Discrepancy in Expression of β-Secretase and Amyloid-β Protein Precursor in Alzheimer-Related Genes in the Rat Medial Temporal Lobe Cortex Following Transient Global Brain Ischemia
Abstract
Brain ischemia may be causally related with Alzheimer’s disease. Presumably, β-secretase and amyloid-β protein precursor gene expression changes may be associated with Alzheimer’s disease neuropathology. Consequently, we have examined quantitative changes in both β-secretase and amyloid-β protein precursor genes in the medial temporal lobe cortex with the use of quantitative rtPCR analysis following 10-min global brain ischemia in rats with survival of 2, 7, and 30 days. The greatest significant overexpression of β-secretase gene was noted on the 2nd day, while on days 7–30 the expression of this gene was only modestly downregulated. Amyloid-β protein precursor gene was downregulated on the 2nd day, but on days 7–30 postischemia, there was a significant reverse tendency. Thus, the demonstrated alterations indicate that the considerable changes of expression of β-secretase and amyloid-β protein precursor genes may be connected with a response of neurons in medial temporal lobe cortex to transient global brain ischemia. Finally, the ischemia-induced gene changes may play a key role in a late and slow onset of Alzheimer-type pathology.
INTRODUCTION
The relation of an increased risk of post-ischemia dementia to age, hippocampus atrophy, and medial temporal lobe neurodegenerative atrophy with hemorrhages has been reported [1–3]. Neuropathological alterations are somewhat similar in Alzheimer’s disease (AD) and, moreover, these lesions are associated with cognitive dysfunction [4]. Medial temporal lobe atrophy among ischemic brain survivors creates higher risks for memory dysfunction and the diagnosis of dementia with Alzheimer phenotype. The patients with moderate to severe medial temporal lobe atrophy demonstrated poor performance on measures of learning, story recall, visual reproduction, visual spatial reasoning, and processing speed [5]. The authors of the above data concluded that among older patients with brain ischemia-induced medial temporal lobe atrophy was very well correlated with memory and visual spatial functioning [5]. Some reports suggest that patients with ischemic brain alterations with severe medial temporal lobe atrophy might have preclinical AD, which is clinically revealed by ischemic brain lesions [2, 6].
Biopsies, autopsies, and other diagnostic studies are in line with clinical examinations of patients indicating that neuropathological processes underlying AD begin within the brain structures involved in memory and learning [7]. This brings the puzzling question why the development of amyloid-β protein precursor and tau protein pathology selectively damages neuronal fields associated with memory and learning? Another ambiguous question emerges: Where does the earliest process or do the processes leading to dementia initially begin? AD patients demonstrate neurodegenerative pathology in temporal lobe (disturbances in language) with diagnosed AD dementia [8]. It has been suggested that the primary event of neurodegenerative pathology in AD starts in the medial temporal lobe and proceeds to entorhinal cortex and hippocampus [9, 10]. Some studies underpin the fact that memory impairment is directly linked to the atrophy of medial temporal lobe during development of AD [11, 12]. On the other hand, a different study has implied that pathological alterations start in the hippocampus [13] and next spread to associate neocortical region of temporal lobe, which is involved in the consolidation of declarative memory [7].
The hippocampus is considered the major neuronal structure underlying episodic memory impairments that are the earliest and the most prominent clinical manifestation of post-ischemic dementia of Alzheimer phenotype [14–16]. Additionally, ischemia is also responsible for a severe injury to the temporal cortex [1] being the main axonal output network from the hippocampus. Both structures are reciprocally interconnected and are necessary for fundamental processes in learning and memory. At the synaptic level, dysfunction induced by ischemia in hippocampus and amyloid-β (Aβ) peptide [17] cause aberrant patterns of activity in the associated neural circuits, destabilize neuronal networks, and impair oscillatory activity. This scenario, ultimately, seems responsible for the early alteration of the processes implicated in learning and memory tasks observed in AD patients [13] and post-ischemic dementia with Alzheimer phenotype [14–16, 18].
However, the specific mechanisms involving pathological neurotransmission at the molecular level, synaptic circuits, or systems that consistently explain soluble Aβ peptide neurotoxic effects and associated neurological deficits remain unknown. Different parts of the amyloid-β protein precursor impair neuronal function and are found in AD brain and in human brains following ischemia [19, 20], as well as in animal brains [21–25]. Brain ischemia is suggested as an etiological factor for AD [21, 24–28]. Some evidence indicates that it is a two-way street as the incidence rate of brain ischemia is significantly higher in AD patients and the disease itself is more severe [4, 29] and often co-exists with typical ischemic neuropathology [26, 30]. At least one-third of postmortem AD brains exhibited micro- and macroinfarctions and ischemic diffuse alterations in the white matter [26, 30] with a weighted average of 43% [31]. Moreover, clinical and experimental studies imply that the interaction of AD pathology and ischemic brain insults might be bidirectional [32].
The results indicate that the AD neuropathology with ischemic pathological metabolism of amyloid-β protein precursor (Aβ peptide generation) increases ischemic vulnerability in various brain areas [33]. The exact mechanism how these conditions contribute to the progression of AD is unknown. To investigate the effect of ischemic brain insult on spreading of AD neuropathology and overall interaction of AD pathology with ischemic brain insults, we used a neuropathologically and behaviorally well-defined model of global brain ischemia [15, 34]. Unfortunately, the relationship between hippocampus neuronal injury and medial temporal lobe injury in brain ischemia and AD has received little attention. On the basis of the known anatomical connectivity between the hippocampus and medial temporal lobe, we hypothesized that in ischemic brain injury of hippocampus there would be an association with neuronal pathology in temporal cortex as measured by regional related metabolism of amyloid-β protein precursor genes [35, 36]. What is more, we intended to understand how Aβ peptide deposition induces ischemic remodeling in medial temporal lobe cortex.
In this pioneer study, we presented the time course of amyloid-β protein precursor and β-secretase gene expression levels, all of which are implicated in AD, by quantitative reverse-transcriptase PCR assay protocol in the medial temporal lobe cortex subjected to transient global brain ischemia due to cardiac arrest.
MATERIALS AND METHODS
Animals and global brain ischemia
Two-month-old female Wistar rats (n = 54, 160–180 g) were submitted to 10-min global brain ischemia due to cardiac arrest [34]. The rats were maintained in pairs per cage in a room temperature of 24±2°C, with 55±5% humidity, and with a 12 h light-dark cycle. All animals had free access to commercial laboratory chow and tap water ad libitum. All experimental procedures were performed during the light phase, between 13:00 and 17:00 under identical conditions. The rats used for experiments were treated in strict accordance with the NIH Guide for Care and Use of Laboratory Animals (1985) and European Communities Council Directive 142 (86/609/EEC), as well as with the approval of the local Ethical Committee. All efforts were made to minimize animal suffering and to reduce the number of rats used.
Our animal model of global brain ischemia in rats clinically represents reversible cardiac arrest. Global brain ischemia was performed by cardiac arrest of 10-min duration [34]. The animals were allowed to survive 2 (n = 11), 7 (n = 18) days, and 1 month (n = 25) after recirculation. Sham-operated rats (n = 54) were exposed to the same procedures as ischemic animals but without induced cardiac arrest and thus served as controls.
Determination of gene expression
Prior to sampling, the brains were perfused with cold 0.9% NaCl via the left ventricle in order to flush blood vessels. After removing the brain from the skull, the brain was put on an ice chilled Petri dish. The ischemic and control pooled samples circa 1 mm3 volume of medial temporal lobe cortex left and right (all cortical layers) were taken with a narrow scalpel. Pooled left and right cortical areas were immediately placed in RNALater solution (Life Technologies, USA). Gene expression of amyloid-β protein precursor (App) and β-secretase (Bace 1) was determined by the reverse-transcription quantitative PCR (RT-qPCR) method. Genes were quantitatively assessed in each sample taken from the ischemic group and referred to gene expression determined in the corresponding samples in the control group –1:1.
Isolation of total cellular RNA
Total cellular RNA isolation was performed according to the method described by Chomczynski and Sacchi [37] with own modification, using TRI-Reagent Solution (Ambion, USA). During this process, the samples of medial temporal lobe cortex were homogenized in 1 ml TRI-Reagent buffer (Ambion, USA) to obtain homogenous suspensions. The suspensions were incubated for 5 min at room temperature until complete dissociation. At the next stage, 200μl chloroform (Sigma-Aldrich, USA) per 1 ml of TRI-Reagent buffer was added to the sample and shaken for 15 s to completely mix the phases. Next, the samples were left for 15 min to incubate at room temperature after which they were centrifuged for 15 min at 14,000 rpm at 4°C in 5415R Eppendorf centrifuges. After the separation, the aqueous phase was placed in a new tube and 500μl 2-propanol (Sigma-Aldrich, USA) was added per 1 ml of TRI-Reagent buffer. The samples were thoroughly mixed and incubated for 20 min at room temperature. Following that, the mixtures were centrifuged for 20 min at 14,000 rpm at 4°C in 5415R Eppendorf centrifuges. Aqueous phase containing RNA was removed from the above precipitate. The RNA phase was washed in cool 80% ethanol and next, obtained RNA samples were stored in 80% ethanol at –20°C for further analysis.
Quantitative and qualitative analysis of RNA
The RNA concentration and purity were measured by spectrophotometry on NanoDrop 2000 (Thermo Scientific, USA). Precipitate of RNA in 80% ethanol was taken out of –20°C and next centrifuged for 15 min at 14,000 rpm at 4°C in 5415R Eppendorf centrifuges. The liquid part was removed, and RNA pellets were left to dry completely at room temperature. Subsequently, the precipitate was dissolved in DNase-, RNase-, and protease-free water (Sigma-Aldrich, USA) at 4°C, the volume depending on RNA concentration.
The cDNA synthesis
The cDNA was synthesized using High-Capacity cDNA Reverse Transcription Kit, according to manufacturer’s instructions (Applied Biosystems, USA). Each reactive mixture contained the following set of reagents: 1×RT buffer, 20 U RNase inhibitor, 50 U reverse transcriptase (Multiscribe Reverse Transcriptase), 1×RT Random Primers, 4 mM of each deoxynucleotide: dATP, dGTP, dTTP, and dCTP plus examined 1μg RNA in DNase-, RNase-, and protease-free water (Sigma-Aldrich, USA) to complete the volume required for reaction. The final volume of reactive mixture was 20μl. Afterwards, the reactive components were thoroughly mixed and centrifuged to fuse them well. The cDNA was synthesized on Veriti Dx (Applied Biosystems, USA) under the following conditions: Stage I: 25°C, 10 min, stage II: 37°C, 120 min, stage III: 85°C, 5 min, stage IV: 4°C.
The qPCR protocol
The cDNA, which was obtained by reverse transcription (RT) procedure, was amplified by real-time gene expression analysis (qPCR) on 7900HT Real-Time Fast System (Applied Biosystems, USA), using the manufacturer’s SDS software. Triplicate qPCR reactions were conducted for each sample. To exclude reagent contamination by foreign DNA, a blind trial was always performed without DNA target. Reaction components included: 1.25μl mixture of probe and oligonucleotide starters specific for genes examined, 12.5μl buffer TaqMan Universal PCR Master Mix, 1μl cDNA, DNase-, RNase- and protease-free water (Sigma Aldrich, USA) to complete the required reactive volume. The reaction was performed on an optic 25μl-well reaction plate, using probe sets of TaqMan Gene Expression Assays (Applied Biosystems, USA) with FAM-NFQ markers and oligonucleotide starters for rat genes: App, Bace 1, and the housekeeping gene: Rpl13a was used as an internal control gene [38]. Amplification protocol included the following cycles: Initial denaturation: 95°C, 10 min and 40 cycles, each composed of two temperatures: 95°C, 15 s and 60°C, 1 min. The number of copies of DNA molecules was monitored and calculated on 7900HT Real-Time Fast System (Applied Biosystems, USA) in each amplification cycle. To calculate the number of examined DNA molecules present in the mixture at the onset of reaction, the number of PCR cycles after which the level of fluorescence exceeded the defined threshold cycle (CT) RQ Study Software (Applied Biosystems, USA) was used. The CT value for each sample of endogenous control gene (Rpl13a) was used to normalize the level of the examined gene expression. The relative level of gene expression was calculated according to the formula [39]: 1. Calculating CT differences between the target gene and the reference gene are presented below: For the examined after ischemia of medial temporal lobe cortex: ΔCTischemic sample = CT target gene from ischemic sample – CT reference gene,ischemic sample and for the calibrator (control sample): ΔCT calibrator = CT target gene from control sample – CT reference gene, control sample . 2. Normalizing ΔCT of the ischemic sample to the ΔCT of the calibrator ΔΔCT =ΔCTischemic sample– ΔCT calibrator . 3. Relative expression (RQ) of rat genes was calculated by the formula: RQ = 2–ΔΔCT . The RQ defines the expression of the examined gene in the medial temporal lobe cortex taken from an ischemic rat with reference to the gene expression in the same structure of a control rat. Finally, the RQs were analyzed after their logarithmic conversion into logarithm of RQ (LogRQ) [40]. Thus, the obtained results were more legible. LogRQ = O means that gene expression in the calibrated sample and the ischemic one are the same. LogRQ < 0 points to decreased gene expression in the ischemic sample, whereas LogRQ > 0 points to signal increased gene expression in the ischemic sample compared to the calibrated one.
Statistical analyses
The results were statistically analyzed by SPSS v. 17 software by means of the non-parametric Mann-Whitney U test and Spearman rho correlation analysis. Data were presented as means±SEM. The level of statistical significance was set at p≤0.01.
RESULTS
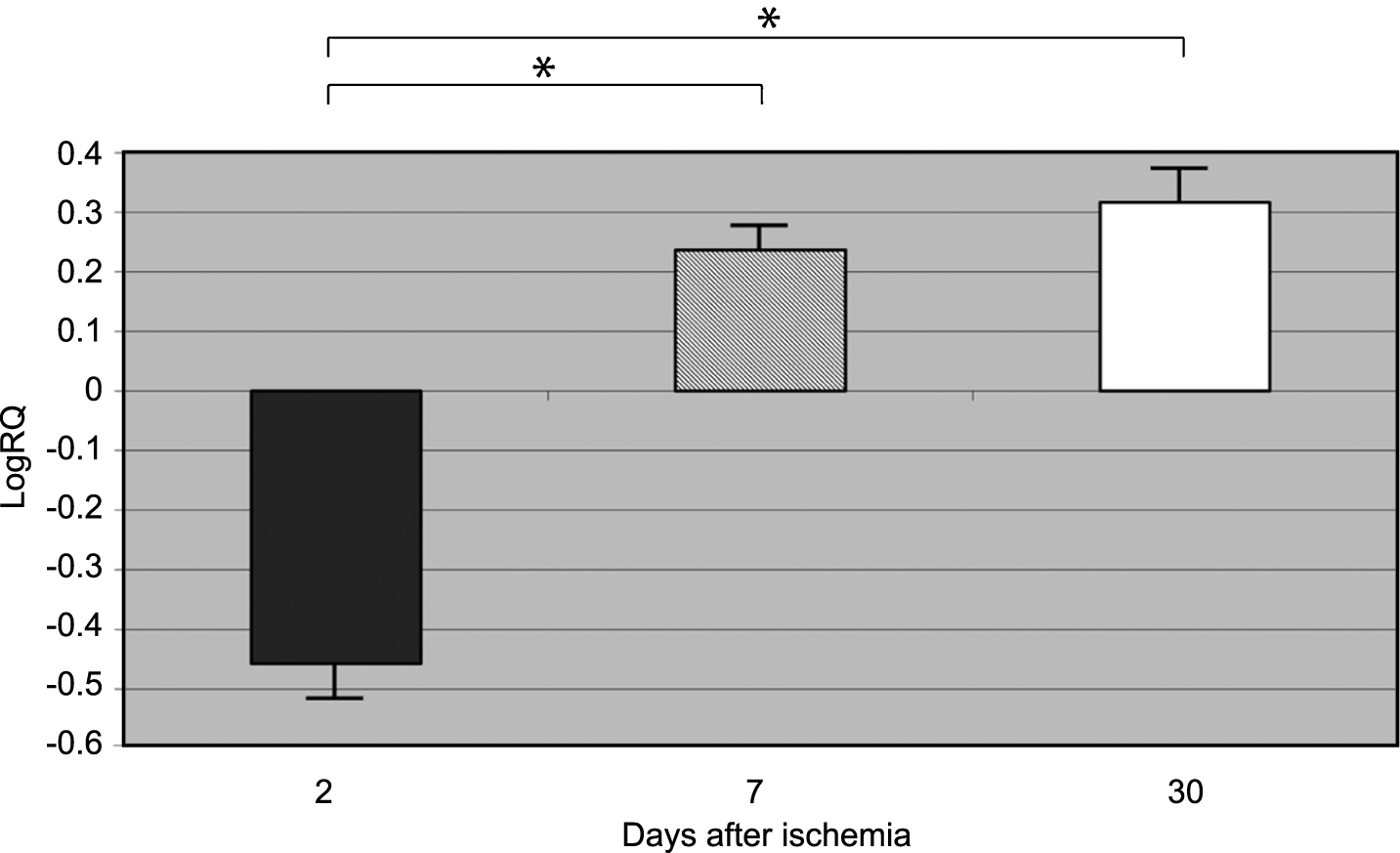
β-Secretase gene expression increased to a maximum of 4.039-fold, and the expression of amyloid-β protein precursor gene decreased to a minimum of –0.678 two days after 10-min complete brain ischemia. The median values of gene expression at 2-day survival, following ischemia, were as follows: β-secretase 1.002 and amyloid-β protein precursor –0.564. The mean expression levels of genes at that time were as follows: β-secretase 1.785±0.518, and amyloid-β protein precursor –0.457±0.069 (Figs. 1 and 2).
β-Secretase gene expression decreased to a minimum –0.710, and the amyloid-β protein precursor gene increased - to 0.457 seven days after 10-min complete brain ischemia. The median values of gene expression at 7-day survival, following ischemia, were as follows: β-secretase –0.159 and amyloid-β protein precursor 0.217. The mean expression levels of genes at that time were the following: β-Secretase –0.049±0.125, and amyloid-β protein precursor 0.236±0.039 (Figs. 1 and 2).
β-secretase gene expression decreased to a minimum of –0.894, and the amyloid-β protein precursor gene expression increased to 1.010 30 days after 10-min complete brain ischemia. The median values of gene expression at 30 days, following ischemia, were as follows: β-secretase 0.02 and amyloid-β protein precursor 0.269. The mean expression levels of genes at that time were as follows: β-secretase –0.079±0.084 and amyloid-β protein precursor 0.316±0.061 (Figs. 1 and 2).
Figures 1 and 2 show changes in the mean expression levels of genes with statistical significance: β-secretase (Fig. 1) and the amyloid-β protein precursor (Fig. 2) after 10-min total cerebral ischemia in rats with survival of 2, 7, and 30 days. Significant (negative) correlations between the levels of Bace 1 and App gene expression in rat medial temporal lobe cortex 2, 7, and 30 days after global brain ischemia were evident (Spearman rho correlation test p < 0.01).
DISCUSSION
In this paper, we evaluated alterations in the expression of amyloid-β protein precursor and β-secretase Alzheimer-related genes in rats subjected to a brief transient global brain ischemia. Our pioneer study shows the time course of amyloid-β protein precursor and β-secretase genes dysregulation in post-ischemic medial temporal lobe cortex. Our data provide the first known in vivo role of the Alzheimer-related genes dysregulation in cortical neurons during recirculation time. Additionally, our results demonstrate that brain ischemia activates delayed neuronal changes in temporal cortex in an Aβ peptide-dependent manner, thus defining a new and important mode of regulation of ischemic cortical neuronal cell damage/death.
After global brain ischemia, some cortical layers of the brain become more susceptible to neuronal damage/death, a phenomenon known as a selective delayed neuronal death and/or selective vulnerability [23]. Selective vulnerability of cortical layers 3, 5, and 6 [23] is subject to intensive studies, and these are the cortical layers of the brain most sensitive to ischemic pathology in rodents and humans where no final mechanisms involved have been explained. The molecular processes of events engaged in the specific disappearance of cortical layers 3, 5, and 6 have been disappointingly limited. To the best of our knowledge, our data constitute the first link between ischemic injury and acute, quantitative expression-selected Alzheimer-related genes (amyloid-β protein precursor and β-secretase), which might support the hypothesis that the Aβ peptide generated by ischemia may be well involved in the ischemia-related AD development and progressive damage/death of neurons in cortical layers [14, 25–28, 32, 41–46].
Our analysis has evaluated for the first time parallel ischemic alterations in gene expression of amyloid-β protein precursor (substrate) and amyloid-β protein precursor metabolism-related genes β-secretase in the medial temporal lobe cortex of rat submitted to transient global brain ischemia. In the present study, we have demonstrated that 2 days post-ischemia resulted in a quick and huge overexpression of β-secretase (maximal increase of 4.039 fold) while at the same period of time, amyloid-β protein precursor was downregulated (respectively –0.678 fold). The investigated protease was impressively changed following 2 days after ischemia (β-secretase). The level of expression of amyloid-β protein precursor investigated gene did not coincide with the earlier data concerning staining of different parts of amyloid-β protein precursor in the rat cortex [17, 21]. Our results indicate that there is a discrepancy between both β-secretase and amyloid-β protein precursor, which was regulated down in the ischemic temporal cortex following 2 days after ischemia. It is clear now that this time necrotic neuronal cell death is associated with post-ischemic neuropathology [47, 48]. During necrotic neuronal death, a loss of all cell membranes integrity was finally noted [49, 50]. Amyloid-β protein precursor is present in large quantities in all cell membranes [51], and, in the above situation, there may be an excess of protease substrate, which is the amyloid-β protein precursor. Loss of membrane integrity allows necrotic cells the ability to trigger an amyloid-β protein precursor uncontrolled release and metabolism [52].
Our data also proved that in the following 7 days, the β-secretase gene expression dropped below its normal value (–0.710 fold). In parallel, the results indicate an increase in amyloid-β protein precursor gene expression (0.457 fold) in the medial temporal lobe cortex after 10-min global brain ischemia. These figures are not complementary to the increase noted in different parts of amyloid-β precursor protein immunostaining after 7 days of recirculation [21]. However, after 30 days gene expression of β-secretase was below normal level (–0.894 fold) and amyloid-β protein precursor was overexpressed (1.010 fold). During 30 days post-ischemia, there was a huge cortical damage and disappearance of layers 3, 5, and 6 of neuronal population in the medial temporal lobe cortex and, therefore, amyloid-β protein precursor gene expression was increased at that time. The dysregulation of all studied genes at this time negatively correlates with intra- and extracellular immunostaining of the respective gene products [17, 21, 41, 43]. Intra- and extracellular Aβ peptide immunoreactivity increases for as long as a year after injury [43], indicating that Aβ peptide accumulation may continue long after amyloid-β protein precursor metabolism processing gene expression drops to below normal value [17, 35, 53].
β-Secretase activity can lead to the generation of Aβ peptide, which in turn may cause secondary damage to the ischemic neurons in the brain. Our results indicate that ischemia of medial temporal lobe cortex may not influence the expression of β-secretase, for all time periods after ischemia and may not potentiate the course of amyloidogenesis in cortex during the whole post-ischemic time. The obtained data seem to provide a new complicated role of the investigated Alzheimer-related genes in ischemic medial temporal lobe cortex. Presumably, we can distinguish local and global disturbances in amyloid-β protein precursor processing. This phenomenon is probably related to movement of soluble Aβ peptide from blood into brain tissue [54, 55]. Finally, it was noted that RAGE mediated additionally delayed neuronal death by enhancing vascular wall injury and neuroinflammation [56].
Probably early post-ischemic Alzheimer-related gene dysregulation triggers delayed gene dysfunction in post-stroke patients and old ischemic rats [57–59]. Buga et al. [57–59] have identified an age-related gene expression pattern following brain ischemia. These studies indicated that genes specific for anxiety, depression, stress, and neuropathic syndrome had harmful response to ischemia in aged rats. Additionally, upregulation of DNA damage and downregulation of anti-apoptotic and neurogenesis genes were observed. Also, genes involved in neuroinflammation, wound healing, scar formation, angiogenesis, and vascular remodeling were upregulated in the aged ischemic rats. The considerable majority of genes associated with angiogenesis, formation of a new basal lamina and involved in maturation had a delayed overexpression in these rats. Buga et al. [57–59] have suggested that the angiogenesis response, in above circumstances, was diminished by the continual overexpression of neuroinflammatory and scar formation genes. The ischemia-induced remodeling of brain tissue presented above seems an important event for the final development of neurodegenerative changes.
The most prevalent pathological hallmark of AD is the accumulation of plaques formed by Aβ peptide, commencing in the hippocampus, and spreading progressively throughout the brain [35, 60, 61]. It is likely that this accumulation in temporal cortex is caused by both increased inflow from blood [54, 55] and impaired clearance of Aβ peptide [62]. These data corroborate earlier immunohistochemical observations from animal models of global brain ischemia and from brains after global brain ischemia in humans, which all suggest a direct relationship between ischemia and increased level of Aβ peptide accumulation in the brain tissue [19–21]. The findings presented above highlight a different and/or delayed regulatory mode in ischemia-induced cell death in temporal cortex as compared to hippocampus through an Aβ peptide-dependent manner [35, 36]. These observations will help to understand the gradual postischemic damage in the brain, delayed Aβ peptide deposition and slow and long-term ischemic spreading neuropathology of AD from hippocampus into medial temporal lobe cortex and other parts of the brain [25, 35, 36, 60, 61]. The present data may partly help to define the molecular mechanism of higher occurrence of neuronal death in ischemic layers 3, 5, and 6 of medial temporal lobe cortex. Our study indicates that both transcription processes negatively correlate with the ischemic condition of neurons in the medial temporal lobe cortex and Aβ peptide accumulation. We may identify more new processes and individual molecules that contribute to the death and complete disappearance of layers 3, 5, and 6 in the ischemic temporal cortex. The detection of these lesions in vivo with the use of rat model of global brain ischemia opens up the possibility of obtaining a better understanding of the ischemic contribution of Alzheimer-related genes to the neuropathophysiology and neuropathology of AD and ischemic injury.
ACKNOWLEDGMENTS
The authors acknowledge support by the Polish National Science Centre (DEC-2013/09/B/NZ7/01345-RP) and by the Mossakowski Medical Research Centre, Polish Academy of Sciences, Poland (T3-RP). Also, this study was supported by a grant from Medical University of Lublin, Poland (DS 475-SJC and DS 222/14-JK). The research was developed using the equipment purchased within the Project “The equipment of innovative laboratories doing research on new medicines used in the therapy of civilization and neoplastic diseases” within the Operation Program Development of Eastern Poland 2007–2013, Priority Axis I Modern Economy, Operations I.3 Innovation Promotion (JK, SJC).
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/15-1102r1).
REFERENCES
[1] | Pluta R ((1985) ) Influence of prostacyclin on early morphological changes in the rabbit brain after complete 20– min ischemia. J Neurol Sci 70: , 305–316. |
[2] | Leys D , Henon H , Mackowiak-Cordoliani MA , Pasquier F ((2005) ) Poststroke dementia. Lancet Neurol 4: , 752–759. |
[3] | Pluta R , Ułamek M , Januszewski S ((2006) ) Micro-blood-brain barrier openings and cytotoxic fragments of amyloid precursor protein accumulation in white matter after ischemic brain injury in long lived rats. Acta Neurochir 96: (Suppl), 267–271. |
[4] | van Rooden S , Goos JDC , van Opstal AM , Versluis MJ , Webb AG , Blauw GJ , van der Flier WM , Scheltens P , Barkhof F , van Buchem MA , van der Grond J ((2014) ) Increased number of microinfarcts in Alzheimer disease at 7-T MR imaging. Radiology 270: , 205–211. |
[5] | Jokinen H , Kalska H , Ylikoski R , Hietanen M , Mäntylä R , Pohjasvaara T , Kaste M , Erkinjuntti T ((2004) ) Medial temporal lobe atrophy and memory deficits in elderly stroke patients. Eur J Neurol 11: , 825–832. |
[6] | Pasquier F , Leys D ((1997) ) Why are stroke patients prone to develop dementia?. J Neurol 244: , 135–142. |
[7] | Arshavsky YI ((2010) ) Why Alzheimer’s disease starts with a memory impairment: Neurophysiological insight. J Alzheimers Dis 20: , 5–16. |
[8] | Pinkston JB , Alekseeva N , Gonzalez Toledo E ((2009) ) Stroke and dementia. Neurol Res 31: , 824–831. |
[9] | Braak H , Braak E ((1995) ) Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging 16: , 271–278. |
[10] | Magnin B , Mesrob L , Kinkingnéhun S , Pélégrini-Issac M , Colliot O , Sarazin M , Dubois B , Lehéricy S , Benali H ((2009) ) Support vector machine-based classification of Alzheimer’s disease from whole-brain anatomical MRI. Neuroradiology 51: , 73–83. |
[11] | Peterson RC , Jack CR Jr , Xu YC , Waring SC , O’Brien PC , Smith GE , Ivnik RJ , Tangalos EG , Boeve BF , Kokmen E ((2000) ) Memory and MRI-based hippocampal volumes in aging and AD. Neurology 54: , 581–587. |
[12] | Jauhiainen AM , Pihlajamäki M , Tervo S , Niskanen E , Tanila H , Hänninen T , Vanninen RL , Soininen H ((2009) ) Discriminating accuracy of medial temporal lobe volumetry and fMRI in mild cognitive impairment. Hippocampus 19: , 166–175. |
[13] | Huang Y , Mucke L ((2012) ) Alzheimer mechanisms and therapeutic strategies. Cell 148: , 1204–1222. |
[14] | Yang SH , Simpkins JW ((2007) ) Ischemia-reperfusion promotes tau and beta-amyloid pathology and a progressive cognitive impairment, In: Ischemia-Reperfusion Pathways in Alzheimer’s Disease, Pluta R, ed. Nova Science Publishers, Inc., New York, pp. 113–138 |
[15] | Kiryk A , Pluta R , Figiel I , Mikosz M , Ułamek M , Niewiadomska G , Jabłoński M , Kaczmarek L ((2011) ) Transient brain ischemia due to cardiac arrest causes irreversible long-lasting cognitive injury. Behav Brain Res 219: , 1–7. |
[16] | Li J , Wang YJ , Zhang M , Fang CQ , Zhou HD ((2011) ) Cerebral ischemia aggravates cognitive impairment in a rat model of Alzheimer’s disease. Life Sci 89: , 86–92. |
[17] | Pluta R , Ułamek M , Jabłoński M ((2009) ) Alzheimer’s mechanisms in ischemic brain degeneration. Anat Rec 292: , 1863–1881. |
[18] | Barra de la Tremblaye P , Plamondon H ((2011) ) Impaired conditioned emotional response and object recognition are concomitant to neuronal damage in the amygdale and perirhinal cortex in middle-aged ischemic rats. Behav Brain Res 219: , 227–233. |
[19] | Qi J , Wu H , Yang Y , Wand D , Chen Y , Gu Y , Liu T ((2007) ) Cerebral ischemia and Alzheimer’s disease: The expression of amyloid-β and apolipoprotein E in human hippocampus. J Alzheimers Dis 12: , 335–341. |
[20] | Maślińska D , Laure-Kamionowska M , Taraszewska A , Deręgowski K , Maśliński S ((2011) ) Immunodistribution of amyloid beta protein (Aβ) and advanced glycation end-product receptors (RAGE) in choroid plexus and ependyma of resuscitated patients. Folia Neuropathol 49: , 295–300. |
[21] | Pluta R , Kida E , Lossinsky AS , Golabek AA , Mossakowski MJ , Wisniewski HM ((1994) ) Complete cerebral ischemia with short-term survival in rats induced by cardiac arrest. I. Extracellular accumulation of Alzheimer’s β-amyloid protein precursor in the brain. Brain Res 649: , 323–328. |
[22] | Popa-Wagner A , Buga AM , Kokaia Z ((2011) ) Perturbed cellular response to brain injury during aging. Ageing Res Rev 10: , 71–79. |
[23] | Pluta R , Jabłoński M , Czuczwar SJ. ((2012) ) Postischemic dementia with Alzheimer phenotype: Selectively vulnerable versus resistant areas of the brain and neurodegeneration versus β-amyloid peptide. Folia Neuropathol 50: , 101–109 |
[24] | Pluta R , Furmaga-Jabłońska W , Maciejewski R , Ułamek-Kozioł M , Jabłoński M ((2013) ) Brain ischemia activates β- and γ-secretase cleavage of amyloid precursor prote, Significance in soradic Alzheimer’s disease Mol Neurobiol 47: , 425–434. |
[25] | Pluta R , Jabłoński M , Ułamek-Kozioł M , Kocki J , Brzozowska J , Januszewski S , Furmaga-Jabłońska W , Bogucka-Kocka A , Maciejewski R , Czuczwar SJ ((2013) ) Sporadic Alzheimer’s disease begins as episodes of brain ischemia and ischemically dysregulated Alzheimer’s disease genes. Mol Neurobiol 48: , 500–515. |
[26] | Kalaria RN ((2000) ) The role of cerebral ischemia in Alzheimer’s disease. Neurobiol Aging 21: , 321–330. |
[27] | De la Torre JC ((2006) ) How do heart disease and stroke become risk factors for Alzheimer’s disease?. Neurol Res 28: , 637–644. |
[28] | Bell RD , Zlokovic BV ((2009) ) Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol 118: , 103–113. |
[29] | Snowdon DA , Greiner LH , Mortimer JA , Riley KP , Greiner PA , Markesbery WR ((1997) ) Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 277: , 813–817. |
[30] | Kalaria RN ((2002) ) Small vessel disease and Alzheimer’s dementia: Pathological considerations. Cerebrovasc Dis 13: (Suppl 2), 48–52. |
[31] | Brundel M , de Bresser J , van Dillen JJ , Kappelle LJ , Biessels GJ ((2012) ) Cerebral microinfarcts: A systematic review of neuropathological studies. JCBFM 32: , 425–436. |
[32] | Pluta R ((2007) ) Ischemia-Reperfusion Pathways in Alzheimer’s Disease Nova Science Publishers, Inc New York. |
[33] | Koistinaho M , Kettunen MI , Goldsteins G , Keinanen R , Salminen A , Ort M , Bures J , Liu D , Kauppinen RA , Higgins LS , Koistinaho J ((2002) ) Beta-amyloid precursor protein transgenic mice that harbor diffuse A beta deposits but do not form plaques show increased ischemic vulnerability: Role of inflammation. Proc Natl Acad Sci U S A 99: , 1610–1615. |
[34] | Pluta R , Lossinsky AS , Mossakowski MJ , Faso L , Wisniewski HM ((1991) ) Reassessment of a new model of complete cerebral ischemia in rats. Method of induction of clinical death, pathophysiology and cerebrovascular pathology. Acta Neuropathol 83: , 1–11. |
[35] | Kocki J , Ułamek-Kozioł M , Bogucka-Kocka A , Januszewski S , Jabłoński M , Gil-Kulik P , Brzozowska J , Petniak A , Furmaga-Jabłońska W , Bogucki J , Czuczwar SJ , Pluta R ((2015) ) Dysregulation of amyloid precursor protein, β-secretase, presenilin 1 and 2 genes in the rat selectively vulnerable CA1 subfield of hippocampus following transient global brain ischemia. J Alzheimers Dis 47: , 1047–1056. |
[36] | Pluta R , Kocki J , Ułamek-Kozioł M , Bogucka-Kocka A , Gil-Kulik P , Januszewski S , Jabłoński M , Petniak A , Brzozowska J , Bogucki J , Furmaga-Jabłońska W , Czuczwar SJ ((2016) ) Alzheimer-associated presenilin 2 gene is dysregulated in rat medial temporal lobe cortex after complete brain ischemia due to cardiac arrest. Pharmacol Rep 68: , 155–161. |
[37] | Chomczynski P , Sacchi N ((1987) ) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 62: , 156–159. |
[38] | Yao L , Chen X , Tian Y , Lu X , Zhang P , Shi Q , Zhang J , Liu Y ((2012) ) Selection of housekeeping genes for normalization of RT-PCR in hypoxic neural stem cells of rat in vitro. Mol Biol Rep 39: , 569–576. |
[39] | Livak KJ , Schmittgen TD ((2001) ) Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔ method. Methods 25: , 402–408. |
[40] | Applied Biosystems (2007) Relative quantitation using comparative CT: Getting started guide, https://www3.appliedbiosystems.com/cms/groups/mcb_support/documents/general-documents/cms_042115.pdf. |
[41] | Pluta R ((2004) ) From brain ischemia-reperfusion injury to possible sporadic Alzheimer’s disease. Curr Neurovasc Res 1: , 441–453. |
[42] | Koistinaho M , Koistinaho J ((2005) ) Interactions between Alzheimer’s disease and cerebral ischemia - focus on inflammation. Brain Res Rev 48: , 240–250. |
[43] | Jabłoński M , Maciejewski R , Januszewski S , Ułamek M , Pluta R ((2011) ) One year follow up in ischemic brain injury and the role of Alzheimer factors. Physiol Res 60: (Suppl 1), S113–S119. |
[44] | Zhang J , Zhang Y , Li J , Xing S , Li C , Li Y , Dang C , Fan Y , Yu J , Pei Z , Zeng J ((2012) ) Autophagosomes accumulation is associated with β-amyloid deposits and secondary damage in the thalamus after focal cortical infarction in hypertensive rats. J Neurochem 120: , 564–573. |
[45] | Zhang Q , Gao T , Luo Y , Chen X , Gao G , Gao X , Zhou Y , Dai J ((2012) ) Transient focal cerebral ischemia/reperfusion induces early and chronic axonal changes in rats: Its importance for the risk of Alzheimer’s disease. PLoS One 7: , e33722. |
[46] | Heikkinen R , Malm T , Heikkilä J , Muona A , Tanila H , Koistinaho M , Koistinaho J ((2014) ) Susceptibility to focal and global brain ischemia of Alzheimer mice displaying Aβ deposits: Effect of immunoglobulin. Aging Dis 5: , 76–87. |
[47] | Wang JY , Xia Q , Chu KT , Pan J , Sun LN , Zeng B , Zhu YJ , Wang Q , Wang K , Luo BY ((2011) ) Severe global cerebral ischemia induced programmed necrosis of hippocampal CA1 neurons in rat is prevented by 3-methyladenine: A widely used inhibitor of autophagy. J Neuropathol Exp Neurol 70: , 314–322. |
[48] | Jouan-Lanhouet S , Riquet F , Duprez L , Vanden Berghe T , Takahashi N , Vandenabeele P ((2014) ) Necroptosis, in vivo detection in experimental disease models. Semin Cell Dev Biol 35: , 2–13. |
[49] | Galluzzi L , Vitale I , Abrams JM , Alnemri ES , Baehrecke EH , Blagosklonny MV , Dawson TM , Dawson VL , El-Deiry WS , Fulda S , Gottlieb E , Green DR , Hengartner MO , Kepp O , Knight RA , Kumar S , Lipton SA , Lu X , Madeo F , Malorni W , Mehlen P , Nuñez G , Peter ME , Piacentini M , Rubinsztein DC , Shi Y , Simon HU , Vandenabeele P , White E , Yuan J , Zhivotovsky B , Melino G , Kroemer G ((2012) ) Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 19: , 107–120. |
[50] | Müller GJ , Hasseldam H , Rasmussen RS , Johansen FF ((2014) ) Dexamethasone enhances necrosis-like neuronal death in ischemic rat hippocampus involving μ-calpain activation. Exp Neurol 261: , 711–719. |
[51] | Groemer TW , Thiel CS , Holt M , Riedel D , Hua Y , Hüve J , Wilhelm BG , Klingauf J ((2011) ) Amyloid precursor protein is trafficked and secreted via synaptic vesicles. PLoS One 6: , e18754. |
[52] | Kaczmarek A , Vandenabeele P , Krysko DV ((2013) ) Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 38: , 209–223. |
[53] | Masel BE , DeWitt DS ((2010) ) Traumatic brain injury: A disease process, not an event. J Neurotrauma 27: , 1529–1540. |
[54] | Pluta R , Barcikowska M , Januszewski S , Misicka A , Lipkowski AW ((1996) ) Evidence of blood— brain barrier permeability/leakage for circulating human Alzheimer’s β– amyloid– (1-42)– peptide. Neuroreport 7: , 1261–1265. |
[55] | Zetterberg H , Mörtberg E , Song L , Chang L , Provuncher GK , Patel PP , Ferrell E , Fournier DR , Kan CW , Campbell TG , Meyer R , Rivnak AJ , Pink BA , Minnehan KA , Piech T , Rissin DM , Duffy DC , Rubertsson S , Wilson DH , Blennow K ((2011) ) Hypoxia due to cardiac arrest induces a time-dependent increase in serum amyloid β levels in humans. PLoS One 6: , e28263. |
[56] | Kamide T , Kitao Y , Takeichi T , Okada A , Mohri H , Schmidt AM , Kawano T , Munesue S , Yamamoto Y , Yamamoto H , Hamada J , Hori O ((2012) ) RAGE mediates vascular injury and inflammation after global cerebral ischemia. Neurochem Int 60: , 220–228. |
[57] | Buga AM , Sascau M , Pisoschi C , Herdon JG , Kessler C , Popa-Wagner A ((2008) ) The genomic response of the ipsilateral and contralateral cortex to stroke in aged rats. J Cell Mol Med 12: , 2731–2753. |
[58] | Buga AM , Scholz CJ , Kumar S , Herdon JG , Alexandru D , Cojocaru GR , Dandekar T , Popa-Wagner A ((2012) ) Identification of new therapeutic targets by genome-wide analysis of gene expression in the ipsilateral cortex of aged rats after stroke. PloS One 7: , e50985. |
[59] | Buga AM , Margaritescu C , Scholz CJ , Radu E , Zelenak C , Popa-Wagner A ((2014) ) Transcriptomic of post-stroke angiogenesis in the aged brain. Front Aging Neurosci 6: , 44. |
[60] | Ballard C , Gauthier S , Corbett A , Brayne C , Aarsland D , Jones E ((2011) ) Alzheimer’s disease. Lancet 377: , 1019–1031. |
[61] | Selkoe D , Mandelkow E , Holtzman D ((2012) ) Deciphering Alzheimer disease. Cold Spring Harb Perspec Med 2: , a011460. |
[62] | Pluta R , Jolkkonen J , Cuzzocrea S , Pedata F , Cechetto D , Popa-Wagner A ((2011) ) Cognitive impairment with vascular impairment and degeneration. Curr Neurovasc Res 8: , 342–350. |
Figures and Tables
Fig.1
The mean gene expression levels of β-secretase in the medial temporal lobe cortex in rats 2, 7, and 30 days after 10-min of global brain ischemia. Marked SEM - standard error of the mean. Indicated statistically significant differences in levels of gene expression between 2 and 7 and between 2 and 30 days after 10-min of global brain ischemia (Mann-Whitney U test). *p≤0.01.
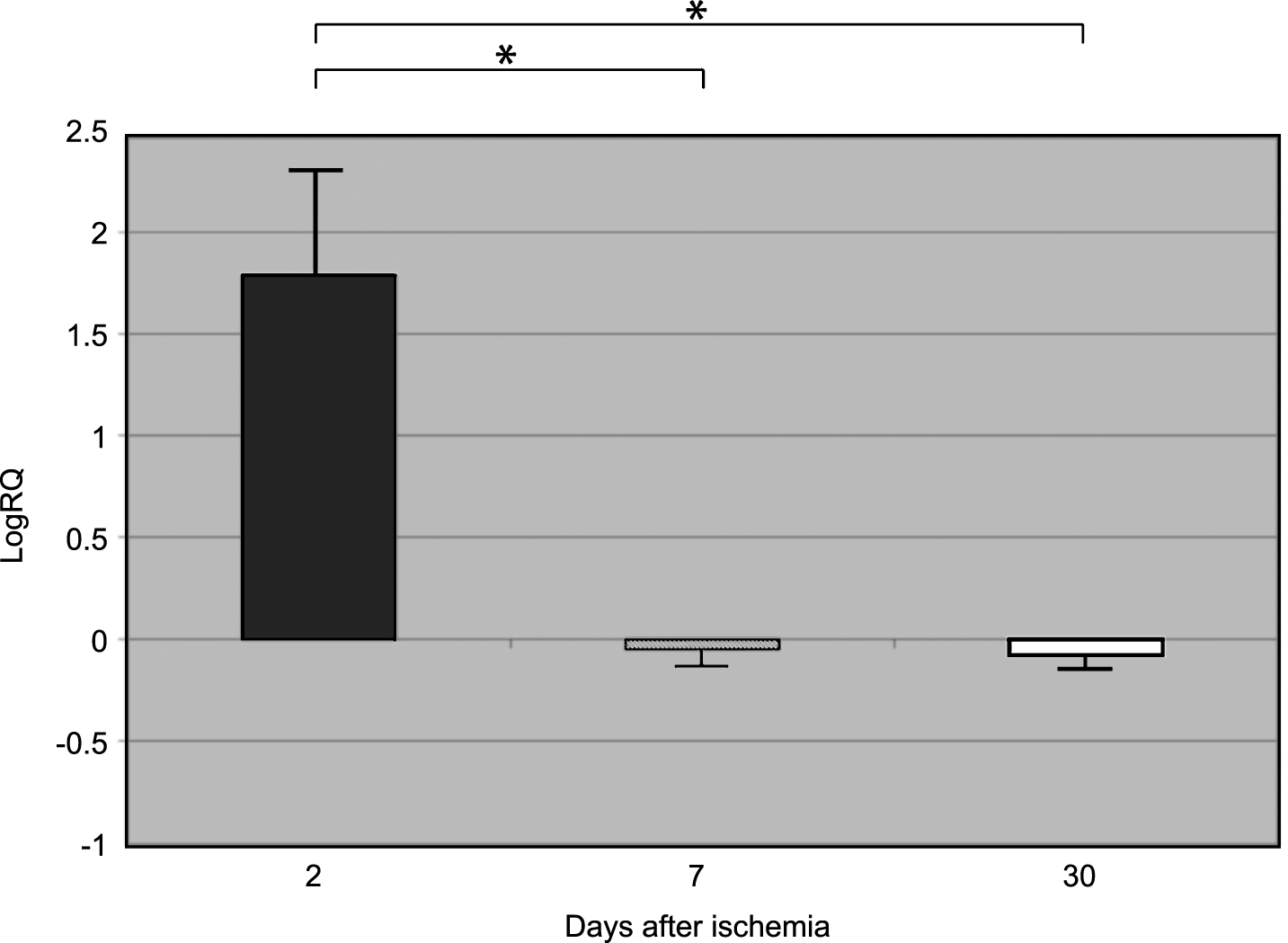
Fig.2
The mean gene expression levels of amyloid-β protein precursor in the medial temporal lobe cortex in rats 2, 7, and 30 days after 10-min of global brain ischemia. Marked SEM - standard error of the mean. Indicated statistically significant differences in levels of gene expression between 2 and 7 and between 2 and 30 days after 10-min of global brain ischemia (Mann-Whitney U test). *p≤0.01.