
Calmodulin Binding Proteins and Alzheimer’s Disease
Abstract
The small, calcium-sensor protein, calmodulin, is ubiquitously expressed and central to cell function in all cell types. Here the literature linking calmodulin to Alzheimer’s disease is reviewed. Several experimentally-verified calmodulin-binding proteins are involved in the formation of amyloid-β plaques including amyloid-β protein precursor, β-secretase, presenilin-1, and ADAM10. Many others possess potential calmodulin-binding domains that remain to be verified. Three calmodulin binding proteins are associated with the formation of neurofibrillary tangles: two kinases (CaMKII, CDK5) and one protein phosphatase (PP2B or calcineurin). Many of the genes recently identified by genome wide association studies and other studies encode proteins that contain putative calmodulin-binding domains but only a couple (e.g., APOE, BIN1) have been experimentally confirmed as calmodulin binding proteins. At least two receptors involved in calcium metabolism and linked to Alzheimer’s disease (mAchR; NMDAR) have also been identified as calmodulin-binding proteins. In addition to this, many proteins that are involved in other cellular events intimately associated with Alzheimer’s disease including calcium channel function, cholesterol metabolism, neuroinflammation, endocytosis, cell cycle events, and apoptosis have been tentatively or experimentally verified as calmodulin binding proteins. The use of calmodulin as a potential biomarker and as a therapeutic target is discussed.
INTRODUCTION
Alzheimer’s disease (AD) is the leading cause of dementia, with close to 50 million people worldwide currently suffering from dementia and the number expected to increase to 1 in 85 people by the year 2050 [1]. First described by the German psychiatrist Alois Alzheimer, it manifests through progressive memory loss ultimately encompassing a loss of recognition of people, places, and things, eventually leading to death [2]. Currently, the cure for AD remains elusive, with treatments focusing on managing symptoms and attempts to slow neurodegeneration. While the initiating events leading to late onset AD (LOAD) remain to be discovered, the formation of amyloid plaques and neurofibrillary tangles remain as central culprits in the neurodegenerative aspects of the disease. More recently, numerous genes and several metabolic events have been identified as either central or contributing factors in AD.
A decade ago, O’Day and Myre [3] proposed the “Calmodulin Hypothesis” as an extension of the well-established “calcium hypothesis” of LOAD. In that study, a number of proteins linked to the production of amyloid-β (Aβ) were shown to possess putative calmodulin binding domains (CaMBDs) suggesting that they could be regulated by the small regulatory, calcium-binding protein calmodulin (CaM). Subsequently, two of the central proteins in this event, amyloid-β precursor protein (AβPP) and beta-secretase (BACE1; beta-site AβPP cleaving enzyme 1) were each shown to bind to and be regulated by CaM [4, 5]. Non-amyloidogenic processing of AβPP in platelets was induced in a time- and dose-dependent manner by the calmodulin antagonist W7 [6]. Over the past 10 years, advancements have been made into understanding the pathways involved in both amyloid plaque and neurofibrillary tangle formation. Based on these new insights, here we first re-examine the role of calmodulin binding proteins (CaMBPs) in the formation of the two primary culprits linked to the symptoms and progression of AD: amyloid plaques and neurofibrillary tangles (Fig. 1). As summarized in Fig. 1, we then look at the function of calmodulin as a regulator of other events and recently identified proteins linked to the disease before looking at CaM as a potential therapeutictarget.
THE CALCIUM HYPOTHESIS AND CALMODULIN
The “calcium hypothesis” is one prominent model for AD [7]. It suggests that an imbalance of calcium levels in cells precedes the signaling pathway malfunctions and neuronal deterioration observed in neurodegenerative diseases. It has long been known that calcium ions (Ca2 +) play a substantial role in normal physiology. In the brain, Ca2 + functions in neurotransmitter synthesis and release as well as in the control of membrane excitability [8]. The intracellular concentration of Ca2 + is kept within a tight range of 10 −7 to 10 −8 mol; variation from the norm, even if slight, is detrimental when it persists [8].
Calcium ions perform their function primarily through binding to membrane or cytoplasmic proteins. Arguably the primary calcium-binding protein and effector of calcium function is the small protein CaM. Highly conserved and ubiquitously expressed in all eukaryotes and in all cell types, it is a member of the EF-hand family of calcium sensors [9]. CaM possesses four E-F hands, two at the C-terminal and two at the N-terminal, allowing it to bind up to four Ca2 + ions [9, 10]. Each lobe (N, C) can bind up to two calcium ions (Fig. 2). The number and location of binding of this cation affects CaM’s conformation and how it interacts with its target CaMBPs. Proteins do not bind to Ca2 +/CaM via traditional targeting sequences. In the absence of Ca2 +, the apo-CaM conformation allows the binding of the sub-group of Ca2 +-independent CaMBPs (Table 1). Ca2 +-independent binding occurs via well-defined IQ [(FILV)Qxxx(RK)Gxxx(RK)xx(FILVWY)] andIQ-like [(FILV)Qxxx(RK)xxxxxxxx] motifs. Upon Ca2 + binding, CaM undergoes significant conformational changes allowing it to bind to Ca2 +-dependent CaMBPs. Ca2 +-dependent binding involves a diversity of motifs and sub-classes that are defined primarily by the sequence and positioning of hydrophobic amino acids sometimes in conjunction with basic residues within the motif (e.g., 1–5–10 motif: xxx(FILVW)xxxx(FAILVW)xxxx(FILVW) where x = any a.a.) [11]. The “Calmodulin Target Database” categorizes the various types of CaM-binding motifs (Table 1; http://calcium.uhnres.utoronto.ca/ctdb/ctdb/home.html). In addition to the major CaMBD categories (e.g., 1–8–14, 1–5–10, etc.), there are also a large number of others that do not fit into any of these specific categories (e.g., 1–12 and basic motifs) among other more recently discovered binding non-canonical motifs [12].
To add to this non-conventional binding interaction, Villarroel et al. [13] have presented evidence indicating that while CaM binds its calcium-dependent targets via these CaMBDs in their normal orientation it can also bind to those sequences in the opposite direction. Furthermore, their data also suggests that upon binding to its target, CaM can undergo a conformational rotation further altering its structural relationship with that CaMBP. Considering the central importance of CaM to essential life processes, this complex protein interaction may be part of the reason CaM historically has eluded serving as a primary target in various diseases. Once the interactions between CaM and specific CaMBPs linked to AD are fully clarified, it may become possible to develop target-specific peptides to fight different aspects of the disease.
CALMODULIN AND AMYLOID PLAQUE FORMATION
The Calmodulin Target Database identifies the presence and types of CaMBDs in protein sequences with over 90% accuracy [11]. Using this database, O’Day and Myre [3] first identified a diversity of potential CaMBPs linked to the formation of amyloid plaques. The “amyloid hypothesis” is arguably the predominant hypothesis for the symptoms and progression of AD [14]. It is based on the aggregation of Aβ peptides plus a multitude of other components to form extracellular amyloid plaques in the brains of AD sufferers [15–17]. Aβ peptides vary slightly in length and are formed through the regulated intramembrane proteolytic (RIP) cleavage of AβPP at the carboxy-terminal fragment (CTF) by β-secretase and γ-secretase enzymes (Fig. 3) [18]. The main products of this pathway are Aβ40 and Aβ42 with Aβ42 being the more toxic fragment [19]. Aβ peptides interfere with normal signaling leading to neuronal malfunction and, eventually, synapse loss [20]. On the other hand, there exists a non-amyloidogenic pathway of cleavage, where AβPP is cleaved in the middle of the Aβ sequence by α-secretase thereby preventing the formation of the neurodegenerative Aβ peptide [18].
CaM is significantly decreased in the brains of AD individuals [21]. In spite of this, the existing CaM can interact with several proteins in the amyloid pathway. Two independent groups have demonstrated experimentally that AβPP binds to CaM [4, 6]. The main β-secretase enzyme in the brain that begins the amyloidogenic processing of AβPP is BACE1 (Fig. 3). Its activity and expression are both increased in AD brains and its ablation completely prevents AβPP cleavage through the amyloidogenic pathway in mouse models [22, 23]. BACE1 cleaves AβPP at CTF-99 (and sometimes CTF-89), as opposed to cleavage at CTF-83 which is observed in the non-amyloidogenic pathway [18]. Most of the action of BACE1 on AβPP occurs in endocytotic vesicles as opposed to when AβPP is localized at the cell membrane [24]. BACE1 binds CaM with its activity being increased up to 2.5-fold by CaM in a dose dependent manner in vitro [5]. Sequence analysis of the enzyme indicates that binding most likely occurs via a 1-16 motif ((FILVW)xxxxxxxxxxxxxx(FILVW)).
After BACE1, γ-secretase performs the final cleavage to produce Aβ. γ-Secretase consists of four main subunits: nicastrin (Nic), anterior pharynx-defective 1 (APH-1), presenilin (PSEN), and presenilin enhancer 2 (PEN-2), which assemble in the order listed (Fig. 3) [25]. PEN-2, the final subunit to be attached, leads to the activation of the enzyme complex [25]. Sequence analysis of the four γ-secretase subunits using the Calmodulin Database revealed that they all possess presumptive CaMBDs, with some having more than one binding motif [3]. Of these subunits, only PSEN has been experimentally verified to bind CaM [26]. Michno et al. [26] also showed that the loss of function of a single CaM copy inhibits Ca2 + dysregulation induced by PSEN in Drosophila neurons.
Several enzymes can degrade Aβ including neprilysin, endothelin-converting enzymes (ECE), insulin degrading enzymes (IDE), and BACE1 (Fig. 3) [18, 27]. Aside from BACE1, none of the other Aβ degrading enzymes have yet been experimentally confirmed to bind CaM, although all of them were found to possess putative CaMBDs [28]. In addition to binding to the enzymes which generate Aβ, CaM was found to directly bind to Aβ42 itself through a 1-8-14 motif ((FILVW)xxxxxx(FAILVW)xxxxx(FILVW)) [29]. When bound to CaM, Aβ cannot inhibit the brain plasma membrane Ca2 +-ATPase (PMCA) and prevent Ca2 + entry into the cell. Thus the accumulated data presents a clear link between amyloid plaque formation, Ca2 +, and CaM revealing how the dysfunction in Ca2 + levels and Aβ production contribute to each other with CaM functioning at many levels during this interaction.
As mentioned above AβPP binds to CaM in vitro in a Ca2 +-independent manner [6]. The non-amyloidogenic pathway involves α-secretase cleavage of AβPP at CTF-83 precluding the formation of Aβ (Fig. 3). AβPP processed in this non-amyloidogenic pathway appears to have a neuroprotective effect. The ADAM (A Disintegrin And Metalloproteinase) family of proteases, specifically ADAM9, 10, 17, and 19 are all thought to exhibit α-secretase activity in neurons [18]. ADAM10, the predominant α-secretase, binds CaM through an IQ-motif [30–32]. In contrast, ADAM17 does not bind CaM [30]. Importantly, W7 an antagonist of CaM stimulates cleavage of AβPP via the non-amyloidogenic pathway [6, 33].
CALMODULIN AND NEUROFIBRILLARY TANGLE FORMATION
Neurofibrillary tangles constitute the second major hallmark of AD. Consisting primarily of hyperphosphorylated tau, they have long been known to be a neuropathological feature of the disease; although their role in neurodegeneration has come into question [34]. Tau belongs to the family of microtubule-associated proteins which function in microtubule assembly and stability [35]. Several early studies provided evidence for an in vitro association between tau and CaM that is Ca2 + dependent (Fig. 4) [36–38]. Tau’s association with CaM prevents its binding to microtubules [36]. Furthermore, tau cannot be phosphorylated by protein kinase C in vitro when it is in a complex with CaM [37]. In spite of this extensive early association, no recent studies have been conducted on the direct interaction between CaM and tau.
Tau can undergo many post-translational modifications including phosphorylation, acetylation, glycation, and oxidation [39]. Phosphorylation is the best studied and important tau modification related to AD, with several kinases and some phosphatases working in concert to regulate phosphorylation at several specific serine, threonine, and tyrosine residues [39]. Of the kinases, two have been found to interact with CaM: Ca2 +/CaM-dependent protein kinase II (CaMKII) and cyclin-dependent kinase 5 (CDK5). In addition, protein phosphatase 2B (PP2B or calcineurin) is a well-established CaMBP that has been historically linked to tau dephosphorylation [40].
CaMKII phosphorylates tau in vitro at different sites including Ser262 which is in the microtubule binding domain of tau (Fig. 3) [41–44]. The enzyme was also found to phosphorylate tau in neurons at Ser416 [45]. CaM is responsible for activating CaMKII by binding to its regulatory segment through a 1–5–10 binding motif [10, 46]. However, CaMKII is capable of acquiring autonomy from CaM in the presence of high frequency Ca-2 + spikes, despite it being able to phosphorylate tau more efficiently in the presence of CaM [43, 46].
It should also be noted that CaMKII has an established role in neuronal apoptosis which is observed throughout the course of the disease. Phosphorylation by CaMKII is a central pro-apoptotic event. Normally a murine double mutant 2 (MDM2) pathway mediates the degradation of CaMKII but this process is defective in AD lymphocytes [47]. The role of CaM in apoptosis has been reviewed by Berchtold and Villalobo [48].
CDK5 is another kinase with a well-established role in tau phosphorylation (Fig. 4). Various studies indicate that it phosphorylates a number of specific residues including Ser202, Thr205, Ser369, and others, both in vitro and in vivo. Additionally, reducing CDK5 activity using RNA interference was found to reduce tau phosphorylation and the number of neurofibrillary tangles formed in transgenic mouse models, making it a possible target for AD treatment [54]. CDK5 has several activators, one of which is p35 [50]. Truncation of p35 into a shorter p25 fragment is observed in AD [50]. Unlike p35, p25 leads to constitutive CDK5 activation [50, 53]. The p10 fragment lost from p35 upon conversion to p25 is involved in the mutually exclusive binding of p35 to microtubules or CDK5 [55, 56]. The p10 segment also facilitates Ca2 +-dependent binding between p35 and CaM, which not observed with p25 (Fig. 4) [55]. It is possible that p35 contains a novel CaMBD since no canonical binding motifs were identified upon sequence analysis of the p35 peptide sequence [55]. Moreover, in the model organism Dictyostelium discoideum, CDK5 itself was found to bind CaM in a Ca2 +-independent manner [57]. The two CaMBDs found in D. discoideum CDK5 are highly conserved in human CDK5 indicating that the same interaction likely occurs in the human brain [57].
The role of the well documented CaMBP PP2B in tau dephosphorylation was established early but recently has been relegated to a less important role with the discovery of other phosphatases linked to the process. More to the point, studies on the function of PP2B in tau dephosphorylation have been contradictory. PP2B is a heterodimer made of A and B subunits. The A subunit is catalytic and known to be activated by CaM in a Ca2 +-dependent manner through a 1–8–14 binding motif while the B subunit is regulatory and is itself largely homologous to CaM and able to bind Ca2 + ions [58, 59]. Early studies in vitro showed PP2B’s ability to dephosphorylate tau at positions including Ser262 and Ser369 (Fig. 4) [60–62]. However, when compared to other phosphatases such as protein phosphatase 2A (PP2A) and protein phosphatase 1 (PP1), it was found that PP2B is only responsible for 7% of the dephosphorylation activity which has been attributed to its low affinity toward tau [63]. Recent studies have reported an increased enzyme activity level of PP2B in AD brains [63]. While this seems contradictory to previous findings, this increase was ascribed to the increased levels of calpain I [64]. Calpain I is Ca2 + activated and its activity is increased in AD brains. It cleaves the A subunit of PP2B and renders it CaM-independent; the truncated form of PP2B has higher activity than the full length protein [64]. Therefore, the persistent high level of phosphorylation observed should indicate that PP2B does not play a major role in tau dephosphorylation. On the other hand, in vivo studies using PP2B-specific inhibitors resulted in hyperphosphorylated tau at positions including Ser262, Ser369, Thr181, and Thr231 [65–67]. To complicate matters further, CaM binding to PP2B, which normally activates the enzyme, was found to inhibit its association with tau [40].
CaMKII AND PP2B ARE INVOLVED IN MEMORY STORAGE
Furthermore, the two enzymes CaMKII and PP2B have been implicated as central players in deficient memory storage that occurs due to the Ca2 + deregulation that occurs in AD individuals. These enzymes often act antagonistically by phosphorylating (CaMKII) and dephosphorylating (PP2B) the same proteins. Long-term potentiation (LTP) has been well studied in the mammalian hippocampus as it is the region of the brain that is critical in the formation and retrieval of various forms of memory. The morphology of hippocampal neurons including dendritic spine structures and their density are essential for memory storage. In the case of AD, progressive damage to the hippocampus results in an inability to form and store certain types of new memories concomitant with alterations in neuronal morphology, dendritic spine density, synapse formation, and deficits in LTP [68, 69]. Importantly, hyperactivation of the Ca2 +/CaM-dependent phosphatase calcineurin in the hippocampus has been reported for various models of AD and suggests that aberrant Ca2 + signaling contributes to deregulated interactions between CaM and calcineurin which impairs memory, increases tau phosphorylation and neuronal death [69, 70]. Short-term inhibition of calcineurin using the inhibitor FK506 was found to effectively reverse object recognition deficits in Tg2576 mice and improved dendritic spine loss, neuronal structure, and morphology in plaque bearing YFP-APP/PS1 transgenic mice [71]. Other work further demonstrated a direct connection between CaMKII activity in spines and spine loss in AD and aging brains [72]. Taken together, these data strongly suggest that the inhibition of calcineurin might protect hippocampal neurons and that the mechanism by which this occurs should be explored further. Recently, Berridge [73] has reviewed the positive role of CaMKII in memory formation and of PP2B in memory erasure. He has presented evidence arguing that the rapid memory erasure that occurs during wakefulness in AD individuals due to the action of PP2B prevents the ability of consolidating these memories during sleep which occurs in those not suffering from the disease. While much remains to be learned about this model, this again puts CaM and two of its primary targets— CaMKII and PP2B— at the center of issues facing those suffering from AD.
CaM AND THE ACETYLCHOLINE RECEPTOR
The oldest of all the models attempting to explain the causes and effects of AD, the “cholinergic hypothesis” is based on the decrease in acetylcholine neurocommunication [74, 75]. In AD, the levels of acetylcholine, acetylcholine receptors (AchR), and cholinergic neurons decrease [76]. Acetylcholine activates either inotropic nicotinic (nAchR) or metabotropic muscarinic (mAchR) receptors. The five subtypes (M1-M5) of mAchR are G-protein coupled receptors (GPCR) involved in Ca2 + signaling. M1 mAchR has been a primary therapeutic focus in the treatment of AD [77]. Like a number of verified GPCRs, M1 mAchR has a CaMBD on the cytoplasmic i3 loop [78]. Peptides derived from this intracellular loop bind to CaM. To date these peptides have not been used therapeutically. It should be noted that CaM also has indirect but major effects on the function of the mAchR first through regulation via phosphorylation (CaMKII) and dephosphorylation (PP2B) and second through more complex processes such as the formation of multiprotein complexes including the binding and activation of TRPC6 channels that are also involved in Ca2 + signaling [79].
NMDAR, A CALMODULIN-BINDING RECEPTOR
The N-methyl-D-aspartate receptor (NMDAR) is a major Ca2 + channel involved in synaptic plasticity and memory [80, 81]. The receptor binds glutamate allowing cations to enter the cell [80]. NMDAR levels are generally reduced in AD, likely as an adaptive mechanism in response to the increased receptor activity due to Ca2 + dysregulation [82]. Likewise, Aβ can cause overactivation of the receptor leading to increased intracellular Ca2 + levels, an effect which can be reduced through NMDAR antagonists [83]. In addition to affecting Ca2 + homeostasis, NMDAR plays a role in the cell’s antioxidant response, where it can augment the cell’s defenses under conditions of oxidative stress [84]. On the other hand, overstimulation of the receptor can also lead to free radical generation which can be detrimental [85].
NMDAR is a heteromultimeric complex with the required presence of at least one monomer of each of NR1 and NR2. Different combinations of NR1 splice products and NR2 subunits vary among different cell types offering the receptor varied pharmacological and physiological properties [81]. CaM is capable of binding the NR1 subunit both in vitro and in vivo [86–88]. Binding occurs at two locations, both of which are in the cytoplasmic domain of NR1, reducing the mean time of the channel being open and the frequency at which it opens thus inhibiting Ca2 + entry [81]. While its specific regulatory role is being elucidated, Apo-CaM can associate with the NR1 subunit at a different site than Ca2 + dependent CaM binding. Upon Ca2 + entry activated Ca2 +/CaM induces a rapid structural shift in the NMDAR allowing for an efficient negative feedback cycle [89]. Binding of NR1 to CaM occurs through a novel 1–7 motif which is highly unusual and only observed in myristoylated alanine-rich C-kinase substrate (MARCKS) [90].
OTHER ROLES FOR CALMODULIN IN ALZHEIMER’S DISEASE
Recent genome wide association studies (GWAS) have linked certain gene polymorphisms with LOAD [91–94]. Although the specific functions of each gene and its encoded protein remain to be elucidated, most are involved either in cholesterol metabolism, neuroinflammation or endocytosis [95]. Using the Calmodulin Target Database, the proteins discussed below were found to be putative CaMBPs but most remain to be experimentally verified (Table 2).
Cholesterol metabolism
The brain is rich in cholesterol, containing approximately 25% of the body’s cholesterol content which is essential for myelin formation [96]. An association between cholesterol and AD was made when statins, which are used to treat elevated cholesterol levels, were found to reduce disease incidence [97]. Furthermore, levels of 24S-hydroxycholesterol, one of the first intermediates of cholesterol elimination, were higher in AD individuals than in healthy controls [98]. Since it cannot cross the blood-brain barrier, the brain’s cholesterol is internally synthesized and must be shuttled around by lipoproteins containing ApoE [96, 99]. ApoE contains two putative CaMBDs each with 2-3 potential binding motifs (Table 2). While the role of CaM binding in ApoE function remains to be clarified, it is well established that the ApoEɛ4 variant is a significant risk factor for LOAD.
Clusterin (CLU) and ATP-binding cassette transporter A7 (ABCA7) are two other proteins involved in cholesterol metabolism with polymorphisms which are risk factors for LOAD [91–93]. Each possesses a single putative CaMBD as revealed through Calmodulin Target Database analysis (Table 2). The CLU protein is multifunctional affecting immunity and apoptosis in addition to playing a role in lipid metabolism [100]. With its expression increased in AD, CLU (also termed apolipoprotein J) is a component of the lipid particles that transport cholesterol [101]. ABCA7 belongs to the ABC transporter superfamily, which regulates cholesterol transport across the cell membrane [102]. However, it is likely that the highly homologous protein ABCA1, which is a proven CaMBP, plays a more prominent role in this process than ABCA7, according to mouse models. Binding through a 1–5–8–14 motif, CaM affects the stability of ABCA1 in vivo, preventing its degradation [103].
Neuroinflammation
Inflammation of neurons and dysregulation of different aspects of the immune response are characteristic of AD brains (reviewed in [104]). GWAS identified several proteins that affect the immune response. All were found through the Calmodulin Target Database to contain presumptive CaMBDs, although none have so far been experimentally tested to confirm the interaction with CaM (Table 2). Cluster of differentiation 33 (CD33), which is upregulated in microglial cells of AD brains, impairs Aβ clearance propagating plaque pathology [91, 105]. A second protein, the complement receptor 1 (Cr1) connects the complement system and CaM to AD although its exact role in propagation of pathology remains unknown [91, 106]. Moreover, polymorphisms in the membrane spanning 4-subfamily A (MS4A) proteins, MS4A4E and MS4A6A, were also found as risk factors for LOAD [91]. The function of the MS4A family remains poorly characterized, although, they may be components of Ca2 + channels and thus, affect Ca2 + homeostasis in neurons [107].
AβPP endocytosis
The AβPP protein is synthesized in the rough endoplasmic reticulum and travels through the trans-Golgi network to the cell membrane where it can be brought back into the cell through receptor-mediated endocytosis [108]. The exact subcellular location where Aβ is generated remains a topic of debate although endocytotic vesicles seem to be likely candidates since inhibition of endocytosis reduces the levels of Aβ produced [109]. A number of putative CaMBPs related to vesicle assembly were also identified including the phosphatidylinositol-binding clathrin assembly protein (PICALM), the scaffolding protein CD2-associated protein (CD2AP), the sortilin-related receptor L (SORL1), ephrin type-A receptor 1 (EPHA1), and the bridging integrator 1 (BIN1) (Table 2) [28]. To date, only BIN1 has been experimentally confirmed as a CaMBP[110].
AD, calmodulin, and the cell cycle
Although the amyloid cascade hypothesis dominates the AD research landscape, a number of alternative theories have been suggested in attempts to understand the pathogenesis of AD. In the developing brain neurons are integrated into complex synaptic networks after they have completed their proliferation, migration, and differentiation. The cellular signals that tightly regulate neuronal connectivity and plasticity also serve to ensure neurons are maintained in a differentiated state while simultaneously preventing reactivation of signaling pathways that control proliferation and cell cycle progression. The protection from aberrant neuronal cell cycle re-entry occurs through strict regulatory mechanisms at specific cell cycle control checkpoints. However, neuronal plasticity and formation of neuritic extensions allows for progression of the cycle early in G1 phase which is followed by opposing signals for re-differentiation back into G0. The mechanisms controlling transient re-entry into the cell cycle is considered to be an essential event for synaptic remodeling (reviewed in [111]).
In AD, the G1/S phase check-point regulatory mechanisms break down to the point where individual neurons can proceed through S-phase and undergo full or partial DNA re-replication with consequent entry into the G2 phase of the cell cycle [112–114]. Normally, in cycling non-neuronal cells, the G2 phase prepares the cell for mitosis, but in the case of post-mitotic neurons, improper cycling halts during the G2 phase and triggers neuronal death by apoptosis. Evidence from a number of animal models of human neurodegenerative disorders strongly suggests that atypical cell cycle re-entry events precede neuronal apoptosis ultimately leading to cell death [114–117]. Upregulation or improper protein degradation alters the normal level of a number of cell cycle proteins, including proliferating cell nuclear antigen (PCNA), cyclin D1, CDK4, and cyclin B1, within the hippocampus and other AD-diseased brain regions. These markers of cell cycle re-entry are not found randomly dispersed throughout the AD diseased brain nor are they detected in age-matched control patient brains [118, 119]. This ectopic cell cycle re-entry (CCR) is believed to account for a significant fraction of cortical neurons that are lost in AD [120]. Even more intriguing is that cell cycle markers are not only one of the earliest cellular abnormalities detected in AD, but theoretically may contribute to AD pathology including tau phosphorylation, Aβ formation, and neuronal calcium-ion dysfunction [121–123]. Seward et al. [124] further showed that CCR requires soluble Aβ, tau, and concomitant activation of kinases including the Ca2 +-calmodulin kinase II (CaMKII). This irregular cell cycle control within specific neuronal populations in the AD brain likely plays an early, yet crucial role for abnormalities associated with AD pathogenesis [117, 125, 126].
Studies in many cell types using a variety of approaches have implicated both Ca2 + and CaM as key regulators of distinct checkpoints in the cell cycle, including early G1, the G1 to S phase transition and G2/M transition. Calmodulin interactions with the CaMK family of CaMBPs act as important regulators of cell cycle progression (reviewed in [128]). Putative CaM-binding motifs have also been detected in a large number of cell cycle proteins including the cyclins (reviewed in [129]). Mitogenic stimulation leads to a variety of Ca2 +-CaM-mediated responses yet when the stimulation is permanent it causes changes in the sub-cellular distribution of CaM, which leads to changes in the total amount of CaM, alters cellular sensitivity to Ca2 + signals, and presents the potential for aberrant interactions between CaM and CaMBPs [130]. Although the deregulation of Ca2 +/calmodulin signaling in AD brains is not well understood, increased levels of CaM and decreased levels of phosphorylated CaMKII have been reported in the hippocampus of APP/PS1 mice [131]. These changes might be reflective of an aberrant involvement of CaM/CaMKII in the impairment of cell cycle control in AD. Indeed many different kinds of signaling pathways are changed in AD, and the relevance of the mitogenic upregulation that may induce cell cycle re-entry in the disease process is far from clear. Future studies will need to focus on identifying the mechanisms that regulate CaM and CaMKII expression in the hippocampus, as well as the downstream effector molecules involved, as this will potentially uncover new pathways for understanding the dysregulation of cell cycle control in ADbrains.
CALMODULIN AS A BIOMARKER
Since AD cannot be fully verified until autopsy, the search for minimally invasive biomarkers for the disease continues [132]. While cerebrospinal levels of Aβ, tau, and phosphorylated tau are effective biomarkers, this approach is fairly invasive and not without problems. Generally blood tests for those same biomarkers have been less than rewarding. Esteras et al. [133] have shown that levels of CaM are significantly increased in lymphoblasts from AD individuals. In a subsequent study, the increased CaM levels in lymphoblasts and peripheral blood mononuclear cells from AD individuals were found to be significantly greater than from non-dementia persons or those suffering from other types of dementia including amyotrophic lateral sclerosis, dementia with Lewy bodies, and frontotemporal dementi, among others [134]. These results suggest that CaM levels in peripheral blood cells have the potential to serve as a biomarker for various dementias including that resultingfrom AD.
CONCLUSION
Considering the multiple regulatory functions that CaM carries out in all cells, it is not surprising that it has many links to the underlying events of AD. CaM is involved in the defining aspects of AD progression and pathogenesis including Aβ generation, tau phosphorylation, Ca2 + homeostasis, cholesterol metabolism, neuroinflammation, AβPP endocytosis, and apoptosis. While it seems to present itself as a primary target in combating the symptoms and progression of the disease, many issues appear to reduce its current appeal as a therapeutic target. It has both positive and negative regulatory roles, making blanket inhibition approaches untenable. Thus currently available pharmaceutically proven CaM antagonists likely would not be an option for medical treatment. However, circumstances could change with the development of new CaM antagonists (e.g., [135]). Since most CaMBPs bind to CaM in unique ways and since some have multiple types of binding sequences, based on the technologies that exist for developing CaM antagonists it should be possible to develop target-specificpharmaceuticals.
The idea of targeting CaM and its CaMBPs is not without precedent. CaM is already a potential target for the treatment of Huntington’s disease, another neurodegenerative disease. Huntington’s disease is an autosomal dominant disorder due to a polyglutamine expansion in the huntingtin protein that is exacerbated by transglutaminase [136]. Huntingtin binds CaM and this binding can be disrupted using a CaM-peptide consisting of amino acids 76–121 of the CaM protein [137]. This peptide was then shown to reduce the level of transglutaminase-modified huntingtin and cytotoxicity in differentiated neuroblastoma (SH-SY5Y) cells. Subsequent treatment of a Huntington’s mouse (R6/2) model with this CaM-peptide led to neuroprotection via the mechanisms established in the tissue culture cells [138]. Thus CaM-Huntingtin binding appears to present a potential therapeutic target in combating this disease. The identification of critical CaMBPs linked to AD may also open a similardoor.
ACKNOWLEDGMENTS
Robert J. Huber is thanked for his constructive comments on a draft of this manuscript. This work was supported by the Natural Sciences and Engineering Research Council of Canada (DHO’D; A6807).
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/14-2772r2).
REFERENCES
1 | Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM(2007) Forecasting the global burden of Alzheimer’s diseaseAlzheimers Dement3: 186191 |
2 | Hippius H, Neundörfer G(2003) The discovery of Alzheimer’s diseaseDialogues Clin Neurosci5: 101108 |
3 | O’Day DH, Myre MA(2004) Calmodulin-binding domains in Alzheimer’s disease proteins: Extending the calcium hypothesisBiochem Biophys Res Commun230: 10511054 |
4 | Myre MA, Tesco G, Tanzi RE, Wasco W(2005) Calmodulin binding to APP and the APLPs.: A Joint Biochemical Society/Neuroscience Ireland Focused Meeting; March 13-16, University College Dublin, Republic of IrelandMolecular Mechanisms of Neurodegeneration |
5 | Chavez SE, O’Day DH (2007) Current Research on Alzheimer’s Disease 37 47 Calmodulin binds to and regulates the activity of beta-secretase (BACE1) Volume 1: Issue 1/2 Nova Scienceublishers, Inc Hauppage, New York |
6 | Canobbio I, Catricalà S, Balduini C, Torti M(2011) Calmodulin regulates the non-amyloidogenic metabolism of amyloid precursor protein in plateletsBiochem Biophys Acta1813: 500506 |
7 | Khachaturian ZS(1994) Calcium hypothesis of Alzheimer’s disease and brain agingAnn N Y Acad Sci747: 111 |
8 | Gareri P, Mattace R, Nava F, De Sarro G(1995) Role of calcium in brain agingGen Pharmacol26: 16511657 |
9 | Chin D, Means AR(2000) Calmodulin: A prototypical calcium sensorTrends Cell Biol10: 322328 |
10 | Rhoads AR, Friedberg F(1997) Sequence motifs for calmodulin recognitionFASEB J11: 331340 |
11 | Yap KL, Kim J, Truong K, Sherman M, Yuan T, Ikura M(2000) Calmodulin target databaseJ Struct Funct Genomics1: 814 |
12 | Tidow H, Nissen P(2013) Structural diversity of calmodulin binding to its target sitesFEBS J280: 55515565 |
13 | Villarroel A, Taglialatela M, Bernardo-Seisdedos G, Aliamo A, Agirre J, Alberdi A, Gomis-Perez C, Soldovieri MV, Ambrosino P, Malo C, Areso P(2014) The ever changing moods of calmodulin: How structurallasticity entails transductional adaptabilityJ Mol Biol426: 27172735 |
14 | Hardy JA, Higgins GA(1992) Alzheimer’s disease: The amyloid cascade hypothesisScience256: 184185 |
15 | Hardy J, Selkoe DJ(2002) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeuticsScience297: 353356 |
16 | Selkoe DJ(2001) Clearing the brain’s amyloid cobwebsNeuron32: 177180 |
17 | Sunde M, Blake CC(1998) From the globular to the fibrous state: Protein structure and structural conversion in amyloid formationQ Rev Biophys31: 139 |
18 | Kojro E, Fahrenholz F(2005) The non-amyloidogenic pathway: Structure and function of alpha-secretasesSubcell Biochem38: 105127 |
19 | Dobson CM(2004) Protein chemistry. In the footsteps of alchemistsScience304: 12591262 |
20 | Chow VW, Mattson MP, Wong PC, Gleichmann M(2010) An overview of APP processing enzymes and productsNeuromolecular Med12: 112 |
21 | McLachlan DR, Wong L, Bergeron C, Baimbridge KG(1987) Calmodulin and calbindin D28K in Alzheimer diseaseAlzheimer Dis Assoc Discord1: 171179 |
22 | Holsinger RM, McLean CA, Beyreyther K, Masters CL, Evin G(2002) Increased expression of the amyloid precursor beta-secretase in Alzheimer’s diseaseAnn Neurol51: 783786 |
23 | Laird FM, Cai H, Savonenko AV, Farah MH, He K, Melnikova T, Wen H, Chiang HC, Xu G, Koliatsos VE, Borchelt DR, Price DL, Lee HK, Wong PC(2005) BACE1, a major determinant of selective vulnerability of the brain to amyloid-beta amyloidogenesis, is essential for cofnitive, emotional, and synaptic functionsJ Neurosci25: 1169311709 |
24 | Saito Y, Sano Y, Vassar R, Gandy S, Nakaya T, Yamamoto Y, Suzuki T(2008) X11 proteins regulate the translocation of amyloid beta-protein precursor (APP) into detergent-resistent membrane and suppress the amyloidogenic cleavage of APP by beta-site-cleaving enzyme in the brainJ Biol Chem283: 3576335771 |
25 | Li H, Wolfe MS, Selkoe DJ(2009) Toward structural elucidation of the γ-secretase complexStructure17: 326334 |
26 | Michno K, Knight D, Campusano JM, van de Hoef D, Boulianne GL(2009) Intracellular calcium deficits incholinergic neurons expressing wild type or FAD-mutant presenilinPLoS One4: e6904 |
27 | Pacheco-Quinto J, Eckman EA(2013) Endothelin-converting enzymes degrade intracellular β-amyloid produced within endosomal/lysosomal pathway and autophagosomesJ Biol Chem288: 56065615 |
28 | O’Day DH, Myre MA (2007) Alzheimer’s Disease Research Trends Chan A Alzheimer’s disease: The calmodulin connection and β-amyloid Nova Biomedical Books NY 1 10 |
29 | Berrocal M, Sepulveda MR, Vazquez-Hernandez M, Mata AM(2012) Calmodulin antagonizes amyloid-β peptides-mediated inhibition of brain plasma membrane Ca(2+)-ATPaseBiochim Biophys Acta1822: 961969 |
30 | Nagano O, Murakami D, Hartmann D, De Strooper B, Saftig P, Iwatsubo T, Nakajima M, Shinohara M, Saya H(2004) Cell-matrix interaction via CD44 is independently regulated by different metalloproteinases activated in response to extraceullular Ca(2+) influx and PKC activationJ Cell Biol165: 893902 |
31 | Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, Kremmer E, Rossner S, Lichtenthaler SF(2010) ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neuronsEMBO J29: 30203032 |
32 | Horiuchi K, Le Gall S, Schulte M, Yamaguchi T, Reiss K, Murphy G, Toyama Y, Hartmann D, Saftig O, Blobel CP(2007) Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influxMol Biol Cell18: 176188 |
33 | Díaz-Rodríguez E, Esparís-Ogando A, Monero JC, Yuste L, Pandiella A(2000) Stimulation of cleavage of membrane proteins by calmodulin inhibitorsBiochem J346: 359367 |
34 | Lee HG, Perry G, Moreira PI, Garrett MR, Liu Q, Zhu X, Takeda A, Nunomura A, Smith MA(2005) Tau phosphorylation in Alzheimer’s disease: Pathogen or protector?Trends Mol Med11: 164169 |
35 | Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin M-L, Yardin C, Terro F(2013) Tau protein kinases; Involvement in Alzheimer’s diseaseAgeing Res Rev12: 289309 |
36 | Padilla R, Maccioni RB, Avila J(1990) Calmodulin binds to a tubulin binding site of the microtubule-associated protein tauMol Cell Biochem97: 3541 |
37 | Baudier J, Mochly-Rosen D, Newton A, Lee SH, Koshland DEJr, Cole RD(1987) Comparison of S100b protein with calmodulin: Interactions with melittin and microtubule-associated tau roteins and inhibition ofhosphorylation of tau poteins by protein kinase CBiochemistry26: 28862893 |
38 | Lee YC, Wolff J(1984) Calmodulin binds to both microtubule-associated protein 2 and tau proteinsJ Biol Chem259: 12261230 |
39 | Mietelska-Porowska A, Wasik U, Goras M, Filipek A, Niewiadomska G(2014) Tau protein modifications and interactions: Their role in function and dysfunctionInt J Mol Sci15: 4674146713 |
40 | Yu DY, Tong L, Song GJ, Lin WL, Zhang LQ, Bai W, Gong H, Yin YX, Wei Q(2008) Tau binds both subunits of calcineurin, and binding is impaired by calmodulinBiochem Biophys Acta1783: 22552261 |
41 | Steiner B, Mandelkow EM, Biernat J, Gustke N, Meyer HE, Schmidt B, Mieskes G, Söling HD, Dreschsel D, Kirschner MW(1990) Phosphorylation of microtubule-associaed protein tau: Identification of the site for Ca2(+)-calmodulin dependent kinase and relationship with tau phosphorylation in Alzheimer tanglesEMBO J9: 35393544 |
42 | Singh TJ, Wang JZ, Novak M, Kontzekova E, Grundke-Iqbal I, Iqbal K(1996) Calcium/calmodulin-dependent protein kinase II phosphorylates au at Ser-262 but only partially inhibits its binding to microtubulesFEBS Lett387: 145148 |
43 | Yamamoto H, Yamauchi E, Taniguchi H, Ono T, Miyamoto E(2002) Phosphorylation of microtubule-associaed protein tau by Ca2+/calmodulin-dependent protein kinase II in its tubulin binding sitesArch Biochem Biophys408: 255262 |
44 | Yoshimura Y, Ichinose T, Yamauchi T(2003) Phosphorylation of tau protein to sites found in Alzheimer’s disease brain is catalyzed by Ca2+/calmodulin-dependent protein kinase II as demonstrated tandem mass spectrometryNeurosci Lett353: 185188 |
45 | Yamamoto H, Hiragami Y, Murayama M, Ishizuka K, Kawahara M, Takashima A(2005) Phosphorylation of tau at serine 416 by Ca2+/calmodulin-dependent protein kinase II in neuronal soma in brainJ Neurochem94: 14381447 |
46 | Straton MM, Chao LH, Schilman H, Kuriyan J(2013) Structural studies on the regulation of Ca2+/calmodulin dependent protein kinase IICurr Opin Sruct Biol23: 292301 |
47 | Esteras N, Alquézar C, Bermejo-Pareja F, Bialopiotrowicz E, Wojda U, Martin-Requero A(2013a) Downregulation of extracellular signal-regulated kinase 1/2 activity by calmodulin KII modulates p21Cip1 levels and survival of immortalized lymphocytes from Alzheimer’s disease patientsNeurobiol Aging34: 10901100 |
48 | Berchtold MW, Villalobo A(2014) The many faces of calmodulin in cell proliferation, programmed cell death, autophagy and cancerBiochim Biopys Acta1843: 398435 |
49 | Lund ET, McKenna R, Evans DB, Sharma SK, Matthews WR(2001) Characterization of the in vitro phosphorylation of human tau by tau protein kinase II (cdk5/p20) using mass spectrometryJ Neurochem76: 12211232 |
50 | Hashiguchi M, Saito T, Hisanaga S, Hashiguchi T(2002) Truncation of CDK5 activator p35 induces intensive phosphorylation of Ser202/Thr205 of human tauJ Biol Chem277: 4452544530 |
51 | Noble W, Olm V, Takata K, Casey E, Mary O, Meyerson J, Gaynor K, Lafrancois J, Wang L, Kondo T, Davies P, Burns M, Nixon R, Dickson D(2003) Cdk5 is a key factor in tau aggregation and tangle formation in vivoNeuron38: 555565 |
52 | Götz J, Nitsch RM(2001) Compartmentalized tau hyperphosphorylation and increased levels of kinases in transgenic miceNeuroreport12: 20072016 |
53 | Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH(1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegenerationNature402: 615622 |
54 | Piedrahita D, Hernández I, López-Tobón A, Fedorov D, Obara B, Manjuath BS, Boudreau RL, Davidson B, LaFerla F, Gallego-Gómez JC, Kosik KS, Cardona-Gómez GP(2010) Silencing of CDK5 reduces neurofibrillary tangles in transgenic Alzheimer’s miceJ Neurosci30: 1396613976 |
55 | He L, Hou Z, Qi RZ(2008) Calmodulin binding and Cdk5 phosphorylation of p35 regulate its effect on microtubulesJ Biol Chem283: 1325213260 |
56 | Hou Z, Li Q, He L, Lim HY, Fu X, Cheung NS, Qi DX, Qi RZ(2007) Microtubule association of the neuronal p35 activator of Cdk5J Biol Chem282: 1866618670 |
57 | Huber RJ, Catalano A, O’Day DH(2013) Cyclin-dependent kinase 5 is a calmodulin-binding protein that associates with puromycin-sensitive aminopeptidase in the nucleus of DictyosteliumBiochem Biophys Acta1833: 1120 |
58 | Hoekman JD, Tokheim AM, Spannaus-Martin DJ, Martin BL(2006) Molecular modeling of the calmodulin binding region of calcineurinProtein J25: 175182 |
59 | Klee CB, Ren H, Wang X(1998) Regulation of the calmodulin-stimulated protein phosphatase, calcineurinJ Biol Chem273: 1336713370 |
60 | Drewes G, Mandelkow EM, Baumann K, Goris J, Merlevedede W, Mandelkow E(1993) Dephosphorylation of tau protein and Alzheimer paired helical filaments by calcineurin and phosphatase-2AFEBS Lett336: 425432 |
61 | Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K(1994) Alzheimer’s disease abnormally phosphorylated tau is dephosphorylated by protein phosphatase-2B (calcineurin)J Neurochem62: 803806 |
62 | Rahman A, Grundke-Iqbal I, Iqbal K(2006) PP2B isolated from human brain preferentially dephosphorylates Ser-262 and Ser-396 of the Alzheimer disease abnormally hyperphosphorylated tauJ Neural Transm113: 219230 |
63 | Liu F, Grundke-Iqbal I, Iqbal K(2005) Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylationEur J Neurosci22: 19421950 |
64 | Liu F, Grundke-Iqbal I, Iqbal K, Oda K, Tomizawa Y, Gong K, CX (2005) Truncation and activation of calcineurin A by calpain I in Alzheimer disease brainJ Biol Chem280: 3775537762 |
65 | Garver TD, Kincaid RL, Conn RA, Billingsley ML(1999) Reduction of calcineurin activity in brain by antisense oligonucleotides leads to persistent phosphorylation of tau protein at Thr181 and Thr231Mol Pharmacol55: 632641 |
66 | Yu DY, Luo J, Bu F, Song GJ, Zhang LQ, Wei Q(2006) Inhibition of calcineurin by infusion of CsA causes hyperphosphorylation of tau and is accompanied by abnormal behavior in miceBiol Chem387: 977983 |
67 | Luo J, Ma J, Yu DY, Bu F, Zhang W, Tu LH, Wei Q(2008) Infusion of FK506, a specific inhibitor of calcineurin, induces potent tau hyperphosphorylation in mouse brainBrain Res Bull76: 464468 |
68 | Koffie RM, Hyman BT, Spires-Jones TL(2011) Alzheimer’s disease: Synapses gone coldMol Neurodegener6: 63 |
69 | Reese LC, Taglialatela G(2011) A role for calcineurin in Alzheimer’s diseaseCurr Neuropharmacol9: 685692 |
70 | Wu HY, Hudry E, Hashimoto T, Kuchibhotla K, Rozkalne A, Fan Z, Spires-Jones T, Xie H, Arbel-Ornath M, Grosskreutz CL, Bacskai BJ, Hyman BT(2010) Amyloid beta induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activationJ Neurosci30: 26362649 |
71 | Rozkalne A, Hyman BT, Spires-Jones TL(2011) Calcineurin inhibition with FK506 ameliorates dendritic spine density deficits in plaque-bearing Alzheimer model miceNeurobiol Dis41: 650654 |
72 | Sun S, Zhang H, Liu J, Popugaeva E, Xu NJ, Feske S, White CL3rd, Bezprozvanny I(2014) Reduced synaptic STIM2 expression and impaired store-operated calcium entry cause destabilization of mature spines in mutant presenilin miceNeuron82: 7993 |
73 | Berridge MJ(2014) Calcium regulation of neural rhythms, memory and Alzheimer’s diseaseJ Physiol592: 281293 |
74 | Richter JA, Perry EK, Tomlinson BE(1980) Acetylcholine and choline levels in post-mortem human brain tissue: Preliminary observations in Alzheimer’s diseaseLife Sci26: 16831689 |
75 | Francis PT, Palmer AM, Snape M, Wilcock GK(1999) The cholinergic hypothesis of Alzheimer’s disease: A review of progressJ Neurol Neurosurg Psychol66: 137147 |
76 | Jiang S, Li Y, Zhang C, Zhao Y, Bu G, Xu H, Zhang Y-W(2014) M1 muscarinic acetycholine receptor in Alzheimer’s diseaseNeurosci Bull30: 295307 |
77 | Anand P, Singh B(2013) A review on cholinesterase inhibitors for Alzheimer’s diseaseArch Pharm Res36: 375399 |
78 | Lucas JL, Wang D, Sadée W(2006) Calmodulin binding to peptides derived from the i3 loop of muscarinic receptorsPharmaceutical Res23: 647653 |
79 | Kim JY, Saffen D(2005) Activation of M1 muscarinic acetylcholine receptors stimulates the formation of a multiprotein complex centered on TRPC6 channelsJ Biol Chem2080: 3203532047 |
80 | Zito K, Scheuss V(2009) NMDA receptor function and physiological modulationSquire LThe New Encyclopedia of NeuroscienceElsevier |
81 | Ehlers MD, Zhang S, Bernhadt JP, Hunganir RL(1996) Inactivation of NMDA receptors by direct interaction of calmodulin with the NR1 subunitCell84: 745755 |
82 | Danysz W, Parsons CG(2012) Alzheimer’s disease, β-amyloid, glutamate, NMDA receptors and memantine— searching for the connectionsBrit J Pharmacol167: 324352 |
83 | Alberdi E, Sánchez-Gómez MV, Cavaliere F, Pérez-Samartin A, Zugaza JL, Trullas R, Domercg M, Matute C(2010) Amyloid beta oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptorsCell Calcium47: 264272 |
84 | Papadia S, Soriano FX, Láveillé F, Martel MA, Dakin KA, Hansen HH, Kaindl A, Sifringer M, Fowler J, Stefovska V, McKenzie G, Craigon M, Corriveau R, Ghazal P, Horsburgh K, Yankner BA, Wyllie DJ, Ikonomidou C, Hardingham GE(2008) Synaptic NMDA receptor activity boosts intrinsic antioxidant defensesNat Neurosci11: 476478 |
85 | Gonzalez J, Jurado-Coronel JC, Avila MF, Sabogal A, Capani F, Barreto GE(2014) NMDARs in neurological diseases: A potential therapeutic targetInt J Neurosci124: 717723 |
86 | Hisatsune C, Umemori H, Inoue T, Michikawa T, Kohda K, Mikoshiba K, Yamamoto T(1997) Phosphorylation-dependent regulation of N-methyl-D-aspartate receptors by calmodulinJ Biol Chem272: 2080520810 |
87 | Rycroft BK, Gibb AJ(2002) Direct effects of calmodulin on NMDA receptor single-channel gating in rat hippocampal granule cellsJ Neurosci22: 88608868 |
88 | Zhang S, Huganir RL(1999) Calmodulin modification of NMDA receptorsMethods Mol Biol128: 103111 |
89 | Akyol Z, Bartos JA, Merrill MA, Faga LA, Jaren OR, Shea MA, Hell JW(2004) Apo-calmodulin binds with its C-terminal domain to the N-methyl-D-aspartate receptor NR1 C0 regionJ Biol Chem279: 21662175 |
90 | Ataman ZA, Gakhar L, Sorensen BR, Hell JW, Shea MA(2007) The NMDA receptor NR1 C1 region bound to calmodulin: Structural insights into functional differences between homologous domainsStructure15: 16031617 |
91 | Harold D, Abraham R, Hollingworth R, Sims R, Gerrish A, Hamshere M, Pahwa JS, Moskvina V, Dowzell K, Williams A, Jones N, Thomas C, Stretton A, Morgan A, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Morgan K, Brown K, Passmore P, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Love S, Kehoe PG, Hardy J, Mead S, Fox N, Rossor M, Collinge J, Maier W, Jessen F, Schürmann B, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frölich M, Hampel H, Hüll M, Rujescu D, Goate A, Kauwe JSK, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn P, van Broeckhoven C, Livingston G, Bass NJ, Gurling H, McQuillin Q, Gwilliam R, Deloukas P, Al-Chalabi A, Shaw CE, Tsolaki M, Singleton A, Guerreiro R, Mühleisen TW, Nöthen MM, Moebus S, Jöckel K-H, Klopp N, Wichmann H-E, Carrasquillo MM, Pankratz VS, Younkin SG, Holmans P, O’Donovan M, Owen MJ, Williams J(2009) Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s diseaseNat Genet41: 10881093 |
92 | Naj AC, Jun G, Beecham GW, Wang L-S, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, Graff-Radford NR, Philip L, De Jager PL, Denis Evans D, Julie A, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JSK, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, St George-Hyslop P, Arnold SE, Barber R, Beach T, Bigio EH, Bowen J, Boxer JD, Burke A, Cairns JR, Carlson NJ, Carney CS, Carrol RM, Chui SL, Clark HC, Corneveaux DG, Cotman J, Cummings CW, DeCarli JL, DeKosky C, Diaz-Arrastia ST, Dick R, Dickson M, Ellis DW, Faber WG, Fallon KM, Farlow KB, Ferris MR, Frosch S, Galasko MP, May Ganguli DR, Gearing M, Geschwind M, Ghetti DH, Gilbert B, Gilman JR, Giordani S, Glass B, Growdon JD, Hamilton JH, Harrell RL, Head LE, Honig E, Hulette LS, Hyman CM, Jicha BT, Jin GA, Johnson L-W, Karlawish N, Karydas J, Kaye A, Kim JA, Koo R, Kowall EH, Lah NW, Levey JL, Lieberman AI, Lopez AP, Mack OL, Marson WJ, Martiniuk DC, Mash F, Masliah DC, McCormick E, McCurry WC, McDavid SM, McKee AN, Mesulam AC, Miller M, Miller BL, Miller CA, Parisi JW, Perl JE, Peskind DP, Petersen E, Poon RC, WW , Quinn J, Rajbhandary JF, Raskind RA, Reisberg M, Ringman B, Roberson JM, Rosenberg ED, Sano RN, Schneider M, Seeley LS, Shelanski W, Slifer ML, Smith MA, Sonnen CD, Spina JA, Stern S, RA , Tanzi R, Trojanowski RE, Troncoso JQ, Van Deerlin JC, Vinters VM, Vonsattel HV, Weintraub JP, Welsh-Bohmer S, Williamson KA, Woltjer J, Cantwell RL, Dombroski LB, Beekly BA, Lunetta DB, Martin KL, Kamboh ER, Saykin MI, Reiman AJ, Bennett EM, Morris DA, Montine JC, Goate TJ, Blacker AM, Tsuang D, Hakonarson DW, Kukull H, Foroud WA, Haines TM, JL , Pericak-Vance1 R, Farrer MA, Schellenberg LA, GD (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s diseaseNat Genet43: 436441 |
93 | Hollingworth P, Harold D, Sims R, Gerrish A, Lambert J-C, Carrasquillo MM, Abraham R, Hamshere ML, Pahwa JS, Moskvina V, Dowzel K, Jones N, Stretton A, Thomas C, Richards A, Ivanov D, Widdowson C, Chapman J, Lovestone S, Powell J, Proitsi P, Lupton MK, Brayne C, Rubinsztein DC, Gill M, Lawlor B, Lynch A, Brown KS, Passmore PA, Craig D, McGuinness B, Todd S, Holmes C, Mann D, Smith AD, Beaumont H, Warden D, Wilcock G, Love S, Kehoe PG, Hooper NM, Vardy ERLC, Hardy J, Mead S, Fox NC, Rossor M, Collinge J, Maier W, Jessen F, Schürmann B, Rüther E, Heun R, Kölsch H, van den Bussche H, Heuser I, Kornhuber J, Wiltfang J, Dichgans M, Frölich L, Hampel H, Hüll M, Gallacher J, Rujescu D, Giegling I, Goate AM, Kauwe JSK, Cruchaga C, Nowotny P, Morris JC, Mayo K, Sleegers K, Bettens K, Engelborghs S, De Deyn PP, Van Broeckhoven C, Livingston G, Bass J, Gurling NJ, McQuillin H, Gwilliam A, Deloukas R, Al-Chalabi P, Shaw A, Tsolaki CE, Singleton M, Guerreiro AB, Mühleisen R, Nöthen TW, Moebus MM, Jöckel S, Klopp K-H, Wichmann N, Pankratz H-E, Sando VS, Aasly SB, Barcikowska JO, Wszolek M, Dickson ZK, Graff-Radford DW, Petersen NR, the Alzheimer’s Disease Neuroimaging Initiative RCvan Duijn CM, Breteler MMB, Ikram MA, DeStefano AL, Fitzpatrick AL, Lopez O, Launer LJ, Seshadri S, consortium CHARGEBerr C, Campion D, Epelbaum J, Dartigues J-F, Tzourio C, Alpérovitch A, Lathrop M, EADI1 consortiumFeulner TM, Friedrich P, Riehle C, Krawczak M, Schreiber S, Mayhaus M, Nicolhaus S, Wagenpfeil S, Steinberg S, Stefansson H, Stefansson K, Snædal J, Björnsson S, Jonsson PV, Chouraki V, Genier-Boley B, Hiltunen M, Soininen H, Combarros O, Zelenika D, Delepine M, Bullido MJ, Pasquier F, Mateo I, Frank-Garcia AF, Porcellini E, Hanon O, Coto E, Alvarez V, Bosco P, Siciliano G, Mancuso M, Panza F, Solfrizzi V, Nacmias B, Sorbi S, Bossù P, Piccardi P, Arosio B, Annoni G, Seripa D, Pilotto A, Scarpini E, Galimberti D, Brice A, Hannequin D, Licastro F, Jones L, Holmans PA, Jonsson T, Riemenschneider M, Morgan K, Younkin SG, Owen MJ, O’Donovan M, Amouyel P, Williams J(2011) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s diseaseNat Genet43: 429435 |
94 | Lambert J-C, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, Jun G, DeStefano AL, Bis JC, Beecham GW, Grenier-Boley B, Russo G, Thornton-Wells TA, Jones N, Smith AV, Chouraki V, Thomas C, Ikram MA, Zelenika D, Vardarajan BN, Kamatani Y, Lin C-F, Gerrish A, Schmidt H, Kunkle B, Dunstan ML, Ruiz A, Bihoreau MT, Choi S-H, Reitz C, Pasquier F, Hollingworth P, Ramirez A, Hanon O, Fitzpatrick AL, Buxbaum JD, Campion D, Crane PK, Baldwin C, Becker T, Gudnason V, Cruchaga C, Craig D, Amin N, Berr C, Lopez OL, De Jager PL, Deramecourt V, Johnston JA, Evans D, Lovestone S, Letenneur L, Morón FJ, Rubinsztein DC, Eiriksdottir G, Sleegers K, Goate AM, Fiévet N, Huentelman MJ, Gill M, Brown K, Kamboh MI, Keller L, Barberger-Gateau P, McGuinness B, Larson EB, Green R, Myers AJ, Dufouil C, Todd S, Wallon D, Love S, Rogaeva E, Gallacher J, St George-Hyslop P, Clarimon J, Lleo A, Bayer A, Tsuang DW, Yu L, Tsolaki M, Bossù P, Spalletta G, Proitsi P, Collinge PJ, Sorbi S, Sanchez-Garcia F, Fox NC, Hardy J, Naranjo MCD, Bosco P, Clarke R, Brayne C, Galimberti D, Mancuso M, Matthews F, European Alzheimer’s disease Initiative (EADI)Genetic, Environmental Risk in Alzheimer’s Disease (GERAD)Alzheimer’s Disease Genetic Consortium (ADGC)Cohorts for HeartAging Research in Genomic Epidemiology (CHARGE)Moebus S, Mecocci P, Del Zompo M, Maier W, Hampel H, Pilotto A, Bullido M, Panza F, Caffarra P, Nacmias B, Gilbert JR, Mayhaus M, Lannfelt L, Hakonarson H, Pichler S, Carrasquillo MM, Ingelsson M, Beekly D, Alvarez V, Zou F, Valladares O, Younkin SG, Coto E, Hamilton-Nelson K-L, Gu W, Razquin C, Pastor P, Mateo I, Owen MJ, Faber KM, Jonsson PV, Combarros O, O’Donovan MC, Cantwell LB, Soininen H, Blacker D, Mead S, Mosley TH, Bennett DA, Harris TB, Fratiglioni L, Holmes C, de Bruijn RFAG, Passmore P, Montine TJ, Bettens K, Rotter JI, Brice A, Morgan K, Foroud TM, Kukull WA, Hannequin D, Powell JF, Nalls MA, Ritchie KR, Lunetta KL, Kauwe1 JSK, Boerwinkle E, Riemenschneider M, Boada M, Hiltunen M, Martin ER, Schmidt R, Rujescu D, Wang L-S, Dartigues J-F, Mayeux R, Tzourio C, Hofman A, Nöthen MM, Graff C, Psaty BM, Jones L, Haines JL, Holmans PA, Lathrop M, Pericak-Vance MA, Launer LJ, Farrer LA, van Duijn CM, Van Broeckhoven C, Moskvina V, Seshadri S, Williams J, Schellenberg GD, Amouyel P(2013) Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s diseaseNat Genet45: 14521458 |
95 | Karch CM, Goate AM(2015) Alzheimer’s disease risk genes and mechanisms of disease pathogenesisBiol Psychiatry77: 4351 |
96 | Kang J, Rivest S(2012) Lipid metabolism and neuroinflammation in Alzheimer’s disease: A role for liver X receptorsEndocr Rev33: 715746 |
97 | Fassbender K, Simons M, Bergmann C, Stroick M, Lutjohan D, Keller P, Runz H, Kuhl S, Bertsch T, von Gergmann K, Hennerici M, Meyreuther K, Hartman T(2001) Simvastatin strongly reduces levels of Alzheimer’s disease beta-amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivoProc Nat Acad Sci U S A98: 58565861 |
98 | Schönknecht P, Lütjohann D, Pantel J, Bardenheuer H, Hartmann T, von Bergmann K, Beyreuther K, Schröder J(2002) Cerebrospinal fluid 24S-hydroxycholesterol is increased in patients with Alzheimer’s disease compared to healthy controlsNeurosci Lett324: 8385 |
99 | Jurevics H, Morell P(1995) Cholesterol for synthesis of myelin is made locally, not imported into brainJ Neurochem64: 895901 |
100 | Jones SE, Jomary C(2002) ClusterinInt J Biochem Cell Biol34: 427431 |
101 | Nuutinen T, Suuronen T, Kauppinen A, Salminen A(2009) Clusterin: A forgotten player in Alzheimer’s diseaseBrain Res Rev61: 89104 |
102 | Kim WS, Weickert CS, Garner B(2008) Role of ATP-binding cassette transporters in brain lipid transport and neurological diseaseJ Neurochem104: 11451166 |
103 | Iwamoto N, Lu R, Abe-Dohmae S, Yokoyama S(2010) Calmodulin interacts with ATP binding cassette transporter A1 to protect from calpain-mediated degradation and upregulates high-density lipoprotein generationArterioscler Thromb Vasc Biol30: 14461452 |
104 | Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T(2000) Inflammation and Alzheimer’s diseaseNeurobiol Aging21: 383421 |
105 | Jiang T, Yu JT, Hu N, Tan MS, Zhu XC, Tan L(2014) CD33 in Alzheimer’s diseaseMol Neurobiol49: 529535 |
106 | Crehan H, Holton P, Wray S, Pocock J, Guerreiro R, Hardy J(2012) Complement receptor 1 (CR1) and Alzheimer’s diseaseImmunobiology217: 244250 |
107 | Ma J, Yu JT, Tan L(2014) MS4A cluster in Alzheimer’s diseaseMol Neurobiol10.1007/s12035-014-8800-z |
108 | Choy RW, Cheng Z, Schekman R(2012) Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid β (Aβ) production in the trans-Golgi networkProc Natl Acad Sci U S A109: E2077E2082 |
109 | Carey RM, Balcz BA, Lopez-Coviella I, Slack BE(2005) Inhibition of dynamin-dependent endocytosis increases shedding of the amyloid precursor protein ectodomain and reduces generation of amyloid beta proteinBMC Cell Biol6: 30 |
110 | Berggård T, Arrigoni G, Olsson O, Fex M, Linse S, James P(2006) 140 mouse brain proteins identified by Ca2+-calmodulin affinity chromatography and tandem mass spectrometryJ Proteome Res5: 669687 |
111 | Arendt T(2005) Alzheimer’s disease as a disorder of dynamic brain self-organizationProg Brain Res147: 355378 |
112 | Nagy Z(2000) Cell cycle regulatory failure in neurones: Causes and consequencesNeurobiol Aging21: 761769 |
113 | Zhu X, McShea A, Harris PL, Raina AK, Castellani RJ, Funk JO, Shah S, Atwood C, Bowen R, Bowser R, Morelli L, Perry G, Smith MA(2004) Elevated expression of a regulator of the G2/M phase of the cell cycle, neuronal CIP-1-associated regulator of cyclin B, in Alzheimer’s diseaseJ Neurosci Res75: 698703 |
114 | Yang Y, Geldmacher DS, Herrup K(2001) DNA replication precedes neuronal cell death in Alzheimer’s diseaseJ Neurosci21: 26612668 |
115 | Nagy ZS, Esiri MM(1997) Apoptosis-related protein expression in the hippocampus in Alzheimer’s diseaseNeurobiol Aging18: 565571 |
116 | Raina AK, Hochman A, Zhu X, Rottkamp CA, Nunomura A, Siedlak SL, Boux H, Castellani RJ, Perry G, Smith MA(2001) Abortive apoptosis in Alzheimer’s diseaseActa Neuropathol101: 305310 |
117 | Herrup K, Neve R, Ackerman SL, Copani A(2004) Divide and die: Cell cycle events as triggers of nerve cell deathJ Neurosci24: 92329239 |
118 | Busser J, Geldmacher DS, Herrup K(1998) Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer’s disease brainJ Neurosci18: 28012807 |
119 | Khurana V, Lu Y, Steinhilb ML, Oldham S, Shulman JM, Feany MB(2006) TOR-mediated cell-cycle activation causes neurodegeneration in atauopathy modelCurr Biol16: 230241 |
120 | Arendt T, Bruckner MK, Mosch B, Losche A(2010) Selective cell death of hyperploid neurons in Alzheimer’s diseaseAm J Pathol177: 1520 |
121 | Walsh DM, Selkoe DJ(2004) Deciphering the molecular basis of memory failure in Alzheimer’s diseaseNeuron44: 181193 |
122 | Zhu X, Rottkamp CA, Boux H, Takeda A, Perry G, Smith MA(2000) Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer diseaseJ Neuropathol Exp Neurol59: 880888 |
123 | Mattson MP, Chan SL(2003) Neuronal and glial calcium signaling in Alzheimer’s diseaseCell Calcium34: 385397 |
124 | Seward ME, Swanson E, Norambuena A, Reimann A, Cochran JN, Li R, Roberson ED, Bloom GS(2013) Amyloid-β signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer’s diseaseJ Cell Sci126: 12781286 |
125 | Yang Y, Varvel NH, Lamb BT, Herrup K(2006) Ectopic cell cycle events link human Alzheimer’s disease and amyloid precursor protein transgenic mouse modelsJ Neurosci26: 775784 |
126 | Lee HG, Casadesus G, Zhu X, Castellani RJ, McShea A, Perry G, Petersen RB, Bajic V, Smith MA(2009) Cell cycle re-entry mediated neurodegeneration and its treatment role in the pathogenesis of Alzheimer’s diseaseNeurochem Int 548488 |
127 | Kahl CR, Means AR(2003) Regulation of cell cycle progression by calcium/calmodulin-dependent pathwaysEndocr Rev24: 719736 |
128 | Skelding KA, Rostas JA, Verrills NM(2011) Controlling the cell cycle: The role of calcium/calmodulin-stimulated protein kinases I and IICell Cycle4: 631639 |
129 | Choi J, Husain M(2006) Calmodulin-mediated cell cycle regulation: New mechanisms for old observationsCell Cycle5: 21832186 |
130 | Gnegy ME(1993) Calmodulin in neurotransmitter and hormone actionAnnu Rev Pharmacol Toxicol33: 4570 |
131 | Min D, Guo F, Zhu S, Xu X, Mao X, Cao Y, Lv X, Gao Q, Wang L, Chen T, Shaw C, Hao L, Cai J(2013) The alterations of Ca2+/calmodulin/CaMKII/CaV1.2 signaling in experimental models of Alzheimer’s disease and vascular dementiaNeurosci Lett538: 6065 |
132 | Young AL, Oxtoby NP, Daga P, Cash DM, Fox NC, Ourselin S, Schott JM, Alexander DC(2014) A data-driven model of biomarker changes in sporadic Alzheimer’s diseaseBrain137: 25642577 |
133 | Esteras N, Munoz U, Alquézar C, Bartolome F, Bermejo-Pareja F, Martin-Requero A(2012) Altered calmodulin degradation and signaling in non-neuronal cells from Alzheimer’s disease patientsCurr Alzheimer Res9: 267277 |
134 | Esteras N, Alquézar C, de la Encarnación A, Vallarejo A, Bermejo-Pareja F, Martin-Requero A(2013) Calmodulin levels in blood cells as a potential marker of Alzheimer’s diseaseAlzheimers Res Ther5: 55 |
135 | Audran E, Dagher R, Gioria S, Tsvetkov PO, Kulikova AA, Didier B, Villa P, Makarov AA, Kilhoffer M-C, Haiech J(2013) A general framework to characterize inhibitors of calmodulin: Use of calmodulin inhibitors to study the interaction between calmodulin and its calmodulin binding domainsBiochim Biophys Acta1833: 17201731 |
136 | Walling HW, Baldassare JJ, Westfall TC(1998) Molecular aspects of Huntington’s diseaseJ Neurosci Res50: 301308 |
137 | Dudek NL, Dai Y, Muma NA(2008) Protective effects of interrupting the binding of calmodulin to mutant huntingtinJ Neuropathol Exp Neurol67: 355365 |
138 | Dudek NL, Dai Y, Muma NA(2010) Neuroprotective effects of calmodulin peptide 76-121aa: Disruption of calmodulin binding to mutant huntingtinBrain Pathol20: 176189 |
Figures and Tables
Fig.1
Calcium-bound calmodulin (CaM) and its calcium-free form (Apo-CaM) are involved to many central events linked to Alzheimer’s disease as discussed in this review.
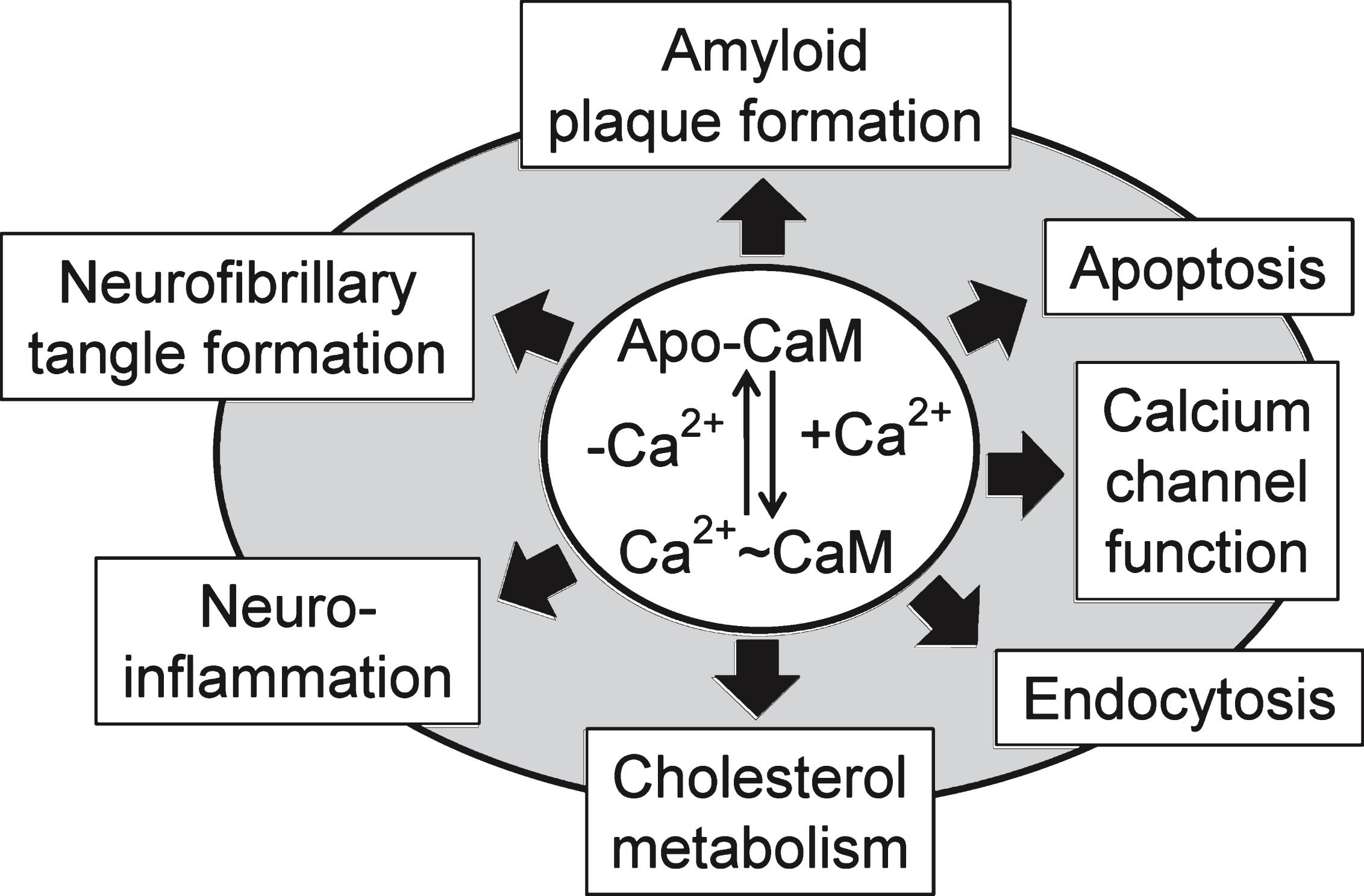
Fig.2
Apo-Calmodulin (Apo-CaM) can bind up to two calcium ions in each of its N-term (pink) or C-term lobes (green). These binding options affect the conformation of CaM and its ability to bind to specific target CaM-binding proteins.
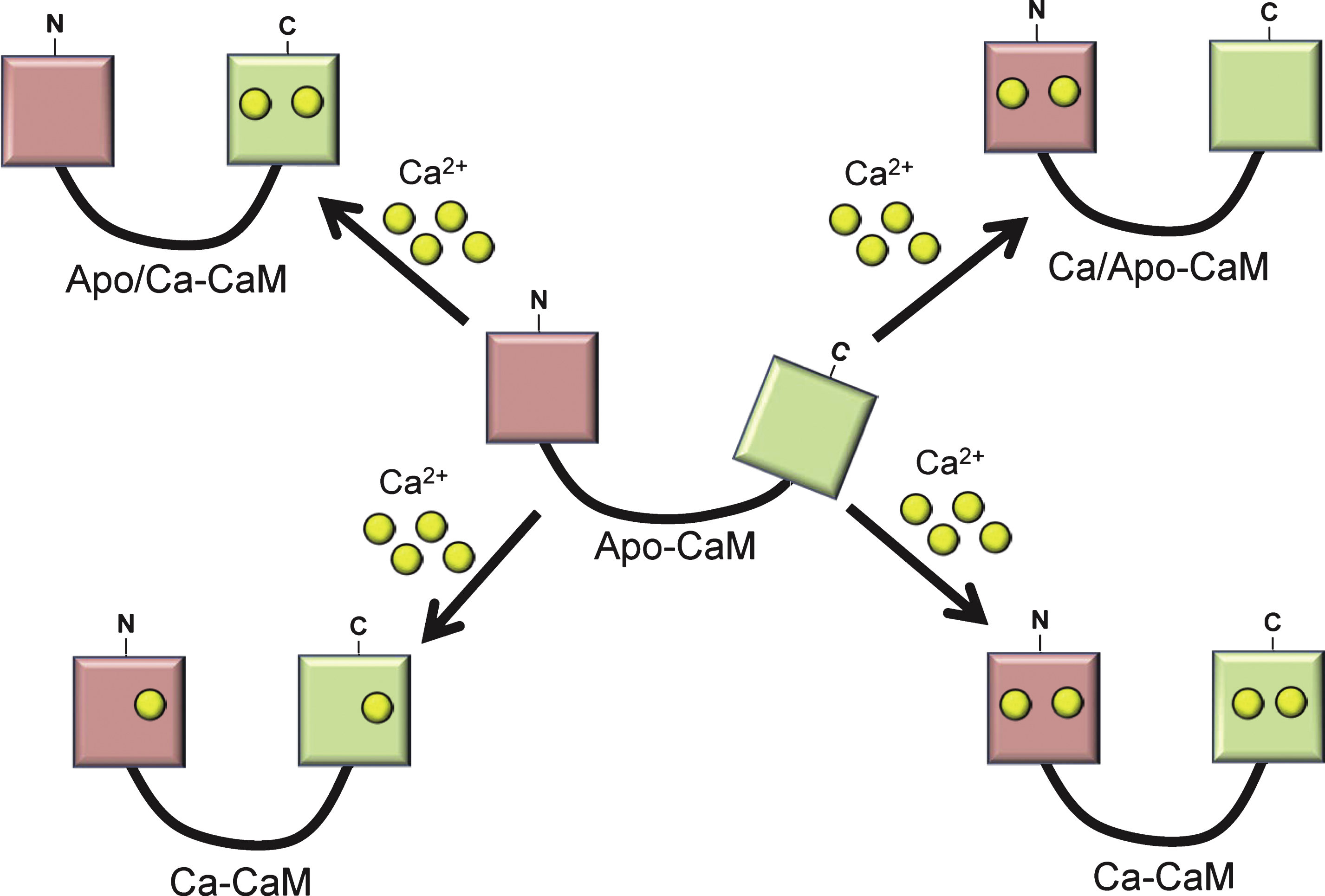
Fig.3
Calmodulin binding proteins linked to the amyloidogenenic and non-amyloidogenic pathways. amyloid-β precursor protein (AβPP); anterior pharynx defective 1 (APH-1); β site-amyloid converting enzyme 1 or β-secretase (BACE1); A Disintegrin And Metalloproteinase Family (ADAM 9,10,17, 19); endothelin-converting enzyme (ECE); insulin-degrading enzyme (IDE); nicastrin (Nic); presenilin enhancer protein 2 (PEN-2); and presenilin-1 (PSN-1); superscripts: *putative CaMBD detected; vCaMBD experimentally verified.
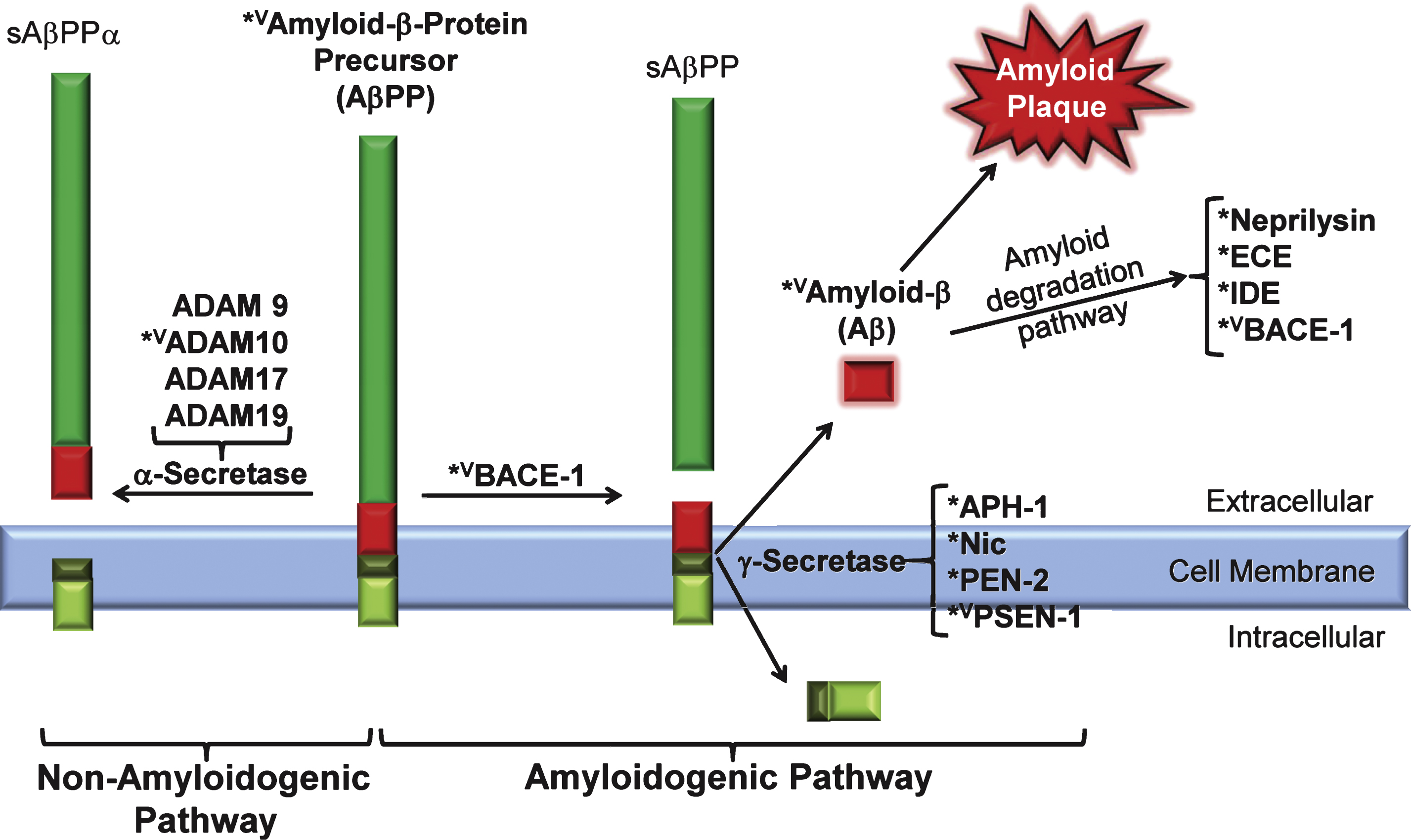
Fig.4
Calmodulin binding proteins linked to tau phosphorylation. Calmodulin-dependent kinase II (CaMKII); cyclin-dependent kinase 5 (CDK5); calmodulin (CaM); neurofibrillary tangles (NFTs); CDK5 activator (P35); protein phosphatase 2B (PP2B); colors: green, stimulated/activated; red, inhibited.
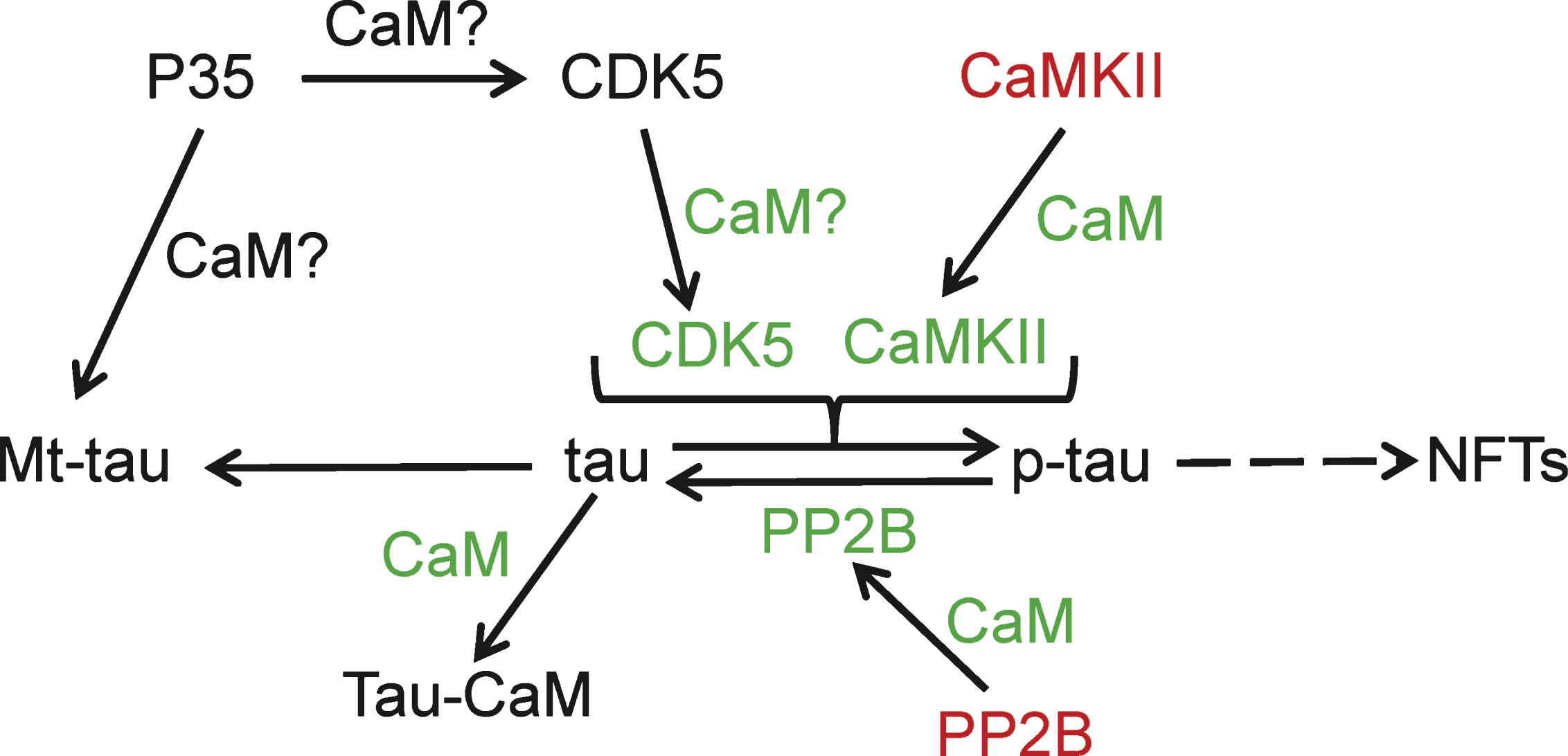
Table 1
Calmodulin binding domain (CaMBD) subclasses that have been identified and experimentally verified
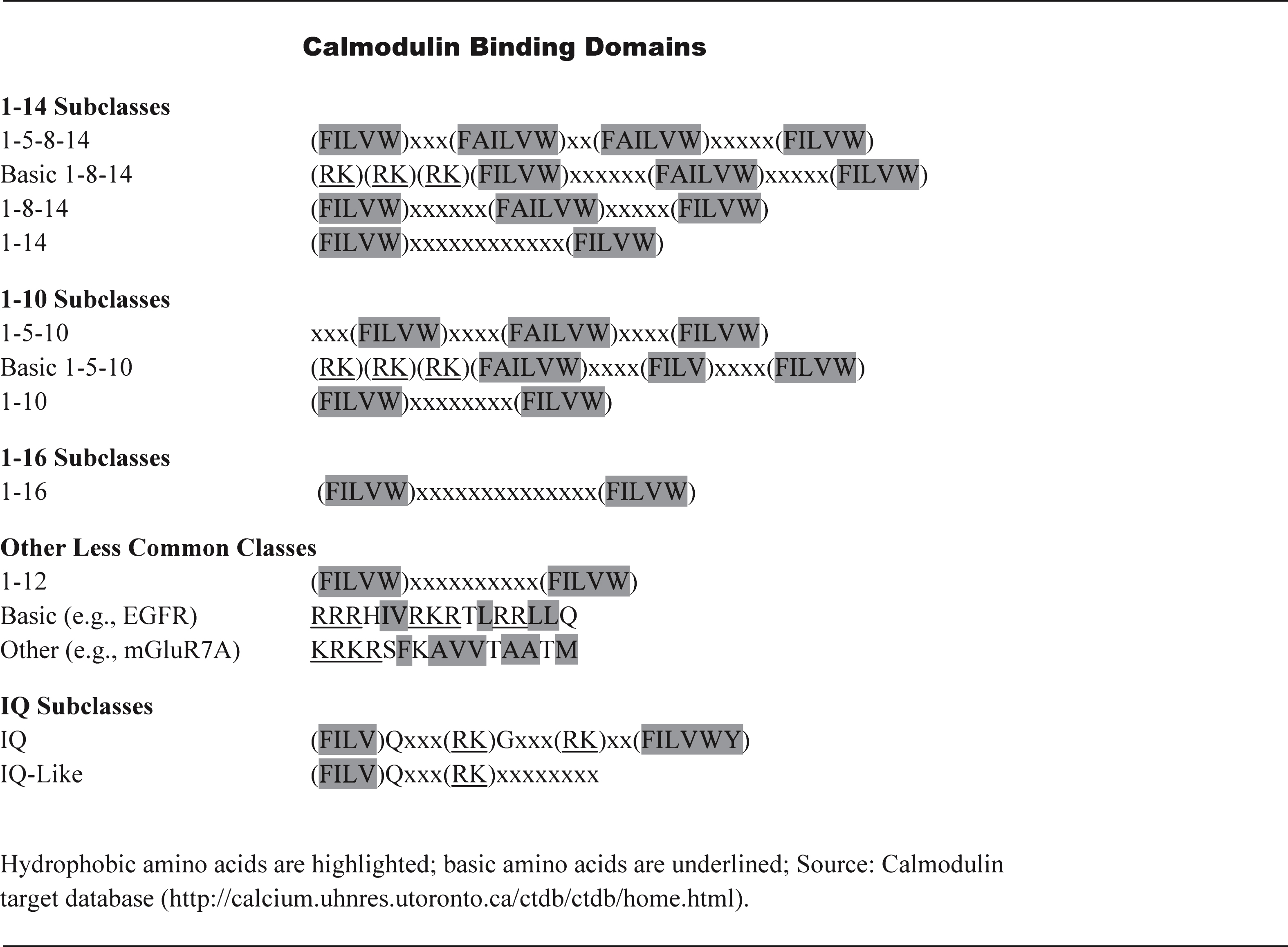 |
Table 2
Putative calmodulin binding domain (CaMBD) classes in GWAS proteins linked to Alzheimer’s disease. Appropriate hydrophobic (green), acidic (yellow), and IQ (cyan) amino acids are highlighted.
 |


