
Vitamin D, Cognition and Alzheimer’s Disease: The Therapeutic Benefit is in the D-Tails
Abstract
Since its discovery during the epidemic of rickets in the early 1920s, the physiological effects of vitamin D on calcium/phosphorus homeostasis have been thoroughly studied. Along with the understanding of its actions on skeletal diseases and advances in cellular and molecular biology, this misnamed vitamin has gained attention as a potential player in a growing number of physiological processes and a variety of diseases. During the last 25 years, vitamin D has emerged as a serious candidate in nervous system development and function and a therapeutic tool in a number of neurological pathologies. More recently, experimental and pre-clinical data suggest a link between vitamin D status and cognitive function. Human studies strongly support a correlation between low levels of circulating 25-hydroxyvitamin D (25(OH)D) and cognitive impairment or dementia in aging populations. In parallel, animal studies show that supplementation with vitamin D is protective against biological processes associated with Alzheimer’s disease (AD) and enhances learning and memory performance in various animal models of aging and AD. These experimental observations support multiple mechanisms by which vitamin D can act against neurodegenerative processes. However, clinical interventional studies are disappointing and fail to associate increased 25(OH)D levels with improved cognitive outcomes. This review collects the current available data from both animal and human studies and discusses the considerations that future studies examining the effects of vitamin D status on neurocognitive function might consider.
VITAMIN D METABOLISM AND MODES OF ACTION
Vitamin D was first discovered during the industrial revolution of the late 1800s during which England was facing an unprecedented epidemic of rickets. In 1918, Sir Edward Mellanby demonstrated that a nutritional deficiency was causing the disease and, soon after, young rickety patients were successfully treated using cod liver oil. 1α,25-dihydroxyvitamin D3 (1,25(OH)2D3), the active form of vitamin D, was isolated for the first time in 1922 by Elmer McCollum and named vitamin D since its discovery followed that of vitamins A, B, and C. Two years later, researchers from three different universities simultaneously discovered that the ultraviolet rays (UVR) from sunlight are a major source of vitamin D [1–3]. For historical and epidemiological reasons, this metabolite was named vitamin. Nowadays, it is considered a major steroid hormone synthesized in the skin under UV light and metabolized several times before acting on a large number of tissues.
Although UVB sunlight is the major source of vitamin D for humans, about 20% of our supply can be obtained from diet. Among the various forms of vitamin D, vitamin D2 (ergocalciferol) and vitamin D3 (cholecalciferol) dominate the scene. Vitamin D2 is produced photochemically by plants and fungi while vitamin D3 is synthesized in the deep layers of the skin in animals (Fig. 1). Both forms, once ingested, are metabolized in the same manner. Cholimicrons from the intestines bind vitamin D and transport it to the lymphatic tissue before entering blood circulation. Once it has reached blood circulation, vitamin D is activated, stored or distributed to its target tissues. In humans and rats, the primary storage sites of unmetabolized vitamin D are adipose tissue and skeletal muscle [4]. The classic vitamin D endocrine system is characterized by two successive hydroxylations, the first being catalyzed by 25-hydroxylases such as CYP2R1 or CYP27A1 in the liver to produce 25(OH)D and the second by CYP27B1 in the kidneys, ultimately producing the active form of vitamin D, 1,25(OH)2D (Fig. 1). Thus, both non-hydroxylated and hydroxylated forms of vitamin D can be found in blood. Like other steroid hormones, transport of vitamin D metabolites in blood circulation is dependent on binding to serum proteins such as vitamin D binding protein (VDBP) or albumin. VDBP binds vitamin D and other related metabolites such as 25(OH)D, 1,25(OH)D and 24,25(OH)D, with high affinity. 25(OH)D bound to VDBP accounts for 99.9% of total circulating 25(OH)D and allows for increased half-life of the metabolite [5]. The association between 25(OH)D and VDBP is essential for the endocrine renal synthesis of active vitamin D metabolites. Indeed, the uptake of 25(OH)D bound to VDBP into the renal proximal tubule cells has been shown to engage the endocytic receptor megalin [6]. This large transmembrane protein acts as a cell surface receptor for VDBP which results in internalization of the 25(OH)D-VDBP complex and conversion of precursor 25(OH)D into the active form by action of CYP27B1. We also now know that many tissues other than the kidney are capable of paracrine/autocrine regulation of 25(OH)D metabolism with the presence of both hydroxylating enzymes and the receptor in many cell types. The mechanism by which 25(OH)D is distributed to these various target tissues is not yet fully elucidated. Since protein bound vitamin D metabolites have limited access to target cells, unless endocytic receptors such as megalin or cubilin are present, it is generally believed that it is the remaining free fractions of 25(OH)D or 1,25(OH)2D that enter target cells by diffusion [5]. However, due to the disparity between likely amounts of free hormone available for passive diffusion and the levels required to efficiently occupy intracellular target receptors, this “free hormone hypothesis” needs furtherinvestigation.
There is some debate regarding the equivalent bioavailability of vitamin D2 versus vitamin D3. Some authors conclude that vitamin D2 is less efficient in raising serum concentrations of 25(OH)D or producing the active metabolite 1,25(OH)2D while others show no difference [7–9]. Because supplementation or fortification of food with vitamin D is the center of an important debate due to the high prevalence of worldwide insufficiency, it is important to assess the efficacy of each form. Several studies have reported that the administration of vitamin D2 leads to a reduction of total 25(OH)D and, more specifically, of circulating 25(OH)D3, leading to a reduction in the active form 1,25(OH)2D [10, 11]. This is potentially due to a competition between both forms for available 25-hydroxylase activity, the first hydroxylation step necessary in obtaining active vitamin D, although the in vivo regulation of the different 25-hydroxylases is not yet fully understood [10]. Furthermore, it seems that the route of administration (bolus versus daily) may affect the comparison of both vitamin D forms in maintaining plasma 25(OH)D, mainly due to a difference of half-life for each metabolite, theiraffinity for their transporters, hydroxylating enzymes or receptors.
According to the traditional model, steroid hormones bind to nuclear receptors and initiate genomic responses through modulation of transcription. Similarly, the vitamin D receptor (VDR) elicits physiological regulation of gene transcription following binding of its ligand, 1,25(OH)2D and, like other steroid hormone receptors, it can regulate a diverse range of biological activities across many tissues (Fig. 1). Advances in transcriptional research provide data that genomic effects are not the only mechanism of action for vitamin D activity [12, 13]. Rapid effects are also observed after treatment with vitamin D, which can be exerted through both the VDR and other membrane-based receptors such as MARRS (Fig. 1). The biological outcome of these two modes of action is relatively inter-related, and the multiplicity of mechanisms of action associated to the diversity of target tissues render this steroid hormone a complex pleitropicmolecule.
VITAMIN D STATUS AND PREVALENCE OF VITAMIN D DEFICIENCY
Vitamin D nutritional status is currently reflected by serum concentrations of 25(OH)D. Regulation of the circulating levels of 25(OH)D depends mainly on sun exposure and nutritional intake. There are several considerations relating to the topic of an “optimal” vitamin D level. Reaching a consensus on optimal vitamin D requirements is in constant debate as measurement methodologies may differ but primarily due to the diverging views on threshold determination. According to the Endocrine Society, vitamin D deficiency is defined by 25(OH)D levels below 20 ng/mL (50 nmol/L) and vitamin D insufficiency as a 25(OH)D of 21–29 ng/mL (52.5–72.5 nmol/L). According to the Institute of Medicine (IOM), seric 25(OH)D level of 16 ng/mL (40 nmol/L) is the median population concentration for maximal calcium absorption and overall bone health, while levels of 20 ng/mL (50 nmol/L) covers the needs of 97.5% of the population [14]. However, others consider 25(OH)D values of 30 ng/mL (75 nmol/L) or higher to be adequate [15, 16]. At the higher end of the spectrum, vitamin D intoxication is observed when seric levels of 25(OH)D are greater than 150 ng/mL (374 nmol/L) [15]. Recommendations for vitamin D requirements are established on the basis of optimal bone health and not on the extraskeletal functions of this steroid hormone. The assays used for measuring circulating hydroxylated vitamin D do not generally discriminate between the two forms present in the serum or the fractions of free and bound metabolites. Moreover, the recommended doses to obtain adequate levels are also under debate. For example, the IOM recommends for healthy adults 15 to 20 μg of daily vitamin D supplementation [14] while the Endocrine Society suggests 50 μg daily [16]. However there are inter-individual differences in the amount of vitamin D supplementation needed, in order to maintain adequate 25(OH)D levels between individuals of different age, body mass, or ethnicity [17]. A study evaluating vitamin D deficiency among different groups found that 96% of black Americans would be classified as deficient using a threshold of 25(OH)D concentration of 30 ng/mL (75 nmol/L). However, these participants had higher bone mineral density and higher calcium levels than white participants. It is suggested that a polymorphism in VDBP results in sufficient bioavailability of 25(OH)D despite its insufficient levels [18]. It therefore remains unanswered what is the optimal 25(OH)D level and if such a level even exists. Furthermore, it remains unexplained to what extent this level is affected by different factors, such as genetic polymorphisms in the VDR, metabolic enzymes or binding proteins, or other environmental determinants and if 25(OH)D serum concentration truly reflects the functional outcome of vitamin D action in other areas than its skeletalfunctions.
On the basis of such definitions, it has been estimated that 1 billion people worldwide suffer vitamin D deficiency or insufficiency, although this prevalence is still uncertain and difficult to account for [15]. According to a recent systematic review, this global public health issue varies strongly depending on geographical location, age, and gender, and it appears that all subgroups of population are at risk for vitamin D deficiency [19]. Another systematic review of 195 vitamin D studies involving more than 168,000 individuals from 44 countries reported that 37% of the participants had 25(OH)D seric concentrations below 20 ng/mL (50 nmol/L), and only 11.9% were above 30 ng/mL (75 nmol/L) [20]. Within these 1 billion people affected by vitamin D insufficiency, elderly people seem particularly at risk, especially when institutionalized or in hospitals. For instance, in Europe and the USA, at latitudes higher than 40°, between 40% and 90% of elderly people, depending on the definition of deficiency considered, suffer from hypovitaminosis D [21]. This high prevalence in the elderly community can be explained by several factors: decreased sun exposure and diminished levels of 7-dehydrocholesterol within the epidermis leading to reduced dermal production of vitamin D, poorer dairy and vitamin D intake, alteration of vitamin D metabolism due to renal or hepatic failure, and increased catabolism due to medication (immunosuppressors, glucocorticoids, antiepileptics) [22, 23]. A deficiency in vitamin D has been linked to many human diseases, particularly those that are age-related, such as Alzheimer’s disease (AD), cancer, cardiovascular disease, type II diabetes, multiple sclerosis, and various inflammatory disorders [24]. Hypovitaminosis D is therefore not to be undermined, not only for optimal bone health, but also in regards of all cause mortality, as pointed out by a recent systematicreview [25].
VITAMIN D AND ALZHEIMER’S DISEASE
A major public health problem is the progression of dementia and noncommunicable diseases such as AD. AD is a neurodegenerative condition characterized clinically by progressive cognitive decline and, histologically, by senile plaques and neurofibrillary tangles. The major component of senile plaques is the amyloid-β protein (Aβ), which is produced by the sequential proteolysis of a ubiquitous transmembrane protein, amyloid-β protein precursor (AβPP). Accumulation of Aβ, accompanied by increased inflammatory responses in the brain, is now viewed as a direct cause of neurodegeneration and cognitive decline [26, 27]. It is estimated that over 25 million people worldwide suffer from dementia, with a predicted 5 million new cases per year [28]. The etiology of the disease is still not fully understood. The first paper suggesting a link between vitamin D and AD dates back to 1992 when Sutherland and colleagues reported decreased VDR mRNA levels in the hippocampus of AD patients [29]. It is now relatively well established that patients with AD present lower concentrations of circulating 25(OH)D when compared to matched controls. Moreover, a consequential number of genetic studies have identified polymorphisms in the VDR or megalin genes that are associated to increased risks of cognitive decline or AD. The first study suggesting a possible genetic association between the VDR and AD dates back to 2007, where the authors indicate that a polymorphism in the VDR region increases the risk of AD by 2.3 times. Additionally, it appears that single nucleotide polymorphisms (SNPs) in the VDR gene might be a cause for some of the alterations in the vitamin D-VDR pathway [30, 31]. Genotyping of 563 participants over 85 years old for 5 different polymorphisms in the VDR gene revealed an association between gene variance and age-related changes in cognitive functioning. More specifically, carriers of BamI and TaqI polymorphisms presented with worse cognitive functioning unlike carrier of the ApaI variant [32]. Examination of ApaI and TaqI gene polymorphisms in 255 AD cases and 260 cognitively screened elderly controls revealed that the presence of each of these haplotypes was associated with the risk of AD [33]. More recently, the frequency of VDR polymorphisms TaqI, ApaI, FokI and BsmI were investigated in 108 AD patients versus 77 healthy controls of the Lower Silesian population cohort. The study did not reveal any significant difference between the two groups for frequency of the TaqI, Fok1 or BsmI polymorphisms. However, the frequency for allele A of ApaI was higher in the control group, which was later associated with a 30% lower risk of AD in Polish and British populations study. The authors note an important difference of risk alleles for these VDR polymorphisms depending on the population studied, suggesting a dependency on ethnic origin and climatic conditions [34]. Another study by Wang and colleagues demonstrated a strong association between a SNP within the transcription factor Cdx-2 binding site and late-onset AD (492 AD cases versus 496 controls). The authors show that the risk allele for CDX2 is associated with lower VDR promoter activity [35]. It was found that the frequency of “TaubF” haplotype (alleles of TaqI, ApaI, Tru9I, BsmI and FokI, respectively) was significantly higher in the patient group with AD compared to the controls and can be considered a risk factor for the disease [30]. Finally, sex-specific gene variations in the VDR and megalin genes have been shown to modify age-related cognitive decline in a cohort of US adults aged 50 years and older [36].
VITAMIN D METABOLISM AND ACTION IN THE BRAIN
Beyond the well-studied endocrine actions of this steroid hormone, it is increasingly recognized that several tissues and cell types display autocrine and/or paracrine activities and that these include the nervous system [37, 38]. The pioneer work of Stumpf et al., Wion et al., and Eyles et al., mapping out the presence and localization of vitamin D metabolites, related enzymes, and their receptor in the brain, has prompted the hypothesis that vitamin D acts as a neurosteroid in specific brain regions, particularly in those related to learning and memory [39–41]. The VDR is present throughout the embryonic and adult brain but at relatively low levels compared to other target organs such as gut and kidney [42]. Stumpf and coworkers were the first to describe target sites for 1,25(OH)2D3 in the brain, primarily in the neuroepithelium and proliferation areas [40]. VDR is also found in the temporal lobe, orbitofrontal cortex, cingulate gyrus of the thalamus, nucleus accumbens, amygdala and the entire olfactory system of adult brain. It is also expressed in hippocampal pyramidal neurons of the CA1, CA2, CA3 and CA4 layers of rodent and human brains [39, 40]. Furthermore, 1,25(OH)2D3 and related metabolites are present in human cerebrospinal fluid [43] and certain genes coding for its synthesis and degradation are expressed in the brain [39, 44–47]. Within the nervous system, VDR has been reported in many cell types including neurons [39, 47–50], glial cells [39, 45, 46], oligodendrocytes [51] and brain endothelial cells[52, 53].
Experimental studies in animal models or cell culture systems have largely contributed evidence linking 1,25(OH)D to neuroprotection. The numerous functions of vitamin D in the nervous system include regulation of neurotrophic factor production, neurotransmitter release, calcium homeostasis, oxidative stress mechanisms and modulation of immune system and inflammatory processes [38, 45, 46, 54–56]. These various processes are known to be altered during aging and in AD pathology, making vitamin D an extremely interesting therapeutic or preventive interventional tool [57]. Importantly, these in vivo and in vitro studies already demonstrate the pleiotropic action of vitamin D in the brain and the versatility by which this neurosteroid might act in a cell-, time-, and context-specific manner. There are clearly a number of ways in which vitamin D can affect brain development and function and these tend to underline a beneficial role for vitamin D against aging processes and neurodegenerative diseases that ultimately lead to cognitivedecline.
ANIMAL STUDIES: A NEUROPROTECTIVE ROLE OF VITAMIN D AND ENHANCED COGNITIVE ABILITIES
A clear result linking vitamin D action to aging processes was the generation of VDR knock out mice that presented with signs of premature aging [58]. Since then, an increasing number of experimental studies in animal or cellular models concur to establish that disruption of vitamin D metabolism and action leads to a perturbation of pathways known to be altered in AD and that supplementation with vitamin D rescues a number of AD markers in rodent models.
An unambiguous action on cognitive decline in aging or AD-like animals
Data obtained with vitamin D supplementation of animal models unambiguously show an improvement in memory and cognitive function along with a decrease in several markers of AD pathology. Interestingly, available experimental data points to a more pronounced effect of vitamin D when delivered to aging or AD-like animals. In a recent study, Latimer et al. showed that a high vitamin D3 diet (10,000 IU/Kg/day) lasting 5 to 6 months prevented cognitive decline in aging rats. More specifically, the authors described the superiority of a high diet compared to a low and medium one (100 or 1,000 IU/Kg, respectively) in significantly improving performance in the Morris water maze (MWM), a hippocampal-dependent learning and memory task [59]. At approximately the same time, Briones and Darwish performed subcutaneous injections of 1,25(OH)2D3, for 21 days, to both young and aged rats (6 months and 20 months, respectively) and observed an attenuation of cognitive impairment only in older animals [60, 61]. Both these studies underlie a role for vitamin D supplementation in the preservation of cognitive function in aging rats. Concerning AD, an important number of animal models now allow for understanding and compound testing in order to better apprehend and possibly treat this debilitating disease. Several groups have used these models to test the hypothesis that vitamin D has protective effects in AD. Taghizadeh et al. demonstrated that the absence of vitamin D in the diet of an adult AD rat model intensifies learning and memory deficits measured by the MWM [62]. A second study by Yu and colleagues used young AβPP transgenic (Tg) mice, under a 12 IU/g vitamin D diet and compared their behavior to those under a control (2.4 IU/g) or deficient (0 IU/g) diet from weaning to 6 months of age. They found an amelioration of performance in the MWM for the animals under the higher vitamin D diet compared to the other groups. However, the group does not present data on the effect of a vitamin D enriched diet in non-AD animals [63]. Our group has observed that a 4-month vitamin D3 supplementation in an AD mouse model (5xFAD mice) does not impact memory function at a young age (5 months) when no cognitive deficits are present (unpublished data). Conversely, vitamin D3 rescues working memory deficits at 8 months of age but this effect is only observed in Tg animals and not in aging wild-type (Wt) mice [64]. Similarly, intraperitoneal injections of 1,25(OH)2D3 for 8 weeks in 8-week-old animals restores learning and memory deficits in an AD mouse model but does not affect cognitive function in their Wt counterparts [65]. To our knowledge, the only study demonstrating an effect of vitamin D treatment preferentially in Wt animals is that of Bennett and colleagues. The authors conducted a study based on a vitamin D2-enriched button mushroom diet in Wt and AD Tg mice. Behavioral analysis was carried out after 7 months of supplementation and revealed subtle changes in cognitive function. Vitamin D2 improved cognitive performance more reliably in Wt mice rather than Tg [66]. The main difference with other studies is the use of vitamin D2 rather than D3. Animal studies therefore support the hypothesis that vitamin D action is likely to be age-dependent and show a clear effect of vitamin D supplementation in restoring cognitive deficits in an AD-like context. The mechanisms by which vitamin D can impact AD pathology are yet to be fully elucidated although a number of processes at play during the development of the pathology have been shown to be targeted by the vitamin D signaling system. Considering the mode of action of this steroid hormone, it is likely that its biological function will be multiple, ranging from direct action on amyloidogenesis to restoration of neurotransmission, modulation of inflammation, regulation of apoptosis, oxidative stress, and vascular processes (Fig. 2). Several studies have made important contributions to the understanding of vitamin D mechanism of action in an AD-like brain.
Vitamin D and amyloidogenesis
On top of the observation that vitamin D supplementation leads to improved cognitive function, all the studies in an AD-like context have also shown that vitamin D treatment, regardless of the model tested, the dosage, the molecule chosen, and the time of treatment decreases the amyloid burden, suggesting a link between vitamin D function and amyloidogenesis [60–66]. The behavioral changes reported in the study by Yu and colleagues were accompanied by a diminished Aβ load, along with an increase in astrocytic reactivity, NGF levels, and decreased TNFα in the brain of treated mice. Interestingly, the authors show a change in AβPP processing, with a variation in AβPP end products depending on the vitamin D regimen [63]. Grimm and collaborators recently studied the impact of a relatively low hypovitaminosis D on AβPP processing in mice and cell cultures. The data reveal an increase in Aβ caused by an increase in β-secretase level and activity, concomitant with a decrease in neprilysin levels [67]. Our team has recently demonstrated that a 5-month vitamin D3 enriched diet decreases the number of amyloid plaques in the frontal cortex, neocortex, and hippocampus of female 5xFAD mice. This reduction is accompanied by decreased astrocytic reactivity and increased microglial activation in the frontal cortex. Moreover, transcriptomic profiling of these brain regions reveals that vitamin D3 supplementation regulates the expression of a large number of genes associated to AD in both Wt and Tg animals [64]. Other clues indicating direct interactions between vitamin D and the amyloid pathway arise from several studies. For instance, overexpression of the VDR or treatment with vitamin D was shown to suppress AβPP transcription in neuroblastoma cells [35]. VDBP, which is increased in thecerebrospinal fluid of AD patients, was also shown to interact directly with Aβ in the brain of both AD patients and animal models. Binding of VDBP with Aβ was associated with reduced aggregation of the amyloid peptide and cellular death in vitro. Overall, the presence of VDBP rescues animal models from synaptic loss and memory deficits induced by Aβ [68]. A team provided extensive work on the crosstalk operated between vitamin D and Aβ signaling pathways. After demonstrating that Aβ treatment of hippocampal and cortical neurons induces VDR repression, they showed that VDR silencing in vitro leads to similar effects to Aβ treatment. Their studies further indicate that vitamin D treatment of cortical neurons reverses the cytotoxic effects induced by Aβ mentioned above as well as prevents upregulation of iNOS [48, 49, 69, 70]. Further in vitro studies examined the effect of vitamin D in combination with memantine on cortical axon degeneration. These two compounds act in a synergistic manner to protect against glutamate or Aβ-induced toxicity in cortical neuronal cultures [71]. Altogether, these findings strongly support a role for vitamin D signaling in reducing amyloidogenesis, one of the hallmarks of AD pathology. Moreover, the putative crosstalk between Aβ pathology and VDR signaling appears to provide neuroprotective mechanisms againstAβ-induced toxicity potentially limiting the decline in cognitive function. This underlies a specific action of vitamin D in preventing or halting cognitive impairment in AD, but what other mechanisms are at play in a “healthy” aging brain?
Synaptic transmission and calcium homeostasis
Brain aging is partly characterized at the cellular level by neuronal death, decreased neuronal excitability, impaired synaptic transmission and altered calcium homeostasis. Along with regulating amyloid processing and deposition, ultimately modulating neurotransmission, vitamin D has also been shown to act on calcium channels and neurotransmitter release in aging brains. Analysis of electrophysiological recordings after vitamin D treatment reveals a modulation of L-type calcium channel (L-VGCC) current and density in CA1 hippocampal neurons of aged rats [72] but no change in long term potentiation (LTP) in CA1 neurons of normal rats [59, 73]. However, a significant increase in neuronal excitability in aging rats [59] and synaptic potentiation in the CA3-CA1 pathway of an AD rat model has been described [74]. Vitamin D-VDRpathway disruption or activation of calcium channels in hippocampal neurons has been reported in several studies. VDR silencing in vitro leads to upregulation of L-type voltage sensitive calcium channels (LVSCC) and NGF, a process typically observed in aging neurons while vitamin D treatment of cultured neurons decreases LVSCC expression, conferring neuroprotection in an aging nervous system [47–49, 69, 72, 75]. Conversely, absence of 1,25(OH)2D3 in mice induces an upregulation of LVGCCs in the dentate gyrus of 8-week-old mice, which results in increased cell proliferation and neurogenesis [76]. It appears that depending on the time frame for treatment, vitamin D may modulate both neuroprotection and neurogenesis by finely regulating the expression of calcium channels on a timely basis. The study by Latimer and colleagues further reveals that vitamin D3 treatment directly modulates the expression of genes and proteins involved in synaptic vesicle trafficking and neurotransmitter release in aging rats [59]. A result confirmed by the transcriptomic study performed in AD female mice after 5 months of vitamin D3 supplementation [64]. Interestingly, earlier work on the effects of vitamin D deficiency during development had already demonstrated that the adult brain later displayed altered expression of a number of genes and proteins involved in calcium regulation, synaptic plasticity and neurotransmission [77, 78]. This consolidates the hypothesis that vitamin D deficiency at earlier ages has repercussions on nervous system processes such as neurotransmission, implicated in aging and AD-like pathology.
Regulation of oxidative stress, brain energy metabolism, and vascular processes
Another possible mechanism for Aβ clearance operates through passage of the blood-brain barrier (BBB). This physical, relatively impermeable, barrier is composed mainly of endothelial cells, astrocytes, and pericytes. In 2011, Ito et al. demonstrated that, 24 hours following an intraperitoneal injection of 1,25(OH)D in mice, elimination of radioactive human Aβ was increased 1.3 times and that the total level of brain Aβ was reduced. This brain-to-blood efflux likely involves internalization of Aβ peptides within brain capillaries [53]. This hypothesis is further confirmed by the work of Durk and colleagues who report increased expression and transport activity of P-glycoprotein in brain capillaries and endothelial cells after incubation with 1,25(OH)D3, resulting in reduced accumulation of human Aβ within these cells [52]. These vitamin D-related activities likely occur via activation of the nuclear receptor VDR present on the cells comprising the BBB [52, 53, 79, 80]. Moreover, action of 1,25(OH)D on brain pericytes elicits anti-inflammatory responses at the transcriptomic level, likely adding to the direct protective role of vitamin D within brain capillaries under chronic inflammation constraints such as those present in AD [80]. In line with these results, the transcriptomic response of cultured mixed neuron-glial cells or whole brain regions such as the neocortex and hippocampus to 1,25(OH)D in normal conditions or in AD mice, reveals dysregulation of genes potentially capable of limiting the progression of neurodegenerative diseases with particular emphasis on AD [64, 79]. Vitamin D can also inhibit the production of inducible nitric oxide synthase (iNOS) an enzyme necessary for synthesis of nitric oxide (NO). During aging or neurodegenerative diseases such as AD, reactive nitrogen and oxygen species produced by damaged neurons, microglia and astrocytes can increase the expression of iNOS. As a result, higher levels of NO can cause cell death through inhibition of mitochondrial and neuronal respiration, further leading to neuronal excitotoxicity [81–83]. Low vitamin D diet has been shown to enhance oxidative stress through transcription of iNOS. More specifically, low levels of vitamin D result in increased tyrosine nitration of brain proteins, alterations in glucose metabolism and mitochondrial changes in the brain of rats aged 16-17 months [84]. Conversely, 1,25(OH)2D3 injections of elderly rats increases brain energy metabolism and modulates levels of reactive oxygen species in hippocampal tissues [61]. Vitamin D treatment of cortical neurons reverses the cytotoxic effects induced by Aβ and prevents upregulation of iNOS [49, 69, 70]. In activated microglia, 1,25(OH)2D3 suppresses the production of NO and the inflammatory mediators, TNFα and interleukin-6 (IL-6), in a dose-dependent manner suggesting direct anti-inflammatory roles for vitamin D in the brain [85]. Local conversion of vitamin D to the active 1,25(OH)2D3 in the brain may be a direct neuroprotective response to CNS inflammation followed by inhibition of NF-κB related iNOS induction [86].
Modulation of inflammatory processes
Another very important component of neurodegenerative diseases such as AD is the establishment of chronic inflammatory processes. Vitamin D is now known as an effective immune modulator and as such has the potential to regulate the inflammatory state in AD pathology. Vitamin D can i) upregulate expression of several neurotrophins, ii) increase secretion of the anti-inflammatory cytokine IL-4, iii) reduce secretion of pro-inflammatory cytokines TNF-α and interleukin-1 beta (IL-1β), and iv) inhibit differentiation of dendritic cells [44–46, 87]. The VDR has been observed on human and rodent microglia, the brain macrophages responsible for immune surveillance and phagocytosis [88]. Supplementation with vitamin D3 for 5 months largely impacts inflammatory and immune gene expression profiles translating into improved functional outcomes in an AD mouse model [64]. Administration of 1,25(OH)D for 21 days induced a change in the inflammatory state and amyloid burden of aged rats (20 months) but had no significant effect in younger ones (6 months). More specifically, the increase in pro-inflammatory cytokine Il-1β and the decrease of anti-inflammatory cytokine Il-10, observed in aged compared to young rats, is reversed after subcutaneous injections of vitamin D [89]. Lee at al. showed that 12-month-old mice injected with vitamin D displayed a change in retinal macrophage morphology and number, indicative of increased phagocytic activity. This process was accompanied by a reduction in complement component C3, a chronic inflammation marker. Overall, this resulted in decreased Aβ load in the retina of these mice, improving visual function [90]. In AD, macrophage phagocytosis of Aβ is defective resulting in decreased Aβ clearance and increased Aβ deposition and inflammation in the brain. Mizwicki et al. demonstrated that vitamin D3 is capable of promoting recovery of Aβ phagocytosis in cultured human macrophages from AD patients possibly by rebalancing the inflammatory state through the modulated transcription of TLRs, IL-1, IL1R1, and chemokines [89, 91].
On top of the mechanisms discussed above concerning the neuroprotective activities of vitamin D during aging or in an AD-like context (Fig. 2), a certain number of hypotheses need to be tested. These are based on the common pathways altered in AD and known to be targets of 1,25(OH)D in the brain or elsewhere. These include action on miRNAs, TLRs, angiogenesis, glycation, and oxidative stress, all these processes being altered in aging brains and particularly in AD [92]. The road to unraveling the mechanisms of action of vitamin D in an aging brain is a long one, as it is now clear that this misnamed vitamin is a potent neurosteroid with pleiotropic actions that are tissue-, time-, individual-, dose-, and pathological context-specific. Due to its capacity to interact with both nuclear and membrane receptors, the downstream effects of vitamin D supplementation are multiple and crosstalks between various signaling pathways render its study rather complex. It is therefore not surprising that clinical observations lead to contradictory results as we are only starting to discover the full potential of this steroid hormone in the nervous system.
HUMAN STUDIES: A CONTROVERSIAL LINK BETWEEN VITAMIN D STATUS AND COGNITIVE OUTCOME
Linking low seric vitamin D concentration with cognitive outcomes in humans has been done through both cross-sectional and longitudinal studies but, to this date, very few interventional studies have been performed. Most works assessed blood vitamin D status, mainly in aging population groups, while only a few used dietary intake as an indicator of vitamin D level. The observed outcomes of these studies are either measures of cognitive performance, incidence of dementia or AD. The main findings from these various studies are reported in Table 1. Overall, observational studies underline an association between vitamin D status and cognitive outcome although this appears to be dependent on a number of factors such as aging or gender. A meta-analysis by Etgen et al. suggested a more than doubled risk of cognitive impairment in patients with vitamin D deficiency [122]. Another systematic review that included 25 cross-sectional and 6 prospective studies demonstrated that in 72% of the studies, lower 25(OH)D was associated to a significantly worse outcome in different cognitive function tests or was linked to a higher incidence of dementia [123]. A meta-analysis by Balion et al. found that AD patients had vitamin D blood concentrations 6.2 nmol/L lower than controls [124]. This difference was found to be ‘clinically relevant’ in another meta-analysis [125]. Moreover, such association is not only for the severe stages of AD [126], but from the prodromal stage (also known as mild cognitive impairment, MCI) [101], which corresponds to an isolated minor neurocognitive disorder with no impact on functional autonomy in daily life activities. Interestingly, it is also reported that hypovitaminosis D is associated with subjective cognitive complaint, a sign that occurs much before the first objective signs of AD and predicts future cognitive decline and dementia [127]. Both these latter findings underline that hypovitaminosis D accompanies the onset of the first AD symptoms and could therefore contribute to the initiation of dementia. However, to date, interventional studies fail to show improvement in cognitive function after vitamin D supplementation. Here, we discuss the specificity by which vitamin D could be linked to cognitive performance in humans.
A potential gender effect
Interestingly, studies performed with female, male, or mixed populations show contrasting results. All studies based on women cohorts report a clear association between hypovitaminosis D and cognitive impairment, while the unique study in men reports no association and reports from both male and female populations are controversial (Table 2). In one study, Annweiler et al. found that low dietary intake of vitamin D (defined as <35 μ/week, or 400 IU/year) in a large cohort of women of mean age 80.5, was significantly associated with global cognitive performance as measured by the Pfeiffer Short Portable Mental State Questionnaire (SPMSQ) [96]. In a second cross-sectional study, they found that vitamin D deficiency, defined as <10 ng/mL (<25 nmol/L), was associated with lower SPMSQ scores in elderly women aged 75 years and older [95]. Conversely, a study among healthy, older Europeans aged 55 to 87 years, demonstrated that seric 25(OH)D concentrations of >32.1 ng/mL (>80 nmol/L), considered optimal, were associated with significantly fewer errors in tests of spatial working memory, particularly in women [111]. One 4-year longitudinal study of 6,257 elderly women reports that lower 25(OH)D blood concentrations at baseline were associated with increased incidence of global cognitive decline measured by the Mini-Mental State Examination (MMSE) test in elderly women [97]. Importantly, this was not observed in a similar study performed in men exclusively [99]. Association of baseline serum 25(OH)D deficiency with the onset of dementia or AD was also observed in two cohorts of women over 75 years old [93, 94]. However, a post-hoc analysis of the largest clinical trial on this topic, the Women’s Health Initiative Calcium and Vitamin D trial, which included over 4,000 women aged 65 and older, found no effect of a 7.8 year supplementation with 100 mg calcium carbonate and 400 IU of vitamin D3 on the incidence of dementia, MCI, or other cognitive measures [98]. Of important note, supplementation with vitamin D during this clinical trial was associated with calcium supplementation. This renders interpretation of results more difficult as it is known that high calcium levels are associated with increased risk of cognitive decline in humans and animals [128, 129]. The dual supplementation could therefore be counterproductive [130]. No interventional study has been performed in men exclusively and 8 pre-post or randomized control trials have been performed in a mixed population (Table 2). Among them, cognitive improvements were found after 8 weeks of supplementation for global cognitive performance [121] and after 4 weeks [120] to 16 months of supplementation [115] for executive functioning. However, these studies were pilot nonrandomized pre-post studies performed in small cohorts of patients. Despite this limitation, the latter findings were consistent with the result of one placebo-controlled randomized clinical trial that reported after 6 months an improvement of information processing speed in the supplementation arm compared to the placebo arm [119]. Nevertheless, a meta-analysis concluded that, although cognition favorably evolves in patients receiving vitamin D supplementation (“before and after” approach), the between-group comparison of the cognitive scores at the end of the follow-up (“comparative” approach) do not show better cognitive scores in the supplementation arm compared to the comparative arm [131]. Thus, although, there appears to be a clear link between vitamin D status reflected either by 25(OH)D serum levels or nutritional intake of vitamin D and cognitive function in women, these studies do not allow for comparison of both genders and might not be representative of a general population. Moreover, some studies have shown such associations in mixed population while others report no link (Table 2), forcing us to consider other possible confounders.
A possible age threshold
The latest study on the topic of vitamin D and cognitive decline was performed in individuals of different ages obtained from The Tromso study. The report highlights a positive association between serum 25(OH)D and cognitive function in individuals aged 65 years and over [103]. The interesting result from this study is that levels of 25(OH)D appear to be predictive of cognitive outcome only in older individuals. Memory and cognitive performance tests were performed at different time points, over a period of 7–13 years, and only the latest time points were significantly correlated with baseline vitamin D levels [103]. We therefore decided to compare studies that allowed for a clear distinction between ages, i.e., studies performed on individuals strictly under 65 or over 65 years of age. As reported in Table 2, this criterion allowed us to discriminate 6 studies conducted in individuals less than 65 years of age and 14 studies comprising of individuals aged over 65 exclusively. The results are quite pronounced as all studies, except one, investigating individuals under 65 years of age report no association between 25(OH)D serum levels and cognitive performance, and conversely nearly all studies in older individuals report a link between vitamin D status and cognitive outcome (Table 2). Cross-sectional studies in adolescents (age 12–16.9 years), based on the data from the NHANES III cohort, found no association of 25(OH)D seric levels and performance in Intelligence Scale and Wide-range Achievement Test [110, 113]. Similar reports, carried out in young adults (20–59 years), showed once again no significant association between 25(OH)D concentrations and measures of cognitive function [110, 113]. Another study examined the link between cognition and vitamin D in earlier stages of life. It demonstrated no correlation between seric levels of both 25(OH)D2 and 25(OH)D3 at mean age 9.8 years, with academic performance outcomes at ages 13-14 and 15-16. This study included a cohort of 3,171 individuals [112]. The only study, to our knowledge, describing an association between cognitive performance and seric 25(OH)D levels in non-aged, non-demented individuals was carried out in a lifespan approach by Maddock and colleagues in 2014 [109]. This study included 6,496 mid-life individuals from a British cohort whose information on childhood cognitive ability, educational attainment, and vitamin D-related behaviors was collected at several points from birth to age 50. At the age of 45, seric vitamin D concentration was measured and, at age 50, verbal memory, verbal fluency, and speed of processing were tested. The results showed a non-linear U-shaped association between 25(OH)D concentrations at age 45 years and immediate word recall at age 50 years, i.e., people with both lower and higher 25(OH)D concentrations performed worse on the cognitive task [109], but this result was likely due to the recent supplementation of those with prior hypovitaminosis D. The controversial link between low vitamin D status and cognitive performance in non-aged individuals has led to only one randomized control trial during which 5,000 IU of vitamin D3 was given daily to young adults for 6 weeks. Thecontrol trial concluded that vitamin D supplementation had no effect on cognitive and emotional measures, such as working memory and depression although enhanced working memory performance over time was observed in both the supplemented and comparative group [118]. The short period of supplementation and the relatively high serum 25(OH)D concentrations at baseline, possibly above the cerebral needs for vitamin D (if any), could have masked the cognitive effect of vitamin D supplementation. However, the null findings concerning vitamin D status and cognitive outcome in young individuals indicates that the time-frame considered for associating 25(OH)D to cognitive performance is of prime concern and that longer follow-up times from longitudinal studies might help decipher a link between hypovitaminosis D at younger ages and cognitive decline in elderly populations. Among the studies investigating the link between vitamin D status and cognitive outcome in elderly populations, two large population-based reports indicate no association between 25(OH)D levels and cognitive performance [110, 114]. Interestingly, this contrasts largely with a study by Llewellyn and colleagues, which performed analysis on the same NHANES III cohort than the above-mentioned studies [106]. The two major differences are i) Llewellyn et al. focused on individuals aged strictly 65 years and older whereas McGrath et al. and Tolppannen et al. included a larger age group, from 60 to 90 years old, and ii) the cognitive measures considered for assessment of performance differed. It appears that the effects of hypovitaminosis D on brain function and cognition do not emerge until later in life. Most importantly, it is possible that vitamin D status does not impact cognitive measures in healthy individuals but is rather linked to the onset of cognitive decline during aging. Finally, the type of cognitive test used for analysis might impact results as vitamin D status could potentially be associated to domain-specific functions.
AD versus non-pathological aging
A review of 37 studies from Balion et al. allowed the authors to compare MMSE scores with levels of 25(OH)D. They found that higher average MMSE scores were associated with 25(OH)D concentrations >50 nmol/L and ultimately that lower vitamin D levels increased the risk for AD [124]. Another meta-analysis concluded that an individual without AD shows a 40% increased chance of having higher vitamin D levels than matched controls [125]. Despite a growing amount of studies linking lower vitamin D to cognitive decline in an associative manner, data specific to hypovitaminosis D and incidence of AD is scarce. However, in every study, a link between vitamin D status and dementia has been observed while studies investigating vitamin D deficiency in the case of non-pathological aging is more controversial (Table 2). Two 7-year follow-ups, the first in 40 women of mean age 78.4 years [93] and the second in 498 women aged 75 years and older [94] aimed at studying the association between levels of vitamin D and the risk of AD. The first report, performed on a relatively small, restricted cohort showed that baseline serum 25(OH)D deficiency associates with the onset of non-AD dementias but not with AD [93]. However, the second study, based on vitamin D dietary intake and including a larger population found that baseline dietary intake of vitamin D was inversely associated with the onset of AD but not with non AD-dementia [94]. These apparent contradictory results could be due to the difference in cohort size for both studies or to the measure used for assessment of vitamin D status. In line with the second observation, two recent longitudinal studies have put forward an association between low levels of seric 25(OH)D and the risk of AD [100, 105]. One study followed 1,658 men and women of mean age 73.6 years for 5-6 years [105], while the second was performed on 10,168 individuals over a period of 30 years [100] and both report an increased risk for AD with decreased levels of 25(OH)D. The latter study, due to the long period of time considered and the size of the cohort, provides a solid basis to the hypothesis that vitamin D deficiency at younger ages is associated to increased risk of cognitive decline, dementia, or AD later on.
To our knowledge, only three prospective studies have investigated the effects of vitamin D supplementation in AD patients. A first study by Stein and colleagues consisted, for phase 1, in the administration of 3,000 IU per day of vitamin D2, for 8 weeks, in 13 older patients. The group found that the increased seric levels of 25(OH)D were accompanied by a significant improvement in the Alzheimer’s Disease Assessment Scale (ADAS-cog) score. However, the second phase of Stein et al.’s study, consisting of a randomized control study in which participants received either physiological doses (1,000 IU per day) or supra-physiological doses (6,000 IU per day) of vitamin D2 for 8 weeks, showed no difference between groups in terms of the ADAS-cog score or the MMSE [121]. However, the very small number of participants in this study along with the shortduration of treatment and possibly the use of vitamin D2 impedes general conclusions to be drawn. The second pre-post study proposed the combination of memantine, a modulator of NMDA receptors, and vitamin D as an effective treatment for improving cognition in AD patients. This pilot study demonstrated the efficacy of combining both treatments for 6 months measured by an increase of MMSE scores compared to the administration of vitamin D or memantine alone [116]. Other clinical trials, based on vitamin D supplementation and assessing cognition decline, as a primary or secondary outcome, are currently performed in aging patients (VITAL-cog, DO HEALTH) or with pathologies such as amnesia(D-cog), Alzheimer’s disease (AD-IDEA), Parkinson’s disease, and type II diabetes (THINK-D). Details can be found on the website clinicaltrials.gov.
A positive effect on executive functions and not episodic memory
As described above, there is robust evidence that vitamin D insufficiency is associated with cognitive impairment as a whole among older adults [122]. However, it is not fully elucidated which domain-specific cognitive functions are disturbed in the case of vitamin D insufficiency and explain the association of serum 25(OH)D concentration with the composite cognitive scores. For instance, as mentioned previously, the contradictory studies by McGrathet al., Tolppanen et al., and Llewellyn et al. based on the cohort from the NHANES III survey might be representative of the different cognitive measures incorporated for analysis. In the work by Llewellyn et al., the authors found an association of 25(OH)D with cognitive impairment calculated as a composite score, i.e., a global measure of cognitive performance assessing both memory, learning, orientation, and attention [106]. In contrast, in the initial observations by McGrath et al. and Tolppanen et al., the authors used only a “memory and learning score”, and found inconclusive results [110, 114], making it unlikely that episodic memory is the domain-specific cognitive function primarily altered in the case of vitamin D insufficiency. A meta-analysis was consistent with this assumption, and showed that the association between vitamin D insufficiency and episodic memory remains uncertain due to modest and clinically non-relevant results [131]. In contrast, this meta-analysis provided compelling evidence that lower serum 25(OH)D concentrations predict executive dysfunction [131]. This hypothesis is corroborated by the findings of Buell and colleagues investigating elders (aged >65) receiving home health services. The authors found that cognitive performance, particularly executive function and not memory, was associated with circulating vitamin D of 50 to 75 nmol/L [102]. An interventional study by Annweiler and colleagues also demonstrated that supplementing elderly individuals with vitamin D3 during 16 months improved executive function [115]. Executive functions refer to a heterogeneous set of high-level processes that control and regulate other abilities and behaviors [132]. To override this heterogeneity, a more recent study aimed to determine which executive subfunction was disturbed in the case of vitamin D insufficiency and found a decrease in mental shifting, but neither in cognitive inhibition nor in information updating, among community-dwelling older adults with vitamin D insufficiency [133]. Mental shifting is the ability to adjust thinking or attention in response to changing goals and/or environmental stimuli in order to behave appropriately [132]. This may thus help to better understand the involvement of vitamin D in the cognitive decline and in the course of dementia. However, to make firm conclusions about the domain-specific cognitive functions affected by vitamin D insufficiency, further studies using a universal battery of cognitive measures will help decipher the subtle specificities by which vitamin D levels are associated to cognitive outcome.
CONSIDERATIONS FOR FUTURE STUDIES
The usual course of action for implementing a drug in clinical trials is to study it thoroughly at the preclinical level before moving to human studies. However, as pointed out by the number of clinical and preclinical studies on vitamin D in health and disease, it appears that clinical observations are now accumulating at a faster pace than animal evidence which somewhat impedes the understanding of the basic neurobiology of vitamin D action in the brain. Therefore, future studies, in both animal and cellular models, might consider several factors in order to better understand the mode of action of this steroid hormone in aging and AD pathology.
So far, very few animal studies have concentrated on models of vitamin D deficiency to better mimic what happens in humans. Especially since vitamin D deficiency in early stages of life has been suggested to strongly impact brain function during aging [77, 78, 134], developing or refining existing models of vitamin D deficiency at different time-points, for varying periods of time, perhaps followed by vitamin D supplementation at various ages might help clarify the impact of such deficiency and when to intervene in relation to cognitive function and AD. Moreover, no animal study, to our knowledge, has compared the effects of vitamin D2 versus vitamin D3 in the context of cognitive impairment nor has assessed possible gender differences in response to vitamin D treatment. Due to the complex pathogenesis of neurodegenerative diseases such as AD, it is unlikely that a single event such as vitamin D deficiency is responsible for disease occurrence. However, hypovitaminosis D might represent a real aggravating factor in this context and this deficiency could be easily corrected. Lastly, the use of animal or cellular models with ongoing efforts will undoubtedly help cast light on the mechanisms by which this potent neurosteroid acts on an aging or AD-like brain.
However, although the human pathological and functional characteristics appear similar in animals, it is difficult to model the various causes, insidious onset, and potentially long-term progression of complex diseases such as AD in mice with a lifespan of months rather than decades. As a result, several issues relating to the translation of animal results to human studies can only be addressed through human studies. Keeping in mind the necessity to move toward more personalized interventional medicine, we can cite here some of the key points that need to be assessed in future trials.
The genetic background
One major difference between animal models and humans is the controlled genetic and environment that offers laboratory work. The possibility that individual differences in vitamin D metabolism exist is not to be undermined as polymorphisms in VDR have been reported as potential risk factors for AD [30, 32, 33]. VDR is not the only metabolite required for proper vitamin D action, as the several enzymes involved in metabolism are key to drive functional vitamin D outcome. If polymorphisms or simply decreased activity of these enzymes is present in certain individuals, vitamin D treatment might not be effective. Moreover, it still remains unknown how vitamin D metabolites enter the nervous system, whether it be by passive diffusion or the use of energy-dependent transporters such as megalin. A recent report suggests that sex-specific gene variations in both the VDR and megalin receptor associate with age-related cognitive decline in a relatively large cohort of US citizens [36]. Thus, defects in transport, metabolism, or action of vitamin D metabolites in an individual-dependent manner need to be taken into considerations when implementing adaptive clinical trials possibly based on Bayesian models rather than classical randomized control trials.
Assessing a possible gender effect
Inter-individual differences, especially inter-gender dissimilarities, should also be taken into account. Sex differences in observed associations of megalin SNPs may be related to interaction of megalin with both estrogen and vitamin D and competitive binding of both steroid hormones to this shared endocytic receptor. Our team has recently observed in a mouse model of AD that vitamin D3 treatment impacts male and female mouse differently, at the behavioral level, according to the time frame for supplementation (unpublished data). Moreover, we have shown that vitamin D likely interacts with the estrogen receptor, Esr1, to regulate molecular pathways relevant to AD pathogenesis [64]. A number of studies suggest a strong link between the drop in estrogen levels occurring during menopause and the development of AD [135–137]. Animal studies could help clarify this matter; however, future clinical trials might consider stratifying results in a gender-specific manner.
Assessing vitamin D status and responsiveness for nervous system related functions
Whether 25(OH)D serum levels appropriately reflect an individual’s capacity to use vitamin D metabolites, particularly in relation to nervous system functions, is of great importance. For instance, conditions such as liver disease and nephrotic syndrome result in reduced VDBP and albumin levels that lead to a reduction in total 25(OH)D and 1,25(OH)D levels without necessarily affecting the free concentrations of these metabolites [138]. As an example, ethnic differences in 25(OH)D levels have been reported. African Americans present lower VDBP levels, hence lower total 25(OH)D levels but without evidence of vitamin D deficiency, potentially due to the action of stable levels of free fractions of 25(OH)D or 1,25(OH)D [18]. If the free fractions are the ones to enter the nervous system, total 25(OH)D serum levels might not reflect the levels of vitamin Dmetabolites entering the brain. Moreover, megalin has been shown to be present at the BBB, in choroid plexus, suggesting a possible role for endocytic uptake of vitamin D or 25(OH)D linked to VDBP into nervous system tissues. The presence of CYP27B1 in the brain allows for local synthesis of 1,25(OH)2D3 in this tissue creating an autocrine/paracrine system for metabolism of active vitamin D compounds. In such systems, metabolites are produced, act and are degraded locally without affecting serum 25(OH)D levels. Maybe alternative approaches to complement the measurement of circulating 25(OH)D are now required to evaluate more precisely an individual’s response and needs for vitamin D in the context of brain function. One approach might be to consider CSF levels of vitamin D metabolites rather than serum levels as a reflector of vitamin D status in the nervous system. 25(OH)D, 24,25(OH)D and 1,25(OH)D have been shown to be present in human CSF, although in lower concentrations than in sera [43] and are reduced in AD patients [139]. A complementary approach to measuring either serum or CSF levels of vitamin D metabolites could be to measure genomic effects of vitamin D on its target genes for profiling an individual’s vitamin D status and capacity to respond to supplementation [140].
An appropriate supplementation according to baseline levels
Since recommendations regarding the use of cholecalciferol supplementation to reach optimal levels of 25(OH)D in humans are under debate, possible conversion of doses from animals to humans might be considered in the context of neurological diseases. Indeed, animal studies show improvement of cognitive function in animal models of aging or AD with specific doses of vitamin D. However, the method for translating doses from animal models to human clinical trials should be carefully considered. Deriving interspecies equivalents for therapeutic interventions has usually been done using Body Surface Area scaling; however, it appears that this might not be as adequate as advanced allometric and physiologically based pharmacokinetic modeling for animal-to-human dosage conversion [141]. This is particularly relevant in the case of vitamin D supplementation, as assessing the “pharmacologically active dose” required in regards to brain function is of great need.
Daily intake of 800 IU is the current recommendation of the IOM for people aged 70 years and older based on estimates that this dose will produce or exceed a threshold of 25(OH)D of 20 ng/mL in 97.5% of people [14]. A randomized double blind study of elderly nursing home residents showed that after 16 weeks of 800 IU/day of vitamin D3, 25(OH)D levels were 33 ng/mL. There was little difference between an 800 IU/day and 2,000 IU/day supplementation except for one participant who did not reach 20 ng/mL under the 800 IU/day regimen indicating that some individuals need higher doses. With 2,000 IU/day, no one reached 50 ng/ml or higher. This study also underlined the complex relationship between baseline 25(OH)D status and responses to supplemental vitamin D. When using doses of 800 IU, 2,000 IU, or 4,000 IU/day, increases in 25(OH)D were consistently higher in those with the lowest baseline [142].
It is noticeable that most trials, showing no cognitive difference in the supplementation arms compared to the comparative arms, have actually included participants with relatively high serum 25(OH)D concentrations at baseline, possibly above the cerebral needs for vitamin D (if any). Moreover, the comparative arm of several trials continued to receive vitamin D supplements, either as part of the study [121] or as part of routine care [120]. So it will be important in the future to examine the cognitive effect of vitamin D supplementation in populations exhibiting vitamin D insufficiency at baseline. The definition of vitamin D insufficiency is consensually not based on what is called a “population-based reference value” (i.e., normality is the reference range involving 95% of the population) but rather on “health-based reference values” (i.e., normality is the 25(OH)D concentration range that prevents adverse health effects) [143]. Defining a “cognition-based reference value” is still the matter of debate.
The nature of the delivered molecule also needs to be considered. The difference in binding affinity of vitamin D2 metabolites to VDBP being weaker than that of vitamin D3, this would lead to a shorter half-life and an increased rate of clearance from circulation. The apparent inefficiency of 25(OH)D2 binding to VDBP may be advantageous in the setting of extra-renal metabolism of 25(OH)D where decreased binding to VDBP may increase the uptake of 25(OH)D2 by target cells. Future studies in animal models may aid in evaluating the relative efficacy of each metabolite in reference to cognitive outcomes. In parallel, human studies should incorporate separate measures for 25(OH)D2 and 25(OH)D3 in serum and CSF along with total versus free concentrations of each molecule after supplementation with either metabolite.
It will also be important to determine whether vitamin D insufficiency triggers AD (i.e., results in a pathological dysfunction of the brain leading to AD) or whether it removes protection of the CNS against AD. Just enough supplements to correct vitamin D insufficiency should be sufficient if vitamin D is a neuroprotectant, although high to very high dose of supplements should be used if vitamin D controls the CNS, with the aim of boosting mental faculties. Also, if vitamin D insufficiency explains partially the pathological process of AD, it may also enhance the efficacy of standard anti-dementia treatments or account for the resistance to these treatments. In line with this, a 6-month controlled trial showed that the combination of memantine+vitamin D was superior to memantine alone and vitamin D alone in preventing cognitive decline among AD patients [116]. Even preliminary, this result certainly engages clinicians to replenish vitamin D before starting anti-dementia treatments or at least to use vitamin D as an adjunct to these treatments.
CONCLUSION
Vitamin D, produced by the skin under UV stimulation or ingested from food, is now described as a steroid hormone, essential for human health. More recently, accumulating evidence indicate that this hormone is a neurosteroid that may be important in aging and age-related cognitive decline. More precisely, numerous preclinical and clinical studies suggest that hypovitaminosis D may be associated with increased risk of developing AD and dementia, without being a causal agent. Inducing genomic and non genomic effects, vitamin D plays a role on calcium homeostasis, neurotransmission, vascularization, Aβ and Tau accumulation, oxidative stress, and inflammation, all of which are disturbed in AD. However, vitamin D’s pleiotropic action is cell-, tissue-, time-, individual-, dose-, pathological context- and, maybe, gender-dependent. As a result, in view of personalized medicine, future randomized trials should take into account the D-tails of every individual included in the cohorts.
DISCLOSURE STATEMENT
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/15-0943r2).
REFERENCES
[1] | Hess AF ((1924) ) On the induction of antirachitic properties in rations by exposure to light. Science 60: , 269. |
[2] | Hume EM , Smith HH ((1924) ) The effect of irradiation of the environment with ultra-violet light upon the growth and calcification of rats, fed on a diet deficient in fat-soluble vitamins. The part played by irradiated sawdust. Biochem J 18: , 1334–1348. |
[3] | Steenbock H ((1924) ) The induction of growth promoting and calcifying properties in a ration by exposure to light. Science 60: , 224–225. |
[4] | Mawer EB , Backhouse J , Holman CA , Lumb GA , Stanbury SW ((1972) ) The distribution and storage of vitamin D and its metabolites in human tissues. Clin Sci 43: , 413–431. |
[5] | Chun RF , Peercy BE , Orwoll ES , Nielson CM , Adams JS , Hewison M ((2014) ) Vitamin D and DBP: The free hormone hypothesis revisited. J Steroid Biochem Mol Biol 144: (Pt A), 132–137. |
[6] | Nykjaer A , Dragun D , Walther D , Vorum H , Jacobsen C , Herz J , Melsen F , Christensen EI , Willnow TE ((1999) ) An endocytic pathway essential for renal uptake and activation of the steroid 25-(OH) vitamin D3. Cell 96: , 507–515. |
[7] | Biancuzzo RM , Clarke N , Reitz RE , Travison TG , Holick MF ((2013) ) Serum concentrations of 1,25-dihydroxyvitamin D2 and 1,25-dihydroxyvitamin D3 in response to vitamin D2 and vitamin D3 supplementation. J Clin Endocrinol Metab 98: , 973–979. |
[8] | Holick MF , Biancuzzo RM , Chen TC , Klein EK , Young A , Bibuld D , Reitz R , Salameh W , Ameri A , Tannenbaum AD ((2008) ) Vitamin D2 is as effective as vitamin D3 in maintaining circulating concentrations of 25-hydroxyvitamin D. J Clin Endocrinol Metab 93: , 677–681. |
[9] | Tripkovic L , Lambert H , Hart K , Smith CP , Bucca G , Penson S , Chope G , Hypponen E , Berry J , Vieth R , Lanham-New S ((2012) ) Comparison of vitamin D2 and vitamin D3 supplementation in raising serum 25-hydroxyvitamin D status: A systematic review and meta-analysis. Am J Clin Nutr 95: , 1357–1364. |
[10] | Lehmann U , Hirche F , Stangl GI , Hinz K , Westphal S , Dierkes J ((2013) ) Bioavailability of vitamin D(2) and D(3) in healthy volunteers, a randomized placebo-controlled trial. J Clin Endocrinol Metab 98: , 4339–4345. |
[11] | Swanson CM , Nielson CM , Shrestha S , Lee CG , Barrett-Connor E , Jans I , Cauley JA , Boonen S , Bouillon R , Vanderschueren D , Orwoll ES ((2014) ) Higher 25(OH)D2 is associated with lower 25(OH)D3 and 1,25(OH)2D3. J Clin Endocrinol Metab 99: , 2736–2744. |
[12] | Haussler MR , Jurutka PW , Mizwicki M , Norman AW ((2011) ) Vitamin D receptor (VDR)-mediated actions of 1alpha,25(OH)(2)vitamin D(3): Genomic and non-genomic mechanisms. Best Pract Res Clin Endocrinol Metab 25: , 543–559. |
[13] | Hii CS , Ferrante A ((2016) ) The non-genomic actions of vitamin D. Nutrients 8: , pii: E135. |
[14] | Ross AC , Manson JE , Abrams SA , Aloia JF , Brannon PM , Clinton SK , Durazo-Arvizu RA , Gallagher JC , Gallo RL , Jones G , Kovacs CS , Mayne ST , Rosen CJ , Shapses SA ((2011) ) The 2011 Dietary Reference Intakes for Calcium and Vitamin D: What dietetics practitioners need to know. J Am Diet Assoc 111: , 524–527. |
[15] | Holick MF ((2007) ) Vitamin D deficiency. N Engl J Med 357: , 266–281. |
[16] | Holick MF , Binkley NC , Bischoff-Ferrari HA , Gordon CM , Hanley DA , Heaney RP , Murad MH , Weaver CM ((2011) ) Evaluation, treatment, and prevention of vitamin D deficiency: An Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 96: , 1911–1930. |
[17] | Mazahery H , von Hurst PR ((2015) ) Factors affecting 25-hydroxyvitamin D concentration in response to vitamin D supplementation. Nutrients 7: , 5111–5142. |
[18] | Powe CE , Evans MK , Wenger J , Zonderman AB , Berg AH , Nalls M , Tamez H , Zhang D , Bhan I , Karumanchi SA , Powe NR , Thadhani R ((2013) ) Vitamin D-binding protein and vitamin D status of black Americans and white Americans. N Engl J Med 369: , 1991–2000. |
[19] | Palacios C and Lilliana G ((2014) ) Is vitamin D deficiency a major global public health problem? J Steroid Biochem Mol Biol 144: , 138–145. |
[20] | Hilger J , Friedel A , Herr R , Rausch T , Roos F , Wahl DA , Pierroz DD , Weber P , Hoffmann K ((2014) ) A systematic review of vitamin D status in populations worldwide. Br J Nutr 111: , 23–45. |
[21] | Fardellone P , Sebert JL , Garabedian M , Bellony R , Maamer M , Agbomson F , Brazier M ((1995) ) Prevalence and biological consequences of vitamin D deficiency in elderly institutionalized subjects. Rev Rhum Engl Ed 62: , 576–581. |
[22] | Holick MF , Matsuoka LY , Wortsman J ((1989) ) Age, vitamin D, and solar ultraviolet. Lancet 2: , 1104–1105. |
[23] | MacLaughlin J , Holick MF ((1985) ) Aging decreases the capacity of human skin to produce vitamin D3. J Clin Invest 76: , 1536–1538. |
[24] | Pines A ((2014) ) Vitamin D and health issues–questioned benefits. Climacteric 17: , 657–659. |
[25] | Rush L , McCartney G , Walsh D , MacKay D ((2013) ) Vitamin D and subsequent all-age and premature mortality: A systematic review. BMC Public Health 13: , 679. |
[26] | Kumar A , Singh A , Ekavali ((2015) ) A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol Rep 67: , 195–203. |
[27] | Zhang F , Jiang L ((2015) ) Neuroinflammation in Alzheimer’s disease. Neuropsychiatr Dis Treat 11: , 243–256. |
[28] | Prince M , Bryce R , Albanese E , Wimo A , Ribeiro W , Ferri CP ((2013) ) The global prevalence of dementia: A systematic review and metaanalysis. Alzheimers Dement 9: , 62–75. |
[29] | Sutherland MK , Somerville MJ , Yoong LK , Bergeron C , Haussler MR , McLachlan DR ((1992) ) Reduction of vitamin D hormone receptor mRNA levels in Alzheimer as compared to Huntington hippocampus: Correlation with calbindin-28k mRNA levels. Brain Res Mol Brain Res 13: , 239–250. |
[30] | Gezen-Ak D , Dursun E , Bilgic B , Hanagasi H , Ertan T , Gurvit H , Emre M , Eker E , Ulutin T , Uysal O , Yilmazer S ((2012) ) Vitamin D receptor gene haplotype is associated with late-onset Alzheimer’s disease. Tohoku J Exp Med 228: , 189–196. |
[31] | Gezen-Ak D , Dursun E , Ertan T , Hanagasi H , Gurvit H , Emre M , Eker E , Ozturk M , Engin F , Yilmazer S ((2007) ) Association between vitamin D receptor gene polymorphism and Alzheimer’s disease. Tohoku J Exp Med 212: , 275–282. |
[32] | Kuningas M , Mooijaart SP , Jolles J , Slagboom PE , Westendorp RG , van Heemst D ((2009) ) VDR gene variants associate with cognitive function and depressive symptoms in old age. Neurobiol Aging 30: , 466–473. |
[33] | Lehmann DJ , Refsum H , Warden DR , Medway C , Wilcock GK , Smith AD ((2011) ) The vitamin D receptor gene is associated with Alzheimer’s disease. Neurosci Lett 504: , 79–82. |
[34] | Laczmanski L , Jakubik M , Bednarek-Tupikowska G , Rymaszewska J , Sloka N , Lwow F ((2015) ) Vitamin D receptor gene polymorphisms in Alzheimer’s disease patients. Exp Gerontol 69: , 142–147. |
[35] | Wang L , Hara K , Van Baaren JM , Price JC , Beecham GW , Gallins PJ , Whitehead PL , Wang G , Lu C , Slifer MA , Zuchner S , Martin ER , Mash D , Haines JL , Pericak-Vance MA , Gilbert JR ((2012) ) Vitamin D receptor and Alzheimer’s disease: A genetic and functional study. Neurobiol Aging 33: , 1844 e1841–1849. |
[36] | Beydoun MA , Ding EL , Beydoun HA , Tanaka T , Ferrucci L , Zonderman AB ((2012) ) Vitamin D receptor and megalin gene polymorphisms and their associations with longitudinal cognitive change in US adults. Am J Clin Nutr 95: , 163–178. |
[37] | DeLuca GC , Kimball SM , Kolasinski J , Ramagopalan SV , Ebers GC ((2013) ) Review: The role of vitamin D in nervous system health and disease. Neuropathol Appl Neurobiol 39: , 458–484. |
[38] | Garcion E , Wion-Barbot N , Montero-Menei CN , Berger F , Wion D ((2002) ) New clues about vitamin D functions in the nervous system. Trends Endocrinol Metab 13: , 100–105. |
[39] | Eyles DW , Smith S , Kinobe R , Hewison M , McGrath JJ ((2005) ) Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J Chem Neuroanat 29: , 21–30. |
[40] | Stumpf WE , O’Brien LP ((1987) ) 1,25 (OH)2 vitamin D3 sites of action in the brain. An autoradiographic study. Histochemistry 87: , 393–406. |
[41] | Wion D , MacGrogan D , Neveu I , Jehan F , Houlgatte R , Brachet P ((1991) ) 1,25-Dihydroxyvitamin D3 is a potent inducer of nerve growth factor synthesis. J Neurosci Res 28: , 110–114. |
[42] | Eyles DW , Liu PY , Josh P , Cui X ((2014) ) Intracellular distribution of the vitamin D receptor in the brain: Comparison with classic target tissues and redistribution with development. Neuroscience 268: , 1–9. |
[43] | Balabanova S , Richter HP , Antoniadis G , Homoki J , Kremmer N , Hanle J , Teller WM ((1984) ) 25-Hydroxyvitamin D, 24, 25-dihydroxyvitamin D and 1,25-dihydroxyvitamin D in human cerebrospinal fluid. Klin Wochenschr 62: , 1086–1090. |
[44] | Naveilhan P , Neveu I , Baudet C , Ohyama KY , Brachet P , Wion D ((1993) ) Expression of 25(OH) vitamin D3 24-hydroxylase gene in glial cells. Neuroreport 5: , 255–257. |
[45] | Neveu I , Naveilhan P , Baudet C , Brachet P , Metsis M ((1994) ) 1,25-dihydroxyvitamin D3 regulates NT-3, NT-4 but not BDNF mRNA in astrocytes. Neuroreport 6: , 124–126. |
[46] | Neveu I , Naveilhan P , Jehan F , Baudet C , Wion D , De Luca HF , Brachet P ((1994) ) 1,25-dihydroxyvitamin D3 regulates the synthesis of nerve growth factor in primary cultures of glial cells. Brain Res Mol Brain Res 24: , 70–76. |
[47] | Dursun E , Gezen-Ak D , Yilmazer S ((2013) ) Beta amyloid suppresses the expression of the vitamin d receptor gene and induces the expression of the vitamin d catabolic enzyme gene in hippocampal neurons. Dement Geriatr Cogn Disord 36: , 76–86. |
[48] | Gezen-Ak D , Dursun E , Yilmazer S ((2011) ) The effects of vitamin D receptor silencing on the expression of LVSCC-A1C and LVSCC-A1D and the release of NGF in cortical neurons. PLoS One 6: , e17553. |
[49] | Gezen-Ak D , Dursun E , Yilmazer S ((2013) ) Vitamin D inquiry in hippocampal neurons: Consequences of vitamin D-VDR pathway disruption on calcium channel and the vitamin D requirement. Neurol Sci 34: , 1453–1458. |
[50] | Taniura H , Ito M , Sanada N , Kuramoto N , Ohno Y , Nakamichi N , Yoneda Y ((2006) ) Chronic vitamin D3 treatment protects against neurotoxicity by glutamate in association with upregulation of vitamin D receptor mRNA expression in cultured rat cortical neurons. J Neurosci Res 83: , 1179–1189. |
[51] | Baas D , Prufer K , Ittel ME , Kuchler-Bopp S , Labourdette G , Sarlieve LL , Brachet P ((2000) ) Rat oligodendrocytes express the vitamin D(3) receptor and respond to 1,25-dihydroxyvitamin D(3). Glia 31: , 59–68. |
[52] | Durk MR , Chan GN , Campos CR , Peart JC , Chow EC , Lee E , Cannon RE , Bendayan R , Miller DS , Pang KS ((2012) ) 1alpha,25-Dihydroxyvitamin D3-liganded vitamin D receptor increases expression and transport activity of P-glycoprotein in isolated rat brain capillaries and human and rat brain microvessel endothelial cells. J Neurochem 123: , 944–953. |
[53] | Ito S , Ohtsuki S , Nezu Y , Koitabashi Y , Murata S , Terasaki T ((2011) ) 1alpha,25-Dihydroxyvitamin D3 enhances cerebral clearance of human amyloid-beta peptide(1-40) from mouse brain across the blood-brain barrier. Fluids Barriers CNS 8: , 20. |
[54] | Bouillon R , Carmeliet G , Daci E , Segaert S , Verstuyf A ((1998) ) Vitamin D metabolism and action. Osteoporos Int 8: (Suppl 2), S13–S19. |
[55] | Cornet A , Baudet C , Neveu I , Baron-Van Evercooren A , Brachet P , Naveilhan P ((1998) ) 1,25-Dihydroxyvitamin D3 regulates the expression of VDR and NGF gene in Schwann cells in vitro. J Neurosci Res 53: , 742–746. |
[56] | Naveilhan P , Neveu I , Baudet C , Funakoshi H , Wion D , Brachet P , Metsis M ((1996) ) 1,25-Dihydroxyvitamin D3 regulates the expression of the low-affinity neurotrophin receptor. Brain Res Mol Brain Res 41: , 259–268. |
[57] | Gezen-Ak D , Yilmazer S , Dursun E ((2014) ) Why vitamin D in Alzheimer’s disease? The hypothesis. J Alzheimers Dis 40: , 257–269. |
[58] | Keisala T , Minasyan A , Lou YR , Zou J , Kalueff AV , Pyykko I , Tuohimaa P ((2009) ) Premature aging in vitamin D receptor mutant mice. J Steroid Biochem Mol Biol 115: , 91–97. |
[59] | Latimer CS , Brewer LD , Searcy JL , Chen KC , Popovic J , Kraner SD , Thibault O , Blalock EM , Landfield PW , Porter NM ((2014) ) Vitamin D prevents cognitive decline and enhances hippocampal synaptic function in aging rats. Proc Natl Acad Sci U S A 111: , E4359–E4366. |
[60] | Briones TL , Darwish H ((2012) ) Vitamin D mitigates age-related cognitive decline through the modulation of pro-inflammatory state and decrease in amyloid burden. J Neuroinflammation 9: , 244. |
[61] | Briones TL , Darwish H ((2014) ) Decrease in age-related tau hyperphosphorylation and cognitive improvement following vitamin D supplementation are associated with modulation of brain energy metabolism and redox state. Neuroscience 262: , 143–155. |
[62] | Taghizadeh M , Djazayery A , Salami M , Eshraghian MR , Zavareh SA ((2011) ) Vitamin-D-free regimen intensifies the spatial learning deficit in Alzheimer’s disease. Int J Neurosci 121: , 16–24. |
[63] | Yu J , Gattoni-Celli M , Zhu H , Bhat NR , Sambamurti K , Gattoni-Celli S , Kindy MS ((2011) ) Vitamin D3-enriched diet correlates with a decrease of amyloid plaques in the brain of AbetaPP transgenic mice. J Alzheimers Dis 25: , 295–307. |
[64] | Landel V , Millet P , Baranger K , Loriod B , Feron F ((2016) ) Vitamin D interacts with Esr1 and Igf1 to regulate molecular pathways relevant to Alzheimer’s disease. Mol Neurodegener 11: , 22. |
[65] | Durk MR , Han K , Chow EC , Ahrens R , Henderson JT , Fraser PE , Pang KS ((2014) ) 1alpha,25-Dihydroxyvitamin D3 reduces cerebral amyloid-beta accumulation and improves cognition in mouse models of Alzheimer’s disease. J Neurosci 34: , 7091–7101. |
[66] | Bennett L , Kersaitis C , Macaulay SL , Munch G , Niedermayer G , Nigro J , Payne M , Sheean P , Vallotton P , Zabaras D , Bird M ((2013) ) Vitamin D2-enriched button mushroom (Agaricus bisporus) improves memory in both wild type and APPswe/PS1dE9 transgenic mice. PLoS One 8: , e76362. |
[67] | Grimm MO , Lehmann J , Mett J , Zimmer VC , Grosgen S , Stahlmann CP , Hundsdorfer B , Haupenthal VJ , Rothhaar TL , Herr C , Bals R , Grimm HS , Hartmann T ((2014) ) Impact of Vitamin D on amyloid precursor protein processing and amyloid-beta peptide degradation in Alzheimer’s disease. Neurodegener Dis 13: , 75–81. |
[68] | Moon M , Song H , Hong HJ , Nam DW , Cha MY , Oh MS , Yu J , Ryu H , Mook-Jung I ((2013) ) Vitamin D-binding protein interacts with Abeta and suppresses Abeta-mediated pathology. Cell Death Differ 20: , 630–638. |
[69] | Dursun E , Gezen-Ak D , Yilmazer S ((2011) ) A novel perspective for Alzheimer’s disease: Vitamin D receptor suppression by amyloid-beta and preventing the amyloid-beta induced alterations by vitamin D in cortical neurons. J Alzheimers Dis 23: , 207–219. |
[70] | Dursun E , Gezen-Ak D , Yilmazer S ((2013) ) A new mechanism for amyloid-beta induction of iNOS: Vitamin D-VDR pathway disruption. J Alzheimers Dis 36: , 459–474. |
[71] | Annweiler C , Brugg B , Peyrin JM , Bartha R , Beauchet O ((2014) ) Combination of memantine and vitamin D prevents axon degeneration induced by amyloid-beta and glutamate. Neurobiol Aging 35: , 331–335. |
[72] | Brewer LD , Porter NM , Kerr DS , Landfield PW , Thibault O ((2006) ) Chronic 1alpha,25-(OH)2 vitamin D3 treatment reduces Ca2+ -mediated hippocampal biomarkers of aging. Cell Calcium 40: , 277–286. |
[73] | Salami M , Talaei SA , Davari S , Taghizadeh M ((2012) ) Hippocampal long term potentiation in rats under different regimens of vitamin D: An in vivo study. Neurosci Lett 509: , 56–59. |
[74] | Taghizadeh M , Talaei SA , Djazayeri A , Salami M ((2014) ) Vitamin D supplementation restores suppressed synaptic plasticity in Alzheimer’s disease. Nutr Neurosci 17: , 172–177. |
[75] | Brewer LD , Thibault V , Chen KC , Langub MC , Landfield PW , Porter NM ((2001) ) Vitamin D hormone confers neuroprotection in parallel with downregulation of L-type calcium channel expression in hippocampal neurons. J Neurosci 21: , 98–108. |
[76] | Zhu Y , Zhou R , Yang R , Zhang Z , Bai Y , Chang F , Li L , Sokabe M , Goltzman D , Miao D , Chen L ((2012) ) Abnormal neurogenesis in the dentate gyrus of adult mice lacking 1,25-dihydroxy vitamin D3 (1,25-(OH)2 D3). Hippocampus 22: , 421–433. |
[77] | Almeras L , Eyles D , Benech P , Laffite D , Villard C , Patatian A , Boucraut J , Mackay-Sim A , McGrath J , Feron F ((2007) ) Developmental vitamin D deficiency alters brain protein expression in the adult rat: Implications for neuropsychiatric disorders. Proteomics 7: , 769–780. |
[78] | Eyles D , Almeras L , Benech P , Patatian A , Mackay-Sim A , McGrath J , Feron F ((2007) ) Developmental vitamin D deficiency alters the expression of genes encoding mitochondrial, cytoskeletal and synaptic proteins in the adult rat brain. J Steroid Biochem Mol Biol 103: , 538–545. |
[79] | Nissou MF , Brocard J , El Atifi M , Guttin A , Andrieux A , Berger F , Issartel JP , Wion D ((2013) ) The transcriptomic response of mixed neuron-glial cell cultures to 1,25-dihydroxyvitamin d3 includes genes limiting the progression of neurodegenerative diseases. J Alzheimers Dis 35: , 553–564. |
[80] | Nissou MF , Guttin A , Zenga C , Berger F , Issartel JP , Wion D ((2014) ) Additional clues for a protective role of vitamin D in neurodegenerative diseases: 1,25-dihydroxyvitamin D3 triggers an anti-inflammatory response in brain pericytes. J Alzheimers Dis 42: , 789–799. |
[81] | Bal-Price A , Brown GC ((2001) ) Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci 21: , 6480–6491. |
[82] | Brown GC ((2007) ) Nitric oxide and mitochondria. Front Biosci 12: , 1024–1033. |
[83] | Brown GC , Borutaite V ((2001) ) Nitric oxide, mitochondria, and cell death. IUBMB Life 52: , 189–195. |
[84] | Keeney JT , Forster S , Sultana R , Brewer LD , Latimer CS , Cai J , Klein JB , Porter NM , Butterfield DA ((2013) ) Dietary vitamin D deficiency in rats from middle to old age leads to elevated tyrosine nitration and proteomics changes in levels of key proteins in brain: Implications for low vitamin D-dependent age-related cognitive decline. Free Radic Biol Med 65: , 324–334. |
[85] | Lefebvre d’Hellencourt C , Montero-Menei CN , Bernard R , Couez D ((2003) ) Vitamin D3 inhibits proinflammatory cytokines and nitric oxide production by the EOC13 microglial cell line. J Neurosci Res 71: , 575–582. |
[86] | Garcion E , Sindji L , Montero-Menei C , Andre C , Brachet P , Darcy F ((1998) ) Expression of inducible nitric oxide synthase during rat brain inflammation: Regulation by 1,25-dihydroxyvitamin D3. Glia 22: , 282–294. |
[87] | Hewison M , Freeman L , Hughes SV , Evans KN , Bland R , Eliopoulos AG , Kilby MD , Moss PA , Chakraverty R ((2003) ) Differential regulation of vitamin D receptor and its ligand in human monocyte-derived dendritic cells. J Immunol 170: , 5382–5390. |
[88] | Lue LF , Kuo YM , Beach T , Walker DG ((2010) ) Microglia activation and anti-inflammatory regulation in Alzheimer’s disease. Mol Neurobiol 41: , 115–128. |
[89] | Mizwicki MT , Liu G , Fiala M , Magpantay L , Sayre J , Siani A , Mahanian M , Weitzman R , Hayden EY , Rosenthal MJ , Nemere I , Ringman J , Teplow DB ((2013) ) 1alpha,25-dihydroxyvitamin D3 and resolvin D1 retune the balance between amyloid-beta phagocytosis and inflammation in Alzheimer’s disease patients. J Alzheimers Dis 34: , 155–170. |
[90] | Lee V , Rekhi E , Hoh Kam J , Jeffery G ((2012) ) Vitamin D rejuvenates aging eyes by reducing inflammation, clearing amyloid beta and improving visual function. Neurobiol Aging 33: , 2382–2389. |
[91] | Mizwicki MT , Menegaz D , Zhang J , Barrientos-Duran A , Tse S , Cashman JR , Griffin PR , Fiala M ((2012) ) Genomic and nongenomic signaling induced by 1alpha,25(OH)2-vitamin D3 promotes the recovery of amyloid-beta phagocytosis by Alzheimer’s disease macrophages. J Alzheimers Dis 29: , 51–62. |
[92] | Luong KV , Nguyen LT ((2013) ) The role of vitamin d in autoimmune hepatitis. J Clin Med Res 5: , 407–415. |
[93] | Annweiler C , Rolland Y , Schott AM , Blain H , Vellas B , Beauchet O ((2011) ) Serum vitamin D deficiency as a predictor of incident non-Alzheimer dementias: A7-year longitudinal study. Dement Geriatr Cogn Disord 32: , 273–278. |
[94] | Annweiler C , Rolland Y , Schott AM , Blain H , Vellas B , Herrmann FR , Beauchet O ((2012) ) Higher vitamin D dietary intake is associated with lower risk of alzheimer’s disease: A 7-year follow-up. J Gerontol A Biol Sci Med Sci 67: , 1205–1211. |
[95] | Annweiler C , Schott AM , Allali G , Bridenbaugh SA , Kressig RW , Allain P , Herrmann FR , Beauchet O ((2010) ) Association of vitamin D deficiency with cognitive impairment in older women: Cross-sectional study. Neurology 74: , 27–32. |
[96] | Annweiler C , Schott AM , Rolland Y , Blain H , Herrmann FR , Beauchet O ((2010) ) Dietary intake of vitamin D and cognition in older women: A large population-based study. Neurology 75: , 1810–1816. |
[97] | Slinin Y , Paudel M , Taylor BC , Ishani A , Rossom R , Yaffe K , Blackwell T , Lui LY , Hochberg M , Ensrud KE ((2012) ) Association between serum 25(OH) vitamin D and the risk of cognitive decline in older women. J Gerontol A Biol Sci Med Sci 67: , 1092–1098. |
[98] | Rossom RC , Espeland MA , Manson JE , Dysken MW , Johnson KC , Lane DS , LeBlanc ES , Lederle FA , Masaki KH , Margolis KL ((2012) ) Calcium and vitamin D supplementation and cognitive impairment in the women’s health initiative. J Am Geriatr Soc 60: , 2197–2205. |
[99] | Slinin Y , Paudel ML , Taylor BC , Fink HA , Ishani A , Canales MT , Yaffe K , Barrett-Connor E , Orwoll ES , Shikany JM , Leblanc ES , Cauley JA , Ensrud KE ((2010) ) 25-Hydroxyvitamin D levels and cognitive performance and decline in elderly men. Neurology 74: , 33–41. |
[100] | Afzal S , Bojesen SE , Nordestgaard BG ((2014) ) Reduced 25-hydroxyvitamin D and risk of Alzheimer’s disease and vascular dementia. Alzheimers Dement 10: , 296–302. |
[101] | Annweiler C , Fantino B , Schott AM , Krolak-Salmon P , Allali G , Beauchet O ((2012) ) Vitamin D insufficiency and mild cognitive impairment: Cross-sectional association. Eur J Neurol 19: , 1023–1029. |
[102] | Buell JS , Scott TM , Dawson-Hughes B , Dallal GE , Rosenberg IH , Folstein MF , Tucker KL ((2009) ) Vitamin D is associated with cognitive function in elders receiving home health services. J Gerontol A Biol Sci Med Sci 64: , 888–895. |
[103] | Jorde R , Mathiesen EB , Rogne S , Wilsgaard T , Kjaergaard M , Grimnes G , Schirmer H ((2015) ) Vitamin D and cognitive function: The Tromso Study. J Neurol Sci 355: , 155–161. |
[104] | Leedahl DD , Cunningham JL , Drake MT , Mundis CB , Kung S , Frye MA , Lapid MI ((2013) ) Hypovitaminosis D in psychiatric inpatients: Clinical correlation with depressive symptoms, cognitive impairment, and prescribing practices. Psychosomatics 54: , 257–262. |
[105] | Littlejohns TJ , Henley WE , Lang IA , Annweiler C , Beauchet O , Chaves PH , Fried L , Kestenbaum BR , Kuller LH , Langa KM , Lopez OL , Kos K , Soni M , Llewellyn DJ ((2014) ) Vitamin D and the risk of dementia and Alzheimer disease. Neurology 83: , 920–928. |
[106] | Llewellyn DJ , Lang IA , Langa KM , Melzer D ((2011) ) Vitamin D and cognitive impairment in the elderly U.S. population. J Gerontol A Biol Sci Med Sci 66: , 59–65. |
[107] | Llewellyn DJ , Lang IA , Langa KM , Muniz-Terrera G , Phillips CL , Cherubini A , Ferrucci L , Melzer D ((2010) ) Vitamin D and risk of cognitive decline in elderly persons. Arch Intern Med 170: , 1135–1141. |
[108] | Llewellyn DJ , Langa KM , Lang IA ((2009) ) Serum 25-hydroxyvitamin D concentration and cognitive impairment. J Geriatr Psychiatry Neurol 22: , 188–195. |
[109] | Maddock J , Geoffroy MC , Power C , Hypponen E ((2014) ) 25-Hydroxyvitamin D and cognitive performance in mid-life. Br J Nutr 111: , 904–914. |
[110] | McGrath J , Scragg R , Chant D , Eyles D , Burne T , Obradovic D ((2007) ) No association between serum 25-hydroxyvitamin D3 level and performance on psychometric tests in NHANES III. Neuroepidemiology 29: , 49–54. |
[111] | Seamans KM , Hill TR , Scully L , Meunier N , Andrillo-Sanchez M , Polito A , Hininger-Favier I , Ciarapica D , Simpson EE , Stewart-Knox BJ , O’Connor JM , Coudray C , Cashman KD ((2010) ) Vitamin D status and measures of cognitive function in healthy older European adults. Eur J Clin Nutr 64: , 1172–1178. |
[112] | Tolppanen AM , Sayers A , Fraser WD , Lawlor DA ((2012) ) Association of serum 25-hydroxyvitamin D(3) and D(2) with academic performance in childhood: Findings from a prospective birth cohort. J Epidemiol Community Health 66: , 1137–1142. |
[113] | Tolppanen AM , Williams D , Lawlor DA ((2011) ) The association of circulating 25-hydroxyvitamin D and calcium with cognitive performance in adolescents: Cross-sectional study using data from the third National Health and Nutrition Examination Survey. Paediatr Perinat Epidemiol 25: , 67–74. |
[114] | Tolppanen AM , Williams DM , Lawlor DA ((2011) ) The association of serum ionized calcium and vitamin D with adult cognitive performance. Epidemiology 22: , 113–117. |
[115] | Annweiler C , Fantino B , Gautier J , Beaudenon M , Thiery S , Beauchet O ((2012) ) Cognitive effects of vitamin D supplementation in older outpatients visiting a memory clinic: A pre-post study. J Am Geriatr Soc 60: , 793–795. |
[116] | Annweiler C , Herrmann FR , Fantino B , Brugg B , Beauchet O ((2012) ) Effectiveness of the combination of memantine plus vitamin D on cognition in patients with Alzheimer disease: A pre-post pilot study. Cogn Behav Neurol 25: , 121–127. |
[117] | Corless D , Dawson E , Fraser F , Ellis M , Evans SJ , Perry JD , Reisner C , Silver CP , Beer M , Boucher BJ , et al. ((1985) ) Do vitamin D supplements improve the physical capabilities of elderly hospital patients? Age Ageing 14: , 76–84. |
[118] | Dean AJ , Bellgrove MA , Hall T , Phan WM , Eyles DW , Kvaskoff D , McGrath JJ ((2011) ) Effects of vitamin D supplementation on cognitive and emotional functioning in young adults–a randomised controlled trial. PLoS One 6: , e25966. |
[119] | Dhesi JK , Jackson SH , Bearne LM , Moniz C , Hurley MV , Swift CG , Allain TJ ((2004) ) Vitamin D supplementation improves neuromuscular function in older people who fall. Age Ageing 33: , 589–595. |
[120] | Przybelski R , Agrawal S , Krueger D , Engelke JA , Walbrun F , Binkley N ((2008) ) Rapid correction of low vitamin D status in nursing home residents. Osteoporos Int 19: , 1621–1628. |
[121] | Stein MS , Scherer SC , Ladd KS , Harrison LC ((2011) ) A randomized controlled trial of high-dose vitamin D2 followed by intranasal insulin in Alzheimer’s disease. J Alzheimers Dis 26: , 477–484. |
[122] | Etgen T , Sander D , Bickel H , Sander K , Forstl H ((2012) ) Vitamin D deficiency, cognitive impairment and dementia: A systematic review and meta-analysis. Dement Geriatr Cogn Disord 33: , 297–305. |
[123] | van der Schaft J , Koek HL , Dijkstra E , Verhaar HJ , van der Schouw YT , Emmelot-Vonk MH ((2013) ) The association between vitamin D and cognition: A systematic review. Ageing Res Rev 12: , 1013–1023. |
[124] | Balion C , Griffith LE , Strifler L , Henderson M , Patterson C , Heckman G , Llewellyn DJ , Raina P ((2012) ) Vitamin D, cognition, and dementia: A systematic review and meta-analysis. Neurology 79: , 1397–1405. |
[125] | Annweiler C , Llewellyn DJ , Beauchet O ((2013) ) Low serum vitamin D concentrations in Alzheimer’s disease: A systematic review and meta-analysis. J Alzheimers Dis 33: , 659–674. |
[126] | Annweiler C , Fantino B , Le Gall D , Schott AM , Berrut G , Beauchet O ((2011) ) Severe vitamin D deficiency is associated with advanced-stage dementia in geriatric inpatients. J Am Geriatr Soc 59: , 169–171. |
[127] | Tot Babberich Ede N , Gourdeau C , Pointel S , Lemarchant B , Beauchet O , Annweiler C ((2015) ) Biology of subjective cognitive complaint amongst geriatric patients: Vitamin D involvement. Curr Alzheimer Res 12: , 173–178. |
[128] | Schram MT , Trompet S , Kamper AM , de Craen AJ , Hofman A , Euser SM , Breteler MM , Westendorp RG ((2007) ) Serum calcium and cognitive function in old age. J Am Geriatr Soc 55: , 1786–1792. |
[129] | Tilvis RS , Kahonen-Vare MH , Jolkkonen J , Valvanne J , Pitkala KH , Strandberg TE ((2004) ) Predictors of cognitive decline and mortality of aged people over a 10-year period. J Gerontol A Biol Sci Med Sci 59: , 268–274. |
[130] | Annweiler C , Beauchet O ((2013) ) Vitamin D and cognition: Recommendations for future trials. J Am Geriatr Soc 61: , 1049–1050. |
[131] | Annweiler C , Montero-Odasso M , Llewellyn DJ , Richard-Devantoy S , Duque G , Beauchet O ((2013) ) Meta-analysis of memory and executive dysfunctions in relation to vitamin D. J Alzheimers Dis 37: , 147–171. |
[132] | Crombie I ((1996) ) The Pocket Guide to Critical Appraisal, BMJ Publishing Group, London, England. |
[133] | Annweiler C , Maby E , Meyerber M , Beauchet O ((2014) ) Hypovitaminosis D and executive dysfunction in older adults with memory complaint: A memory clinic-based study. Dement Geriatr Cogn Disord 37: , 286–293. |
[134] | Cui X , Gooch H , Groves NJ , Sah P , Burne TH , Eyles DW , McGrath JJ ((2015) ) Vitamin D and the brain: Key questions for future research. J Steroid Biochem Mol Biol 148: , 305–309. |
[135] | Blair JA , McGee H , Bhatta S , Palm R , Casadesus G ((2015) ) Hypothalamic-pituitary-gonadal axis involvement in learning and memory and Alzheimer’s disease: More than “just” estrogen. Front Endocrinol (Lausanne) 6: , 45. |
[136] | Lan YL , Zhao J , Li S ((2015) ) Update on the neuroprotective effect of estrogen receptor alpha against Alzheimer’s disease. J Alzheimers Dis 43: , 1137–1148. |
[137] | Li R , Cui J , Shen Y ((2014) ) Brain sex matters: Estrogen in cognition and Alzheimer’s disease. Mol Cell Endocrinol 389: , 13–21. |
[138] | Bikle D ((2000) ) Vitamin D: Production, metabolism, and mechanisms of action. In, Endotext, De Groot LJ , Beck-Peccoz P , Chrousos G , Dungan K , Grossman A , Hershman JM , Koch C , McLachlan R , New M , Rebar R , Singer F , Vinik A , Weickert MO , eds. South Dartmouth (MA). |
[139] | Johansson P , Almqvist EG , Johansson JO , Mattsson N , Andreasson U , Hansson O , Wallin A , Blennow K , Zetterberg H , Svensson J ((2013) ) Cerebrospinal fluid (CSF) 25-hydroxyvitamin D concentration and CSF acetylcholinesterase activity are reduced in patients with Alzheimer’s disease. PLoS One 8: , e81989. |
[140] | Ryynanen J , Neme A , Tuomainen TP , Virtanen JK , Voutilainen S , Nurmi T , de Mello VD , Uusitupa M , Carlberg C ((2014) ) Changes in vitamin D target gene expression in adipose tissue monitor the vitamin D response of human individuals. Mol Nutr Food Res 58: , 2036–2045. |
[141] | Blanchard OL , Smoliga JM ((2015) ) Translating dosages from animal models to human clinical trials–revisiting body surface area scaling. FASEB J 29: , 1629–1634. |
[142] | Schwartz JB , Kane L , Bikle D ((2016) ) Response of vitamin D concentration to vitamin D3 administration in older adults without sun exposure: A randomized double-blind trial. J Am Geriatr Soc 64: , 65–72. |
[143] | Annweiler C , Beauchet O ((2014) ) Vitamin d in older adults: The need to specify standard values with respect to cognition. Front Aging Neurosci 6: , 72. |
Figures and Tables
Fig.1
Vitamin D acquisition, metabolism and modes of action. Cholecalciferol or vitamin D3 and ergocalciferol or vitamin D2 follow the same metabolic pathway. Blood metabolites include 25(OH)D produced by the liver, which is bound to vitamin D binding protein. Renal and extra-renal CYP27B1 (1α-hydroxylase) produces the active metabolite 1,25(OH)2D. 25(OH)D or 1,25(OH)2D enters the cell through passive diffusion or megalin-dependent transport. Once inside the cell, 1,25(OH)2D binds to its nuclear receptor VDR and after dimerization with RXR, forms a regulatory complex which can bind target genes that contain a vitamin D responsive element (VDRE). 1,25(OH)2D can also induce rapid non-genomic responses by binding to its membrane receptor MARRS or a membrane VDR and regulate the activity of adenylate cyclase, PLC and PKC proteins. 1,25(OH)2D also induces modulation of calcium release from intracellular stores and can interact with TGF and EGF receptors to modulate cell cycle processes. These different modes of action and the crosstalks operated by vitamin D signaling ultimately lead to transcription modulation of hundreds of genes, depending on the cell-type considered.
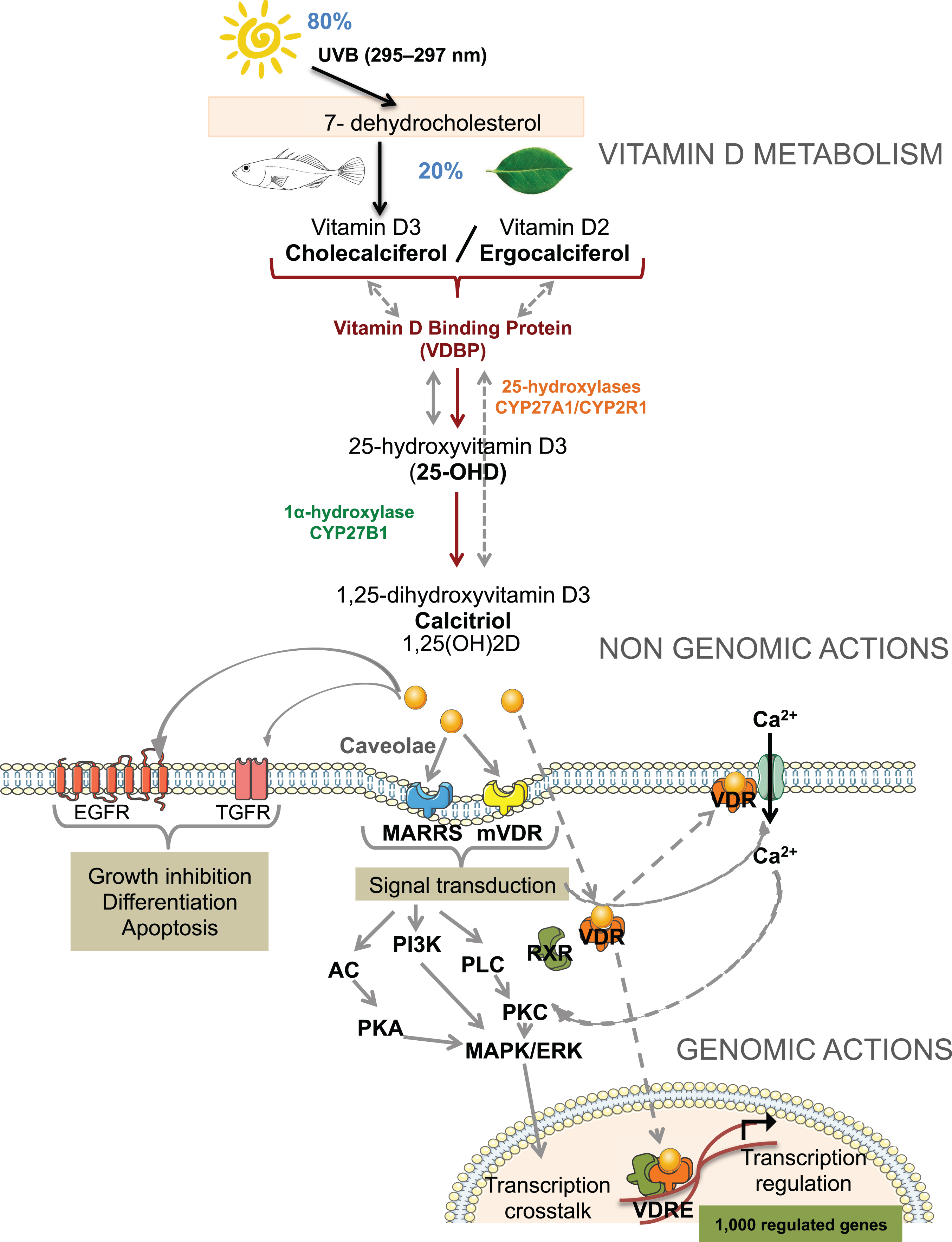
Fig.2
Proposed mechanisms of vitamin D-mediated multi-targeted effects in AD. Vitamin D imbalance is proposed to alter mechanisms implicated in aging and AD pathogenesis. Suggested protective effects of vitamin D supplementation concern regulation of vascular processes and oxidative stress, calcium homeostasis, neurotransmission, modulation of immune and inflammatory processes, and direct impact on amyloidogenesis, ultimately improving cognitive functions.
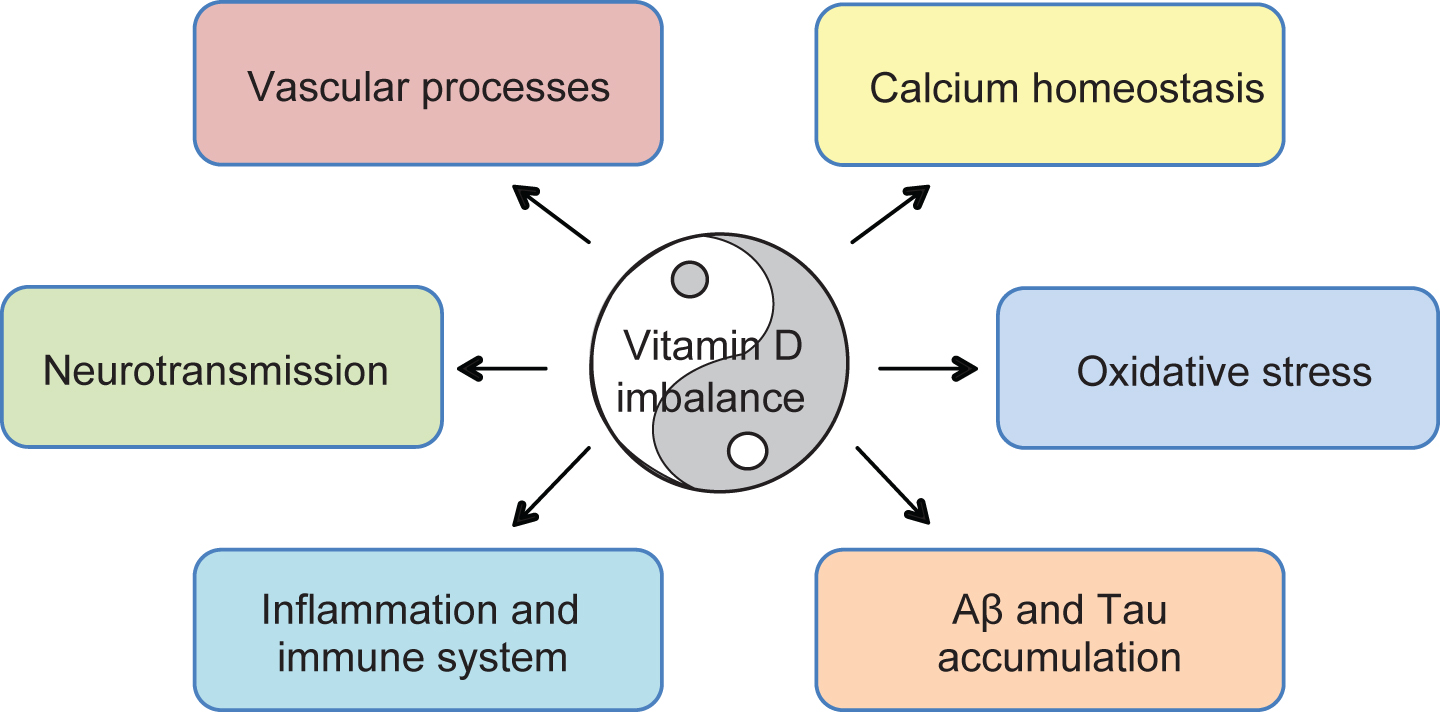
Table 1
Human and animal studies examining the link between vitamin D and cognitive outcome
| REFERENCE | POPULATION STUDIED | VITAMIN D STATUS | FOLLOW UP | DOSE | MEASURED OUTCOME | RESULTS |
| HUMAN STUDIES | ||||||
| Cross-sectional studies | ||||||
| McGrath et al. [110] | Adolescent group (12–17 years); Adult group (20–60 years); Elderly group (60–90 years) | serum 25(OH)D | NA | NA | Psychometric measures | No association between lower levels of 25(OH)D and impaired performance in either group |
| Llewellyn et al. [108] | 65 years and older | serum 25(OH)D | NA | NA | Abbreviated Mental Test Score | Low serum 25 OH)D associates with increased odd of cognitive impairment |
| Buell et al. [102] | 65–99 years | serum 25(OH)D | NA | NA | Battery of neuropsychological tests | 25(OH)D levels associate with executive function and attention but not memory |
| Seamans et al. [111] | Healthy older individuals aged 55–87 years | serum 25(OH)D | NA | NA | CANTAB | Inverse correlation between serum 25(OH)D concentrations and spatial working memory test parameters, an effect more pronounced in women than men |
| Annweiler et al. [95] | Elderly women aged 75 years and older | serum 25(OH)D | NA | NA | SPMSQ | No linear association between serum 25(OH)D levels and SPMSQ scores but vitamin D deficiency associates with cognitive impairment |
| Annweiler et al. [96] | Elderly women of mean age 80.5 years | weekly vitamin D dietary intake | NA | NA | SPMSQ | Association between weekly vitamin D dietary intake and SPMSQ scores |
| Tolppanen et al. [113] | Adolescents (12–16.9 years) | serum 25(OH)D | NA | NA | WISC-R and WRAT-R | No association between serum levels of 25(OH)D and cognitive function |
| Tolppanen et al. [114] | Younger (20–59 years) and older adults (60–90 years) | serum 25(OH)D | NA | NA | Neurobehavioral evaluation system and memory recall tests | No association between serum levels of 25(OH)D and cognitive function in either group |
| Llewellyn et al. [106] | 65 years and older | serum 25(OH)D | NA | NA | MMSE; East Boston Memory test | Levels of 25(OH)D significantly associate with odds of cognitive impairment |
| Annweiler et al. [101] | Elderly individuals of mean age 71.1 years with MCI | serum 25(OH)D | NA | NA | MMSE; FAB | Low serum 25(OH)D levels associate with MCI diagnosis in non demented individuals |
| Leedahl et al. [104] | Psychiatric patients aged 18–99 years | serum 25(OH)D | NA | NA | MMSE; Patient Health Questionnaire | No association between hypovitaminosis D and depressive symptoms or cognitive function |
| Jorde et al. [103] | All age groups | serum 25(OH)D | NA | NA | MMSE; word recall; digit-symbol coding; finger taping | Positive association between levels of 25(OH)D and cognitive performance in individuals aged 65 years and older |
| Longitudinal studies | ||||||
| Slinin et al. [99] | Men aged 65 years and older | serum 25(OH)D | 4.6 years on average | NA | 3MS | No evidence for association between low levels of 25(OH)D and incidence of cognitive decline |
| Llewellyn et al. [107] | 65 years and older | serum 25(OH)D | 6 years (follow-ups every 3 years) | NA | MMSE | Severely vitamin D deficient individuals display increased cognitive decline |
| Annweiler et al. [93] | Elderly women of mean age 78.4 years | serum 25(OH)D | 7 years | NA | DSM-IV and NINCDS-ADRDA | Baseline serum 25(OH)D deficiency associated with the onset of non-Alzheimer’s dementias but not with the onset of AD |
| Tolpannen et al. [112] | Children of mean age 9.8 years | serum 25(OH)D | Academic performance measured at 13-14 and 15-16 years | NA | Performance in general certificates of education examinations | No association between serum 25(OH)D levels and educational outcome;, higher 25(OH)D concentrations associated with worse academic performance at 15-16 years. |
| Annweiler et al. [94] | Elderly women of mean age 79.8 years | vitamin D dietary intake | 7 years | NA | DSM-IV and NINCDS-ADRDA | Baseline vitamin D dietary intakes associate with a lower risk of developing AD but not other dementias |
| Slinin et al. [97] | Women of 65 years and older | serum 25(OH)D | 4 years | NA | MMSE | Low levels of 25(OH)D associate with cognitive impairment at baseline and with increased risk for global cognitive decline |
| Maddock et al. [109] | Adults aged 45 years | serum 25(OH)D | 5 years | NA | Verbal memory, fluency and speed of processing | Non-linear association of 25(OH)D levels with immediate word recall; both low and high serum concentrations are associated with worse cognitive performance |
| Afzal et al. [100] | All age groups | serum 25(OH)D | 30 years | NA | Diagnosis of AD and vascular dementia | Increased risk of AD and vascular dementia associated with reduced plasma 25(OH)D |
| LittleJohns et al. [105] | Elderly individuals of mean age 73.7 years | serum 25(OH)D | 5.6 years | NA | Diagnosis of dementia or AD NINCDS-ADRDA | Association between baseline vitamin D concentrations and the risk of incident all-cause dementia and AD |
| Jorde et al. [103] | All age groups | serum 25(OH)D | 7–13 years | NA | MMSE; word recall; digit-symbol coding; finger taping | 25(OH)D level is predictive of age-induced cognitive decline |
| Pre-post studies | ||||||
| Przybelski et al. [120] | Nursing home participants | serum 25(OH)D2 and 25(OH)D3 | 4 weeks | 50,000 IU of vitamin D2, 3 times a week | NPI | Increasing serum levels of total 25OHD does not improve cognition or behavior |
| Dean et al. [118] | Young adults of mean age 21.45 years | serum 25(OH)D | 6 weeks | 5,000 IU of vitamin D3 daily | Working memory, response inhibition and cognitive flexibility | No effect of vitamin D supplementation on cognitive and emotional functioning in healthy young adults |
| Stein et al. [121] | 60 years and older | serum 25(OH)D | 8 weeks | 3,000 IU of vitamin D2 daily | ADAS-cog and DAD | Increased serum levels of 25(OH)D associates with improvement in ADAS-cog score |
| Annweiler et al. [115] | Elderly individuals of mean age 80.6 years | serum 25(OH)D | 16 months | 800 IU daily or 10,000 IU monthly | MMSE; CAB and FAB | Vitamin D3 supplementation improves executive function |
| Annweiler et al. [116] | AD patients of mean age 84.7 years | NA | 6 months | Between 400 and 1,000 IU daily or 100,000 and 200,000 monthly of vitamin D3 with memantine | MMSE | Combination of memantine and vitamin D is associated to increased MMSE scores |
| Randomized Control Trials | ||||||
| Corless et al. [117] | Elderly geriatric patients | serum 25(OH)D | 2 to 9 months | 9,000 units of vitamin D2 | ADL assessment | No significant difference in the performance of the control or treated group |
| Dhesi et al. [119] | 65 years and older | serum 25(OH)D | 6 months post intervention | Single intramuscular injection of 600,000 IU of vitamin D2 | CRT | Improvement of information processing speed in supplemented arm compared to placebo group |
| Stein et al. [121] | 60 years and older | serum 25(OH)D | 8 weeks | 1,000 IU or 6,000 IU of vitamin D2 daily combined to an intranasal dose of insulin | MMSE; WMS-R LM; ADAS-cog and DAD | No difference between groups concerning the ADAS-cog or MMSE scores |
| Rossom et al. [98] | Women of 65 years and older | serum 25(OH)D | 7.8 years on average | 400 IU of vitamin D3 daily combined to calcium carbonate | DSM-IV | No association between treatment groups and incidence of cognitive decline |
| ANIMAL STUDIES | ||||||
| Taghizadeh et al. [62] | Adult male AD rat model | serum 25(OH)D | 8 weeks | Vitamin D free or vitamin D enriched with 1,25OH2D3 (1,000 ng/100 g dry food) diets | MWM | The absence of vitamin D intensifies spatial learning memory deficits |
| Yu et al. [63] | Young 1 month old AD mouse model | serum 25(OH)D | 5 months | 12 IU/g versus 2,4 or 0 IU/g of vitamin D3 containing diets | MWM | Enhanced performance in the high vitamin D diet group |
| Briones et al. [60] | Young (6 months) and old (20 months) rats | serum 25(OH)D | 21 days | Daily subcutaneous injections of 1,25(OH)D3 | MWM and Y-maze | Preservation of cognitive performance in aged rats only |
| Bennett et al. [66] | 2 month old AD mouse model | serum 25(OH)D | 7 months | Vitamin D2 containing diets | MWM | Enhanced cognitive performance in Wt mice rather than in Tg |
| Latimer et al. [59] | Middle-aged rats | serum 25(OH)D | 5-6 months | 10,000 IU/Kg versus 100 or 1,000 IU/KG of vitamin D3 containing diets | MWM | Dose dependent association of 25(OH)D levels and cognitive performance |
| Durk et al. [65] | 8 week old AD mouse models | serum 25(OH)D | 8 weeks | Intra-peritoneal injections of 1,25(OH)D3, 3 times a week | Fear conditioning paradigm | Restoration of learning and memory deficits in Tg treated mice |
| Landel et al. [64] | 4 month old female AD mouse model | NA | 4 months | 7,500 IU/Kg versus 1,000 IU/Kg of vitamin D3 | 6-arm radial water maze and Y-maze | Improved learning and working memory |
3MS, modified MMSE; ADAS-cog, Alzheimer’s disease Assessment Scale-cognitive subscale; ADL, Activity of Daily Living; CANTAB, Cambridge Neuropsychological Testing Automated Battery; CRT, Choice Reaction Time; DAD, Disability Assessment in Dementia Questionnaire; FAB, Frontal Assessment Battery score; MMSE, Mini Mental State Examination; MWM, Morris Water Maze; NINCDS-ADRDA, National Institute of Neurological and Communicative Disorders – Alzheimer’s Disease and Related Disorders Association; NPI, Neuropsychiatric Inventory; SPMSQ, Pfeiffer Short Portable Mental State Questionnaire; WISC-R, Wechsler Intelligence Scale for Children-revised; WMS-R LM, Wechsler Memory Scale-Revised Logical Memory; WRAT-R, Wide-range Achievement Test-revised.
Table 2
Comparison of observed outcomes in human studies linking vitamin D status to cognitive measures, according to potential confounding factors: gender, age and presence of pathology
| Potential confounder | ASSOCIATIVE STUDIES | INTERVENTIONAL STUDIES | |||||
| Number of studies | Results | Number of studies | Results | ||||
| Association (Number of studies) | No association (Number of studies) | Improvement (Number of studies) | No improvement (Number of studies) | ||||
| GENDER | Women | 5 | 5 | 0 | 1 | 0 | 1 |
| [93–97] | [93–97] | [98] | [98] | ||||
| Men | 1 | 0 | 1 | 0 | 0 | 0 | |
| [99] | [99] | ||||||
| Both | 15 | 12 | 4 | 8 | 2 | 6 | |
| [100–114] | [100–109, 111] | [110, 112–114] | [115–121] | [116, 119] | [115, 117, 118, 120, 121] | ||
| AGE | Under 65 | 5 | 1 | 4 | 1 | 0 | 1 |
| [109, 110, 112–114] | [109] | [110, 112–114] | [118] | [118] | |||
| Over 65 | 13 | 12 | 1 | 8 | 2 | 6 | |
| [93–97, 99, 101–103, 105–108] | [93–97, 101–103, 105–108] | [99] | [98, 115–117, 119–121] | [115, 119] | [98, 116, 117, 120, 121] | ||
| PATHOLOGY | Cognitively healthy elderly | 13 | 9 | 4 | 5 | 2 | 3 |
| [95–97, 99, 102–104, 106–108, 110, 111, 114] | [95–97, 102, 103, 106–108, 111] | [99, 104, 110, 114] | [98, 115, 117, 119, 120] | [115, 119] | [98, 117, 120] | ||
| MCI, AD, non-AD dementia | 5 | 5 | 0 | 3 | 0 | 3 | |
| [93, 94, 100, 101, 105] | [93, 94, 100, 101, 105] | [116, 121] | [116, 121] | ||||


