Immunization with Small Amyloid-β-derived Cyclopeptide Conjugates Diminishes Amyloid-β-Induced Neurodegeneration in Mice
Abstract
Background: Soluble oligomeric (misfolded) species of amyloid-β (Aβ) are the main mediators of toxicity in Alzheimer’s disease (AD). These oligomers subsequently form aggregates of insoluble fibrils that precipitate as extracellular and perivascular plaques in the brain. Active immunization against Aβ is a promising disease modifying strategy. However, eliciting an immune response against Aβ in general may interfere with its biological function and was shown to cause unwanted side-effects. Therefore, we have developed a novel experimental vaccine based on conformational neo-epitopes that are exposed in the misfolded oligomeric Aβ, inducing a specific antibody response.
Objective: Here we investigate the protective effects of the experimental vaccine against oligomeric Aβ1-42-induced neuronal fiber loss in vivo.
Methods: C57BL/6 mice were immunized or mock-immunized. Antibody responses were measured by enzyme-linked immunosorbent assay. Next, mice received a stereotactic injection of oligomeric Aβ1-42 into the nucleus basalis of Meynert (NBM) on one side of the brain (lesion side), and scrambled Aβ1-42 peptide in the contralateral NBM (control side). The densities of choline acetyltransferase-stained cholinergic fibers origination from the NBM were measured in the parietal neocortex postmortem. The percentage of fiber loss in the lesion side was determined relative to the control side of the brain.
Results: Immunized responders (79%) showed 23% less cholinergic fiber loss (p = 0.01) relative to mock-immunized mice. Moreover, fiber loss in immunized responders correlated negatively with the measured antibody responses (R2 = 0.29, p = 0.02).
Conclusion: These results may provide a lead towards a (prophylactic) vaccine to prevent or at least attenuate (early onset) AD symptoms.
INTRODUCTION
With the worldwide aging populations growing, the burden and prevalence of Alzheimer’s disease (AD) is rapidly increasing. Over the next years, AD may constitute an epidemic with high levels of individual suffering and staggering health care costs [1]. AD is characterized by its pathological features of extracellular amyloid-β (Aβ) plaques and tau protein aggregates known as intracellular neurofibrillary tangles. Although characteristic for AD, it is generally believed that both Aβ plaques and neurofibrillary tangles are relatively non-toxic. Indeed, Aβ plaque load does not directly correlate with clinical symptoms of patients [2, 3]. Instead, soluble oligomeric forms of Aβ are believed to be the main mediators of toxicity in AD [4–8]. These oligomeric species subsequently form aggregates of insoluble fibrils that precipitate as extracellular and perivascular plaques in the brain. In addition, intracellular aggregates of phosphorylated tau protein are formed at a later stage of the disease [9, 10]. Based on this sequence of events it seems consequent to consider an immunization strategy directed against epitopes specific for the misfolded oligomeric forms of Aβ. Since 1999 [11], there has been considerable interest in passive or active immunization with Aβ or Aβ-derived constructs against AD, although a breakthrough has not yet been reached[12–19].
In a previous study, we showed that tetanus toxoid (TTd) conjugates of small cyclic peptides derived from Aβ, in particular cyclo[Aβ22-28-YNGK’]-TTd, induced antibodies in Balb/c mice that cross-react in vitro with oligomeric and fibrillar Aβ1-42 and amyloid plaques in mouse and human brain tissue. The antibodies did not recognize the homologous unmodified linear peptide or the amyloid-β protein precursor, indicating a high specificity for misfolded Aβ [20].
Testing the neuroprotective effect of AD-related vaccines or compounds in in vivo models is rather complicated and in most cases indirect. Mutant mice with phenotypes resembling AD pathology often do not show an overt neurotoxicity, and the protective function of compounds or vaccines can only be evaluated by indirectly measuring the postmortem plaque load or by analyses in living mice such as electrophysiological changes and/or cognitive/behavioral changes. In the present study, we have investigated if immunization can induce protection against acute oligomeric Aβ1-42-induced neurodegeneration of the cholinergic nucleus basalis magnocellularis ofMeynert (NBM) in C57BL/6 mice. This in vivo model combines several advantages for testing compounds in proof of concept studies and reflects some essential hallmarks of AD pathology. The NBM is one of the early regions to be affected during AD and the overall neuropathology in this model, including cholinergic denervation and microglial activation, features much of the pathology found in AD patients [21]. The NBM lesion model provides the means to test directly the neuroprotective effect of the active immunization using cyclo-Aβ peptides. Cholinergic fibers emanating from the NBM innervate cortical and hippocampal brain areas. Loss of this innervation can be quantified by measuring fiber density and used to determine neuroprotective strategies in AD [21–23]. Injecting a validated amount of neurotoxic oligomeric Aβ1-42 into the NBM creates an acute lesion with ∼40% loss of cholinergic neurons [21]. Here we provide evidence that active immunization using a trivalent vaccine consisting of cyclo[Aβ22-28-YNGK’], cyclo[Aβ23-29-YNGK’], and cyclo[Aβ22-29-YNGK’] peptide TTd conjugates provides significant protection in the Aβ1-42-induced NBM lesion model.
MATERIALS AND METHODS
Materials
Lyophilized human Aβ1-42 (DAEFRHDSGY10EVHHQKLVFF20AEDVGSNKGA30IIGLMVGGVV40IA) was purchased from AnaSpec (Freemont, CA, USA) or rPeptide (USA). Scrambled-sequence Aβ1-42 was purchased from rPeptide (USA). Monophosphoryl lipid A (MPL) was purchased from Sigma-Aldrich (Lipid A, monophosphoryl from Salmonella enterica serotype minnesota Re 595, Re mutant). An aqueous suspension of aluminum hydroxide (Alhydrogel 2%, 10.3 mg Al3 +/ml) was purchased from Brenntag (Frederikssund, Denmark), and monoclonal antibody 6E10 against Aβ1-17 from Covance Research Products (Dedham, MA, USA). Tetanus toxoid (TTd) was produced by the former Vaccine Division of the National Institute for Public Health and the Environment (Bilthoven, The Netherlands).
Animals and housing
Experiments were performed using 33 male C57BL/6JRj mice (Janvier, Le Genest-St-Isle, France), 6–8 weeks old at arrival. All mice were housed individually in macrolon type II cages (length 35 cm, width 15 cm, height 13.5 cm, Bayer, Germany), with sawdust as bedding and shredded cardboard as nesting material. The mice were kept in a climate room with controlled temperature (22 ± 1°C) and humidity (55 ± 10%). A light/dark (LD) schedule (12 h light –12 h dark; lights on at 07:00 h GMT+1 h; ± 50 lux) was maintained. Food (standard rodent chow: RMHB/2180, Arie Block BV, Woerden, NL) and normal tap water were available ad libitum. Cages were cleaned at least every two weeks. All mice were checked daily for food/water/health/activity/abnormal behavior. All procedures were in accordance with the regulation of the ethical committee for the use of experimental animals of the University of Groningen, The Netherlands (License number DEC 6579A).
Experimental outline
Mice were habituated to the climate room and housing conditions for seven days before receiving the first round of immunization. The mice were immunized by subcutaneous injection in the groin with either our trivalent vaccine (referred to as ‘immunized’ mice, n = 24) or mock vaccine (referred to as ‘mock-immunized’ or ‘mock’ mice, n = 9). Three, four, or five immunizations were performed with three-weekly intervals. An overview of the experimental procedures is provided in Fig. 1. Twelve days after all mice had received the third immunization, blood samples were collected from the tail vein and the derived serum samples were analyzed the next day (day 55) by enzyme-linked immunosorbent assay (ELISA). Based on the anti-oligomeric Aβ1-42 endpoint titers obtained (ELISA 1), eight immunized and three mock-immunized mice were selected for stereotactic Aβ1-42-induced NBM lesion surgery which was performed the next day (group A, day 56, two weeks after the third immunization). Remaining mice received a fourth immunization (on day 63) and another tail puncture after twelve days (on day 75). Based on the titers obtained (ELISA 2), eight immunized and four mock-immunized mice were selected for Aβ1-42-induced NBM lesion surgery (group B, day 77, two weeks after the fourth immunization). Remaining mice (group C: eight immunized, two mock-immunized mice) received a fifth immunization (on day 84) and received Aβ1-42-induced NBM lesion surgery two weeks later (day 98). Mice from each group were transcardially perfused ten days after NBM lesion surgery. Final blood samples were collected from the heart just before perfusion and derived serum samples were analyzed later (ELISA 3). Brains were isolated for further immunohistochemicalanalysis.
Peptide synthesis and purification
The synthesis and purification of the cyclic peptides cyclo[Aβ22–28-YNGK’], cyclo[Aβ23-29-YNGK’], and cyclo[Aβ22-29-YNGK’], in which K’ is Nɛ-(S-acetylmercaptoacetyl)lysyl for conjugation purposes, was performed as described earlier[20, 24].
Preparation of conjugates and vaccines
The peptides were coupled to either bromoacetylated TTd (to obtain vaccine components) or maleimidyl-modified bovine serum albumin (BSA) (to obtain ELISA coating antigens) [25]. Bromoacetylated TTd was also capped with 2-aminoethanethiol, without prior incubation with peptide, to obtain the protein for the mock vaccine (capped TTd).
Vaccine preparation
Aliquots (300 or 120 μl) of a solution of MPL (1.0 mg/ml) in chloroform/methanol/water, 74/23/3, were evaporated to dryness in round bottom polypropylene tubes. MPL films obtained from 300 μg samples were reconstituted with 0.60 ml of each cyclopeptide-TTd conjugate (0.50 mg/ml in physiological salt), followed by 0.58 ml Alhydrogel. MPL films obtained from 120 μg samples were reconstituted with 0.72 ml of capped TTd (0.50 mg/ml in physiological salt), followed by 0.23 ml Alhydrogel. The tubes containing the suspensions were left on a roller mixer for 1 h at room temperature and thereafter stored overnight at 4°C). Then, the concentrated vaccine was diluted with 6.62 ml of physiological salt and the concentrated mock vaccine with 2.65 ml physiological salt. One dose of 0.3 ml of vaccine contained 10 μg of each cyclopeptide-TTd conjugate, 10 μg MPL, and 0.2 mg Al3 +. One dose of 0.3 ml of mock vaccine contained 30 μg capped TTd, 10 μg MPL, and 0.2 mg Al3 +.
ELISA
Wells of microtiter plates (Greiner 655092) were coated with oligomeric Aβ1-42, peptide-BSA conjugates or TTd. Freshly prepared Aβ1-42 oligomers were diluted to a final concentration of 2.5 μM (11.3 μg/ml) in 0.04 M sodium carbonate/bicarbonate buffer, pH 9.7. Peptide-BSA conjugates and TTd were diluted to a total concentration of 0.5 μg/ml in the same carbonate/bicarbonate buffer. Aliquots (100 μl) of these solutions were transferred into wells of the plates. The plates were incubated for 90 min at 37°C, emptied and washed twice with tap water containing 0.05% Tween 80. The plates were incubated with threefold serialdilutions of the serum samples in phosphate buffered saline containing 0.1% Tween 80. Monoclonal antibody 6E10 was used as positive control on plates coated with oligomeric Aβ1-42. The starting dilution for determination of anti-oligomeric Aβ1-42 titers and anti-linear[Aβ1-15-C] titers was 1/150. For determination of anti-cyclopeptide and anti-TTd titers, a starting dilution of 1/1500 was used. The plates were further processed as described earlier [26]. Endpoint titers were defined as the dilution, which gave an optical density (OD) reading identical to the mean OD plus three times standard deviation of sera of mice immunized with mock vaccine [27].
Preparation of oligomeric Aβ1-42 for stereotactic NBM lesions
Oligomeric Aβ1-42 was prepared as described before [28–30]. In short, lyophilized Aβ1-42 peptide (rPeptide, USA) was dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP, Sigma-Aldrich, St. Louis, MO, USA) at a concentration of 1 mM. The peptide solution was aliquoted and the HFIP removed by evaporation at room temperature in a flow cabinet for 3 h. The dry peptide films were stored at –20°C until further processing. Before use, Aβ1-42 films were dissolved in anhydrous DMSO to 5 mM and subsequently diluted in neurobasal medium to a final concentration of 250 μM. The solution was incubated at 4°C for 24 h to enable Aβ1-42 oligomerization. Scrambled Aβ1-42 control peptide was processed using exactly the same procedure.
Stereotactic Aβ1-42-induced NBM lesions
Mice were anaesthetized with an isoflurane/oxygen gas mixture and received 60 μl finadyne s.c. (1 mg/ml, Schering-Plough NV/SA, Brussels, Belgium). Mice were then placed in a stereotact (Kopf instruments model 900, Tujunga, CA, USA) secured with cheek bars and a tooth-bar/nose clamp. The stereotact was equipped to support maintenance of the isoflurane/oxygen anesthesia. Eyes were protected against dehydration by applying Vita-Pos® salve (Ursapharm) and the shaved skin was disinfected with 70% ethanol. A small medial incision was made from bregma to lambda. The periost was gently scraped away with a scalpel and the scalp was cleaned using cotton swaps. A hole was drilled in the skull and the dura was punctured with a needle. A 1 μl Hamilton syringe was slowly lowered into the brain (coordinates: 0.6 mm posterior to bregma; 2.1 mm lateral to the sagittal suture). 0.2 μl of 250 μM oligomeric Aβ1-42 was slowly (0.1 μl/min) injected at 4.6 and 4.4 mm ventral to the dura (total unilateral injection amount: 0.4 μl, 100 pmol). The stereotactic coordinates had been confirmed by several pilot experiments in which trypan blue was injected into the NBM and traced (postmortem) to the NBM. After injection, the syringe was kept in place for 2 min at each injection site to optimize diffusion into the brain and to limit spread of the solution during withdrawal of the needle. The same procedure was followed on the contralateral side of the brain, where scrambled Aβ1-42 control peptide was injected. Left/right injection of the peptides were randomized between mice. After contralateral injection, mice were immediately removed from the stereotact. The head wound was sutured (Ethicon perma-hand N266 5-0), disinfected with Povidine-iodine (Betadine®) and mice received an i.p. injection of 0.5 ml warm ( ± 25°C) saline/glucose (0.45% NaCl+2.5% glucose). Mice were placed back in their homecage and kept under a IR heat lamp for 24 h.
Transcardial perfusion
At postoperative day 10, under deep pentobarbital anesthesia, mice were perfused transcardially for 1 min with 0.9% NaCl+0.5% heparin (400U) in H2O (15 ml/min), followed by 150 ml 4% paraformaldehyde (PF) in 0.1 M phosphate buffer (PB) for fixation. Brains were collected, post fixated for 24 h in 4% PF in 0.1 M PB, rinsed for one day in 0.01 M phosphate buffered saline (PBS, pH 7.4) and then kept overnight in 30% sucrose in PBS cryoprotectant at room temperature. Brains were frozen using liquid nitrogen and stored at –80°C. The brains were cut in 20 μm coronal sections using a cryotome, collected in PBS (0.01 M, pH = 7.4) containing 0.1% sodium azide and kept at 4°C until choline acetyltransferase (ChAT) immunostaining.
Immunohistochemical ChAT staining
The free floating brain sections were rinsed three times (rinsing was always performed in 0.01 M PBS, pH = 7.4, for 5 min on a shaker) before being incubated for 30 min in 0.3% H2O2 in PBS. Next, the brain slices were rinsed five times before pre-incubation for 1 h at room temperature (RT) in PBS containing 5% normal rabbit serum (NRS, Zymed, San Francisco, CA, USA) and 0.4% Triton X-100. Subsequently, sections were incubated for 3 days at 4°C in the primary antibody solution containing 1:333 diluted goat anti-ChAT IgG (Millipore, Billerica, MA, USA), 1% NRS, 0.5% BSA, and 0.4% Triton X-100 in PBS. Next, brain sections were rinsed five times and thereafter incubated at room temperature for 4 h in the secondary antibody solution containing rabbit anti-goat IgG 1:500 (Sigma), 1% NRS, 0.2% Triton X-100, and 0.5% BSA in PBS. Afterwards, sections were rinsed five times before being incubated at RT for 2 h with Vectastain Elite ABC Kit (Vector Laboratories, CA, USA). Both the ‘A’ and ‘B’ solution were diluted to 1:500. The staining was completed with nickel-enhanced (BDH Chemicals Ltd., UK) diaminobenzidine (DAB) reaction in the presence of H2O2. The next day, slices were mounted on glass from a 1% gelatin+0.01% aluin solution, dried overnight and defatted/dehydrated through respectively 100% EtOH, 100% EtOH, 70% EtOH+30% Xylol, 30% EtOH+70% Xylol, 100% Xylol, 100% Xylol, 100% Xylol. Glass preparations were cover slipped using DPX mountant, dried for two days and then cleaned.
Quantitative image analysis
The parietal neocortex is topographically the target of the afferent cholinergic pathway from the NBM sites where the Aβ1-42 oligomers were injected. The quantification procedure was established in our laboratory and described in detail in a series of previous publications [21, 22, 31–33]. Briefly, exact measurement took place in the superficial sublayer of the layer V cortical area representing the densest zone of cortical cholinergic innervation (coordinates: 0.6 mm posterior to bregma [34]. After background subtraction and gray-scale threshold determination, the surface area of ChAT-positive fibers (percentage of the area covered by ChAT-positive cholinergic fibers relative to the total sampling area) was measured on both contralateral sides of eight coronal sections per animal using a Quantimet 600HR Image Analysis System (Leica, Rijswijk, the Netherlands). The percentage of fiber loss relative to the control side was calculated within each of the slices and these values were averaged as an indicator of cholinergic degeneration per animal. One immunized mouse showed over 40% fiber loss and was excluded as an outlier from all analyses.
Statistics
Statistical analyses were performed using GraphPad Prism 5.01 (GraphPad software, Inc.) and SigmaPlot 12.5 (Systat software, inc.). Fiber densities in control and lesion sides of the brains of mock and immunized mice were analyzed using a 2-wayrepeated measures (RM) analysis of variance (ANOVA). Differences between groups were tested by two-tailed unpaired t-tests. Pre-post differences between groups were tested by two-tailed paired t-test. Correlations were performed using Pearson correlations. p < 0.05 was consideredsignificant.
RESULTS
ELISA: Results before Aβ1-42-induced nucleus basalis lesions
Mice were immunized with either trivalent vaccine (immunized, n = 24) or mock vaccine (mock, n = 9). Three, four or five (mock-) immunizations were performed with three-weekly intervals while intermediate blood samples were taken from the tail vain (see Fig. 1). The derived serum samples were analyzed by enzyme-linked immunosorbent assay (ELISA) using oligomeric Aβ1-42 as coating antigen. Fig. 2 shows the 10log[Aβ1-42 endpoint titers] obtained. Vertical axis categories are numbered between square brackets for easy reference.
After three immunizations (ELISA 1 in Fig. 1), a significant difference was found between all mock and immunized mice (Fig. 2 [1]: two-tailed unpaired t-test: p < 0.001).
Mice (n = 8) with high titers were selected for the first round of Aβ1-42-induced NBM lesion surgery, forming ‘group A’ together with randomly selected mock animals (n = 3; Fig. 2 [2]: two-tailed unpaired t-test mock versus immunized mice: p < 0.001). Remaining animals still showed a significant difference between mock and immunized mice (Fig. 2 [3]: two-tailed unpaired t-test: p < 0.05), but received a fourth (mock-) immunization followed by a second tail vein puncture. Aβ titers (ELISA 2) showed that the difference between mock and immunized mice was maintained (Fig. 2 [4]: two-tailed unpaired t-test: p < 0.05), but that the fourth immunization did not significantly increase Aβ titers (Fig. 2 [4] versus [3]: two-tailed paired t-test before versus after fourth immunization: mock: p = 0.98, immunized: p = 0.81). Again, immunized mice with high titers (n = 8) and randomly selected mock mice (n = 4) were selected for the second round of Aβ1-42-induced NBM lesion surgery, forming group B (Fig. 2 [5]: two-tailed unpaired t-test mock versus immunized mice: p < 0.05). The remaining mock (n = 2) and immunized (n = 8) mice formed group C. As measured from the serum samples after the fourth immunization (ELISA 2), no significant difference between mock and immunized animals was present in group C mice (Fig. 2 [6]: two-tailed unpaired t-test: p = 0.46). These mice received a fifth round of (mock-)immunization before Aβ1-42-induced NBM lesion surgery.
ELISA: Results after Aβ1-42-induced nucleus basalis lesions
After the Aβ1-42-induced NBM lesions, blood samples from all mice were obtained by a heart puncture just after the mice were sacrificed, before transcardial perfusion. ELISA analysis (ELISA 3) showed that considering all animals, a significant difference was again found between mock and immunized mice (Fig. 2 [7]: two-tailed unpaired t-test: p < 0.001). Similarly, a significant difference was again found between mock and immunized mice of group A and B, but not of group C (Fig. 2 [8–10]: two-tailed unpaired t-test: group A: p < 0.001; group B: p < 0.05; group C: p = 0.46). These results confirm the earlier intermediate titers before the Aβ1-42-induced NBM lesions (ELISA 1 and 2).
No significant differences were found between the titers before versus after the Aβ1-42-induced NBM lesions of mock and immunized mice (statistical comparisons not indicated in Fig. 2: two-tailed paired t-test, before versus after NBM lesion: group A mock mice: p = 0.42; group A immunized mice: p = 0.33; group B mock mice: p = 0.94; group B immunized mice: p = 0.30; group C mock mice: p = 0.67; group C immunized mice: p = 0.96). These results indicate that injecting oligomeric Aβ1-42 in the NBM, did not significantly affect antibody responses. Moreover, these results indicate that the fifth round of (mock-) immunizations, as performed on the group C animals, did not significantly increase their average antibody response. Together, the results show that three rounds of immunizations are very much effective, while a fourth and fifth round of immunizations have only minor added effect.
ELISA: Responders and non-responders
The ELISA results revealed a degree of variation in the measured antibody responses. Although most mice responded well to the immunizations, others showed a lesser response. Supplementary Table 1 shows the 10log[Aβ1-42 endpoint titers] obtained for individual mice. We used a rigorous criterion to identify responders and non-responders. ‘Responders’ were defined as mice which showed higher titers than the average of the mock animals plus three standard deviations. After three immunizations, 14 out of 24 mice (56%) were identified as responders (ELISA 1).Measurements on serum samples after the fourth immunization (ELISA 2) revealed both increases- and decreases in titers. Measurements from serum samples obtained after the heart puncture before transcardial perfusion (ELISA 3) showed 16 out of 24 responders (67%). Overall, 79% of the mice were identified as responders in at least one measurement, leaving five mice (21%) identified as consistent non-responders.
ELISA: Investigating vaccine specificity
The sera obtained by heart puncture were analyzed more extensively (ELISA 3). In addition to oligomeric Aβ1-42, BSA conjugates of cyclo[Aβ22-28-YNGK’] and its 23–29 and 22–29 analogues and linear [Aβ1-15-C], as well as plain TTd, were used as coating antigens. As expected, all serum samples obtained after the heart puncture had high anti-TTd antibody titers (all mice showed a 10log[endpoint titer] > 5.52 on a TTd ELISA coating, data not shown). The serum samples of immunized mice showed high antibody titers against each of the three amyloid cyclopeptides (Supplementary Table 1), but not to linear Aβ1-15 which contains the immunodominant epitope of Aβ1-42 (data not shown).
Immunohistochemistry: Aβ1-42-induced nucleus basalis lesions
An excitotoxic damage model was used to assess whether immunized mice were more protected against oligomeric Aβ1-42 insult to the brain compared to mock mice. The NBM is the origin of many cholinergic projections to the cortex. Oligomeric Aβ1-42 was unilaterally injected in the NBM and the neurotoxic impact assessed by determining cholinergic fiber loss in the parietal neocortex relatively to the control hemisphere which was injected with scrambled Aβ1-42 peptide (Aβ-scr) (Fig. 3).
In the mock mice, average fiber density on the control and lesion side was 10.30 ± 0.36 SEM and 7.95 ± 0.28 SEM, respectively. In the immunized mice, average fiber density on the control and lesion side was 9.91 ± 0.35 SEM and 8.17 ± 0.33 SEM, respectively. A 2-way RM ANOVA with independent variables ‘treatment’ (mock-immunized or immunized) and ‘brain side’ (oligomeric Aβ1-42-injected or scrambled Aβ1-42-injected), revealed a significant difference between brain sides. The side of the brain injected with oligomeric Aβ1-42 consistently showed less fiber density than the scrambled-Aβ injected sides of the brains (F (1,30) = 398.83, p < 0.001; Fig. 4A). An ‘All Pairwise Multiple Comparison’ posthoc test (Holm-Sidak method) revealed that this difference was present within both the mock- (t = 13.54, p < 0.001) and immunized mice (t = 16.02, p < 0.001). These results indicate that the oligomeric Aβ1-42-induced NBM lesion model was highly effective. A significant interaction effect was found between treatment and brain side (F (1,30) = 8.88, p = 0.006), but no significant effect of treatment was detected (F (1,30) = 0.02, p = 0.88) within either the oligomeric Aβ1-42-injected- (t = 0.38, p = 0.71), or scrambled Aβ1-42-injected (t = 0.67, p = 0.51) brain sides. However, differences between mock and immunized mice became apparent when comparing lesion sides with control sides within each of the brain slices and calculating the average percentage of fiber loss (Fig. 4B). On average, immunized mice showed significantly less loss of cholinergic fibers in the parietal neocortex than mock mice (immunized: 16.77 % ± 0.99 SEM versus mock: 21.05 % ± 1.43 SEM, two-tailed unpaired t-test: p = 0.02, effect size: Cohen’s d = 0.95). As expected, these statistics slightly improve when excluding the five identified non-responders from the immunized group (immunized responders: 16.22% ± 1.09 SEM versus mock: 21.05% ± 1.43 SEM, two-tailed unpaired t-test: p = 0.01, effect size: Cohen’s d = 1.08). Immunized mice showed 20.3% reduced fiber loss relative to the mock mice, or 22.9% when excluding the five non-responders from the immunized mice.
Correlation between cholinergic fiber loss and antibody response
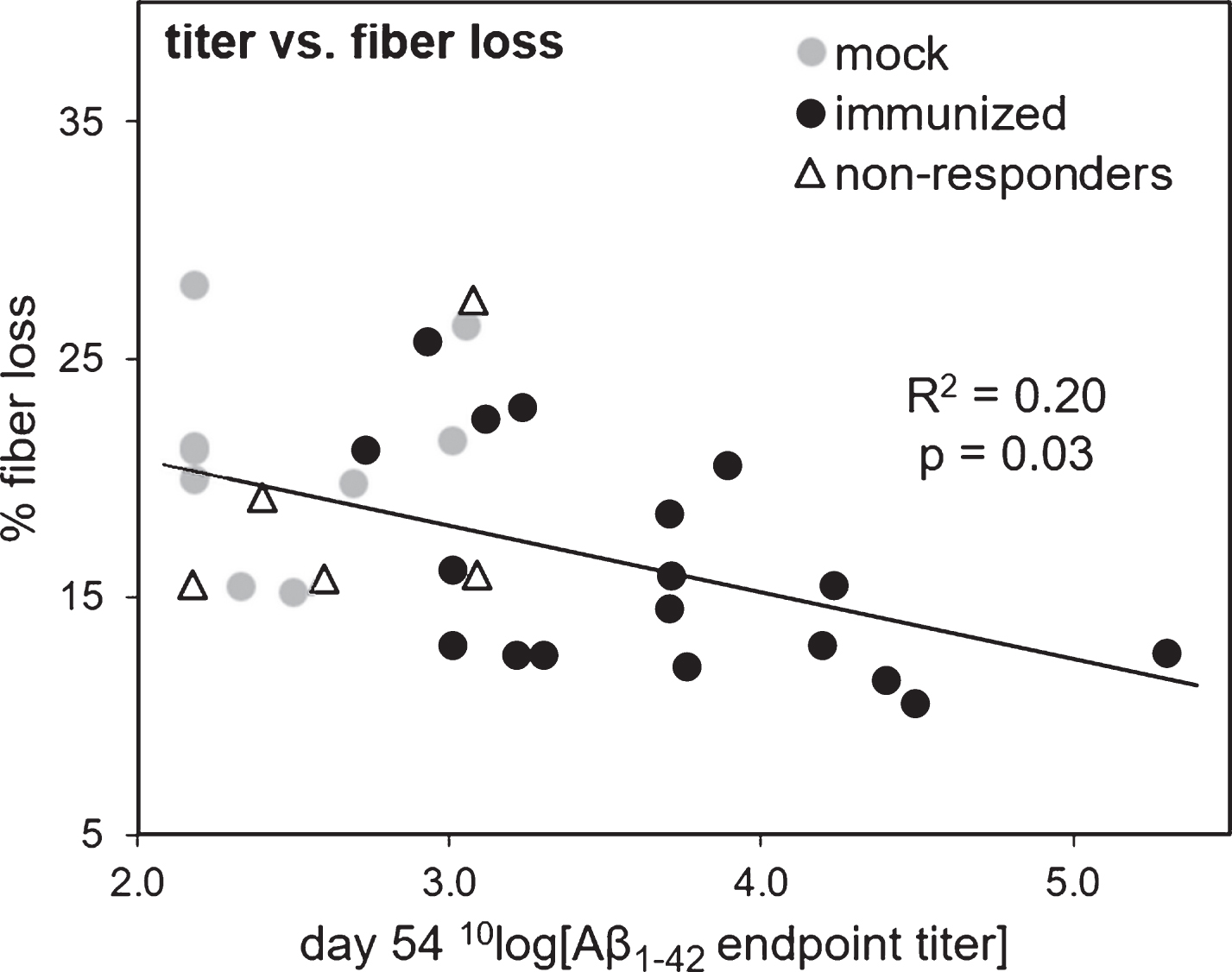
We investigated whether the loss of cholinergic fiber density in the cortex correlated negatively with the measured antibody responses. When using the Aβ1-42 endpoint titers based on the serum samples obtained after the heart puncture (ELISA 3), a significant negative correlation was found between the titers and the percentage of fiber loss when considering all mice (Pearson r = –0.38, R2 = 0.15, p = 0.03). However, this correlation was not significant when considering only the immunized mice, with or without the five non-responders (p = 0.49 and p = 0.91, respectively). The titers obtained from the tail vain puncture before Aβ1-42-induced NBM lesions, after all mice had equally received 3 immunizations (ELISA 1), showed similar results with a better correlation. Using these titers, a significant negative correlation was found between fiber loss and antibody response (considering all mice: Pearson r = –0.49, R2 = 0.24, p = 0.004; considering only the immunized mice: Pearson r = –0.44, R2 = 0.20, p = 0.03; considering immunized mice without the non-responders: Pearson r = –0.54, R2 = 0.29, p = 0.02). Results are shown in Fig. 5.
DISCUSSION
In recent years it has become more and more apparent that amyloid plaques do not primarily correlate with AD pathology. This seems to be rather true for the amount of tangles consisting of hyperphosphorylated tau proteins [35–40]. A possible explanation for that may lie in the fact that Aβ plaques per se are not neurotoxic and that tangles in contrast represent dead neuronal cell bodies and therefore neuronal cell loss. In fact the most neurotoxic molecules in AD pathology turned out to be soluble oligomeric forms of misfolded Aβ [6, 41–44]. Since even under normal physiological conditions Aβ is produced [45], a vaccination against non-oligomeric or non-fibrillary Aβ might result in unwanted side effects [46]. Indeed, non-aggregated Aβ peptide at low doses have been found to exhibit neurotrophic effects including enhancement of neuronal survival, neurite-promoting and neural stem cell differentiation effects [47–51]. Using cyclic peptides mimicking conformational neo-epitopes of oligomeric Aβ forms could circumvent the problems that would arise if peptides would be used which represent epitopes related to Aβ in general.
In a previous study, we have shown that TTd conjugates of small cyclic peptides derived from Aβ, in particular cyclo[Aβ22-28-YNGK’]-TTd can be used for active immunization in mice. Two immunizations effectively produced antibodies in Balb/c mice that cross-react in vitro with oligomeric and fibrillar Aβ1-42 and amyloid plaques in mouse and human brain tissue. Importantly, the antibodies did not recognize the homologous linear peptide or the amyloid-β protein precursor, indicating a high specificity for misfolded Aβ [20]. In preliminary experiments it was found that C57BL/6 mice responded rather poorly to the original vaccine formulation, that is, cyclo[Aβ22-28-YNGK’]-TTd conjugate with aluminum phosphate as adjuvant. The response in C57BL/6 was improved by immunization with the trivalent vaccine as used in the current study (a mixture of TTd conjugates of three cyclopeptides, cyclo[Aβ22-28-YNGK’] and its 23–29 and 22–29 analogues, with aluminum hydroxide gel and monophosphoryl lipid A (MPL) as adjuvant). In addition, a third immunization significantly improved the antibody response (data not shown). Our current data confirm these earlier results and show that a subsequent fourth and fifth round of immunizations had little added effect on the antibody response. It is noteworthy that in the course of our investigation it was reported that the MPL adjuvant by itself may have beneficial effects on AD pathology, as measured in AβPPswe/PS1 transgenic mice [52, 53]. In the current study, both immunized and mock-immunized mice received the same amounts and dosages of MPL adjuvant. Therefore, the mock mice might have had some level of protection, induced by the MPL they received, indicating that the effect of immunization might be underestimated in the current study. Future studies may lead to further optimization of the vaccine formula, dosage, and administration scheme to enhance its efficacy.
We used the established NBM lesion model in C57Bl/6 mice [21–23], to test directly whether the specific antibodies produced by the active immunization have a protective potential against the cytotoxic effects of oligomeric Aβ1-42. We found that immunized mice showed 20% less cholinergic fiber loss in the parietal neocortex relative to mock-immunized mice (or 23% when excluding five non-responders). These results are very promising.
Injecting substantial amounts of neurotoxic oligomeric Aβ1-42 into the NBM creates an acute lesion. It was not certain whether the used Aβ concentration and incubation time (10 days) were optimal to show an effect at all. Similarly, it was uncertain if there would be enough neutralizing antibodies produced by the immune system and if these antibodies would reach the lesion site effectively enough. Although we did not attempt to detect (in brain slices) the specificanti-Aβ antibodies produced by the immune system, it has been shown that peripheral antibodies are able to cross the blood-brain barrier [54]. Additionally, due to the administration of Aβ1-42 via stereotactic injection, a local microglial response may have led to a local leakage of the blood brain barrier, allowing anti-Aβ antibodies to enter the brain parenchyma from blood vessels.
A main source of variation was the observed difference in antibody response of the mice. This variation allowed us to correlate the antibody responses of individual mice to the measured fiber loss. As expected, fiber loss significantly correlated negatively with the antibody titers induced by immunization. When considering only the immunized mice, this was however only true when using the titers obtained after all mice had received three rounds of immunizations (ELISA 1, before Aβ1-42-induced NBM lesions), but not when using the titers obtained from the serum samples after the NBM lesions (ELISA 3). Note that the blood samples taken after three rounds of immunizations (ELISA 1) were collected from all mice at the same time, while the heart punctures (ELISA 3) were acquired and processed at different days for the three groups (see Supplemental Table 1). The serum samples obtained after three rounds of immunizations (ELISA 1) are therefore comparable measures and this initial assessment of the antibody response showed to correlate significantly with the measured fiber loss, even within the immunized group only and excluding consistent non-responders. These results are promising given that titers are not an accurate measure for effective antibody passage over the blood brain barrier and for the effectiveness of the immune system to neutralize Aβ.
Taken together, the tested trivalent vaccine has now been demonstrated to effectively induce a specific antibody response against misfolded Aβ in Balb/c as well as C57BL/6 mice without noticeable side effects. Furthermore, in the present study we show that this active immunization protects against acute oligomeric Aβ1-42 insult to the brain in vivo. A clear next step is to immunize transgenic AD model mice and investigate the effect of immunization on cognition and amyloid plaque load with aging. Together, these results may open up the way to vaccination at an early stage of AD symptoms or before.
ACKNOWLEDGMENTS
We thank Dr Germie van den Dobbelsteen for advice on the immunization of the mice and Hans Strootman and Vibeke Bruinenberg for practical guidance of this part of the study. We thank Wanda Douwenga for help with the tail vein punctures and perfusions and Jan Keijser for help with the immunohistochemical analysis.
Authors’ disclosures available online (http://www.j-alz.com/manuscript-disclosures/15-1136r1).
Appendices
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-151136.
REFERENCES
[1] | Brookmeyer R , Johnson E , Ziegler-Graham K , Arrighi HM ((2007) ) Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement 3: , 186–191. |
[2] | Nelson PT , Alafuzoff I , Bigio EH , Bouras C , Braak H , Cairns NJ , Castellani RJ , Crain BJ , Davies P , Del TK , Duyckaerts C , Frosch MP , Haroutunian V , Hof PR , Hulette CM , Hyman BT , Iwatsubo T , Jellinger KA , Jicha GA , Kovari E , Kukull WA , Leverenz JB , Love S , Mackenzie IR , Mann DM , Masliah E , McKee AC , Montine TJ , Morris JC , Schneider JA , Sonnen JA , Thal DR , Trojanowski JQ , Troncoso JC , Wisniewski T , Woltjer RL , Beach TG ((2012) ) Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J Neuropathol Exp Neurol 71: , 362–381. |
[3] | Terry RD ((1996) ) The pathogenesis of Alzheimer disease: An alternative to the amyloid hypothesis. J Neuropathol Exp Neurol 55: , 1023–1025. |
[4] | Wisniewski T , Goni F ((2015) ) Immunotherapeutic appro-aches for Alzheimer’s disease. Neuron 85: , 1162–1176. |
[5] | Klein WL , Stine WB Jr , Teplow DB ((2004) ) Small assemblies of unmodified amyloid beta-protein are the proximate neurotoxin in Alzheimer’s disease. Neurobiol Aging 25: , 569–580. |
[6] | Haass C , Selkoe DJ ((2007) ) Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol 8: , 101–112. |
[7] | Glabe CG ((2008) ) Structural classification of toxic amyloid oligomers. J Biol Chem 283: , 29639–29643. |
[8] | Ashe KH , Aguzzi A ((2013) ) Prions, prionoids and pathogenic proteins in Alzheimer disease. Prion 7: , 55–59. |
[9] | Choi SH , Kim YH , Hebisch M , Sliwinski C , Lee S , D’Avanzo C , Chen H , Hooli B , Asselin C , Muffat J , Klee JB , Zhang C , Wainger BJ , Peitz M , Kovacs DM , Woolf CJ , Wagner SL , Tanzi RE , Kim DY ((2014) ) A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515: , 274–278. |
[10] | Oddo S , Caccamo A , Tran L , Lambert MP , Glabe CG , Klein WL , LaFerla FM ((2006) ) Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J Biol Chem 281: , 1599–1604. |
[11] | Schenk D , Barbour R , Dunn W , Gordon G , Grajeda H , Guido T , Hu K , Huang J , Johnson-Wood K , Khan K , Kholodenko D , Lee M , Liao Z , Lieberburg I , Motter R , Mutter L , Soriano F , Shopp G , Vasquez N , Vandevert C , Walker S , Wogulis M , Yednock T , Games D , Seubert P ((1999) ) Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400: , 173–177. |
[12] | Agadjanyan MG , Petrovsky N , Ghochikyan A ((2015) ) A fresh perspective from immunologists and vaccine researchers: Active vaccination strategies to prevent and reverse Alzheimer’s disease. Alzheimers Dement 11: , 1246–1259. |
[13] | Delrieu J , Ousset PJ , Voisin T , Vellas B ((2014) ) Amyloid beta peptide immunotherapy in Alzheimer disease. Rev Neurol (Paris) 170: , 739–748. |
[14] | Fettelschoss A , Zabel F , Bachmann MF ((2014) ) Vaccination against Alzheimer disease: An update on future strategies. Hum Vaccin Immunother 10: , 847–851. |
[15] | Giacobini E , Gold G ((2013) ) Alzheimer disease therapy–moving from amyloid-beta to tau. Nat Rev Neurol 9: , 677–686. |
[16] | Lambracht-Washington D , Rosenberg RN ((2013) ) Advances in the development of vaccines for Alzheimer’s disease. Discov Med 15: , 319–326. |
[17] | Lemere CA ((2013) ) Immunotherapy for Alzheimer’s disease: Hoops and hurdles. Mol Neurodegener 8: , 36. |
[18] | Mandler M , Santic R , Gruber P , Cinar Y , Pichler D , Funke SA , Willbold D , Schneeberger A , Schmidt W , Mattner F ((2015) ) Tailoring the antibody response to aggregated Aβ using novel Alzheimer-vaccines.e. PLoS One 10: , 0115237–. |
[19] | Panza F , Logroscino G , Imbimbo BP , Solfrizzi V ((2014) ) Is there still any hope for amyloid-based immunotherapy for Alzheimer’s disease?. Curr Opin Psychiatry 27: , 128–137. |
[20] | Hoogerhout P , Kamphuis W , Brugghe HF , Sluijs JA , Timmermans HA , Westdijk J , Zomer G , Boog CJ , Hol EM , van den Dobbelsteen GP ((2011) ) A cyclic undecamer peptide mimics a turn in folded Alzheimer amyloid β and elicits antibodies against oligomeric and fibrillar amyloid and plaques. PLoS One 6: , e19110. |
[21] | Granic I , Nyakas C , Luiten PG , Eisel UL , Halmy LG , Gross G , Schoemaker H , Moller A , Nimmrich V ((2010) ) Calpain inhibition prevents amyloid-beta-induced neurodegeneration and associated behavioral dysfunction in rats. Neuropharmacology 59: , 334–342. |
[22] | Dolga AM , Granic I , Nijholt IM , Nyakas C , van der Zee EA , Luiten PG , Eisel UL ((2009) ) Pretreatment with lovastatin prevents N-methyl-D-aspartate-induced neurodegeneration in the magnocellular nucleus basalis and behavioral dysfunction. J Alzheimers Dis 17: , 327–336. |
[23] | Luiten PG , Douma BR , van der Zee EA , Nyakas C ((1995) ) Neuroprotection against NMDA induced cell death in rat nucleus basalis by Ca2+antagonist nimodipine, influence of aging and developmental drug treatment. Neurodegeneration 4: , 307–314. |
[24] | Brugghe HF , Timmermans HA , Van Unen LM , Ten Hove GJ , Van de Werken G , Poolman JT , Hoogerhout P ((1994) ) Simultaneous multiple synthesis and selective conjugation of cyclized peptides derived from a surface loop of a meningococcal class 1 outer membrane protein. Int J Pept Protein Res 43: , 166–172. |
[25] | Drijfhout JW , Hoogerhout P ((2000) ) Methods of preparing peptide-carrier conjugates. In Fmoc Solid Phase Peptide Synthesis: A Practical Approach, Chan WC , White PD, eds. Oxford University Press, pp. 229–241. |
[26] | Westdijk J , van den Ijssel , Thalen M , Beuvery C , Jiskoot W ((1997) ) Quantification of cell-associated and free antigens in Bordetella pertussis suspensions by antigen binding ELISA. J Immunoassay 18: , 267–284. |
[27] | Frey A , Di CJ , Zurakowski D ((1998) ) A statistically defined endpoint titer determination method for immunoassays. J Immunol Methods 221: , 35–41. |
[28] | Dahlgren KN , Manelli AM , Stine WB Jr , Baker LK , Krafft GA , LaDu MJ ((2002) ) Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem 277: , 32046–32053. |
[29] | Granic I , Masman MF , Kees MC , Nijholt IM , Naude PJ , de HA , Borbely E , Penke B , Luiten PG , Eisel UL ((2010) ) LPYFDa neutralizes amyloid-beta-induced memory impairment and toxicity. J Alzheimers Dis 19: , 991–1005. |
[30] | Stine WB Jr , Dahlgren KN , Krafft GA , LaDu MJ ((2003) ) In vitro characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis. J Biol Chem 278: , 11612–11622. |
[31] | Horvath KM , Abraham IM , Harkany T , Meerlo P , Bohus BG , Nyakas C , Luiten PG ((2000) ) Postnatal treatment with ACTH-(4-9) analog ORG 2766 attenuates N-methyl-D-aspartate-induced excitotoxicity in rat nucleus basalis in adulthood. Eur J Pharmacol 405: , 33–42. |
[32] | Harkany T , Mulder J , Horvath KM , Keijser J , van der Meeberg EK , Nyakas C , Luiten PG ((2001) ) Oral post-lesion administration of 5-HT(1A) receptor agonist repinotan hydrochloride (BAY x 3702) attenuates NMDA-induced delayed neuronal death in rat magnocellular nucleus basalis. Neuroscience 108: , 629–642. |
[33] | Harkany T , O’Mahony S , Kelly JP , Soos K , Toro I , Penke B , Luiten PG , Nyakas C , Gulya K , Leonard BE ((1998) ) Beta-amyloid(Phe(SO3H)24)25-35 in rat nucleus basalis induces behavioral dysfunctions, impairs learning and memory and disrupts cortical cholinergic innervation. Behav Brain Res 90: , 133–145. |
[34] | Franklin KBJ , Paxinos G ((1997) ) The mouse brain in stereotaxic coordinates, Academic Press. |
[35] | Alafuzoff I , Iqbal K , Friden H , Adolfsson R , Winblad B ((1987) ) Histopathological criteria for progressive dementia disorders: Clinical-pathological correlation and classification by multivariate data analysis. Acta Neuropathol 74: , 209–225. |
[36] | Arriagada PV , Growdon JH , Hedley-Whyte ET , Hyman BT ((1992) ) Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology 42: , 631–639. |
[37] | Gomez-Isla T , Hollister R , West H , Mui S , Growdon JH , Petersen RC , Parisi JE , Hyman BT ((1997) ) Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol 41: , 17–24. |
[38] | Herrup K ((2015) ) The case for rejecting the amyloid cascade hypothesis. Nat Neurosci 18: , 794–799. |
[39] | Musiek ES , Holtzman DM ((2015) ) Three dimensions of the amyloid hypothesis: Time, space and ‘wingmen’. Nat Neurosci 18: , 800–806. |
[40] | Wilcock GK , Esiri MM ((1982) ) Plaques, tangles and dementia. A quantitative study. J Neurol Sci 56: , 343–356. |
[41] | Lue LF , Kuo YM , Roher AE , Brachova L , Shen Y , Sue L , Beach T , Kurth JH , Rydel RE , Rogers J ((1999) ) Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer’s disease. Am J Pathol 155: , 853–862. |
[42] | McLean CA , Cherny RA , Fraser FW , Fuller SJ , Smith MJ , Beyreuther K , Bush AI , Masters CL ((1999) ) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol 46: , 860–866. |
[43] | Naslund J , Haroutunian V , Mohs R , Davis KL , Davies P , Greengard P , Buxbaum JD ((2000) ) Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA 283: , 1571–1577. |
[44] | Wang J , Dickson DW , Trojanowski JQ , Lee VM ((1999) ) The levels of soluble versus insoluble brain Abeta distinguish Alzheimer’s disease from normal and pathologic aging. Exp Neurol 158: , 328–337. |
[45] | Moghekar A , Rao S , Li M , Ruben D , Mammen A , Tang X , O’Brien RJ ((2011) ) Large quantities of Abeta peptide are constitutively released during amyloid precursor protein metabolism in vivo and in vitro. J Biol Chem 286: , 15989–15997. |
[46] | Tabira T ((2010) ) Immunization therapy for Alzheimer disease: A comprehensive review of active immunization strategies. Tohoku J Exp Med 220: , 95–106. |
[47] | Heo C , Chang KA , Choi HS , Kim HS , Kim S , Liew H , Kim JA , Yu E , Ma J , Suh YH ((2007) ) Effects of the monomeric, oligomeric, and fibrillar Abeta42 peptides on the proliferation and differentiation of adult neural stem cells from subventricular zone. J Neurochem 102: , 493–500. |
[48] | Pike CJ , Burdick D , Walencewicz AJ , Glabe CG , Cotman CW ((1993) ) Neurodegeneration induced by beta-amyloid peptides in vitro: The role of peptide assembly state. J Neurosci 13: , 1676–1687 . |
[49] | Whitson JS , Selkoe DJ , Cotman CW ((1989) ) Amyloid beta protein enhances the survival of hippocampal neurons in vitro. Science 243: , 1488–1490. |
[50] | Whitson JS , Glabe CG , Shintani E , Abcar A , Cotman CW ((1990) ) Beta-amyloid protein promotes neuritic branching in hippocampal cultures. Neurosci Lett 110: , 319–324. |
[51] | Yankner BA , Duffy LK , Kirschner DA ((1990) ) Neurotrophic and neurotoxic effects of amyloid beta protein: Reversal by tachykinin neuroeptides. Science 250: , 279–282 . |
[52] | Michaud JP , Halle M , Lampron A , Theriault P , Prefontaine P , Filali M , Tribout-Jover P , Lanteigne AM , Jodoin R , Cluff C , Brichard V , Palmantier R , Pilorget A , Larocque D , Rivest S ((2013) ) Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer’s disease-related pathology. Proc Natl Acad Sci U S A 110: , , 1941–1946. |
[53] | Wang DB ((2013) ) Monophosphoryl lipid A is an lipopolys-accharide-derived Toll-like receptor 4 agonist which may improve Alzheimer’s disease pathology. Expert Opin Biol Ther 13: , 1639–1641. |
[54] | Sas A , Jones R , Tyor W ((2008) ) Intra-peritoneal injection of polyclonal anti-interferon alpha antibodies cross the blood brain barrier and neutralize interferon alpha. Neurochem Res 33: , 2281–2287. |
Figures and Tables
Fig.1
Experimental overview. Mice were randomly assigned to be mock-immunized (mock) or immunized. All mice received three rounds of subcutaneous injections in the groin with a 3-week interval. Mock mice received mock vaccine, immunized mice received our trivalent vaccine. Blood samples were collected from the tail vein and the sera was analyzed by ELISA. Based on the anti-oligomeric Aβ1-42 endpoint titers obtained (ELISA 1), eight well-responding immunized mice and three mock mice were selected for oligomeric Aβ1-42-induced NBM lesion surgery (group A). Remaining mice received a fourth immunization and another tail puncture. Based on the titers obtained (ELISA 2),eight immunized and four mock mice were selected for NBM lesion surgery (group B). Remaining mice (group C: eight immunized, two mock mice) received a fifth immunization before NBM lesion surgery. Mice from each group were transcardially perfused ten days after NBM lesion surgery. Final blood samples were collected from the heart just before perfusion and sera obtained were analyzed later (ELISA 3).Experimental days are indicated on the left.
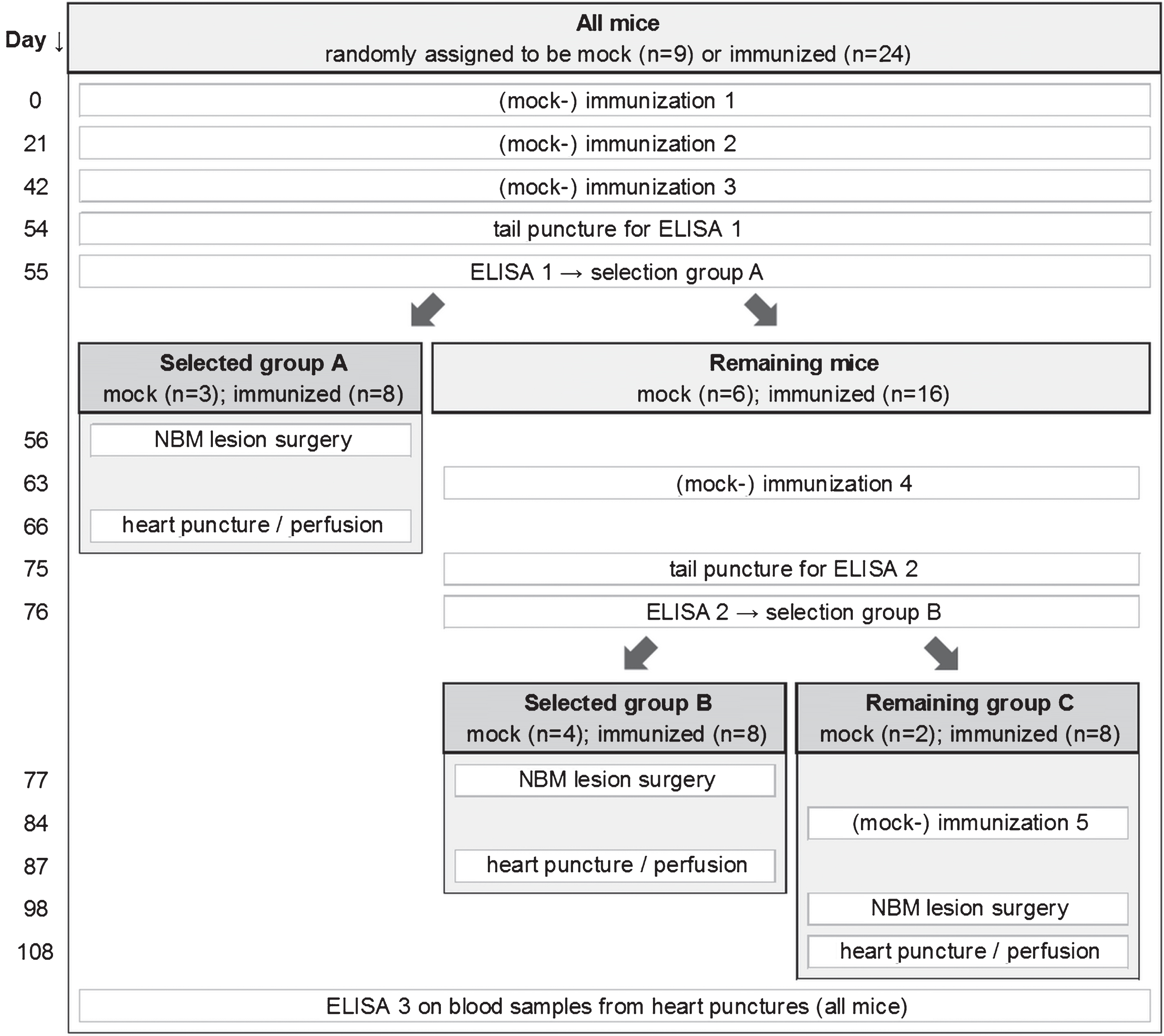
Fig.2
Average of the 10log value of anti-oligomeric Aβ1-42 ELISA endpoint titers of immunized and mock mice before and after Aβ1-42-induced NBM lesions. After all mice had received three (mock-) immunizations, mice with high titers (ELISA 1) and randomly selected mock mice were selected as the first group to receive NBM lesion surgery (group A). Remaining mice received a fourth (mock-) immunization. Again, mice with high titers (ELISA 2) and randomly selected mock mice were selected as the second group to receive NBM lesion surgery (group B). Remaining mice (group C) received a fifth (mock-) immunization before lesion surgery. Measurements before NBM lesions (ELISA 1 and 2) were performed on sera from blood samples collected from the tail vein. Measurements after NBM lesions (ELISA 3) were performed on sera from blood samples taken from the heart, just before transcardial perfusion. The results show that three rounds of immunizations are highly effective, while a fourth and fifth round of immunizations have only minor added effect. Vertical axis categories are numbered between square brackets for easy reference. Error bars represent SEM. Statistical indicators: *p < 0.05, ***p < 0.001, ns indicates p > 0.05.
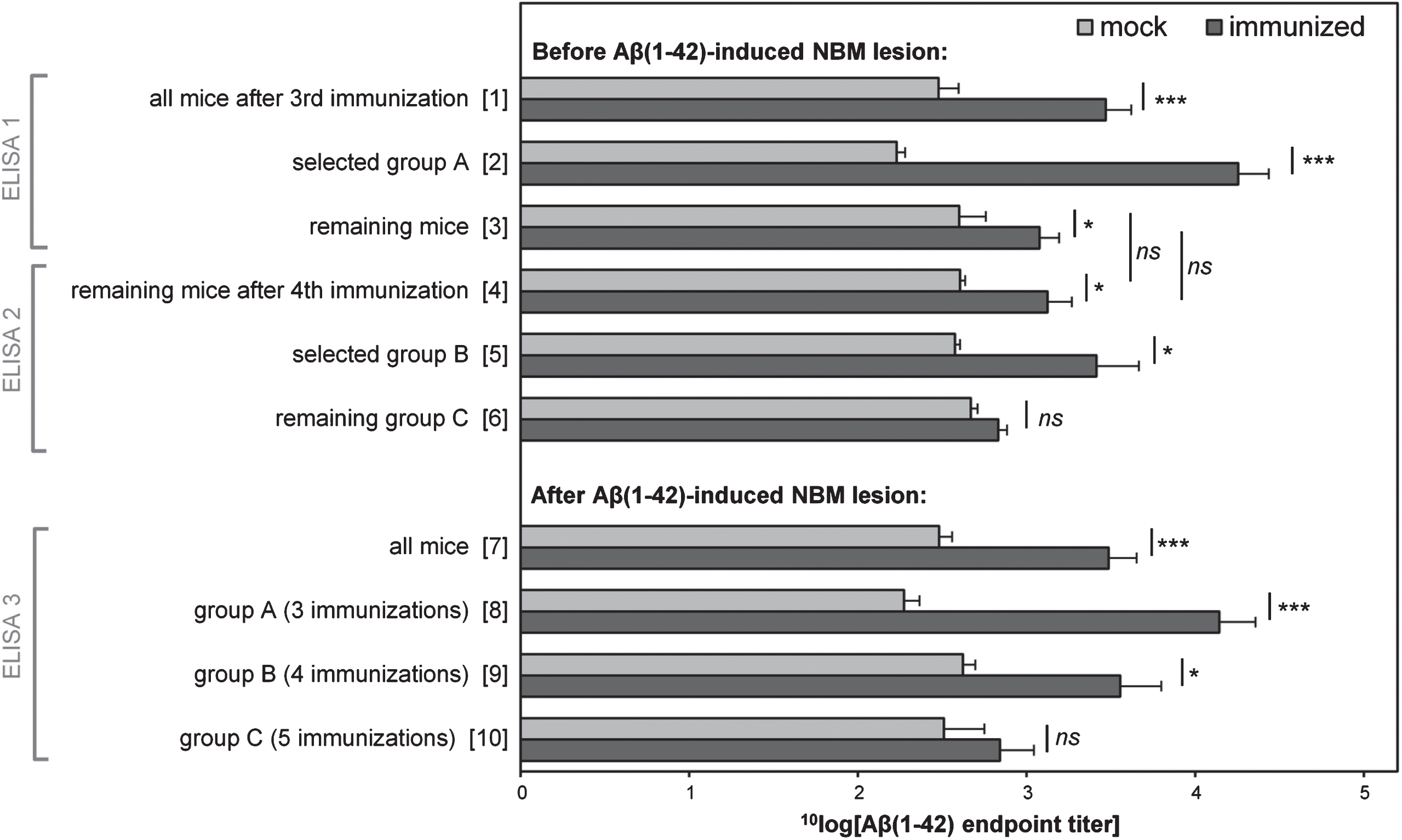
Fig.3
Active immunization prevents the loss of cholinergic innervations induced by oligomeric Aβ1-42 injected into the NBM. Shown are representative images of ChAT-positive fibers in the parietal neocortex of mock (A and B) and immunized mice (C and D). Images on the left (A and C) show the side of the brain injected with scrambled Aβ1-42 control peptide (Aβ-scr). Images on the right (B and D) show the contralateral lesion-sides of the brain injected with oligomeric Aβ1-42 (Aβ-oligo). Note that in practice the compounds were randomly injected in the left- or right side of the brain. Comparing control and lesion sides of the brain, immunized mice show reduced fiber loss compared to mock mice. In each image the area between the parallel bars indicate the quantified area (layer V of the somatosensory cortex). The horizontal scale bar shown in (D) applies to all images and represents 100 μm.
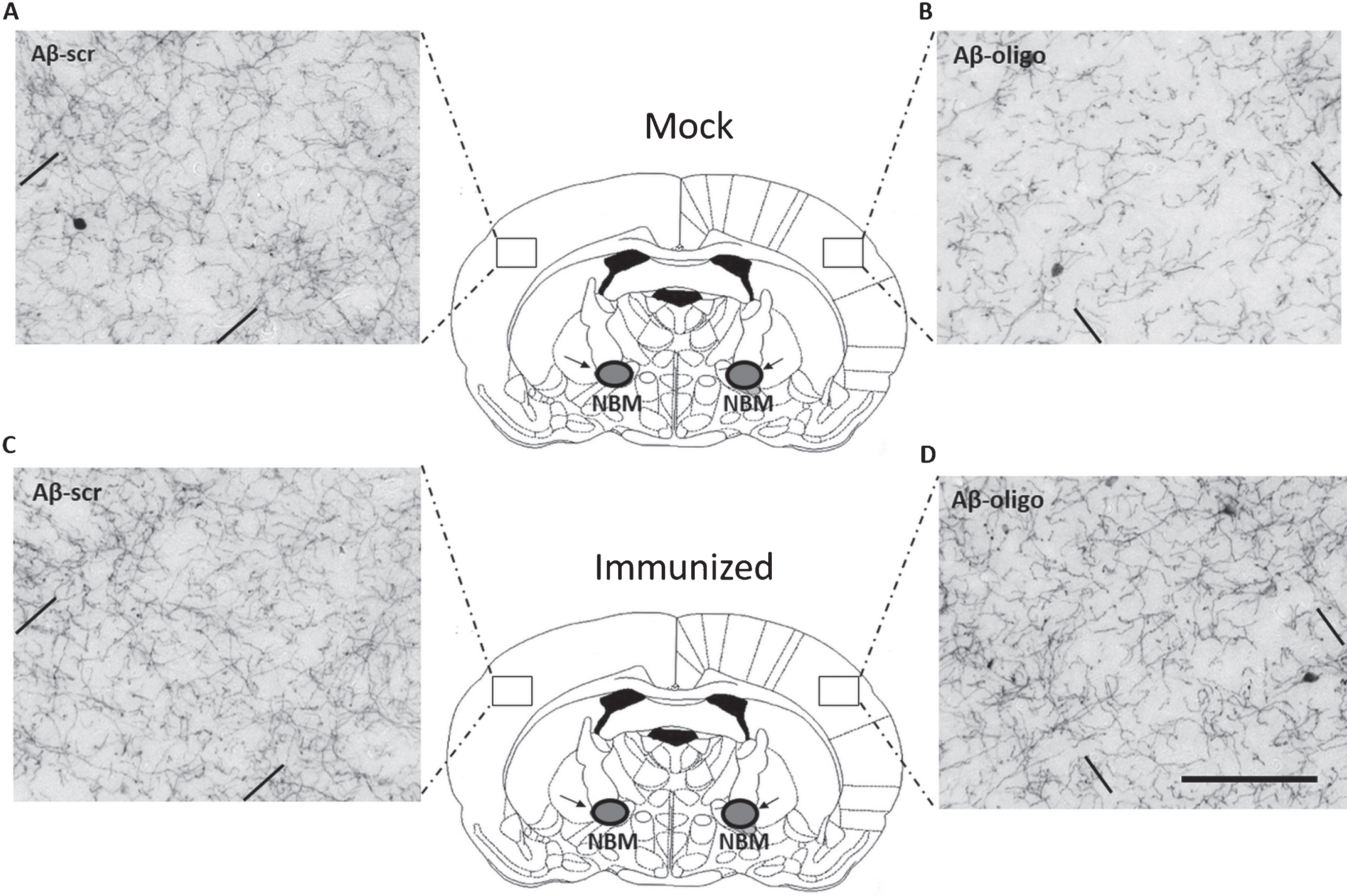
Fig.4
Effectiveness of the oligomeric Aβ1-42-induced NBM lesion model and effects in mock and immunized mice. All mice consistently showed less cortical cholinergic fiber density on the Aβ injected side of the brain, as compared to the scrambled-Aβ (Aβscr) injected control side of the brain (A). Compared to mock mice, immunized mice showed significantly reduced cholinergic fiber loss in the cortex, as measured by optical fiber density after ChAT immunostaining (B). In both panels the error bars represent SEM.
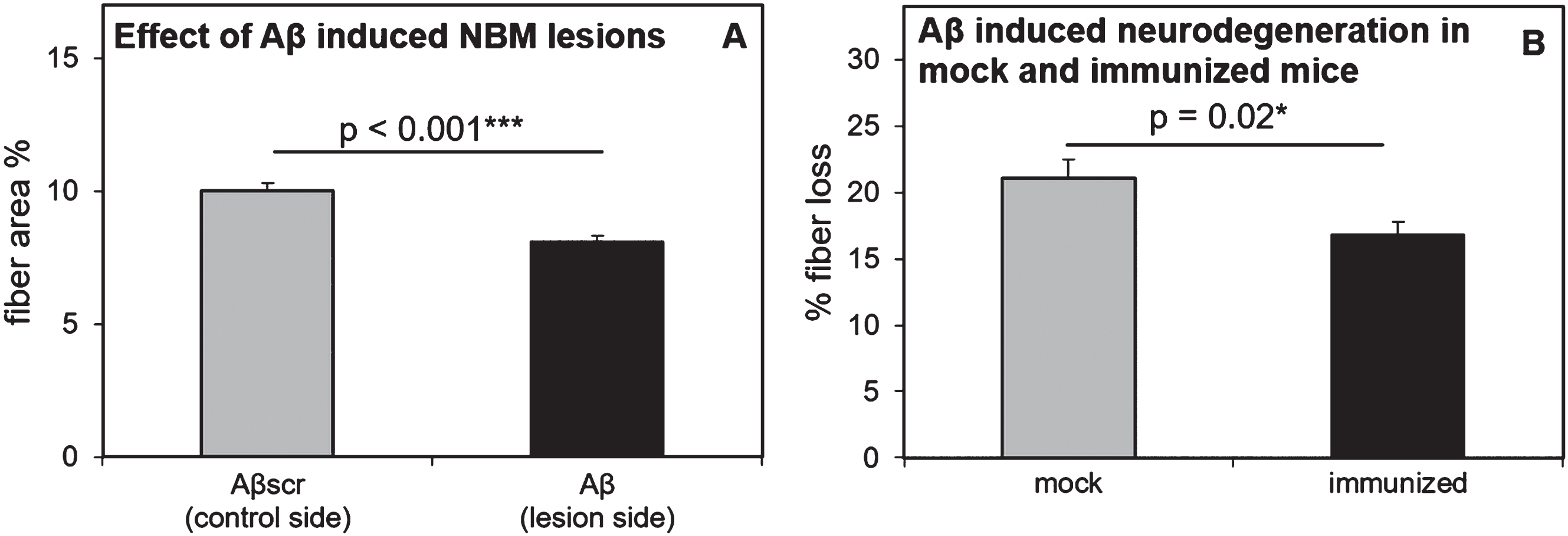
Fig.5
Correlation of the 10log value of anti-oligomeric Aβ1-42 endpoint titers (x-axis) versus cholinergic fiber loss (y-axis). Titers were obtained by ELISA of sera obtained from blood samples taken from the tail vein on day 54 (ELISA 1), after which all mice had received three immunization injections. Percentage of fiber loss was calculated for each animal based on ChAT-stained cholinergic fiber density in the bilateral parietal neocortex, which is the target of afferent cholinergic pathways from the bilateral NBM sites. The Aβ1-42 injected lesion side of the brain was compared to the scrambled-Aβ1-42 injected control side of the brain to determine the percentage of cholinergic fiber loss. The linear regression line and Pearson correlation is based on the immunized mice (indicated by black circles) including the non-responders (separately indicated by open triangles), but not including the mock mice (indicated by grey circles). The R-squared and two-tailed p-value is indicated in the figure.