masters CL , McLean C , Tanzi RE , Cappai R , Barnham KJ , Ciccotosto GD , Rogers JT , Bush AI ((2010) ) Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 142: , 857–867. |
[126] | Joshi P , Liang JO , DiMonte K , Sullivan J , Pimplikar SW ((2009) ) Amyloid precursor protein is required for convergent-extension movements during Zebrafish development. Dev Biol 335: , 1–11. |
[127] | Liao HK , Wang Y , Watt KEN , Wen Q , Breitbach J , Kemmet CK , Clark KJ , Ekker SC , Essner JJ , McGrail M ((2012) ) Tol2 gene trap integrations in the zebrafish amyloid precursor protein genes appa and aplp2 reveal accumulation of secreted APP at the embryonic veins. Dev Dyn 241: , 415–425. |
[128] | Luna S , Cameron DJ , Ethell DW ((2013) ) Amyloid-beta and APP deficiencies cause severe cerebrovascular defects: Important work for an old villain. PLoS One 8: , e75052. |
[129] | Sobol A , Galluzzo P , Liang S , Rambo B , Skucha S , Weber MJ , Alani S , Bocchetta M ((2015) ) Amyloid precursor protein (APP) affects global protein synthesis in dividing human cells. J Cell Physiol 230: , 1064–1074. |
[130] | Song P , Pimplikar SW ((2012) ) Knockdown of amyloid precursor protein in zebrafish causes defects in motor axon outgrowth. PLoS One 7: , e34209. |
[131] | Nikolaev A , McLaughlin T , O’Leary DD , Tessier-Lavigne M ((2009) ) APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 457: , 981–989 . |
[132] | Carrano A , Das P ((2015) ) Altered innate immune and glial cell responses to inflammatory stimuli in amyloid precursor protein knockout mice. PLoS One 10: , e0140210. |
[133] | Grimm MO , Mett J , Stahlmann CP , Grosgen S , Haupenthal VJ , Blumel T , Hundsdorfer B , Zimmer VC , Mylonas NT , Tanila H , Muller U , Grimm HS , Hartmann T ((2015) ) APP intracellular domain derived from amyloidogenic beta- and gamma-secretase cleavage regulates neprilysin expression. Front Aging Neurosci 7: , 77. |
[134] | Kerridge C , Belyaev ND , Nalivaeva NN , Turner AJ ((2014) ) The Abeta-clearance protein transthyretin, like neprilysin, is epigenetically regulated by the amyloid precursor protein intracellular domain. J Neurochem 130: , 419–431. |
[135] | Greenough MA , Volitakis I , Li QX , Laughton K , Evin G , Ho M , Dalziel AH , Camakaris J , Bush AI ((2011) ) Presenilins promote the cellular uptake of copper and zinc and maintain copper chaperone of SOD1-dependent copper/zinc superoxide dismutase activity. J Biol Chem 286: , 9776–9786. |
[136] | Agrawal V , Sawhney N , Hickey E , McCarthy JV ((2015) ) Loss of presenilin 2 function is associated with defective LPS-mediated innate immune responsiveness. Mol Neurobiol, doi: 10.1007/s12035-015-9285-0 |
[137] | Kummer MP , Heneka MT ((2014) ) Truncated and modified amyloid-beta species. Alzheimers Res Ther 6: , 28. |
[138] | Gandy S ((2005) ) The role of cerebral amyloid beta accumulation in common forms of Alzheimer disease. J Clin Invest 115: , 1121–1129. |
[139] | Kogel D , Deller T , Behl C ((2012) ) Roles of amyloid precursor protein family members in neuroprotection, stress signaling and aging. Exp Brain Res 217: , 471–479. |
[140] | Kontush A ((2001) ) Amyloid-beta: An antioxidant that becomes a pro-oxidant and critically contributes to Alzheimer’s disease. Free Radic Biol Med 31: , 1120–1131. |
[141] | Kontush A , Donarski N , Beisiegel U ((2001) ) Resistance of human cerebrospinal fluid to in vitro oxidation is directly related to its amyloid-beta content. Free Radic Res 35: , 507–517. |
[142] | Nadal RC , Rigby SE , Viles JH ((2008) ) Amyloid beta-Cu2+ complexes in both monomeric and fibrillar forms do not generate H2O2 catalytically but quench hydroxyl radicals. Biochemistry 47: , 11653–11664. |
[143] | Smith MA , Casadesus G , Joseph JA , Perry G ((2002) ) Amyloid-beta and tau serve antioxidant functions in the aging and Alzheimer brain. Free Radic Biol Med 33: , 1194–1199. |
[144] | Baruch-Suchodolsky R , Fischer B ((2009) ) Abeta40, either soluble or aggregated, is a remarkably potent antioxidant in cell-free oxidative systems. Biochemistry 48: , 4354–4370. |
[145] | Atwood CS , Obrenovich ME , Liu T , Chan H , Perry G , Smith MA , Martins RN ((2003) ) Amyloid-beta: A chameleon walking in two worlds: A review of the trophic and toxic properties of amyloid-beta. Brain Res Brain Res Rev 43: , 1–16. |
[146] | Cameron DJ , Galvin C , Alkam T , Sidhu H , Ellison J , Luna S , Ethell DW ((2012) ) Alzheimer’s-related peptide amyloid-beta plays a conserved role in angiogenesis. PLoS One 7: , e39598. |
[147] | Dietrich HH , Xiang C , Han BH , Zipfel GJ , Holtzman DM ((2010) ) Soluble amyloid-beta, effect on cerebral arteriolar regulation and vascular cells. Mol Neurodegener 5: , 15. |
[148] | Walsh DM , Klyubin I , Fadeeva JV , Cullen WK , Anwyl R , Wolfe MS , Rowan MJ , Selkoe DJ ((2002) ) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416: , 535–539. |
[149] | Soscia SJ , Kirby JE , Washicosky KJ , Tucker SM , Ingelsson M , Hyman B , Burton MA , Goldstein LE , Duong S , Tanzi RE , Moir RD ((2010) ) The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 5: , e9505. |
[150] | Bailey JA , Maloney B , Ge YW , Lahiri DK ((2011) ) Functional activity of the novel Alzheimer’s amyloid beta-peptide interacting domain (AbetaID) in the APP and BACE1 promoter sequences and implications in activating apoptotic genes and in amyloidogenesis. Gene 488: , 13–22. |
[151] | Maloney B , Lahiri DK ((2011) ) The Alzheimer’s amyloid beta-peptide (Abeta) binds a specific DNA Abeta-interacting domain (AbetaID) in the APP, BACE1, and APOE promoters in a sequence-specific manner: Characterizing a new regulatory motif. Gene 488: , 1–12. |
[152] | Piccini A , Borghi R , Guglielmotto M , Tamagno E , Cirmena G , Garuti A , Pollero V , Cammarata S , Fornaro M , Messa M , Colombo L , Salmona M , Perry G , Tabaton M ((2012) ) beta-amyloid 1-42 induces physiological transcriptional regulation of BACE1. J Neurochem 122: , 1023–1031. |
[153] | Barucker C , Harmeier A , Weiske J , Fauler B , Albring KF , Prokop S , Hildebrand P , Lurz R , Heppner FL , Huber O , Multhaup G ((2014) ) Nuclear translocation uncovers the amyloid Peptide abeta42 as a regulator of gene transcription. J Biol Chem 289: , 20182–20191. |
[154] | Sun X , He G , Qing H , Zhou W , Dobie F , Cai F , Staufenbiel M , Huang LE , Song W ((2006) ) Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci U S A 103: , 18727–18732. |
[155] | Guglielmotto M , Aragno M , Autelli R , Giliberto L , Novo E , Colombatto S , Danni O , Parola M , Smith MA , Perry G , Tamagno E , Tabaton M ((2009) ) The up-regulation of BACE1 mediated by hypoxia and ischemic injury: Role of oxidative stress and HIF1alpha. J Neurochem 108: , 1045–1056. |
[156] | Tamagno E , Guglielmotto M , Aragno M , Borghi R , Autelli R , Giliberto L , Muraca G , Danni O , Zhu X , Smith MA , Perry G , Jo DG , Mattson MP , Tabaton M ((2008) ) Oxidative stress activates a positive feedback between the gamma- and beta-secretase cleavages of the beta-amyloid precursor protein. J Neurochem 104: , 683–695. |
[157] | Moussavi Nik SH , Wilson L , Newman M , Croft K , Mori TA , Musgrave I , Lardelli M ((2012) ) The BACE1-PSEN-AbetaPP regulatory axis has an ancient role in response to low oxygen/oxidative stress. J Alzheimers Dis 28: , 515–530. |
[158] | Li Y , Zhou W , Tong Y , He G , Song W ((2006) ) Control of APP processing and Abeta generation level by BACE1 enzymatic activity and transcription. FASEB J 20: , 285–292. |
[159] | Jonsson T , Atwal JK , Steinberg S , Snaedal J , Jonsson PV , Bjornsson S , Stefansson H , Sulem P , Gudbjartsson D , Maloney J , Hoyte K , Gustafson A , Liu Y , Lu Y , Bhangale T , Graham RR , Huttenlocher J , Bjornsdottir G , Andreassen OA , Jonsson EG , Palotie A , Behrens TW , Magnusson OT , Kong A , Thorsteinsdottir U , Watts RJ , Stefansson K ((2012) ) A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488: , 96–99. |
[160] | Shebl FM , Hsing AW , Park Y , Hollenbeck AR , Chu LW , Meyer TE , Koshiol J ((2014) ) Non-steroidal anti-inflammatory drugs use is associated with reduced risk of inflammation-associated cancers: NIH-AARP study. PLoS One 9: , e114633. |
[161] | Le Provost GS , Pullar CE ((2015) ) beta2-adrenoceptor activation modulates skin wound healing processes to reduce scarring. J Invest Dermatol 135: , 279–288. |
[162] | Deng Y , Wang Z , Wang R , Zhang X , Zhang S , Wu Y , Staufenbiel M , Cai F , Song W ((2013) ) Amyloid-beta protein (Abeta) Glu11 is the major beta-secretase site of beta-site amyloid-beta precursor protein-cleaving enzyme 1(BACE1), and shifting the cleavage site to Abeta Asp1 contributes to Alzheimer pathogenesis. Eur J Neurosci 37: , 1962–1969. |
[163] | Villa JC , Chiu D , Brandes AH , Escorcia FE , Villa CH , Maguire WF , Hu CJ , de Stanchina E , Simon MC , Sisodia SS , Scheinberg DA , Li YM ((2014) ) Nontranscriptional role of Hif-1alpha in activation of gamma-secretase and notch signaling in breast cancer. Cell Rep 8: , 1077–1092. |
[164] | Daulatzai MA ((2015) ) Evidence of neurodegeneration in obstructive sleep apnea: Relationship between obstructive sleep apnea and cognitive dysfunction in the elderly. J Neurosci Res 93: , 1778–1794. |
[165] | Oresic M , Hyotylainen T , Herukka SK , Sysi-Aho M , Mattila I , Seppanan-Laakso T , Julkunen V , Gopalacharyulu PV , Hallikainen M , Koikkalainen J , Kivipelto M , Helisalmi S , Lotjonen J , Soininen H ((2011) ) Metabolome in progression to Alzheimer’s disease. Transl Psychiatry 1: , e57. |
[166] | Fang Y , Tan J , Zhang Q ((2015) ) Signaling pathways and mechanisms of hypoxia-induced autophagy in the animal cells. Cell Biol Int 39: , 891–898. |
[167] | Gama Sosa MA , Gasperi RD , Rocher AB , Wang AC , Janssen WG , Flores T , Perez GM , Schmeidler J , Dickstein DL , Hof PR , Elder GA ((2010) ) Age-related vascular pathology in transgenic mice expressing presenilin 1-associated familial Alzheimer’s disease mutations. Am J Pathol 176: , 353–368. |
[168] | Arendash GW , Su GC , Crawford FC , Bjugstad KB , Mullan M ((1999) ) Intravascular beta-amyloid infusion increases blood pressure: Implications for a vasoactive role of beta-amyloid in the pathogenesis of Alzheimer’s disease. Neurosci Lett 268: , 17–20. |
[169] | Zhang X , Zhou K , Wang R , Cui J , Lipton SA , Liao FF , Xu H , Zhang YW ((2007) ) Hypoxia-inducible factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J Biol Chem 282: , 10873–10880. |
[170] | Zetterberg H , Mortberg E , Song L , Chang L , Provuncher GK , Patel PP , Ferrell E , Fournier DR , Kan CW , Campbell TG , Meyer R , Rivnak AJ , Pink BA , Minnehan KA , Piech T , Rissin DM , Duffy DC , Rubertsson S , Wilson DH , Blennow K ((2011) ) Hypoxia due to cardiac arrest induces a time-dependent increase in serum amyloid beta levels in humans. PLoS One 6: , e28263. |
[171] | Villemagne VL , Burnham S , Bourgeat P , Brown B , Ellis KA , Salvado O , Szoeke C , Macaulay SL , Martins R , Maruff P , Ames D , Rowe CC , Masters CL , Australian Imaging B, Lifestyle Research G ((2013) ) Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol 12: , 357–367. |
[172] | Di Marco LY , Venneri A , Farkas E , Evans PC , Marzo A , Frangi AF ((2015) ) Vascular dysfunction in the pathogenesis of Alzheimer’s disease - A review of endothelium-mediated mechanisms and ensuing vicious circles. Neurobiol Dis 82: , 593–606. |
[173] | Duering M , Grimm MO , Grimm HS , Schroder J , Hartmann T ((2005) ) Mean age of onset in familial Alzheimer’s disease is determined by amyloid beta 42. Neurobiol Aging 26: , 785–788. |
[174] | Kumar-Singh S , Theuns J , Van Broeck B , Pirici D , Vennekens K , Corsmit E , Cruts M , Dermaut B , Wang R , Van Broeckhoven C ((2006) ) Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum Mutat 27: , 686–695. |
[175] | Cook DG , Forman MS , Sung JC , Leight S , Kolson DL , Iwatsubo T , Lee VM , Doms RW ((1997) ) Alzheimer’s A beta(1-42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat Med 3: , 1021–1023. |
[176] | Hartmann T , Bieger SC , Bruhl B , Tienari PJ , Ida N , Allsop D , Roberts GW , Masters CL , Dotti CG , Unsicker K , Beyreuther K ((1997) ) Distinct sites of intracellular production for Alzheimer’s disease A beta40/42 amyloid peptides. Nat Med 3: , 1016–1020. |
Figures and Tables
Fig.1
Disease-causing mutations in PSEN1, PSEN2 and AβPP. A) Cleavages of AβPP. β-Secretase cleavage at the β-site, not the β’-site, followed by γ-secretase cleavage is required to produce Aβ peptides. Panels B, C, and D are modifications of diagrams taken from the Alzheimer Disease & Frontotemporal Dementia Mutation Database [47]. Red amino acid residues are sites of pathogenic mutation causing dementia. Green residues are non-pathogenic variants. The pathogenic nature of orange residues is unclear. Transmembrane domains (TM) and residues encoded by particular exons (Ex) are indicated and numbered. B) Mutations and cleavage sites in AβPP in the region of Aβ. Mutations are thought to affect the level of production of Aβ by changing cleave site preference or affect the structure of Aβ, or both. The production of AICD may also be affected. The position of the “protective” A673T mutation [159] is highlighted with a yellow background. C) Mutations in PSEN1. Residues affected by mutations causing Pick Disease, Familial Acne Inversa and Dilated Cardiomyopathy are highlighted with a yellow background. Residues deleted by fAD mutations causing loss of exon 9 are highlighted with a pink background. The L271V mutation causes increased formation of a naturally-occurring PSEN1 isoform lacking residues encoded by exon 8 (highlighted by a blue background) but it is the simultaneous expression of full-length PSEN1 containing the valine residue at position 271 that apparently causes fAD [51, 91]. D) Mutations in PSEN2. Residues included in the hypoxia-induced isoform PS2V are highlighted with a green background. The position of the unique frame-shift fAD mutation K115Efs and of the S130L mutation causing Dilated Cardiomyopathy are highlighted with yellow background. The “mutations” putatively identified by Kadmiri et al. [121, 122] are upstream of the region shown in B or are indicated with brown arrows and text in C and D.
![Disease-causing mutations in PSEN1, PSEN2 and AβPP. A) Cleavages of AβPP. β-Secretase cleavage at the β-site, not the β’-site, followed by γ-secretase cleavage is required to produce Aβ peptides. Panels B, C, and D are modifications of diagrams taken from the Alzheimer Disease & Frontotemporal Dementia Mutation Database [47]. Red amino acid residues are sites of pathogenic mutation causing dementia. Green residues are non-pathogenic variants. The pathogenic nature of orange residues is unclear. Transmembrane domains (TM) and residues encoded by particular exons (Ex) are indicated and numbered. B) Mutations and cleavage sites in AβPP in the region of Aβ. Mutations are thought to affect the level of production of Aβ by changing cleave site preference or affect the structure of Aβ, or both. The production of AICD may also be affected. The position of the “protective” A673T mutation [159] is highlighted with a yellow background. C) Mutations in PSEN1. Residues affected by mutations causing Pick Disease, Familial Acne Inversa and Dilated Cardiomyopathy are highlighted with a yellow background. Residues deleted by fAD mutations causing loss of exon 9 are highlighted with a pink background. The L271V mutation causes increased formation of a naturally-occurring PSEN1 isoform lacking residues encoded by exon 8 (highlighted by a blue background) but it is the simultaneous expression of full-length PSEN1 containing the valine residue at position 271 that apparently causes fAD [51, 91]. D) Mutations in PSEN2. Residues included in the hypoxia-induced isoform PS2V are highlighted with a green background. The position of the unique frame-shift fAD mutation K115Efs and of the S130L mutation causing Dilated Cardiomyopathy are highlighted with yellow background. The “mutations” putatively identified by Kadmiri et al. [121, 122] are upstream of the region shown in B or are indicated with brown arrows and text in C and D.](https://content.iospress.com:443/media/jad/2016/52-3/jad-52-3-jad151186/jad-52-jad151186-g001.jpg)
Fig.2
Hypothetical model of action of PSEN1 holoprotein including multimerization and a possible interaction with HIF1α. PSEN holoproteins can form multimers and these multimers may be necessary for the endoproteolysis that activates γ-secretase activity. Mutations that preserve the open reading frames of PSENs may allow mutant holoproteins to bind to wild type holoproteins and inhibit holoprotein-dependent activities that are critical to cellular homeostasis in ageing brains. Under hypoxic conditions HIF1α may normally interact with multimeric holoprotein complexes to stimulate formation of active γ-secretase. Normal lysosome acidification may be required for correct metabolism and/or secretion of AβPP, AICD and Aβ.