
Reduced Incidence of Dementia in Solid Organ Transplant Patients Treated with Calcineurin Inhibitors
Abstract
Experimental evidence suggests that the protein phosphatase calcineurin mediates the action of amyloid-β (Aβ) oligomers, the most toxic amyloid species thought to drive initial cognitive decline in Alzheimer’s disease (AD). However, there is currently no evidence that inhibition of calcineurin could prevent the onset of AD in humans. Here, we report for the first time that individuals chronically treated with calcineurin inhibitors to prevent solid organ transplant rejection have a significantly lower incidence of AD/dementia as compared to the general population. This result prompts further clinical development of calcineurin inhibition as a viable treatment for AD.
Alzheimer’s disease (AD) is the most common age-associated neurodegenerative disorder for which there is no resolving cure. Compelling evidence indicates that small soluble oligomers of amyloid-β (Aβ) protein that precede senile plaque formation are the most toxic Aβ species in the AD brain [1, 2], known to selectively target synaptic integrity and function [2–6]. Consequently, there is large consensus that preventing Aβ oligomer synaptotoxicity would be an effective treatment strategy [5, 7, 8].
Calcineurin (CN) is an important phosphatase modulating synaptic activity and memory formation [9–11]. Excess CN activity disrupts synaptic architecture and impairs memory [10, 12, 13], but also its complete suppression negatively affects memory [10]. This suggests that normalization and careful modulation of CN activity is critical to allow proper memory processing and cognition [12, 14]. Notably, we and others have shown that CN mediates both the neurotoxic and cognitive effects of Aβ oligomers [15–24], and elevated CN has been also shown in the CNS of AD patients, suggesting a central role of CN in AD onset and/or clinical progression [25–28]. Indeed, our previous studies in mouse models demonstrated that an acute treatment with the CN inhibitor (CNI) FK506/Tacrolimus (TAC) in Tg2576 mice (a well-established model of AD) as well as in wt mice acutely injected icv with Aβ oligomers restores memory function in these cognitively impaired animals [16, 19, 29]. We further showed that biochemical markers and electrophysiological measurements of synaptic efficiency, that are known to be disrupted by Aβ oligomers [2, 3, 5, 6, 30, 31], are restored following TAC treatment [29]. It is important to notice that the beneficial effects of TAC were not due to immunosuppression but rather to a direct inhibition of CN. In fact, another clinically used immunosuppressant, Rapamycin, which does not inhibit CN was ineffective in preventing Aβ-induced behavioral, biochemical, and electrophysiological deficits under acute treatment conditions [29] (however, see [32, 33] as reviewed in [34, 35] for CN-independent, autophagy-driven effects of a chronic Rapamycyn treatment on AD mouse models).
While these results strongly indicate that inhibition of CN by TAC may protect neurons from the damaging effects of Aβ oligomers, the question as to whether such strategy may be effective in preventing the onset and progression of AD in humans remains unresolved. It is particularly challenging to preliminary test this hypothesis directly on AD patients since the implementation of a de novo treatment with CNI is associated with systemic immunosuppression and consequently with increased risk of infections and malignancies. An innovative alternative approach asking whether CNI could be a promising treatment for preventing (and possibly halting or reversing) AD in humans may come from the post hoc analysis of patients who received organ transplants. Organ transplant recipients represent a cohort of patients mostly maintained on CNI based chronic immunosuppression (using either TAC or cyclosporine) where the hypothesis that CNI reduces the prevalence of symptomatic AD can be tested without adding unnecessary risk to the patients. Patients receiving organ transplantation in any age group, including ages (>65 years) that are at higher risk of developing AD, are followed for many years (usually until death) in transplant centers with all their comorbidities carefully monitored and recorded by a multidisciplinary team. Any manifestation of memory impairment or dementia is immediately noted and monitored since it can limit compliance with treatment. Furthermore, younger individuals in lower risk age for AD, are kept for many years on CNI. These subjects are expected to have long survival after transplantation and the effect of long-term chronic CNI treatment on the development of AD could be assessed through the analysis of their follow-up medical records.
With this goal in mind, according to approved IRB protocols we retrospectively studied a population of 2,644 patients who received a total of 3,167 organ transplants at our institution. These transplant recipients were maintained on chronic CNI immunosuppressive therapy to prevent allograft rejection. Patients were then stratified for age at the time of transplant into the following groups: <65; 65–74; 75–84; >85 years of age. The gender and ethnicity demographic of this patient cohort is shown in Supplementary Table 1. The number of patients with CNI-based immunosuppression was evaluated at age of first transplant, age at the time of analysis (August 2013) or at time of death (if deceased). All cognitive impairments indicated in the physician note were marked as positive hit for dementia while acute/transient conditions diagnosed as secondary to infection or drug toxicity and subsequently resolved with proper treatment were excluded. From this analysis patients in different age groups were transplanted respectively at <65 (95.3% ); 65–74 (4.1% ); 75–84 (0.3% ); >85 (0.03% ). At time of data collection, 22.2% of patients (n = 587) were collectively >65 years of age. Of these patients, 438 (16.5% ) were 65–74 years old; 135 (5.1% ) were 75–84 years old, and 14 (0.5% ) were >85 years old. We observed evidence of dementia in 2 subjects (2/2,057; 0.09% ) in the cohort <65 years old, while in the cohort >65 years of age, we identified 6 demented patients (6/587; 1.02% ). Total patients were 60% males and 40% females, with 6 males and 2 females demented. When analyzed by age group, 5 patients with dementia were in the 65–74 years old group (5/438; 1.14% ); 1 was in the 75–84 years old group (1/135; 0.7% ); and none in the >85 years old group (0/14). This data is summarized in Table 1. Of note, 4 of these demented patients (1 in the <65 and 3 in the 64–74 age groups) had no mention of dementia in follow-up visits years later (average 5.25 years; range from 1 to 7 years after initial diagnosis) while they were receiving CNI. All other demented patients were described to have only mild dementia and one patient diagnosed at 81 years of age with mild dementia had been on CNI for 19 years prior to it.
The number of patients with a diagnosis of dementia obtained from medical records in our patient cohort was then compared to national data obtained from the 2014 Alzheimer’s Association Facts and Figures dataset on age group-matched patients to compare the prevalence of AD (Fig. 1). These data clearly show that the prevalence of dementia and AD in our patient cohort is significantly lower (in fact, almost absent) as compared to national data from the general population (p < 0.0001 for each age group except <65 that was not significant; Chi-Square test). Despite the encouraging findings observed in the >85 years old these were potentially limited by the small number of patients available in that group. Since our institution mostly serves patients from the State of Texas, we further compared our >65 years cohort with the prevalence of AD in the general population of Texas (Alzheimer Association Facts and Figures 2014) obtaining similar results (1.02% versus 10.13% , respectively; p < 0.0001; Chi-Square Test). Collectively, these results suggest that inhibition of CN prevented the occurrence of dementia in chronically treated patients.
While this patient population is encouraged to adhere to diet/exercise regimens and are instructed not to use recreational drugs, alcohol, or tobacco that could also favor neuronal function, the compliance rate is limited and unlikely to explain the findings observed here. On the other hand, several diseases leading to kidney transplantation (by far the most common solid organ transplant in our patient population; >80% ) such as diabetes and hypertension are also well-established risk factors to develop dementia and AD [36–39]. For example diabetes and hypertension are the leading causes (over 70% ) of end-stage-renal-disease leading to kidney transplant [40, 41]. Moreover, other risk factors for end-stage-renal-disease are obesity and advanced age (American Kidney Foundation) that are also well-recognized risk factors for AD [42–46]. Consequently, one should expect our transplant patient population (that includes a majority of kidney transplant patients) to be skewed toward high risk of developing dementia rather than being protected from it, as suggested by our data. This consideration further supports the predicted protective effect of CNI treatment in this population. Lastly, over several decades CNIs have been the common denominator of our immunosuppressive protocols in association with other medications (steroids, mycophenolate mofetil, rapamycin, etc.). Since we did not observe differences in protection over any particular time period covering our patient database, it is unlikely that the observed beneficial effects may be due to drugs different than CNIs.
In conclusion, our findings confirm for the first time in humans the data obtained from animal models and supports our hypothesis that CNI has a protective effect on the development (and possibly progression and even reversal) of AD. This result holds true even when the patient cohort is stratified according to age, indicating that the beneficial effects of CNI treatment is not affected by increasing age. Furthermore, our transplanted patient cohort should be at risk of AD (mostly kidney transplant patients with strong risk factors for AD), making our results even more suggestive of a remarkable protective effect of CNI treatment. Whether CNIs may be effective in clinically diagnosed MCI or early AD subjects remains to be determined.
ACKNOWLEDGMENTS
Supported in part by NIH/NIA grant 1R01AG042890 and the Mitchell Center for Neurodegenerative Diseases to GT.
The authors wish to thank Ms. Cathy Cantieri, Applications Specialist at UTMB, for her valuable support in data mining for this study.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/15-0065r1).
Appendices
The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JAD-150065.
REFERENCES
1 | Larson ME, Lesne SE(2012) Soluble Abeta oligomer production and toxicityJ Neurochem120: Suppl 1125139 |
2 | Walsh DM, Selkoe DJ(2007) A beta oligomers - a decade of discoveryJ Neurochem101: 11721184 |
3 | Ma T, Klann E(2012) Amyloid beta: Linking synaptic plasticity failure to memory disruption in Alzheimer’s diseaseJ Neurochem120: Suppl 1140148 |
4 | Bjorklund NL, Reese LC, Sadagoparamanujam VM, Ghirardi V, Woltjer RL, Taglialatela G(2012) Absence of amyloidbeta oligomers at the postsynapse and regulated synaptic Zn2+ in cognitively intact aged individuals withAlzheimer’s disease neuropathologyMol Neurodegener7: 23 |
5 | Wilcox KC, Lacor PN, Pitt J, Klein WL(2011) Abeta oligomer-induced synapse degeneration in Alzheimer’s diseaseCell Mol Neurobiol31: 939948 |
6 | Arendt T(2009) Synaptic degeneration in Alzheimer’s diseaseActa Neuropathol118: 167179 |
7 | Selkoe DJ(2008) Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behaviorBehav Brain Res192: 106113 |
8 | Klein WL(2013) Synaptotoxic amyloid-beta oligomers: A molecular basis for the cause, diagnosis, and treatment ofAlzheimer’s disease?J Alzheimers Dis33: Suppl 1S49S65 |
9 | Mansuy IM(2003) Calcineurin in memory and bidirectional plasticityBiochem Biophys Res Commun311: 11951208 |
10 | Malleret G, Haditsch U, Genoux D, Jones MW, Bliss TV, Vanhoose AM, Weitlauf C, Kandel ER, Winder DG, Mansuy IM(2001) Inducible and reversible enhancement of learning, memory, and long-term potentiation by genetic inhibition of calcineurinCell104: 675686 |
11 | Mansuy IM, Mayford M, Jacob B, Kandel ER, Bach ME(1998) Restricted and regulated overexpression reveals calcineurin as a key component in the transition from short-term to long-term memoryCell92: 3949 |
12 | Weitlauf C, Winder D(2001) Calcineurin, synaptic plasticity, andmemoryScientificWorldJournal1: 530533 |
13 | Zhuo M, Zhang W, Son H, Mansuy I, Sobel RA, Seidman J, Kandel ER(1999) A selective role of calcineurin aalpha in synaptic depotentiation in hippocampusProc Natl Acad Sci U S A96: 46504655 |
14 | Sharma SK, Bagnall MW, Sutton MA, Carew TJ(2003) Inhibition of calcineurin facilitates the induction of memory for sensitization in Aplysia: Requirement of mitogen-activated protein kinaseProc Natl Acad Sci U S A100: 48614866 |
15 | Taglialatela G, Hogan D, Zhang WR, Dineley KT(2009) Intermediate- and long-term recognition memory deficits in Tg2576 mice are reversed with acute calcineurin inhibitionBehav Brain Res200: 9599 |
16 | Reese LC, Zhang W, Dineley KT, Kayed R, Taglialatela G(2008) Selective induction of calcineurin activity and signaling by oligomeric amyloid betaAging Cell7: 824835 |
17 | Agostinho P, Lopes JP, Velez Z, Oliveira CR(2008) Overactivation of calcineurin induced by amyloid-beta andprion proteinsNeurochem Int52: 12261233 |
18 | Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL(2007) Natural oligomers of the Alzheimeramyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependentsignaling pathwayJ Neurosci27: 28662875 |
19 | Dineley KT, Hogan D, Zhang WR, Taglialatela G(2007) Acute inhibition of calcineurin restores associativelearning and memory in Tg2576 APP transgenic miceNeurobiol Learn Mem88: 217224 |
20 | Agostinho P, Oliveira CR(2003) Involvement of calcineurin in the neurotoxic effects induced by amyloid-beta andprion peptidesEur J Neurosci17: 11891196 |
21 | Yao W, Zou HJ, Sun D, Ren SQ(2013) Abeta induces acute depression of excitatory glutamatergic synaptictransmission through distinct phosphatase-dependent mechanisms in rat CA1 pyramidal neuronsBrain Res1515: 8897 |
22 | Cavallucci V, Berretta N, Nobili A, Nistico R, Mercuri NB, D’Amelio M(2013) Calcineurin inhibition rescues earlysynaptic plasticity deficits in a mouse model of Alzheimer’s diseaseNeuromolecular Med15: 541548 |
23 | Rozkalne A, Hyman BT, Spires-Jones TL(2011) Calcineurin inhibition with FK506 ameliorates dendritic spinsity deficits in plaque-bearing Alzheimer model miceNeurobiol Dis41: 650654 |
24 | Ramser EM, Gan KJ, Decker H, Fan EY, Suzuki MM, Ferreira ST, Silverman MA(2013) Amyloid-beta oligomers inducetau-independent disruption of BDNF axonal transport via calcineurin activation in cultured hippocampal neuronsMol Biol Cell24: 24942505 |
25 | Reese LC, Laezza F, Woltjer R, Taglialatela G(2011) Dysregulated phosphorylation of Ca(2+)/calmodulin-dependentprotein kinase II-alpha in the hippocampus of subjects with mild cognitive impairment and Alzheimer’s diseaseJ Neurochem119: 791804 |
26 | Abdul HM, Sama MA, Furman JL, Mathis DM, Beckett TL, Weidner AM, Patel ES, Baig I, Murphy MP, LeVine H3rd, Kraner SD, Norris CM(2009) Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signalingJ Neurosci29: 1295712969 |
27 | Reese LC, Taglialatela G(2011) A role for calcineurin in Alzheimer’s diseaseCurr Neuropharmacol9: 685692 |
28 | Braithwaite SP, Stock JB, Lombroso PJ, Nairn AC(2012) Protein phosphatases and Alzheimer’s diseaseProg Mol Biol Transl Sci106: 343379 |
29 | Dineley KT, Kayed R, Neugebauer V, Fu Y, Zhang W, Reese LC, Taglialatela G(2010) Amyloid-beta oligomers impair fear conditioned memory in a calcineurin-dependent fashion in miceJ Neurosci Res88: 29232932 |
30 | Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ(2008) Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memoryNat Med14: 837842 |
31 | Klyubin I, Betts V, Welzel AT, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Peng Y, Wisniewski T, Selkoe DJ, Anwyl R, Walsh DM, Rowan MJ(2008) Amyloid beta protein dimer-containing human CSF disrupts synapticplasticity: Prevention by systemic passive immunizationJ Neurosci28: 42314237 |
32 | Majumder S, Caccamo A, Medina DX, Benavides AD, Javors MA, Kraig E, Strong R, Richardson A, Oddo S(2012) Lifelong rapamycin administration ameliorates age-dependent cognitive deficits by reducing IL-1beta and enhancingNMDA signalingAging Cell11: 326335 |
33 | Majumder S, Richardson A, Strong R, Oddo S(2011) Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficitsPLoS One6: e25416 |
34 | Oddo S(2012) The role of mTOR signaling in Alzheimer diseaseFront Biosci (Schol Ed)4: 941952 |
35 | Richardson A, Galvan V, Lin AL, Oddo S(2014) How longevity research can lead to therapies for Alzheimer’s disease: The rapamycin storyExp Gerontol10.1016/j.exger.2014.12.002 |
36 | Correia SC, Santos RX, Perry G, Zhu X, Moreira PI, Smith MA(2011) Insulin-resistant brain state: The culprit in sporadic Alzheimer’s disease?Ageing Res Rev10: 264273 |
37 | Zhao WQ, Townsend M(2009) Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer’s diseaseBiochim Biophys Acta1792: 482496 |
38 | Purnell C, Gao S, Callahan CM, Hendrie HC(2009) Cardiovascular risk factors and incident Alzheimer disease: A systematic review of the literatureAlzheimer Dis Assoc Disord23: 110 |
39 | Luchsinger JA, Gustafson DR(2009) Adiposity, type 2 diabetes, and Alzheimer’s diseaseJ Alzheimers Dis16: 693704 |
40 | Rodger RS(2012) Approach to the management of endstage renal diseaseClin Med12: 472475 |
41 | Rettig RA, Levinsky NG(1991) Kidney Failure and the Federal GovernmentWashington, DCNational Academies Press |
42 | Emmerzaal TL, Kiliaan AJ, Gustafson DR(2015) 2003-2013: A decade of body mass index, Alzheimer’s disease, and dementiaJ Alzheimers Dis43: 739755 |
43 | Priyadarshini M, Kamal MA, Greig NH, Realef M, Abuzenadah AM, Chaudhary AG, Damanhouri GA(2012) Alzheimer’s disease and type 2 diabetes: Exploring the association to obesity and tyrosine hydroxylaseCNS Neurol Disord Drug Targets11: 482489 |
44 | Hildreth KL, Van Pelt RE, Schwartz RS(2012) Obesity, insulin resistance, and Alzheimer’s diseaseObesity (Silver Spring)20: 15491557 |
45 | Swaab DF, Dubelaar EJ, Hofman MA, Scherder EJ, van Someren EJ, Verwer RW(2002) Brain aging and Alzheimer’s disease; use it or lose itProg Brain Res138: 343373 |
46 | Morris JC(2005) Dementia update 2005Alzheimer Dis Assoc Disord19: 100117 |
Figures and Tables
Fig.1
Prevalence of clinically diagnosed dementia (including Alzheimer’s disease) in the general population as compared to transplanted patients treated with immunosuppressive calcineurin inhibitors (CNI). Patients were grouped according to age at time of last follow up medical examination or death. Table underneath figure shows actual numbers in the transplanted patient cohort studied for the present report. ***p < 0.0001 as compared to age-matched group in the general population (χ 2 test).
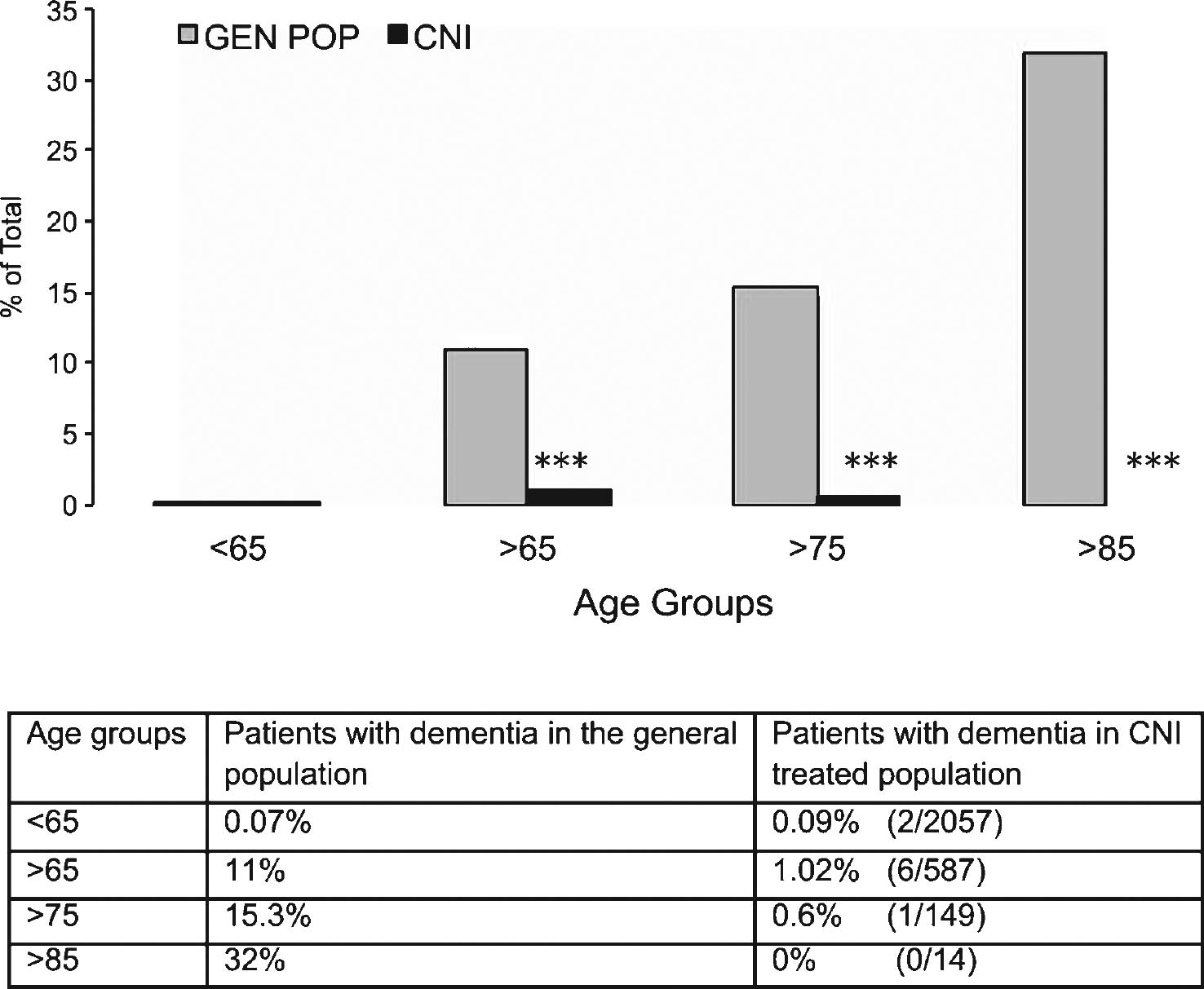
Table 1
Rate and age distribution of calcineurin inhibitor-treated patients with dementia
| Age Groups | Number of Patients | Number of patients with dementia | Percentage of patients with dementia |
| <65 | 2057 | 2 | 0.09% |
| 65–74 | 438 | 5 | 1.14% |
| 75–84 | 135 | 1 | 0.7% |
| >85 | 14 | 0 | 0% |
| Total | 2644 | 8 | 0.3% |

