The Effects of Latrepirdine on Amyloid-β Aggregation and Toxicity
Abstract
Latrepirdine (Dimebon™) has been demonstrated to be a neuroprotective and cognition improving agent in neurodegenerative diseases that feature protein aggregation and deposition, such as Alzheimer’s disease (AD). The accumulation of amyloid-β (Aβ) protein aggregates is a key event in the neurodegenerative process in AD. This study explores if latrepirdine modulation of protein aggregation contributes to its neuroprotective mechanism of action. Assessment of neuronal cell death showed that there was a significant reduction in lactate dehydrogenase release at an equimolar ratio of Aβ:latrepirdine and with lower concentrations of latrepirdine. The ability of latrepirdine to alter the formation of Aβ42 aggregates was assessed by thioflavin-T fluorescence, western immunoblotting and atomic force microscopy (AFM). Despite showing a reduction in thioflavin-T fluorescence with latrepirdine treatment, indicating a decrease in aggregation, immunoblotting and AFM showed a modest increase in both the formation and size of Aβ aggregates. The discrepancies between thioflavin-T and the other assays are consistent with previous evidence that cyclic molecules can interfere with thioflavin-T binding of amyloid protein preparations. The ability of latrepirdine to modulate Aβ aggregation appears to be independent of its neuroprotective effects, and is unlikely to be a mechanism by which latrepirdine offers protection. This study investigates the effect of latrepirdine on Aβ aggregation, and presents evidence suggesting that caution should be applied in the use of thioflavin-T fluorescence based assays as a method for screening compounds for protein aggregation altering properties.
INTRODUCTION
Alzheimer’s disease (AD) is the most common cause of senile dementia. It is clinically characterized by a progressive deterioration of memory, cognitive function, and the ability to perform daily tasks. Pathologically AD is characterized by severe cerebral atrophy due to a loss of hippocampal and neocortical neurons [1]. Major neuropathological hallmarks include neurofibrillary tangles (consisting of aggregates of the tau protein), and extracellular deposition of amyloid plaques, comprised mainly of aggregates of a protein known as amyloid-β (Aβ) [2]. The accumulation of Aβ is thought to occur early in the disease process, prior to onset of clinically relevant symptoms [3]. Increases in cerebral levels Aβ have key roles in downstream events that lead to neurodegeneration, including tau hyperphosphorylation and accumulation [4].
The Aβ protein is generated from the processing of its parent protein, the amyloid-β protein precursor (AβPP), (reviewed [5]). Enzymatic cleavage of AβPP via β-secretase, followed by γ-secretase, generates multiple Aβ species, including the more common soluble monomeric peptides of 40 amino acids and the more insoluble Aβ peptide of 42 amino acids (Aβ42). In AD, Aβ42 monomers aggregate into progressively larger species under various physiological conditions, and these different forms can differentially affect neuronal function [6]. Both soluble and insoluble Aβ forms exist in brain [7], and it is now recognized that an aberrant increase in soluble Aβ correlates to a disease phenotype and is correlated with disease severity [7, 8].
Oligomer and fibril forms of Aβ are the most common species found in the brains of AD patients. Both synthetic and naturally secreted human Aβ oligomers have been shown to reduce long-term potentiation [9]. Furthermore, the effects of Aβ oligomers on long-term potentiation, and the accumulation of aggregates, can be reduced in vivo through an application of anti-Aβ antibodies [10, 11]. These results strongly support a causative role for Aβ aggregation in the cognitive dysfunction observed in AD. Enhancing effective clearance of Aβ aggregates, and the modulation of Aβ aggregation to reduce deposition and toxicity in the brain, have been considered as potential therapeutic strategies in AD [12].
One of the more recent drugs to be tested for its efficacy against AD is latrepirdine. Latrepirdine (DimebonTM) has been shown in vitro and in vivo to be neuroprotective [13]. However, its mode of action is largely uncharacterized and poorly understood. Latrepirdine is an orally-available, small molecule previously approved in Russia as a non-selective antihistamine [14]. Preclinical trials in rats with induced cognitive impairment showed that latrepirdine improved their learning skills and memory when compared to both untreated rats and rats treated with anticholinesterase inhibitors [14]. Furthermore, latrepirdine has been shown to protect neuronal cultures against Aβ42-induced toxicity [14]. Clinical trials for latrepirdine as a treatment for AD, however, have produced mixed results. The initial clinical study on a Russian cohort showed that patients receiving latrepirdine had improved measures of cognitive ability, function, and behavior when compared to both baseline and placebo patients [15]. However, in subsequent phase 3 clinical trials, a 6-month US-based replication trial (CONNECTION) and a 12-month trial with patients enrolled in the US, Australia, New Zealand, and Western Europe (CONCERT), latrepirdine treatment showed no benefits [16–18]. The exact causes for the mixed results in the AD clinical trials are unknown, in addition to trial design and targeting patients with late stage symptoms and significant neuronal damage, a lack of understanding of the exact mechanism of action of the drug, could be contributing factors [19].
Latrepirdine has been shown to have effects on a number of cellular functions (reviewed [20, 21]) as well as protect neuronal cultures from Aβ toxicity [14], and significantly reduce intracellular Aβ42 levels in in vitro cell based and in vivo animal based AD studies [22]. A protective role of latrepirdine has been shown by the clearance of α-synuclein and γ-synuclein in mouse brain and neuronal cells [23, 24]. Latrepirdine has also been shown to inhibit the aggregation of the TDP-43 protein involved in the pathology of amyotrophic lateral sclerosis [25]. Our own findings in a yeast model have shown that latrepirdine can reduce Aβ42 aggregates [26]. These findings suggest that the ability of latrepirdine to modulate protein aggregation may account for its action in promoting the clearance of protein aggregates and thus contribute to the underlying neuroprotective mechanism of action. This study addressed this by investigating the effect of latrepirdine on Aβ aggregation.
MATERIALS AND METHODS
Materials
High-performance liquid chromatography (HPLC) purified (95% purity) synthetic human beta amyloid (Aβ; Lot 2534), was purchased from WM Keck Foundation (Yale University, New Haven, CT). Latrepirdine (Dimebolin dihydrochloride MW: 392.37) was purchased from Biotrend AG (Zurich). Thioflavin-T was purchased from Sigma-Aldrich (St. Louis, MO, USA). Mouse monoclonal antibody WO2, raised against amino acid residues 5–8 of N-terminal Aβ sequence [27] was kindly provided by Professor Colin Masters (University of Melbourne, VIC, Australia). Horseradish peroxidase (HRP) conjugated anti-mouse antibodies were purchased from GE (Rydalmere, NSW, Australia). NuPage Novex 4–12% Bis-Tris gels, lithium dodecyl sulfate (LDS) sample buffer (40% glycerol 4% LDS, 0.025% phenol red, 0.025% serva blue G250, 2 mM ethylenediaminetetraacetic acid (EDTA) disodium, pH 7.6), 2–(N–morpholino) ethanesulfonic acid (MES) running buffer (50 mM Tris base, 50 mM MES, 1 mM EDTA, 0.1% SDS at pH 7.3), and the iBlot Western transfer kit were all purchased from Invitrogen (Mulgrave, VIC, Australia). 3–(N–morpholino propanesulfonic acid (MOPS) sample buffer (50 mM MOPS, 50 mM Tris, 20% glycerol, 0.05% Coomassie), MOPS/Coomassie running buffer (50 mM MOPS, 50 mM 50 mM Tris, 0.002% Coomassie), and MOPS running buffer (50 mM MOPS, 50 mM Tris) were used for blue native gel electrophoresis.
The SHSY5Y neuronal cell line was obtained from ATCC Global Bioresource Centre, and was cultured at 37°C in a 5% CO2 atmosphere. Cell culture medium used was Dulbecco’s Modified Eagle Medium, purchased from Gibco (Mulgrave, VIC, Australia), supplemented with 10% fetal bovine serum (FBS). Hams F12 media was purchased from PromoCell (Heidelberg, Germany). CytoTox One Homogenous Membrane Integrity assay (LDH assay), and Cell Titer 96® AQueous One solution Cell Proliferation assays (MTS Assay) were purchased from Promega(Madison, WI, USA).
Preparation of Aβ aggregates
Aβ aggregates were prepared according to the protocol outlined by Stine et al., with some modification [28]. Synthetic Aβ was first dissolved in hexafluoroisopropanol (HFIP), to produce a 1 mM solution. Varying volumes of this solution was then aliquoted into 1.6 mL Eppendorf tubes and left to evaporate overnight. The films produced were stored at –80°C until required. For both oligomeric and tris buffered saline (TBS) preparations of Aβ, a 5 mM stock was prepared with dimethylsulfoxide (DMSO). To ensure the Aβ was completely resuspended the tube was vortexed and bath sonicated. For the oligomeric preparation the Aβ/DMSO solution was diluted to 100 μM with Hams F12 media and incubated for 24 h at 4°C. For the TBS preparation, the Aβ/DMSO solution was diluted to 100 μM with TBS and incubated for 24 h at room temperature.
Thioflavin-T (thioT) fluorescence assay
The effect of latrepirdine on Aβ aggregation was measured using the thioT amyloid-binding fluorescence assay. Fluorescence was measured using a FLUOStar Optima (BMG Labtech, Ortenberg,Germany), excitation and emission maxima were set to 450 nm and 490 nm respectively. The Aβ was prepared in the presence or absence of various concentrations of latrepirdine (20, 50, 100, and 200 μM) and incubated with 5 μM thioT for 16 h with fluorescence readings made at 10 min intervals. Experiments were performed in quadruplicate wells and each experiment was repeated a minimum of three times. Control background thioT fluorescence was subtracted from all sample results for each assay.
Western immunoblotting and atomic force microscopy (AFM) analysis of Aβ aggregates in the presence of absence of latrepirdine
Samples of 100 ng and 200 ng quantities were analyzed by 4–12% Bis-Tris gels (MES/Blue Native Polyacrylamide gel electrophoresis (PAGE) MOPS buffering). Gels were electro-transferred to nitrocellulose or polyvinylidene fluoride (PVDF) membranes and analyzed by immunoblotting using WO2 as previously described [26, 27]. Membranes were blocked in 5% skim milk in TBS solution for 1 h, primary antibody (WO2) was diluted 1/5000 in TBS with 0.05% Tween 20 (TBST) and 0.5% skim milk solution for 2 h, followed by three washes with TBST. HRP conjugated anti-mouse secondary antibody was diluted by 1/5000 in TBST and skim milk solution and incubated for 1 h. After washing the membranes were incubated for 2 min with enhanced chemiluminescence reagent and exposed to Hyperfilm (GE) for periods ranging from 15 s to 16 h.
For AFM, Aβ (100 μM) was prepared in the presence or absence of 1 mM latrepirdine. Samples were diluted to 25 μM and 5 μL was loaded onto freshly cleaved V1 grade muscovite mica for AFM. After 5 min incubation of the sample on the mica it was washed 3 times with deionized water, and dried under nitrogen gas. Samples were visualized using a NT-MDT microscope in semi-contact mode with the following parameters: a minimum contact force, amplitude between 0.5–2 V (dependent on cantilever) magnitude 20 nA, and scan rates 0.5–1 Hz. All data was processed using Nova NT-MDT software v1.1.0.1780. 4 10 μm2 fields of view were examined for each sample, and the software was used to magnify areas of interest. The images were produced by the probes movement across the mica, and the measurements used in the cross sectional analyses were made from the displacement of the probe as it moved over the sample.
Neuronal cell viability
The SHSY5Y neuronal cell line was cultured at 37°C in a 5% CO2 atmosphere in Dulbecco’s Modified Eagle Medium (DMEM), supplemented with 10% FBS. Cells were cultured and treated with oligomeric preparations of Aβ. Prior to treatment Aβ was incubated for 16 h in the presence, or absence, of a latrepirdine concentration range (3–300 μM). Cells were plated in 96-well plates, 10,000 cells per well and incubated for 24 h at 37°C, 5% CO2. Prior to cell treatment the incubated Aβ samples were desalted using 3KDa molecular weight cut off, Amicon Bioseparations Centricon columns (Millipore). These samples were diluted to 30 μM Aβ in DMEM containing 1% FBS and Hams F12 media and used to treat the cells. The cells were incubated for 4 days at 37°C, with 5% CO2, and cell viability assayed for release of lactate dehydrogenase (LDH), as per the manufacturer’s instructions (Promega, USA).
Statistical analysis
Data obtained from neuronal cell viability and thioT assays were evaluated using a one-way ANOVA with planned contrasts followed by a post hoc Bonferroni correction for multiple comparisons. Data was graphically represented using means (±standard deviation) of; % LDH release when compared to a total lysis control, and thioT fluorescence measurements. The level of statistical significance was set at 0.05. All statisticswere performed using IBM SPSS Statistics for Macintosh, Version 22.0 (Armonk, NY).
RESULTS
Latrepirdine has been shown to be protective in serum starved SHSY5Y cells [29] and SHSY5Y cells expressing α-synuclein [24]. To determine if latrepirdine can protect against Aβ induced neuronal death, LDH release from SHSY5Y cells was assessed in cells treated with Aβ oligomers in the presence of vehicle and 0.05 to 20 μM of latrepirdine (Fig. 1). Treatment with 20 μM Aβ in the presence of vehicle led to 60% LDH release. A one-way ANOVA was conducted comparing the effect of latrepirdine treatment on % LDH released. Latrepirdine significantly reduced LDH release from 1 μM latrepirdine (p < 0.005) reaching a 35% reduction at 20 μM (p < 0.005, 1:1 Aβ-latrepirdine molar ratio, Fig. 1, [F(7,16) = 10.583, p < 0.0005]). A similar reduction in cell death at this concentration has been reported previously [30]. LDH release from cells treated with latrepirdine only was similar to vehicle only treated cells, indicating that at the concentrations used, latrepirdine did not cause significant cell death. Overall, consistent with previousstudies we have shown that latrepirdine can offer protection against Aβ toxicity [26, 31]. We next investigated whether inhibiting Aβ aggregation could be a mechanism underlying latrepirdine neuroprotective effects.
To assess whether latrepirdine can alter Aβ aggregation, monomeric Aβ42 was incubated at 4°C for 24 h in the presence of increasing doses of latrepirdine. Following incubation, samples were mixed with thioT solution, and fluorescence readings were measured. With reference to the Aβ only control, a significant dose dependent reduction in thioT fluorescence was observed upon the addition of latrepirdine (Fig. 2A), indicating a reduction in Aβ42 aggregation.
To investigate whether the reduction in thioT fluorescence was time dependent, a time course thioT assay was performed. Assay conditions were modified slightly to accommodate the instrument constraints. In particular, latrepirdine was added to Aβ42 just after the addition of thioT and incubated at room temperature (RT) with the fluorescence readings recorded every 10 min for the 16 h time course. The Aβ42 preparation was also prepared in a buffered solution (TBS) which was necessary for the prolonged incubations at RT. Direct comparisons by SDS-PAGE of oligomeric preparations with TBS preparations reveals similar aggregates were formed (Supplementary Fig. 1A). The TBS preparation contained similar sized aggregates to the oligomer preparation (0.05–0.2 μm in diameter and heights ranging from 2–15 nm) but also contained larger aggregates (from 0.2–1 μm in diameter, and their height ranged from 20–50 nm) (Supplementary Fig. 2B, C). Assaying the TBS preparations revealed a time dependent aggregation during the 16 h incubation period (Fig. 2B). However, a dose-dependent reduction in fluorescence units was observed in samples incubated with latrepirdine, where at an Aβ:latrepirdine molar ratio of 1:10, a 1.5 fold reduction was observed. It was noted that at time 0 (t0, 0–10 min) an almost 2-fold reduction at the 1:10 ratio compared to Aβ only was observed. Several possibilities could account for this observation, including a direct interaction between latrepirdine and Aβ thereby reducing the binding of thioT at t0. This would result in a decrease in fluorescence at the endpoint (16 h). Alternatively, latrepirdine may bind and displace thioT from binding to the Aβ aggregates either through binding to latrepirdine directly or by binding directly to the Aβ aggregates.
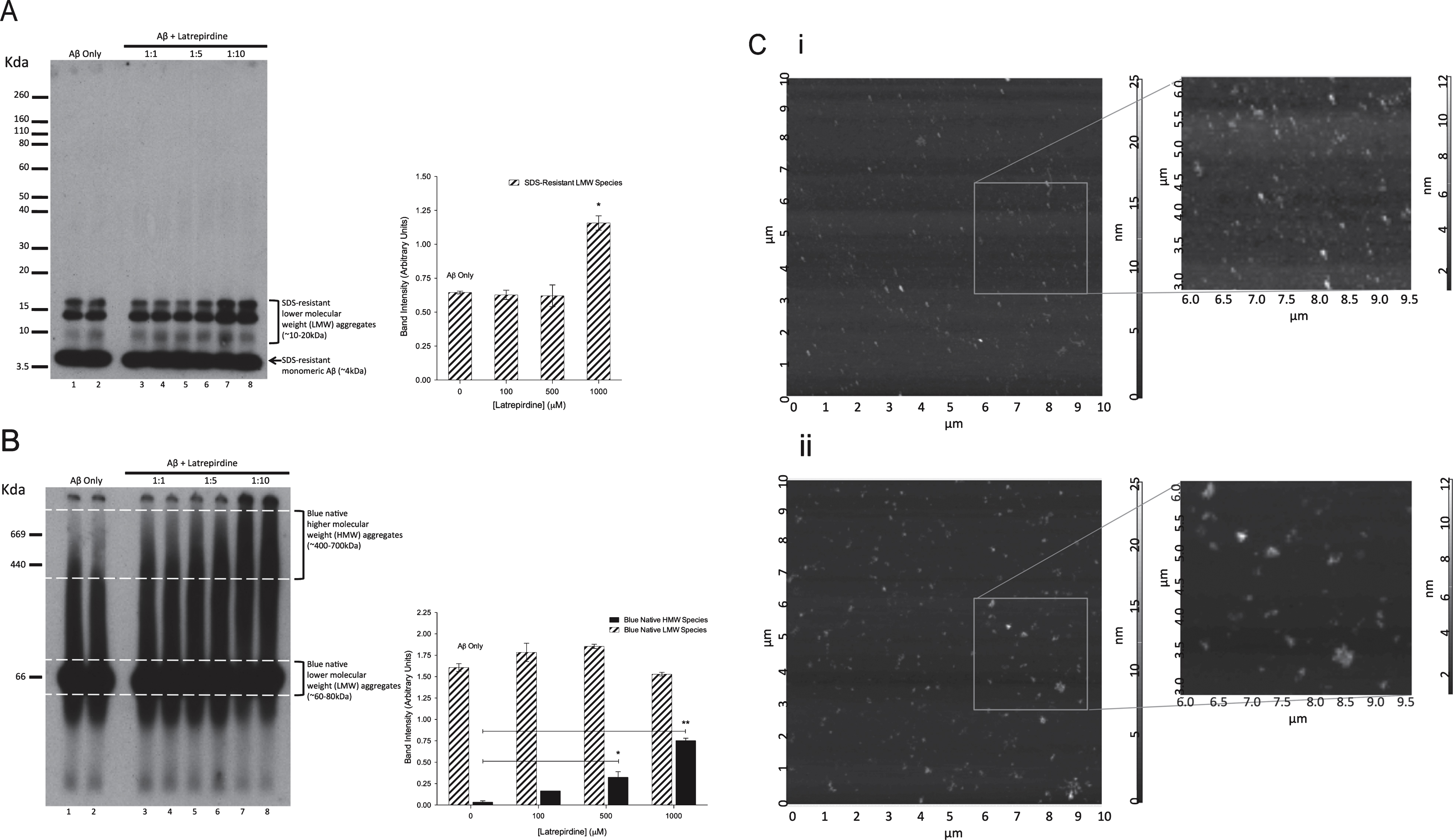
To obtain a better understanding of this phenomenon and to determine if latrepirdine has similar effects when added after Aβ aggregation has commenced, the previous time course experiment was repeated with modification. Latrepirdine or vehicle was added after Aβ aggregation had been proceeding for 6 h (Fig. 2A). As shown before, fluorescence steadily increased prior to the addition of latrepirdine resulting in a sharp decrease in observed fluorescence. These results indicate that the observed time course experiment data could be either, a result of direct displacement of thioT binding or alternatively, latrepirdine reducing/influencing the rate of aggregation. To determine if the observed changes in aggregation detected by thioT are reflecting the progression of Aβ aggregation, an analysis of the formed products on SDS-PAGE or BN-PAGE was performed. Aβ preparations were incubated in the presence or absence of latrepirdine (at a 1:1 or 1:10 molar ratio) for 0 h (t0), 6 h (t6), and 16 h (t16) and then subjected to the gel electrophoresis under either denaturing conditions SDS-PAGE or non-denaturing BN-PAGE (Fig. 3B, C). At t0, lower molecular weight aggregates were particularly prominent in both SDS-PAGE (∼4 kDa) and BN-PAGE (∼70 kDA). However, at t6 and t16, higher molecular weight aggregates (40–160 kDa, 670 kDa) were observed, being most prominent at t16. Latrepirdine did not appear to reduce (Fig. 3B, C, lanes 9–12 and 15–16) the formation of these higher molecular weight aggregates.
Latrepirdine modestly effects the formation of Aβ aggregates
End point thioT analysis of Aβ oligomers (Fig. 2A) supported that latrepirdine dose dependently reduced oligomer formation. Similar results were also observed when latrepirdine was added after Aβ oligomers had been formed (Supplementary Fig. 2). Due to the observed disparity in results described previously by thioT analysis and gel electrophoresis, the formation of Aβ oligomers in the presence or absence of latrepirdine was analyzed using gel electrophoresis and AFM.
Western immunoblotting showed an increased amount of the lower and higher molecular weight species, most prominently observed at a 1:10 molar ratio of Aβ:latrepirdine (Fig. 4A, B, lanes 7 and 8). Collectively, these results suggest that latrepirdine may induce either more Aβ oligomer formation or promote the formation of larger Aβ oligomer species. To determine if the size and shape of the Aβ oligomers was altered, Aβ oligomers or Aβ:latrepirdine mixtures (1:10 molar ratio) were subjected to analysis using AFM. In the Aβ oligomer preparation, the size of Aβ aggregates was observed to range from 0.05–0.2 μm in diameter with heights from 2–15 nm (Fig. 4C, i, Supplementary Fig. 3A). Similar sized oligomers were also observed in Aβ:latrepirdine mixture, however, larger Aβ aggregates with diameters ranging from 0.2–0.5 μm and heights from 10–35 nm were also present (Fig. 3C, ii, Supplementary Fig. 3B). These results suggest that latrepirdine leads to the formation of larger aggregates. Overall, these results show that latrepirdine had modest effects on the formation of Aβ aggregates, which were only observed when latrepirdine is in 10-fold excess. Treating cells at a 1:10 Aβ:latrepirdine molar ratio or treating with a control containing the equivalent concentration of latrepirdine (300 μM) resulted in total cell death (data not shown). These findings indicated that latrepirdine at these supraphysiological doses was probably a major contributing factor leading to the observed cell death. Thus, the consequence of latrepirdine promoting the formation of larger aggregates could not be evaluated.
DISCUSSION
The accumulation and aggregation of Aβ is a key event in the pathogenesis of AD, and preventing the formation or disrupting Aβ aggregation, thereby reducing neuronal dysfunction in the brain has been proposed as a therapeutic strategy. Inhibitors and modulators of Aβ aggregation, including peptide, protein and small molecular classes of natural and synthetic origin, have been shown to modulate toxicity of Aβ in vitro and in vivo [32, 33]. Latrepirdine is a small polycyclic molecule belonging to a class of pro-neurogenic compounds [13]. Findings from previous studies suggest that the ability of latrepirdine to modulate protein aggregation may explain its action in promoting the clearance of protein aggregates and could contribute to its neuroprotective mechanism of action [23, 25]. This study has addressed whether latrepirdine alters Aβ oligomer formation and if these effects were related to the amelioration of Aβ neurotoxicity in vitro.
The Aβ protein has a natural tendency to self assemble into multimeric forms on the basis of cross-beta sheets which resemble a common folding pathway for amyloidogenic proteins. The thioT fluorescence assay is a commonly used technique to monitor this self-assembly/aggregation of amyloidogenic proteins and is a rapid method to determine the efficacy of inhibitors and modulators of aggregation. Molar ratios of Aβ:latrepirdine ranging from 1:1–1:10, showed a dose dependent reduction in thioT fluorescence in both end point and time course assays. In the time-course assay, however, the dose dependent reduction was observed at the start of the assay (t0), before fluorescence increased in a time dependent manner. Similarly, a sharp reduction in fluorescence, followed by a steady increase, was observed when latrepirdine was added 6 h after Aβ commenced aggregation, despite no observed reduction in aggregate formation by SDS-PAGE or BN-PAGE. A possible explanation for this observation is that latrepirdine displaces or prevents thioT binding to Aβ aggregates either through direct binding to thioT or by binding to the Aβ aggregates. Support for this hypothesis comes from a recent study that showed small molecular weight cyclic compounds with similar structure to thioT reduce fluorescence in amyloid protein preparations [34]. This study suggested that such compounds could actively compete for the same binding site, thereby showing decreased fluorescence in the absence of any apparent structural change [34]. In conjunction with the findings presented in this study, this indicates that more attention to the interpretation of using fluorescent dyes such as thioT in measuring protein aggregation kinetics is required. As we found that thioT could not reliably assess the effects of latrepirdine on Aβ aggregation, the direct characterization of the formation of aggregates, with particular focus on Aβ oligomers, was performed by gel electrophoresis and AFM.
Oligomer and fibrillar species represent the majority of the diverse structural assemblies formed by amyloidogenic proteins like Aβ. There is increasing evidence that supports a causative role of the soluble oligomeric forms in neurodegeneration observed in AD rather than the insoluble fibrillar Aβ counterparts [7]. The focus of this study was therefore to assess the effects of latrepirdine on oligomeric Aβ structures. Oligomers can be reliably generated in vitro, can exist as smaller or higher molecular weight species, less than 20 kDa and greater than 35 kDa respectively (Supplementary Fig. 1A, B), and are 0.05–0.2 μm in size (Supplementary Fig. 1C, D) [36]. The addition of latrepirdine during the generation of oligomers appeared to modestly increase their formation, however only when added in 10-fold excess. This was observed following immunoblotting where increased amounts of lower and higher molecular weight species were present. Furthermore, AFM analysis showed that in addition to 0.2 μm oligomer aggregates, larger 0.5 μm aggregates were also present and these may represent extensions of smaller oligomer species [36]. The significance of an effect of promoting oligomer formation by latrepirdine remains unclear. At higher molar ratio of Aβ:latrepirdine, latrepirdine could promote the extension of oligomers further to protofibrils (0.5–1 μm) [6, 36] or fibrils (3-4 μm) [36]. Cyclic small molecules have been shown previously to promote the formation of larger Aβ species resulting in reduced toxicity [37], and this is consistent with the proposed hypothesis that ‘amyloid’ structuring of proteins is a detoxification strategy to mask the promiscuous surface of the oligomeric building block [35].
Although the use of higher molar ratios of laterpirdine:Aβ could be investigated to provide further insight into what type of aggregates are generated, the findings of this study suggest that the neuroprotective effect of latrepirdine may be independent of its ability to modulate Aβ aggregation. Treating cells at a molar ratio of 1:10 resulted in complete cell death, due to the supraphysiological levels of latrepirdine required to show changes in Aβ aggregation. However, assessment of the neuronal cell death showed that there was a significant reduction in LDH release at lower ratios (1:0.05 to 1:1) where influences on Aβ aggregation were not evident. Zhang et al. showed that under non-stressed conditions, latrepirdine can increase mitochondrial membrane potential and cellular ATP levels in SH-SY5Y cells, but not alter mitochondria DNA content. This indicates that mitochondrial function is enhanced [29]. In serum-starved cells, mitochondrial membrane potential of latrepirdine treated cells was maintained under conditions of increased intracellular calcium concentrations, indicating that mitochondrial function is preserved even under external stresses. Further evidence supporting the concept that latrepirdine improves mitochondrial function comes from recent reports that have shown that latrepirdine can decrease calcium retention capacity of rat brain mitochondria [38] and protect against Aβ induced changes in mitochondria morphology and respiration [39].
The findings of this study indicate that latrepirdine can modulate Aβ oligomer formation, but this effect appears to be independent of its ability to reduce the resulting toxicity induced by oligomers. This finding is further supported by studies showing latrepirdine has no significant effect on the in vitro assembly of recombinant human α-synuclein [24]. The reduction in protein aggregates with latrepirdine treatment reported by others [22] may be associated with its ability to promote clearance of toxic protein aggregates via catabolic pathways, such as autophagy [24, 26, 40], rather than its direct effects on the amyloid structure. This, in combination with evidence indicating that latrepirdine maintains mitochondrial function in the presence of external stresses, may explain its neuroprotective activity.
Although, in the initial Russian clinical trials, latrepirdine was shown to improve cognition in patients with mild to moderate AD [15], recent phase IIIclinical trials have shown that the drug is of limited benefit to moderate-severe AD cases [16]. Differences in trial design, population bias (Russian phase II trial versus multinational phase III trials) and targeting end-stage AD patients, have all been proposed as reasons for the different outcomes [19]. It is also important to consider the mechanism by which latrepirdine can target neurotoxicity induced by Aβ aggregation, particularly if more potent analogues are to be developed for AD. This study has provided an investigation into the effect of latrepirdine on Aβ aggregation, and the methods used will be useful in assessing other amyloid disrupting compounds. However, the widely used thioT fluorescence assay should be used with caution when investigating compounds thought to possess aggregation altering capabilities.
ACKNOWLEDGMENTS
The authors acknowledge the support of NH&MRC APP1009295 to RM and GV. GV is supported by the NH&MRC (APP1045507 and APP1105698) and the Curtin University Senior Research Fellowship (CRF140196). We thank Linda Wijaya for assisting in AFM.
Authors’ disclosures available online (http://www.j-alz.com/manuscript-disclosures/15-0790r2).
Appendices
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-150790.
REFERENCES
[1] | Terry RD ((2006) ) Alzheimer’s disease and the aging brain, J Geriatr Psychiatry Neurol 19: , 125–128. |
[2] | Masters CL , Multhaup G , Simms G , Pottgiesser J , Martins RN , Beyreuther K ((1985) ) Neuronal origin of a cerebral amyloid: Neurofibrillary tangles of Alzheimer’s disease contain the same protein as the amyloid of plaque cores and blood vessels, EMBO J 4: , 2757–2763. |
[3] | Villemagne VL , Burnham S , Bourgeat P , Brown B , Ellis KA , Salvado O , Szoeke C , Macaulay SL , Martins R , Maruff P , Ames D , Rowe CC , Masters CL , Australian Imaging B, Lifestyle Research G ((2013) ) Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study, Lancet Neurol 12: , 357–367. |
[4] | Lewis J , Dickson DW , Lin WL , Chisholm L , Corral A , Jones G , Yen SH , Sahara N , Skipper L , Yager D , Eckman C , Hardy J , Hutton M , McGowan E ((2001) ) Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP, Science 293: , 1487–1491. |
[5] | Krishnaswamy S , Verdile G , Groth D , Kanyenda L , Martins RN ((2009) ) The structure and function of Alzheimer’s gamma secretase enzyme complex, Crit Rev Clin Lab Sci 46: , 282–301. |
[6] | Dahlgren KN , Manelli AM , Stine WB Jr , Baker LK , Krafft GA , LaDu MJ ((2002) ) Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability, J Biol Chem 277: , 32046–32053. |
[7] | McLean CA , Cherny RA , Fraser FW , Fuller SJ , Smith MJ , Beyreuther K , Bush AI , Masters CL ((1999) ) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease, Ann Neurol 46: , 860–866. |
[8] | Fonte J , Miklossy J , Atwood C , Martins R ((2001) ) The severity of cortical Alzheimer’s type changes is positively correlated with increased amyloid-beta Levels: Resolubilization of amyloid-beta with transition metal ion chelators, J Alzheimers Dis 3: , 209–219. |
[9] | Walsh DM , Klyubin I , Fadeeva JV , Cullen WK , Anwyl R , Wolfe MS , Rowan MJ , Selkoe DJ ((2002) ) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo, Nature 416: , 535–539. |
[10] | Cleary JP , Walsh DM , Hofmeister JJ , Shankar GM , Kuskowski MA , Selkoe DJ , Ashe KH ((2005) ) Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function, Nat Neurosci 8: , 79–84. |
[11] | Shankar GM , Bloodgood BL , Townsend M , Walsh DM , Selkoe DJ , Sabatini BL ((2007) ) Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway, J Neurosci 27: , 2866–2875. |
[12] | Mangialasche F , Solomon A , Winblad B , Mecocci P , Kivipelto M ((2010) ) Alzheimer’s disease: Clinical trials and drug development, Lancet Neurol 9: , 702–716. |
[13] | Pieper AA , Xie S , Capota E , Estill SJ , Zhong J , Long JM , Becker GL , Huntington P , Goldman SE , Shen CH , Capota M , Britt JK , Kotti T , Ure K , Brat DJ , Williams NS , MacMillan KS , Naidoo J , Melito L , Hsieh J , De Brabander J , Ready JM , McKnight SL ((2010) ) Discovery of a proneurogenic, neuroprotective chemical, Cell 142: , 39–51. |
[14] | Bachurin S , Bukatina E , Lermontova N , Tkachenko S , Afanasiev A , Grigoriev V , Grigorieva I , Ivanov Y , Sablin S , Zefirov N ((2001) ) Antihistamine agent Dimebon as a novel neuroprotector and a cognition enhancer, Ann N Y Acad Sci 939: , 425–435. |
[15] | Doody RS , Gavrilova SI , Sano M , Thomas RG , Aisen PS , Bachurin SO , Seely L , Hung D , dimebon I ((2008) ) Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: A randomised, double-blind, placebo-controlled study, Lancet 372: , 207–215. |
[16] | Doody R , Winblad B , Cummings J , Tariot P , Sano M , Aisen P , Selby B , Seely L ((2012) ) Dimebon in Alzheimer’s disease: Summary and contrast of three efficacy trials, Alzheimers Dement 8: , 456. |
[17] | Chau S , Herrmann N , Ruthirakuhan MT , Chen JJ , Lanctot KL ((2015) ) Latrepirdine for Alzheimer’s disease, Cochrane Database Syst Rev 4: , CD009524. |
[18] | Cano-Cuenca N , Solis-Garcia del Pozo JE , Jordan J ((2014) ) Evidence for the efficacy of latrepirdine (Dimebon) treatment for improvement of cognitive function: A meta-analysis, J Alzheimers Dis 38: , 155–164. |
[19] | Miller G ((2010) ) Pharmacology. The puzzling rise and fall of a dark-horse Alzheimer’s drug, Science 327: , 1309. |
[20] | Bharadwaj PR , Bates KA , Porter T , Teimouri E , Perry G , Steele JW , Gandy S , Groth D , Martins RN , Verdile G ((2013) ) Latrepirdine: Molecular mechanisms underlying potential therapeutic roles in Alzheimer’s and other neurodegenerative diseases, Transl Psychiatry 3: , e332. |
[21] | Ustyugov A , Shevtsova E , Bachurin S ((2015) ) Novel sites of neuroprotective action of Dimebon (latrepirdine), Mol Neurobiol 52: , 970–978. |
[22] | Steele JW , Lachenmayer ML , Ju S , Stock A , Liken J , Kim SH , Delgado LM , Alfaro IE , Bernales S , Verdile G , Bharadwaj P , Gupta V , Barr R , Friss A , Dolios G , Wang R , Ringe D , Fraser P , Westaway D , St George-Hyslop PH , Szabo P , Relkin NR , Buxbaum JD , Glabe CG , Protter AA , Martins RN , Ehrlich ME , Petsko GA , Yue Z , Gandy S ((2013) ) Latrepirdine improves cognition and arrests progression of neuropathology in an Alzheimer’s mouse model, Mol Psychiatry 18: , 889–897. |
[23] | Bachurin SO , Shelkovnikova TA , Ustyugov AA , Peters O , Khritankova I , Afanasieva MA , Tarasova TV , Alentov II , Buchman VL , Ninkina NN ((2012) ) Dimebon slows progression of proteinopathy in gamma-synuclein transgenic mice, Neurotox Res 22: , 33–42. |
[24] | Steele JW , Ju S , Lachenmayer ML , Liken J , Stock A , Kim SH , Delgado LM , Alfaro IE , Bernales S , Verdile G , Bharadwaj P , Gupta V , Barr R , Friss A , Dolios G , Wang R , Ringe D , Protter AA , Martins RN , Ehrlich ME , Yue Z , Petsko GA , Gandy S ((2012) ) Latrepirdine stimulates autophagy and reduces accumulation of alpha-synuclein in cells and in mouse brain, Mol Psychiatry 18: , 882–888. |
[25] | Yamashita M , Nonaka T , Arai T , Kametani F , Buchman VL , Ninkina N , Bachurin SO , Akiyama H , Goedert M , Hasegawa M ((2009) ) Methylene blue and dimebon inhibit aggregation of TDP-43 in cellular models, FEBS Lett 583: , 2419–2424. |
[26] | Bharadwaj PR , Verdile G , Barr RK , Gupta V , Steele JW , Lachenmayer ML , Yue Z , Ehrlich ME , Petsko G , Ju S , Ringe D , Sankovich SE , Caine JM , Macreadie IG , Gandy S , Martins RN ((2012) ) Latrepirdine (dimebon) enhances autophagy and reduces intracellular GFP-Abeta42 levels in yeast, J Alzheimers Dis 32: , 949–967. |
[27] | Ida N , Hartmann T , Pantel J , Schroder J , Zerfass R , Forstl H , Sandbrink R , Masters CL , Beyreuther K ((1996) ) Analysis of heterogeneous βA4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay, J Biol Chem 271: , 22908–22914. |
[28] | Stine WB , Jungbauer L , Yu C , LaDu MJ ((2011) ) Preparing synthetic Abeta in different aggregation states, Methods Mol Biol 670: , 13–32. |
[29] | Zhang S , Hedskog L , Petersen CA , Winblad B , Ankarcrona M ((2010) ) Dimebon (latrepirdine) enhances mitochondrialfunction and protects neuronal cells from death, J Alzheimers Dis 21: , 389–402. |
[30] | Perez SE , Nadeem M , Sadleir KR , Matras J , Kelley CM , Counts SE , Vassar R , Mufson EJ ((2012) ) Dimebon alters hippocampal amyloid pathology in 3xTg-AD mice, Int J Physiol Pathophysiol Pharmacol 4: , 115–127. |
[31] | Lermontova NN , Redkozubov AE , Shevtsova EF , Serkova TP , Kireeva EG , Bachurin SO ((2001) ) Dimebon and tacrine inhibit neurotoxic action of beta-amyloid in culture and block L-type Ca(2+) channels, Bull Exp Biol Med 132: , 1079–1083. |
[32] | Amijee H , Madine J , Middleton DA , Doig AJ ((2009) ) Inhibitors of protein aggregation and toxicity. Biochem Soc Trans 37: , 692–696. |
[33] | Bastianetto S , Krantic S , Quirion R ((2008) ) Polyphenols as potential inhibitors of amyloid aggregation and toxicity: Possible significance to Alzheimer’s disease, Mini Rev Med Chem 8: , 429–435. |
[34] | Noormagi A , Primar K , Tougu V , Palumaa P ((2012) ) Interference of low-molecular substances with the thioflavin-T fluorescence assay of amyloid fibrils, J Pept Sci 18: , 59–64 . |
[35] | Carrell RW , Mushunje A , Zhou A ((2008) ) Serpins show structural basis for oligomer toxicity and amyloid ubiquity, FEBS Lett 582: , 2537–2541. |
[36] | Stine WB Jr , Dahlgren KN , Krafft GA , LaDu MJ ((2003) ) characterization of conditions for amyloid-beta peptide oligomerization and fibrillogenesis, J Biol Chem 278: , 11612–11622. |
[37] | Wong HE , Qi W , Choi HM , Fernandez EJ , Kwon I ((2011) ) A safe, blood-brain barrier permeable triphenylmethane dye inhibits amyloid-beta neurotoxicity by generating nontoxic aggregates, ACS Chem Neurosci 2: , 645–657. |
[38] | Shevtsova EF , Vinogradova DV , Kireeva EG , Reddy VP , Aliev G , Bachurin SO ((2014) ) Dimebon attenuates the Abeta-induced mitochondrial permeabilization, Curr Alzheimer Res 11: , 422–429. |
[39] | Eckert GP , Renner K , Eckert SH , Eckmann J , Hagl S , Abdel-Kader RM , Kurz C , Leuner K , Muller WE ((2012) ) Mitochondrial dysfunction–a pharmacological target in Alzheimer’s disease, Mol Neurobiol 46: , 136–150. |
[40] | Khritankova IV , Kukharskiy MS , Lytkina OA , Bachurin SO , Shorning BY ((2012) ) Dimebon activates autophagosome components in human neuroblastoma SH-SY5Y cells, Dokl Biochem Biophys 446: , 251–253. |
Figures and Tables
Fig.1
Neuronal cell viability assays measured by LDH release. Cells were treated for 4 days with oligomeric Aβ after its co-incubation in the presence or absence of various concentrations of latrepirdine. Samples % LDH release when compared to a total lysis control (values represent means±standard deviation (n = 4), *p < 0.05 after correction for multiple testing, when compared to total lysis control).
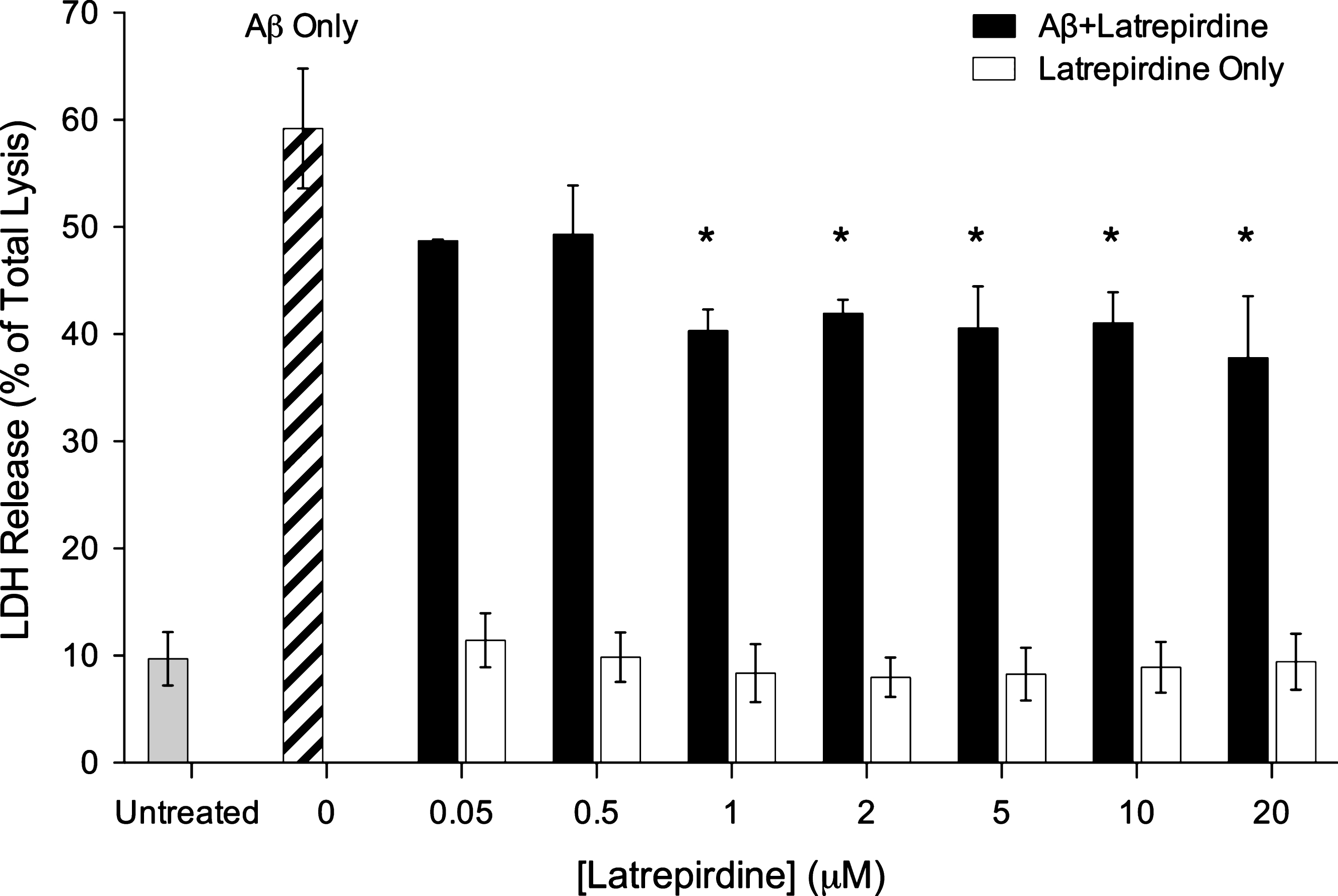
Fig.2
A) Endpoint thioflavin T assay on Aβ42 oligomerisation in the absence or presence of latrepirdine. ThioT was added to oligomer Aβ42 preparations incubated with different concentrations of latrepirdine, followed by measurement of fluorescent intensity. Significant reductions in fluorescence intensity was observed at all latrepirdine concentrations (values represent means±standard deviation (n = 3), *p < 0.05, **p < 0.001 after correction for multiple testing, when compared to Aβ42). B) Thioflavin T assay on Aβ42 aggregation over 16 h in the presence or absence of latrepirdine. Fluorescent intensity of Aβ incubated with latrepirdine and thioT was measured every 10 min for 16 h. A dose dependent reduction in the maximum fluorescence was observed with latrepirdine treatment.
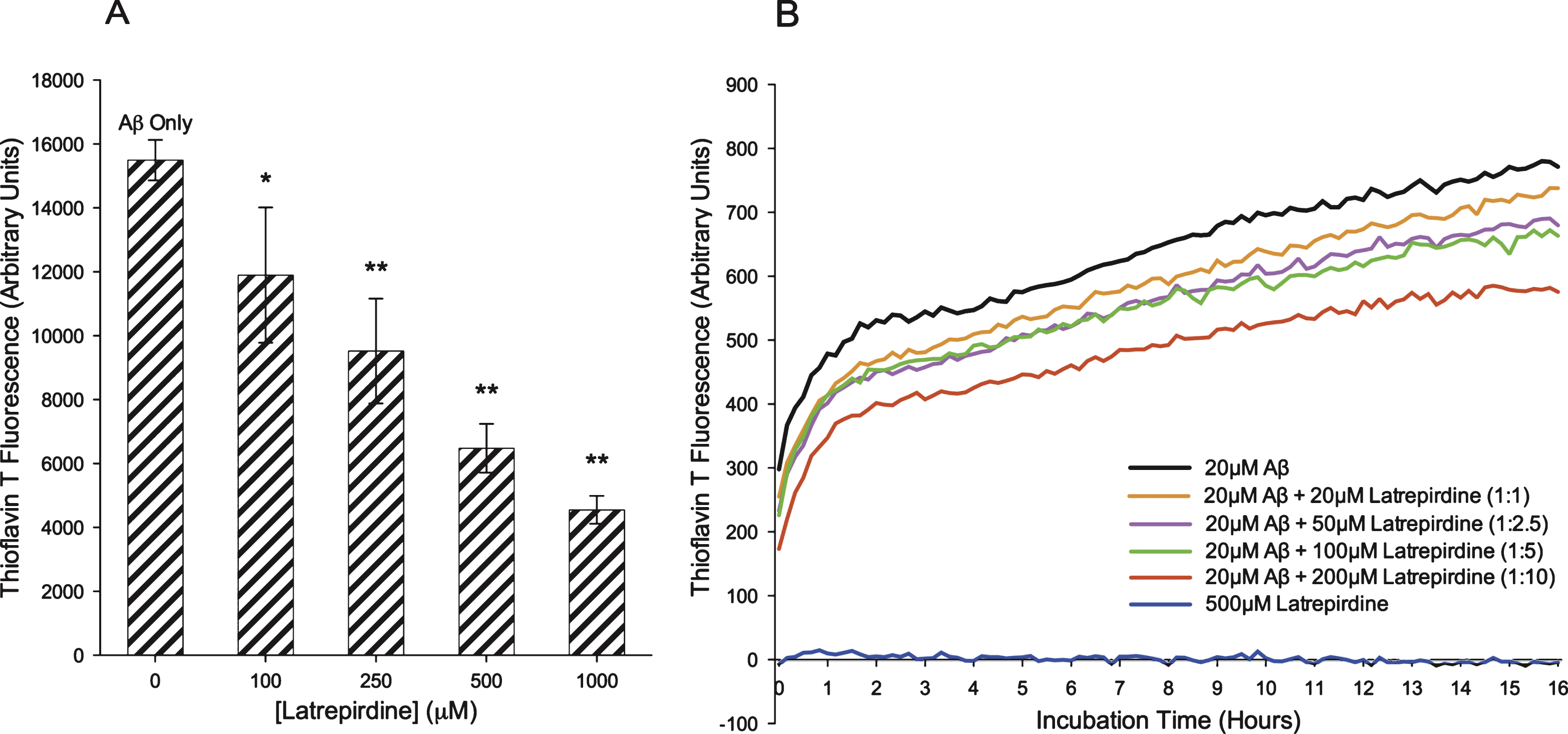
Fig.3
Addition of latrepirdine at 6 h post Aβ aggregation reduces Thioflavin T fluorescence but has minimal impact on formation of aggregates. A) Fluorescent intensity of Aβ42, with latrepirdine addition after 6 h of Aβ42 aggregation, and thioT was measured every 10 min for 16 h fluorescence increased in a steady fashion prior to the addition of latrepirdine when a sharp decrease in fluorescence was observed. SDS-PAGE (B) and blue native PAGE (C) separation and western blot of 200 ng Aβ samples before aggregation (lanes 1–6), after 6 h of aggregation and the addition of latrepirdine (lanes 7–12), and after a total of 20 h aggregation (lanes 13–18).
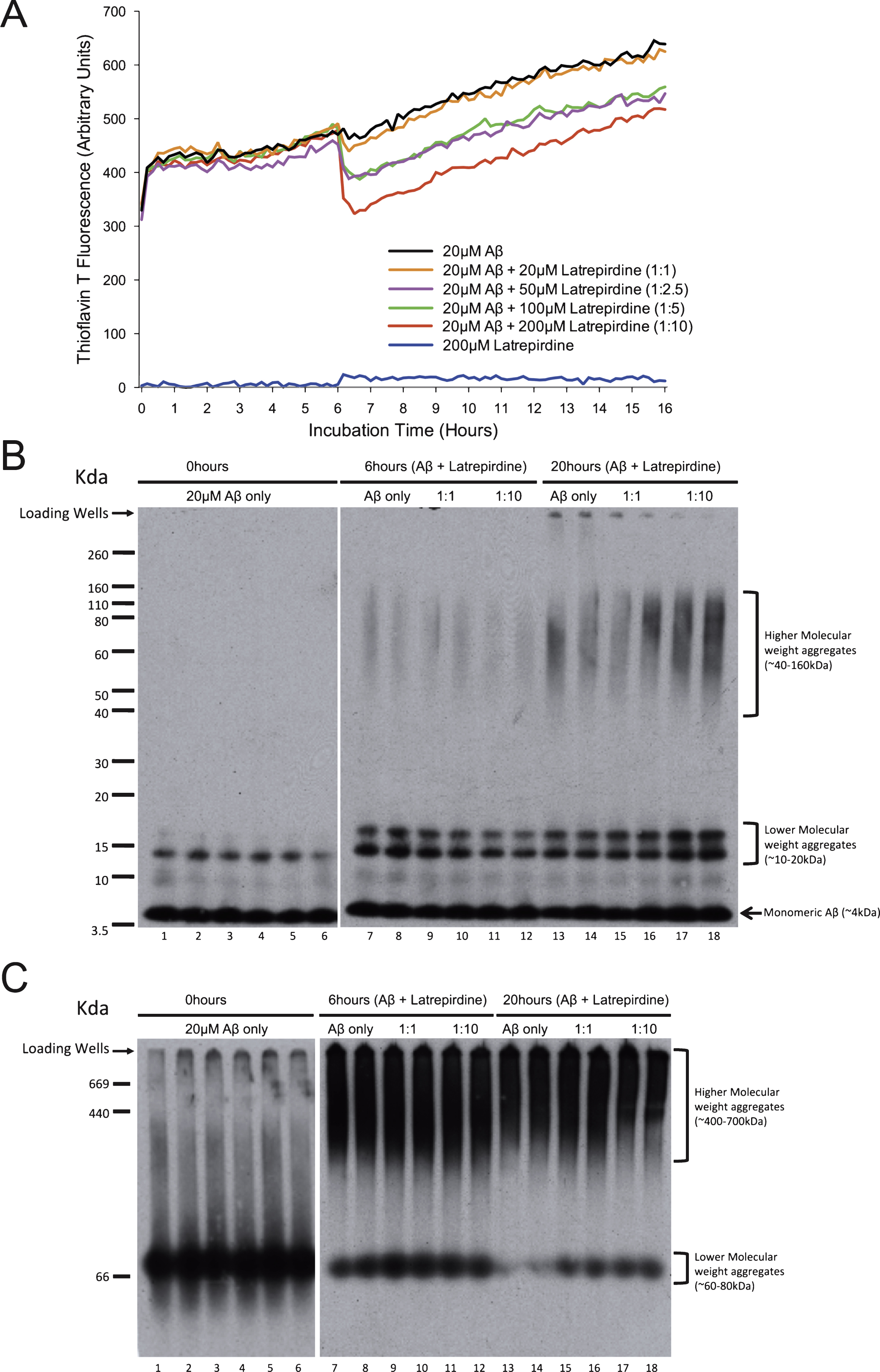
Fig.4
SDS-PAGE (A), BN- PAGE (B), and AFM (C) analysis of Aβ42 oligomerized in the presence or absence of latrepirdine. A,B) Aβ42 (lanes 1, 2, 9, 10) was incubated in the presence of latrepirdine at a molar ratio 1:1 (lanes 3, 4, 11, 12), 1:5 (lanes 5, 6, 13, 14), or 1:10 (lanes 7, 8, 15, 16) for 24 h. 200 ng underwent gel electrophoresis, prior to undergoing western immunoblotting. Quantification of higher and lower molecular weight species from western blots displayed as band intensity (values represent means±standard deviation (n = 4), *p < 0.05, **p < 0.01 after correction for multiple testing). (C) A 10 μm2 representative sections of Aβ42 alone (i) or incubated with latrepirdine in a 1:10 ratio (ii) observed by AFM. Magnified field of views and cross-sectional analyses were made using the Nova AFM software. Aβ oligomer preparations (i) contain aggregates 0.05–0.2 μM in diameter with heights from 2–15 nm Aβ oligomer: latrepirdine preparations (ii), contain larger aggregates with diameters of 0.2–0.5 μm and heights from 10–35 nm.