
Non-Catalytic Roles of Presenilin Throughout Evolution
Abstract
Research into Alzheimer’s disease pathology and treatment has often focused on presenilin proteins. These proteins provide the key catalytic activity of the γ-secretase complex in the cleavage of amyloid-β precursor protein and resultant amyloid tangle deposition. Over the last 25 years, screening novel drugs to control this aberrant proteolytic activity has yet to identify effective treatments for the disease. In the search for other mechanisms of presenilin pathology, several studies have demonstrated that mammalian presenilin proteins also act in a non-proteolytic role as a scaffold to co-localize key signaling proteins. This role is likely to represent an ancestral presenilin function, as it has been described in genetically distant species including non-mammalian animals, plants, and a simple eukaryotic amoeba Dictyostelium that diverged from the human lineage over a billion years ago. Here, we review the non-catalytic scaffold role of presenilin, from mammalian models to other biomedical models, and include recent insights using Dictyostelium, to suggest that this role may provide an early evolutionary function of presenilin proteins.
INTRODUCTION
The membrane protease γ-secretase has been the subject of intense study since it was linked to the etiology of Alzheimer’s disease (AD) [1]. It is composed of four transmembrane proteins, which includes a Presenilin catalytic subunit and three other proteins that are involved in complex stabilization and substrate binding, namely Anterior pharynx defective 1 (Aph1), Nicastrin, and Presenilin enhancer 2 (Pen2). In mammals, there are two presenilin proteins, Presenilin 1 (Psen1) and Presenilin 2 (Psen2), and mutations in these subunits are commonly found in patients with the early onset, autosomal dominant AD [2–4]. Presenilins are 9-pass transmembrane proteins that undergo an endoproteolytic cleavage between the sixth and seventh transmembrane domains, yielding N-terminal and C-terminal fragments that remain bound to each other [5, 6].
The well-recognized central role of presenilin proteins in AD pathology is through the cleavage of the amyloid-β precursor protein (AβPP), to produce a short amyloid peptide, Aβ40. Mutations in Psen1, Psen2, or AβPP that are associated with familial AD alter the cleavage of AβPP to increase the production of a more hydrophobic form of the amyloid peptide, Aβ42 [7]. This longer peptide is much more likely to form oligomers than the Aβ40 peptide, resulting in the production of extracellular plaques. These amyloid plaques comprise the hallmark of AD pathology [8], and the amyloid oligomers are thought to initiate a cascade of molecular consequences including tau phosphorylation, diminished increases in postsynaptic Ca2 +, and postsynaptic depression and dendritic spine loss that results in neuronal death [6, 9, 10]. This critical proteolytic function has focused research and therapeutic drug development toward the identification of presenilin small molecule inhibitors [11] and more recently monoclonal antibodies [12] as possible treatments for the disease. However, we are yet to identify new treatments that show efficacy in blocking AD progression.
Presenilin proteins, as part of the γ-secretase complex, also cleave a wide range of other transmembrane proteins [13, 14], including proteins involved in both development and normal cell function. One of these substrates, Notch, must be cleaved by presenilin to perform a critical developmental role, shown both in in vitro models and in vivo studies where mice lacking presenilin function show embryonic lethality [15], and is also evident in simple models such as the nematode C. elegans [15–17]. Interference with γ-secretase cleavage of these and other substrates is a major problem in developing presenilins as targets for therapeutic intervention in AD.
In addition to an extensive range of proteolytic substrates, presenilins have also been implicated in other cellular functions, including regulating calcium homeostasis [18], cell-cell adhesion [20], and membrane trafficking [19, 20]. A disruption in intracellular calcium signaling as a result of presenilin 1 and 2 mutations has also been linked to familial dilated cardiomyopathy [21], although whether this defect is related to the catalytic or non-catalytic roles of presenilin is unknown. Presenilin function in calcium homeostasis in C. elegans is independent of γ-secretase proteolytic activity [22]. Mutations in presenilin 1 and the γ-secretase components Nicastrin and Pen2 have also been linked to familial acne inversa (also known as hidradenitis suppurativa), an autosomal dominant, chronic inflammatory disease of hair follicles (reviewed in [23]). Most of the hidradenitis suppurativa mutations deleteriously affect one allele, which most likely leads to haploinsuffiency, whereas the predominantly missense mutations associated with familial AD most likely result in production of a mutant protein alongside wild-type protein that is produced from the unaffected allele. Again, it is unclear whether the role of the γ-secretase complex in this context is dependent on proteolytic activity or other non-catalytic roles.
Studies in mammalian cells and mice have demonstrated a presenilin function in the regulation of signaling pathways that is independent of their catalytic activity in proteolysis. Instead, the cytosolic regions of presenilin act as a scaffold that brings together key protein kinases and their substrates [24–26]. A non-proteolytic function for presenilin in multicellular development has also been described in other kingdoms, for example in early plants such as the moss Physcomitrella patens [27], and in a protist, the social amoeba Dictyostelium discoideum [28, 29]. This non-catalytic aspect of presenilin function in development in evolutionarily diverse species suggests that it represents an early role for presenilins. In this review, we outline studies using mammalian and non-mammalian models to elucidate non-catalytic function as a scaffold for presenilin proteins that are conserved from mammals to amoebae.
A NON-CATALYTIC FUNCTION OF PRESENILIN AS A SCAFFOLD IN MAMMALS
The first indication of a scaffold role for presenilins was the demonstration that presenilin binds to both a central kinase involved in Wnt signaling, glycogen synthase kinase 3β (GSK3β), and a key GSK3β substrate, the transcriptional regulator β-catenin [24–26]. In mammals, β-catenin has two roles that are mediated by distinct pools, a membrane-bound pool of the protein that functions in adhesion together with members of the cadherin family including E- and N-cadherins, and a cytosolic pool of the protein that can translocate to the nucleus and act as a transcriptional co-factor when canonical Wnt/wingless signaling is active. In the absence of Wnt/wingless, β-catenin is phosphorylated by GSK3β, promoting the ubiquitination of β-catenin and its subsequent degradation by the proteasome (Fig. 1). However, it was initially unclear whether an interaction between presenilin and GSK3β/β-catenin stabilized β-catenin or enhanced its degradation, since early studies used protein overexpression instead of manipulating endogenous proteins or their levels. Murayama and colleagues [26] showed that overexpression of wild-type Psen1 in a mammalian cell line, COS7, reduced cytoplasmic β-catenin levels in a GSK3β-dependent manner, and that different mutations in presenilin 1 associated with familial AD (M146L, A246L, or L392V) did not alter this reduction. However, Zhang and colleagues [24] showed the opposite relationship, where following overexpression of Psen1 and β-catenin in a human HEK293 cell line, elevated presenilin 1 levels correlated with enhanced β-catenin stability, and that familial AD mutations in presenilin 1 (M146V, C410Y, or I143T) reduced this ability to stabilize β-catenin (Fig. 1). A similar role was suggested by comparing neurons from wild-type mice and from mice overexpressing human Psen1, together with fibroblasts from wild-type and Psen1-null mice [24]. To resolve this discrepancy, Kang and colleagues [25] examined the turnover and steady state levels of endogenous β-catenin in tissue culture cells and transgenic mice. In tissue culture, they found that the complex between Psen1 and β-catenin did not contain APC or E-cadherin, and so was presumably independent of the Wnt signaling-related cytosolic β-catenin degradation complex and the membrane-bound adhesion related pool of β-catenin, and thus represented a third pool of regulated β-catenin. They also showed that GSK3β interacted with the N-terminal fragment of Psen1 (amino acids 1-298), and that β-catenin bound to the C-terminal fragment of Psen1 (amino acids 299–427) (Fig. 1). This GSK3β binding was reduced by mutations in Psen1 associated with familial AD (M146L and ΔX9 (lacking amino acid 291–319)) compared to wild-type Psen1, whereas β-catenin binding was unaffected. Importantly, wild-type Psen1 facilitated the turnover of β-catenin, whereas the M146L and ΔX9 mutations slowed this rate, even though no difference in the steady-state level of β-catenin was detectable. Thus, initial discrepancies in data from experiments using artificially high protein levels were resolved by using endogenous proteins to demonstrate a role for presenilin as a scaffold that binds both GSK3β and β-catenin to facilitate β-catenin phosphorylation and turnover (Fig. 1).
The scaffold role of presenilin was subsequently confirmed, in vivo and in vitro, by examining brain tissue from transgenic mice expressing wild-type or M146L human Psen1 [25]. In these experiments, the steady state levels of β-catenin were lower in the brains of mice overexpressing wild-type human Psen1 compared to control littermates, whereas β-catenin levels were stabilized by expression of the Psen1 M146L mutant protein. Additionally, fibroblasts from psen1-/- mice had reduced turnover and increased steady state levels of β-catenin when compared to fibroblasts from hemizygous psen1+/- mice. Overexpression of wild-type Psen1 in psen1-/- fibroblasts restored the turnover of β-catenin, whereas it was only partially restored by the ΔX9 familial AD mutant.
Kang and colleagues then examined the mechanism by which the level of β-catenin was modulated by Psen1 [30]. They found that Psen1 was required for a stepwise phosphorylation of β-catenin by separate kinases to ensure its ubiquitination and consequent degradation. First, β-catenin is phosphorylated at serine 45, which can be performed by protein kinase A (PKA) or another unidentified kinase, which is a prerequisite for the subsequent phosphorylation at serine 33/serine 37/threonine 41 by GSK3β [30].Presenilin 1 functioned as a scaffold to bind all three proteins, PKA, GSK3β, and β-catenin. This regulation of β-catenin function is separable from the Notch and γ-secretase cleavage activity of Psen1, since a mutated Psen1 protein unable to bind β-catenin, Psen1Δcat (lacking residues 330–360), still retains Notch and γ-secretase cleavage activity and so rescues the embryonic lethality of Psen1-null mice [31]. However, Psen1Δcat cannot rescue the hyper-proliferation of psen1-/- fibroblasts that is a result of dysregulated β-catenin/Wnt signaling [32]. Thus, the Psen1 function as a scaffold that binds GSK3β, β-catenin, and possibly PKA, is independent of γ-secretase proteolytic activity toward Notch or AβPP. These kinases are widely recognized to play a key role in AD pathology, and as potential targets for therapeutic intervention [33, 34].
Research in this area using mammalian models is complicated by the embryonic lethality of Psen1-null animals, making understanding presenilin functions in the adult organism and tissues difficult without using inducible or tissue-specific gene ablation. To circumvent the embryonic lethality of Psen1 ablation, Xia and colleagues [31] expressed human presenilin 1 in the neuronal cells of psen1-/- mice, which enabled these mice to complete embryonic development, and so they could examine the role of dysregulated β-catenin signaling in the remaining psen1-/- tissues of these mice. Surprisingly, the mice showed higher cytosolic levels of β-catenin in primary keratinocytes compared to wild-type littermates, which could be rescued by expression of wild-type Psen1 but not the Psen1Δcat protein that is unable to bind β-catenin. The psen1-/- mice expressing neuronal human Psen1 also had numerous skin tumors that contained nuclear β-catenin. Thus, altered levels of presenilin clearly regulate cellular function, with likely effects on scaffold function and β-catenin signaling. Later studies also showed that a single homozygous knock-in presenilin mutation (L435F) caused a complete loss of γ-secretase activity, and age-dependent neurodegeneration, suggesting AD pathology arises from loss of presenilin function [35]. It remains unclear if this mutation alters the scaffold role of presenilins in this model.
A range of non-mammalian biomedical models have also been used to investigate presenilin function, with some of these studies providing supportive data on their role broadly across genetically distant species. Clear homologues of mammalian presenilins are present in both multicellular organisms like the metazoans Drosophila melanogaster (fruit fly) and Caenorhabditis elegans (nematode worm), and in plants, and in unicellular organisms like the social amoeba Dictyostelium discoideum (Fig. 2). In the yeast Saccharomyces cerevisiae, however, a single gene, ypf1, encodes a protein with a presenilin fold that is unlikely to represent a presenilin orthologue, since it shares greater similarity with an intramembrane protease related to presenilins, the signal peptide peptidase [36]. In addition, clear homologues of the remaining components of γ-secretase are not present in the genome of S. cerevisiae, although homologues of all four γ-secretase components are identifiable in other sequenced fungal genomes.
PRESENILIN FUNCTION IN FLIES
The fruit fly, Drosophila melanogaster, provides a useful genetic model to study presenilin function. Flies possess only a single copy of presenilin (dps) and the other core components of the γ-secretase complex. In addition to its well-established role in Notch processing, Dps appears also to regulate β-catenin in a similar manner to that observed in mammalian models, where presenilin regulates the levels of the β-catenin orthologue armadillo (Arm) [30]. In addition, Drosophila presenilin is a negative regulator of wingless (Wnt) signaling, and, critically, this involves GSK3β and β-catenin [37]. However, dps knockdown has no effect on total Arm levels [38], but it is possible that, as in mammals, only a membrane-bound pool of Arm/β-catenin is affected by presenilin. Dps has also been shown to regulate axonal transport of the Drosophila APP homologue, Dappl, although inhibiting γ-secretase activity had an effect similar to reduction of dps levels [39], suggesting that this is a proteolytic function of presenilin. This regulation of axonal transport appears to be through GSK3β activity that affects the membrane binding of the motor proteins kinesin-1 and dynein; Dps positively regulates GSK3β activity and membrane association [40]. These data contrast to those in mice, where Psen1 negatively regulates GSK3β activity to control the phosphorylation of kinesin light chain (KLC), and thus the membrane association of kinesin I and axonal transport of multiple cargoes [41]. Interestingly, Psen controls the level of Cyclic-AMP-Response Element-Binding (CREB)-Binding Protein (CBP) in the adult fly CNS, a gene that is required for cognitive function in flies and humans [42]. This regulation by Psen is independent of its proteolysis-dependent activation of the co-transcriptional activity of Notch, plausibly by affecting the activity of an alternate Psen substrate or through a non-catalytic activity of Psen.
PRESENILIN FUNCTION IN WORMS
Much of the early characterization of presenilin structure and function came from studies of the C. elegans homologue Sel-12 that was identified in a screen for genes regulating the activity of the C. elegans Notch homologue Lin-12 [43]. Subsequent studies showed that Sel-12 is composed of 8 transmembrane domains, and is cleaved into N- and C-terminal fragments [44]. Expression of human presenilins could rescue the defects in sel-12-mutants, and human presenilins containing familial AD mutations had a reduced ability to rescue these defects [16, 45]. Sel-12 was required for the processing and trafficking of Lin-12 [46]. C. elegans also contains a second presenilin homologue, HOP-1, that is functionally redundant with Sel-12 [47]. Recent research in C. elegans has also identified a role for Sel-12 in calcium homeostasis, mitochondrial function, and apoptosis [22, 48], independent of the role of Sel-12 in γ-secretase proteolytic activity, suggesting a non-catalytic function for presenilins in C. elegans [22].
A NON-CATALYTIC FUNCTION OF PRESENILIN IN AN EARLY PLANT
Although presenilin and the other components of γ-secretase are present in higher plants [49], in depth functional analysis has only been conducted in the moss Physcomitrella patens. Presenilin ablation in this organism produced pleiotropic phenotypic abnormalities, including development and differentiation changes, and defects in light or gravity sensing, which stemmed in part from cytoskeletal abnormalities [27]. Importantly, these defects were rescued by the expression of human Psen1. Conversely, expression of P. patens psen in mouse psen1-/- fibroblasts rescued the hyperproliferative phenotype of these cells resulting from dysregulated β-catenin/Wnt signaling. These results demonstrate that the human and moss proteins have conserved functions. However, this study found that the activity of the human Psen1 in rescuing the moss mutant phenotype was not through its catalytic activity, as Psen1 with either of the catalytic aspartic acids mutated to alanine (D257A or D385A) also rescued the growth effect. This result suggested that the role of presenilins in this model is likely through a scaffold function. Interestingly, P. patens Psen could not rescue Notch cleavage in psen1-/- fibroblasts, possibly because it is unable to assemble with the remaining mouse γ-secretase components.
A NON-CATALYTIC FUNCTION OF PRESENILINS IN DICTYOSTELIUM DEVELOPMENT
The social amoeba Dictyostelium discoideum is a valuable model to investigate presenilin function [28, 29]. Dictyostelium is one of the simplest eukaryotic model organisms that contains two presenilin genes, psenA and psenB [28] (Fig. 2). Dictyostelium lacks the vital proteolytic substrates Notch and APP, providing a simple system for research that is not complicated by the lethal effects of a loss of function resulting in a disruption of Notch processing. Furthermore, gene ablation and biochemical and developmental studies can rapidly provide information on presenilin function in these models.
In this organism, the developmental cycle (Fig. 3A) begins when single cells undergo starvation, inducing a highly regulated pattern of cell aggregation in response to the release of extracellular cAMP, leading to the formation of mounds, which undergo complex morphogenesis and cell-type differentiation to produce a fruiting body composed of a stalk holding aloft a ball of spores [50]. Dictyostelium is a useful model organism to study intercellular signaling and human diseases [51–53], since it retains many signaling pathways that have been lost in yeast [54], it is simple to grow and maintain in the laboratory, and isogenic knockout and overexpression lines can be used to identify the role of individual proteins.
In Dictyostelium, as in mammals, the two presenilins have partially redundant functions in controlling multicellular development [28, 29]. Ablation of either psenA or psenB had no effect on the formation of mature fruiting bodies in Dictyostelium development, whereas a psenA−/psenB− double mutant showed a block at the mound stage [28] (Fig. 3B). This developmental role of presenilins has been conserved throughout evolution, as expression of either Dictyostelium psenB or human psen1 in psenA−/psenB− cells rescues this developmental block.
The Dictyostelium presenilin proteins also share a similar structure and localization to mammalian presenilins. The proteins show similar tertiary structure (Fig. 4A) and both mammalian and Dictyostelium presenilin proteins localize to the endoplasmic reticulum (Fig. 4B and [28]). Both human and Dictyostelium presenilin proteins also share the key catalytic aspartic acids and PAL sequences found in their human counterparts (Fig. 4C).
To investigate the mechanism of this developmental function of Dictyostelium presenilin proteins, a series of experiments employed mutated presenilin proteins. Expressing either human Psen1 D257A/D385A or Dictyostelium PsenB D348A/D394A proteins lacking both catalytic aspartic acids in psenA−/psenB− cells restored fruiting body formation (Fig. 3B) [28]. These data indicate a non-catalytic role for presenilin proteins in Dictyostelium development. This developmental activity is likely to be independent of the γ-secretase complex, since ablation of nicastrin, which is required for γ-secretase activity in mammals [55], does not cause a similar block in development at the mound stage [28]. Furthermore, the Dictyostelium PsenB protein is likely to have proteolytic activity, as it cleaves Notch-related targets in a mouse psen1-/- blastocyst cell line [28]; however, the endogenous targets in Dictyostelium remain to be identified. It is intriguing that presenilin ablation in Dictyostelium reduces stalk cell differentiation, a process that is regulated by GSKA (the Dictyostelium orthologue of GKS3) activity in this organism [56], implicating a direct regulatory role of presenilins in inhibiting GSKA activity in this model. Also, psenA−/psenB− cells show defects in both streaming behavior during the aggregation phase of development and in spore cell production, processes that are both controlled by PKA [57, 58]. These two observations hint that the non-catalytic presenilin function in Dictyostelium development may be via GSKA and PKA. These results, together with those from moss, strongly support a conserved non-catalytic role of presenilin proteins in development that is independent of γ-secretase proteolytic activity. Since AD pathology has been linked to both GSK3 and PKA activity [33, 34], further investigation of this non-catalytic presenilin function may provide mechanisms to reduce pathogenic AD-related signaling.
MOVING FORWARD
Understanding the full range of presenilin functions has important implications in research focused on both development and medicine. In development, the loss of Notch signaling following presenilin ablation has over-shadowed other developmental roles for these proteins. In medical research, in particular regarding AD, research has tended to focus on the proteolytic function of presenilin proteins (and the γ-secretase complex), and attempts to regulate this activity has provided scant evidence of new therapeutic approaches. New insights into the roles of presenilin proteins have been provided through the use of a 3Rs approach (animal replacement, reduction, and refinement [59]) where models such as Drosophila, C. elegans, and P. patens have enabled the advance of new methodologies to study presenilin and γ-secretase function, without the use of mammalian models. Using this approach with a social amoeba, Dictyostelium, a key cellular function of presenilin proteins has been shown to regulate development through a non-catalytic role [28], involving GSKA and PKA signaling perturbation, and is consistent with that demonstrated in mammalian models in AD pathology [33, 34]. Further research using both 3Rs and mammalian models relating to non-catalytic presenilin roles is likely to provide new insights into the cellular functions of these important proteins and may provide alternative approaches for therapeutic intervention to reverse the pathology associated with presenilin-dependent diseases.
ACKNOWLEDGMENTS
GPO was supported by an NC3Rs Grant (NC/M001504/1) and a Dr Hadwen Trust for Humane Research (DHT) grant, which is the UK leading medical research charity that funds and promotes exclusively human-relevant research that encourages the progress of medicine with the replacement of the use of animals in research.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/15-0940r2).
REFERENCES
[1] | De Strooper B , Iwatsubo T , Wolfe MS ((2012) ) Presenilins and gamma-secretase: Structure, function, and role in Alzheimer Disease. Cold Spring Harb Perspect Med 2: , a006304. |
[2] | Levy-Lahad E , Wijsman EM , Nemens E , Anderson L , Goddard KA , Weber JL ((1995) ) A familial Alzheimer’s disease locus on chromosome 1. Science 269: , 970–973. |
[3] | Rogaev EI , Sherrington R , Rogaeva EA , Levesque G , Ikeda M , Liang Y , Chi H , Lin C , Holman K , Tsuda T , Mar L , Sorbi S , Nacmias B , Piacentini S , Amaducci L , Chumakov I , Cohen D , Lannfelt L , Fraser PE , Rommens JM , St George-Hyslop PH ((1995) ) Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 376: , 775–778. |
[4] | Sherrington R , Rogaev EI , Liang Y , Rogaeva EA , Levesque G , Ikeda M , Chi H , Lin C , Li G , Holman K , Tsuda T , Mar L , Foncin JF , Bruni AC , Montesi MP , Sorbi S , Rainero I , Pinessi L , Nee L , Chumakov I , Pollen D , Brookes A , Sanseau P , Polinsky RJ , Waso W , Da Silva HA , Haines JL , Perkicak-Vance MA , Tanzi RE , Roses AD , Fraser PE , Rommens JM , St George-Hyslop PH ((1995) ) Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 375: , 754–760. |
[5] | Thinakaran G , Borchelt DR , Lee MK , Slunt HH , Spitzer L , Kim G , Ratovitsky T , Davenport F , Nordstedt C , Seeger M , Hardy J , Levey AI , Gandy SE , Copeland NG , Price DL , Sisodia SS ((1996) ) Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron 17: , 181–190. |
[6] | Capell A , Grunberg J , Pesold B , Diehlmann A , Citron M , Nixon R , Beyreuthers K , Selkoe DJ , Haass C ((1998) ) The proteolytic fragments of the Alzheimer’s disease-associated presenilin-1 form heterodimers and occur as a 100-150-kDa molecular mass complex. J Biol Chem 273: , 3205–3211. |
[7] | De Strooper B ((2007) ) Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep 8: , 141–146. |
[8] | McKhann G , Drachman D , Folstein M , Katzman R , Price D , Stadlan EM ((1984) ) Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34: , 939–944. |
[9] | Wille H , Drewes G , Biernat J , Mandelkow EM , Mandelkow E ((1992) ) Alzheimer-like paired helical filaments and antiparallel dimers formed from microtubule-associated protein tau in vitro. J Cell Biol 118: , 573–584. |
[10] | Palop JJ , Mucke L ((2010) ) Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: From synapses toward neural networks. Nat Neurosci 13: , 812–818. |
[11] | Dominguez DI , De SB , Annaert W ((2001) ) Secretases as therapeutic targets for the treatment of Alzheimer’s disease. Amyloid 8: , 124–142. |
[12] | Takagi-Niidome S , Osawa S , Tomita T , Iwatsubo T ((2013) ) Inhibition of gamma-secretase activity by a monoclonal antibody against the extracellular hydrophilic loop of presenilin 1. Biochemistry 52: , 61–69. |
[13] | Parks AL , Curtis D ((2007) ) Presenilin diversifies its portfolio. Trends Genet 23: , 140–150. |
[14] | Haapasalo A , Kovacs DM ((2011) ) The many substrates of presenilin/gamma-secretase. J Alzheimers Dis 25: , 3–28. |
[15] | Xue Y , Gao X , Lindsell CE , Norton CR , Chang B , Hicks C , Gendron-Maguire M , Rand EB , Weinmaster G , Gridley T ((1999) ) Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet 8: , 723–730. |
[16] | Levitan D , Doyle TG , Brousseau D , Lee MK , Thinakaran G , Slunt HH , Sisodia SS , Greenwald I ((1996) ) Assessment of normal and mutant human presenilin function in Caenorhabditis elegans. Proc Natl Acad Sci U S A 93: , 14940–14944. |
[17] | De Strooper B , Saftig P , Craessaerts K , Vanderstichele H , Guhde G , Annaert W , Von Figura K , Van Leuven F ((1998) ) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature, 391: , 387–390. |
[18] | Berridge MJ ((2011) ) Calcium signalling and Alzheimer’s disease. Neurochem Res 36: , 1149–1156. |
[19] | Georgakopoulos A , Marambaud P , Efthimiopoulos S , Shioi J , Cui W , Li HC , Shütte M , Gordon R , Holstein GR , Martinelli G , Mehta P , Friedrich VL Jr , Robakis NK ((1999) ) Presenilin-1 forms complexes with the cadherin/catenin cell-cell adhesion system and is recruited to intercellular and synaptic contacts. Mol Cell 4: , 893–902. |
[20] | Rajendran L , Annaert W ((2012) ) Membrane trafficking pathways in Alzheimer’s disease. Traffic 13: , 759–770. |
[21] | Li D , Parks SB , Kushner JD , Nauman D , Burgess D , Ludwigsen S , Partain J , Nixon RR , Allen CN , Irwin RP , Jakobs PM , Litt M , Hershberger RE ((2006) ) Mutations of presenilin genes in dilated cardiomyopathy and heart failure. Am J Hum Genet 79: , 1030–1039. |
[22] | Sarasija S , Norman KR ((2015) ) A gamma-secretase independent role for presenilin in calcium homeostasis impacts mitochondrial function and morphology in Caenorhabditis elegans. Genetics, 201: , 1453–1466. |
[23] | Setta-Kaffetzi N , Navarini AA , Patel VM , Pullabhatla V , Pink AE , Choon SE , Allen MA , Burden AD , Griffiths CE , Seyger MM , Kirty B , Trembath RC , Simpson MA , Smith CH , Capon F , Barker JN ((2013) ) Rare pathogenic variants in IL36RN underlie a spectrum of psoriasis-associated pustular phenotypes. J Invest Dermatol 133: , 1366–1369. |
[24] | Zhang Z , Hartmann H , Do VM , Abramowski D , Sturchler-Pierrat C , Staufenbiel M , Sommer B , van de Wetering M , Clevers H , Saftig P , De Strooper B , He X , Yankner BA ((1998) ) Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature 395: , 698–702. |
[25] | Kang DE , Soriano S , Frosch MP , Collins T , Naruse S , Sisodia SS , Leibowitz G , Levine F , Koo EH ((1999) ) Presenilin 1 facilitates the constitutive turnover of beta-catenin: Differential activity of Alzheimer’s disease-linked PS1 mutants in the beta-catenin-signaling pathway. J Neurosci 19: , 4229–4237. |
[26] | Murayama M , Tanaka S , Palacino J , Murayama O , Honda T , Sun X , Yasutake K , Nihonmatsu N , Wolozin B , Takashima A ((1998) ) Direct association of presenilin-1 with beta-catenin. FEBS Lett 433: , 73–77. |
[27] | Khandelwal A , Chandu D , Roe CM , Kopan R , Quatrano RS ((2007) ) Moonlighting activity of presenilin in plants is independent of gamma-secretase and evolutionarily conserved. Proc Natl Acad Sci U S A 104: , 13337–13342. |
[28] | Ludtmann MH , Otto GP , Schilde C , Chen ZH , Allan CY , Brace S , Beesley PW , Kimmel AR , Fisher P , Killick R , Williams RS ((2014) ) An ancestral non-proteolytic role for presenilin proteins in multicellular development of the social amoeba. J Cell Sci 127: , 1576–1584. |
[29] | McMains VC , Myre M , Kreppel L , Kimmel AR ((2010) ) possesses highly diverged presenilin/gamma-secretase that regulates growth and cell-fate specification and can accurately process human APP: A system for functional studies of the presenilin/gamma-secretase complex. Dis Model Mech 3: , 581–594. |
[30] | Kang DE , Soriano S , Xia X , Eberhart CG , De SB , Zheng H , Koo EH ((2002) ) Presenilin couples the paired phosphorylation of beta-catenin independent of axin: Implications for beta-catenin activation in tumorigenesis. Cell 110: , 751–762. |
[31] | Xia X , Qian S , Soriano S , Wu Y , Fletcher AM , Wang XJ , Koo EH , Wu X , Zheng H ((2001) ) Loss of presenilin 1 is associated with enhanced beta-catenin signaling and skin tumorigenesis. Proc Natl Acad Sci U S A 98: , 10863–10868. |
[32] | Soriano S , Kang DE , Fu M , Pestell R , Chevallier N , Zheng H , Koo EH ((2001) ) Presenilin 1 negatively regulates beta-catenin/T cell factor/lymphoid enhancer factor-1 signaling independently of beta-amyloid precursor protein and notch processing. J Cell Biol 152: , 785–794. |
[33] | Myeku N , Clelland CL , Emrani S , Kukushkin NV , Yu WH , Goldberg AL , Duff Ke ((2016) ) Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat Med 22: , 46–53. |
[34] | Medina M , Avila J ((2013) ) Understanding the relationship between GSK-3 and Alzheimer’s disease: A focus on how GSK-3 can modulate synaptic plasticity processes. Expert Rev Neurother 13: , 495–503. |
[35] | Xia D , Watanabe H , Wu B , Lee SH , Li Y , Tsvetkov E , Bolshakov VY , Shen J , Kelleher RJ 3rd ((2015) ) Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron 85: , 967–981. |
[36] | Avci D , Fuchs S , Schrul B , Fukumori A , Breker M , Frumkin I , Chen C , Biniossek ML , Kremmer E , Schilling O , Steiner H , Shuldiner M , Lemberg MK ((2014) ) The yeast -intramembrane protease Ypf1 refines nutrient sensing by regulating transporter abundance. Mol Cell 56: , 630–640. |
[37] | Cox RT , McEwen DG , Myster DL , Duronio RJ , Loureiro J , Peifer MA ((2000) ) screen for mutations that suppress the phenotype of Drosophila armadillo, the beta-catenin homolog. Genetics 155: , 1725–1740. |
[38] | Matsubayashi H , Sese S , Lee JS , Shirakawa T , Iwatsubo T , Tomita T , Yanagawa S ((2004) ) Biochemical characterization of the Drosophila wingless signaling pathway based on RNA interference. Mol Cell Biol 24: , 2012–2024. |
[39] | Gunawardena S , Yang G , Goldstein LS ((2013) ) Presenilin controls kinesin-1 and dynein function during APP-vesicle transport in vivo. Hum Mol Genet 22: , 3828–3843. |
[40] | Dolma K , Iacobucci GJ , Hong ZK , Shandilya J , Toska E , White JA , Spina E , Gunawardena S ((2014) ) Presenilin influences glycogen synthase kinase-3 beta (GSK-3beta) for kinesin-1 and dynein function during axonal transport. Hum Mol Genet 23: , 1121–1133. |
[41] | Pigino G , Morfini G , Pelsman A , Mattson MP , Brady ST , Busciglio J ((2003) ) Alzheimer’s presenilin 1 mutations impair kinesin-based axonal transport. J Neurosci 23: , 4499–4508. |
[42] | Boyles RS , Lantz KM , Poertner S , Georges SJ , Andres AJ ((2010) ) Presenilin controls CBP levels in the adult Drosophila central nervous system. PLoS One 5: , e14332. |
[43] | Levitan D , Greenwald I ((1995) ) Facilitation of lin-12-mediated signalling by sel-12, a Caenorhabditis elegans S182 Alzheimer’s disease gene. Nature 377: , 351–354. |
[44] | Li X , Greenwald I ((1998) ) Additional evidence for an eight-transmembrane-domain topology for Caenorhabditis elegans and human presenilins. Proc Natl Acad Sci U S A 95: , 7109–7114. |
[45] | Baumeister R , Leimer U , Zweckbronner I , Jakubek C , Grunberg J , Haass C ((1997) ) Human presenilin-1, but not familial Alzheimer’s disease (FAD) mutants, facilitate Caenorhabditis elegans Notch signalling independently of proteolytic processing. Genes Funct 1: , 149–159. |
[46] | Levitan D , Greenwald I ((1998) ) Effects of SEL-12 presenilin on LIN-12 localization and function in Caenorhabditis elegans. Development 125: , 3599–3606. |
[47] | Li X , Greenwald I ((1997) ) HOP-1, a Caenorhabditis elegans presenilin, appears to be functionally redundant with SEL-12 presenilin and to facilitate LIN-12 and GLP-1 signaling. Proc Natl Acad Sci U S A 94: , 12204–12209. |
[48] | Kitagawa N , Shimohama S , Oeda T , Uemura K , Kohno R , Kuzuya A , Shibasaki H , Ishii N ((2003) ) The role of the presenilin-1 homologue gene sel-12 of Caenorhabditis elegans in apoptotic activities. J Biol Chem 278: , 12130–12134. |
[49] | Smolarkiewicz M , Skrzypczak T , Michalak M , Lesniewicz K , Walker JR , Ingram G , Wojtaszek P ((2014) ) Gamma-secretase subunits associate in intracellular membrane compartments in Arabidopsis thaliana. J Exp Bot 65: , 3015–3027. |
[50] | Williams RS , Boeckeler K , Graf R , Muller-Taubenberger A , Li Z , Isberg RR , Wessels D , Soll Dr , Alexander H , Alexander S ((2006) ) Towards a molecular understanding of human diseases using Dictyostelium discoideum. Trends Mol Med 12: , 415–424. |
[51] | Williams RSB ((2005) ) Pharmacogenetics in model systems: Defining a common mechanism of action for mood stabilisers. Prog Neuropsychopharmacol Biol Psychiatry 29: , 1029–1037. |
[52] | Waheed A , Ludtmann MH , Pakes N , Robery S , Kuspa A , Dinh C , Baines D , Williams RS , Carew MA ((2014) ) Naringenin inhibits the growth of and MDCK-derived cysts in a TRPP2 (polycystin-2)-dependent manner. Br J Pharmacol 171: , 2659–2670. |
[53] | Chang P , Orabi B , Deranieh RM , Dham M , Hoeller O , Shimshoni JA , Yagen B , Bialer M , Greenberg ML , Walker MC , Williams RS ((2012) ) The antiepileptic drug valproic acid and other medium-chain fatty acids acutely reduce phosphoinositide levels independently of inositol in Dictyostelium. Dis Model Mech 5: , 115–124. |
[54] | Eichinger L , Pachebat JA , Glockner G , Rajandream MA , Sucgang R , Berriman M , Song J , Olsen R , Szafranksi K , Xu Q , Tunggal B , Kummerfeld S , Madera M , Konfortov BA , Rivero F , BAnkier AT , Lehmann R , Hamlin N , Davies R , Gaudet P , Frey P , Pilcher K , Chen G , Saunders D , Sodergren E , Davis P , Kerhornou A , Nie X , Hall N , Anjard C , Hemphill L , Bason N , Farbrother P , Desany B , Just E , Morio T , Rost R , Churcher C , Cooper J , Haydock S , van Driessche N , Cronin A , goodhead I , Muzny D , Mourier T , Pain A , Lu M , Harper D , Lindsay R , Hauser H , James K , Quiles M , Madan Babu M , Saito T , Buchrieser C , Wardroper A , Felder M , Thangavelu M , Johnson D , Kinghts A , Loulseged H , Mungall K , Oliver K , Price C , Quail MA , Urushihara H , Hernandez J , Rabbinowitsch E , Steffen D , Sanders M , Ma J , Kohara Y , Sharp S , Simmonds M , Spiegler S , Tivey A , Sugano S , White B , Walker D , Woodward J , Winckler T , Tanaka Y , Shaulsky G , Schleicher M , Weinstock G , Rosenthal A , Cox EC , Chisholm RL , Gibbs R , Loomis WF , Platzer M , Kay RR , Williams J , Dear PH , Noegel AA , Barrel B , Kuspa A ((2005) ) The genome of the social amoeba. Nature 435: , 43–57. |
[55] | Edbauer D , Winkler E , Regula JT , Pesold B , Steiner H , Haass C ((2003) ) Reconstitution of gamma-secretase activity. Nat Cell Biol 5: , 486–488. |
[56] | Harwood AJ , Plyte SE , Woodgett J , Strutt H , Kay RR ((1995) ) Glycogen synthase kinase 3 regulates cell fate in. Cell 80: , 139–148. |
[57] | Mann SK , Firtel RA ((1993) ) cAMP-dependent protein kinase differentially regulates prestalk and prespore differentiation during development. Development 119: , 135–146. |
[58] | Kriebel PW , Barr VA , Parent CA ((2003) ) Adenylyl cyclase localization regulates streaming during chemotaxis. Cell 112: , 549–560. |
[59] | Graham ML , Prescott MJ ((2015) ) The multifactorial role of the 3Rs in shifting the harm-benefit analysis in animal models of disease. Eur J Pharmacol 759: , 19–29. |
[60] | Kelley LA , Mezulis S , Yates CM , Wass MN , Sternberg MJ ((2015) ) The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc 10: , 845–858. |
Figures and Tables
Fig.1
A proposed model for the scaffolding role of the presenilin proteins. Under normal conditions (A), GSK3β binds to the Psen1 N-terminal fragment in the cytoplasmic loop (residues 250–290) and β-catenin binds to the C-terminal fragment (residues 330–360). This binding results in the phosphorylation of β-catenin at S33/S37/T41 by GSK3β, and therefore increased turnover of β-catenin. B) Mutations in Psen1 affecting binding of either GSK3β or β-catenin reduce the phosphorylation, and therefore ubiquitination, of β-catenin, leading to increased levels of nuclear β-catenin and enhanced β-catenin-dependent signaling.
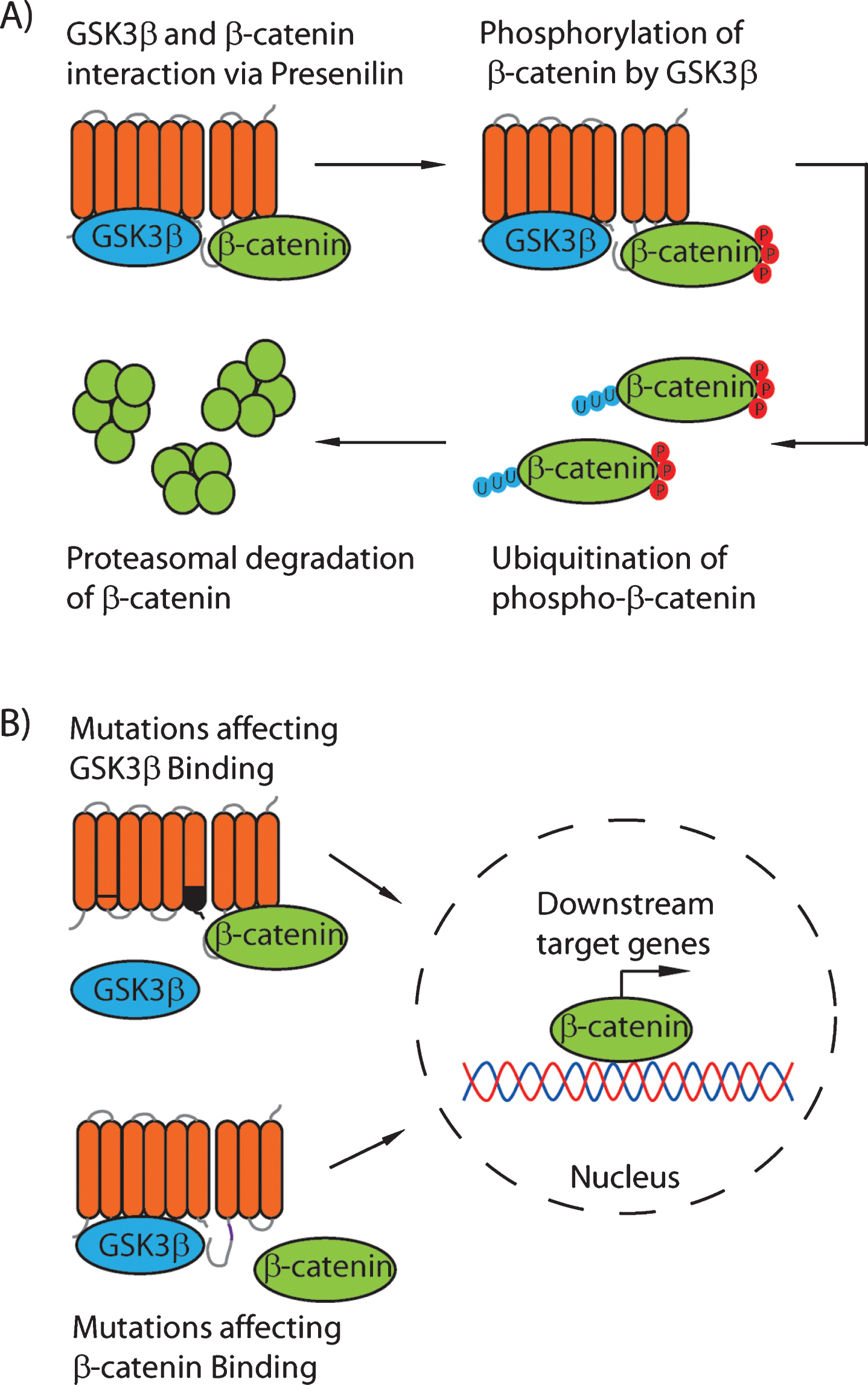
Fig.2
Presenilin proteins are distributed widely in evolutionarily distant species. A phylogenetic tree showing the conservation of the presenilin proteins from various species from different kingdoms including Animalia, Plantae, and Protista. Accession numbers for the proteins were: H. sapiens Psen1: P49768, M. musculus Psen1: P49769, R. norvegicus Psen1: P97887, X. laevis Psen1: O12976, D. rerio Psen1: Q9W6T7, H. sapiens Psen2: P49810, M. musculus Psen2: Q61144, R. norvegicus Psen2: Q88777, X. laevis Psen2: O12977, D. rerio Psen2: Q90ZE4, C. elegans Psen: P52166, D. discoideum PsenA: Q54ET2, D. discoideum PsenB: Q54DE8, P. pallidum PsenA: D3B4U3, B. prasinos Psen1: K8EKK4, O. sativa Psen: C7J054, P. patens Psen1: A9S846, A. thaliana Psen1: O64668, A. thaliana Psen2: Q9SIK7, S. cerevisiae YPF1: P34248, D. melanogaster Psen: O02194.
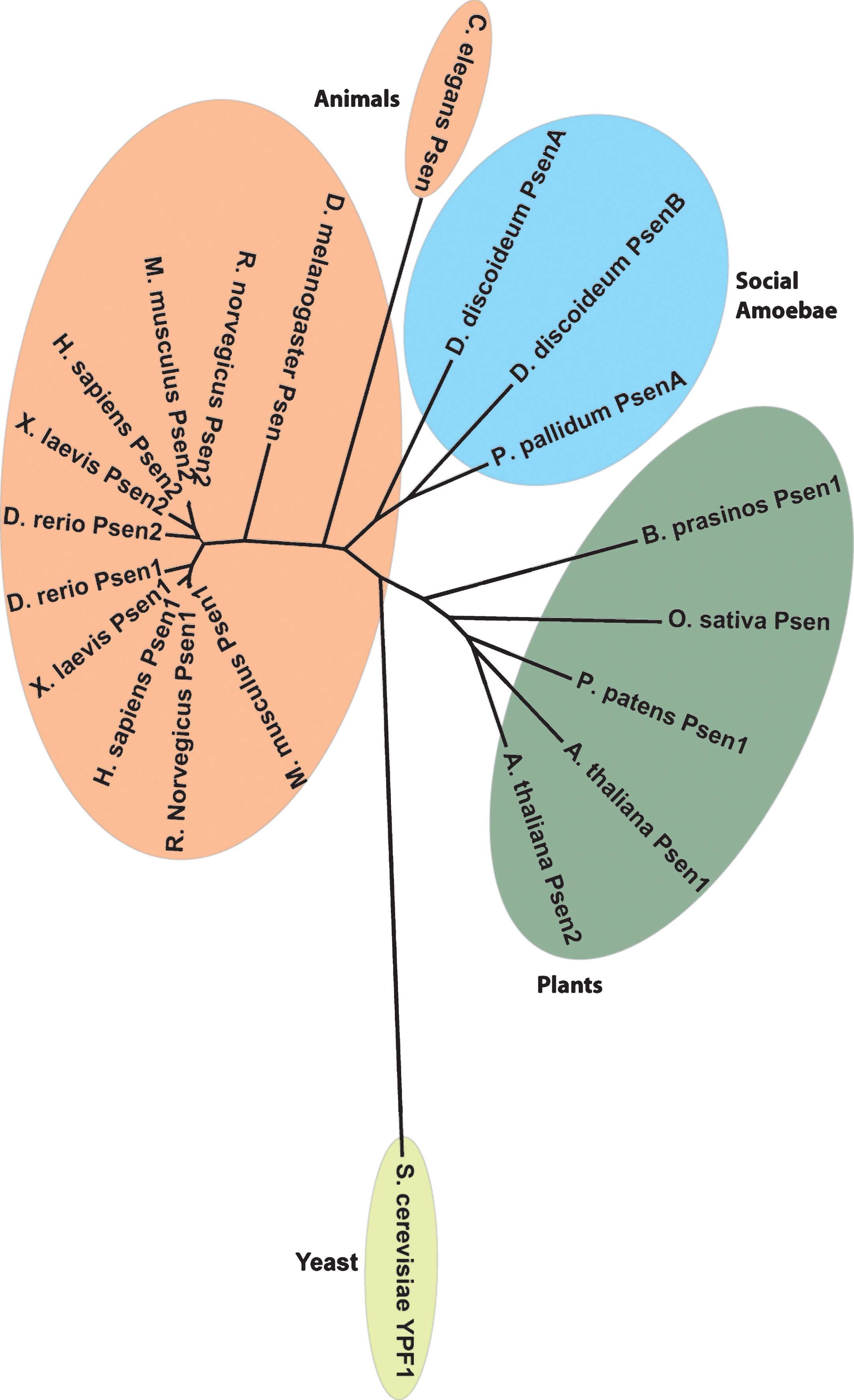
Fig.3
Presenilin activity controls Dictyostelium multicellular development through a non-catalytic mechanism for both the Dictyostelium PsenB and Human Psen1 proteins. A) Schematic diagram of the lifecycle of Dictyostelium discoideum. Free-living amoebae feed on bacteria, and when starved initiate multicellular development by aggregating towards cAMP. The cells form a mound (tight aggregate) that can differentiate into a motile slug that migrates toward heat and light, eventually halting and forming a Mexican hat structure. This structure culminates to form the mature fruiting body composed of a ball of spores held aloft by a stalk composed of dead cells. B) Dictyostelium cells were starved on nitrocellulose membranes for 24 h. Wild-type cells form mature fruiting bodies, whereas loss of both presenilin genes (psenA−/psenB−) arrests development at the mound stage. Normal development is restored by expressing wild-type Dictyostelium PsenB or human Psen1, suggesting a conserved function for both proteins. This conserved developmental role is through a non-catalytic mechanism, since expression of either Dictyostelium PsenB or human Psen1 mutated at the two catalytic aspartate residues (PsenB D348A/D394A or Psen1 D257A/D385A) also restores multicellular development. Bar = 0.5 mm.
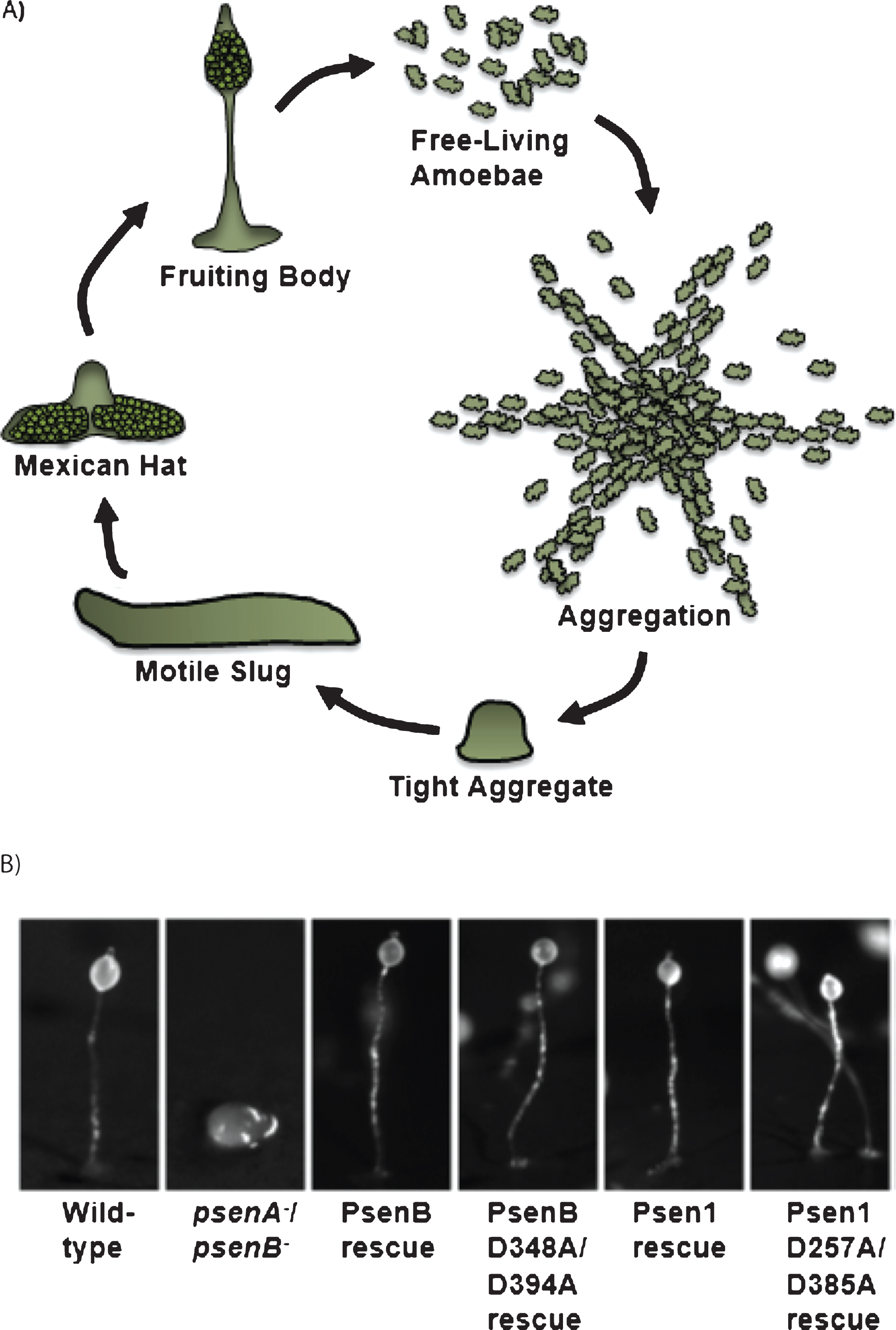
Fig.4
Comparison of the human and Dictyostelium presenilin proteins. A) Comparison of the predicted structure of the human presenilin 1 and the Dictyostelium presenilin B proteins viewed from the side and from above (from Phyre 2) [60]. B) Subcellular localization of presenilin proteins. Psen1 and PsenB tagged with GFP both localize to the endoplasmic reticulum (compare to the endoplasmic reticulum protein Calnexin tagged with RFP) when expressed in psenA−/psenB− cells. C) Alignments of the PALP region of human and Dictyostelium presenilin proteins (upper panel), as well as regions surrounding the two catalytic aspartic acids (lower panel). Identical amino acids are boxed (light blue) and critical amino acid residues are highlighted (dark blue).
![Comparison of the human and Dictyostelium presenilin proteins. A) Comparison of the predicted structure of the human presenilin 1 and the Dictyostelium presenilin B proteins viewed from the side and from above (from Phyre 2) [60]. B) Subcellular localization of presenilin proteins. Psen1 and PsenB tagged with GFP both localize to the endoplasmic reticulum (compare to the endoplasmic reticulum protein Calnexin tagged with RFP) when expressed in psenA−/psenB− cells. C) Alignments of the PALP region of human and Dictyostelium presenilin proteins (upper panel), as well as regions surrounding the two catalytic aspartic acids (lower panel). Identical amino acids are boxed (light blue) and critical amino acid residues are highlighted (dark blue).](https://content.iospress.com:443/media/jad/2016/52-4/jad-52-4-jad150940/jad-52-jad150940-g004.jpg)


