Dihydropyridine Derivatives Modulate Heat Shock Responses and have a Neuroprotective Effect in a Transgenic Mouse Model of Alzheimer’s Disease
Abstract
Heat shock proteins (Hsps) have chaperone activity and play a pivotal role in the homeostasis of proteins by preventing misfolding, by clearing aggregated and damaged proteins from cells, and by maintaining proteins in an active state. Alzheimer’s disease (AD) is thought to be caused by amyloid-β peptide that triggers tau hyperphosphorylation, which is neurotoxic. Although proteostasis capacity declines with age and facilitates the manifestation of neurodegenerative diseases such as AD, the upregulation of chaperones improves prognosis. Our research goal is to identify potent Hsp co-inducers that enhance protein homeostasis for the treatment of AD, especially 1,4-dihydropyridine derivatives optimized for their ability to modulate cellular stress responses. Based on favorable toxicological data and Hsp co-inducing activity, LA1011 was selected for the in vivo analysis of its neuroprotective effect in the APPxPS1 mouse model of AD. Here, we report that 6 months of LA1011 administration effectively improved the spatial learning and memory functions in wild type mice and eliminated neurodegeneration in double mutant mice. Furthermore, Hsp co-inducer therapy preserves the number of neurons, increases dendritic spine density, and reduces tau pathology and amyloid plaque formation in transgenic AD mice. In conclusion, the Hsp co-inducer LA1011 is neuroprotective and therefore is a potential pharmaceutical candidate for the therapy of neurodegenerative diseases, particularly AD.
INTRODUCTION
Protein quality control requires constant surveillance to prevent proteins misfolding, aggregating and their subsequent loss of function. Maintenance of proteome integrity and protein homeostasis (proteostasis) critically depends on a complex network of heat shock proteins (Hsps) that act as molecular chaperones [1]. Small Hsps, together with other members of the chaperone network such as the Hsp70 protein family, help maintain normal protein homeostasis that is essential for cellular function [2]. However, Hsps decline with age [3, 4] and this correlates with a loss in the capacity of cells to maintain protein homeostasis. It is therefore not surprising that the prevalence of neurodegenerative diseases (NDDs) such as Alzheimer’s disease (AD) and Parkinson’s disease increases with age [2, 5]. Hsps play a role in extending longevity in animal models, but it is unclear whether this is true in humans [6].
Despite great diversity in the primary amino-acid sequence of proteins, the oligomers of different misfolded amyloidogenic proteins (hyperphosphorylated tau, amyloid-β, α-synuclein) elicit toxicity by common mechanisms [7]. This toxicity involves membrane perturbations, calcium dysregulation, the accumulation of reactive oxygen species, endoplasmic reticulum (ER)-stress, impairment of the ubiquitin-proteasome system, and the activation of apoptotic signaling pathways [8–11]. Indeed, it is hypothesized that the initiation of NDDs is a result of ER-stress, damaged mitochondria, and loss of Ca2+- and protein homeostasis [12–16]. Additionally, ER-stress caused by ER Ca2+-depletion, hypoxia, and/or hypoglycemia, initiates the unfolded protein response (UPR) [17] and prolongs UPR activation thus triggering apoptotic pathways that result in cell death [18]. Furthermore, mitochondrial energy production failure and the mitochondria-associated ER membrane may play key roles in the generation of amyloid proteins and the initiation of NDDs [19, 20].
Maintenance of protein homeostasis by the modulation of protein processing and folding systems by chaperone induction are key therapeutic targets of drug discovery for AD treatment [21]. The induction of Hsps also offers a potential strategy for the treatment of other neurodegenerative disorders [22, 23]. Small Hsps (such as Hsp27) bind to improperly folded, partially denatured proteins, preventing their aggregation and promoting cell survival. Recovery is further enhanced when sHsp-bound proteins are transferred to ATP-dependent chaperones like Hsp70, which facilitates refolding [24] or transfer to protein degradation machinery. It was reported that Hsp27, Hsp70, proteasomes, and autophagosomes form a complex network that maintains protein homeostasis [25, 26]. Hsp27 and Hsp70 are usually co-expressed in response to stress stimuli [27]. Sequestration of misfolded proteins into chaperone-enriched aggregates is a protective strategy that slows the decline of proteostasis during aging of the nematode Caenorhabditis elegans [28]. Because Hsp27 and 70 have a central role in protein homeostasis, their induction may prevent and disassemble toxic protein aggregates, which might represent a novel target for the treatment of NDDs [2, 29–37].
Regulation of intracellular Ca2+ levels contributes to neuronal defense against protein misfolding. Thus, Ca2+ channel blockers (CCBs) may be putative drugs for the prevention and treatment of NDDs such as AD [38–40]. The family of 1,4-dihydropyridines (DHPs) represents one of the most important groups of CCBs. DHPs were originally identified as calcium antagonists (nifedipine) and are widely used to treat hypertension and heart failure [41]. Substitution of the CCB molecular nucleus with variety of groups is the most commonly tested scaffold among L-type CCBs. These CCB variations result in a plethora of diverse activities at receptors, channels, transporters, and metabolizing enzymes [42]. In particular, DHPs interact with ion channels and G-protein coupled receptors [43]. CCBs have also been used in dementia therapy and nilvadipine is well tolerated by AD patients and is a promising candidate for AD treatment [38, 44, 45].
Clinical data suggest that the underlying therapeutic mechanism in AD is independent of the anti-hypertensive action of the CCB. Longitudinal studies of elderly people with AD support the use of CCBs as potential disease progression therapy. An ongoing advanced clinical trial (CT number 02017340) for the treatment of AD with nilvadipine is due to close in 2017 [39]. Clinical evidence from epidemiological studies, including a cohort of 3,000 people over 74 years of age, supports the efficacy of DHP CCBs in reducing or delaying the development of AD [44]. The recent Systolic Hypertension in Europe randomized control trial reported a 55% reduction in the incidence of dementia in people taking nitrendipine over a 5-year follow-up period [46].
The neuroprotective mechanism of CCBs is poorly understood. In the presence of CCBs, amyloid-β (Aβ) production, aggregation, and neurotoxicity is decreased, improving neuronal function both in vitro and in vivo [38, 40]. BAY w 9798 is a DHP structurally related to the Ca-antagonist nifedipine with anti-inflammatory and anti-oxidative properties but with no calcium antagonistic effects [47]. Because lipophilic compounds DHPs bind to the membrane lipid bilayer, they potentially act indirectly as allo-network drugs via the propagation of changes in cellular networks. In fact, lipid bilayer partitioning of DHPs occurs prior to the drug binding to receptors [48, 49]. Changes in the lipid phase of membranes alter cell signaling [50, 51], which is also associated with a variety of diseases including NDDs. Modulation of the cell membrane composition and structure as a molecular base for drug discovery and new disease treatment was recently reviewed and the term “membrane lipid therapy” was introduced [52].
Hsp co-inducer hydroxamic acid derivatives can normalize the dysregulated expression of Hsps in pathological conditions and interact with membranes, resulting in specific alterations in the membrane nanostructure [53–55]. We searched for novel substances that do not directly exert stress on cells, but have long-term effects on cellular stress responses and viability (co-inducing activity). Such Hsp co-inducer compounds potentiate the response to a pre-existing stress without exhibiting effects in non-stressed environments. These compounds provide a higher degree of selectivity affecting only the diseased/damaged tissue compared with non-specific stressors. They also augment normal physiologic stress response systems rather than just blocking or activating a single enzyme that likely leads to a compensatory feedback loop, counteracting its long-term benefits.
The aim of this study was to investigate the beneficial effect of Hsp co-inducers in a mouse model of AD. A family of novel co-inducer compounds was synthesized [56] from which three were selected for further investigations (LA1011, LA1030, and LA1044). The co-inducing effect of Hsp27, Hsp40, and Hsp70 was verified. The neuroprotective effect of the optimized compound LA1011 was demonstrated in the APPxPS1 AD transgenic mouse model using a spatial navigation test. Histology studies of mouse brain slices were performed to investigate neuronal viability, changes in dendritic spine density and the presence of Aβ and tau-pathology-characteristic abnormalities in AD [57].
MATERIALS AND METHODS
Preparation and optimization of 1,4-dihydropyridines
BAY w 9798 [58] was selected as a starting compound in our study [56]. During optimization using standard dihydropyridine synthetic protocols, the substituents at positions 1, 2, 4, and 6 were modified systematically, resulting in LA1030, a more lipophilic compound with improved activity. To augment the oral bioavailability of the compounds, one or two cationic centers were introduced to substituents at positions 2 and 6. LA1011 and LA1044 were the most active compounds. The compounds were synthesized using conventional techniques from commercially available starting materials [59–63] (Fig. 1). Compounds were characterized by melting point and 1H-NMR data (recorded on a DRX 400 MHz Bruker spectrometer).
Synthesis of LA1011
Dihydropyridine (1 mmol), 5 mmol dimethylamine-hydrochloride, 5 mmol paraformaldehyde, and 1 ml acetic acid were mixed and heated at 95°C for 5 h. The mixture was evaporated, dissolved in water and extracted with ether. After separation, the aqueous phase was neutralized with NaHCO3 and extracted with ethyl acetate. The organic phase was dried and evaporated, and the residue was purified by column chromatography on silica with a mixture of hexane/ethyl acetate. The pure fractions were collected, transferred to hydrochloride or to other salts, and recrystallized from methanol diethyl ether. The melting point (Mp) was 273–276°C. 1HNMR data: D2O, 400 MHz, δ: 7.51 (d, J = 8.3 Hz, 2 H, ArH), 7.28 (d, J = 8.6 Hz, 2 H, ArH), 5.07 (s, 1 H, CH), 3.67 (s, 6 H, COOCH3), 3.14–3.59 (m, 11 H, -CH2-, -CH3), 2.87 (s, 12 H, -CH3).
Synthesis of LA1030
A mixture of 0.05 mol p-trifluoromethyl benzaldehyde, 0.1 mol methyl 3-oxovaleroate, and 0.05 mol methylamine hydrochloride was refluxed in 25 ml pyridine for 5 h. The residue was dissolved in dichloromethane and washed with water after evaporation of the solvent. The organic phase was dried and evaporated. The residue was crystallized from methanol. The Mp was 133–138°C; 1HNMR data: CDCl3, 400 MHz, δ: 7.39 (d, J = 8.6 Hz, 2 H, ArH), 7.21 (d, J = 8.6 Hz, 2 H, ArH), 5.08 (s, 1 H, CH), 3.67 (s, 6 H, COOCH3), 3.14 (s, 3 H, N-CH3), 2.99–3.11 (m, 2 H, -CH2-), 2.71–2.84 (m, 2 H, -CH2-), 1.06–1.12 (m, 6 H, CH3).
Synthesis of LA1044
Compound LA1044 was transformed to the 2-bromomethyl derivative in pyridine with pyridiniumbromoperbromide [62]. One mmol of this product was dissolved in 10 ml acetonitrile and 2 eq. of K2CO3 and 1.1 eq. of 1,2,3,4-tetrahydroisoquinoline was then added. The mixture was stirred until the reaction was complete, then filtered and evaporated, and the residue was purified by chromatography. The Mp was 175–177°C; 1HNMR data: CDCl3, 400 MHz, δ: 7.47 (d, J = 8.2 Hz, 2 H, ArH), 7.29 (d, J = 8.2 Hz, 2 H, ArH), 7.06–7.16 (m, 3 H, ArH), 6.91–6.97 (m, 1 H, ArH), 5.22 (s, 1 H, CH), 4.20 (d, J = 14.2 Hz, 1 H, C-CH2-N), 3.80 (d, J = 14.2 Hz, 1 H, C-CH2-N), 3.73 (s, 3 H, COOCH3), 3.72 (s, 3 H, COOCH3), 3.58–3.69 (m, 2 H, CH2), 3.35 (s, 3 H, N-CH3), 2.67–2.85 (m, 4 H, CH2), 2.48 (s, 3 H, CH3).
Measurement of Hsp co-inducing activity
The effect of drug candidates on the stress response was tested with the SH-SY5Y (ATCC® CRL-2266trademark) human neuroblastoma cell line stably transfected with a plasmid containing the Hsp70 promoter fused to yellow fluorescent protein (YFP) (pEYFP-N1, Clontech), a kind gift of Lea Sistonen (Åbo Akademi University, Turku, Finland) (SH-SY5Y-pHsp70-YFP). Cells were maintained in a logarithmic-phase of growth on 100 mm dishes (Orange Scientific, Braine-l’Alleud, Belgium) in DMEM-F12 (1:1) medium (Life Technologies, NY, USA) supplemented with 10% heat-inactivated fetal bovine serum (Life Technologies). Cultures were maintained at 37°C in a humidified atmosphere incubator with 5% CO2. When cells reached 80% confluence, they were harvested in Trypsin/EDTA Solution (Lonza, Basel, Switzerland) and were plated at a density of 5×105 cells/6-cm culture dishes (Orange Scientific). After 24 h, cells were pre-incubated either with or without LA1011 (5μM) or LA1044 (40μM) for 30 min. Following pre-incubation, plates were divided into two groups: The control group was maintained at 37°C for 17 h and the other was heat-shocked at 42°C for 1 h and then placed back at 37°C for a 16 h recovery period (HS group). After recovery the expression of YFP, which is directly correlated to the activity of its Hsp70 promoter, was followed by measuring the fluorescence intensity of the cells with a BD Accuri C6 flow cytometer (Becton, Dickinson and Company, NJ, USA) [64].
Western blot analysis
SH-SY5Y cells were grown and treated with the drug candidates and heat exactly as described above. Following the recovery, cells were extracted in RIPA (radioimmunoprecipitation assay) buffer. Cell lysates were constantly agitated at 4°C for 2 h then centrifuged at 12,000×g for 1 min and supernatants were saved. After determining the protein concentration (BCA assay kit, Thermo Scientific, Rockford, IL, USA), 20μg of total protein was separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis under reducing conditions according to the method of Laemmli [65]. Proteins were blotted onto polyvinylidene difluoride membranes (Immobilon-P; Millipore, MA, USA). The blot was incubated overnight at 4°C in the presence of α-Hsp27 (ADI-SPA-803), α-Hsp70 (ADI-SPA-810), α-Hsp60 (ADI-SPA-806), α-Hsp90 (ADI-SPA-830) (Enzo Life Sciences, NY, USA), and α-Hsp40 (C-20:sc-1800, Santa Cruz Biotechnology, Santa Cruz, CA, USA), antibodies diluted 1:1000 in phosphate-buffered saline solution (PBS). After overnight incubation, the membrane was washed three times in PBS–0.05% Tween and incubated for 1 h in peroxidase-conjugated anti-mouse, anti-rabbit or anti-goat IgG antibody (Sigma-Aldrich, St. Louis, MO, USA) diluted 1:50,000 in PBS containing 3% non-fat dry milk powder. The immune complexes were detected using enhanced chemiluminescence (ECL) (Amersham, Little Chalfont, Buckinghamshire, UK) according to the supplier’s instructions and analyzed using AlphaEase FC software (Alpha Innotech, San Leandro, Canada). Immunoreactive bands were normalized to the density α-GAPDH (1:20,000) (G9545; Sigma-Aldrich) from the same sample.
Cytotoxicity assays
In vitro test: SH-SY5Y cells were homogenously distributed on 96-well plates in 5×103 cells/well density and incubated at 37°C. Then, 24 h later, cells were treated with LA1011, LA1030 or LA1044 at a 100 nM-200μM concentration range for 24 h. The viability of the cells was measured using alamarBlue® assay kit (Life Technologies) according to the supplier’s instructions. The in vivo acute toxicity of drugs selected for animal experiments were assayed in 2-month-old BALB/c mice (n = 10) by injecting the compound in PBS solution intraperitoneally (ip) at a 300 mg/kg dose.
Measurement of neuroprotection
Animals and treatments
B6C3-Tg (APPswe/PS1dE9)85Dbo/Mmjax mice (APPxPS1) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA) and maintained on the C57BL/6 genetic background (tg). The mice were kept in individual ventilated cages (Acéllabor Ltd., Hungary) on a 12/12 h dark/light cycle (lights on from 7 a.m. to 7 p.m.) with food and water available ad libitum and were treated ip with 3 mg/kg LA1011 once a day for 6 months. Four groups (8 animals/group) of 3 months old male mice were used: 1) wild type (C57BL/6) mice (wt) treated with saline; 2) wt treated with LA1011; 3) tg treated with saline; and 4) tg treated with LA1011.
Animal care followed the Communities Council Directive of 22 September 2010 (2010/63/EU on the protection of animals used for scientific purposes). Formal approval to conduct the experiments described was obtained from the Animal Experimentation Committee of the University of Szeged.
Behavioral test
The Morris water maze (MWM) is one of the most commonly used experimental rodent models to measure spatial learning and memory [66–73]. Animals were trained in an open-field water maze (diameter: 130 cm) filled with water (23±1°C) made opaque with milk. The pool was divided into four virtual quadrants and the invisible platform (diameter: 10 cm) was submerged in the middle of one of the four quadrants. Around the pool there was a black curtain. The animals swam twice a day for 5 consecutive days, and were launched from four different starting points. They were placed into the water facing the wall of the pool and were given 90 s to find the platform and 15 s to stay on it. Animals that did not find the platform were gently guided and placed on it. The escape latency data were calculated automatically by a video tracking system (EthoVision 2002, Noldus Information Technology, Netherlands).
Histology studies
After the MWM test, the mice were deeply anesthetized and transcardially perfused with 10 ml of 4°C PBS, followed by 30 ml of 4°C paraformaldehyde solution (4% in phosphate buffer, pH 7.4). Mouse brains were removed and post-fixed for 24 h in the same fixative (4°C), and subsequently placed in a 30% sucrose solution for 72 h at 4°C. Hippocampal coronal sections were cut to 30μm on a cryostat, and the slices were collected and stored at 4°C in PBS for free floating histochemistry.
Cresyl violet (Nissl) staining
Cresyl violet readily binds to the acidic components of RNA-rich ribosomes, as well as the nuclei and nucleoli of nerve cells and is used to determine neuron density. Slices (n = 4, 2 slice/animal) were mounted on slides and stained in filtered 1% cresyl violet solution for 5 min and dehydrated subsequently in 50% 70%, 95%, and twice in 100% ethanol for 1 min each. Slides were finally placed in xylene for another 10 min and coverslipped. Digital images were captured by a digital slide scanner (Mirax Midi, Carl Zeiss, Hungary), and analysis was performed using the Histoquant program (3DHistech, Hungary).
Tau immunohistochemistry
After quenching of endogenous peroxidase activity and non-specific blocking, the sections (n = 4, 4 slices/animals) were incubated overnight at 4°C with paired helical filament (PHF)-tau monoclonal antibody (clone AT100, Pierce Biotechnology, USA) at a 1:800 dilution in PBS (pH 7.4) in the presence of 20% goat serum and 0.2% Triton X-100. On the following day, the sections were washed in PBS and incubated for 1 h at room temperature with biotinylated goat anti-mouse antibody (1:400) followed by 1 h incubation with avidin-biotin complex (Vectastain Elite ABC Kit, Vector Laboratories, Burlingame, CA, USA; 1:400). After washing, sections were incubated in peroxidase substrate solution (nickel-enhanced 3,3′-diaminobenzidine) until the desired stain intensity developed. Following rinsing in tap water, sections were mounted on gelatin-coated slides, air-dried, dehydrated, and coverslipped with DPX mountant for histology (Fluka BioChemika, Buchs, Switzerland). Digital images were captured by a digital slide scanner (Mirax Midi), and analysis was performed using the Histoquant program (3DHistech).
Quantification of dendritic spine density using Golgi impregnation
The FD Rapid GolgiStaintrademark Kit (FD NeuroTechnologies, Consulting & Services, Inc., Columbia, MD, USA) was used (n = 3, 1 slice per animal and 2 neurons per slice) to measure dendritic spine density in the hippocampal CA1 area. Experimental animals were deeply anesthetized before brains were removed as quickly as possible and then carefully handled to avoid damage or pressing of the tissue. The tissue was immersed in an impregnation solution (A + B solution) and stored at room temperature for 2 weeks in the dark. The brains were transferred into another solution (C) and stored at 4°C in the dark for at least 48 h. Then, 100-μm thick coronal sections were cut with a microtome (Zeiss Microm HM 650 V). Sections were mounted on gelatin coated glass slides. After the staining and dehydration procedure, the slides were covered with DPX (VWR International, Radnor, PA, USA).
Golgi sections were studied by inverse light microscopy using oil-immersion objectives. The spine density of the proximal apical dendrite area was analyzed (100–200μm from soma). One segment (100μm in length) from a second-third-order dendrite protruding from its parent apical dendrite was chosen in each examined neuron for spine density quantification, as described by Nagy et al. [74]. The dendrites were selected under a 100×oil immersion lens and the images (600×) of these apical dendrites were captured through a CCD camera (1,600×1,200 pixel) connected to a light microscope (Olympus Vanox-T AH-2) and a computer. Serial images were captured from each dendrite in the analyzed segment. The captured multiple photomicrographs from one dendrite were then stacked into one file. To stack the images Image- Pro-Plus image analysis software (IPP; Media Cybernetics, Silver Spring, MD, USA) was used. Measurement of the spine density was performed by two independent experimenters to blind the analysis.
Statistical analysis
For statistical evaluation of western blots (Fig. 3), one-way ANOVA with post hoc Games-Howell test was used, which was performed by R 3.1.1 for Windows software. Data represent the mean±standard deviation (SD) (n = 6) (Figs. 2 and 3). A p < 0.05 value was considered statistically significant. Statistical analysis of the Morris water maze experiment (Fig. 4) was performed using SPSS software and Python’s Lifelines library (Davidson-Pilon, C., Lifelines, 2016), Github repository. The latency time to attain the platform (with a 90-s limit) was measured to determine which of the four groups had the highest learning rate. The longer the latency time to the platform is, the lower the learning capacity of the mice. “Survival curves” using the Cox Proportional Hazard model [75] were fitted, and the days and the treatments were selected as covariates. The comparison of treatment groups was performed using log-rank tests.
One-way ANOVA followed by Fisher’s LSD post hoc test was used for histological analysis and for dendritic spine density measurements (Fig. 5) using SPSS statistical software. Differences with a p-value of less than 0.05 were considered significant unless indicated otherwise.
RESULTS
Hsp co-inducing activity of LA1011, LA1030, and LA1044
We used SH-SY5Y human neuroblastoma cells stably expressing YFP under the control of an Hsp70 promoter to select the non-toxic concentrations of active Hsp co-inducing compounds (data not shown). In addition to the original lead compound (BAY w 9798), we investigated the effects of three novel drug candidates (LA1011, LA1030, and LA1044) on Hsp70 promoter activity (Fig. 2).
The drug candidates were added to SH-SY5Y-pHsp70-YFP cells at the four indicated concentrations 30 min prior to heat treatment (42°C, 1 h), which was followed by a 16 h recovery period at 37°C. After recovery, the expression of YFP was determined by flow cytometry. At 37°C none of the compounds had a significant effect on Hsp70 promoter activity (data not shown); however, at an elevated temperature (42°C) pronounced expression of YFP was observed (2-3 fold increase in intensity), which was further increased by administering the LA compounds, thus indicating the co-induction property of these drug candidates (Fig. 2). From these highly active “hit” molecules we chose water soluble LA1011 and water insoluble LA1044 for further in vitro and in vivo investigations.
The Hsp co-inducing effect of LA1011 and LA1044 on different Hsp-classes in SH-SY5Y cells was studied by western blot. Representative immunoblots (Fig. 3) demonstrate that heat shock induced Hsp27, Hsp40, and Hsp70 proteins; however, the level of Hsp60 and Hsp90 remained unchanged (Supplementary Figure 1). The administration of LA1011 (5μM) and LA1044 (40μM) further increased the level of Hsp27, Hsp40, and Hsp70. Importantly, in the absence of heat shock (cellular stress), these compounds did not alter the level of any Hsps measured. Results summarized in Fig. 3 demonstrate the unique Hsp co-inducer properties of LA1011 and LA1044, as they act only during stress. Inhibition of the ATPase activity of Hsp90 by naturally identified antibiotics and synthetic molecules is well-documented [76, 77] and is a known mechanism for the upregulation of the heat shock response. We therefore tested the ability of LA1011 to modulate the ATPase activity of yeast Hsp90. We found that LA1011 did not modify the ATPase activity of yeast Hsp90 at concentrations of 5 and 10μM (Supplementary Figure 2).
In vivo neuroprotective effect of LA1011
After the acute toxicity test (at 300 mg/kg) was negative, the neuroprotective effect of the Hsp co-inducer LA1011 was studied in tg mice using behavioral and histological methods. The applied chronic dosage of 3 mg/kg for the in vivo experiments was selected based on the in vitro results.
Spatial navigation in MWM
After 6 months daily ip treatment of mice with either LA1011 (3 mg/kg) or saline, the MWM test was applied. The animals were placed into the water facing the sidewalls of the pool at different starting positions across trials, and the swim time to find the platform (escape latency) was followed with a tracking system. The animals learned to swim to the correct location with decreasing escape latencies across days (Supplementary Figure 3). Pairwise comparison of daily performances was made using the log-rank test and included all mice. Compared to day one of testing, mice were more likely to find the platform on day two (p = 1.65e-5), day three (p = 5.24e-6), day four (p = 1.88e-11), and day five (p = 4.55e-12). The probability to reach the platform was determined using the Cox Proportional Hazard model (Fig. 4). Significant differences were found between wt+saline versus wt+LA1011 (p = 0.047), wt+saline versus tg+saline (p = 0.036), and tg+saline versus tg+LA1011 (p = 0.001) treatment groups. Importantly, mice in the tg+saline group required significantly more time to find the platform compared to the wt+saline group; however, the 6 months daily ip LA1011 treatment tg mice showed a significantly improved learning ability (tg+saline versus tg+LA1011 p = 0.001). Interestingly, we found LA1011 improved the memory potency (procognitive) of mice as wt+LA1011 treated mice required less time to complete the task compared with controls (wt+saline) (p = 0.047).
Histology
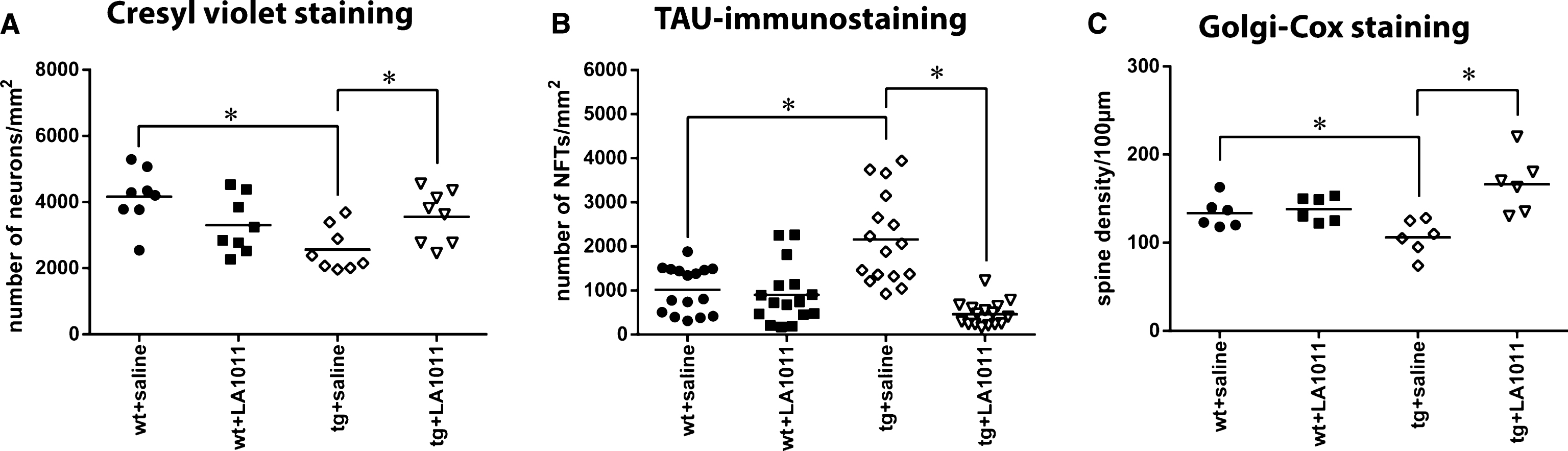
The animals were treated as described in “Animals and treatments”. Brain sections (prepared as described in “Materials and Methods”) were stained with cresyl violet, which stain Nissl substance in the cytoplasm of neurons in paraformaldehyde or formalin-fixed tissue. Digital images were analyzed by HistoQuant program. The difference in the number of neurons was significant between the groups (Fig. 5A–D). Significantly fewer neurons were observed in the tg+saline group compared with the wt group (tg+saline versus wt+saline group, p = 0.002). Cresyl-violet staining confirmed that LA1011 was neuroprotective in the transgenic AD mouse model. Treating mice with LA1011 for 6 months significantly prevented the loss of neurons in this group (tg+saline versus tg+LA1011, p = 0.041).
Tau-pathology studies used immunohistochemistry to detect tau in PHF, a major component of the neurofibrillary tangles (NFTs) involved in the pathology of AD that are not present in normal biopsies [78]. Digital images were analyzed by HistoQuant program. There were significant differences in the number of abnormally accumulated NFTs in the hippocampus (HC) between the groups (p = 0.001) (Fig. 5B; Supplementary Figure 4E–H). There were more NFTs in the HC slices of the tg+saline group compared with the wt+saline group (p = 0.001). Most importantly, the administration of Hsp co-inducer LA1011 decreased the number of NFTs in tg mice HC to the level found in the HC of wt mice (p = 0.001) (Fig. 5B).
Long-term memory is mediated in part by the growth of new dendritic spines (or the enlargement of pre-existing spines) to reinforce a particular neural pathway. According to the literature, APPxPS1 mice undergo spine loss already by the age of 4 months [79]. One of the best neurohistological methods to observe detailed morphology of neurons is Golgi staining. The Golgi-Cox protocol can be used to study the experimental effects of different pharmacological manipulations on the spatial distribution of neurons, dendrite density, spine number, and morphology. After the Golgi-Cox staining of the HC CA1 area, digital images were used to count dendritic spines. The tg+saline group had fewer dendritic spines compared with the wt+saline group (p = 0.045). The 6-month LA1011 treatment of transgenic animals resulted in a considerably greater number of spines in the HC slices (tg+saline versus tg+LA1011; p = 0.001). The treatment did not affect the spine number of wt animals. This could be explained by the molecule’s “HSP co-modulating activity” namely it acts like “smart molecules” by selective interactions with only those cells, which are under acute or chronic stress.
Aβ peptide initiates inflammation in the HC of the human brain in AD. Aβ is chemically “sticky” and gradually builds up into plaques [57], which can be identified by immunohistochemistry using anti-Aβ antibody. We observed amyloid plaque-like structures in tg mice (Supplementary Figure 4I–L). The representative examples of sections show a nos amount of plaque formation in the HC of transgenic saline treated animals compared to that of wt samples. Administering LA1011 to tg mice decreased plaque formation (Supplementary Figure 4K, L). There were no signs of amyloid plaques in sections from wt animals (Supplementary Figure 4I, J).
DISCUSSION
At present, there are few drugs that improve the memory deficits associated with normal aging and none that prevent cognitive decline in chronic neurodegenerative conditions such as AD. Except for rare cases of familial AD, the cause of AD is not known, but disease is highly correlated with aging, a process involving a wide variety of physiological changes. Therefore, it is likely that cells in the aging brain are compromised not from a single cause but from the convergence of multiple insults. Unfortunately, drug candidates for AD have all failed in clinical trials, perhaps because one target is not sufficient or because the targets are also critical for normal brain function. Molecular chaperones have central roles in each of the arms of cellular proteostasis (synthesis, folding, disaggregation, and degradation of proteins); therefore, the approach described in this study, the administration of Hsp co-inducer 1,4 DHPs, might have therapeutic benefits for NDDs including AD.
The most important finding of this study is that the novel Hsp co-inducer is broadly neuroprotective and has the ability to enhance memory in normal animals as well as to prevent memory deficits in AD transgenic mice. The neurotrophic and memory-enhancing activities of LA1011 might be associated with an increase in the levels of Hsp70, Hsp40, and Hsp25.
The Hsp co-inducer effect of the novel 1,4-DHP compounds LA1011 and LA1044 was demonstrated in vitro by a promoter probing assay (Fig. 2), and by western blot in a SH-SY5Y human neuroblastoma cell line (Fig. 3). The inhibition of the ATPase activity of Hsp90 is a known mechanism for the upregulation of the heat shock response. We therefore tested the ability of LA1011 to modulate the ATPase activity of yeast Hsp90 and found that LA1011 did not modify the ATPase activity of yeast Hsp90 at concentrations of 5 and 10μM (Supplementary Figure 2).
The observed Hsp modulating activity of the compounds was separable from the Ca2+ channel antagonist effect responsible for the antihypertensive activity of many known 1,4-DHPs. For the known Ca2+ channel antagonist nilvadipine, the EC50 for Hsp-modulation and for Ca2+ channel inhibition is equal [56, 80]. The EC50 for the novel compounds described in this study was above 10; thus, the separation of a potential antihypertensive effect allows the selected compounds to be used to treat pathophysiological conditions mediated by Hsps, including NDDs, without the risk of hypotension. The best co-inducer, LA1011 was selected for in vivo experiments in the tg mouse model of AD and proved to be neuroprotective. LA1011 prevented neuron loss in the HC of tg mice (Fig. 5A), decreased NFT accumulation (Fig. 5B), and saved dendritic spine numbers (Fig. 5C). In the MWM task, both wt groups showed a progressive decrease in latency to reach the platform, a measure of spatial learning. Tg animals, however, showed significantly higher latencies compared to all other experimental groups suggestive of delayed learning. In line with the histological results, 6 months treatment with LA1011 significantly restored the spatial learning capability of tg mice (p = 0.001) and further improved the memory of wt animals (Fig. 4). These results can be explained in the context of our knowledge of the pathogenetic mechanisms of AD. Furthermore, maintenance of protein homeostasis by chaperone induction predicts the induction of a beneficial effect of LA1011 on brain function and morphology.
AD is a complex, multifactorial syndrome with a number of leading mechanisms, including astrocyte hyperactivity (reactive astrogliosis), neuroinflammation, mitochondrial dysfunction, and the accumulation of misfolded proteins (Aβ, NFT, α-synuclein) in the brain [57]. A net loss of dendritic spines is observed in AD and cases of intellectual disability. Because dendritic spines are plastic structures whose lifespan is influenced by input activity [81], spine dynamics may play an important role in the maintenance of memory over a lifetime.
The original Aβ hypothesis of Hardy states that an imbalance exists between Aβ production and clearance resulting in Aβ accumulation [82, 83]. During the last 20 years, intensive research has targeted the biosynthesis of Aβ (β-and γ-secretases), as well as the clearance of Aβ and amyloid plaques from the brain [84]. However, these trials were unsuccessful and no efficient drug treatment of AD exists currently, although a number of novel AD drug trials are in progress.
Recent data on the molecular basis of AD pathophysiology suggest a number of leading mechanisms [85]. Mitochondrial DNA mutations leading to oxidative stress-induced mitochondria dysfunction triggers a chain of cellular events leading to neurodegeneration and a “vicious circle” that ends in dementia [86, 87]. Molecular chaperones and the ubiquitin-proteasome system represent the most important defensive mechanisms against misfolded proteins in AD [88]. Each protein aggregation disease can be characterized by a different set of Hsps that can rescue specific types of aggregation [89]. Some of these Hsps have demonstrated effectiveness in vivo, in mouse models of protein-aggregation diseases. Hsp70 and Hsp90 promote tau solubility and tau binding to microtubules, reduce insoluble tau, and reduce tau hyperphosphorylation in a transgenic mouse model of AD [90]. In addition, therapeutic inducers of the Hsp70/Hsp110 system protected Hsp-deficient mice against traumatic brain injury [30] and the upregulation of Hsp by geldanamycin reduced brain injury in a mouse model of intracerebral hemorrhage [33]. A molecular chaperone, the human BRICHOS domain, can specifically inhibit the Aβ-catalytic cycle (Aβ1 - 42 fibrils effectively catalyze the formation of neurotoxic Aβ-oligomers) and limit human Aβ1 - 42 toxicity [91]. In addition, some molecular chaperones forcibly untangle protein aggregates (disaggregases) [92]. Omi/HtrA2, a mammalian chaperone, selectively binds and detoxifies oligomeric Aβ by disaggregation [93]. Exogenous human Hsp70 had dramatic neuroprotective effects in olfactory bulbectomy mice and 5xFAD mouse models of neurodegeneration [29] based on its anti-apoptotic and anti-inflammatory effects. Consequently, small molecule activators of the heat shock response provide great therapeutic promise [94]. Ca2+-dependent activation of heat shock factor 1 suppressed protein misfolding and reprograming of protein homeostasis [95]. Arimoclomol, an Hsp co-inducer, may protect motor neurons by enhancing Hsp expression, thus directly affecting protein aggregation and clearance of misfolded assemblies via the proteasome-ubiquitin system [32]. Recent experiments have demonstrated the dual role of Hsp70 [96], which functions as a molecular chaperone for damaged proteins, as well as simultaneously stabilizing lysosomes, which can revert NDD-associated lysosomal pathology [96–98].
In summary, unregulated Hsp induction could cause the excessive overexpression of Hsps leading to instability of the stress response and unwanted side effects [99]. Because LA1011 and LA1044 increase Hsp expression only under stressed conditions, the therapeutic use of DHPs in this study is novel and perhaps an optimal strategy for the treatment of AD and other NDDs.
ACKNOWLEDGMENTS
We are grateful to László Siklós for the measurements of dendritic spine density and to Zsolt Datki and Mónika Mihók for animal treatment. This work was supported by the EC Health Program “Memoload” (FP-7 project N° 201.159), the Hungarian National Development Agency (TAMOP-4.1.1.C-13/1/KONV-2014-0001), the Hungarian Basic Research Fund (OTKA, NK100857, NN111006) and LipidArt Ltd. Chrisostomos Prodromou and Katie L.I.M Blundell are supported by the Wellcome Trust, 095605/Z11/Z.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/15-0860r2).
Appendices
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-150860.
REFERENCES
[1] | Kim YE , Hipp MS , Bracher A , Hayer-Hartl M , Hartl FU ((2013) ) Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem 82: , 323–355. |
[2] | Toth ME , Santha M , Penke B , Vígh L ((2015) ) How to stabilize both the proteins and the membranes: Diverse effects of sHsps in neuroprotection. In The Big Book on Small Heat Shock Proteins, Heat Shock Proteins, 8: , Tanguay RM, Hightower LE, eds. Springer, Cham, pp. 527–562. |
[3] | Calderwood SK , Murshid A , Prince T ((2009) ) The shock of aging: Molecular chaperones and the heat shock response in longevity and aging–a mini-review. Gerontology 55: , 550–558. |
[4] | Kayani AC , Morton JP , McArdle A ((2008) ) The exercise-induced stress response in skeletal muscle: Failure during aging. Appl Physiol Nutr Metab 33: , 1033–1041. |
[5] | Valastyan JS , Lindquist S ((2014) ) Mechanisms of protein-folding diseases at a glance. Dis Model Mech 7: , 9–14. |
[6] | Murshid A , Eguchi T , Calderwood SK ((2013) ) Stress proteins in aging and life span. Int J Hyperther 29: , 442–447. |
[7] | Bemporad F , Chiti F ((2012) ) Protein misfolded oligomers: Experimental approaches, mechanism of formation, and structure-toxicity relationships. Chem Biol 19: , 315–327. |
[8] | Resenberger UK , Harmeier A , Woerner AC , Goodman JL , Muller V , Krishnan R , Vabulas RM , Kretzschmar HA , Lindquist S , Hartl FU , Multhaup G , Winklhofer KF , Tatzelt J ((2011) ) The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. EMBO J 30: , 2057–2070. |
[9] | Bucciantini M , Giannoni E , Chiti F , Baroni F , Formigli L , Zurdo JS , Taddei N , Ramponi G , Dobson CM , Stefani M ((2002) ) Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 416: , 507–511. |
[10] | Demuro A , Mina E , Kayed R , Milton SC , Parker I , Glabe CG ((2005) ) Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem 280: , 17294–17300. |
[11] | Bence NF , Sampat RM , Kopito RR ((2001) ) Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292: , 1552–1555. |
[12] | Endres K , Reinhardt S ((2013) ) ER-stress in Alzheimer’s disease: Turning the scale?. Am J Neurodegener Dis 2: , 247–265. |
[13] | Hetz C , Mollereau B ((2014) ) Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat Rev Neurosci 15: , 233–249. |
[14] | Li JQ , Yu JT , Jiang T , Tan L ((2015) ) Endoplasmic reticulum dysfunction in Alzheimer’s disease. Mol Neurobiol 51: , 383–395. |
[15] | Li SH , Yang L , Selzer ME , Hu Y ((2013) ) Neuronal endoplasmic reticulum stress in axon injury and neurodegeneration. Ann Neurol 74: , 768–777. |
[16] | Tadic V , Prell T , Lautenschlaeger J , Grosskreutz J ((2014) ) The ER mitochondria calcium cycle and ER stress response as therapeutic targets in amyotrophic lateral sclerosis. Front Cell Neurosci 8: , 147. |
[17] | Hetz C , Chevet E , Harding HP ((2013) ) Targeting the unfolded protein response in disease. Nat Rev Drug Discov 12: , 703–719. |
[18] | Placido AI , Pereira CMF , Duarte AI , Candeias E , Correia SC , Santos RX , Carvalho C , Cardoso S , Oliveira CR , Moreira PI ((2014) ) The role of endoplasmic reticulum in amyloid precursor protein processing and trafficking: Implications for Alzheimer’s disease. Biochim Biophys Acta 1842: , 1444–1453. |
[19] | Schon EA , Area-Gomez E ((2013) ) Mitochondria-associated ER membranes in Alzheimer disease. Mol Cell Neurosci 55: , 26–36. |
[20] | Schreiner B , Hedskog L , Wiehager B , Ankarcrona M ((2015) ) Amyloid-beta peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J Alzheimers Dis 43: , 369–374. |
[21] | Alavez S , Vantipalli MC , Zucker DJS , Klang IM , Lithgow GJ ((2011) ) Amyloid-binding compounds maintain protein homeostasis during ageing and extend lifespan. Nature 472: , 226–229. |
[22] | Nagai Y , Fujikake N , Popiel HA , Wada K ((2010) ) Induction of molecular chaperones as a therapeutic strategy for the polyglutamine diseases. Curr Pharm Biotechnol 11: , 188–197. |
[23] | Klettner A ((2004) ) The induction of heat shock proteins as a potential strategy to treat neurodegenerative disorders. Drug News Perspect 17: , 299–306. |
[24] | Hartl FU , Bracher A , Hayer-Hartl M ((2011) ) Molecular chaperones in protein folding and proteostasis. Nature 475: , 324–332. |
[25] | Ehrnsperger M , Graber S , Gaestel M , Buchner J ((1997) ) Binding of non-native protein to Hsp25 during heat shock creates a reservoir of folding intermediates for reactivation. EMBO J 16: , 221–229. |
[26] | Mymrikov EV , Seit-Nebi AS , Gusev NB ((2011) ) Large potentials of small heat shock proteins. Physiol Rev 91: , 1123–1159. |
[27] | Finka A , Mattoo RUH , Goloubinoff P ((2011) ) Meta-analysis of heat- and chemically upregulated chaperone genes in plant and human cells. Cell Stress Chaperones 16: , 15–31. |
[28] | Walther DM , Kasturi P , Zheng M , Pinkert S , Vecchi G , Ciryam P , Morimoto RI , Dobson CM , Vendruscolo M , Mann M , Hartl FU ((2015) ) Widespread proteome remodeling and aggregation in aging C. elegans. Cell 161: , 919–932. |
[29] | Bobkova NV , Garbuz DG , Nesterova I , Medvinskaya N , Samokhin A , Alexandrova I , Yashin V , Karpov V , Kukharsky MS , Ninkina NN , Smirnov AA , Nudler E , Evgen’ev M ((2014) ) Therapeutic effect of exogenous Hsp70 in mouse models of Alzheimer’s disease. J Alzheimers Dis 38: , 425–435. |
[30] | Eroglu B , Kimbler DE , Pang JF , Choi J , Moskophidis D , Yanasak N , Dhandapani KM , Mivechi NF ((2014) ) Therapeutic inducers of the HSP70/HSP110 protect mice against traumatic brain injury. J Neurochem 130: , 626–641. |
[31] | Franklin TB , Krueger-Naug AM , Clarke DB , Arrigo AP , Currie RW ((2005) ) The role of heat shock proteins Hsp70 and Hsp27 in cellular protection of the central nervous system. Int J Hyperther 21: , 379–392. |
[32] | Kalmar B , Lu CH , Greensmith L ((2014) ) The role of heat shock proteins in amyotrophic lateral sclerosis: The therapeutic potential of Arimoclomol. Pharmacol Therapeut 141: , 40–54. |
[33] | Hoshino T , Murao N , Namba T , Takehara M , Adachi H , Katsuno M , Sobue G , Matsushima T , Suzuki T , Mizushima T ((2011) ) Suppression of Alzheimer’s disease-related phenotypes by expression of heat shock protein 70 in mice. J Neurosci 31: , 5225–5234. |
[34] | Wang H , Tan MS , Lu RC , Yu JT , Tan L ((2014) ) Heat shock proteins at the crossroads between cancer and Alzheimer’s xisease. Biomed Res Int 2014: , 239164. |
[35] | Morimoto RI ((2008) ) Proteotoxic stress and inducible chaperone networks in neurodegenerative disease and aging. Genes Dev 22: , 1427–1438. |
[36] | Soti C , Csermely P ((2002) ) Chaperones and aging: Role in neurodegeneration and in other civilizational diseases. Neurochem Int 41: , 383–389. |
[37] | Toth ME , Szegedi V , Varga E , Juhasz G , Horvath J , Borbely E , Csibrany B , Alfoldi R , Lenart N , Penke B , Santha M ((2013) ) Overexpression of Hsp27 ameliorates symptoms of Alzheimer’s disease in APP/PS1 mice. Cell Stress Chaperones 18: , 759–771. |
[38] | Iwasaki K , Egashira N , Takagaki Y , Yoshimitsu Y , Hatip-Al-Khatib I , Mishima K , Fujiwara M ((2007) ) Nilvadipine prevents the impairment of spatial memory induced by cerebral ischemia combined with beta-amyloid in rats. Biol Pharm Bull 30: , 698–701. |
[39] | Broadstock M , Ballard C , Corbett A ((2014) ) Latest treatment options for Alzheimer’s disease, Parkinson’s disease dementia and dementia with Lewy bodies. Expert Opin Pharmacother 15: , 1797–1810. |
[40] | Corbett A , Ballard C ((2013) ) Is a potential Alzheimer’s therapy already in use for other conditions? Can medications for hypertension, diabetes and acne help with the symptoms?. Expert Opin Investig Drugs 22: , 941–943. |
[41] | Cattaruzza M , Wachter R , Wagner AH , Hecker M ((2000) ) Modulation by dihydropyridine-type calcium channel antagonists of cytokine-inducible gene expression in vascular smooth muscle cells. Br J Pharmacol 129: , 1155–1162. |
[42] | Carosati E , Ioan P , Micucci M , Broccatelli F , Cruciani G , Zhorov BS , Chiarini A , Budriesi R ((2012) ) 1,4-dihydropyridine scaffold in medicinal chemistry, the story so far and perspectives (part 2): Action in other targets and antitargets. Curr Med Chem 19: , 4306–4323. |
[43] | Ioan P , Carosati E , Micucci M , Cruciani G , Broccatelli F , Zhorov BS , Chiarini A , Budriesi R ((2011) ) 1,4-dihydropyridine scaffold in medicinal chemistry, the story so far and perspectives (part 1): Action in ion channels and GPCRs. Curr Med Chem 18: , 4901–4922. |
[44] | Kennelly S , Abdullah L , Kenny RA , Mathura V , Luis CA , Mouzon B , Crawford F , Mullan M , Lawlor B ((2012) ) Apolipoprotein E genotype-specific short-term cognitive benefits of treatment with the antihypertensive nilvadipine in Alzheimer’s patients–an open-label trial. Int J Geriatr Psychiatry 27: , 415–422. |
[45] | Kennelly SP , Abdullah L , Paris D , Parish J , Mathura V , Mullan M , Crawford F , Lawlor BA , Kenny RA ((2011) ) Demonstration of safety in Alzheimer’s patients for intervention with an anti-hypertensive drug Nilvadipine: Results from a 6-week open label study. Int J Geriatr Psychiatry 26: , 1038–1045. |
[46] | Forette F , Seux ML , Staessen JA , Thijs L , Babarskiene MR , Babeanu S , Bossini A , Fagard R , Gil-Extremera B , Laks T , Kobalava Z , Sarti C , Tuomilehto J , Vanhanen H , Webster J , Yodfat Y , Birkenhäger WH , Systolic Hypertension in Europe Investigators ((2002) ) The prevention of dementia with antihypertensive treatment. Arch Intern Med 162: , 2046–2052. |
[47] | Matsui T , Yamagishi S , Nakamura K , Inoue H ((2007) ) Bay w 9798, a dihydropyridine structurally related to nifedipine with no calcium channel-blocking properties, inhibits tumour necrosis factor-alpha-induced vascular cell adhesion molecule-1 expression in endothelial cells by suppressing reactive oxygen species generation. J Int Med Res 35: , 886–891. |
[48] | Herbette LG , Vanterve YMH , Rhodes DG ((1989) ) Interaction of 1,4 dihydropyridine calcium-channel antagonists with biological-membranes - lipid bilayer partitioning could occur before drug-binding to receptors. J Mol Cell Cardiol 21: , 187–201. |
[49] | Mason RP , Chester DW ((1989) ) Diffusional dynamics of an active rhodamine-labeled 1,4-dihydropyridine in sarcolemmal lipid multibilayers. Biophys J 56: , 1193–1201. |
[50] | Balogh G , Peter M , Glatz A , Gombos I , Torok Z , Horvath I , Harwood JL , Vigh L ((2013) ) Key role of lipids in heat stress management. FEBS Lett 587: , 1970–1980. |
[51] | Torok Z , Crul T , Maresca B , Schutz GJ , Viana F , Dindia L , Piotto S , Brameshuber M , Balogh G , Peeter M , Porta A , Trapani A , Gombos I , Glatz A , Gungor B , Peksel B , Vigh L , Csoboz B , Horvath I , Vijayan MM , Hooper PL , Harwood JL , Vigh L ((2014) ) Plasma membranes as heat stress sensors: From lipid-controlled molecular switches to therapeutic applications. Biochim Biophys Acta 1838: , 1594–1618. |
[52] | Escriba PV , Busquets X , Inokuchi JI , Balogh G , Torok Z , Horvath I , Harwood JL , Vigh L ((2015) ) Membrane lipid therapy: Modulation of the cell membrane composition and structure as a molecular base for drug discovery and new disease treatment. Prog Lipid Res 59: , 38–53. |
[53] | Torok Z , Tsvetkova NM , Balogh G , Horvath I , Nagy E , Penzes Z , Hargitai J , Bensaude O , Csermely P , Crowe JH , Maresca B , Vigh L ((2003) ) Heat shock protein coinducers with no effect on protein denaturation specifically modulate the membrane lipid phase. Proc Natl Acad Sci U S A 100: , 3131–3136. |
[54] | Gombos I , Crul T , Piotto S , Gungor B , Torok Z , Balogh G , Peter M , Slotte JP , Campana F , Pilbat AM , Hunya A , Toth N , Literati-Nagy Z , Vigh L , Glatz A , Brameshuber M , Schutz GJ , Hevener A , Febbraio MA , Horvath I , Vigh L ((2011) ) Membrane-lipid therapy in operation: The HSP co-inducer BGP-15 activates stress signal transduction pathways by remodeling plasma membrane rafts. PLoS One 6: , e28818. |
[55] | Vigh L , Literati PN , Horvath I , Torok Z , Balogh G , Glatz A , Kovacs E , Boros I , Ferdinandy P , Farkas B , Jaszlits L , Jednakovits A , Koranyi L , Maresca B ((1997) ) Bimoclomol: A nontoxic, hydroxylamine derivative with stress protein-inducing activity and cytoprotective effects. Nat Med 3: , 1150–1154. |
[56] | Fulop F , Vigh L , Torok Z , Penke B , Horvath I , Balogh G , Bernath S , Hunya Á ((2013) ) 1,4 dihydropyridine derivatives with HSP modulating activity. PCT. Int Appl WO2013076516 A1. |
[57] | Duyckaerts C , Delatour B , Potier MC ((2009) ) Classification and basic pathology of Alzheimer disease. Acta Neuropathol 118: , 5–36. |
[58] | Behner O , Wollweber H , Rosen B , Zaiss S , Goldmann S ((1991) ) Use of N-alkylated 1,4-dihydropyridine carboxylic acid esters as drugs, new compounds and process for their preparation. European Patent EP 0451654 A2, B1. |
[59] | Aritomi J , Ueda S , Nishimura H ((1980) ) Mannich reaction of dihydropyridine derivatives. 1. Reactions with secondary-amines. Chem Pharm Bull 28: , 3163–3171. |
[60] | Dekamin MG , Ilkhanizadeh S , Latifidoost Z , Daemi H , Karimi Z , Barikani M ((2014) ) Alginic acid: A highly efficient renewable and heterogeneous biopolymeric catalyst for one-pot synthesis of the Hantzsch 1,4-dihydropyridines. RSC Adv 4: , 56658–56664. |
[61] | Desai B , Dallinger D , Kappe CO ((2006) ) Microwave-assisted solution phase synthesis of dihydropyrimidine C5 amides and esters. Tetrahedron 62: , 4651–4664. |
[62] | Ebiike H , Maruyama K , Ozawa Y , Yamazaki Y , Achiwa K ((1997) ) Asymmetric synthesis of (R)-nilvadipine and (S)-NB 818 via regioselective bromination of chiral 1,4-dihydropyridines as a key step and enzymatic resolution of racemic 2-hydroxymethyl-1,4-dihydropyridine derivatives. Chem Pharm Bull 45: , 869–876. |
[63] | Hantzsch A ((1881) ) Condensationsprodukte aus aldehydammoniak und ketonartigen verbindungen. Chem Ber 14: , 1637–1638. |
[64] | Wysocka A , Krawczyk Z ((2000) ) Green fluorescent protein as a marker for monitoring activity of stress-inducible hsp70 rat gene promoter. Mol Cell Biochem 215: , 153–156. |
[65] | Laemmli UK ((1970) ) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: , 680–685. |
[66] | Morris RGM , Garrud P , Rawlins JNP , Okeefe J ((1982) ) Place navigation impaired in rats with hippocampal-lesions. Nature 297: , 681–683. |
[67] | D’Hooge R , De Deyn PP ((2001) ) Applications of the Morris water maze in the study of learning and memory. Brain Res Rev 36: , 60–90. |
[68] | Mendez-Lopez M , Mendez M , Sampedro-Piquero P , Arias JL ((2013) ) Spatial learning-related changes in metabolic activity of limbic structures at different posttask delays. J Neurosci Res 91: , 151–159. |
[69] | Morris R ((1984) ) Developments of a water-maze procedure for studying spatial-learning in the rat. J Neurosci Methods 11: , 47–60. |
[70] | Morris RGM , Anderson E , Lynch GS , Baudry M ((1986) ) Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, Ap5. Nature 319: , 774–776. |
[71] | Moser MB , Moser EI , Forrest E , Andersen P , Morris RGM ((1995) ) Spatial-learning with a Minislab in the dorsal hippocampus. Proc Natl Acad Sci U S A 92: , 9697–9701. |
[72] | Sipos E , Kurunczi A , Kasza A , Horvath J , Felszeghy K , Laroche S , Toldi J , Parducz A , Penke B , Penke Z ((2007) ) beta-amyloid pathology in the entorhinal cortex of rats induces memory deficits: Implications for Alzheimer’s disease. Neuroscience 147: , 28–36. |
[73] | Vorhees CV , Williams MT ((2006) ) Morris water maze: Procedures for assessing spatial and related forms of learning and memory. Nat Protoc 1: , 848–858. |
[74] | Nagy D , Kocsis K , Fuzik J , Marosi M , Kis Z , Teichberg VI , Toldi J , Farkas T ((2011) ) Kainate postconditioning restores LTP in ischemic hippocampal CA1: Onset-dependent second pathophysiological stress. Neuropharmacology 61: , 1026–1032. |
[75] | Jahn-Eimermacher A , Lasarzik I , Raber J ((2011) ) Statistical analysis of latency outcomes in behavioral experiments. Behav Brain Res 221: , 271–275. |
[76] | Zhao H , Michaelis ML , Blagg BS ((2012) ) Hsp90 modulation for the treatment of Alzheimer’s disease. Adv Pharmacol 64: , 1–25. |
[77] | Roe SM , Prodromou C , O’Brien R , Ladbury JE , Piper PW , Pearl LH ((1999) ) Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J Med Chem 42: , 260–266. |
[78] | Zheng-Fischhofer Q , Biernat J , Mandelkow EM , Illenberger S , Godemann R , Mandelkow E ((1998) ) Phosphorylation of tau protein at Ser214 regulates the binding of tau to microtubules and is part of the epitope of the Alzheimer-tau specific antibody AT100. Mol Biol Cell 9: , 395a. |
[79] | Smith DL , Pozueta J , Gong B , Arancio O , Shelanski M ((2009) ) Reversal of long-term dendritic spine alterations in Alzheimer disease models. Proc Natl Acad Sci U S A 106: , 16877–16882. |
[80] | Hoffmann A , Kraul H , Burkardt I ((1997) ) Nilvadipine in hypertension - Experience in ambulatory treatment. Int J Clin Pharmacol Ther 35: , 195–203. |
[81] | De Roo M , Klauser P , Muller D ((2008) ) LTP promotes a selective long-term stabilization and clustering of dendritic spines. PLoS Biol 6: , 1850–1860. |
[82] | Hardy J , Allsop D ((1991) ) Amyloid deposition as the central event in the etiology of Alzheimer’s disease. Trends Pharmacol Sci 12: , 383–388. |
[83] | Hardy J ((2009) ) The amyloid hypothesis for Alzheimer’s disease: A critical reappraisal. J Neurochem 110: , 1129–1134. |
[84] | Anand R , Gill KD , Mahdi AA ((2014) ) Therapeutics of Alzheimer’s disease: Past, present and future. Neuropharmacology 76: , 27–50. |
[85] | van Dijk G , van Heijningen S , Reijne AC , Nyakas C , van der Zee EA , Eisel UL ((2015) ) Integrative neurobiology of metabolic diseases, neuroinflammation, and neurodegeneration. Front Neurosci 9: , 173. |
[86] | Cha MY , Kim DK , Mook-Jung I ((2015) ) The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp Mol Med 47: , e150. |
[87] | Simoncini C , Orsucci D , Ienco EC , Siciliano G , Bonuccelli U , Mancuso M ((2015) ) Alzheimer’s pathogenesis and its link to the mitochondrion. Oxid Med Cell Longev 2015: , 803942. |
[88] | Sulistio YA , Heese K ((2016) ) The ubiquitin-proteasome system and molecular chaperone deregulation in Alzheimer’s disease. Mol Neurobiol 53: , 905–931. |
[89] | Kakkar V , Meister-Broekema M , Minoia M , Carra S , Kampinga HH ((2014) ) Barcoding heat shock proteins to human diseases: Looking beyond the heat shock response. Dis Model Mech 7: , 421–434. |
[90] | Dou F , Netzer WJ , Tanemura K , Li F , Hartl FU , Takashima A , Gouras GK , Greengard P , Xu HX ((2003) ) Chaperones increase association of tau protein with microtubules. Proc Natl Acad Sci U S A 100: , 721–726. |
[91] | Cohen SIA , Arosio P , Presto J , Kurudenkandy FR , Biverstal H , Dolfe L , Dunning C , Yang XT , Frohm B , Vendruscolo M , Johansson J , Dobson CM , Fisahn A , Knowles TPJ , Linse S ((2015) ) A molecular chaperone breaks the catalytic cycle that generates toxic A beta oligomers. Nat Struct Mol Biol 22: , 207–213. |
[92] | Doyle SM , Genest O , Wickner S ((2013) ) Protein rescue from aggregates by powerful molecular chaperone machines. Nat Rev Mol Cell Biol 14: , 617–629. |
[93] | Liu ML , Liu MJ , Shen YF , Ryu H , Kim HJ , Klupsch K , Downward J , Hong ST ((2009) ) Omi is a mammalian heat-shock protein that selectively binds and detoxifies oligomeric amyloid-beta. J Cell Sci 122: , 1917–1926. |
[94] | West JD , Wang YY , Morano KA ((2012) ) Small molecule activators of the heat shock response: Chemical properties, molecular targets, and therapeutic promise. Chem Res Toxicol 25: , 2036–2053. |
[95] | Silva MC , Amaral MD , Morimoto RI ((2013) ) Neuronal reprograming of protein homeostasis by calcium-dependent regulation of the heat shock response. PLoS Genet 9: , e1003711. |
[96] | Yamashima T ((2013) ) Reconsider Alzheimer’s disease by the ‘calpain-cathepsin hypothesis’-A perspective review. Prog Neurobiol 105: , 1–23. |
[97] | Zhu H , Yoshimoto T , Yamashima T ((2014) ) Heat shock protein 70.1 (Hsp70.1) affects neuronal cell fate by regulating lysosomal acid sphingomyelinase. J Biol Chem 289: , 27432–27443. |
[98] | Horvath I , Vigh L ((2010) ) Cell biology: Stability in times of stress. Nature 463: , 436–438. |
[99] | Lamech LT , Haynes CM ((2015) ) The unpredictability of prolonged activation of stress response pathways. J Cell Biol 209: , 781–787. |
[100] | Prodromou C , Siligardi G , O’Brien R , Woolfson DN , Regan L , Panaretou B , Ladbury JE , Piper PW , Pearl LH ((1999) ) Regulation of Hsp90 ATPase activity by tetratricopeptide repeat (TPR)-domain co-chaperones. EMBO J 18: , 754–762. |
Figures and Tables
Fig.1
Chemical structure of the synthesized 1,4-dihydropyridine derivatives.
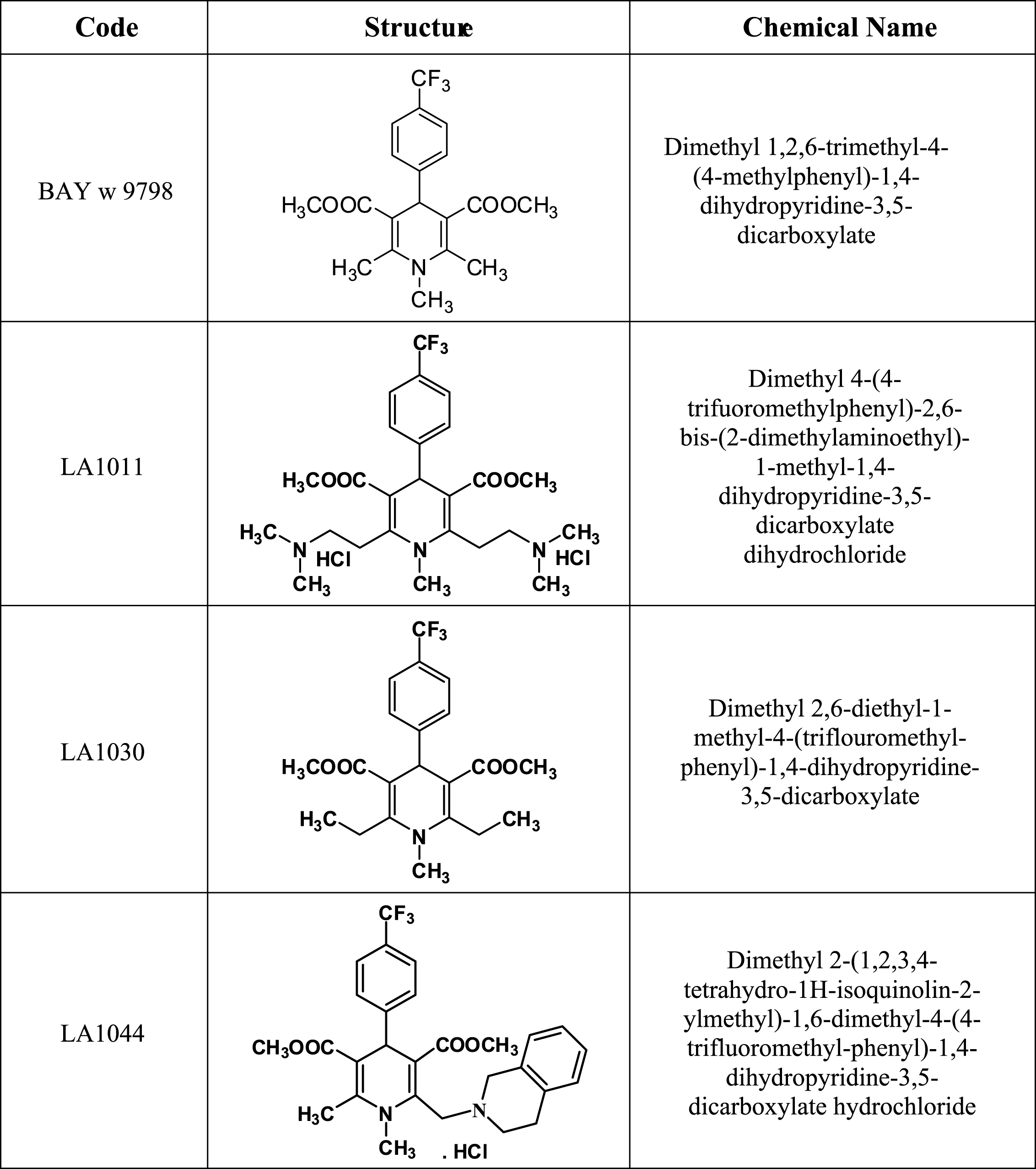
Fig.2
The Hsp70 co-inducer effect of various LA drug candidates was screened by using a promoter-probing assay. Bars representing co-induction were calculated by the following formula: Fold change = [(Fx(42)-F0(37)]/[F0(42)-F0(37)], where “F” represents the fluorescent intensity of cells incubated in the absence “0” or presence of drug candidates of concentration “x” at 37°C or 42°C for 1 h. The missing bars for LA1011 (20 and 40μM) indicate that the drug was toxic at these concentrations. Data are means±SD.
![The Hsp70 co-inducer effect of various LA drug candidates was screened by using a promoter-probing assay. Bars representing co-induction were calculated by the following formula: Fold change = [(Fx(42)-F0(37)]/[F0(42)-F0(37)], where “F” represents the fluorescent intensity of cells incubated in the absence “0” or presence of drug candidates of concentration “x” at 37°C or 42°C for 1 h. The missing bars for LA1011 (20 and 40μM) indicate that the drug was toxic at these concentrations. Data are means±SD.](https://content.iospress.com:443/media/jad/2016/53-2/jad-53-2-jad150860/jad-53-jad150860-g002.jpg)
Fig.3
Effect of heat and LA1011 or LA1044 treatment on Hsp70, Hsp27 and Hsp40 levels in SH-SY5Y human neuroblastoma cell line measured by western blotting (A, B) and quantitative densitometric analysis (C, D). Cells were untreated or treated with LA1011 (5μM) or LA1044 (40μM) at 37°C or 42°C. Box plots represent quantitative data normalized to GAPDH. All data represent the mean±SD (n = 6) and p < 0.05 was considered statistically significant. HS = heat shock at 42°C, 1 h. *p < 0.05, when data are compared to LA compound (–), HS (–) sample. #p < 0.05, when data are compared to LA compound (–), HS (+) sample. One way ANOVA followed by a Tukey post hoc test was used for statistical comparisons.
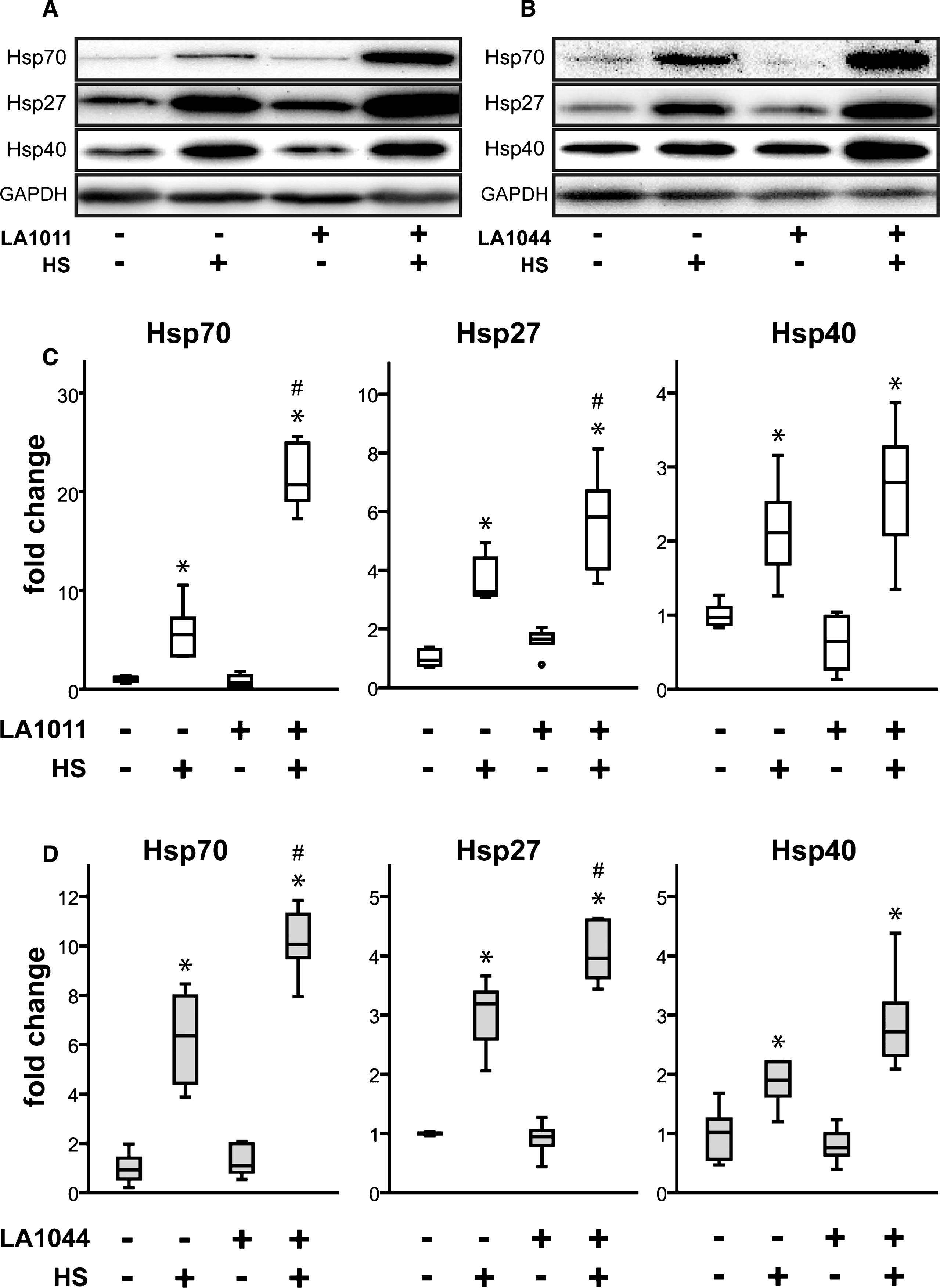
Fig.4
Effect of LA1011 treatment on Morris water maze performance. The fitted survival curves using the Cox Proportional Hazard model represents the probability that animals find the platform during a trial, capped at 90 s. The effect of day has been modeled in. We compared wt+saline versus wt+LA1011 (p = 0.047), wt+saline versus tg+saline (p = 0.036), and tg+saline versus tg+LA1011 (p = 0.001) treatment groups using log-rank tests (n = 8/group).
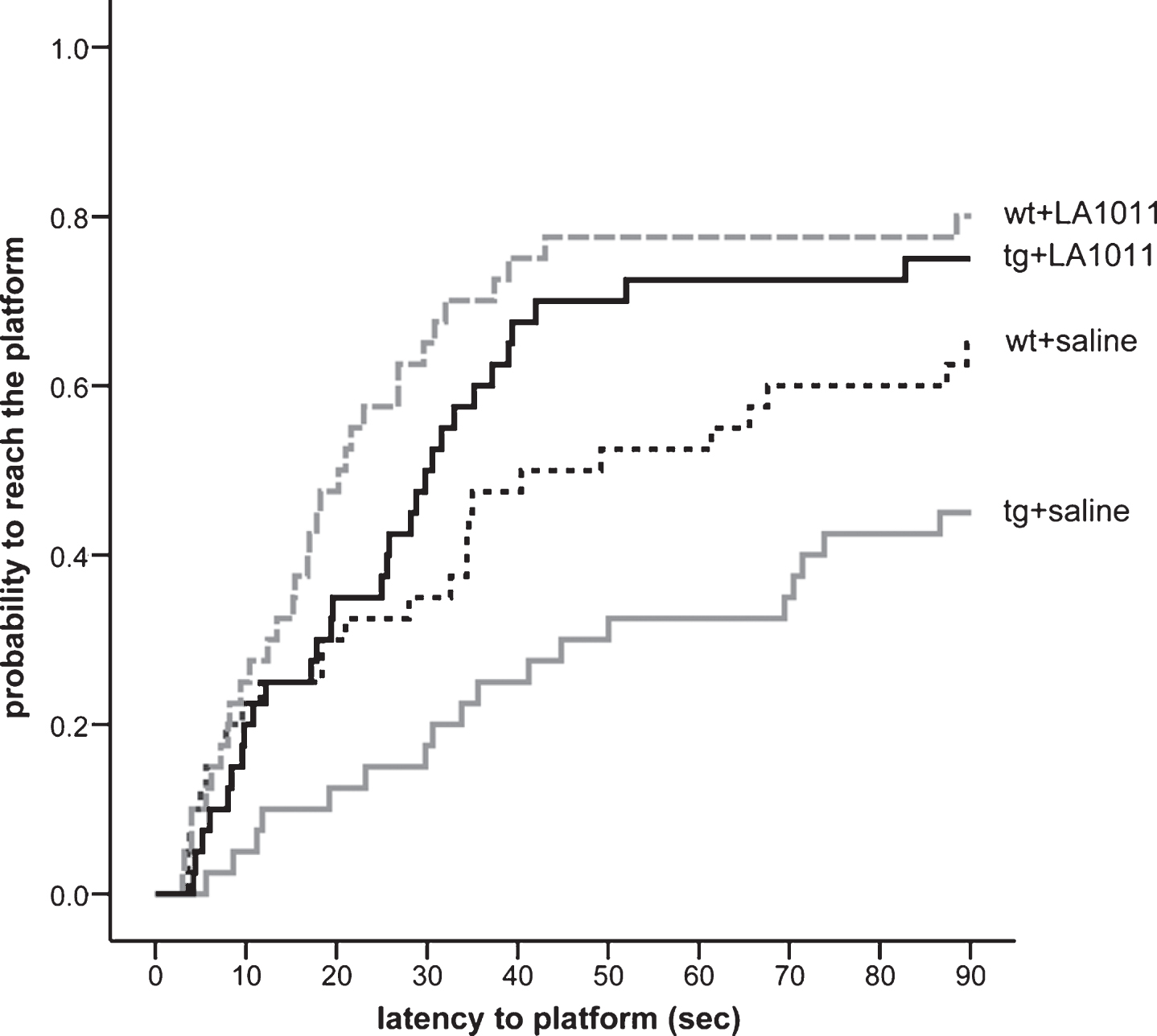
Fig.5
Cresyl-violet staining, tau-immunostaining, and Golgi-Cox staining of hippocampal slices of wt and tg animals after 6 months treatment. (A) Number of counted neurons/mm2; n = 4, 2 slices/animal, (B) number of counted neurofibrillary tangles/mm2; n = 4, 4 slices/animal and (C) measured spine density/100μm; n = 3, 2 slices/animal. Each dot represents the counted raw data, while horizontal bars indicate mean values. Statistical significance was determined by one-way ANOVA, followed by Fisher’s LSD post hoc test. Differences with a p-value <0.05 were considered significant; n refers to the number of animals.