
Unraveling Alzheimer’s: Making Sense of the Relationship between Diabetes and Alzheimer’s Disease1
Abstract
Numerous studies have documented a strong association between diabetes and Alzheimer’s disease (AD). The nature of the relationship, however, has remained a puzzle, in part because of seemingly incongruent findings. For example, some studies have concluded that insulin deficiency is primarily at fault, suggesting that intranasal insulin or inhibiting the insulin-degrading enzyme (IDE) could be beneficial. Other research has concluded that hyperinsulinemia is to blame, which implies that intranasal insulin or the inhibition of IDE would exacerbate the disease. Such antithetical conclusions pose a serious obstacle to making progress on treatments. However, careful integration of multiple strands of research, with attention to the methods used in different studies, makes it possible to disentangle the research on AD. This integration suggests that there is an important relationship between insulin, IDE, and AD that yields multiple pathways to AD depending on the where deficiency or excess in the cycle occurs. I review evidence for each of these pathways here. The results suggest that avoiding excess insulin, and supporting robust IDE levels, could be important ways of preventing and lessening the impact of AD. I also describe what further tests need to be conducted to verify the arguments made in the paper, and their implications for treating AD.
Alzheimer’s disease (AD) is a devastating, fatal disease that affects an estimated 5.2 million Americans, and 44 million people worldwide. It is a disease that is often harder on the families and caregivers of the patient than on the patient themselves. The direct costs to the US of AD in 2014 were estimated to total $214 billion [1]. Adding in the informal costs (the costs of family members and friends providing unpaid care to those with dementia) doubles those figures. The bulk of the financial burden of AD is not due to the cost of drugs or doctor’s visits; instead, the vast majority (75–84%) of the expenses go to nursing home care, plus formal and informal home care [2]. The drugs currently available for AD (primarily cholinesterase inhibitors and memantine) offer only incremental improvements in symptoms— they do not stop the progression of the disease [3–6]. Analysts estimate that AD will cost $1.5 trillion per year US by 2050 if a more effective treatment is not developed [7].
AD has been a difficult puzzle to solve, in part because of an abundance of seemingly incongruent findings. For example, though many studies have identified a significant association between type 2 diabetes mellitus (T2DM) and AD (see the review provided in Table 1), these studies have often come to different conclusions about the causal mechanism and its implications for treatment. Some studies, for example, have concluded that insulin deficiency is primarily at fault, suggesting that intranasal insulin or inhibiting the insulin-degrading enzyme (IDE) could be beneficial [8–11]. Other research has concluded that hyperinsulinemia is to blame, which implies that intranasal insulin or the inhibition of IDE could exacerbate the disease [12–16]. Similarly, recent studies have found a strong association between AD and amylin, a peptide hormone co-secreted with insulin [17–20]. Jackson et al. and Srodulski et al. found that amylin, like the amyloid-β (Aβ) protein thought to be central to the pathology of AD, forms amyloid plaques in the brain, leading them to conclude that amylin might be another amyloid with a causal role in dementia. Oskarsson et al. similarly found that amylin and Aβ were colocalized in plaques in the brain, leading them to draw similar conclusions. However, two other recent studies came to an opposite conclusion: they obtained results suggesting that amylin provides a neuroprotective effect that reduces the symptoms of AD [21, 22].
Despite these seemingly incongruent results, careful integration of multiple strands of research (with attention to the methods used in different studies) makes it possible to disentangle the research on AD. Such an integration points to the likelihood of a relationship between insulin, IDE, and AD that yields multiple pathways to the disease depending on the where deficiency or excess in the cycle occurs.
I posit that the weight of the evidence suggests that under normal circumstances, insulin upregulates the expression of IDE, which subsequently breaks insulin down [23, 24]. IDE, however, also plays important roles in breaking down Aβ and other amyloidogenic peptides. This system can malfunction and lead to AD in at least four different ways as outlined below (see Fig. 1) Evidence for each of these will be reviewed in turn in the sections that follow:
1) An individual with severe insulin deficiency (e.g., undertreated type 1 diabetes mellitus, T1DM) may have inadequate IDE activity, resulting in the accumulation of Aβ (and other protein aggregates) in the brain [23, 25, 26].
2) An individual with diminished IDE (or similar protease) production may end up with accumulation of Aβ and amylin plaques in the brain [27–32].
3) If an individual has exceptionally high levels of insulin (and amylin) in the body (e.g., due to early stages of T2DM), the insulin and amylin may competitively inhibit the breakdown of Aβ, resulting in AD [30, 33–35].
4) An individual may produce more than a typical level of an amyloidogenic peptide, or experience synergistic interactions between amyloidogenic peptides, leading to deposition that outpaces clearing by IDE and similar proteases [17–19, 36–42].
INSULIN, INSULIN DEGRADING ENZYME, AND ALZHEIMER’S DISEASE
The potential roles of insulin and insulin degrading enzyme in AD and other dementias came under scrutiny because of the strong association between T2DM and AD. I thus begin here by briefly reviewing the research on the relationship between T2DM and AD, and I will then review the evidence (summaries are provided in Table 1) for the pathway proposed in Fig. 1 and its possible malfunctions.
Type 2 diabetes and Alzheimer’s disease
There is an abundance of studies suggesting that T2DM has a positive association with AD. Some of the most well known research in this area was based on a large population study in Rotterdam [43, 44]. The longitudinal results from this study suggested that diabetes increased the risk of dementia by 1.9 fold, and that diabetic patients treated with insulin were at even greater risk (4.3 fold). Another longitudinal cohort study from the same time frame found that T2DM was associated with an increased risk for all dementias (1.66), and AD in particular (2.27 fold for men, 1.37 fold for women) [45]. Several other longitudinal cohort studies followed, with nearly all concluding that T2DM increased the risk of AD between 1.3 fold and 1.8 fold [46–49].
When cognitive testing is used to assess AD, there is a risk that other types of dementias (notably vascular dementia) will be labeled AD. A diagnosis of AD may often be the result of multiple pathologies [50]. Using autopsies, Schneider et al., for example, found that of 179 probable AD cases, almost half exhibited mixed pathologies— most commonly vascular dementia as a result of cerebral infarctions or neocortical Lewy bodies [51]. Notably, many studies have also implicated T2DM in an increased risk of vascular dementia [46, 52, 53], and, as I will explain later, we have reason to suspect that T2DM could also increase the risk of Lewy bodies.
As noted previously, there is considerable disagreement about the mechanism by which T2DM increases the risk of AD. One line of research posits that it is the excess of insulin or glucose from T2DM that leads to AD. Several studies have found that people with AD have significantly higher levels of insulin and glucose than healthy controls [13, 15, 54]. In a large sample longitudinal study of elderly subjects, Luchsinger et al. conclude that 39% of AD in their sample was attributable to hyperinsulinemia or diabetes [55]. This is also consistent with earlier findings of Ott et al. that diabetics that were treated with insulin had far greater risk of AD [43, 44].
Another line of research, however, suggests that it is insulin deficiency (either by the relative deficiency that results from insulin resistance in early stages of T2DM, or absolute deficiency that occurs when beta cell dysfunction occurs in full-blown T2DM) that causes AD by impairing insulin’s ability to perform its roles in the brain [8, 56–61]. Insulin and its signaling pathways have been found to affect learning and memory [62, 63]. For example, some research has found that administering insulin growth factors in rats and mice enhances memory retention [63, 64], and depleting it impairs learning [65]. However, the mechanisms of insulin’s roles in the brain remain poorly understood [66]. The potential roles of insulin deficiency in AD has led some researchers to conclude that administration of intranasal insulin, or insulin sensitizers, would attenuate neurological deficits. For example, in several studies, Reger et al. found that moderate doses of intranasal insulin improved some memory symptoms in older adults with AD or mild cognitive impairment [57–59]. However, it is important to note first that patients with diabetes were excluded from the study, and second that the results were nonmonotonic in the dosages of insulin. That is, at higher levels, insulin administration impaired performance.
If insulin deficiency is one of the main culprits in AD, then we would expect a very strong relationship between undertreated T1DM (whereby an individual does not produce insulin or amylin at all) and AD. However, T1DM is much rarer than T2DM, and in the past it often resulted in much earlier mortality. Furthermore, survival of T1DM requires individuals to be treated with insulin, confounding our ability to analyze its relationship with AD. In a few studies, T1DM was induced in mice via administration of streptozotocin, and these mice exhibited AD (or AD-like) pathology [11, 24, 67, 68]1. For example, in the study by Wang et al., inducing T1DM in mice via administration of streptozotocin resulted in increased Aβ generation, neuritic plaque formation, and spatial memory deficits in mice engineered to exhibit AD [11]. Similarly, a study by Clodfelder-Miller et al. found that within three days of insulin depletion by streptozotocin, mice exhibited a five-fold increase in tau phosphorylation [68]. There is also some evidence to suggest that having a severe insulin deficiency, as in T1DM, could result in inadequate upregulation of proteases that break down Aβ [71]. This leads, then, to the first malfunction pathway proposed above.
MALFUNCTION 1. SEVERE INSULIN DEFICIENCY MAY LEAD TO INADEQUATE PROTEASE PRODUCTION OR ACTIVITY
There is a paucity of information about what stimulates IDE and other amyloid proteases, however, the studies that do exist suggest that insulin is a primary regulator of IDE [23, 24, 26]. For example, in one study, Zhao et al. conclude that IDE upregulation requires insulin-mediated Akt (also known as protein kinase B) activation [23], and mice induced to have T1DM (and thus do not produce insulin) expressed significantly less IDE [24]. Rezende et al. also found that when insulin secretion levels were dramatically reduced in mice through protein secretion, there was a concomitant dramatic decrease in IDE expression in the liver [72]. This suggests that a severe insulin deficiency could possibly be accompanied by an IDE deficiency that could lead to or exacerbate AD. However, because severe insulin deficiency is typically fatal if untreated, this malfunction pathway is probably the least likely to be observed in situ. Furthermore, there are other proteases known to act upon Aβ (e.g., cathepsin D, endothelin-converting enzyme, neprilysin [73–77]) that may not be as reliant upon insulin, and we know relatively little about how these proteases are stimulated, which ones act on Aβ in vivo, and how they may compensate for each other. This area thus requires much further research.
MALFUNCTION 2. DIMINISHED PROTEASE PRODUCTION OR ACTIVITY RESULTS IN AMYLOID ACCUMULATION
IDE is one of the most widely studied proteases in studies of the relationship between insulin and Aβ. It is a ubiquitously expressed zinc metalloprotease that breaks down or irreversibly binds insulin [27, 33, 36, 78, 79], amylin [36], Aβ [27–31, 33, 80–86], and other amyloidogenic peptides [34, 36], including alpha synuclein [87, 88], which is implicated in Parkinson’s disease and Lewy body dementia. An individual with diminished IDE production (for example, due to the excess of an IDE inhibitor such as nitric oxide [89], or an inadequacy of zinc) might thus end up with accumulation of Aβ, amylin plaques, and alpha synuclein in the brain.
There are numerous studies showing that diminished IDE can result in accumulation of Aβ and memory deficits [27, 90, 91]. For example, Farris et al. found that IDE knockout mice had a greater than 50% decrease in Aβ degradation in both brain membrane fractions and primary neuronal cultures. [27] The mice exhibited an increase in cerebral accumulation of endogenous Aβ (consistent with AD), hyperinsulinemia, and glucose intolerance. Madani et al. found that neprilysin knockout mice had increased Aβ deposition [91], and Miller et al. found that IDE knockout mice had significantly increased levels of Aβ in the brain [92]. Vingtdeux et al. found that CALHM1 potentiates IDE activity, and CALHM1 knockout mice exhibited roughly a 50% increase in endogenous Aβ concentrations in the whole brain and primary neurons [85].
There is also research suggesting that deficiency in IDE might lead to an accumulation of insulin in the brain (hyperinsulinemia), leading to insulin resistance and glucose intolerance [27, 32]. Though the possibility of IDE regulating insulin in the brain remains to be directly tested, this possibility suggests that one of the reasons that AD and T2DM are closely associated might be because they can be caused by the same mechanism.
Diminished IDE production may also account for at least part of the association between the ApoE4 allele and AD. ApoE4 is a well-established risk factor for AD; most estimates suggest that roughly 20–25% of the population has the ApoE4 allele, yet roughly 50% of the AD population has the ApoE4 allele [21, 93, 94]. Furthermore, in their longitudinal study of 2,574 Japanese-American men (and 216 autopsies), Peila et al. found that the combination of diabetes and having one or more copies of the ApoE4 allele resulted in a dramatically higher risk of AD— 5.5 fold— than either risk alone [46]. Having a single copy of the allele is thought to triple the risk for late-onset AD; having two copies of the allele is thought to increase the risk of AD 12 fold [94]. Importantly, Cook et al. found that AD patients with the ApoE4 allele had a 50% reduction in expression of IDE compared to AD patients without the ApoE4 allele, suggesting that the ApoE4 allele may play a role in IDE deficiency leading to AD [95]. The mechanisms that underlie ApoE4’s relationship to AD remain to be more fully explored, however.
Qiu et al.’s study, and a review by Qiu and Folstein, suggest that IDE is likely to be an important protease involved in Aβ degradation [30, 35]. Furthermore, since the IDE gene plays a significant role also in diabetes, this would help to explain the strong relationship between the two pathologies. However, it is important to note that are other proteases that appear to play roles in breaking down or binding Aβ such as neprilysin, endothelin-converting enzyme, cathepsin B, or cathepsin D [73, 83, 96]. The role of these proteases in Aβ degradation and AD pathology have thus far received far less attention and warrant further study.
MALFUNCTION 3. EXCESS INSULIN OR AMYLIN COMPETITIVELY INHIBITS BREAKDOWN OF Aβ
Though IDE can break down a range of amyloidogenic peptides [34], it has a preferential affinity for insulin [33, 36]. This suggests the possibility that, consistent with Qiu et al.’s hypothesis, one of the causal mechanisms leading to sporadic AD may be the competitive inhibition of Aβ by insulin or amylin [30]. Numerous studies have shown that insulin completely competitively inhibits the breakdown of both Aβ [30, 33, 35] and amylin [36]. Furthermore, though insulin upregulates the expression of IDE, it has been shown that its ability to increase IDE diminishes at high levels [23]. This suggests that in the hyperinsulinemia of early T2DM (or obesity) [97, 98], insulin levels could outstrip IDE’s ability to break it down. If IDE preferentially targets insulin and insulin levels are outpacing IDE production, IDE’s ability to breakdown other amyloidogenic proteins will be markedly reduced. As eloquently stated in Bennett et al., “Normally, a balance exists between deposition and degradation of the amyloidogenic peptide. When the levels of the peptide exceed the capacity to degrade them, either by increased expression of the peptide, or decreased expression of the enzymatic activity of IDE, the balance is shifted from degradation to deposition.” [36] The chronic hyperinsulinemia of early T2DM and its potential to competitively inhibit the breakdown of Aβ thus likely explains a large portion of the association between T2DM and AD.
This competitive inhibition of degradation among different IDE substrates brings us to the recent amylin studies. In 2013, Jackson et al. found patients with AD had large amylin plaque deposits and mixed amylin and Aβ deposits in their brains [17]. They concluded that amylin may play a causal role in AD. Then in 2014, Srodulski et al. found that rats that were genetically modified to express human amylin accumulated amylin oligomers in their brains, and exhibited neurological deficits [18]. Oskarsson et al. also found that amylin and Aβ were colocalized in the brains of human AD patients [19]. If amylin, which is co-secreted with insulin, competes with Aβ for degradation, promotes the deposition of Aβ, or accumulates in plaques on the brain itself, this is another strong mechanism by which T2DM and dementia may be linked.
Remarkably, another set of recent studies appeared to directly counter this conclusion by finding that amylin exhibited a neuroprotective effect [21, 99, 100]. These studies found that injection of pramlintide (an amylin analog), or rat/mouse amylin, appeared to improve memory symptoms in mice engineered to exhibit AD or accelerated senescence. Furthermore, administration of pramlintide to humans appeared to improve some learning and memory measures. However, two important features of these recent studies must be noted. First, the studies used rat/mouse amylin, or pramlintide (synthetic amylin), both of which have proline substitutions at positions 25, 28, and 29 that limit amylin’s self-aggregating properties, thus rendering it non-amyloidogenic. Second, in all the studies except for Qiu et al.’s study of homebound elderly [22], the subjects were diabetes free, and thus likely had no deficiency in their IDE system or excess of competing insulin. In the Qiu et al. study of homebound elderly, the researchers found some differences in memory scores across quartiles of amylin levels across all subjects (including those with diabetes), however these differences were not monotonically increasing. Furthermore, the significant differences they report in amylin levels across AD patients, mild cognitive impairment patients, and healthy persons were only for patients without diabetes.
The preceding suggests that pramlintide might have beneficial effects for people without diabetes. This could be due to its role in preventing post-prandial glucose spikes or by inhibiting glucagon, both of which would collectively lessen insulin demand. These mechanisms of potential benefit, however, are speculative; the mechanism of pramlintide’s potential neuroprotective effect is unclear. For people with diabetes, however, pramlintide seems far less likely to be beneficial in AD. In people with diabetes, insulin and amylin levels are already high, and may have already exhausted their capacity for stimulating IDE production and activity.
MALFUNCTION 4. EXCESS PRODUCTION OF AN AMYLOIDOGENIC PROTEIN, OR POSITIVE INTERACTIONS BETWEEN AMYLOIDOGENIC PROTEINS, RESULT IN AMYLOID ACCUMULATION
Excess production of an amyloidogenic protein can result in amyloid deposition outpacing amyloid degradation, resulting in amyloid accumulation [36]. For example, mutations in the amyloid precursor protein gene (the gene associated with early-onset AD) can increase the relative production of Aβ42, leading to amyloid deposition [37, 38]. Similarly, excess production of amylin (as in T2DM) may directly cause a build-up of amylin plaques on the brain [17]. There is also growing evidence that some amyloidogenic proteins can foster the formation of amyloids of other proteins, in a process known as “cross-seeding” [39–41]. For example, alpha synuclein can initiate tau amyloid formation, and then the two peptides can increase the polymerization of each other [42]. Similarly, amylin may interact with Aβ to increase its formation of plaques [17–19].
IMPLICATIONS
The implications of the preceding are very important, because the mechanism by which AD arises strongly determines what treatments are likely to be effective. If insulin deficiency is the main culprit, for example, then intranasal insulin might be of significant benefit. If, however, hyperinsulinemia and/or competitive inhibition of degradation of Aβ by a degrading protease is the main cause, then administering intranasal insulin could exacerbate the disease. Furthermore, there is evidence that hyperinsulinemia precedes the hyperglycemia of T2DM by many years and it is likely the chronic overstimulation of insulin receptors that leads to insulin resistance [35, 97, 101]. This suggests that extreme caution should be exercised in giving a diabetic or pre-diabetic individual extra insulin— it may just add fuel to the fire.
The evidence overwhelmingly suggests that testing for glucose tolerance should occur early and often. There is no downside to glucose tolerance testing other than minor cost, and the upside could be the earlier identification and treatment of pre-diabetes and diabetes. Given the evidence of a very strong association between hyperinsulinemia and AD (and other forms of dementia), every patient with AD or mild cognitive impairment should be regularly tested for glucose intolerance. Furthermore, obesity is also strongly associated with hyperinsulinemia [97] and diabetes [102], and thus, not surprisingly, increases the risk of AD [103]. Given the widespread prevalence of obesity, pre-diabetes, and diabetes, glucose intolerance testing should become a standard part of regular health screening in the broader population. According to the American Diabetes Association, of the 29.1 million Americans with diabetes in 2012, 8.1 million (28%) were undiagnosed. Furthermore, 86 million Americans are estimated to be pre-diabetic (i.e., have impaired glucose tolerance) and most will have no symptoms that are recognizable without medical testing [104]. Early identification of, and intervention for, diabetes and prediabetes could significantly lessen the impact of both diabetes and AD.
The review and arguments here suggest a few necessary future studies:
1. Insulin Deficiency versus Hyperinsulinemia
First, to test the competing hypotheses that it is insulin deficiency versus hyperinsulinemia that create or exacerbate AD (and thus the appropriateness of administering intranasal insulin) mice (including wild type, mice altered to develop AD, and mice altered to develop both AD and hyperinsulinemia) should be given chronic intranasal insulin and then subjected to both cognitive testing and quantification of amyloid burden and tauopathy. If the pathways to AD posed in this paper are correct, intranasal insulin should make AD in hyperinsulinemic mice worse. Its effects on the other mice should be very informative: if it yields moderate neurological improvements in the AD mice, this would support its use in non-hyperinsulinemic AD patients. It might, however, induce hyperinsulinemia and insulin resistance leading to T2DM in the wild type mice, and both T2DM and worsening of AD in the AD mice by competitively inhibiting the breakdown of Aβ. Ideally a range of doses of insulin will be used to assess potential curvilinear effects.
2. Neuroprotective versus Competitive Inhibition Effects of Amylin and Pramlintide
Next, to assess the neuroprotective versus competitive inhibition effects of amylin and pramlintide, in vitro studies should first be conducted to assess the degree to which pramlintide (rather than human amylin) competitively inhibits the breakdown of Aβ. If IDE responds primarily to the amyloidogenic motif rather than particular amino acid sequences, as suggested by Kurochkin [34] and others, perhaps pramlintide competes less for breakdown of IDE compared to human amylin. Next different groups of mice (wild type, mice altered to develop AD and hyperinsulinemia, mice altered to develop AD but without hyperinsulinemia, and IDE knockout mice altered to develop AD) should be chronically treated with different amylins (pramlintide, human amylin), at different dosage levels to observe the potential differential effects of amylin and pramlintide depending on the state of the insulin-IDE system of the subject. IDE inhibitors that can decrease (rather than eliminate) IDE could also be used to more precisely explore the relationships between intranasal insulin, pramlintide, IDE, and AD [32, 105]. This should be followed by cognitive testing and quantification of amyloid burden and tauopathy. If the arguments about competitive inhibition presented earlier are correct, the administration of human amylin should exacerbate AD in all but the wild-type mice. I was unable to find any studies of whether pramlintide competitively inhibits the breakdown of Aβ, so it is impossible to predict the outcome of the tests with pramlintide. If, however, it turns out that pramlintide does competitively inhibit the breakdown of Aβ, then its administration should also exacerbate AD in all but the wild-type mice.
If the results of the first set of studies above find that hyperinsulinemia rather than insulin deficiency exacerbates AD, then this implies that we should be able to significantly lower rates of AD through educating people about how to better manage their blood sugar and insulin. A very large portion of the population has little understanding of the glycemic index of carbohydrates. Furthermore, very few people know that some artificial sweeteners produce an insulin response. This also yields a potential policy implication that perhaps food should be labeled with a measure of its effect on insulin. The results of the second study should help us to identify any AD risks of administering pramlintide (which is already given to many diabetic patients in addition to insulin sensitizers) and help us to assess the overall reliability, efficacy, and safety of administering these drugs.
If the results of future studies support the arguments made in this paper, then this suggests that much more research should be done to identify the different proteases that degrade Aβ in vivo, to better understand their promoters and inhibitors, and to evaluate the safety and efficacy of increasing the availability of such proteases in both AD and diabetes. Relatedly, it would be valuable to develop easily administered diagnostic tests to identify protease deficiencies.
Finally, the work here hints at some very fruitful opportunities for extending the findings in AD to better understand other neurodegenerative diseases that involve protein aggregation in the brain. For example, many of the studies cited here with IDE knockout or IDE upregulated mice should be adapted for studies of Parkinson’s disease, Lewy body dementia, and Huntington’s disease. These latter diseases have longer peptide sequences than IDE is thought to degrade (IDE has been shown to only cleave peptides of 50 amino acids or shorter [106]), but recent studies indicate that IDE nonproteolytically inhibits alpha synuclein fibril formation by binding to alpha synuclein oligomers and preventing them from forming amyloids [88], suggesting there could be great promise in this research pathway.
DISCLOSURE STATEMENT
The author’s disclosure is available online (http://j-alz.com/manuscript-disclosures/15-0980r2).
REFERENCES
[1] | ((2015) ) 2015 Alzheimer’s disease facts and figures. Alzheimers Dement 11: , 332–384. |
[2] | Hurd MD , Martorell P , Delavande A , Mullen KJ , Langa KM ((2013) ) Monetary costs of dementia in the United States. N Engl J Med 368: , 1326–1334. |
[3] | Kavirajan H , Schneider LS ((2007) ) Efficacy and adverse effects of cholinesterase inhibitors and memantine in vascular dementia: A meta-analysis of randomised controlled trials. Lancet Neurol 6: , 782–792. |
[4] | Korczyn AD ((2012) ) Why have we failed to cure Alzheimer’s disease?. J Alzheimers Dis 29: , 275–282. |
[5] | Trinh NH , Hoblyn J , Mohanty SU , Yaffe K ((2003) ) Efficacy of cholinesterase inhibitors in the treatment of neuropsychiatric symptoms and functional impairment in Alzheimer disease - A meta-analysis. JAMA 289: , 210–216. |
[6] | Lanctot KL , Herrmann N , Yau KK , Khan LR , Liu BA , Loulou MM , Einarson TR ((2003) ) Efficacy and safety of cholinesterase inhibitors in Alzheimer’s disease: A meta-analysis. Can Med Assoc J 169: , 557–564. |
[7] | Zissimopoulos J , Crimmins E , Clair P St. ((2014) ) The value of delaying Alzheimer disease onset. Conference: Forum for Health Economics and Policy |
[8] | de la Monte SM ((2012) ) Brain insulin resistance and deficiency as therapeutic targets in Alzheimer’s disease. Curr Alzheimer Res 9: , 35–66. |
[9] | de la Monte SM ((2012) ) Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs 72: , 49–66. |
[10] | Devi L , Alldred MJ , Ginsberg SD , Ohno M ((2012) ) Mechanisms underlying insulin deficiency-induced acceleration of beta-amyloidosis in a mouse model of Alzheimer’s Disease.e. PLoS One 7: , e32792. |
[11] | Wang X , Zheng W , Xie J-W , Wang T , Wang S-L , Teng W-P , Wang Z-Y ((2010) ) Insulin deficiency exacerbates cerebral amyloidosis and behavioral deficits in an Alzheimer transgenic mouse model. Mol Neurodegener 5: , 46. |
[12] | Luchsinger JA ((2010) ) The relationship between the continuum of elevated adiposity, hyperinsulinemia, and type 2 diabetes and late-onset Alzheimer’s disease: An epidemiological perspective. In Diabetes, Insulin and Alzheimer’s Disease Craft S, Christen Y, eds., Springer-Verlag Berlin Heidelberg, pp. 89–107. |
[13] | Carantoni M , Zuliani G , Munari MR , D’Elia K , Palmieri E , Fellin R ((2000) ) Alzheimer disease and vascular dementia: Relationships with fasting glucose and insulin levels. Dement Geriatr Cogn Disord 11: , 176–180. |
[14] | Young SE , Mainous AG , Carnemolla M ((2006) ) Hyperinsulinemia and cognitive decline in a middle-aged cohort. Diabetes Care 29: , 2688–2693. |
[15] | Razay G , Wilcock GK ((1994) ) Hyperinsulinemia and Alzheimer’s disease. Age Ageing 23: , 396–399. |
[16] | Fishel MA , Watson GS , Montine TJ , Wang Q , Green PS , Kulstad JJ , Cook DG , Peskind ER , Baker LD , Goldgaber D , Nie W , Asthana S , Plymate SR , Schwartz MW , Craft S ((2005) ) Hyperinsulinemia provokes synchronous increases in central inflammation and beta-amyloid in normal adults. Arch Neurol 62: , 1539–1544. |
[17] | Jackson K , Barisone G , Diaz E , Lin L , deCarli C , despa F ((2013) ) > Amylin deposition in the brain: A second amyloid in Alzheimer disease?. Ann Neurol 74: , 517–526. |
[18] | Srodulski S , Sharma S , Bachstetter AB , Brelsfoard JM , Pascual C , Xie XS , Saatman KE , Van Eldik LJ , Despa F ((2014) ) Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Mol Neurodegener 9: , 30. |
[19] | Oskarsson ME , Paulsson JF , Schultz SW , Ingelsson M , Westermark P , Westermark GT ((2015) ) In vivo seeding and cross-seeding of localized amyloidosis: A molecular link between type 2 diabetes and Alzheimer disease. Am J Pathol 185: , 834–846. |
[20] | Lutz TA , Meyer U ((2015) ) Amylin at the interface between metabolic and neurodegenerative disorders. Front Neurosci 9: , 216. |
[21] | Adler BL , Yarchoan M , Hwang HM , Louneva N , Blair JA , Palm R , Smith MA , Lee H-g , Arnold SE , Casadesus G ((2014) ) Neuroprotective effects of the amylin analogue pramlintide on Alzheimer’s disease pathogenesis and cognition. Neurobiol Aging 35: , 793–801. |
[22] | Qiu WQ , Au R , Zhu H , Wallack M , Liebson E , Li H , Rosenzweig J , Mwamburi M , Stern RA ((2014) ) Positive association between plasma amylin and cognition in a homebound elderly population. J Alzheimers Dis 42: , 555–563. |
[23] | Zhao LX , Teter B , Morihara T , Lim GP , Ambegaokar SS , Ubeda OJ , Frautschy SA , Cole GM ((2004) ) Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: Implications for Alzheimer’s disease intervention. J Neurosci 24: , 11120–11126. |
[24] | Jolivalt CG , Hurford R , Lee CA , Dumaop W , Rockenstein E , Masliah E ((2010) ) Type 1 diabetes exaggerates features of Alzheimer’s disease in APP transgenic mice. Exp Neurol 223: , 422–431. |
[25] | Jiang Y , Mizisin AP , Rearden A , Jolivalt CG ((2010) ) Diabetes induces changes in ILK, PINCH and components of related pathways in the spinal cord of rats. Brain Res 1332: , 100–109. |
[26] | Pivovarova O , Goegebakan O , Pfeiffer AFH , Rudovich N ((2009) ) Glucose inhibits the insulin-induced activation of the insulin-degrading enzyme in HepG2 cells. Diabetologia 52: , 1656–1664. |
[27] | Farris W , Mansourian S , Chang Y , Lindsley L , Eckman EA , Frosch MP , Eckman CB , Tanzi RE , Selkoe DJ , Guenette S ((2003) ) Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A 100: , 4162–4167. |
[28] | McDermott JR , Gibson AM ((1997) ) Degradation of Alzheimer’s beta-amyloid protein by human and rat brain peptidases: Involvement of insulin-degrading enzyme. Neurochem Res 22: , 49–56. |
[29] | Qiu WQ , Borth W , Ye Z , Haass C , Teplow DB , Selkoe DJ ((1996) ) Degradation of amyloid beta-protein by a serine protease-alpha(2)-macroglobulin complex. J Biol Chem 271: , 8443–8451. |
[30] | Qiu WQ , Walsh DM , Ye Z , Vekrellis K , Zhang JM , Podlisny MB , Rosner MR , Safavi A , Hersh LB , Selkoe DJ ((1998) ) Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J Biol Chem 273: , 32730–32738. |
[31] | Farris W , Mansourian S , Leissring MA , Eckman EA , Bertram L , Eckman CB , Tanzi RE , Selkoe DJ ((2004) ) Partial loss-of-function mutations in insulin-degrading enzyme that induce diabetes also impair degradation of amyloid beta-protein. Am J Pathol 164: , 1425–1434. |
[32] | Deprez-Poulain R , Hennuyer N , Bosc D , Liang WG , Enee E , Marechal X , Charton J , Totobenazara J , Berte G , Jahklal J , Verdelet T , Dumont J , Dassonneville S , Woitrain E , Gauriot M , Paquet C , Duplan I , Hermant P , Cantrelle F-X , Sevin E , Culot M , Landry V , Herledan A , Piveteau C , Lippens G , Leroux F , Tang W-J , van Endert P , Staels B , Deprez B ((2015) ) Catalytic site inhibition of insulin-degrading enzyme by a small molecule induces glucose intolerance in mice. Nat Commun 6: , 8250. |
[33] | Perez A , Morelli L , Cresto JC , Castano EM ((2000) ) Degradation of soluble amyloid beta-peptides 1-40, 1-42, and the Dutch variant 1-40Q by insulin degrading enzyme from Alzheimer disease and control brains. Neurochem Res 25: , 247–255. |
[34] | Kurochkin IV ((1998) ) Amyloidogenic determinant as a substrate recognition motif of insulin-degrading enzyme. FEBS Lett 427: , 153–156. |
[35] | Qiu WQ , Folstein MF ((2006) ) Insulin, insulin-degrading enzyme and amyloid-beta peptide in Alzheimer’s disease: Review and hypothesis. Neurobiol Aging 27: , 190–198. |
[36] | Bennett RG , Duckworth WC , Hamel FG ((2000) ) Degradation of amylin by insulin-degrading enzyme. J Biol Chem 275: , 36621–36625. |
[37] | Hardy J , Selkoe DJ ((2002) ) Medicine - The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: , 353–356. |
[38] | Yin YI , Bassit B , Zhu L , Yang X , Wang C , Li Y-M ((2007) ) gamma-secretase substrate concentration modulates the A beta 42/A beta 40 ratio. J Biol Chem 282: , 23639–23644. |
[39] | Morales R , Chen B , Soto C ((2009) ) Are amyloids infectious? In Current Hypotheses and Research Milestones in Alzheimer’s Disease, Maccioni RB, Perry G, eds. Springer Science+Business Media, LCC, New York, pp. 171–180 . |
[40] | Morales R , Green KM , Soto C ((2009) ) Cross currents in protein misfolding disorders: Interactions and therapy. CNS Neurol Disord Drug Targets 8: , 363–371. |
[41] | Morales R , Moreno-Gonzalez I , Soto C ((2013) ) Cross-seeding of misfolded proteins: Implications for etiology and pathogenesis of protein misfolding diseases. PLoS Pathog 9: , e1003537. |
[42] | Giasson BI , Lee VMY , Trojanowski JQ ((2003) ) Interactions of amyloidogenic proteins. Neuromolecular Med 4: , 49–58. |
[43] | Ott A , Breteler MMB , Van Harskamp F , Grobbee DE , Hofman A ((1996) ) Incidence of Alzheimer’s disease and vascular dementia in the Rotterdam study. Neurobiol Aging 17: , S79–S79. |
[44] | Ott A , Stolk R , Van Harskamp F , Pols H , Hofman A , Breteler M ((1999) ) Diabetes mellitus and the risk of dementia: The Rotterdam study. Neurology 10: , 1937–1942. |
[45] | Leibson CL , Rocca WA , Hanson VA , Cha R , Kokmen E , O’Brien PC , Palumbo PJ ((1997) ) The risk of dementia among persons with diabetes mellitus: A population-based cohort study. Ann N Y Acad Sci 826: , 422–427. |
[46] | Peila R , Rodriguez BL , Launer LJ ((2002) ) Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies - The Honolulu-Asia Aging Study. Diabetes 51: , 1256–1262. |
[47] | Arvanitakis Z , Wilson RS , Bienias JL , Evans DA , Bennett DA ((2004) ) Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol 61: , 661–666. |
[48] | Luchsinger JA , Tang MX , Stern Y , Shea S , Mayeux R ((2001) ) Diabetes mellitus and risk of Alzheimer’s disease and dementia with stroke in a multiethnic cohort. Am J Epidemiol 154: , 635–641. |
[49] | Xu WL , Qiu CX , Wahlin A , Winblad B , Fratiglioni L ((2004) ) Diabetes mellitus and risk of dementia in the Kungsholmen project - A 6-year follow-up study. Neurology 63: , 1181–1186. |
[50] | Schneider JA , Arvanitakis Z , Leurgans SE , Bennett DA ((2009) ) The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol 66: , 200–208. |
[51] | Schneider JA , Arvanitakis Z , Bang W , Bennett DA ((2007) ) Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69: , 2197–2204. |
[52] | Xu WL , Qiu CX , Wahlin A , Winblad B , Fratiglioni L ((2004) ) Diabetes mellitus and risk of dementia in the kungsholmen project: A 6-year follow-up study. Neurobiol Aging 25: , S484–S484. |
[53] | MacKnight C , Rockwood K , Awalt E , McDowell I ((2002) ) Diabetes mellitus and the risk of dementia, Alzheimer’s disease and vascular cognitive impairment in the Canadian Study of Health and Aging. Dement Geriatr Cogn Disord 14: , 77–83. |
[54] | Fujisawa Y , Sasaki K , Akiyama K ((1991) ) Increased insulin levels after OGTT load in peripheral blood and cerebrospinal fluid of patients with dementia of Alzheimer type. Biol Psychiatry 30: , 1219–1228. |
[55] | Luchsinger JA , Tang MX , Shea S , Mayeux R ((2004) ) Hyperinsulinemia and risk of Alzheimer disease. Neurology 63: , 1187–1192. |
[56] | Craft S , Baker LD , Montine TJ , Minoshima S , Watson GS , Claxton A , Arbuckle M , Callaghan M , Tsai E , Plymate SR , Green PS , Leverenz J , Cross D , Gerton B ((2012) ) Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment a pilot clinical trial. Arch Neurol 69: , 29–38. |
[57] | Reger MA , Watson GS , Frey WH , Baker LD , Cholerton B , Keeling ML , Belongia DA , Fishel MA , Plymate SR , Schellenberg GD , Cherrier MM , Craft S ((2006) ) Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype. Neurobiol Aging 27: , 451–458. |
[58] | Reger MA , Watson GS , Green PS , Baker LD , Cholerton B , Fishel MA , Plymate SR , Cherrier MM , Schellenberg GD , Frey WH II , Craft S ((2008) ) Intranasal insulin administration dose-dependently modulates verbal memory and plasma amyloid-beta in memory-impaired older adults. J Alzheimers Dis 13: , 323–331. |
[59] | Reger MA , Watson GS , Green PS , Wilkinson CW , Baker LD , Cholerton B , Fishel MA , Plymate SR , Breitner JCS , DeGroodt W , Mehta P , Craft S ((2008) ) Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 70: , 440–448. |
[60] | Freiherr J , Hallschmid M , Frey WH II , Bruenner YF , Chapman CD , Hoelscher C , Craft S , De Felice FG , Benedict C ((2013) ) Intranasal Insulin as a treatment for Alzheimer’s disease: A review of basic research and clinical evidence. Cns Drugs 27: , 505–514. |
[61] | Talbot K , Wang H-Y , Kazi H , Han L-Y , Bakshi KP , Stucky A , Fuino RL , Kawaguchi KR , Samoyedny AJ , Wilson RS , Arvanitakis Z , Schneider JA , Wolf BA , Bennett DA , Trojanowski JQ , Arnold SE ((2012) ) Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest 122: , 1316–1338. |
[62] | Zhao W-Q , Alkon DL ((2001) ) Role of insulin and insulin receptor in learning and memory. Mol Cell Endocrinol 177: , 125–134. |
[63] | Stern SA , Chen DY , Alberini CM ((2014) ) The effect of insulin and insulin-like growth factors on hippocampus- and amygdala-dependent long-term memory formation. Learn Mem 21: , 556–563. |
[64] | Chen DY , Stern SA , Garcia-Osta A , Saunier-Rebori B , Pollonini G , Bambah-Mukku D , Blitzer RD , Alberini CM ((2011) ) A critical role for IGF-II in memory consolidation and enhancement. Nature 469: , 491–497. |
[65] | Lupien SB , Bluhm EJ , Ishii DN ((2003) ) Systemic insulin-like growth factor-I administration prevents cognitive impairment in diabetic rats, and brain IGF regulates learning/memory in normal adult rats. J Neurosci Res 74: , 512–523. |
[66] | Dou J-T , Chen M , Dufour F , Alkon DL , Zhao W-Q ((2005) ) Insulin receptor signaling in long-term memory consolidation following spatial learning. Learn Mem 12: , 646–655. |
[67] | de la Monte SM , Wands JR ((2008) ) Alzheimer’s disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol 2: , 1101–1113. |
[68] | Clodfelder-Miller BJ , Zmijewska AA , Johnson GVW , Jope RS ((2006) ) Tau is hyperphosphorylated at multiple sites in mouse brain in vivo after streptozotocin-induced insulin deficiency. Diabetes 55: , 3320–3325. |
[69] | Rai S , Kamat PK , Nath C , Shukla R ((2014) ) Glial activation and post-synaptic neurotoxicity: The key events in Streptozotocin (ICV) induced memory impairment in rats. Pharmacol Biochem Behav 117: , 104–117. |
[70] | Davidson E , Coppey L , Lu B , Arballo V , Calcutt NA , Gerard C , Yorek M ((2009) ) The roles of streptozotocin neurotoxicity and neutral endopeptidase in murine experimental diabetic neuropathy. Experimental Diabetes Research 2009: , 9. |
[71] | Kochkina EG , Plesneva SA , Vasilev DS , Zhuravin IA , Turner AJ , Nalivaeva NN ((2015) ) Effects of ageing and experimental diabetes on insulin-degrading enzyme expression in male rat tissues. Biogerontology 16: , 473–484. |
[72] | Rezende LF , Camargo RL , Branco RCS , Cappelli APG , Boschero AC , Carneiro EM ((2014) ) Reduced insulin clearance and lower insulin-degrading enzyme expression in the liver might contribute to the thrifty phenotype of protein-restricted mice. Br J Nutr 112: , 900–907. |
[73] | Hamazaki H ((1996) ) Cathepsin D is involved in the clearance of Alzheimer’s β-amyloid protein. FEBS Lett 396: , 139–142. |
[74] | McDermott JR , Gibson AM ((1996) ) Degradation of Alzheimer’s beta-amyloid protein by human cathepsin D. Neuroreport 7: , 2163–2166. |
[75] | Saido T , Leissring MA ((2012) ) Proteolytic degradation of amyloid beta-protein. Cold Spring Harb Perspect Med 2: , a006379. |
[76] | Eckman CB , Watson M , Eckman EA ((2002) ) Degradation of the Alzheimer’s amyloid beta peptide by endothelin converting enzyme. Abstract No. 19. Soc Neurosci Abstr19. |
[77] | Eckman EA , Eckman CB ((2005) ) A beta-degrading enzymes: Modulators of Alzheimer’s disease pathogenesis and targets for therapeutic intervention. Biochem Soc Trans 33: , 1101–1105. |
[78] | Duckworth WC ((1988) ) Insulin degradation: Mechanisms, products, and significance. Endocr Rev 9: , 319–345. |
[79] | Kuo WL , Gehm BD , Rosner MR ((1991) ) The insulin degrading enzyme regulates both intracellular and extracellular insulin levels via an insulin shunt. J Cell Biochem Suppl 78. |
[80] | Kurochkin I , Goto S ((1994) ) Beta-amyloid peptide is specifically recognized and cleaved by insulin-degrading enzyme. Neurobiol Aging 15: , S83. |
[81] | Vekrellis K , Chiu S , Mansourian S , Selkoe D ((2000) ) Insulin-degrading enzyme is the major Abeta-degrading protease in human control and Alzheimer’s disease brains. Soc Neurosci Abstr 26: , Abstract No.-588.585. |
[82] | Leissring MA , Farris W , Chang AY , Walsh DM , Wu XN , Sun XY , Frosch MP , Selkoe DJ ((2003) ) Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 40: , 1087–1093. |
[83] | Miners JS , Barua N , Kehoe PG , Gill S , Love S ((2011) ) A beta-Degrading Enzymes: Potential for Treatment of Alzheimer Disease. J Neuropathol Exp Neurol 70: , 944–959. |
[84] | de Tullio MB , Morelli L , Castano EM ((2008) ) The irreversible binding of amyloid peptide substrates to insulin-degrading enzyme A biological perspective. Prion 2: , 51–56. |
[85] | Vingtdeux V , Chandakkar P , Zhao H , Blanc L , Ruiz S , Marambaud P ((2015) ) CALHM1 ion channel elicits amyloid-beta clearance by insulin-degrading enzyme in cell lines and in vivo in the mouse brain. J Cell Sci 128: , 2330–2338. |
[86] | Quan Q , Wang J , Li X , Wang Y ((2013) ) Ginsenoside Rg1 Decreases A beta(1-42) level by upregulating PPAR gamma and IDE expression in the hippocampus of a rat model of Alzheimer’s disease. PLoS One 8: e59155. |
[87] | Steneberg P , Bernardo L , Edfalk S , Lundberg L , Backlund F , Ostenson C-G , Edlund H ((2013) ) The type 2 diabetes-associated gene ide is required for insulin secretion and suppression of alpha-synuclein levels in beta-cells. Diabetes 62: , 2004–2014. |
[88] | Sharma SK , Chorell E , Steneberg P , Vernersson-Lindahl E , Edlund H , Wittung-Stafshede P ((2015) ) Insulin-degrading enzyme prevents alpha-synuclein fibril formation in a nonproteolytical manner. Sci Rep 5: , 12531. |
[89] | Cordes CM , Bennett RG , Siford GL , Hamel FG ((2009) ) Nitric oxide inhibits insulin-degrading enzyme activity and function through S-nitrosylation. Biochem Pharmacol 77: , 1064–1073. |
[90] | Pedersen WA , McMillan PJ , Kulstad JJ , Leverenz JB , Craft S , Haynatzki GR ((2006) ) Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp Neurol 199: , 265–273. |
[91] | Madani R , Poirier R , Wolfer DP , Welzl H , Groscurth P , Lipp H-P , Lu B , El Mouedden M , Mercken M , Nitsch RM , Mohajeri MH ((2006) ) Lack of neprilysin suffices to generate murine amyloid-like deposits in the brain and behavioral deficit in vivo. J Neurosci Res 84: , 1871–1878. |
[92] | Miller BC , Eckman EA , Sambamurti K , Dobbs N , Chow KM , Eckman CB , Hersh LB , Thiele DL ((2003) ) Amyloid-beta peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc Natl Acad Sci U S A 100: , 6221–6226. |
[93] | Akomolafe A , Beiser A , Meigs PB , Au R , Green RC , Farrer LA , Wolf PA , Seshadri S ((2006) ) Diabetes mellitus and risk of developing Alzheimer disease - Results from the Framingham study. Arch Neurol 63: , 1551–1555. |
[94] | Alonso Vilatela ME , Lopez-Lopez M , Yeseas-Gomez P ((2012) ) Genetics of Alzheimer’s disease. Arch Med Res 43: , 622–631. |
[95] | Cook DG , Leverenz JB , McMillan PJ , Kulstad JJ , Ericksen S , Roth RA , Schellenberg GD , Jin LW , Kovacina KS , Craft S ((2003) ) Reduced hippocampal insulin-degrading enzyme in late-onset Alzheimer’s disease is associated with the apolipoprotein E-epsilon 4 allele. Am J Pathol 162: , 313–319. |
[96] | Leissring MA , Turner AJ ((2013) ) Regulation of distinct pools of amyloid beta-protein by multiple cellular proteases. Alzheimers Res Ther 5: , 37. |
[97] | Shanik MH , Xu Y , Skrha J , Dankner R , Zick Y , Roth J ((2008) ) Insulin resistance and hyperinsulinemia: Is hyperinsulinemia the cart or the horse?. Diabetes Care 31: (Suppl 2), S262–268. |
[98] | Pories WJ , Dohm GL ((2012) ) Diabetes: Have we got it all wrong? Hyperinsulinism as the culprit: Surgery provides the evidence. Diabetes Care 35: , 2438–2442. |
[99] | Zhu H , Wang X , Wallack M , Li H , Carreras I , Dedeoglu A , Hur JY , Zheng H , Li H , Fine R , Mwamburi M , Sun X , Kowall N , Stern RA , Qiu WQ ((2015) ) Intraperitoneal injection of the pancreatic peptide amylin potently reduces behavioral impairment and brain amyloid pathology in murine models of Alzheimer’s disease. Mol Psychiatry 20: , 232–239. |
[100] | Qiu WQ , Zhu H ((2014) ) Amylin and its analogs: A friend or foe for the treatment of Alzheimer’s disease?. Front Aging Neurosci 6: , 186. |
[101] | Weyer C , Hanson R , Tataranni PA , Bogardus C , Pratley RE ((2000) ) Fasting hyperinsulinemia predicts type 2 diabetes independent of insulin resistance: Evidence for a pathogenic role of basal insulin hypersecretion. Diabetes 49: , A25–A26. |
[102] | Leong KS , Wilding JP ((1999) ) Obesity and diabetes. Best Pract Res Clin Endocrinol Metab 13: , 221–237. |
[103] | Chuang YF , An Y , Bilgel M , Wong DF , Troncoso JC , O’Brien RJ , Breitner JC , Ferruci L , Resnick SM , Thambisetty M ((2015) ) Midlife adiposity predicts earlier onset of Alzheimer’s dementia, neuropathology and presymptomatic cerebral amyloid accumulation. Mol Psychiatry doi: 10.1038/mp.2015.129 |
[104] | Centers for Disease Control, and Prevention ((2014) ) National Diabetes Statistics Report: Estimates of Diabetes and its Burden in the United States, 2014. U.S. Department of Health and Human Services Atlanta, GA. |
[105] | Leissring MA , Malito E , Hedouin S , Reinstatler L , Sahara T , Abdul-Hay SO , Choudhry S , Maharvi GM , Fauq AH , Huzarska M , May PS , Choi S , Logan TP , Turk BE , Cantley LC , Manolopoulou M , Tang WJ , Stein RL , Cuny GD , Selkoe DJ ((2010) ) Designed inhibitors of insulin-degrading enzyme regulate the catabolism and activity of insulin. PLoS One 5: , e10504. |
[106] | Shen Y , Joachimiak A , Rosner MR , Tang WJ ((2006) ) Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature 443: , 870–874. |
[107] | Stewart R , Liolitsa D ((1999) ) Type 2 diabetes mellitus, cognitive impairment and dementia. Diabet Med 16: , 93–112. |
[108] | Rivera EJ , Goldin A , Fulmer N , Tavares R , Wands JR , de la Monte SM ((2005) ) Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J Alzheimers Dis 8: , 247–268. |
[109] | Arvanitakis Z , Schneider JA , Wilson RS , Li Y , Arnold SE , Wang Z , Bennett DA ((2006) ) Diabetes is related to cerebral infarction but not to AD pathology in older persons. Neurology 67: , 1960–1965. |
[110] | Kurochkin IV , Goto S ((1994) ) Alzheimer’s β-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett 345: , 33–37. |
[111] | Qiu WQ , Ye Z , Kosik KS , Selkoe DJ ((1996) ) Degradation of amyloid beta-protein by a secreted metalloprotease in neural and non-neural cells. Soc Neurosci Abstr 22: , 1945. |
[112] | Vekrellis K , Ye Z , Qiu WQ , Walsh D , Hartley D , Chesneau V , Rosner MR , Selkoe DJ ((2000) ) Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme. J Neurosci 20: , 1657–1665. |
[113] | Abdul-Hay SO , Kang D , McBride M , Li L , Zhao J , Leissring MA ((2011) ) Deletion of insulin-degrading enzyme elicits antipodal, age-dependent effects on glucose and insulin tolerance. PLoS One 6: , e20818. |
[114] | Zhao J , Li L , Leissring M ((2009) ) Insulin-degrading enzyme is exported via an unconventional protein secretion pathway. Mol Neurodegener 4: , 1–5. |
[115] | Yin F , Zhang Y , Guo L , Kong S , Liu J ((2012) ) Geniposide regulates insulin-degrading enzyme expression to inhibit the cytotoxicity of Abeta(1-42) in cortical neurons. CNS Neurol Disord Drug Targets 11: , 1045–1051. |
Figures and Tables
Fig.1
The insulin-protease-amyloid degradation pathway and its potential malfunctions.
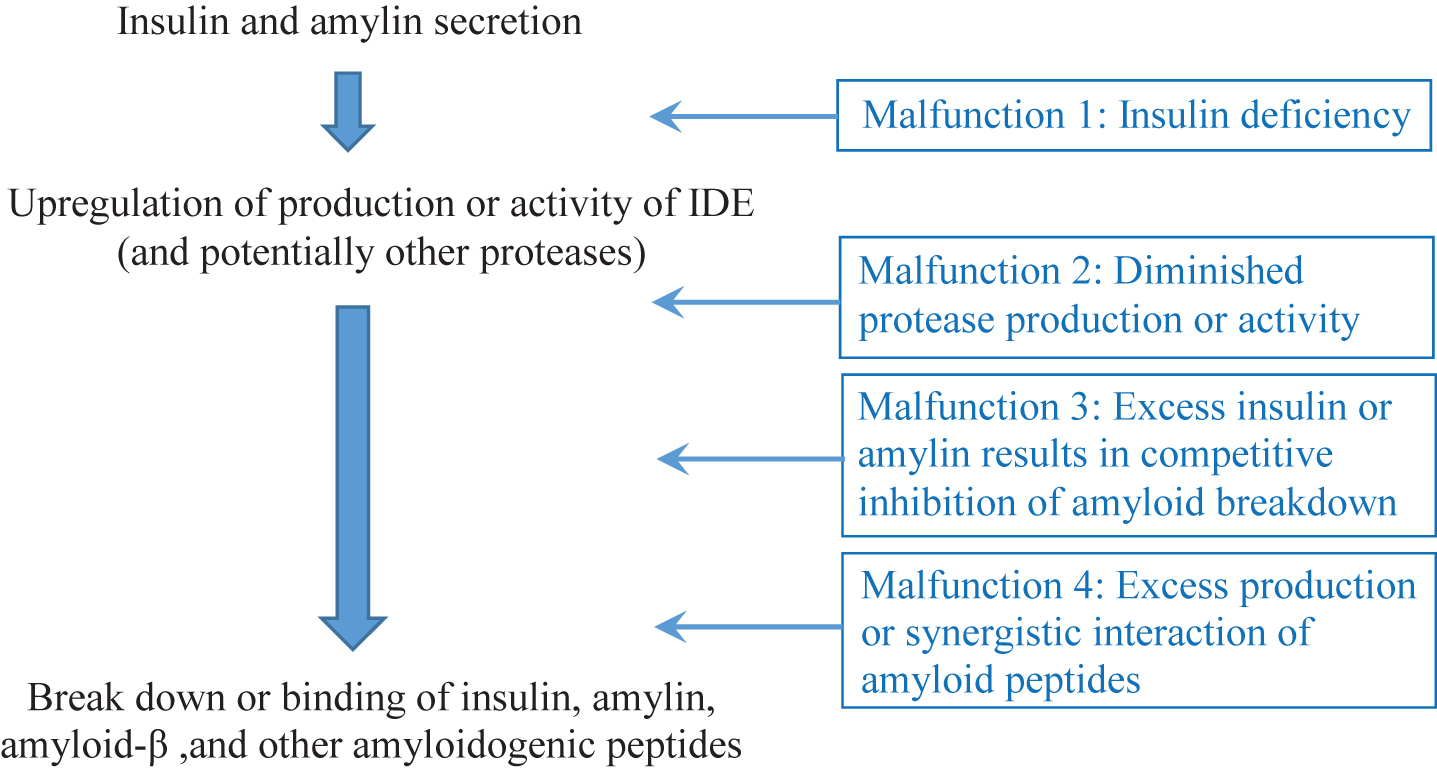
Table 1
Review of literature pertaining to the insulin, insulin degrading enzyme, and Alzheimer’s pathways
| Citation | Year | Type | Findings |
| T2DM and Alzheimer’s disease | |||
| Ott et al., 1996 [43] | 1996 | Large population study | 6330 participants between 55–90, 11.4% had diabetes at baseline. Of those with dementia, 22.3% had diabetes (1.3 fold increase). In particular, a very strong relationship was found between dementia, and diabetes when treated with insulin (3.2 fold increase). |
| Leibson et al., 1997 [45] | 1997 | Longitudinal cohort study | DM was associated with an increased risk of all dementia (1.66 fold), and AD (2.27 for men, 1.37 for women). |
| Ott et al., 1999 [44] | 1999 | Longitudinal cohort study | Finds that diabetes almost double the risk of dementia (1.9 RR) and AD (1.9 RR). Patients treated with insulin were at the highest risk of dementia (4.3 RR) |
| Stewart and Liolitsa, 1999 [107] | 1999 | Review | |
| Luchsinger et al., 2001 [48] | 2001 | Longitudinal cohort study | Study of Black and Hispanic cohort for average of 4.3 years suggested that diabetes (baseline rate of 20%) led to a 1.3 fold increase in AD, a 1.6 fold increase in AD or cognitive impairment without dementia (without stroke). |
| Peila et al., 2002 [46] | 2002 | Longitudinal cohort study and postmortems | 2,574 Japanese-American men enrolled in study and 216 underwent autopsy.35% were diabetic at baseline. Found that Type 2 diabetes was associated with higher rates of total dementia (1.5 fold), AD (1.8 fold), and vascular dementia (2.3 fold). Those that also had ApoE4 allele had a 5.5 fold risk for AD compared to those with neither risk factor. Risk of neurofibrillary tangles was much higher in presence of both diabetes and ApoE4 allele than with either alone, suggesting an interactive effect. |
| MacKnight et al., 2002 [53] | 2002 | Longitudinal cohort study | 5,574 subjects of Canadian Study of Health and Aging, Average age of 74; diabetes (baseline rate of 12.1%) found to increase rate of vascular dementia but not AD. |
| Arvanitakis et al., 2004 [47] | 2004 | Longitudinal cohort study | Uses cognitive testing to determine AD. Finds that diabetes increases risk of AD by 1.65 fold and increases rate of decline. Found no relationship between diabetes and stroke. |
| Xu et al., 2004 [49] | 2004 | Longitudinal cohort study | Diabetes increased the risk of dementia 1.5 fold (1.6 vascular dementia, 1.3 AD). Treatment with oral antidiabetic medications increased risk to 1.7. |
| Rivera et al., 2005 [108] | 2005 | Postmortems | Finds that AD brains exhibit lower levels of mRNA for insulin, IGF-1, and IGFII polypeptides and their receptors |
| Akomolafe et al., 2006 [93] | 2006 | Longitudinal cohort study | Finds that can only detect that DM increases risk of dementia when looking at groups that are not already at high risk due to ApoE4, etc. (At baseline, finds that 9.13 % have diabetes and 22–24% have ApoE4.) |
| Arvanitakis et al., 2007 [109] | 2006 | Longitudinal cohort study | Diabetes was associated with increased risk of cerebral infarction (2.47 fold), but not to increased global AD score. However, subjects had a mean age of 86, “Nearly all subjects had at least some AD pathology: the mean composite measure of AD pathology was 0.71 (SD 0.65; range 0 to 2.88).” Furthermore, sample only had a baseline diabetes rate of 15%. Those without diabetes had a higher ratio of ApoE4 (31%) versus those with diabetes (26%). |
| Schneider et al., 2007 [51] | 2007 | Postmortems compared to diagnoses | The majority of older persons have one or more brain pathologies; those with dementia most often have multiple brain pathologies. |
| Schneider et al., 2009 [50] | 2009 | Postmortems | Of the 179 people with probable AD, 87.7% were pathologically confirmed to have AD, and 45.8% had mixed pathologies. Of the 134 with MCI, 54.4 % were pathologically confirmed to have AD, and 19.4% had mixed pathologies. |
| Talbot et al., 2012 [61] | 2012 | Postmortems | Brain insulin resistance was strongly associated with episodic and working memory deficits, even after controlling for Aβ plaques, neurofibrillary tangles, and ApoE4. |
| Alzheimer’s disease increased by insulin deficiency or hyperinsulinemia? | |||
| Fujisawa et al., 1991 [54] | 1991 | In vivo, humans | People with AD had significantly higher insulin levels in the cerebrospinal fluid both under fasting conditions and in response to a glucose tolerance test. |
| Razay and Wilcock, 1994 [15] | 1994 | In vivo, humans | Women with AD had higher plasma insulin, higher glucose, and higher body mass index than controls. The differences were not significant for men. |
| Ott et al., 1996 [43] | 1996 | Large population study | 6330 participants between 55–90; 11.4% had diabetes at baseline. Of those with dementia, 22.3% had diabetes (1.3 fold increase). In particular, a very strong relationship was found between dementia, and diabetes when treated with insulin (3.2 fold increase). |
| Ott et al., 1999 [44] | 1999 | Longitudinal cohort study | Finds that diabetes almost double the risk of dementia (1.9 RR) and AD (1.9 RR). Patients treated with insulin were at the highest risk of dementia (4.3 RR). |
| Carantoni et al., 2000 [13] | 2000 | In vivo, humans | Non-diabetic patients with vascular dementia or AD had significantly higher fasting glucose and insulin levels than healthy controls. |
| Luchsinger et al., 2004 [55] | 2004 | Longitudinal cohort study | 683 elderly persons were followed over time. Finds that the risk of AD attributable to the presence of hyperinsulinemia or diabetes is 39%. |
| Fishel et al., 2005 [16] | 2005 | In vivo, humans | Concludes that moderate hyperinsulinemia can elevate inflammatory markets and Aβ42 in the periphery and the brain, thereby increasing the risk of AD. |
| Reger et al., 2006 [57] | 2006 | In vivo, humans | Moderate amounts of intranasal insulin improved some measures of memory in individuals without ApoE4 allele, but impaired performance on some measures of memory in individuals with ApoE4 allele. |
| Pedersen et al., 2006 [90] | 2006 | In vivo, mice | Rosiglitazone (an insulin sensitizer) attenuated memory deficits in Tg2576 Alzheimer mice. It appeared to work by lowing glucocorticoid levels (chronically high glucocorticoids have previously been shown to reduce IDE mRNA in the dentate gyrus of the hippocampus, and to reduce IDE activity in the whole hippocampus). |
| Young et al., 2006 [14] | 2006 | In vivo, humans | Finds that hyperinsulinemia was associated with a significantly lower baseline of delayed word recall, digit symbol subtest, and first letter word fluency. Furthermore, those with hyperinsulinemia had a greater decline over six years in delayed word recall and word fluency. |
| Reger et al., 2008 [59] | 2008 | In vivo, humans | 25 adults with AD or mild cognitive impairment (and NOT diabetes) were assigned randomly to receive intranasal insulin or a placebo. Moderate amounts of intranasal insulin (20 IU) improved immediate recall in older adults with AD or mild cognitive impairment, but not longer term recall. At higher levels of intranasal insulin (40 IU or 60 IU) performance declined to levels below those of placebo. Furthermore, subjects who had the ApoE4 allele showed poorer performance than all other subjects, and that performance did not get better with intranasal insulin. |
| de la Monte, 2012 [8] | 2012 | Review | |
| Craft et al., 2012 [56] | 2012 | In vivo, humans | Finds positive effects of intranasal insulin on memory impaired adults. Again, results more reliably positive for moderate dose (20 IU). |
| Freiherr et al., 2013 [60] | 2013 | Review | Concludes that insulin has neuroprotective effect. |
| Insulin and/or amylin stimulate IDE production/activity | |||
| Zhao et al., 2004 [23] | 2004 | In vitro | Higher levels of insulin resulted in higher levels of IDE protein; insulin –>PI3 kinase –>upregulates IDE (**HOWEVER, it appeared that at very high levels of insulin, it did not continue to increase IDE: 20, 200, and 500 nM; 155.81±14.49% relative to control at 20 nM, 167.86±25.02 at 200 nM, and 157.69±29.39 at 500 nM); IDE levels lower in AD brains; IDE lowest with two copies of ApoE4 allele; second lowest with one copy of ApoE4 allele. |
| Jolivalt et al., 2010 [24] | 2010 | In vivo, mice | Streptozotocin was used to induce T1DM in mice. Mice who had T1DM performed worse than wild type mice; mice who had both T1DM and AD (APP mice) performed the worst. Brain assays verified increased plaques. Note: T1DM x APP mice expressed significantly less IDE. “IDE upregulation requires insulin-mediated Akt activation (Zhao et al., 2004a). Therefore, insulin deficiency and a decreased signaling via the PI3K pathway may contribute to decreased IDE expression and thus contribute to increased Aβ protein levels as a result of a reduced degradation.” (pg. 428) |
| Kochkina et al., 2015 [71] | 2015 | In vivo, rats | Inducing Type 1 diabetes via streptozotocin caused IDE to go up in liver and striatum, but down in cortex and hippocampus (cortex and hippocampus are the regions where AD effects first begin to show). |
| IDE breaks down insulin | |||
| Kuo et al., 1991 [79] | 1991 | In vitro | IDE breaks down insulin |
| IDE, neprilysin, and potentially other proteases break down Aβ, amylin, and other amyloidogenic peptides | |||
| Kurochkin and Goto, 1994 [110] | 1994 | In vitro | IDE (rat) breaks down Aβ; acidification significantly inhibits IDE’s breakdown of Aβ; Aβ links to IDE so fast that is hard to label IDE in presence of Aβ. |
| Qiu et al., 1996 [111] | 1996 | In vitro | Aβ is broken down by a metalloprotease. |
| Hamazaki, 1996 [73] | 1996 | In vitro | Cathepsin D purified from rat brain hydrolyzed Aβ. |
| McDermott and Gibson, 1997 [28] | 1997 | In vitro, postmortem human and rat brains | IDE breaks down Aβ. |
| Qiu et al., 1998 [30] | 1998 | In vitro | IDE breaks down Aβ. |
| Kurochkin, 1998 [34] | 1998 | Review | IDE acts on peptides that share an ability to form amyloid fibrils. |
| Bennett et al., 2000 [36] | 2000 | In vitro | IDE degrades amylin; insulin inhibited amylin in a dose-dependent manner and insulin degradation was also inhibited by amylin. Excess insulin also inhibits its own degradation. “Normally, a balance exists between deposition and degradation of the amyloidogenic peptide. When the levels of the peptide exceed the capacity of IDE to degrade them, either by increased expression of the peptide, or decreased expression or enzymatic activity of IDE, the balance is shifted from degradation to deposition. In the case of Type 2 diabetes, both insulin and amylin secretion are increased due to peripheral insulin resistance. Because IDE has approximately 4-fold greater affinity for insulin than for amylin, amylin degradation will be proportionately impaired.” |
| Perez et al., 2000 [33] | 2000 | In vitro, postmortem brain tissue, rat IDE | IDE breaks down Aβ and Aβ analogs; IDE breakdown of Aβ inhibited by excess insulin; Aβ also inhibits breakdown of insulin but is less potent than the reverse. |
| Vekrellis et al., 2000 [112] | 2000 | In vitro | IDE breaks down Aβ. |
| Farris et al., 2003 [27] | 2003 | In vivo, mice | IDE knockout mice had a greater than 50% decrease in Aβ degradation in both brain membrane fractions and primary neuronal cultures and a similar deficit in insulin degradation in liver. The mice showed an increase in cerebral accumulation of endogenous Aβ (characteristic of AD), hyperinsulinemia, and glucose intolerance. Ergo IDE dysfunction may underlie both AD and T2DM. |
| Leissring et al., 2003 [82] | 2003 | In vivo, mice | Upregulation of either neprilysin or IDE (via genetic modification) lessened the mortality of APP mice, and increased Aβ degradation. Furthermore, the proteases acted upon the soluble, monomeric Aβ species prior to over deposition. The authors note that “the vast majority of Aβ-degrading activity in these brain membrane samples is attributable to IDE” IDE decreased both soluble and insoluble Aβ, and “ ... relatively small changes in the activity of IDE and other Aβ-degrading proteases can dramatically impact steady-state cerebral Aβ levels, suggesting that modest increases in proteolytic clearance of Aβ in humans might be sufficient to effect significant changes in the overall economy of brain Aβ” Also, “chronic neuronal overexpression of IDE or NEP beginning postnatally did not itself have obvious deleterious consequences.” |
| Farris et al., 2004 [31] | 2004 | In vivo, mice | Partial loss-of-function mutations in the IDE gene can impair regulation of Aβ levels without early debilitating dementia, consistent with it having a potential role in late onset AD |
| Madani et al., 2006 [91] | 2006 | In vivo, mice | Neprilysin knockout mice exhibit Alzheimer’s-like behavior impairment and increased Aβ deposits. |
| de Tullio et al., 2008 [84] | 2008 | In vitro | Aβ binds to IDE very strongly. |
| Miners et al., 2011 [83] | 2011 | Review | Neprilysin (NEP), insulin-degrading enzyme, and endothelin-converting enzyme reduce Aβ levels and protect against cognitive impairment in mouse models of AD. |
| Steneberg et al., 2013 [87] | 2013 | In vivo, mice | Deficiency in IDE leads to deficiency in insulin because IDE is needed to replenish the pool of insulin granules. “We find that glucose-stimulated insulin secretion (GSIS) is decreased in Ide KO mice due to impaired replenishment of the releasable pool of granules and that the Ide gene is haploinsufficient.” Also finds that alpha synuclein and IDE levels are inversely correlated, and finds evidence that alpha synuclein suppresses insulin secretion. |
| Quan et al., 2013 [86] | 2013 | In vivo, rat | Ginsenoside Rg1 upregulates proliferator-activated receptor y (PPARy); PPARγ can upregulate IDE expression and IDE can degrade Aβ1 - 42. |
| Vingtdeux et al., 2015 [85] | 2015 | In vitro and in vivo, mice | CALHM1 potentiates IDE activity, facilitating degradation of Aβ. Calhm1 knockout mice had roughly a 50% increase in endogenous Aβ concentrations in the whole brain and primary neurons. |
| Sharma et al., 2015 [88] | 2015 | In vitro | IDE binds to alpha synuclein oligomers, thereby preventing them from forming amyloids. Furthermore, the presence of alpha synuclein intensified IDE’s proteolytic activity on another fluorogenic substrate, “substrate V.” The researchers also found that adding APP to the mix competitively inhibited the breakdown of “substrate V.” |
| IDE has preferential affinity for insulin; insulin, amylin, Aβ competitive inhibit each other’s breakdown | |||
| Qui et al., 1998 [30] | 1998 | In vitro | Finds that IDE is secreted endogeneously by the microglial cell line (suggesting that it is IDE that breaks down Aβ rather than other potential proteases). Also demonstrates that 110-kDA IDE was present in fresh lumbar CSF obtained from living patients, indicating that this protease exists extracellularly under in vivo conditions. IDE breaks down Aβ; degradation of Aβ was completely inhibited by competitive IDE substrate, insulin. |
| Perez et al., 2000 [33] | 2000 | In vitro, postmortem brain tissue, rat IDE | IDE breaks down Aβ and Aβ analogs; IDE breakdown of Aβ inhibited by excess insulin; Aβ also inhibits breakdown of insulin but is less potent than the reverse. |
| Bennett, et al., 2000 [36] | 2000 | In vitro | IDE degrades amylin; insulin inhibited amylin in a dose-dependent manner and insulin degradation was also inhibited by amylin. Excess insulin also inhibits its own degradation. “Normally, a balance exists between deposition and degradation of the amyloidogenic peptide. When the levels of the peptide exceed the capacity of IDE to degrade them, either by increased expression of the peptide, or decreased expression or enzymatic activity of IDE, the balance is shifted from degradation to deposition. In the case of Type 2 diabetes, both insulin and amylin secretion are increased due to peripheral insulin resistance. Because IDE has approximately 4-fold greater affinity for insulin than for amylin, amylin degradation will be proportionately impaired.” |
| Qiu and Folstein, 2006 [35] | 2006 | Review | Reviews studies that show that insulin competitively inhibits the breakdown of Aβ. |
| Amylin and Pramlintide and AD | |||
| Jackson et al., 2013 [17] | 2013 | Postmortems | Subjects with AD and diabetes, or AD alone, both showed large amylin plaque deposits in the brain. They also found mixed amylin and Aβ plaques. “Amylin deposition in AD brains suggests undiagnosed insulin resistance in these patients, which is common to aging."(p.521) |
| Srodulski et al., 2014 [18] | 2014 | In vivo, rats | Rats genetically modified to overexpress human amylin accumulated amylin oligomers in their brains and exhibited neurological deficits. |
| Adler et al., 2014 [21] | 2014 | In vivo, humans and mice | AD and MCI patients exhibited significantly lower plasma amylin. Pramlintide injections to accelerated senescence mice improved their memory. |
| Qiu and Zhu, 2014 [100] | 2014 | In vivo, mice | Amylin and pramlintide helped reduce neurological deficits in mice that were engineered to have AD. Also found that in humans, amylin increases the Aβ42 in the blood and CSF. |
| Zhu et al., 2015 [99] | 2015 | In vivo, mice | Pramlintide improved neurological symptoms in APP mice. |
| Oskarsson et al., 2015 [19] | 2015 | In vivo, mice; postmortem, humans | Amylin fibrils promote an increase in the formation of amylin fibrils (self-reinforced seeding). Also found that amylin fibrils cross-seeded Aβ fibrils, and amylin and Aβ were found to be colocalized in plaques in AD patient brains. |
| Lutz and Meyer, 2015 [20] | 2015 | Review | Discusses the similarities between amylin and Aβ, and reviews research suggesting that amyloid may constitute a “second amyloid” that plays an important role in neurodegenerative disorders. |
| Excess production of amyloidogenic proteins or synergistic interactions of amyloidogenic proteins can increase Aβ deposition | |||
| Giasson et al., 2003 [42] | 2003 | Review | Alpha synuclein readily self-polymerizes; tau does not. However, Giasson et al demonstrated (in vitro) that alpha-synuclein could initiate the polymerization of tau in vitro and that tau and alpha synuclein synergistically promote and propagate the polymerization of each other. |
| Yin et al., 2007 [38] | 2007 | In vitro | APP mutations enhance the preference of γ-secretase for the 42-site over the 40-site cleavage; higher γ-secretase substrate concentrations result in higher ratios of Aβ42/Aβ40. |
| Morale et al., 2009 [39] | 2009 | Review | The infectious agent in prion disease is thought to be the misfolded protein structure. Because this misfolded protein structure is also common to the other amyloid proteins, they may be infectious as well. |
| Morales et al., 2009 [40] | 2009 | Review | Morales et al. review considerable evidence (epidemiological, in vitro, in vivo) that different protein misfolding disorders can cross-seed each other. Particular focus is paid to cross-seeding between Aβ, amylin, and alpha-synuclein. |
| Oskarsson et al., 2015 [19] | 2015 | In vivo, mice; postmortem, humans | Amylin fibrils promote an increase in the formation of amylin fibrils (self-reinforced seeding). Also found that amylin fibrils cross-seeded Aβ fibrils, and amylin and Aβ were found to be colocalized in plaques in AD patient brains. |
| IDE inhibition may make T2DM worse | |||
| Farris et al., 2003 [27] | 2003 | In vivo, mice | IDE knockout mice exhibit hyperinsulinemia, glucose intolerance, and increased Aβ. |
| Abdul-Hay et al., 2011 [113] | 2011 | In vivo, mice | Although Ide (-/-) mice have elevated insulin levels, they exhibit impaired, rather than improved, glucose tolerance that may arise from compensatory insulin signaling dysfunction. |
| Deprez-Poulain et al., 2015 [32] | 2015 | In vivo, mice | Inhibiting IDE results in glucose intolerance. |
| Promotors and inhibitors of IDE secretion or IDE breakdown of Aβ | |||
| Kurochkin and Goto, 1994 [110] | 1994 | In vitro | IDE (rat) breaks down Aβ; acidification significantly inhibits IDE’s breakdown of Aβ. |
| Perez et al., 2000 [33] | 2000 | In vitro, postmortem brain tissue, rat IDE | Bacitracin, phenanthroline, and N0ethylmaleimide each inhibit IDE. |
| Cook et al., 2003 [95] | 2003 | ApoE4 associated with reduced IDE expression. | |
| Zhao et al., 2004 [23] | 2004 | In vitro | ApoE4 associated with reduced IDE expression. |
| Zhao et al., 2009 [114] | 2009 | It is not well understood what promotes IDE secretion. | |
| Cordes et al., 2009 [89] | 2009 | Nitric Oxide inhibits insulin degrading enzyme. | |
| Yin et al., 2012 [115] | 2012 | Geniposide promotes IDE breakdown of Aβ; bacitracin inhibits insulin degrading enzyme. | |
| Quan et al., 2013 [86] | 2013 | In vivo, rat | Ginsenoside Rg1 upregulates proliferator-activated receptor y (PPARy); PPARγ can upregulate IDE expression and IDE can degrade Aβ1 - 42. |


