
Increased Number of Plasma B Cells Producing Autoantibodies Against Aβ42 Protofibrils in Alzheimer’s Disease
Abstract
The Alzheimer’s disease (AD)-related peptide amyloid-β (Aβ) has a propensity to aggregate into various assemblies including toxic soluble Aβ protofibrils. Several studies have reported the existence of anti-Aβ antibodies in humans. However, it is still debated whether levels of anti-Aβ antibodies are altered in AD patients compared to healthy individuals. Formation of immune complexes with plasma Aβ makes it difficult to reliably measure the concentration of circulating anti-Aβ antibodies with certain immunoassays, potentially leading to an underestimation. Here we have investigated anti-Aβ antibody production on a cellular level by measuring the amount of anti-Aβ antibody producing cells instead of the plasma level of anti-Aβ antibodies. To our knowledge, this is the first time the anti-Aβ antibody response in plasma has been compared in AD patients and age-matched healthy individuals using the enzyme-linked immunospot (ELISpot) technique. Both AD patients and healthy individuals had low levels of B cells producing antibodies binding Aβ40 monomers, whereas the number of cells producing antibodies toward Aβ42 protofibrils was higher overall and significantly higher in AD compared to healthy controls. This study shows, by an alternative and reliable method, that there is a specific immune response to the toxic Aβ protofibrils, which is significantly increased in AD patients.
INTRODUCTION
In Alzheimer’s disease (AD) brains, extracellular amyloid deposits are found in the parenchyma as plaques and in vessel walls as congophilic amyloid angiopathy. These hallmarks of AD are composed of insoluble fibrils of amyloid-β (Aβ), a hydrophobic and self-aggregating peptide. Prior to plaque deposition, Aβ gradually polymerizes from monomers into larger
soluble molecular forms (oligomers/protofibrils) and eventually into insoluble fibrils. Several of the intermediate Aβ species have been shown to elicit adverse biological effects both in vitro and in vivo [1–5], suggesting that they play a central role in the pathogenesis. We have previously identified a pathogenic mutation in the amyloid-β protein precursor (AβPP) gene, the Arctic mutation (E693G). This mutation, causing early onset AD, is located within the Aβ domain and enhances the formation of Aβ protofibrils, suggesting that this Aβ species may be causative of the disease [6]. Moreover, it has been shown that Aβ protofibrils are present in cerebrospinal fluid (CSF) and that the levels of large soluble Aβ aggregates in CSF correlate with cognitive impairment, implying that the progression of the disease is dependent on Aβ protofibrils [7–10].
Autoantibodies, reactive to self-antigens, are often associated with pathological functions leading to autoimmune diseases. However, some autoantibodies play a physiological role in the body without leading to pathological processes and disease progression. These naturally occurring autoantibodies are believed to have a role in maintaining homeostasis and preventing inflammation [11]. Natural autoantibodies against Aβ (anti-Aβ antibodies) have been found in both AD and non-demented individuals [12, 13]. A vast part of anti-Aβ antibodies are believed to be produced in response to Aβ in the periphery and central nervous system. It has been proposed that anti-Aβ antibodies predominantly show affinity toward the toxic aggregated forms of Aβ, including oligomers and protofibrils, which may be considered nonself-antigens, and not to monomers [14–17]. The function of anti-Aβ antibodies is not understood but they have been implied to interfere with Aβ toxicity and the oligomerization and fibrillization of Aβ [1, 14, 16, 18, 19]. As a therapeutic approach, intravenous infusion of polyvalent IgG antibodies from healthy individuals (IVIG), proposed to contain anti-Aβ antibodies, has been studied in AD with positive effects on mouse behavior [14, 20] and AD cognition [21–23]. Nevertheless, to this date, clinical trials have failed to show any positive treatment effects of IVIG in AD [24, 25]. Aducanumab, originally derived from healthy aged individuals, makes up yet another therapeutic strategy for the use of anti-Aβ antibodies in AD and it has recently entered phase 3 clinical trials. Aducanumab recognizes aggregated forms of Aβ and appears to reduce Aβ deposits in patients with early and mild forms of the disease and to slow cognitive decline [26].
Different studies report discrepancies regarding plasma levels of anti-Aβ antibodies in AD. Numerous reports have demonstrated decreased [12, 13, 15, 27–31], elevated [16, 18, 32, 33], or equal [1, 34, 35] levels in AD patients compared to control subjects. Anti-Aβ42 antibodies levels in Down syndrome patients have been found to be higher in plasma compared to non-Down syndrome mentally retarded subjects with perinatal brain injury and to age-matched healthy controls [36]. Moreover, an autoimmune reaction against Aβ, mediated by anti-Aβ antibodies, has been implied to be directly involved in the pathogenesis of cerebral amyloid-related inflammation with increased levels of anti-Aβ antibodies in CSF [37, 38]. In the current study, instead of studying antibodies in plasma, we utilized the enzyme-linked immunospot (ELISpot) technique to investigate the presence of anti-Aβ antibody secreting cells in the blood. With this technique, first described by Czerkinsky et al. [39], secretion of anti-Aβ antibodies can be analyzed on the cellular level (Fig. 1) avoiding enzyme-linked immunosorbent assay (ELISA) associated problems, e.g. formation of immune complexes between circulating Aβ antibodies and native Aβ, when analyzing plasma samples. In addition, the ELISpot method enabled us to compare the antibody response toward monomeric and protofibrillar forms of Aβ. Here we have found that the number of B cells producing IgG antibodies towards Aβ42 protofibrils was significantly higher in AD patients compared to healthy individuals.
MATERIALS AND METHODS
Study material
Blood sampling from AD patients (n = 50) and age-matched healthy individuals (n = 55) was performed according to standardized procedures at the Memory Disorder Unit, Uppsala University Hospital, Uppsala, Sweden as approved by the Regional ethical committee in Uppsala (decision number 2009/097). Clinical AD diagnosis was determined by the NINCDS-ADRDA criteria, after a comprehensive clinical diagnostic procedure including neuroimaging. Mini-Mental State Examination (MMSE, Folstein – 75) was administered and scores were used as a rough measurement of cognitive function. Age-matched healthy individuals were recruited by local advertisement at the Memory Disorder Unit, Uppsala University. Inclusion of healthy individuals required absence of cognitive impairment or other dementia disorders. Both AD patients and age-matched healthy individuals volunteered to be part of the study and written informed consent was obtained from all subjects or their closest relative.
Preparation of biotinylated Aβ40 monomers and Aβ42 protofibrils
Synthetic Aβ40 monomers and Aβ42 protofibrils were prepared as previously described [18, 40, 41]. Synthetic Aβ40 (Polypeptide Laboratories AB, Limhamn, Sweden) dissolved in 10 mM NaOH, was diluted in 2×phosphate buffered saline (PBS) to 100μM. Synthetic Aβ42 (American Peptide Company Inc., Sunnyvale, CA, USA) dissolved in 10 mM NaOH, diluted in 10×PBS to 443μM (2 mg/ml), was incubated for 30 min at 37°C and centrifuged for 5 min at 17 900×g to remove any insoluble aggregates and then diluted with PBS to a final concentration of 100μM. Both Aβ40 monomers and Aβ42 protofibrils were biotinylated with Sulfo-NHS-LC-Biotin (Thermo Fischer Scientific Inc., Waltham, MA, USA) according to manufacturer’s guidelines with an Aβ/biotin molar ratio of 1/20. The Aβ42 protofibril concentration is expressed in monomer concentration units.
Aβ protofibril ELISA
Aβ protofibril ELISA using the monoclonal antibody 158 (mAb158) was performed according to the protocol described by Englund et al. [42]. In short, 96-well EIA/RIA plates (Corning Inc., Corning, NY, USA) were coated with 200 ng/well of mAb158 antibody in 100μl PBS at 4°C overnight. Plates were blocked with 1% bovine serum albumin (BSA) in PBS with 0.15% Kathon. Standard series of synthetic Aβ42 protofibrils, either biotinylated or not, were diluted in ELISA incubation buffer (0.05% Tween, 0.1% BSA and 0.15% Kathon in PBS at pH 7.4) and added to the plates for incubation 2 h at room temperature (RT). After washing, the plates were incubated for 1 h at RT with 0.5μg/ml mAb158 conjugated to horseradish peroxidase. K-blue aqueous TMB substrate (Neogen Corporation, Lansing, MI, USA) was used as horseradish peroxidase substrate, and the reaction was stopped with 1 M H2SO4. Absorbance was measured by Tecan Infinite M200 PRO (Tecan Group Ltd., Männedorf, Switzerland) spectrophotometer at 450 nm and analyzed with Magellan v7.0 software (Tecan Group Ltd.). Washing was performed by adding 250μl washing buffer (PBS with 0.1% Tween 20 and 0.15% Kathon) repeated three times between each step of the ELISA.
PBMC preparation
Whole blood, sampled in BD Vacutainer CPTtrademark Cell Preparation Tubes with sodium heparin (BD bioscience, Franklin Lakes, NJ, USA), was diluted 1:2 in PBS and gently layered over cold Ficoll-Paque PLUS (GE Healthcare, Little Chalfont, UK) and kept on ice before centrifuged for 20 min at 900×g without brake at 18–20°C. The cell layer on top of the Ficoll-Paque PLUS consisting of peripheral blood mononuclear cells (PBMCs) was collected and diluted in PBS. PBMCs were centrifuged again for 7 min at 450×g at 18–20°C. The supernatant was removed and cells were resuspended in 5 ml PBS and counted before they were centrifuged at 350×g for 7 min at 18–20°C. Finally, cells were resuspended in freezing medium (10% DMSO in heat inactivated fetal bovine serum (FBS) (Nordic Biolabs AB, Täby, Sweden) and placed at – 70°C in a Mr. Frostytrademark Freezing Container (Thermo Fisher) for at least 3 h before being transferred to liquid nitrogen for extended storage.
Flow cytometry
For each individual cell sample, 5×106 PBMCs were quickly thawed at 37°C and diluted in 40 ml cold wash buffer (PBS supplemented with 2.5% FBS (Life Technologies Ltd. Paisley, UK) and 0.1% sodium azide). The cell suspension was centrifuged at 250×g for 5 min and after discarding the supernatant, the pellet was resuspended in 400μl wash buffer. Each sample was stained for 1 h in a light-protected environment at 2°C with titrated amounts of anti CD19 labelled with Alexa Fluor 700 (BD Biosciences). Samples were analyzed using a BD LSR II Special Order System, controlled by the BD FACSDiva 6.0 software (BD Biosciences). A preliminary forward scatter (FSC) versus side scatter (SSC) gate was used to identify lymphocytes and, depending on sample size, a total of up to 100.000 in-gate events were recorded. All datasets were migrated to FlowJo 7.6.5 (Treestar Inc. Ashland, OR, USA) for further gating and analysis.
Activation of B cell antibody production
Five million PBMCs were thawed and diluted in complete medium (RPMI 1640 (Nordic Biolabs), supplemented with a mix of penicillin (100 U/ml) and streptomycin (100μg/ml) (Thermo Fisher), L-glutamine (2 mM) (Life Technologies), Hepes (10 mM) (Sigma-Aldrich, Saint Louis, MO, USA), and FBS (10% ) (Nordic Biolabs). The PBMCs were washed twice in complete medium by centrifugation for 10 min at 200×g before incubation in complete medium at 37°C, 5% CO2 for 1 h. After resuspension, any debris or cell aggregates were allowed to sediment and the cells were transferred to a new centrifuge vial. To activate the IgG production, cells were stimulated for 72 h at 37°C, 5% CO2 in complete medium supplemented with the polyclonal activator R848 (1μg/ml) (Mabtech AB, Nacka Strand, Sweden) and recombinant human Interleukin-2 (10 ng/ml) (Mabtech AB).
ELISpot
EliSpotPLUS kit for human IgG (Mabtech AB) was used to study the IgG producing B cells. First, ELISpot plates were activated with 70% Ethanol (50μl/well) during 1 min and washed with sterile H2O and coated with mouse anti-human IgG mAbs MT91/145 (15μg/ml) overnight at 4°C. Plates were then washed five times with sterile PBS followed by 1 h incubation in complete medium. PBMCs were added in triplicates to the plates at a concentration of 500.000 cells/well for the analysis of anti-Aβ antibodies and 50.000 cells/well for measuring the total number of IgG secreting cells. After incubation for 24 h in 37°C, 5% CO2, cells were removed by washing four times in PBS in a Tecan Hydrospeed plate washer (Tecan Group Ltd.) followed by incubation with biotinylated Aβ42 protofibrils (500 nM) or biotinylated Aβ40 monomers (500 nM) for 2 h at RT to detect anti-Aβ antibodies and with biotinylated α-human IgG mAbs MT78/145 (1μg/ml) for determination of the total number of IgG producing cells. After another round of washing, the plates were incubated for 1 h at RT with the conjugate Streptavidin-Alkaline Phosphatase (Streptavidin-ALP) (1:1000 in PBS supplemented with 0.5% FBS). Finally, plates were washed and developed with filtered 5-bromo-4-chloro-3-indolyl-phosphate in conjunction with nitro blue tetrazolium (BCIP/NBT) substrate until spots were evident. After stopping the substrate reaction by washing extensively in tap water, plates were dried and analyzed with an ELISpot reader (AID, Strassberg, Germany).
Statistical analysis
GraphPad Prism 5 was used for all statistical analyses (GraphPad Software, Inc., La Jolla, USA). Unpaired t test with Welch’s correction was used throughout the study and the results are presented in scattergrams or box plots with mean ± standard deviation. For correlation, Spearman rank correlation was used with the 95% confidence interval, spearman r and p values included in the graph. Level of significance were set at *p < 0.05, **p < 0.01, and ***p < 0.001. The study was blinded until time of statistical analysis.
RESULTS
Blood from age-matched AD patients (n = 50, mean age 77.8 years) and healthy individuals (n = 55, mean age 74.3 years) were used for the preparation of PBMCs. For the analysis of cells secreting anti-Aβ antibodies, 500.000 cells stimulated for 72 h with the polyclonal activator R848 and recombinant IL-2 were added to each well of ELISpot plates coated with anti-IgG. As a positive control, cells (50,000 cells/well) were added in parallel wells but analyzed for the total number of IgG secreting cells. Samples with no or very few IgG producing cells in the positive control wells (AD patients (n = 7), healthy individuals (n = 10)) were excluded from the study. Similarly, samples (AD patients (n = 4), healthy individuals (n = 11)) with too low numbers of PBMCs already after extraction or after thawing were excluded. The remaining samples that were included in the study are displayed in Table 1. In general, more IgG-producing B cells were found in blood from individuals with AD compared to age matched controls (Fig. 2A). However, no differences in B cell numbers were detected when comparing AD patients to healthy individuals by flow cytometry (Fig. 2B).
To investigate the presence of B cells secreting anti-Aβ antibodies, two different biotinylated Aβ preparations were used; synthetic Aβ40 monomers and synthetic Aβ42 protofibrils. Synthetic Aβ42 protofibrils were here defined as soluble Aβ aggregates larger than 100 kDa, i.e., eluting in the void volume on a size exclusion Sephadex 75 column. The Aβ42 protofibrils were first run in an Aβ protofibril specific ELISA to control that the protofibril structure was intact after the biotinylation step. The biotinylated Aβ42 protofibrils were readily detected by a protofibril specific ELISA and there was no significant difference between Aβ42 protofibrils with or without biotin confirming conformational maintenance (Fig. 2C). In general, a higher number of cells produced antibodies recognizing synthetic Aβ42 protofibrils than synthetic Aβ40 monomers (p < 0.0001) (Fig. 3A). While there was no significant difference between AD patients and healthy individuals with regard to the number of plasma B cells producing antibodies against monomeric Aβ40 (Fig. 3B), the number of cells secreting antibodies to Aβ42 protofibrils was significantly higher in AD individuals compared to healthy individuals (p = 0.043) (Fig. 3C). This difference was even more evident for ApoE4 allele homo- or heterozygous AD individuals (p = 0.031). Moreover, in AD patients the number of B cells producing antibodies to Aβ42 protofibrils correlated weakly with the total number of IgG producing B cells (Spearman r = 0.59, p < 0.001) (Fig. 3D).
No statistically significant correlation was found between the frequencies of anti-Aβ antibody secreting cells in the AD group and Mini Mental Status (based on Mini Mental Test) (Fig. 4). Nor was there a correlation between the number of Aβ-specific cells and age or gender except for in the control group, where females had significantly higher frequencies of B cells producing antibodies towards Aβ42 protofibrils than males (p = 0.025).
DISCUSSION
Use of different methods, study setups and various forms of Aβ may explain the conflicting results regarding anti-Aβ antibodies in AD and healthy individuals. The vast majority of earlier publications reporting Aβ antibody levels in human serum/plasma have used ELISA [12, 13, 28, 31– 35, 43– 48]. With ELISA, the reliability of the measurements can be questioned as there is a risk of immune complex formation between circulating Aβ antibodies and native Aβ in the blood. Thus, antibodies may be prevented from binding Aβ in ELISA plates, leading to an underestimation of antibody levels. This problem can be overcome by measuring anti-Aβ antibodies after dissolving the complexes, typically through a brief treatment with acid. Gustaw et al. measured the levels using ELISA before and after dissociation and found that anti-Aβ antibody levels were increased in both AD patients and normal control subjects and that AD patients displayed higher levels of anti-Aβ antibodies compared to normal controls after treatment [29, 30]. Another problem when using ELISA is that Aβ monomers may artificially adopt amyloidogenic epitopes due to surface adsorption [15], potentially causing misinterpretations regarding antibody recognition of specific Aβ forms. Therefore, to distinguish different forms of Aβ in immunoassays, Aβ should preferably be kept in solution. We used the ELISpot assay since this not only eliminates the problem of antibodies occurring in complexes but also allows the testing of Aβ in a soluble form. The ELISpot assay is also very sensitive and enables the detection of the very small populations typically seen in antigen-specific responses. This way ELISpot enabled us to accurately measure the presence of B cells producing anti-Aβ antibodies in AD patients and age-matched healthy individuals and to compare the anti-Aβ antibody selectivity toward Aβ40 monomers and Aβ42 protofibrils. The choice of different Aβ peptides for generation of monomers and protofibrils was motivated by their different propensity to aggregate and thus maintain their conformation throughout the experiment. Since the C-terminus of aggregated Aβ is less accessible for antibody binding [49, 50], we do not expect the two extra C-terminal amino acids in the Aβ42 protofibril preparation to affect the results.
Before measuring the Aβ specific IgG production, flow cytometry was performed to estimate the total B cell numbers in the blood of both AD patients and healthy individuals. Earlier reports have found lower levels of B cells in AD compared to healthy individuals [51– 54]. However, Speciale et al. observed differences in IgG levels in serum between AD and healthy controls [52] and Pellicano et al. observed activation of B cells by Aβ42 by enhanced expression of the chemokine CCR5 [51]. Our data did not show any significant difference between AD and healthy individuals regarding the percentage of B cells (Fig. 2B). Surprisingly, ELISpot revealed the overall number of IgG producing B cells to be higher in the AD patients compared to the healthy individuals, suggesting a larger proportion of more easily activated B cells in AD patients.
Regarding antibodies binding to Aβ40 monomers, we only detected low numbers of B cells producing such antibodies in both AD patients and healthy individuals. As Aβ monomers represent the native form of the peptide in the human body, this was expected and has also been shown before [55]. In contrast, B cells producing antibodies specific for Aβ42 protofibrils were found in higher numbers and were increased in AD patients compared to the age-matched healthy controls. This result is likely to be a consequence of increased levels of Aβ protofibrils in AD patients, which would support their suggested importance in the pathology of the disease [7, 18]. We also found a weak correlation between the number of B cells producing Aβ42 protofibril specific antibodies and the total number of IgG producing cells. There is a growing recognition that immune responses in AD patients are dysregulated, something that may also play a role in AD [56]. The number of IgG producing B cells might be a consequence of enhanced activation of the immune response in AD. Our study did not investigate whether the antibodies recognizing Aβ42 protofibrils also recognized generic epitopes common to amyloid structures on other proteins.
As previously reported for anti-Aβ antibodies in plasma [27, 34], MMSE, age, or gender, except for women in the group of healthy individuals, did not show any correlation to the levels of B cells producing anti-Aβ antibodies, thereby abating the role of such antibodies as a potential biomarker for AD. Nevertheless, it would be interesting to follow AD patients over time to see if and how the number of B cells producing anti-Aβ antibodies correlates with the cognitive decline on an individual level. Conti et al. have previously described that AD patients receiving acetylcholinesterase inhibitors (AChEI) increased the anti-Aβ42 antibody levels in plasma compared to untreated AD patients [57]. AD patients show an increased production of interleukin-4 and monocyte chemotactic protein-1, two positive regulators of Th2 differentiation, following AChEI treatment. Possibly, AChEI treatment can induce a Th2-mediated immune response leading to an increase of immunoglobulin production by B lymphocytes, including anti-Aβ42 antibodies [58]. However, as anti-Aβ42 antibodies levels were evaluated by ELISA, the differences between AChEI treated and untreated AD patients could reflect variations in Aβ/anti-Aβ42 antibody complexes rather than number of anti-Aβ42 antibodies. As all patients in the AD group except for four had been or were still on treatment with AChEI in our study, evaluation regarding AChEI influence on anti-Aβ antibody production from B cells among AD patients was not possible.
In conclusion, we report for the first time that ELISpot can be used to measure anti-Aβ antibodies from B cells in the blood. Low levels of B cells producing antibodies to Aβ40 monomers were seen in both AD and healthy individuals whereas the number of cells producing antibodies to Aβ42 protofibrils was significantly higher in AD compared to healthy individuals. Both IVIG and therapeutic anti-Aβ antibodies originally derived from healthy individuals are evaluated as AD therapies. Thus, the role of anti-Aβ antibodies in AD, as well as their physiological importance need to be further evaluated to learn if they could contribute to new therapeutic approaches for AD.
ACKNOWLEDGMENTS
We are grateful to Anne-Marie Ljungberg for her assistance during PBMC preparation and Käthe Ström for blood sampling. This work was supported by the Swedish Research Council (#2012-2172, LL), Alzheimerfonden (FEP, SS) and Uppsala Berzelii Technology Centre for Neurodiagnostics (DS, SS, LL, FEP).
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/15-0236r1).
REFERENCES
1 | Klyubin I, Cullen WK, Hu NW, Rowan MJ(2012) Alzheimer’s disease Abeta assemblies mediating rapid disruption of synaptic plasticity and memoryMol Brain5: 25 |
2 | Klyubin I, Walsh DM, Cullen WK, Fadeeva JV, Anwyl R, Selkoe DJ, Rowan MJ(2004) Soluble Arctic amyloid beta protein inhibits hippocampal long-term potentiation in vivoEur J Neurosci19: 28392846 |
3 | Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ(2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivoNature416: 535539 |
4 | Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ(1999) Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neuronsJ Neurosci19: 88768884 |
5 | Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL(2007) Abeta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s diseaseJ Neurosci27: 796807 |
6 | Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, Naslund J, Lannfelt L(2001) The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formationNat Neurosci4: 887893 |
7 | Fukumoto H, Tokuda T, Kasai T, Ishigami N, Hidaka H, Kondo M, Allsop D, Nakagawa M(2010) High-molecular-weight beta-amyloid oligomers are elevated in cerebrospinal fluid of Alzheimer patientsFASEB J24: 27162726 |
8 | Santos AN, Ewers M, Minthon L, Simm A, Silber RE, Blennow K, Prvulovic D, Hansson O, Hampel H(2012) Amyloid-beta oligomers in cerebrospinal fluid are associated with cognitive decline in patients with Alzheimer’s diseaseJ Alzheimers Dis29: 171176 |
9 | Tucker S, Moller C, Tegerstedt K, Lord A, Laudon H, Sjodahl J, Soderberg L, Spens E, Sahlin C, Waara ER, Satlin A, Gellerfors P, Osswald G, Lannfelt L(2015) The murine version of BAN2401 (mAb158) selectively reduces amyloid-beta protofibrils in brain and cerebrospinal fluid of tg-ArcSwe miceJ Alzheimers Dis43: 575588 |
10 | Savage MJ, Kalinina J, Wolfe A, Tugusheva K, Korn R, Cash-Mason T, Maxwell JW, Hatcher NG, Haugabook SJ, Wu G, Howell BJ, Renger JJ, Shughrue PJ, McCampbell A(2014) A sensitive abeta oligomer assay discriminates Alzheimer’s and aged control cerebrospinal fluidJ Neurosci34: 28842897 |
11 | Gold M, Pul R, Bach JP, Stangel M, Dodel R(2012) Pathogenic and physiological autoantibodies in the central nervous systemImmunol Rev248: 6886 |
12 | Du Y, Dodel R, Hampel H, Buerger K, Lin S, Eastwood B, Bales K, Gao F, Moeller HJ, Oertel W, Farlow M, Paul S(2001) Reduced levels of amyloid beta-peptide antibody in Alzheimer diseaseNeurology57: 801805 |
13 | Weksler ME, Relkin N, Turkenich R, LaRusse S, Zhou L, Szabo P(2002) Patients with Alzheimer disease have lower levels of serum anti-amyloid peptide antibodies than healthy elderly individualsExp Gerontol37: 943948 |
14 | Dodel R, Balakrishnan K, Keyvani K, Deuster O, Neff F, Andrei-Selmer LC, Roskam S, Stuer C, Al-Abed Y, Noelker C, Balzer-Geldsetzer M, Oertel W, Du Y, Bacher M(2011) Naturally occurring autoantibodies against beta-amyloid: Investigating their role in transgenic animal and in vitro models of Alzheimer’s diseaseJ Neurosci31: 58475854 |
15 | O’Nuallain B, Acero L, Williams AD, Koeppen HP, Weber A, Schwarz HP, Wall JS, Weiss DT, Solomon A(2008) Human plasma contains cross-reactive Abeta conformer-specific IgG antibodiesBiochemistry47: 1225412256 |
16 | Di Malta C, Fryer JD, Settembre C, Ballabio A(2012) Astrocyte dysfunction triggers neurodegeneration in a lysosomal storage disorderProc Natl Acad Sci U S A109: E2334E2342 |
17 | Szabo P, Mujalli DM, Rotondi ML, Sharma R, Weber A, Schwarz HP, Weksler ME, Relkin N(2010) Measurement of anti-beta amyloid antibodies in human bloodJ Neuroimmunol227: 167174 |
18 | Sehlin D, Englund H, Simu B, Karlsson M, Ingelsson M, Nikolajeff F, Lannfelt L, Pettersson FE(2012) Large aggregates are the major soluble Abeta species in AD brain fractionated with density gradient ultracentrifugationPLoS One7: e32014 |
19 | Du Y, Wei X, Dodel R, Sommer N, Hampel H, Gao F, Ma Z, Zhao L, Oertel WH, Farlow M(2003) Human anti-beta-amyloid antibodies block beta-amyloid fibril formation and prevent beta-amyloid-induced neurotoxicityBrain126: 19351939 |
20 | St-Amour I, Pare I, Tremblay C, Coulombe K, Bazin R, Calon F(2014) IVIg protects the 3xTg-AD mouse model of Alzheimer’s disease from memory deficit and Abeta pathologyJ Neuroinflammation11: 54 |
21 | Morgan D(2010) Immunotherapy for Alzheimer’s diseaseJ Intern Med269: 5463 |
22 | Taguchi H, Planque S, Nishiyama Y, Symersky J, Boivin S, Szabo P, Friedland RP, Ramsland PA, Edmundson AB, Weksler ME, Paul S(2008) Autoantibody-catalyzed hydrolysis of amyloid beta peptideJ Biol Chem283: 47144722 |
23 | Relkin NR, Szabo P, Adamiak B, Burgut T, Monthe C, Lent RW, Younkin S, Younkin L, Schiff R, Weksler ME(2009) 18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer diseaseNeurobiol Aging30: 17281736 |
24 | Dodel R, Rominger A, Bartenstein P, Barkhof F, Blennow K, Forster S, Winter Y, Bach JP, Popp J, Alferink J, Wiltfang J, Buerger K, Otto M, Antuono P, Jacoby M, Richter R, Stevens J, Melamed I, Goldstein J, Haag S, Wietek S, Farlow M, Jessen F(2013) Intravenous immunoglobulin for treatment of mild-to-moderate Alzheimer’s disease: A phase 2, randomised, double-blind, placebo-controlled, dose-finding trialLancet Neurol12: 233243 |
25 | Relkin N(2014) Clinical trials of intravenous immunoglobulin for Alzheimer’s diseaseJ Clin Immunol34: Suppl 1S74S79 |
26 | Alzheimer Research Forum, Drugs in Clinical Trials: Aducanumab, http://www.alzforum.org/therapeutics/aducanumab, Accessed April 4, 2015 |
27 | Brettschneider S, Morgenthaler NG, Teipel SJ, Fischer-Schulz C, Burger K, Dodel R, Du Y, Moller HJ, Bergmann A, Hampel H(2005) Decreased serum amyloid beta(1-42) autoantibody levels in Alzheimer’s disease, determined by a newly developed immuno-precipitation assay with radiolabeled amyloid beta(1-42) peptideBiol Psychiatry57: 813816 |
28 | Moir RD, Tseitlin KA, Soscia S, Hyman BT, Irizarry MC, Tanzi RE(2005) Autoantibodies to redox-modified oligomeric Abeta are attenuated in the plasma of Alzheimer’s disease patientsJ Biol Chem280: 1745817463 |
29 | Gustaw KA, Garrett MR, Lee HG, Castellani RJ, Zagorski MG, Prakasam A, Siedlak SL, Zhu X, Perry G, Petersen RB, Friedland RP, Smith MA(2008) Antigen-antibody dissociation in Alzheimer disease: A novel approach to diagnosisJ Neurochem106: 13501356 |
30 | Gustaw-Rothenberg KA, Siedlak SL, Bonda DJ, Lerner A, Tabaton M, Perry G, Smith MA(2010) Dissociated amyloid-beta antibody levels as a serum biomarker for the progression of Alzheimer’s disease: A population-based studyExp Gerontol45: 4752 |
31 | Qu BX, Gong Y, Moore C, Fu M, German DC, Chang LY, Rosenberg R, Diaz-Arrastia R(2014) Beta-amyloid auto-antibodies are reduced in Alzheimer’s diseaseJ Neuroimmunol274: 168173 |
32 | Mruthinti S, Buccafusco JJ, Hill WD, Waller JL, Jackson TW, Zamrini EY, Schade RF(2004) Autoimmunity in Alzheimer’s disease: Increased levels of circulating IgGs binding Abeta and RAGE peptidesNeurobiol Aging25: 10231032 |
33 | Storace D, Cammarata S, Borghi R, Sanguineti R, Giliberto L, Piccini A, Pollero V, Novello C, Caltagirone C, Smith MA, Bossu P, Perry G, Odetti P, Tabaton M(2010) Elevation of beta-amyloid 1-42 autoantibodies in the blood of amnestic patients with mild cognitive impairmentArch Neurol67: 867872 |
34 | Hyman BT, Smith C, Buldyrev I, Whelan C, Brown H, Tang MX, Mayeux R(2001) Autoantibodies to amyloid-beta and Alzheimer’s diseaseAnn Neurol49: 808810 |
35 | Klaver AC, Coffey MP, Smith LM, Bennett DA, Finke JM, Dang L, Loeffler DA(2011) ELISA measurement of specific non-antigen-bound antibodies to Abeta1-42 monomer and soluble oligomers in sera from Alzheimer’s disease, mild cognitively impaired, and noncognitively impaired subjectsJ Neuroinflammation8: 93 |
36 | Conti E, Galimberti G, Piazza F, Raggi ME, Ferrarese C(2010) Increased soluble APPalpha, Abeta 1-42, and anti-Abeta 1-42 antibodies in plasma from down syndrome patientsAlzheimer Dis Assoc Disord24: 96100 |
37 | Boncoraglio GB, Piazza F, Savoiardo M, Farina L, DiFrancesco JC, Prioni S, Tagliavini F, Parati EA, Giaccone G(2015) Prodromal Alzheimer’s disease presenting as cerebral amyloid angiopathy-related inflammation with spontaneous amyloid-related imaging abnormalities and high cerebrospinal fluid anti-abeta autoantibodiesJ Alzheimers Dis45: 363367 |
38 | Piazza F, Greenberg SM, Savoiardo M, Gardinetti M, Chiapparini L, Raicher I, Nitrini R, Sakaguchi H, Brioschi M, Billo G, Colombo A, Lanzani F, Piscosquito G, Carriero MR, Giaccone G, Tagliavini F, Ferrarese C, DiFrancesco JC(2013) Anti-amyloid beta autoantibodies in cerebral amyloid angiopathy-related inflammation: Implications for amyloid-modifying therapiesAnn Neurol73: 449458 |
39 | Czerkinsky CC, Nilsson LA, Nygren H, Ouchterlony O, Tarkowski A(1983) A solid-phase enzyme-linked immunospot (ELISPOT) assay for enumeration of specific antibody-secreting cellsJ Immunol Methods65: 109121 |
40 | Sehlin D, Sollvander S, Paulie S, Brundin R, Ingelsson M, Lannfelt L, Pettersson FE, Englund H(2010) Interference from heterophilic antibodies in amyloid-beta oligomer ELISAsJ Alzheimers Dis21: 12951301 |
41 | Magnusson K, Sehlin D, Syvanen S, Svedberg MM, Philipson O, Soderberg L, Tegerstedt K, Holmquist M, Gellerfors P, Tolmachev V, Antoni G, Lannfelt L, Hall H, Nilsson LN(2013) Specific uptake of an amyloid-beta protofibril-binding antibody-tracer in AbetaPP transgenic mouse brainJ Alzheimers Dis37: 2940 |
42 | Englund H, Sehlin D, Johansson AS, Nilsson LN, Gellerfors P, Paulie S, Lannfelt L, Pettersson FE(2007) Sensitive ELISA detection of amyloid-beta protofibrils in biological samplesJ Neurochem103: 334345 |
43 | Baril L, Nicolas L, Croisile B, Crozier P, Hessler C, Sassolas A, McCormick JB, Trannoy E(2004) Immune response to Abeta-peptides in peripheral blood from patients with Alzheimer’s disease and control subjectsNeurosci Lett355: 226230 |
44 | Gruden MA, Davudova TB, Malisauskas M, Zamotin VV, Sewell RD, Voskresenskaya NI, Kostanyan IA, Sherstnev VV, Morozova-Roche LA(2004) Autoimmune responses to amyloid structures of Abeta(25-35) peptide and human lysozyme in the serum of patients with progressive Alzheimer’s diseaseDement Geriatr Cogn Disord18: 165171 |
45 | Nath A, Hall E, Tuzova M, Dobbs M, Jons M, Anderson C, Woodward J, Guo Z, Fu W, Kryscio R, Wekstein D, Smith C, Markesbery WR, Mattson MP(2003) Autoantibodies to amyloid beta-peptide (Abeta) are increased in Alzheimer’s disease patients and Abeta antibodies can enhance Abeta neurotoxicity: Implications for disease pathogenesis and vaccine developmentNeuromolecular Med3: 2939 |
46 | Sohn JH, So JO, Hong HJ, Kim JW, Na DR, Kim M, Kim H, Nam E, Ha HJ, Kim YH, Mook-Jung I(2009) Identification of autoantibody against beta-amyloid peptide in the serum of elderlyFront Biosci14: 38793883 |
47 | Song MS, Mook-Jung I, Lee HJ, Min JY, Park MH(2007) Serum anti-amyloid-beta antibodies and Alzheimer’s disease in elderly Korean patientsJ Int Med Res35: 301306 |
48 | Jianping L, Zhibing Y, Wei Q, Zhikai C, Jie X, Jinbiao L(2006) Low avidity and level of serum anti-Abeta antibodies in Alzheimer diseaseAlzheimer Dis Assoc Disord20: 127132 |
49 | Stenh C, Englund H, Lord A, Johansson AS, Almeida CG, Gellerfors P, Greengard P, Gouras GK, Lannfelt L, Nilsson LN(2005) Amyloid-beta oligomers are inefficiently measured by enzyme-linked immunosorbent assayAnn Neurol58: 147150 |
50 | Englund H, Degerman Gunnarsson M, Brundin RM, Hedlund M, Kilander L, Lannfelt L, Pettersson FE(2009) Oligomerization partially explains the lowering of Abeta42 in Alzheimer’s disease cerebrospinal fluidNeurodegener Dis6: 139147 |
51 | Pellicano M, Bulati M, Buffa S, Barbagallo M, Di Prima A, Misiano G, Picone P, Di Carlo M, Nuzzo D, Candore G, Vasto S, Lio D, Caruso C, Colonna-Romano G(2010) Systemic immune responses in Alzheimer’s disease: In vitro mononuclear cell activation and cytokine productionJ Alzheimers Dis21: 181192 |
52 | Speciale L, Calabrese E, Saresella M, Tinelli C, Mariani C, Sanvito L, Longhi R, Ferrante P(2007) Lymphocyte subset patterns and cytokine production in Alzheimer’s disease patientsNeurobiol Aging28: 11631169 |
53 | Richartz-Salzburger E, Batra A, Stransky E, Laske C, Kohler N, Bartels M, Buchkremer G, Schott K(2007) Altered lymphocyte distribution in Alzheimer’s diseaseJ Psychiatr Res41: 174178 |
54 | Xue SR, Xu DH, Yang XX, Dong WL(2009) Alterations in lymphocyte subset patterns and co-stimulatory molecules in patients with Alzheimer diseaseChin Med J (Engl)122: 14691472 |
55 | O’Nuallain B, Wetzel R(2002) Conformational Abs recognizing a generic amyloid fibril epitopeProc Natl Acad Sci U S A99: 14851490 |
56 | Britschgi M, Wyss-Coray T(2007) Systemic and acquired immune responses in Alzheimer’s diseaseInt Rev Neurobiol82: 205233 |
57 | Conti E, Galimberti G, Tremolizzo L, Masetto A, Cereda D, Zanchi C, Piazza F, Casati M, Isella V, Appollonio I, Ferrarese C(2010) Cholinesterase inhibitor use is associated with increased plasma levels of anti-Abeta 1-42 antibodies in Alzheimer’s disease patientsNeurosci Lett486: 193196 |
58 | Reale M, Iarlori C, Gambi F, Feliciani C, Isabella L, Gambi D(2006) The acetylcholinesterase inhibitor, Donepezil, regulates a Th2 bias in Alzheimer’s disease patientsNeuropharmacology50: 606613 |
Figures and Tables
Fig.1
B cell ELISpot. A) PBMCs were collected by Ficoll-Paque separation, stimulated with R848 and hIL-2 to start their antibody production and B) added to ELISpot wells coated with anti-IgG capture antibodies. C) Biotinylated Aβ binds to the captured anti-Aβ antibodies secreted from the activated B cells. D) The dots representing cells secreting anti-Aβ antibodies were visualized by Streptavidin-ALP.
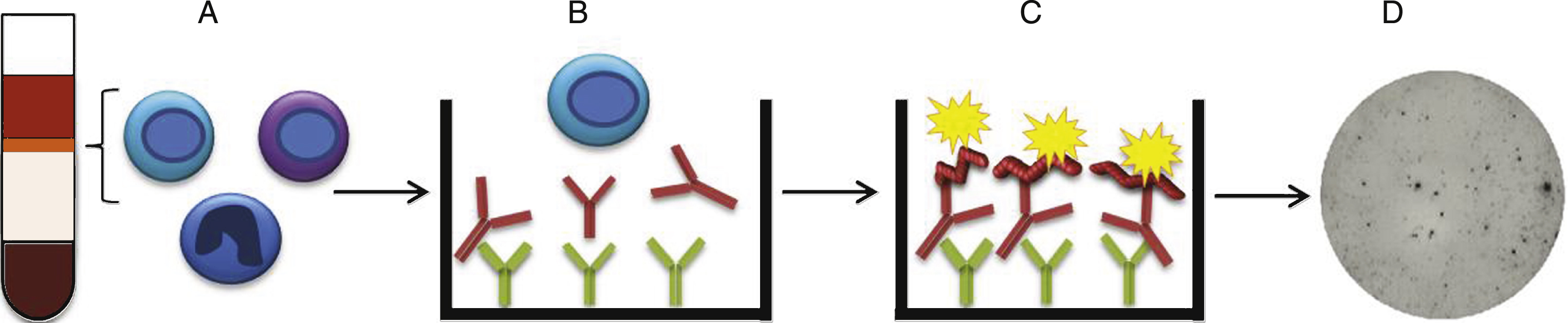
Fig.2
Characterization of PBMCs and validation of the antigen. A) Total IgG production from activated B cells. Number of spots representing IgG producing cells detected in the ELISpot. AD individuals (n = 39) (■) had higher number of spots than healthy controls (n = 30) (•) (p≤0.05). PBMCs from individuals failing activation (spots <20) (AD patients (n = 7), healthy controls (n = 10)) were excluded from the study (below horizontal line). B) B cell levels in AD patients and healthy individuals. The proportion of B cells in PBMCs was defined with flow cytometry by determining the number of CD19+ cells. There was no significant difference in the percentage of B cell in AD patients compared to healthy controls (p = 0.085). C) Aβ42 protofibril evaluation with protofibril specific ELISA. No considerable difference was noticed comparing biotinylated Aβ42 protofibrils with non-biotinylated Aβ42 protofibrils.
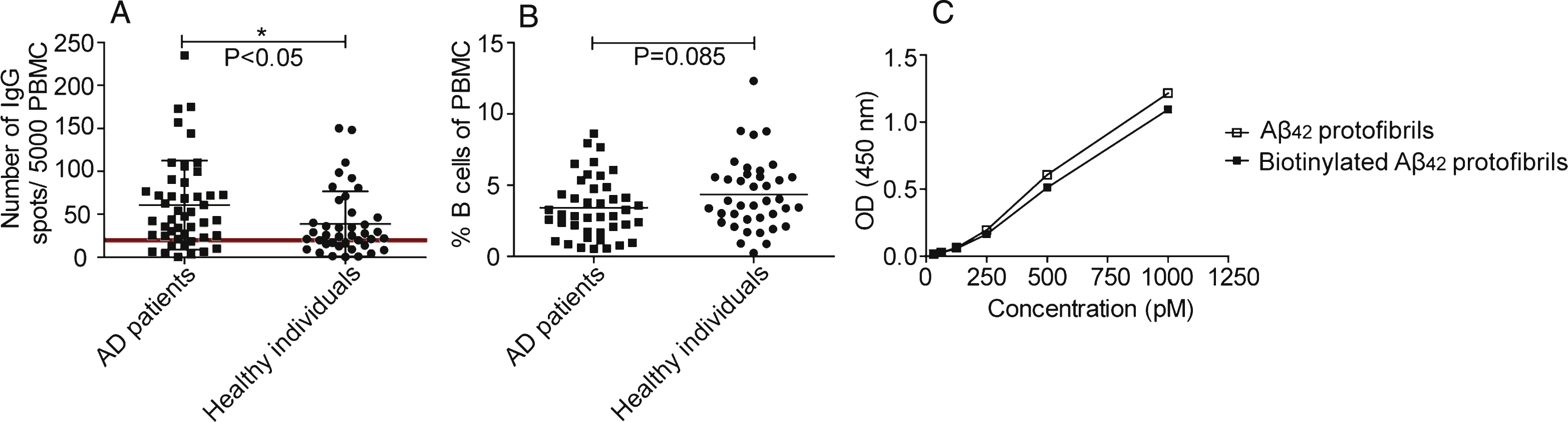
Fig.3
B cell production of anti-Aβ40 monomer and anti-Aβ42 protofibril antibodies. A) Number of spots representing cells producing antibodies to Aβ40 monomers (■) and Aβ42 protofibrils (•). Looking at the whole study material (n = 63), antibodies detected Aβ42 protofibrils to a higher degree than Aβ40 monomers (p = <0.0001). B) B cell production of anti-Aβ40 monomer antibodies. Number of spots representing cells producing antibodies to Aβ40 monomers in AD patients (n = 37) (■) and healthy individuals (n = 26) (•) No significant difference was seen when comparing AD patients with healthy controls (p < 0.23). C) Number of spots representing cells producing antibodies detecting Aβ42 protofibrils in AD individuals (n = 37) (■) and healthy individuals (n = 26) (•). There was a significant difference in number of spots comparing AD patients with healthy individuals (p < 0.05). D) In AD patients, the number of spots representing B cells producing antibodies binding to Aβ42 protofibrils correlated weakly with the number of spots representing the total number of IgG producing B cells (Spearman r = 0.59, p < 0.001).
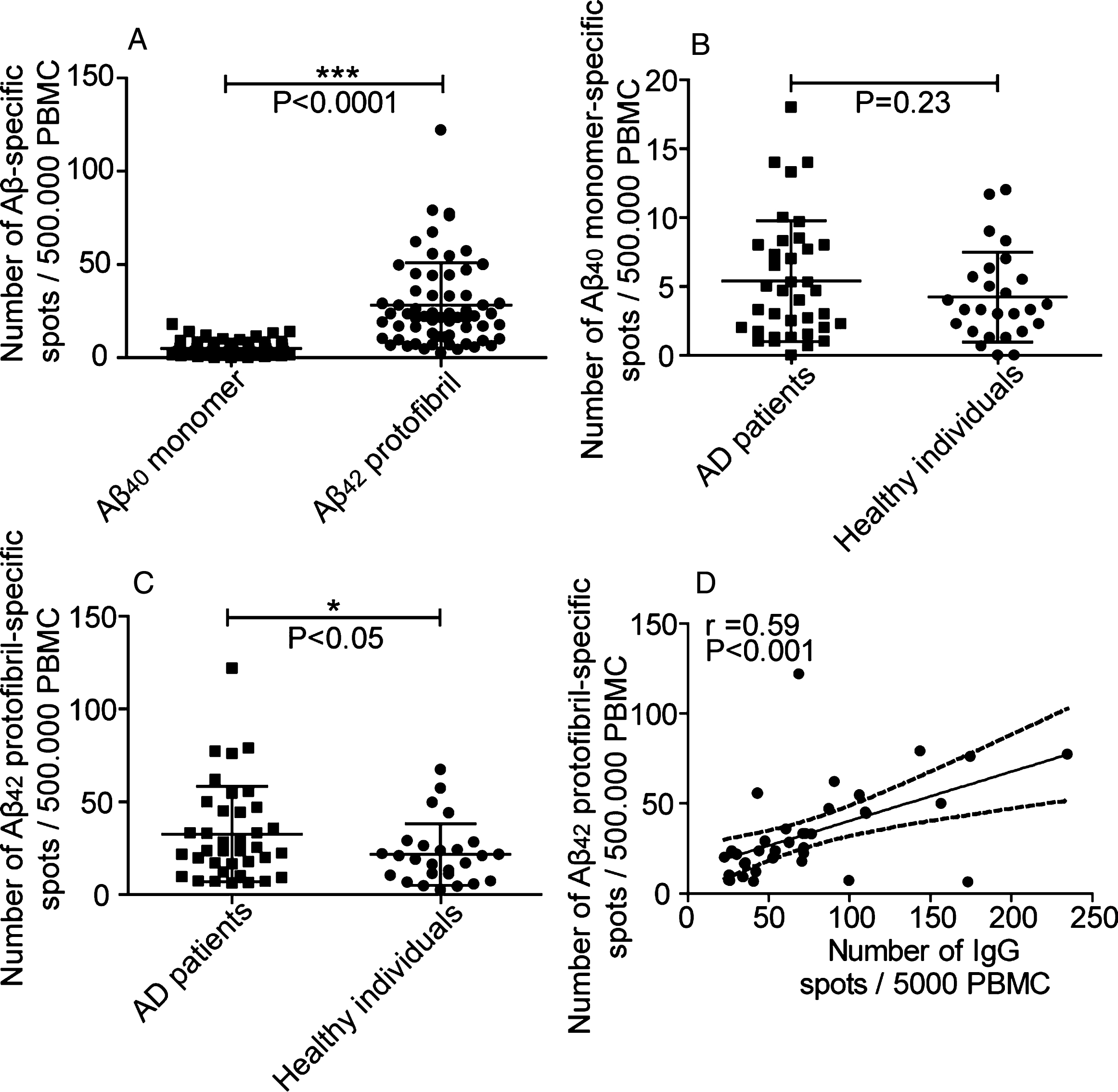
Fig.4
Anti-Aβ42 protofibril antibody response correlated with mini mental state. No correlation was seen when comparing the levels of anti-Aβ42 protofibril selective antibody responses to the state of cognition in AD patients (n = 22).
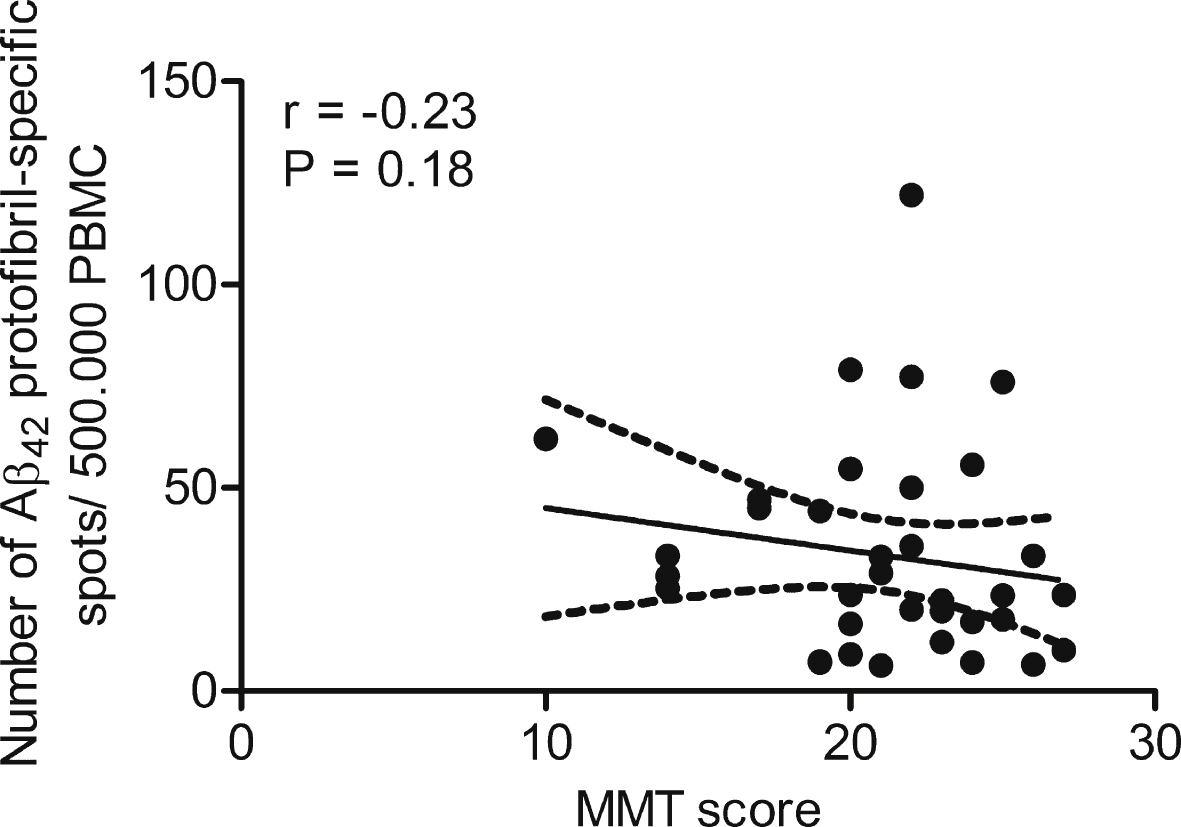
Table 1
AD patients and age-matched healthy individuals included in the study. The PBMCs used in the study were derived from AD patients and age-matched healthy controls. The table presents gender, age and ApoE allele frequencies for AD patients and healthy controls, respectively
| AD patients (n = 46) | Healthy individuals (n = 44) | |
| Age (years) | ||
| Mean ± SD | 78.0 ± 6.5 | 74.8 ± 7.0 |
| Range | 63– 90 | 51– 87 |
| Gender | ||
| Female/Male | 20/26 | 24/20 |
| MMSE score* | ||
| Mean ± SD | 21.0 ± 3.8 | |
| Range | 10– 27 | |
| ApoE allele** | ||
| E2 | 11.1% | 15.9% |
| E3 | 91.1% | 90.9% |
| E4 | 62.2% | 38.6% |
*MMSE score from 1 patient was not available and was therefore not included in the calculations. **ApoE genotype from 1 AD patient was not available and was therefore not included in the calculations.


