Do Microglia Default on Network Maintenance in Alzheimer’s Disease?
Abstract
Although the cause of Alzheimer’s disease (AD) remains unknown, a number of new findings suggest that the immune system may play a critical role in the early stages of the disease. Genome-wide association studies have identified a wide array of risk-associated genes for AD, many of which are associated with abnormal functioning of immune cells. Microglia are the brain’s immune cells. They play an important role in maintaining the brain’s extracellular environment, including clearance of aggregated proteins such as amyloid-β (Aβ). Recent studies suggest that microglia play a more active role in the brain than initially considered. Specifically, microglia provide trophic support to neurons and also regulate synapses. Microglial regulation of neuronal activity may have important consequences for AD. In this article we review the function of microglia in AD and examine the possible relationship between microglial dysfunction and network abnormalities, which occur very early in disease pathogenesis.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive, neurodegenerative disease that primarily affects the regions of the brain that are associated with high functioning. AD is characterized by progressive dementia that begins with mood changes, memory loss, and reduced cognition [1]. The primary pathogenic process in AD is the accumulation of amyloid-β protein (Aβ) [1–3]. Aβ aggregates into extracellular amyloid plaques that are a hallmark pathological feature of the disease. Aβ is cleaved from the larger amyloid-β protein precursor (AβPP) [4–6]. However, it remains unclear why Aβ, a protein fragment normally only present in small amounts within the brain, is able to accumulate in the AD brain and cause toxicity. In a small percentage (5%) of AD sufferers, the cause of the disease is genetic. Inherited mutations within the AβPP gene itself appear to predispose the protein to Aβ production [7]. Mutations within the presenilin 1 and 2 genes, encoding proteins that form part of the secretase complex that cleaves the Aβ peptide from AβPP, also result in inherited AD due to accumulations of Aβ [8–11].
The cause of AD is largely unknown for the remaining 95% of cases of sporadic AD, which typically develops a decade or two later than familial AD [12]. However, the degenerative processes are nearly identical between the two forms of the disease. Therefore, it is reasonable to assume that the underlying disease process is the same between the two forms of the disease. Genetic studies have identified a number of genetic risk factors for AD. An early discovery was that allelic variants of apolipoprotein E (ApoE) carry inherently different risks of AD [13–15]. In particular, the ɛ4 allele carries a high risk of AD, with risk of disease occurring in a dose-dependent manner based onzygosity. The ApoE ɛ4 allele is associated with increased Aβ aggregation, reduced lipid transport, and reduced receptor-mediated Aβ clearance [16]. Interestingly, ApoE is predominantly expressed by non-neuronal cells— astrocytes and microglia, rather than by neurons [17]. These findings suggested that although clinical AD manifests from neuronal degeneration, other cells of the central nervous system (CNS) may be intimately involved in pathogenesis or disease progression.
More recently, genome-wide association studies (GWAS) have been used to identify a large number of risk genes for AD. In 2013, a mutation in the triggering receptor expressed on myeloid cells 2 (TREM2) was identified [18, 19]. TREM2 is almost exclusively expressed by immune cells within the brain, and mutations to TREM2 are associated with decreased phagocytosis and an increased pro-inflammatory reactive phenotype. Individuals heterozygous for TREM2 mutations have a high risk of developing AD, however the mutation is rare [18, 19]. Additional AD risk factor genes that have been identified include genes associated with lipid processing, endocytosis, and the immune response, which have recently been covered in excellent reviews [20, 21]. The common unifying feature of these immune-associated mutations is that they are proposed to interfere with microglial function, in particular, the efficiency of phagocytosis [20]. Specifically, mutations to complement receptor 1 (CR1) and cluster of differentiation 33 (CD33) can result in reduced activity of the complement system and reduced phagocytosis [22, 23]. Phosphatidylinositol binding clathrin assembly protein (PICALM) and bridging integrator 1 (BIN1) mutations affect clathrin-mediated endocytosis [24, 25] and SORL1 mutations reduce intracellular trafficking of AβPP [26]. The function of some of these proteins in relation to phagocytosis is discussed later in this review. The identification of such a wide array of risk genes associated with reduced immune cell function now leads us to believe that abnormal functioning of immune cells may play a more important role in the early stages of disease than previously considered.
MICROGLIA
Microglia are the immune cells of the CNS and account for approximately 10% of the CNS cellpopulation, with regional variation in density [27, 28]. During embryonic development, microglia originate from yolk sac progenitor cells that migrate into the developing CNS during early embryogenesis [29,30].Following construction of the blood-brain barrier (BBB), microglia are renewed by local turnover [31]. In the healthy brain, microglia actively support neurons through the release of insulin-like growth factor 1, nerve growth factor, ciliary neurotrophic factor, and brain-derived neurotrophic factor (BDNF) [32–34]. Microglia also provide indirect support to neurons by clearance of debris to maintain the extracellular environment, and phagocytosis of apoptotic cells to facilitate neurogenesis [35, 36]. In the adult brain, microglia coordinate much of their activity with astrocytes and activate in response to similar stimuli [37, 38]. Dysfunctional signaling between microglia and astrocytes often results in chronic inflammation, a characteristic of many neurodegenerative diseases [39, 40].
Historically, it has been thought that microglia ‘rest’ when not responding to inflammatory stimuli or damage [41, 42]. However, this notion is being increasingly recognized as inaccurate [43]. When not involved in active inflammatory signaling, microglia constantly patrol the neuropil by extension and retraction of their finely branched processes [44]. Microglial activation is often broadly classified into two states; pro-inflammatory (M1) or anti-inflammatory (M2) [36, 45], based on similar phenotypes in peripheral macrophages [46]. M1 activated microglia are characterized by increased expression of pro-inflammatory mediators and cytokines, including inducible nitric oxide synthase, tumor necrosis factor-α, and interleukin-1β, often under the control of the transcription factor nuclear factor-κB [45]. Pro-inflammatory microglia rapidly retract their processes and adopt an amoeboid morphology and often migrate closer to the site of injury [47]. Anti-inflammatory M2 activation of microglia, often referred to as alternative activation, represents the other side of microglial behavior. Anti-inflammatory activation is characterized by increased expression of cytokines including arginase 1 and interleukin-10, and is associated with increased ramification of processes [45]. The polarization of microglia into M1 or M2 throughout the brain is well characterized, especially in neurodegenerative diseases [48]. In the AD brain, microglia expressing markers of M1 activation are typically localized to brain regions such as the hippocampus that are most heavily affected in the disease [49]. However, it is important to note that M1 and M2 classifications of microglia may over-simplify microglial phenotypes and may only represent the extremes of microglial activation [50]. It has been more recently proposed that microglia likely occupy a continuum between these phenotypes [39, 51].
Do microglia have multiple roles in AD?
Classical pro-inflammatory activation of microglia has long been associated with AD [39, 49]. Samples taken from late-stage AD brains contain characteristic signs of inflammation, including amoeboid morphology of microglia, high levels of pro-inflammatory cytokines in the cerebrospinal fluid, and evidence of neuronal damage due to chronic exposure to pro-inflammatory cytokines and oxidative stress [52, 53]. The cause of this inflammation may be in response to direct toxicity of Aβ to neurons resulting in activation of nearby microglia and astrocytes [53, 54]. However, Aβ may also induce inflammatory activation of microglia and astrocytes. Activated immune cells are typically present surrounding amyloid plaques [55–57], with such peri-plaque cells exhibiting strong evidence of pro-inflammatory activation [56, 58–60]. The presence of undigested Aβ particles within these activated microglia may suggest that the Aβ peptide itself is a pro-inflammatory signal for microglia [61–64]. In vitro experiments provide supporting evidence for the in vivo studies, with Aβ promoting pro-inflammatory microglial activation [65, 66], and also acting as a potent chemotactic signal [67].
However, it is important to note that although widespread inflammation is characteristic of late-stage AD, it remains unclear what role inflammation could play in early stages of the disease. Some evidence suggests that reducing inflammation through the long-term use of some non-steroidal anti-inflammatory drugs (NSAIDs) can reduce the risk of AD [68]. However, these findings have not yet been verified in clinical trials [69, 70]. Little is understood about how NSAIDs and related compounds affect the delicate balance of pro- versus anti-inflammatory microglial activity within the brain. Although there is considerable evidence to suggest that chronic inflammation may contribute to pathology in the later stages of AD, it is important to note that inflammation normally only represents a small aspect of microglial function. The non-inflammatory functions of microglia may play a more important role in early disease; specifically, microglial functions relating to maintenance of the CNS.
Phagocytosis: A vital role of microglia that may be lost in AD
Phagocytosis is a complex process involving the recognition, engulfment, and degradation of particles larger than 0.5 μm [71]. Although most cell types have the capacity to phagocytose, it is normally the role of highly specialized cells, predominantly those of the immune cell lineage [72]. Within the brain, microglia perform the bulk of phagocytosis, although astrocytes also contribute [37]. Many peripheral phagocytic cells have a preferred target for phagocytosis; however, microglia are most like their peripheral cousins, macrophages, and readily ingest a wide array of structures, including dead cells, invading pathogens and extracellular proteins [45].
Microglial phagocytosis is triggered by a number of stimuli, for which microglia express a wide variety of receptors. Release of adenosine triphosphate (ATP) from apoptotic or damaged cells is a potent signal for microglial phagocytosis, triggering the purinergic receptor P2Y2 [73]. Similarly, fractalkine, recognized by the microglial receptor CX3CR1, is released by damaged cells and promotes microglial phagocytosis [74]. Microglia also express a wide array of ‘scavenging receptors’, defined by having a relatively broad range of ligand targets [75], and receptors for various components of the complement system, including complement C1q and complement C3, the receptor for which is known by many names including CR3, CD11b, MAC1, and integrin αm [36]. A number of scavenging receptors have been proposed to interact with Aβ peptides, including receptor for advanced glycation products, macrophage scavenger receptor with collagenous structure, scavenger receptor A-1 (SCARA-1), SCARB-1 and SCARB-2/CD36 [76–80]. However, it is important to note that these receptors also interact with a broad range of other phagocytic ligands, suggesting that microglia detect Aβ in a non-specific manner.
Phagocytosis of Aβ by immune cells, including microglia, is proposed to assist with clearance of Aβ from the brain [81]. Other mechanisms of Aβ clearance include extracellular proteolysis by Aβ-degrading enzyme and neprilysin, astrocyte-mediated interstitial bulk-flow [81], and potentially direct clearance into the lymphatic system [82]. Microglia in AD appear to have a reduced capacity for Aβ clearance which is likely to result in additional accumulation of Aβ [83]. The TREM2 mutation that increases the risk of AD reduces the normal inhibitory effect of TREM2 on microglial activation and thereby increases microglial pro-inflammatory signaling [84, 85]. This leads to reduced Aβ uptake. Similarly, mutations in complement receptor CR1 and CD33 are associated with an increased risk of AD [86, 87]. These mutations are proposed to cause a loss of receptor activity, thereby reducing phagocytosis of their respectiveligands. Other putative genetic risk alleles for AD include the lipid transporter ATP-binding cassette subfamily A member 7 (ABCA7) and interleukin-1 receptor accessory protein (IL-1RAP) [88, 89]. Mutations in ABCA7 can cause a loss of receptor activity, resulting in reduced microglial phagocytic function [88, 90]. Similarly, an intronic single-nucleotide polymorphism identified in IL-1RAP is proposed to reduce expression of IL-1RAP, resulting in reduced microglial activation and phagocytosis [89]. The activity of microglia in the CNS is affected by a wide range of stimuli and is also dependent on the composition and function of surface receptors. These results highlight how minor changes to a number of microglial receptors have major implications in the context of disease and lead us to consider how these changes to microglial function increase the risk of AD, a disease that is characterized not only by accumulation of Aβ, but also synapse loss and neuronal dysfunction.
SYNAPTIC PRUNING: MICROGLIA CAN REGULATE NETWORK ACTIVITY
Recently, a new function has been proposed for microglia. A number of studies have provided evidence that microglia prune synapses throughout life. Microglia are known to remove extraneous synapses during development to ensure that only meaningful connections remain [43]. It was, however, thought that differentiated astrocytes performed the majority of synaptic pruning in the adult brain [91]. The discovery that microglial processes are constantly active within the brain and are often positioned near synapses raised the question of whether microglial synaptic pruning continued throughout life [44, 47, 92–94]. This question was answered in 2014 in a study that demonstrated that microglia do prune synapses into adulthood, and that this activity is important for normal brain function [95]. These findings supported those found a year earlier in a study reporting that ablation of microglia from brain slices increases synapse density and results in abnormal firing of hippocampalneurons [96].
Astrocytes have long been known to have important roles in synaptic maintenance and are considered to be as much a part of the synapse as pre- and post-synaptic neurons. Hence the term ‘tripartite synapse’ was coined for the structure [97]. Microglia are increasingly considered to be equally important at the synaptic structure, forming a key component of a ‘quadripartite synapse’, comprising pre- and post-synaptic neurons, astrocytes, and microglia [98]. Microglial synaptic pruning is proposed to occur alongside astrocytic synaptic pruning and occurs in response to astrocyte-derived signals including transforming growth factor-β and the complement proteins C1q and C3 [98–101].
Microglia prune synapses as an extension of normal phagocytosis. Synapses to be cleared are tagged with complement proteins [101], expressed by the neurons under the direction of astrocytes [99]. Microglial synaptic pruning is dependent on neuronal activity [101]. Active synapses are protected by expression of C1q on the membrane, whereas quiet synapses express C3 that triggers C3 cleavage and binding of C3 to CR3 on microglia. CR3 activation results in microglial phagocytosis of the synapse [99, 101]. Deletion of complement C1q or C3 in mice reduces synaptic loss during postnatal development, resulting in increased synaptic density within the cortex and hippocampus [102, 103], an effect that lasts with aging [104]. Conversely, activation of CR3 during inflammation and hypoxia induces long-term depression in the hippocampus [105]. These findings highlight the importance of complement regulation for normal synaptic maintenance; however there are additional mechanisms that may also be utilized by microglia to regulate synapses. Neuronally-derived fractalkine appears to regulate synaptic strength independently of electrical activity. Selective activation of the fractalkine receptor CX3CR1 increases synaptic strength [106], whereas deficiency in CX3CR1 results in impaired long-term potentiation and reduced hippocampal synaptic plasticity [107].
Multiple elements of the complement cascade are expressed in AD brains [108]. The complement system has also been implicated in AD because a number of risk mutations involve complement genes, including CR1 and CD33. Current findings suggest that CR1 mutations are associated with reduced inhibition of phagocytosis [109]. Reducing C3 in AD transgenic mice reduces microgliosis and increases cerebral Aβ load [110]. Elevated complement is also a feature of epilepsy, Huntington’s disease, Parkinson’s disease, and multiple sclerosis [111, 112].
Although the use of the complement system appears to be the primary mechanism for synaptic pruning, other signaling pathways have also been described. Neuronal expression of PirB, a major histocompatibility class I receptor, controls synaptic plasticity by promoting synaptic pruning by microglia [113, 114]. Interestingly, Aβ is proposed to activate PirB resulting in increased synaptic pruning [115], an interaction that may be responsive to pharmacological intervention. Furthermore, microglial release of BDNF promotes synaptic pruning through the induction of tyrosine receptor kinase B autophosphorylation and downstream signaling [116]. Specific depletion of BDNF expression in microglia results in learning and memory impairments [116]. Circadian expression of cathepsin S, a microglial-specific lysosomal cysteine protease, is associated with modulation of cortical neuron activity [117]. Increased cathepsin S secretion by microglia during the awake phase of mice (dark hours) reduces synaptic neurotransmission by cortical neurons to strengthen synapses [117]. Ablation of cathepsin S induces hyperexcitability and loss of coordinated neuronal activity. A common thread to these findings is that disruption of microglial signaling either through deletion of microglia or impairment of key receptors, results in similar functional deficits. In particular, microglial abnormalities dramatically impair normal hippocampal function, which may have direct consequences for the coordination of neuronal activity such as during memory consolidation.
Altered microglial behavior may underlie altered neuronal firing in AD
Although we lack information about how microglia may affect network function in early AD, there is evidence to suggest that altering microglial function has direct consequences for neuronal activity (Fig. 1). Targeted deletion of key microglial receptors reduces the capacity of the mice to learn tasks, specifically those involving hippocampal learning [107, 116]. Reduced developmental synaptic pruning in mice results in overall decreased functional connectivity in the brain and autism-like behaviors including deficits in social interaction and increased repetitive activity [118]. These behavioral effects occur due to reduced functional brain connectivity and weak synaptic neurotransmission as a result of reduced microglial pruning of synapses. In hippocampal tissue slices, depletion of microglia is followed by increased synaptic density and excitatory postsynaptic currents, reversible upon replenishment of cultures with microglia [96].
Altering microglial activity by exposure to pro-inflammatory stimuli results in changes to synaptic activity. Pro-inflammatory activation of microglia with lipopolysaccharide (LPS) in hippocampal slice preparations results in a rapid increase in excitatory postsynaptic currents in neurons, mediated by astrocytic activation [38]. Inflammation has also been shown to trigger a regional-specific increase in neuronal spines, specifically thin dendritic spines that are associated with plasticity [119]. Microglia also increase release of BDNF following LPS exposure [120]. Activation of microglial CR3 during hypoxia has been shown to increase hippocampal long-term depression via nicotinamide adenine dinucleotide phosphate (NADPH) signaling and internalization of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) GluR2 subunits [105]. These findings suggest that activation of microglia can alter both synapses and neuronalactivity.
Altered neuronal activity is an early phenomenon in AD
Increasingly, altered network activity is considered a feature of AD, specifically, early dysfunction within the default mode network (DMN). The DMN coordinates introspective thought such as day-dreaming and self-referential thought and is normally deactivated during focused thought on external tasks [121, 122]. Imaging of white matter using diffusion tensor imaging [123] or measurement of neuronal activity through glucose metabolism [124] demonstrates that the DMN is consistently hypoactive in AD. Such DMN hypoactivity may represent a diagnostic marker for AD [125]. As AD worsens, network abnormalities progress through to interconnected regions including the hippocampus, dorsal attention network, salience network, sensorimotor network, and executive control network [126]. These network abnormalities closely correlate with increasing symptom severity [126]. A small number of studies have demonstrated that these regions that are specifically affected in AD are also characterized by an increase in activated microglia [127, 128]. It must be noted that some of the changes in neuronal activity that are seen in AD may be due to synaptic toxicity of Aβ [129]. Aβ has been demonstrated to cause damage to synapses and predispose neurons to hyperexcitability and excitotoxicity [129, 130]. Specifically, Aβ binds to subunits of AMPA and N-methyl-D-aspartate (NMDA) excitatory receptors and causes increased calcium influx into the neuron [131–133]. Aβ may reduce inhibitory synapses [134], and may directly perturb astrocyte-neuron signaling, on which neurons are typically heavily reliant [135].
The cause of DMN hypoactivity in AD is not yet clear; however studies performed in cohorts that are genetically predisposed to AD suggest that DMN hypoactivity is preceded by a period of hyperactivity and increased functional connectivity [123, 136], often manifesting as an absence of normal DMN deactivation during external tasks [137–140]. DMN hyperactivity may interfere with hippocampal memory encoding, leading to the memory deficits that are present in mild cognitive impairment [141, 142]. It has been proposed that hippocampal hyperexcitability in AD may develop as a protective mechanism against increased input from the DMN [142–144]. As AD progresses, the initial hyperexcitability of the DMN and hippocampus may result in hypoactivity due to exhaustion of compensatory mechanisms [123, 136]. Evidence from both transgenic AD mice and longitudinal human studies supports an exhaustion model of hyperactivation leading to later hypoactivation [143, 145–147]. Interestingly, a number of studies report a lower incidence of AD among those who regularly practice meditation which specifically ‘calms’ the DMN [148].
CONCLUSIONS
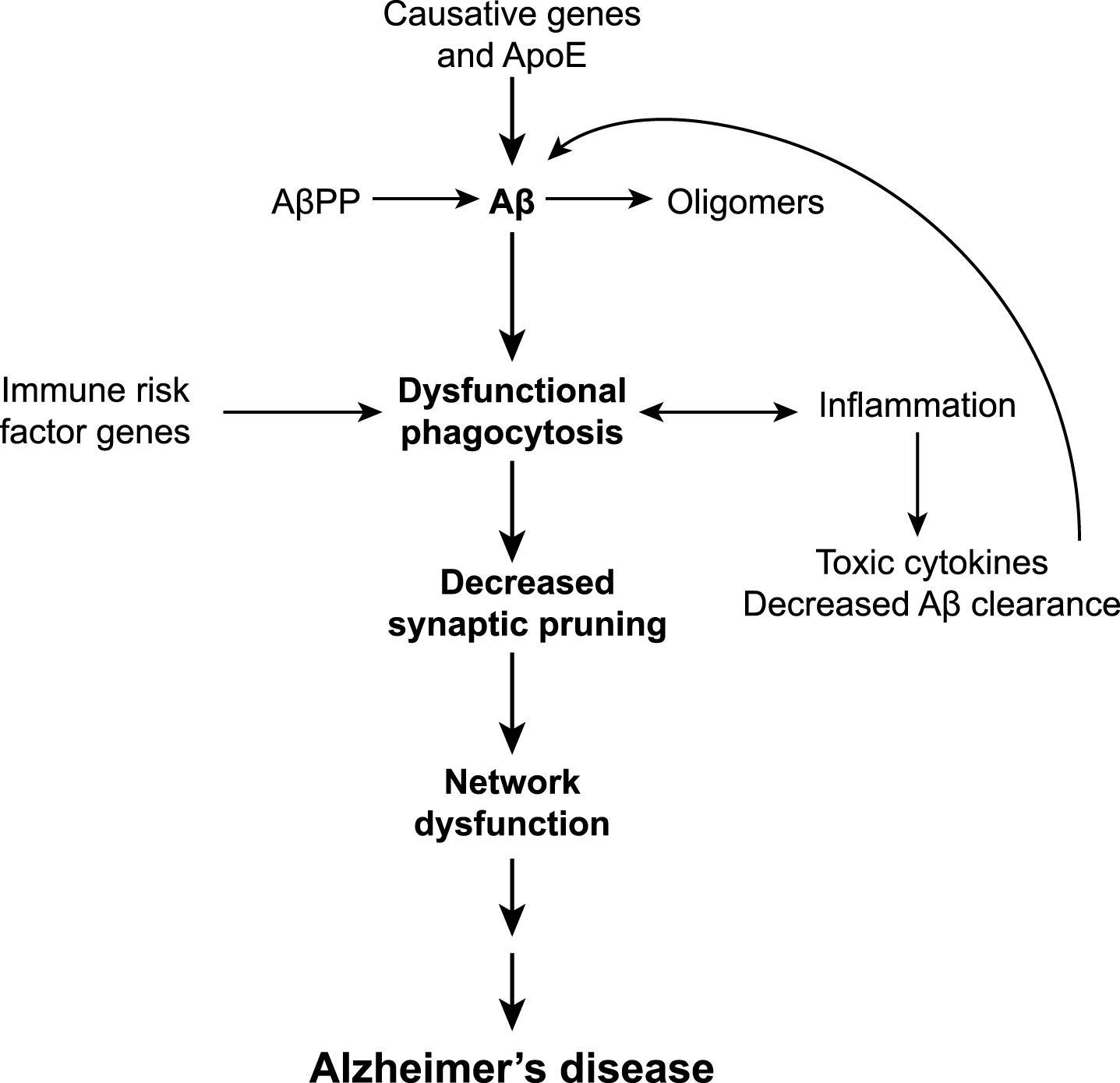
Our understanding of AD as a disease is changing. Historically considered to be primarily a disease of neuronal degeneration, this neurocentric view has widened to encompass non-neuronal cells such as astrocytes into our understanding of the disease process and pathogenesis. A proposed model for microglia in AD is shown in Fig. 2. Microglia perform a wide range of functions in the CNS and although this includes induction of an inflammatory reaction in response to damage, they also have critical roles for maintaining normal function in the brain. Recent evidence shows that microglia regulate neuronal activity through synaptic pruning throughout life as an extension on their normal phagocytosis behavior. The discovery of a large number of AD risk genes associated with reduced immune cell function suggests that perturbed microglial phagocytosis could lead to AD. In our model, altered microglial phagocytosis of synapses results in network dysfunction and onset of AD, occurring downstream of Aβ.
The immune system and microglia represent a novel target for intervention in AD. Importantly, a large number of anti-inflammatory drugs are already in use for other conditions. What is important to know at this stage is exactly how to best target immune cell function. The studies outlined here provide evidence that an indiscriminate dampening down of all microglial activity may result in a worse outcome for individuals by suppressing normal microglial regulatory functions. We currently do not know whether future microglial-based therapies should focus on reducing chronic inflammation or conversely, whether they should be aimed at boosting microglial phagocytosis. It is also likely that future treatment strategies may use a combination of approaches to target Aβ, immune cell phagocytosis and network activity. An increasing view in the AD field is that any drug or therapy needs to be provided very early in the disease process to maximize its beneficial effects. Although we are currently unable to effectively target those at risk of AD at such an early stage, advances in neuroimaging for subtle changes in network activity, or in assays for immune cell function, may provide new avenues for identification of early damage and risk of disease.
ACKNOWLEDGMENTS
DHS is supported by grants from the National Health and Medical Research Council (NHMRC). AJV is supported by a NHMRC postdoctoral fellowship. KAS is the recipient of a part-funded fellowship from the Alzheimer’s Australia Dementia Research Foundation (AADRF).
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/15-1075r1).
REFERENCES
[1] | Selkoe DJ ((2011) ) Alzheimer’s disease. Cold Spring Harb Perspect Biol 3: , pii: a004457. |
[2] | Masters CL , Simms G , Weinman NA , Multhaup G , McDonald BL , Beyreuther K ((1985) ) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A 82: , 4245–4249. |
[3] | Glenner GG , Wong CW ((1984) ) Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120: , 885–890. |
[4] | Goldgaber D , Lerman MI , McBride OW , Saffiotti U , Gajdusek DC ((1987) ) Characterization and chromosomal localization of a cDNA encoding brain amyloid of Alzheimer’s disease. Science 235: , 877–880. |
[5] | Kang J , Lemaire HG , Unterbeck A , Salbaum JM , Masters CL , Grzeschik KH , Multhaup G , Beyreuther K , Muller-Hill B ((1987) ) The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor. Nature 325: , 733–736. |
[6] | Robakis NK , Ramakrishna N , Wolfe G , Wisniewski HM ((1987) ) Molecular cloning and characterization of a cDNA encoding the cerebrovascular and the neuritic plaque amyloid peptides. Proc Natl Acad Sci U S A 84: , 4190–4194. |
[7] | Levy E , Carman MD , Fernandez-Madrid IJ , Power MD , Lieberburg I , van Duinen SG , Bots GT , Luyendijk W , Frangione B ((1990) ) Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 248: , 1124–1126. |
[8] | Levy-Lahad E , Wasco W , Poorkaj P , Romano DM , Oshima J , Pettingell WH , Yu CE , Jondro PD , Schmidt SD , Wang K , et al ((1995) ) Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 269: , 973–977. |
[9] | Rogaev EI , Sherrington R , Rogaeva EA , Levesque G , Ikeda M , Liang Y , Chi H , Lin C , Holman K , Tsuda T , et al ((1995) ) Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376: , 775–778. |
[10] | Sherrington R , Rogaev EI , Liang Y , Rogaeva EA , Levesque G , Ikeda M , Chi H , Lin C , Li G , Holman K , Tsuda T , Mar L , Foncin JF , Bruni AC , Montesi MP , Sorbi S , Rainero I , Pinessi L , Nee L , Chumakov I , Pollen D , Brookes A , Sanseau P , Polinsky RJ , Wasco W , Da Silva HA , Haines JL , Perkicak-Vance MA , Tanzi RE , Roses AD , Fraser PE , Rommens JM , St George-Hyslop PH ((1995) ) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375: , 754–760. |
[11] | Levy-Lahad E , Wijsman EM , Nemens E , Anderson L , Goddard KA , Weber JL , Bird TD , Schellenberg GD ((1995) ) A familial Alzheimer’s disease locus on chromosome 1. Science 269: , 970–973. |
[12] | Small SA , Gandy S ((2006) ) Sorting through the cell biology of Alzheimer’s disease: Intracellular pathways to pathogenesis. Neuron 52: , 15–31. |
[13] | Strittmatter WJ , Weisgraber KH , Huang DY , Dong LM , Salvesen GS , Pericak-Vance M , Schmechel D , Saunders AM , Goldgaber D , Roses AD ((1993) ) Binding of human apolipoprotein E to synthetic amyloid beta peptide: Isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A 90: , 8098–8102. |
[14] | Corder EH , Saunders AM , Strittmatter WJ , Schmechel DE , Gaskell PC , Small GW , Roses AD , Haines JL , Pericak-Vance MA ((1993) ) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261: , 921–923. |
[15] | Schmechel DE , Saunders AM , Strittmatter WJ , Crain BJ , Hulette CM , Joo SH , Pericak-Vance MA , Goldgaber D , Roses AD ((1993) ) Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Natl Acad Sci U S A 90: , 9649–9653. |
[16] | Holtzman DM , Herz J , Bu G ((2012) ) Apolipoprotein E and apolipoprotein E receptors: Normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med 2: , a006312. |
[17] | Xu Q , Bernardo A , Walker D , Kanegawa T , Mahley RW , Huang Y ((2006) ) Profile and regulation of apolipoprotein E (ApoE) expression in the CNS in mice with targeting of green fluorescent protein gene to the ApoE locus. J Neurosci 26: , 4985–4994. |
[18] | Guerreiro R , Wojtas A , Bras J , Carrasquillo M , Rogaeva E , Majounie E , Cruchaga C , Sassi C , Kauwe JS , Younkin S , Hazrati L , Collinge J , Pocock J , Lashley T , Williams J , Lambert JC , Amouyel P , Goate A , Rademakers R , Morgan K , Powell J , St George-Hyslop P , Singleton A , Hardy J ((2013) ) TREM2 variants in Alzheimer’s disease. N Engl J Med 368: , 117–127. |
[19] | Jonsson T , Stefansson H , Steinberg S , Jonsdottir I , Jonsson PV , Snaedal J , Bjornsson S , Huttenlocher J , Levey AI , Lah JJ , Rujescu D , Hampel H , Giegling I , Andreassen OA , Engedal K , Ulstein I , Djurovic S , Ibrahim-Verbaas C , Hofman A , Ikram MA , van Duijn CM , Thorsteinsdottir U , Kong A , Stefansson K ((2013) ) Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368: , 107–116. |
[20] | Van Cauwenberghe C , Van Broeckhoven C , Sleegers K ((2015) ) The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet Med doi: 10.1038/gim.2015.117 [Epub ahead of print] |
[21] | Mhatre SD , Tsai CA , Rubin AJ , James ML , Andreasson KI ((2015) ) Microglial malfunction: The third rail in the development of Alzheimer’s disease. Trends Neurosci 38: , 621–636. |
[22] | Hazrati LN , Van Cauwenberghe C , Brooks PL , Brouwers N , Ghani M , Sato C , Cruts M , Sleegers K , St George-Hyslop P , Van Broeckhoven C , Rogaeva E ((2012) ) Genetic association of CR1 with Alzheimer’s disease: A tentative disease mechanism. Neurobiol Aging 33: , 2949 e2945–2949 e2912. |
[23] | Griciuc A , Serrano-Pozo A , Parrado AR , Lesinski AN , Asselin CN , Mullin K , Hooli B , Choi SH , Hyman BT , Tanzi RE ((2013) ) Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78: , 631–643. |
[24] | Harold D , Abraham R , Hollingworth P , Sims R , Gerrish A , Hamshere ML , Pahwa JS , Moskvina V , Dowzell K , Williams A , Jones N , Thomas C , Stretton A , Morgan AR , Lovestone S , Powell J , Proitsi P , Lupton MK , Brayne C , Rubinsztein DC , Gill M , Lawlor B , Lynch A , Morgan K , Brown KS , Passmore PA , Craig D , McGuinness B , Todd S , Holmes C , Mann D , Smith AD , Love S , Kehoe PG , Hardy J , Mead S , Fox N , Rossor M , Collinge J , Maier W , Jessen F , Schurmann B , Heun R , van den Bussche H , Heuser I , Kornhuber J , Wiltfang J , Dichgans M , Frolich L , Hampel H , Hull M , Rujescu D , Goate AM , Kauwe JS , Cruchaga C , Nowotny P , Morris JC , Mayo K , Sleegers K , Bettens K , Engelborghs S , De Deyn PP , Van Broeckhoven C , Livingston G , Bass NJ , Gurling H , McQuillin A , Gwilliam R , Deloukas P , Al-Chalabi A , Shaw CE , Tsolaki M , Singleton AB , Guerreiro R , Muhleisen TW , Nothen MM , Moebus S , Jockel KH , Klopp N , Wichmann HE , Carrasquillo MM , Pankratz VS , Younkin SG , Holmans PA , O’Donovan M , Owen MJ , Williams J ((2009) ) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat Genet 41: , 1088–1093. |
[25] | Chapuis J , Hansmannel F , Gistelinck M , Mounier A , Van Cauwenberghe C , Kolen KV , Geller F , Sottejeau Y , Harold D , Dourlen P , Grenier-Boley B , Kamatani Y , Delepine B , Demiautte F , Zelenika D , Zommer N , Hamdane M , Bellenguez C , Dartigues JF , Hauw JJ , Letronne F , Ayral AM , Sleegers K , Schellens A , Broeck LV , Engelborghs S , De Deyn PP , Vandenberghe R , O’Donovan M , Owen M , Epelbaum J , Mercken M , Karran E , Bantscheff M , Drewes G , Joberty G , Campion D , Octave JN , Berr C , Lathrop M , Callaerts P , Mann D , Williams J , Buee L , Dewachter I , Van Broeckhoven C , Amouyel P , Moechars D , Dermaut B , Lambert JC , consortium G ((2013) ) Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol Psychiatry 18: , 1225–1234. |
[26] | Young JE , Boulanger-Weill J , Williams DA , Woodruff G , Buen F , Revilla AC , Herrera C , Israel MA , Yuan SH , Edland SD , Goldstein LS ((2015) ) Elucidating molecular phenotypes caused by the SORL1 Alzheimer’s disease genetic risk factor using human induced pluripotent stem cells. Cell Stem Cell 16: , 373–385. |
[27] | Lawson LJ , Perry VH , Dri P , Gordon S ((1990) ) Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39: , 151–170. |
[28] | Mittelbronn M , Dietz K , Schluesener HJ , Meyermann R ((2001) ) Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol 101: , 249–255. |
[29] | Ginhoux F , Greter M , Leboeuf M , Nandi S , See P , Gokhan S , Mehler MF , Conway SJ , Ng LG , Stanley ER , Samokhvalov IM , Merad M ((2010) ) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330: , 841–845. |
[30] | Alliot F , Godin I , Pessac B ((1999) ) Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res 117: , 145–152. |
[31] | Ajami B , Bennett JL , Krieger C , Tetzlaff W , Rossi FM ((2007) ) Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 10: , 1538–1543. |
[32] | Ueno M , Fujita Y , Tanaka T , Nakamura Y , Kikuta J , Ishii M , Yamashita T ((2013) ) Layer V cortical neurons require microglial support for survival during postnatal development. Nat Neurosci 16: , 543–551. |
[33] | Miao J , Ding M , Zhang A , Xiao Z , Qi W , Luo N , Di W , Tao Y , Fang Y ((2012) ) Pleiotrophin promotes microglia proliferation and secretion of neurotrophic factors by activating extracellular signal-regulated kinase 1/2 pathway. Neurosci Res 74: , 269–276. |
[34] | Coull JA , Beggs S , Boudreau D , Boivin D , Tsuda M , Inoue K , Gravel C , Salter MW , De Koninck Y ((2005) ) BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438: , 1017–1021. |
[35] | Hanisch UK , Kettenmann H ((2007) ) Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 10: , 1387–1394. |
[36] | Kettenmann H , Hanisch UK , Noda M , Verkhratsky A ((2011) ) Physiology of microglia. Physiol Rev 91: , 461–553. |
[37] | Liu W , Tang Y , Feng J ((2011) ) Cross talk between activation of microglia and astrocytes in pathological conditions in the central nervous system. Life Sci 89: , 141–146. |
[38] | Pascual O , Ben Achour S , Rostaing P , Triller A , Bessis A ((2012) ) Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci U S A 109: , 197–205. |
[39] | Heneka MT , Carson MJ , El Khoury J , Landreth GE , Brosseron F , Feinstein DL , Jacobs AH , Wyss-Coray T , Vitorica J , Ransohoff RM , Herrup K , Frautschy SA , Finsen B , Brown GC , Verkhratsky A , Yamanaka K , Koistinaho J , Latz E , Halle A , Petzold GC , Town T , Morgan D , Shinohara ML , Perry VH , Holmes C , Bazan NG , Brooks DJ , Hunot S , Joseph B , Deigendesch N , Garaschuk O , Boddeke E , Dinarello CA , Breitner JC , Cole GM , Golenbock DT , Kummer MP ((2015) ) Neuroinflammationin Alzheimer’s disease. Lancet Neurol 14: , 388–405. |
[40] | Lee KM , MacLean AG ((2015) ) New advances on glial activation in health and disease. World J Virol 4: , 42–55. |
[41] | Streit WJ , Walter SA , Pennell NA ((1999) ) Reactive microgliosis. Prog Neurobiol 57: , 563–581. |
[42] | Graeber MB ((2010) ) Changing face of microglia. Science 330: , 783–788. |
[43] | Tremblay ME , Lecours C , Samson L , Sanchez-Zafra V , Sierra A ((2015) ) From the Cajal alumni Achucarro and Rio-Hortega to the rediscovery of never-resting microglia. Front Neuroanat 9: , 45. |
[44] | Nimmerjahn A , Kirchhoff F , Helmchen F ((2005) ) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308: , 1314–1318. |
[45] | Ransohoff RM , Perry VH ((2009) ) Microglial physiology: Unique stimuli, specialized responses. Annu Rev Immunol 27: , 119–145. |
[46] | Mills CD ((2012) ) M1 and M2 macrophages: Oracles of health and disease. Crit Rev Immunol 32: , 463–488. |
[47] | Davalos D , Grutzendler J , Yang G , Kim JV , Zuo Y , Jung S , Littman DR , Dustin ML , Gan WB ((2005) ) ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8: , 752–758. |
[48] | Boche D , Perry VH , Nicoll JA ((2013) ) Review: Activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol 39: , 3–18. |
[49] | Prokop S , Miller KR , Heppner FL ((2013) ) Microglia actions in Alzheimer’s disease. Acta Neuropathol 126: , 461–477. |
[50] | Mosser DM , Edwards JP ((2008) ) Exploring the full spectrum of macrophage activation. Nat Rev Immunol 8: , 958–969. |
[51] | Town T , Nikolic V , Tan J ((2005) ) The microglial “activation” continuum: From innate to adaptive responses. J Neuroinflammation 2: , 24. |
[52] | Streit WJ , Mrak RE , Griffin WS ((2004) ) Microglia and neuroinflammation: A pathological perspective. J Neuroinflammation 1: , 14–17. |
[53] | Butterfield DA , Drake J , Pocernich C , Castegna A ((2001) ) Evidence of oxidative damage in Alzheimer’s disease brain: Central role for amyloid beta-peptide. Trends Mol Med 7: , 548–554. |
[54] | Hensley K , Carney JM , Mattson MP , Aksenova M , Harris M , Wu JF , Floyd RA , Butterfield DA ((1994) ) A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: Relevance to Alzheimer disease. Proc Natl Acad Sci U S A 91: , 3270–3274. |
[55] | Wisniewski HM , Wegiel J , Wang KC , Kujawa M , Lach B ((1989) ) Ultrastructural studies of the cells forming amyloid fibers in classical plaques. Can J Neurol Sci 16: , 535–542. |
[56] | Itagaki S , McGeer PL , Akiyama H , Zhu S , Selkoe D ((1989) ) Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol 24: , 173–182. |
[57] | Terry RD , Gonatas NK , Weiss M ((1964) ) Ultrastructural studies in Alzheimer’s presenile dementia. Am J Pathol 44: , 269–297. |
[58] | Bonaiuto C , McDonald PP , Rossi F , Cassatella MA ((1997) ) Activation of nuclear factor-kappa B by beta-amyloid peptides and interferon-gamma in murine microglia. J Neuroimmunol 77: , 51–56. |
[59] | McGeer PL , Itagaki S , Tago H , McGeer EG ((1987) ) Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett 79: , 195–200. |
[60] | Rogers J , Luber-Narod J , Styren SD , Civin WH ((1988) ) Expression of immune system-associated antigens by cells of the human central nervous system: Relationship to the pathology of Alzheimer’s disease. Neurobiol Aging 9: , 339–349. |
[61] | Combs CK , Johnson DE , Cannady SB , Lehman TM , Landreth GE ((1999) ) Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of beta-amyloid and prion proteins. J Neurosci 19: , 928–939. |
[62] | Combs CK , Johnson DE , Karlo JC , Cannady SB , Landreth GE ((2000) ) Inflammatory mechanisms in Alzheimer’s disease: Inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J Neurosci 20: , 558–567. |
[63] | Klegeris A , McGeer PL ((1997) ) beta-amyloid protein enhances macrophage production of oxygen free radicals and glutamate. J Neurosci Res 49: , 229–235. |
[64] | McDonald DR , Brunden KR , Landreth GE ((1997) ) Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. J Neurosci 17: , 2284–2294. |
[65] | Floden AM , Combs CK ((2006) ) Beta-amyloid stimulates murine postnatal and adult microglia cultures in a unique manner. J Neurosci 26: , 4644–4648. |
[66] | Floden AM , Li S , Combs CK ((2005) ) Beta-amyloid-stimulated microglia induce neuron death via synergistic stimulation of tumor necrosis factor alpha and NMDA receptors. J Neurosci 25: , 2566–2575. |
[67] | Orr AG , Orr AL , Li XJ , Gross RE , Traynelis SF ((2009) ) Adenosine A(2A) receptor mediates microglial process retraction. Nat Neurosci 12: , 872–878. |
[68] | Weggen S , Eriksen JL , Das P , Sagi SA , Wang R , Pietrzik CU , Findlay KA , Smith TE , Murphy MP , Bulter T , Kang DE , Marquez-Sterling N , Golde TE , Koo EH ((2001) ) A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature 414: , 212–216. |
[69] | Adapt Research Group, Martin BK , Szekely C , Brandt J , Piantadosi S , Breitner JC , Craft S , Evans D , Green R , Mullan M ((2008) ) Cognitive function over time in the Alzheimer’s disease anti-inflammatory prevention trial (ADAPT): Results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol 65: , 896–905. |
[70] | Dregan A , Chowienczyk P , Armstrong D ((2015) ) Patterns of anti-inflammatory drug use and risk of dementia: A matched case-control study. Eur J Neurol 22: , 1421–1428. |
[71] | Mukherjee S , Ghosh RN , Maxfield FR ((1997) ) Endocytosis. Physiol Rev 77: , 759–803. |
[72] | Sierra A , Abiega O , Shahraz A , Neumann H ((2013) ) Janus-faced microglia: Beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci 7: , 6. |
[73] | Elliott MR , Chekeni FB , Trampont PC , Lazarowski ER , Kadl A , Walk SF , Park D , Woodson RI , Ostankovich M , Sharma P , Lysiak JJ , Harden TK , Leitinger N , Ravichandran KS ((2009) ) Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461: , 282–286. |
[74] | Noda M, Doi Y, Liang J, Kawanokuchi J, Sonobe Y, Takeuchi H, Mizuno T, Suzumura A ((2011) ) Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression. J Biol Chem 286: , 2308–2319. |
[75] | Wilkinson K , El Khoury J ((2012) ) Microglial scavenger receptors and their roles in the pathogenesis of Alzheimer’s disease. Int J Alzheimers Dis 2012: . |
[76] | El Khoury J , Hickman SE , Thomas CA , Cao L , Silverstein SC , Loike JD ((1996) ) Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature 382: , 716–719. |
[77] | Alarcon R , Fuenzalida C , Santibanez M , von Bernhardi R ((2005) ) Expression of scavenger receptors in glial cells. Comparing the adhesion of astrocytes and microglia from neonatal rats to surface-bound beta-amyloid. J Biol Chem 280: , 30406–30415. |
[78] | Husemann J , Loike JD , Kodama T , Silverstein SC ((2001) ) Scavenger receptor class B type I (SR-BI) mediates adhesion of neonatal murine microglia to fibrillar beta-amyloid. J Neuroimmunol 114: , 142–150. |
[79] | El Khoury JB , Moore KJ , Means TK , Leung J , Terada K , Toft M , Freeman MW , Luster AD ((2003) ) CD36 mediates the innate host response to beta-amyloid. J Exp Med 197: , 1657–1666. |
[80] | Yan SD , Chen X , Fu J , Chen M , Zhu H , Roher A , Slattery T , Zhao L , Nagashima M , Morser J , Migheli A , Nawroth P , Stern D , Schmidt AM ((1996) ) RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 382: , 685–691. |
[81] | Tarasoff-Conway JM , Carare RO , Osorio RS , Glodzik L , Butler T , Fieremans E , Axel L , Rusinek H , Nicholson C , Zlokovic BV , Frangione B , Blennow K , Menard J , Zetterberg H , Wisniewski T , de Leon MJ ((2015) ) Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol 11: , 457–470. |
[82] | Louveau A , Smirnov I , Keyes TJ , Eccles JD , Rouhani SJ , Peske JD , Derecki NC , Castle D , Mandell JW , Lee KS , Harris TH , Kipnis J ((2015) ) Structural and functional features of central nervous system lymphatic vessels. Nature 523: , 337–341. |
[83] | Lee CY , Landreth GE ((2010) ) The role of microglia in amyloid clearance from the AD brain. J Neural Transm (Vienna) 117: , 949–960. |
[84] | Jin JJ , Kim HD , Maxwell JA , Li L , Fukuchi K ((2008) ) Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer’s disease. J Neuroinflammation 5: , 23. |
[85] | Sessa G , Podini P , Mariani M , Meroni A , Spreafico R , Sinigaglia F , Colonna M , Panina P , Meldolesi J ((2004) ) Distribution and signaling of TREM2/DAP12, the receptor system mutated in human polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy dementia. Eur J Neurosci 20: , 2617–2628. |
[86] | Lambert JC , Heath S , Even G , Campion D , Sleegers K , Hiltunen M , Combarros O , Zelenika D , Bullido MJ , Tavernier B , Letenneur L , Bettens K , Berr C , Pasquier F , Fievet N , Barberger-Gateau P , Engelborghs S , De Deyn P , Mateo I , Franck A , Helisalmi S , Porcellini E , Hanon O , European Alzheimer’s Disease Initiative Initiative, de Pancorbo MM , Lendon C , Dufouil C , Jaillard C , Leveillard T , Alvarez V , Bosco P , Mancuso M , Panza F , Nacmias B , Bossu P , Piccardi P , Annoni G , Seripa D , Galimberti D , Hannequin D , Licastro F , Soininen H , Ritchie K , Blanche H , Dartigues JF , Tzourio C , Gut I , Van Broeckhoven C , Alperovitch A , Lathrop M , Amouyel P ((2009) ) Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat Genet 41: , 1094–1099. |
[87] | Naj AC , Jun G , Beecham GW , Wang LS , Vardarajan BN , Buros J , Gallins PJ , Buxbaum JD , Jarvik GP , Crane PK , Larson EB , Bird TD , Boeve BF , Graff-Radford NR , De Jager PL , Evans D , Schneider JA , Carrasquillo MM , Ertekin-Taner N , Younkin SG , Cruchaga C , Kauwe JS , Nowotny P , Kramer P , Hardy J , Huentelman MJ , Myers AJ , Barmada MM , Demirci FY , Baldwin CT , Green RC , Rogaeva E , St George-Hyslop P , Arnold SE , Barber R , Beach T , Bigio EH , Bowen JD , Boxer A , Burke JR , Cairns NJ , Carlson CS , Carney RM , Carroll SL , Chui HC , Clark DG , Corneveaux J , Cotman CW , Cummings JL , DeCarli C , DeKosky ST , Diaz-Arrastia R , Dick M , Dickson DW , Ellis WG , Faber KM , Fallon KB , Farlow MR , Ferris S , Frosch MP , Galasko DR , Ganguli M , Gearing M , Geschwind DH , Ghetti B , Gilbert JR , Gilman S , Giordani B , Glass JD , Growdon JH , Hamilton RL , Harrell LE , Head E , Honig LS , Hulette CM , Hyman BT , Jicha GA , Jin LW , Johnson N , Karlawish J , Karydas A , Kaye JA , Kim R , Koo EH , Kowall NW , Lah JJ , Levey AI , Lieberman AP , Lopez OL , Mack WJ , Marson DC , Martiniuk F , Mash DC , Masliah E , McCormick WC , McCurry SM , McDavid AN , McKee AC , Mesulam M , Miller BL , Miller CA , Miller JW , Parisi JE , Perl DP , Peskind E , Petersen RC , Poon WW , Quinn JF , Rajbhandary RA , Raskind M , Reisberg B , Ringman JM , Roberson ED , Rosenberg RN , Sano M , Schneider LS , Seeley W , Shelanski ML , Slifer MA , Smith CD , Sonnen JA , Spina S , Stern RA , Tanzi RE , Trojanowski JQ , Troncoso JC , Van Deerlin VM , Vinters HV , Vonsattel JP , Weintraub S , Welsh-Bohmer KA , Williamson J , Woltjer RL , Cantwell LB , Dombroski BA , Beekly D , Lunetta KL , Martin ER , Kamboh MI , Saykin AJ , Reiman EM , Bennett DA , Morris JC , Montine TJ , Goate AM , Blacker D , Tsuang DW , Hakonarson H , Kukull WA , Foroud TM , Haines JL , Mayeux R , Pericak-Vance MA , Farrer LA , Schellenberg GD ((2011) ) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet 43: , 436–441. |
[88] | Steinberg S , Stefansson H , Jonsson T , Johannsdottir H , Ingason A , Helgason H , Sulem P , Magnusson OT , Gudjonsson SA , Unnsteinsdottir U , Kong A , Helisalmi S , Soininen H , Lah JJ , DemGene , Aarsland D , Fladby T , Ulstein ID , Djurovic S , Sando SB , White LR , Knudsen GP , Westlye LT , Selbaek G , Giegling I , Hampel H , Hiltunen M , Levey AI , Andreassen OA , Rujescu D , Jonsson PV , Bjornsson S , Snaedal J , Stefansson K ((2015) ) Loss-of-function variants in ABCA7 confer risk of Alzheimer’s disease. Nat Genet 47: , 445–447. |
[89] | Ramanan VK , Risacher SL , Nho K , Kim S , Shen L , McDonald BC , Yoder KK , Hutchins GD , West JD , Tallman EF , Gao S , Foroud TM , Farlow MR , De Jager PL , Bennett DA , Aisen PS , Petersen RC , Jack CR Jr , Toga AW , Green RC , Jagust WJ , Weiner MW , Saykin AJ , Alzheimer’s Disease Neuroimaging Initiative ((2015) ) GWAS of longitudinal amyloid accumulation on 18F-florbetapir PET in Alzheimer’s disease implicates microglial activation gene IL1RAP. Brain 138: , 3076–3088. |
[90] | Kim WS , Li H , Ruberu K , Chan S , Elliott DA , Low JK , Cheng D , Karl T , Garner B ((2013) ) Deletion of Abca7 increases cerebral amyloid-beta accumulation in the J20 mouse model of Alzheimer’s disease. J Neurosci 33: , 4387–4394. |
[91] | Stephan AH , Barres BA , Stevens B ((2012) ) The complement system: An unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci 35: , 369–389. |
[92] | Paolicelli RC , Bolasco G , Pagani F , Maggi L , Scianni M , Panzanelli P , Giustetto M , Ferreira TA , Guiducci E , Dumas L , Ragozzino D , Gross CT ((2011) ) Synaptic pruning by microglia is necessary for normal brain development. Science 333: , 1456–1458. |
[93] | Tremblay ME , Lowery RL , Majewska AK ((2010) ) Microglial interactions with synapses are modulated by visual experience. PLoS Biol 8: , e1000527. |
[94] | Wake H , Moorhouse AJ , Jinno S , Kohsaka S , Nabekura J ((2009) ) Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 29: , 3974–3980. |
[95] | Chen Z , Jalabi W , Hu W , Park HJ , Gale JT , Kidd GJ , Bernatowicz R , Gossman ZC , Chen JT , Dutta R , Trapp BD ((2014) ) Microglial displacement of inhibitory synapses provides neuroprotection in the adult brain. Nat Commun 5: , 4486. |
[96] | Ji K , Akgul G , Wollmuth LP , Tsirka SE ((2013) ) Microglia actively regulate the number of functional synapses. PLoS One 8: , e56293 |
[97] | Araque A , Parpura V , Sanzgiri RP , Haydon PG ((1999) ) Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci 22: , 208–215. |
[98] | Schafer DP , Lehrman EK , Stevens B ((2013) ) The “quad-partite” synapse: Microglia-synapse interactions in the developing and mature CNS. Glia 61: , 24–36. |
[99] | Stevens B , Allen NJ , Vazquez LE , Howell GR , Christopherson KS , Nouri N , Micheva KD , Mehalow AK , Huberman AD , Stafford B , Sher A , Litke AM , Lambris JD , Smith SJ , John SW , Barres BA ((2007) ) The classical complement cascade mediates CNS synapse elimination. Cell 131: , 1164–1178. |
[100] | Bialas AR , Stevens B ((2013) ) TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci 16: , 1773–1782. |
[101] | Schafer DP , Lehrman EK , Kautzman AG , Koyama R , Mardinly AR , Yamasaki R , Ransohoff RM , Greenberg ME , Barres BA , Stevens B ((2012) ) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74: , 691–705. |
[102] | Perez-Alcazar M , Daborg J , Stokowska A , Wasling P , Bjorefeldt A , Kalm M , Zetterberg H , Carlstrom KE , Blomgren K , Ekdahl CT , Hanse E , Pekna M ((2014) ) Altered cognitive performance and synaptic function in the hippocampus of mice lacking C3. Exp Neurol 253: , 154–164. |
[103] | Chu Y , Jin X , Parada I , Pesic A , Stevens B , Barres B , Prince DA ((2010) ) Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci U S A 107: , 7975–7980. |
[104] | Shi Q , Colodner KJ , Matousek SB , Merry K , Hong S , Kenison JE , Frost JL , Le KX , Li S , Dodart JC , Caldarone BJ , Stevens B , Lemere CA ((2015) ) Complement C3-deficient mice fail to display age-related hippocampal decline. J Neurosci 35: , 13029–13042. |
[105] | Zhang J , Malik A , Choi HB , Ko RW , Dissing-Olesen L , MacVicar BA ((2014) ) Microglial CR3 activation triggers long-term synaptic depression in the hippocampus via NADPH oxidase. Neuron 82: , 195–207. |
[106] | Clark AK , Gruber-Schoffnegger D , Drdla-Schutting R , Gerhold KJ , Malcangio M , Sandkuhler J ((2015) ) Selective activation of microglia facilitates synaptic strength. J Neurosci 35: , 4552–4570. |
[107] | Rogers JT , Morganti JM , Bachstetter AD , Hudson CE , Peters MM , Grimmig BA , Weeber EJ , Bickford PC , Gemma C ((2011) ) CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J Neurosci 31: , 16241–16250. |
[108] | Veerhuis R ((2011) ) Histological and direct evidence for the role of complement in the neuroinflammation of AD. Curr Alzheimer Res 8: , 34–58. |
[109] | Villegas-Llerena C , Phillips A , Reitboeck PG , Hardy J , Pocock JM ((2015) ) Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr Opin Neurobiol 36: , 74–81. |
[110] | Maier M , Peng Y , Jiang L , Seabrook TJ , Carroll MC , Lemere CA ((2008) ) Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci 28: , 6333–6341. |
[111] | Finehout EJ , Franck Z , Lee KH ((2005) ) Complement protein isoforms in CSF as possible biomarkers for neurodegenerative disease. Dis Markers 21: , 93–101. |
[112] | Aronica E , Boer K , van Vliet EA , Redeker S , Baayen JC , Spliet WG , van Rijen PC , Troost D , da Silva FH , Wadman WJ , Gorter JA ((2007) ) Complement activation in experimental and human temporal lobe epilepsy. Neurobiol Dis 26: , 497–511. |
[113] | Bochner DN , Sapp RW , Adelson JD , Zhang S , Lee H , Djurisic M , Syken J , Dan Y , Shatz CJ ((2014) ) Blocking PirB up-regulates spines and functional synapses to unlock visual cortical plasticity and facilitate recovery from amblyopia. Sci Transl Med 6: , 258ra140. |
[114] | Colonna M , Samaridis J , Cella M , Angman L , Allen RL , O’Callaghan CA , Dunbar R , Ogg GS , Cerundolo V , Rolink A ((1998) ) Human myelomonocytic cells express an inhibitory receptor for classical and nonclassical MHC class I molecules. J Immunol 160: , 3096–3100. |
[115] | Kim T , Vidal GS , Djurisic M , William CM , Birnbaum ME , Garcia KC , Hyman BT , Shatz CJ ((2013) ) Human LilrB2 is a beta-amyloid receptor and its murine homolog PirB regulates synaptic plasticity in an Alzheimer’s model. Science 341: , 1399–1404. |
[116] | Parkhurst CN , Yang G , Ninan I , Savas JN , Yates JR , Lafaille JJ , Hempstead BL , Littman DR , Gan WB ((2013) ) Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155: , 1596–1609. |
[117] | Hayashi Y , Koyanagi S , Kusunose N , Okada R , Wu Z , Tozaki-Saitoh H , Ukai K , Kohsaka S , Inoue K , Ohdo S , Nakanishi H ((2013) ) The intrinsic microglial molecular clock controls synaptic strength via the circadian expression of cathepsin S. Sci Rep 3: , 2744. |
[118] | Zhan Y , Paolicelli RC , Sforazzini F , Weinhard L , Bolasco G , Pagani F , Vyssotski AL , Bifone A , Gozzi A , Ragozzino D , Gross CT ((2014) ) Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci 17: , 400–406. |
[119] | Chugh D , Nilsson P , Afjei SA , Bakochi A , Ekdahl CT ((2013) ) Brain inflammation induces post-synaptic changes during early synapse formation in adult-born hippocampal neurons. Exp Neurol 250: , 176–188. |
[120] | Gomes C , Ferreira R , George J , Sanches R , Rodrigues DI , Goncalves N , Cunha RA ((2013) ) Activation of microglial cells triggers a release of brain-derived neurotrophic factor (BDNF) inducing their proliferation in an adenosine A2A receptor-dependent manner: A2A receptor blockade prevents BDNF release and proliferation of microglia. J Neuroinflammation 10: , 16. |
[121] | Greicius MD , Krasnow B , Reiss AL , Menon V ((2003) ) Functional connectivity in the resting brain: A network analysis of the default mode hypothesis. Proc Natl Acad Sci U S A 100: , 253–258. |
[122] | Huijbers W , Pennartz CM , Cabeza R , Daselaar SM ((2011) ) The hippocampus is coupled with the default network during memory retrieval but not during memory encoding. PLoS One 6: , e17463. |
[123] | Prescott JW , Guidon A , Doraiswamy PM , Roy Choudhury K , Liu C , Petrella JR , Alzheimer’s Disease Neuroimaging Initiative ((2014) ) The Alzheimer structural connectome: Changes in cortical network topology with increased amyloid plaque burden. Radiology 273: , 175–184. |
[124] | Benzinger TL , Blazey T , Jack CR Jr , Koeppe RA , Su Y , Xiong C , Raichle ME , Snyder AZ , Ances BM , Bateman RJ , Cairns NJ , Fagan AM , Goate A , Marcus DS , Aisen PS , Christensen JJ , Ercole L , Hornbeck RC , Farrar AM , Aldea P , Jasielec MS , Owen CJ , Xie X , Mayeux R , Brickman A , McDade E , Klunk W , Mathis CA , Ringman J , Thompson PM , Ghetti B , Saykin AJ , Sperling RA , Johnson KA , Salloway S , Correia S , Schofield PR , Masters CL , Rowe C , Villemagne VL , Martins R , Ourselin S , Rossor MN , Fox NC , Cash DM , Weiner MW , Holtzman DM , Buckles VD , Moulder K , Morris JC ((2013) ) Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. Proc Natl Acad Sci U S A 110: , E4502–E4509. |
[125] | Ryan NS , Fox NC ((2009) ) Imaging biomarkers in Alzheimer’s disease. Ann N Y Acad Sci 1180: , 20–27. |
[126] | Brier MR , Thomas JB , Snyder AZ , Benzinger TL , Zhang D , Raichle ME , Holtzman DM , Morris JC , Ances BM ((2012) ) Loss of intranetwork and internetwork resting state functional connections with Alzheimer’s disease progression. J Neurosci 32: , 8890–8899. |
[127] | Edison P , Archer HA , Gerhard A , Hinz R , Pavese N , Turkheimer FE , Hammers A , Tai YF , Fox N , Kennedy A , Rossor M , Brooks DJ ((2008) ) Microglia, amyloid, and cognition in Alzheimer’s disease: An [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis 32: , 412–419. |
[128] | Fan Z , Okello AA , Brooks DJ , Edison P ((2015) ) Longitudinal influence of microglial activation and amyloid on neuronal function in Alzheimer’s disease. Brain 138: (Pt 12) 3685–3698. |
[129] | Shankar GM , Walsh DM ((2009) ) Alzheimer’s disease: Synaptic dysfunction and Abeta. Mol Neurodegener 4: , 48. |
[130] | Ferreira ST , Lourenco MV , Oliveira MM , De Felice FG ((2015) ) Soluble amyloid-beta oligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front Cell Neurosci 9: , 191. |
[131] | Shankar GM , Bloodgood BL , Townsend M , Walsh DM , Selkoe DJ , Sabatini BL ((2007) ) Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci 27: , 2866–2875. |
[132] | Chang EH , Savage MJ , Flood DG , Thomas JM , Levy RB , Mahadomrongkul V , Shirao T , Aoki C , Huerta PT ((2006) ) AMPA receptor downscaling at the onset of Alzheimer’s disease pathology in double knockin mice. Proc Natl Acad Sci U S A 103: , 3410–3415. |
[133] | Hsieh H , Boehm J , Sato C , Iwatsubo T , Tomita T , Sisodia S , Malinow R ((2006) ) AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron 52: , 831–843. |
[134] | Ulrich D ((2015) ) Amyloid-beta impairs synaptic inhibition via GABA(A) receptor endocytosis. J Neurosci 35: , 9205–9210. |
[135] | Verkhratsky A , Sofroniew MV , Messing A , deLanerolle NC , Rempe D , Rodriguez JJ , Nedergaard M ((2012) ) Neurological diseases as primary gliopathies: A reassessment of neurocentrism. ASN Neuro 4: , e00082. |
[136] | Myers N , Pasquini L , Gottler J , Grimmer T , Koch K , Ortner M , Neitzel J , Muhlau M , Forster S , Kurz A , Forstl H , Zimmer C , Wohlschlager AM , Riedl V , Drzezga A , Sorg C ((2014) ) Within-patient correspondence of amyloid-beta and intrinsic network connectivity in Alzheimer’s disease. Brain 137: , 2052–2064. |
[137] | Pihlajamaki M , Sperling RA ((2009) ) Functional MRI assessment of task-induced deactivation of the default mode network in Alzheimer’s disease and at-risk older individuals. Behav Neurol 21: , 77–91. |
[138] | Sperling RA , LaViolette PS , O’Keefe K , O’Brien J , Rentz DM , Pihlajamaki M , Marshall G , Hyman BT , Selkoe DJ , Hedden T , Buckner RL , Becker JA , Johnson KA ((2009) ) Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron 63: , 178–188. |
[139] | Lustig C , Snyder AZ , Bhakta M , O’Brien KC , McAvoy M , Raichle ME , Morris JC , Buckner RL ((2003) ) Functional deactivations: Change with age and dementia of the Alzheimer type. Proc Natl Acad Sci U S A 100: , 14504–14509. |
[140] | Petrella JR , Prince SE , Wang L , Hellegers C , Doraiswamy PM ((2007) ) Prognostic value of posteromedial cortex deactivation in mild cognitive impairment. PLoS One 2: , e1104. |
[141] | Daselaar SM , Prince SE , Cabeza R ((2004) ) When less means more: Deactivations during encoding that predict subsequent memory. Neuroimage 23: , 921–927. |
[142] | Miller SL , Celone K , DePeau K , Diamond E , Dickerson BC , Rentz D , Pihlajamaki M , Sperling RA ((2008) ) Age-related memory impairment associated with loss of parietal deactivation but preserved hippocampal activation. Proc Natl Acad Sci U S A 105: , 2181–2186. |
[143] | Celone KA , Calhoun VD , Dickerson BC , Atri A , Chua EF , Miller SL , DePeau K , Rentz DM , Selkoe DJ , Blacker D , Albert MS , Sperling RA ((2006) ) Alterations in memory networks in mild cognitive impairment and Alzheimer’s disease: An independent component analysis. J Neurosci 26: , 10222–10231. |
[144] | Elman JA , Oh H , Madison CM , Baker SL , Vogel JW , Marks SM , Crowley S , O’Neil JP , Jagust WJ ((2014) ) Neural compensation in older people with brain amyloid-beta deposition. Nat Neurosci 17: , 1316–1318. |
[145] | He Y , Wang L , Zang Y , Tian L , Zhang X , Li K , Jiang T ((2007) ) Regional coherence changes in the early stages of Alzheimer’s disease: A combined structural and resting-state functional MRI study. Neuroimage 35: , 488–500. |
[146] | Palop JJ , Mucke L ((2009) ) Epilepsy and cognitive impairments in Alzheimer disease. Arch Neurol 66: , 435–440. |
[147] | Davis KE , Fox S , Gigg J ((2014) ) Increased hippocampal excitability in the 3xTgAD mouse model for Alzheimer’s disease in vivo. PLoS One 9: , e91203. |
[148] | Innes KE , Selfe TK ((2014) ) Meditation as a therapeutic intervention for adults at risk for Alzheimer’s disease - potential benefits and underlying mechanisms. Front Psychiatry 5: , 40. |
Figures and Tables
Fig.1
Increased microglial reactivity or increased protein clearance may reduce microglial synaptic maintenance. (1) Microglia monitor and prune CNS synapses throughout life in conjunction with peri-synaptic astrocytes. Microglia also phagocytose small amounts of extracellular protein such as Aβ, although it is not yet known whether microglia perform both phagocytic tasks simultaneously. Dysfunctional microglial activity at the synapse may produce one of the two following scenarios. (2) Increased pro-inflammatory activation of microglia results in increased production of inflammatory cytokines, co-activating peri-synaptic astrocytes, resulting in neuronal excitability and degeneration. (3) Alternatively, increased demands on microglial phagocytosis, such as elevated Aβ production, may reduce synaptic maintenance and result in increased Aβ at the synapse.
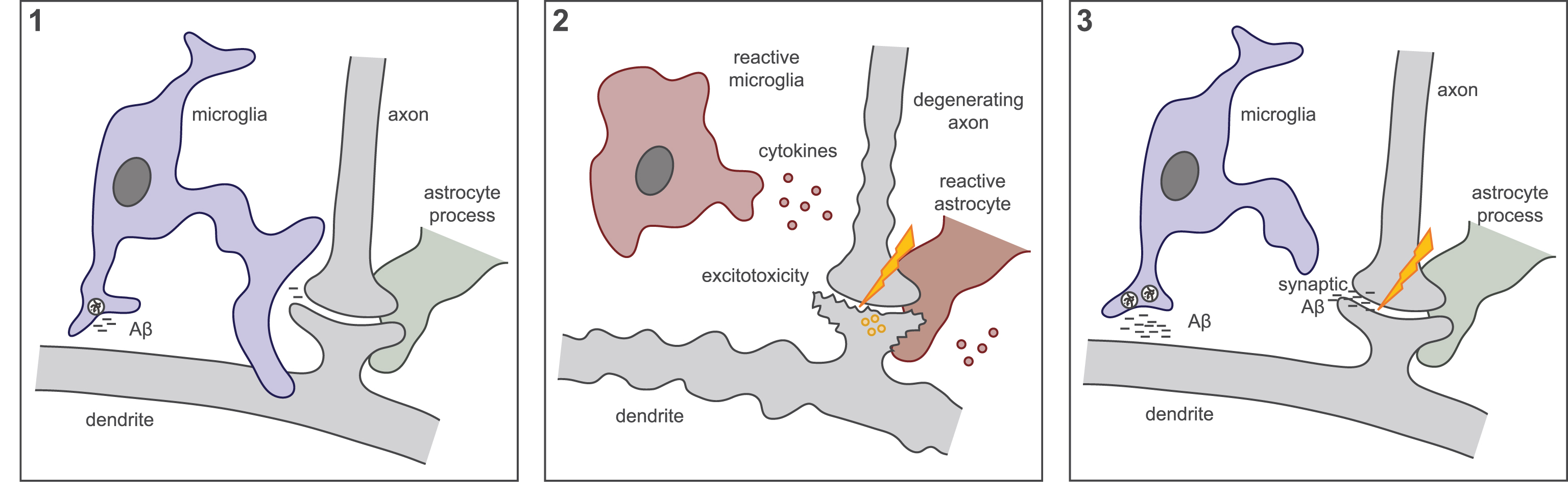
Fig.2
A model for microglial dysfunction that occurs downstream of Aβ, resulting in network dysfunction progressing to AD.