
Microglia as a Possible Alternative Therapeutic for Dementia
Abstract
Dementia is a syndrome in which there is deterioration in memory, behavior, and the ability to perform everyday activities. Alzheimer’s disease and vascular dementia are the most common forms of dementia. There is evidence supporting the hypothesis that inflammatory and immune mechanisms are involved in dementia. Microglia, the resident macrophage tissues in the central nervous system, play a significant role in neuroinflammation and play an important role in amyloid-β clearance in the brain, and impaired microglial clearance of amyloid-β has also been shown to be involved in the pathogenesis of Alzheimer’s disease. However, there is also abundant evidence that microglia have harmful actions in dementia. Once activated, they can mediate uptake at neuronal synapses. They can also exacerbate tau pathology and secrete deleterious inflammatory factors that can directly or indirectly damage neurons. Thus, depending on the stage of the disease, microglia can act both protectively and detrimentally. Therefore, it is still necessary to continue with studies to better understand the role of microglia in the pathology of dementia. Currently available drugs can only improve cognitive symptoms, have no impact on progression and are not curative, so identifying and studying new therapeutic approaches is important. Considering the role played by microglia in this pathology, it has been pointed out as a possible therapeutic approach. This manuscript aims to address the relationship between microglia and dementia and how this relationship could be used for therapeutic purposes.
INTRODUCTION
Dementia is an umbrella term that includes different diseases that are almost all progressive, affecting memory, behavior and other cognitive abilities, and that also greatly interfere with the ability of people to carry out their daily activities [1]. In 2015, dementia affected approximately 5% of the elderly population worldwide; this means that 47 million people lived with dementia, and there are forecasts that indicate that in 2050 there will be nearly 152 million of people with this condition [2, 3].
Dementia is one of the leading causes of disability and dependency among the elderly worldwide; it leads to higher costs for government, communities, families and individuals; moreover it has a great impact on various areas of the lives of patients [1]. It is caused by different diseases and injuries that affect the brain directly or indirectly, such as Alzheimer’s disease (AD) or stroke, related to vascular dementia (VaD). Mixed forms are very common, since the limits between the subtypes are not very clear because the diagnosis is clinical, considering symptoms and patterns of onset and course of the disease [4]. With 60–70% of cases, AD is the most common form [4], the second being VaD, which represents 17–25% of dementia cases [5].
Being a complex disease, there are various factors involved in the pathogenesis of dementia, and although the causes of neurodegeneration remain unknown, the role of the immune system has been a rapidly-expanding field of research [6–8]. Although there are great differences in the neuropathology of AD and other dementias of vascular origin, they share certain characteristics, such as neuroinflammation and mechanisms related to the immune system, which have been significantly implicated in the pathophysiology of both AD and of VaD [9]. Immune system dysfunction has attracted much attention due to its correlation with the progression of AD. Clinical and genetic evidence has shown that immune cells can undergo significant dynamic changes and perform diverse functions at different stages of the disease [10]. On the other hand, neuroinflammation is an immunological cascade reaction that is mediated by glial cells in the central nervous system (CNS) where innate immunity is found. Inflammatory reactions can be triggered by various damaging events, such as infection, ischemia, or hypoxia. Ischemia and hypoxia produced by long-term chronic hypoperfusion can overactivated neuroinflammation, causing apoptosis, damage to the blood-brain barrier (BBB), and other pathologic changes, triggering or aggravating the onset and progression of vascular cognitive impairment [11].
Given the implications for both health and economics, finding effective treatments against dementia has been a priority issue; however, it has not had the desired success. The relationship between microglia and dementia has been pointed out as a new therapeutic target [6–8]. However, further studies are needed to understand the spatiotemporal pattern of neuroinflammation modulated by microglia in the pathogenesis of vascular dementia [12], and also to clarify the role of microglia in AD pathology [13]. This manuscript aims to address the relationship between microglia and dementia, and how this relationship could be used for therapeutic purposes.
THE GLIAL CELLS IN THE BRAIN
While neurons are the highly specialized cells in the brain, they are completely dependent on glial cells. These non-neuronal cells, such as oligodendrocytes, astrocytes, and microglia, support essential brain functions, such as maintaining neurotransmitter communication, metabolism, trophic support, myelin sheath formation, wound healing and immune surveillance [14].
Oligodendrocytes are myelinating brain cells that keep neurons healthy [15]. In addition to providing insulation to the axons, they provide trophic support, affect the structure of the axons, and modify their electrical properties by controlling their diameter and the grouping of specific ion channels in the nodal and paranodal region. They are responsible for the propagation of information at high speed in the axons of vertebrates [14].
Astrocytes, which constitute about 20–40% of the brain cells, are the main secretory cells, they release gliotransmitters, including neurotransmitters. In an activated state, activated microglia and reactive astrocytes release soluble factors (glutamate, reactive oxygen species (ROS) and cytokines) that adversely influence neurons and trigger neuroinflammatory signals in them [15, 16]. Due to their important neuroinflammatory activity, astrocytes are generally considered another important response to neurodegeneration [17].
Astrocytic factors help neurons maintain normal physiological functioning and the maintenance of cerebral vasculature. Furthermore, astrocytes play an important role in synaptic pruning due to their phagocytic property [18]. Recent evidence suggests that astrocytes may aid the development of neurodegenerative diseases due to their primary role in neuronal function and metabolism [17].
Microglia act as the first line of defense; is they are the main brain neuroimmune soldiers, continually identifying changes in their environment and responding to toxins, invading pathogens, and cellular debris [19]. In response to a pathogen, microglia act rapidly, acquiring different phenotypes to defend neurons. During neuroinflammation, reactive microglia and astrocytes secrete soluble mediators that negatively affect neuronal health and promote neurodegeneration [15].
Microglia represent 0.5% to 16% of all glial cells in humans, depending on the region studied [20, 21]. The microglia have great functional diversity; some of their characteristics and functions are listed in Table 1. In addition to defending against infectious pathogens, traumatic brain injury, and neurodegenerative diseases, microglia contribute to brain homeostasis. In recent times, there has been an increase in research to try to elucidate the role of microglia in the healthy and diseased brain, and in aging [19]. Microglial phagocytosis removes damaged neurons that disrupt the brain, dying neurons before they become necrotic and release debris, and dead neurons and their debris, which are pro-inflammatory and cause brain disruption [22, 23], and they also have an important role after a stroke. They have a central role in neuroinflammation and in amyloid-β (Aβ) clearance in the brain [24, 25].
Table 1
Characteristics and functions of microglia
| Characteristic | Description |
| Morphological | Classified into three types: branched at rest (quiescent), activated, and amoeboid (phagocytic) [20]. |
| Main function | Maintaining homeostasis by removing cellular debris, dying cells, or misfolded proteins [89]. |
| Other functions | It participate in the establishment of normal neuronal connectivity and critical regulatory processes for CNS development, such as synaptic pruning [90]. |
| In the developing brain, microglia sculpt neuronal synapses [88]. | |
| In the adult brain, microglia are involved in monitoring changes in neuronal activity, modulating higher cognitive functions such as learning and memory, and acting as local phagocytes and damage sensors in the brain parenchyma [88, 89]. |
CNS, central nervous system.
One of the main questions regarding the role of microglia is whether their function is protective or detrimental against neurodegenerative diseases. With the existing evidence, it has been concluded that this depends on the characteristics of the disease, the specific microglial pathway and the stage of the disease [26].
Likewise, scientific advances in genetics have made it possible to identify many genes associated with neurodegenerative diseases, allowing a better understanding of pathological processes from another perspective. Particularly for AD, genome-wide association studies (GWAS) and gene expression studies have pointed to the pathogenic contributions of microglial cells and prompted studies of microglial function/dysfunction [26].
ROLE OF MICROGLIA IN AGING
Aging leads to abnormal neuronal activity, impaired biological function, decreased synaptic plasticity, deregulation of neuronal Ca2 + homeostasis, and inflammation in brain cells; these characteristics impair mental and physical functions and contribute to neurodegenerative diseases [27]. Specifically, cellular senescence and chronic inflammation in response to aging are considered indicators of brain aging and important factors in neurodegeneration. Cellular senescence is defined as an irreversible arrest of the cell cycle caused by various internal and external factors [28]. Meanwhile, although inflammation is a protective mechanism, inflammation as a response to cellular senescence and aging turns out to be harmful since it promotes the aging of the organism.
During this aging process, damage occurs to the innate and acquired immune system. An increase in the expression of proinflammatory cytokines (including tumor necrosis factor alpha (TNF-α), interleukin (IL)-1β, IL-6, IL-8, ROS, C-C motif chemokine ligands (CCL-2, and CCL-5)), which promote an inflammatory response [29]. This response chronically induces neuroinflammation, causing neuronal damage, which leads to age-related neurodegeneration.
This process is very complex since a vicious circle is generated, in which the chronic inflammatory response limits the beneficial functions of microglia and exerts neurotoxic effects by increasing the release of inflammatory cytokines and inhibiting neuronal regeneration. In aging misfolded proteins, cellular debris and other inflammatory stimuli accumulate in the brain, which induces continuous stimulation of microglia and a decrease in the phagocytic potential and surveillance capacity, triggering an increase in production of harmful inflammatory substances and thus promoting aging [27].
ROLE OF MICROGLIA IN NEURODEGENERATIVE DISEASES
Microglia-mediated neurotoxic neuroinflammation is a common feature of several neurodegenerative diseases, such as AD, Huntington’s disease (HD), and Parkinson’s disease (PD) [27, 30, 31].
During the initial phases of the diseases, microglia have a neuroprotective effect by releasing anti-inflammatory mediators and relieving inflammatory effects induced by neurotoxicants; however, as the abnormal accumulation of proteins such as Aβ, tau oligomers, α-synuclein, mutant Huntingtin continues, microglia exhibit neurotoxic effects, where neuronal apoptosis and neuroinflammation are exacerbated, thereby promoting disease progression [27].
Furthermore, microglia in their neurotoxic state amplify BBB damage by releasing proinflammatory cytokines and chemokines, such as TNF-α, inducible NOS and CCL-5, these chemokines induce the infiltration of peripheral circulating immune cells into the brain, which further amplifies the inflammatory response and causes tissue damage and apoptosis [32, 33].
Therefore, it has been proposed that the regulation of microglial states in different pathological states can improve neuroinflammation, thereby slowing disease progression.
ALZHEIMER’S DISEASE AND MICROGLIA
As mentioned, AD is the most common type of dementia; it is a neurodegenerative and genetically complex disease characterized by progressive memory loss [34]. AD patients show loss of neurons and synapses, Aβ plaques, and intracellular aggregation of hyperphosphorylated tau neurofibrillary tangles in neurons [35]. Aβ plaques are generated by aggregation on Aβ peptide, synthesized from amyloid-β protein precursor (AβPP) [36, 37]. On the other hand, neurofibrillary tangles are the intraneuronal accumulation of insoluble aggregates of tau. Although AD-associated cognitive impairment is strongly related to Aβ accumulation and the severity of tau pathology, the precise relationship between these two pathological features is still controversial [38].
It is important to keep in mind that AD is a heterogeneous neurodegenerative condition with respect to the clinical presentation and progression of the AD pathology. Therefore, it should be considered how the heterogeneity of the disease and the temporal order of the neuropathological characteristics of AD impact on the identification of the objective, the moment of treatment and the development of therapies to reduce AD pathologies and address the cognitive symptoms [34].
In the pathogenesis of AD there are various etiological and pathogenic factors that have been described, such as altered calcium homeostasis, genetic, hormonal, inflammatory or immunological factors, vascular dysfunction and deregulation of the cell cycle, which lead to a dysfunction of the neurotransmitter system and consequently to a decline cognitive [35]. It should also be noted that from the genetic point of view, AD is a multigenic disorder, including a range of genetic effect sizes. These variants range from high penetrance mutations causing familial AD (such as gene variants in APP, PSEN1, and PSEN2) and the common APOE ɛ4 allele that has more impact on sporadic AD risk to variants with relatively small effects, which have been identified in GWAS studies, pointing to many genes related to immune systems, such as SORL1 [39], CLU /ApoJ [40], ABCA7 [41], CD33 [34], MS4A6A [42], IL1RAP [43], PLCG2 [44, 45], and ABI3 [42].
Within the immune system, microglia play a very important role; during the progression of AD, microglia have a complex role, due to their diverse phenotypes and the variety of activation pathways [46]. When damage is perceived, microglia can rapidly proliferate and change morphology to engulf foreign substances and dead cells. Also, activated microglia secrete Il-1α, TNFα, and C1q. These cytokines induce the activation of A1 astrocytes [32], a type of astrocytes that has been classified as harmful, since they lose most of the normal functions of astrocytes, do not have the ability to promote neuronal survival, growth, synaptogenesis, and phagocytosis, and have the capacity to produce the death of neurons and oligodendrocytes [47]. This type of astrocytes is very present in human neurodegenerative diseases, such as AD, HD, PD, and multiple sclerosis [32].
Therefore, in the early stages, microglia are beneficial, eliminating Aβ plaques and secreting trophic factors to promote tissue repair and protect damaged neurons [20]. However, in later stages, the excessive accumulation of abnormal proteins and increased senescence cause changes in the brain microenvironment, microglia become more sensitive to inflammatory stimuli, there is a secretion of large amounts of cytokines and further promoted the production of Aβ and the activation of many inflammatory pathways, such as NOD-type receptor family and pyrin structural domain 3-containing (NLRP3) inflammatory vesicles, again favoring the release of proinflammatory cytokines and accelerating neurodegeneration [48, 49].
As the phagocytic activity of microglia is affected, an abnormal accumulation of Aβ also occurs, which accelerates the processing of AβPP, thereby exacerbating neuronal damage and promoting disease progression [50].
On the other hand, the presence of tau promotes the senescence of microglia, which also causes a reduction in their phagocytic function and after phagocytosing neurons carrying tau aggregates, microglia begin to release insoluble tau aggregates. And begins to damage neighboring microglia [51, 52]. A unique innate and adaptive immune response has also been reported in mice with tauopathy [53]; a notable increase in T cells, especially cytotoxic ones, was observed in areas with tau pathology, correlating with the degree of neuronal loss. Depletion of microglia or T cells blocks tau-mediated neurodegeneration.
As the condition worsens, the inflammatory response persists and overexposure to oxidation creates a neurotoxic environment, excess Aβ and tau proteins cause redox imbalance, neuroinflammation, iron overload, oxidative stress, and reduce the release of neurotrophic factors, which accelerates the reduction of neuronal cells and increases the aggregation of Aβ and tau proteins and accelerate the pathological progression of AD, forming a vicious circle [54, 55].
VASCULAR DEMENTIA AND MICROGLIA
VaD is the result of general and local effects of vascular disease, which cause a direct alteration in the flow of blood vessels in the brain [56]. VaD includes a wide range of clinical symptoms, depending on the causes and location of the damage [57]. The etiology of VaD includes large and small vessel damage, small vessel disease of the brain being the most common cause [5]. Based on imaging and pathologic changes, vascular dementia is classified into several subtypes, including multi-infarct dementia, strategic infarct dementia, small vessel dementia, hypoperfusion dementia, hemorrhagic dementia, hereditary vascular dementia, and AD with vascular disease [12].
Some of the risk factors for cerebrovascular disease, and therefore VaD as well, are circulating factors such as plasma total cholesterol, low-density lipoprotein cholesterol, or diseases such as diabetes mellitus, hypertension, atrial fibrillation, hypercholesterolemia, atherosclerosis, and smoking. Genetic factors may influence the degree to which these risk factors contribute to cognitive decline [5, 56, 57].
Various factors are involved in the pathogenesis of dementia, and although the causes of neurodegeneration have not yet been fully elucidated, the role of the immune system has been a rapidly expanding field of research [58]. During the early phase of vascular injury, chemokines generated by endothelial cells attract microglia into the vessels, thus performing a beneficial function, maintaining the integrity of the BBB. However, sustained inflammation causes the microglia to change from a protective to a deleterious phenotype. The alteration of the BBB is increased by generating proinflammatory cytokines and phagocytosing axons and astrocytic ends [59].
Undoubtedly, microglia play a significant role within the brain microenvironment, perform several functions, have highly dynamic motility, and interact with various cell types in response to environmental stimuli. However, the spatiotemporal pattern of neuroinflammation modulated by microglia within the pathogenesis of vascular dementia is still not clear.
PHARMACOTHERAPY
Until the past decade, there were only two classes of drugs approved for dementia, specifically for AD: cholinesterase inhibitors and N-methyl-D-aspartate receptor antagonists (Table 2) [60, 61]. Although they do not provide a cure, their purpose is to stabilize cognitive and functional status, providing the secondary benefit of potentially reducing behavioral problems associated with dementia [61]. Although some of these drugs are used off-label for other types of dementia, the mainstay of treatment for vascular cognitive impairment is reducing the risk of systemic vascular disease and the use of neuro-drugs such as cholinesterase inhibitors [62]. Reducing the risk of stroke at the population level provides a promising opportunity to also reduce the rate of dementia, given that stroke doubles the risk of developing dementia [63] and 90% of strokes are considered preventable [64–66].
Table 2
Currently approved drugs and drugs in the approval process for Alzheimer’s disease
| Drug | Type of drug and target therapeutic | Regulatory status |
| Donepezil | Acetylcholinesterase inhibitor. It increases the availability of acetylcholine in the synapse [91]. | It is approved for use in mild, moderate, and severe AD. Off-label indications: LBD, traumatic brain injury, VaD and dementia associated with PD [91]. |
| Galantamine | Acetylcholinesterase inhibitor and it is an allosteric potentiator of α4β2 and presynaptic α-7 nicotinic acetylcholine receptors [92]. | It was approved by the FDA in 2001 for the treatment of mild to moderately severe AD. Iff-label uses: VaD, dementia associated with PD, FTD, dementia associated with multiple sclerosis, among others [92]. |
| Rivastigmine | Acetylcholinesterase inhibitors [93]. | It was approved by the FDA in 1997, and it is indicated to treat mild to moderate dementia of the AD type. Other indications: mild to moderate dementia that is associated with PD [94]. |
| Memantine | Memantine exerts its therapeutic effect by being a low to moderate affinity non-competitive NMDA receptor antagonist. | It was approved in 2002 in Europe and in 2003 In the USA to treat moderate-to-severe AD [95]. |
| Aducanumab | It is a monoclonal antibody that attaches to Aβ which forms plaques in the brains of people with AD, and therefore it is intended remove the Aβ plaques [72, 73]. | The FDA approved it through the Accelerated Approval pathway on June 7, 2021 [70, 71] The EMA refused the marketing authorization in December, 2021; the company withdrew the marketing authorization application on April 20, 2022 [73]. |
| Lecanemab | It is a humanized immunoglobulin gamma 1 monoclonal antibody directed against aggregated soluble and insoluble forms of Aβ [75]. | The FDA approved it on Jan 6, 2023 through the Accelerated Approval Program, it is expected to get conventional approval from the FDA and is under review by the EMA [74–76], from the NMPA from China in December, 2022 and by Health Canada [96]. In Japan, it is in priority review by the PMDA [97]. |
Aβ, amyloid-beta; AD, Alzheimer’s disease; EMA, European Medicines Agency; FDA, Food and Drug Administration; FTD, frontotemporal dementia; LBD, Lewy body dementia; NMDA, N-methyl-D-aspartate; NMPA, National Medical Products Administration; PD, Parkinson disease; PMDA, Pharmaceuticals and Medical Devices Agency; VaD, vascular dementia.
Therefore, the search for new treatments and therapeutic strategies with the aim of stopping the course of the disease continues with some urgency; these new treatments have been developed considering the neuropathological complexity, and some are directed at multiple targets and with the intention of being administered in the initial phases of the disease. Thus, it is also important to develop new diagnostic techniques to identify the disease in its early stages [67]. Most of the research has focused on therapies directed against Aβ, mainly immunotherapies, others directed at decreasing the production of Aβ, through the inhibition of β- and γ-secretase, and also therapies directed at the tau protein [68].
Although many challenges remain to be resolved, immunotherapy has emerged as a very promising treatment option, as it includes disease-modifying drugs by targeting Aβ [69], which has been considered the main hallmark of AD and, although controversially, the cause of cognitive decline. Efforts to develop new drugs against the disease appeared to pay off on June 7, 2021, when the Food and Drug Administration (FDA) approved aducanumab (marketed as Aduhelm) through the Accelerated Approval pathway [70, 71]. aducanumab is a type of human monoclonal antibody that can selectively interact with Aβ aggregates, including soluble oligomers and insoluble fibrils, by attaching to Aβ, Aduhelm was expected to help remove the plaques from the brain and delay the worsening of the disease [72, 73]. Since 2003, Aduhelm was the first novel therapy approved for AD, it was the first treatment directed at the underlying pathophysiology in AD, the Aβ plaques in the brain. Clinical trials of Aduhelm have shown a reduction in these plaques, which is expected to lead to a reduction in the clinical deterioration of this devastating form of dementia [70, 71]. Despite FDA approval, the European Medicines Agency (EMA) recommended a denial of marketing authorization in December 2021. The EMA considered that although Aduhelm reduces Aβ in the brain, the link between this effect and clinical improvement had not been established; it mentioned that the results of the studies were conflicting and did not convincingly show the efficacy of Aduhelm for the treatment at early-stage of AD. Also, the studies did not show that the drug was safe enough. For this reason, the company that owns the molecule withdrew the EMA marketing authorization application for Aduhelm on April 20, 2022 [73].
Lecanemab (marketed as Leqembi) following the same pathway of Accelerated Approval as Aduhelm, and got its approval on Jan 6, 2023, and it is expected to get conventional approval from the FDA in the summer of 2023, and is under review by the EMA and other institutions [74–76]. Lecanemab is a humanized immunoglobulin gamma 1 (IgG1) monoclonal therapy that targets both soluble and insoluble aggregated forms of Aβ [75]. Leqembi is indicated for the treatment of AD, in patients with mild cognitive impairment or mild dementia stage of disease [74].
The search for drugs has mainly focused on drugs designed to reduce or remove Aβ plaques that are found around neurons and the tau protein; however, considering the little success obtained, in recent decades research has been promoted to find other possible therapeutic approaches, such as inflammation, infection and immunity, and plasticity and neuroprotection in the clinical stage; these 2 groups represent the largest proportion of pathways that have been explored [54, 72].
MICROGLIA AS A THERAPEUTIC TARGET
Derived from the evidence of the participation of microglia in neurodegenerative diseases, different therapeutic strategies aimed at this objective have been tested. The tests have been carried out at the cellular level, in studies in animal models and even in clinical trials [77]. Modulation of inflammation is the most used strategy and has been tested in conditions such as AD, PD, HD, amyotrophic lateral sclerosis, frontotemporal dementia, multiple system atrophy, and progressive supranuclear palsy. Other strategies have also been tested, such as the inhibition of the synthesis and secretion of microglial exosomes, alterations in microglial metabolism, alterations in the microglial phenotype, and activation of TREM2.
Particularly with dementia, some immunomodulators have been tested; however, some of these small molecules have short retention in the brain [78]. Synthetic macromolecular chemical reagents have difficulty crossing the BBB, which is why their application is limited. Therefore, reagents that are able to cross the BBB and have immunomodulatory functions have significant potential in AD therapeutics [54]. Zhong and colleagues [54], in an APP/PS1 model mice, developed Prussian blue/polyamidoamine (PAMAM) dendrimer/Angiopep-2 (PPA) nanoparticles that can regulate the mitophagy of microglia as a potential AD treatment. PPA nanoparticles show high BBB permeability and are capable of scavenging ROS and restoring mitochondrial function of microglia. The nanoparticles effectively reduce neurotoxic Aβ aggregate and rescue the cognitive functions in the model mice.
In addition to the development of nanoparticles capable of crossing the BBB, other molecules, and other techniques capable of modifying microglial function, have also been tested (Table 3). Until now, most have been preclinical studies. Alternatives include transcranial alternating current stimulation (tACS) [79] and the administration of homotaurine, which is not yet authorized as a new AD drug, but its use in the treatment of cognitive decline symptoms and for prevention of AD is still considered promising, and it is currently under analysis as a nutraceutical [80]. These alternatives have been tested in humans, but trials were intended as pilots and given the limited sample of participants, more studies are needed. Also, there is also a phase 3 study of Masitnib, which has proven to be beneficial for patients with mild to moderate AD, and a confirmatory study is currently underway, which includes biomarker outcomes [81].
Table 3
New microglia-related treatment proposals for AD
| Prussian blue/ polyamidoamine dendrimer/Angio-pep2 nanoparticles | 4D9, a rat IgG2a monoclonal antibody | CBD isolate intraperitoneally | Peripheral IL-33, pleiotropic cytokine | Murine recombinant IL-33 via intraperitoneal | |
| Model | APP/PS1 mouse model | Mouse model | 5xFAD transgenic mice. | AD mouse models | APP/PS1 transgenic mice |
| Result | The nanoparticles can cross the BBB with high efficiency, exerting synergistic effects of eliminating ROS and restoring the mitochondrial function of microglia, thus exhibiting efficient neuroprotection, and rescuing the cognitive functions of AD mouse model [54]. | 4D9 activated TREM2-dependent phospho-SYK signaling in a dose-dependent manner and enhanced TREM2 function resulting in increased phagocytosis of Aβ and myelin in vitro. | CBD was reported to elevate IL-33 and TREM2 expression in glial cells while suppressing proinflammatory IL-6 expression in peripheral blood leukocytes in proportion to cognitive improvement. With increased TREM2 expression, amyloid reduction was observed within brain tissue, suggesting that CBD may reduce amyloid production and/or aid in amyloid removal. Therefore, CBD may represent a safe and effective therapy against cognitive decline, in part by modulating glial cell functions [85]. | It was observed that microglial phagocytosis and Aβ degradation are enhanced. In addition, IL-33 treatment modifies microglia/ macrophages to an alternate activation phenotype, resulting in a reduced proinflammatory response in AD mouse brains, and an enhancing effect on synaptic and memory functions has been observed [82]. | Aβ pathology was reported to be ameliorated by reprogramming microglial epigenetic and transcriptomic profiles to induce a microglial subpopulation with enhanced phagocytic activity, resulting in increased Aβ clearance. Through a PU.1-dependent transcriptional pathway, PU.1 coordinates the transcriptional program during antigen presentation, hematopoiesis, and microglial development [83]. |
| Studies involving human subjects | |||||
| 40 Hz (gamma) tACS administered 1 h daily for 2 or 4 weeks. | Homotaurine tablets (a natural amino acid first identified in different species of marine red algae) | Masitinib, a tyrosine kinase inhibitor, orally administered | |||
| Participants | Mild to moderate AD | Amnestic MCI (14 participants) | Study randomized, controlled, phase 3 trial. | ||
| Results | Repeated sessions of gamma tACS produced a significant increase in cerebral blood flow in the temporal lobes, without showing significant adverse effects. 40 Hz tACS could interfere with the pathophysiological cascade of AD by modulating interneuron activity that contributes to global network dysfunction and by activating microglial waste removal and/or restoring perfusion in cortical areas deteriorated to ensure an adequate amount of nutrients and clearance of toxic products [79]. | This study shows that after 12-month supplementation, the patients increased serum levels of the anti-inflammatory cytokines IL-33 and IL-10, which in turn appears related to improved episodic memory performances. In addition, a decreased circulating level of the pro-inflammatory cytokine IL-18 was observed. Homotaurine is a GABA-receptor agonist and GABA-mediated mechanisms are both crucial for learning and memory tasks and are able to suppress the inflammatory response of astrocytes and microglia [80]. | This study is the first successful randomized, controlled, phase 3 trial in AD of a tyrosine kinase inhibitor, targeting innate immune cells. It has been suggested that mast and/or macrophage/microglia are in mild-to-moderate AD, by switching the neuroimmune system from a neurotoxic state towards a neuroprotective state, changing the neuronal microenvironment. The results indicate that masitinib (4.5 mg/kg/day), as an adjunct to cholinesterase inhibitor and/or memantine, could benefit patients with mild-to-moderate AD, and a confirmatory pivotal study, including biomarker outcomes, has been initiated [81]. | ||
5xFAD, five AD-linked mutations transgenic mice; Aβ, amyloid-beta; AD, Alzheimer’s disease; APP/PS1, mice model that contain human transgenes for both APP and PSEN1; BBB, blood-brain barrier; CBD, cannabidiol; GABA, gamma-aminobutyric acid; IL, interleukin; MCI, mild cognitive impairment; ROS, reactive oxygen species; SIK, spleen associated tyrosine kinase; tACS, transcranial alternating current stimulation; TREM2, triggering receptor expressed on myeloid cells 2.
DISCUSSION
Despite the great economic, social and health impact that dementia has on patients and their caregivers around the world, currently, pharmacotherapeutic options are limited, and are aimed at improving cognitive and behavioral symptoms. Until a couple of years ago, there were only two classes of drugs available, cholinesterase inhibitors and N-methyl-D-aspartate (NMDA) receptor antagonists, neither of which has evidence that they are capable of stopping or reversing the progress of the disease [60, 61]. In addition, the prescription of these medications should be regularly evaluated and discontinued when dementia is considered to have progressed to the point where the patient is totally dependent for all daily activities and it is no longer possible to maintain functional and cognitive status, or when the adverse effects outweigh the beneficial effects [61].
The growing research for the development of new drugs has led to promising immunotherapies that include disease-modifying drugs aimed at Aβ [69], such as Aducanumab and Lecanemab, both of which were approved by accelerated pathway by the FDA [70, 71]. Aducanumab has not been without controversy regarding its efficacy and even its safety, and even the company that owns the molecule withdrew the EMA marketing authorization application for Aduhelm in 2022 [73]. On the other hand, Lecanemab is close to obtaining approval through the conventional route by the FDA and by other regulatory authorities.
However, it is important to emphasize that these new options are indicated for initial stages of the disease, and even considering these new options, the options are limited. Therefore, the priority remains 1) to develop biomarkers or new techniques that allow an earlier diagnosis of the disease and 2) to continue the search for new therapeutic options.
There is another important reason why the development of therapeutic strategies for dementia is complicated, and it is due to the multifactorial and multigenetic nature of the condition itself. With scientific advances and genetic data analysis techniques, it has been possible to more fully understand the role played by the immune system in the development of dementia, especially the importance of the proper functioning of microglia.
Taking this into account, efforts have been made to better understand the role of microglia and thus find strategies that lead to stopping the progress of the pathology. Table 3 lists some of the studies that have sought to influence microglia to find an effective treatment against AD progression (see Fig. 1). One approach that has been evaluated is the direct addition of IL-33, with positive results; it was observed that microglial phagocytosis is enhanced and Aβ clearance increased [82, 83]. Also, a monoclonal antibody, 4D9, and a cannabidiol isolate have been tested in mouse models, as both modify TREM2 and lead to increased Aβ phagocytosis [84, 85].
Fig. 1
Beneficial effects of therapeutic strategies developed against AD related to microglia. Once Aβ plaque begins to proliferate, the microglia rapidly become activated, change shape, and begin to phagocytose harmful substances, but also begin to phagocytose neurons. There is loss of neurons and synapses, accumulation of Aβ, neurofibrillary tangles, and ROS. Different strategies targeting microglia have been tested, which have shown different beneficial effects. Angiopep-2 nanoparticles exerting synergistic effects of eliminating ROS and rescuing the cognitive functions. 4D9, a monoclonal antibody, enhanced TREM2 function resulting in increased phagocytosis of Aβ and myelin in vitro. Cannabidiol elevated IL-33 and TREM2 expression in glial cells while suppressing proinflammatory IL-6 expression in peripheral blood leukocytes in proportion to cognitive improvement and amyloid reduction was observed. With the administration of IL-33 treatment was observed that microglial phagocytosis and Aβ degradation are enhanced. Repetitive sessions of gamma tACS lead to a significant increase in cerebral blood flow in the temporal lobes, which contributes to global network dysfunction and by activating microglial waste removal and/or restoring perfusion in cortical areas deteriorated to ensure an adequate amount of nutrients and clearance of toxic products. Finally, Homotaurine shows increased serum levels of the anti-inflammatory cytokines IL-33 and IL-10 in the patients, which in turn appears related to improved episodic memory performances. Created with BioRender.com.
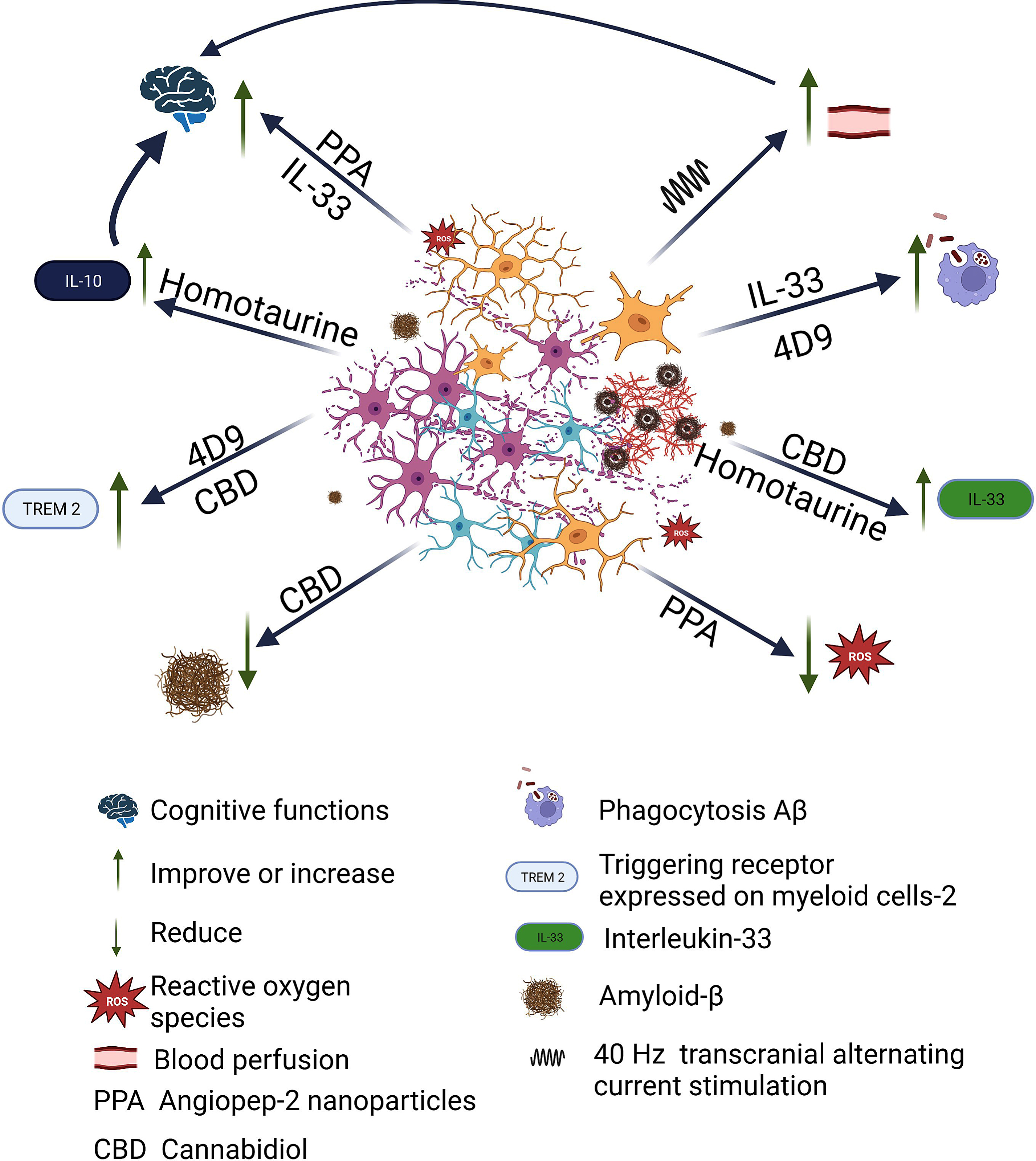
However, these studies have been carried out in animal models, and it is necessary to remember that there are significant differences between human and rodent microglia, especially when comparing microglia in diseased human brains with decades of development of a disease phenotype, while in the case of rodents the disease phenotype in microglia develops in weeks. Therefore, extrapolation of results from rodent models to elderly humans needs to be taken with caution [86]. Also, in table 3, articles of research carried out in humans are cited, where tACS and a nutraceutical are tested [79, 80]. Sprugnoli et al. [79], considering that AD is associated with alterations in cortical perfusion, and that recently, neuronal activity in the gamma band has been identified as a driver of arteriolar vasomotion, coupled with the fact that it has been shown that the exogenously induced increase in gamma oscillations (specifically at 40 Hz) promotes microglial activation and causes subsequent reduction of Aβ and p-tau deposits in a mouse model of AD, described the study in which the objective was to translate into humans the preclinical findings of 40 Hz gamma stimulation in animal models of AD by means of tACS applied to a sample of 15 patients with mild to moderate AD. Nevertheless, both this and the study with homotaurine, which was carried out on 14 participants, are pilot studies with a very small sample, so they should be carried out with more participants.
But there are also studies of drugs in more advanced stages, as is the case of Masitinib [81], a tyrosine kinase inhibitor, that has demonstrated neuroprotective action in neurodegenerative diseases via inhibition of mast cell and microglia/macrophage activity, and in a previous phase 2 trial, a slower progression was observed in mild-to-moderate AD patients [87].
Finally, it is important to mention that although most of the efforts and attention to search for therapeutic targets and alternatives are focused on AD, and although the pathophysiology of the different types of dementia seems very different, it is also known that the immune system and neuroinflammation interact in an important way among different neurodegenerative diseases. However, it is also important to continue investigating the impact that the special characteristics of different diseases have on how the microglia response is triggered.
However, the role played by microglia in the pathogenesis of dementia is still unclear, as their action benefits or harms the evaluation of the disease, and to date, studies have indicated that its effect will depend on the stage in which the disease is found [88].
CONCLUSION
Despite the impact of dementia both in the field of health and in society, the efforts to find a treatment continue without bearing fruit; to date the approved drugs only act on some symptoms, but do not cure or slow down the evolution of the pathological process. Although some promising immunomodulatory therapies have emerged, following conflicting results, some molecules are withdrawing their application for authorization at the EMA or are awaiting conventional approval by the FDA and other institutions.
For these reasons, the search for new strategies continues to be a priority issue and a challenge for the scientific community. Since the immune system, and in particular, the malfunctioning of microglia, has been identified as a key factor in the development of dementia, it has been pointed out as a possible therapeutic strategy for this pathology. Although there are some proposals aimed at microglial regulation, they are still in preclinical stages or require further studies for their evolution; however, with aspects still to be resolved, the results have been promising.
Advances towards understanding the pathological mechanisms of dementia allow us to identify the metabolic pathways involved and their possible link with the design of new therapeutic strategies, recognizing the importance of comprehensive treatment of complex pathologies, such as dementia.
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
References
[1] | Global action plan on the public health response to dementia 2017-2025, https://www.who.int/publicationsdetail-redirect/global-action-plan-on-the-public-healthresponse-to-dementia-2017—2025, Accessed on April 24, 2023. |
[2] | WHO-Dementia thematicbrief epidemiology 2015, https://hal.science/hal-03517019v1/preview/WHO-%20Dementia_thematicbrief_epidemiology_2015.pdf, Accessed on April 25, 2023. |
[3] | Nichols E , Steinmetz JD , Vollset SE , Fukutaki K , Chalek J , Abd-Allah F , Abdoli A , Abualhasan A , Abu-Gharbieh E , Akram TT et al. ((2022) ) Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019, Lancet Public Health 7: , e105–e125. |
[4] | OMS y Alzheimer’s Disease International (ADI) (2013) Dementia: A public health priority, https://www.who.int/publications/i/item/dementia-apublic-health-priority, Last updated August 24, 2012, Accessed on April 24, 2023. |
[5] | Appleton JP , Scutt P , Sprigg N , Bath PM ((2017) ) Hypercholesterolaemia and vascular dementia. Clin Sci 131: , 1561–1578. |
[6] | Zhang B , Gaiteri C , Bodea L-G , Wang Z , McElwee J , Podtelezhnikov AA , Zhang C , Xie T , Tran L , Dobrin R , Fluder E , Clurman B , Melquist S , Narayanan M , Suver C , Shah H , Mahajan M , Gillis T , Mysore J , MacDonald ME , Lamb JR , Bennett DA , Molony C , Stone DJ , Gudnason V , Myers AJ , Schadt EE , Neumann H , Zhu J , Emilsson V ((2013) ) Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153: , 707–720. |
[7] | International Genomics of Alzheimer’s Disease Consortium (IGAP) ((2015) ) Convergent genetic and expression data implicate immunity in Alzheimer’s disease. Alzheimers Dement 11: , 658–671. |
[8] | Efthymiou AG , Goate AM ((2017) ) Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol Neurodegener 12: , 43. |
[9] | García-Culebras A , Cuartero MI , Peña-Martínez C , Moraga A , Vázquez-Reyes S , de Castro-Millán FJ , Cortes-Canteli M , Lizasoain I , Moro MÁ (2023) Myeloid cells in vascular dementia and Alzheimer’s disease: Possible therapeutic targets? Br J Pharmacol, doi:10.1111/bph.16159. |
[10] | Wu K-M , Zhang Y-R , Huang Y-Y , Dong Q , Tan L , Yu J-T ((2021) ) The role of the immune system in Alzheimer’s disease. Ageing Res Rev 70: , 101409. |
[11] | Tian Z , Ji X , Liu J ((2022) ) Neuroinflammation in Vascular cognitive impairment and dementia: Current evidence, advances, and prospects. Int J Mol Sci 23: , 6224. |
[12] | O’Brien JT , Thomas A ((2015) ) Vascular dementia. Lancet 386: , 1698–1706. |
[13] | Zhong L , Chen X-F ((2019) ) The emerging roles and therapeutic potential of soluble TREM2 in Alzheimer’s disease. Front Aging Neurosci 11: , 328. |
[14] | von Bernhardi R , Eugenín-von Bernhardi J , Flores B , Eugenín León J ((2016) ) Glial cells and integrity of the nervous system. Adv Exp Med Biol 949: , 1–24. |
[15] | Ragupathy H , Vukku M , Barodia SK ((2023) ) Cell-typespecific mitochondrial quality control in the brain: A plausible mechanism of neurodegeneration Int J Mol Sci 24: , 14421. |
[16] | Casaril AM , Katsalifis A , Schmidt RM , Bas-Orth C ((2022) ) Activated glia cells cause bioenergetic impairment of neurons that can be rescued by knock-down of the mitochondrial calcium uniporter. Biochem Biophys Res Commun 608: , 45–51. |
[17] | Mulica P , Grünewald A , Pereira SL ((2021) ) Astrocyte-neuron metabolic crosstalk in neurodegeneration: A mitochondrial perspective. Front Endocrinol 12: , 668517. |
[18] | MacVicar BA , Newman EA ((2015) ) Astrocyte regulation of blood flow in the brain, Cold Spring Harb Perspect Biol 7: , a020388. |
[19] | Hickman SE , Kingery ND , Ohsumi T , Borowsky M , Wang L , Means TK , Khoury JE ((2013) ) The microglial sensome revealed by direct RNA sequencing. Nat Neurosci 16: , 1896–1905. |
[20] | Sarlus H , Heneka MT ((2017) ) Microglia in Alzheimer’s disease. J Clin Invest 127: , 3240–3249. |
[21] | Mittelbronn M , Dietz K , Schluesener HJ , Meyermann R ((2001) ) Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol (Berl) 101: , 249–255. |
[22] | Brown GC ((2021) ) Neuronal loss after stroke due to microglial phagocytosis of stressed neurons. Int J Mol Sci 22: , 13442. |
[23] | Cunningham CL , Martínez-Cerdeño V , Noctor SC ((2013) ) Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci 33: , 4216–4233. |
[24] | Hickman SE , Allison EK , Khoury J ((2008) ) Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci 28: , 8354–8360. |
[25] | Griciuc A , Serrano A , Parrado AR , Lesinski AN , Asselin N , Mullin K , Hooli B , Choi SH , Hyman BT , Tanzi RE ((2013) ) Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 78: , 631–643. |
[26] | Bohlen CJ , Friedman BA , Dejanovic B , Sheng M ((2019) ) Microglia in brain development, homeostasis, and neurodegeneration. Annu Rev Genet 53: , 263–288. |
[27] | Han T , Xu Y , Sun L , Hashimoto M , Wei J ((2024) ) Microglial response to aging and neuroinflammation in the development of neurodegenerative diseases. Neural Regen Res 19: , 1241. |
[28] | Rodier F , Campisi J ((2011) ) Four faces of cellular senescence. J Cell Biol 192: , 547–556. |
[29] | Teissier T , Boulanger E , Cox LS ((2022) ) Interconnections between inflammageing and immunosenescence during ageing. Cells 11: , 359. |
[30] | De Marchi F , Munitic I , Vidatic L , Papić E , Racki V , Nimac J , Jurak I , Novotni G , Rogelj B , Vuletic V , Liscic RM , Cannon JR , Buratti E , Mazzini L , Hecimovic S ((2023) ) Overlapping neuroimmune mechanisms and therapeutic targets in neurodegenerative disorders. Biomedicines 11: , 2793. |
[31] | Nayak D , Roth TL , McGavern DB ((2014) ) Microglia development and function. Annu Rev Immunol 32: , 367. |
[32] | Liddelow SA , Guttenplan KA , Clarke LE , Bennett FC , Bohlen CJ , Schirmer L , Bennett ML , Münch AE , Chung W-S , Peterson TC , Wilton DK , Frouin A , Napier BA , Panicker N , Kumar M , Buckwalter MS , Rowitch DH , Dawson VL , Dawson TM , Stevens B , Barres BA ((2017) ) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541: , 481–487. |
[33] | Kim Y , Cho AY , Kim HC , Ryu D , Jo SA , Jung Y-S ((2022) ) Effects of natural polyphenols on oxidative stress-mediated blood-brain barrier dysfunction. Antioxidants 11: , 197. |
[34] | Jorfi M , Maaser-Hecker A , Tanzi RE ((2023) ) The neuroimmune axis of Alzheimer’s disease. Genome Med 15: , 6. |
[35] | Paolo G Di , Kim T ((2012) ) Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat Rev Neurosci 12: , 284–296. |
[36] | Cordy JM , Hooper NM , Turner AJ ((2006) ) The involvement of lipid rafts in Alzheimer’s disease (review). Mol Membr Biol 23: , 111–122. |
[37] | Saxena U ((2009) ) Lipid metabolism and Alzheimer’s disease: Pathways and possibilities. Expert Opin Ther Targets 13: , 331–338. |
[38] | Small SA , Duff K ((2008) ) Linking Aβ and tau in late-onset Alzheimer’s disease: A dual pathway hypothesis. Neuron 60: , 534–42. |
[39] | Yin R-H , Yu J-T , Tan L ((2015) ) The role of SORL1 in Alzheimer’s disease. Mol Neurobiol 51: , 909–918. |
[40] | DeMattos RB , Cirrito JR , Parsadanian M , May PC , O’Dell MA , Taylor JW , Harmony JAK , Aronow BJ , Bales KR , Paul SM , Holtzman DM ((2004) ) ApoE and clusterin cooperatively suppress Aβ levels and deposition: Evidence that ApoE regulates extracellular Aβ metabolism, Neuron 41: , 193–202. |
[41] | Fu Y , Hsiao J-HT , Paxinos G , Halliday GM , Kim WS ((2016) ) ABCA7 mediates phagocytic clearance of amyloid-β in the brain. J Alzheimers Dis 54: , 569–584. |
[42] | Hansen DV , Hanson JE , Sheng M ((2018) ) Microglia in Alzheimer’s disease. J Cell Biol 217: , 459–472. |
[43] | Ghosh S , Wu MD , Shaftel SS , Kyrkanides S , LaFerla FM , Olschowka JA , O’Banion MK ((2013) ) Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J Neurosci 33: , 5053–5064. |
[44] | Milner JD ((2015) ) PLAID: A syndrome of complex patterns of disease and unique phenotypes. J Clin Immunol 35: , 527–530. |
[45] | Hernández-Ostiz S , Prieto-Torres L , Xirotagaros G , Noguera-Morel L , Hernández-Martín Á , Torrelo A ((2017) ) Enfermedades autoinflamatorias en dermatología pediátrica. Parte 1: Síndromes urticariformes, síndromes pustulosos y síndromes con ulceraciones cutáneo-mucosas, Actas Dermo-Sifiliograficas 108: , 609–619. |
[46] | Leng F , Edison P ((2021) ) Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat Rev Neurol 17: , 157–172. |
[47] | Zamanian JL , Xu L , Foo LC , Nouri N , Zhou L , Giffard RG , Barres BA ((2012) ) Genomic analysis of reactive astrogliosis. J Neurosci 32: , 6391–6410. |
[48] | Mata-Martínez E , Díaz-Muñoz M , Vázquez-Cuevas FG ((2022) ) Glial cells and brain diseases: Inflammasomes as relevant pathological entities. Front Cell Neurosci 16: , 929529. |
[49] | Merighi S , Nigro M , Travagli A , Gessi S ((2022) ) Microglia and Alzheimer’s disease. Int J Mol Sci 23: , 12990. |
[50] | Aikawa T , Holm M-L , Kanekiyo T ((2018) ) ABCA7 and pathogenic pathways of Alzheimer’s disease. Brain Sci 8: , 27. |
[51] | Ju Y , Tam KY ((2021) ) Pathological mechanisms and therapeutic strategies for Alzheimer’s disease. Neural Regen Res 17: , 543–549. |
[52] | Brelstaff JH , Mason M , Katsinelos T , McEwan WA , Ghetti B , Tolkovsky AM , Spillantini MG ((2021) ) Microglia become hypofunctional and release metalloproteases and tau seeds when phagocytosing live neurons with P301S tau aggregates., eabg, Sci Adv 7: , 4980. |
[53] | Chen X , Firulyova M , Manis M , Herz J , Smirnov I , Aladyeva E , Wang C , Bao X , Finn MB , Hu H , Shchukina I , Kim MW , Yuede CM , Kipnis J , Artyomov MN , Ulrich JD , Holtzman DM ((2023) ) Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature 615: , 668–677. |
[54] | Zhong G , Long H , Zhou T , Liu Y , Zhao J , Han J , Yang X , Yu Y , Chen F , Shi S ((2022) ) Blood-brain barrier Permeable nanoparticles for Alzheimer’s disease treatment by selective mitophagy of microglia. Biomaterials 288: , 121690. |
[55] | Zhao Y , Zhao B ((2013) ) Oxidative stress and the pathogenesis of Alzheimer’s disease, Oxid Med Cell Longev 2013: , e316523. |
[56] | Kalaria RN ((2016) ) Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer’s disease. Acta Neuropathol 131: , 659–685. |
[57] | Panza F , D’Introno A , Colacicco AM , Capurso C , Pichichero G , Capurso SA , Capurso A , Solfrizzi V ((2006) ) Lipid metabolism in cognitive decline and dementia. Brain Res Rev 51: , 275–292. |
[58] | Jevtic S , Sengar AS , Salter MW , McLaurin J ((2017) ) The role of the immune system in Alzheimer disease: Etiology and treatment. Ageing Res Rev 40: , 84–94. |
[59] | Yang Y , Zhao X , Zhu Z , Zhang L ((2022) ) Vascular dementia: A microglia’s perspective. Ageing Res Rev 81: , 101734. |
[60] | Szeto JYY , Lewis SJG ((2016) ) Current treatment options for Alzheimer’s disease and Parkinson’s disease dementia. Curr Neuropharmacol 14: , 326–338. |
[61] | Kim LD , Factora RM ((2018) ) Alzheimer dementia: Starting, stopping drug therapy. Cleve Clin J Med 85: , 209–214. |
[62] | Malik R , Kalra S , Bhatia S , Harrasi AA , Singh G , Mohan S , Makeen HA , Albratty M , Meraya A , Bahar B , Tambuwala MM ((2022) ) Overview of therapeutic targets in management of dementia. Biomed Pharmacother 152: , 113168. |
[63] | Kuźma E , Lourida I , Moore SF , Levine DA , Ukoumunne OC , Llewellyn DJ ((2018) ) Stroke and dementia risk: A systematic review and meta-analysis. Alzheimers Dement 14: , 1416–1426. |
[64] | Feigin VL , Roth GA , Naghavi M , Parmar P , Krishnamurthi R , Chugh S , Mensah GA , Norrving B , Shiue I , Ng M , Estep K , Cercy K , Murray CJL , Forouzanfar MH ((2016) ) Global burden of stroke and risk factors in 188 countries, during 1990-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet Neurol 15: , 913–924. |
[65] | O’Donnell MJ , Xavier D , Liu L , Zhang H , Chin SL , Rao-Melacini P , Rangarajan S , Islam S , Pais P , McQueen MJ , Mondo C , Damasceno A , Lopez-Jaramillo P , Hankey GJ , Dans AL , Yusoff K , Truelsen T , Diener H-C , Sacco RL , Ryglewicz D , Czlonkowska A , Weimar C , Wang X , Yusuf S , INTERSTROKE investigators ((2010) ) Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the INTERSTROKE study): A case-control study. Lancet 376: , 112–123. |
[66] | Hachinski V , Einhäupl K , Ganten D , Alladi S , Brayne C , Stephan BCM , Sweeney MD , Zlokovic B , Iturria-Medina Y , Iadecola C , Nishimura N , Schaffer CB , Whitehead SN , Black SE , Østergaard L , Wardlaw J , Greenberg S , Friberg L , Norrving B , Rowe B , Joanette Y , Hacke W , Kuller L , Dichgans M , Endres M , Khachaturian ZS ((2019) ) Preventing dementia by preventing stroke: The Berlin Manifesto. Alzheimers Dement 15: , 961–984. |
[67] | Folch J , Ettcheto M , Petrov D , Abad S , Pedrós I , Marin M , Olloquequi J , Camins A ((2015) ) Una revisión de los avances en la terapéutica de la enfermedad de Alzheimer: Estrategia frente a la proteína β-amiloide., Neurologya 33: , 47–58. |
[68] | Godyń J , Jończyk J , Panek D , Malawska B ((2016) ) Therapeutic strategies for Alzheimer’s disease in clinical trials. Pharmacol Rep 68: , 127–138. |
[69] | Söderberg L , Johannesson M , Nygren P , Laudon H , Eriksson F , Osswald G , Möller C , Lannfelt L ((2023) ) Lecanemab, aducanumab, and gantenerumab — binding profiles to different forms of amyloid-beta might explain efficacy and side effects in clinical trials for Alzheimer’s disease. Neurotherapeutics 20: , 195–206. |
[70] | Research C for DE and (2022) FDA’s Decision to Approve New Treatment for Alzheimer’s Disease. FDA, https://www.fda.gov/drugs/news-events-humandrugs/fdas-decision-approve-new-treatment-alzheimersdisease, Accessed on June 6, 2023. |
[71] | FDA, FDA Grants Accelerated Approval for Alzheimer’s Drug, https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-drug, Last updated July 6, 2021, Accessed on July 6, 2021. |
[72] | Yang P , Sun F ((2021) ) Aducanumab: The first targeted Alzheimer’s therapy. Drug Discov Ther 15: , 166–168. |
[73] | European Medicines Agency, Aduhelm: Withdrawn application, https://www.ema.europa.eu/en/medicines/human/withdrawn-applications/aduhelm, Last updated April 22, 2022, Accessed on April 22, 2023 |
[74] | FDA, FDA Grants Accelerated Approval for Alzheimer’s Disease Treatment, https://www.fda.gov/newsevents/press-announcements/fda-grants-acceleratedapproval-alzheimers-disease-treatment, Accessed on June 1, 2023. |
[75] | FDA Approves LEQEMBITM (lecanemab-irmb) Under the Accelerated Approval Pathway for the Treatment of Alzheimer’s Disease | Biogen, https://investors.biogen.com/news-releases/news-release-details/fdaapproves-leqembitm-lecanemab-irmb-under-acceleratedapproval, Accessed on June 7, 2023. |
[76] | European Medicines Agency, Medicines for human use under evaluation. https://www.ema.europa.eu/en/medicines/medicineshuman-use-under-evaluation. Last updated September 17, 2018, Accessed on June 7, 2023. |
[77] | Gao C , Jiang J , Tan Y , Chen S ((2023) ) Microglia in neurodegenerative diseases: Mechanism and potential therapeutic targets. Signal Transduct Target Ther 8: , 1–37. |
[78] | Lee JY , Han SH , Park MH , Song I-S , Choi M-K , Yu E , Park C-M , Kim H-J , Kim SH , Schuchman EH , Jin HK , Bae J ((2020) ) N-AS-triggered SPMs are direct regulators of microglia in a model of Alzheimer’s disease. Nat Commun 11: , 2358. |
[79] | Sprugnoli G , Munsch F , Cappon D , Paciorek R , Macone J , Connor A , El Fakhri G , Salvador R , Ruffini G , Donohoe K , Shafi MM , Press D , Alsop DC , Pascual Leone A , Santarnecchi E ((2021) ) Impact of multisession 40Hz tACS on hippocampal perfusion in patients with Alzheimer’s disease. Alzheimers Res Ther 13: , 203. |
[80] | Toppi E , Sireno L , Lembo M , Banaj N , Messina B , Golesorkhtafti S , Spalletta G , Bossù P ((2022) ) IL-33 and IL-10 serum levels increase in MCI patients following homotaurine treatment. Front Immunol 13: , 813951. |
[81] | Dubois B , López-Arrieta J , Lipschitz S , Doskas T , Spiru L , Moroz S , Venger O , Vermersch P , Moussy A , Mansfield CD , Hermine O , Tsolaki M for the AB09004 Study Group Investigators ((2023) ) Masitinib for mild-to-moderate Alzheimer’s disease: Results from a randomized, placebo-controlled, phase 3, clinical trial. Alzheimers Res Ther 15: , 39. |
[82] | Fu AKY , Hung K-W , Yuen MYF , Zhou X , Mak DSY , Chan ICW , Cheung TH , Zhang B , Fu W-Y , Liew FY , Ip NY ((2016) ) IL-33 ameliorates Alzheimer’s disease-like pathology and cognitive decline., E2705-E, Proc Natl Acad Sci U S A 113: , 2713. |
[83] | Lau S-F , Chen C , Fu W-Y , Qu JY , Cheung TH , Fu AKY , Ip NY ((2020) ) IL-33-PU.1 transcriptome reprogramming drives functional state transition and clearance activity of microglia in Alzheimer’s disease. Cell Rep 31: , 107530. |
[84] | Schlepckow K , Monroe KM , Kleinberger G , Cantuti-Castelvetri L , Parhizkar S , Xia D , Willem M , Werner G , Pettkus N , Brunner B , Sülzen A , Nuscher B , Hampel H , Xiang X , Feederle R , Tahirovic S , Park JI , Prorok R , Mahon C , Liang C-C , Shi J , Kim DJ , Sabelström H , Huang F , Di Paolo G , Simons M , Lewcock JW , Haass C ((2020) ) Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region, EMBO Mol Med 12: , e11227. |
[85] | Khodadadi H , Salles ÉL , Jarrahi A , Costigliola V , Khan M , Yu JC , Morgan JC , Hess DC , Vaibhav K , Dhandapani KM , Baban B ((2021) ) Cannabidiol ameliorates cognitive function via regulation of IL-33 and TREM2 upregulation in a murine model of Alzheimer’s disease. J Alzheimers Dis 80: , 973–977. |
[86] | Walker DG ((2020) ) Defining activation states of microglia in human brain tissue: An unresolved issue for Alzheimer’s disease. Neuroimmunol Neuroinflammation 7: , 194–214. |
[87] | Piette F , Belmin J , Vincent H , Schmidt N , Pariel S , Verny M , Marquis C , Mely J , Hugonot-Diener L , Kinet J-P , Dubreuil P , Moussy A , Hermine O ((2011) ) Masitinib as an adjunct therapy for mild-to-moderate Alzheimer’s disease: A randomised, placebo-controlled phase 2 trial. Alzheimers Res Ther 3: , 16. |
[88] | Bartels T , De Schepper S , Hong S ((2020) ) Microglia modulate neurodegeneration in Alzheimer’s and Parkinson’s diseases. Science 370: , 66–69. |
[89] | Udeochu JC , Shea JM , Villeda SA ((2016) ) Microglia communication: Parallels between aging and Alzheimer’s disease. Clin Exp Neuroimmunol 7: , 114–125. |
[90] | Pierre WC , Smith PLP , Londono I , Chemtob S , Mallard C , Lodygensky GA ((2017) ) Neonatal microglia: The cornerstone of brain fate. Brain Behav Immun 59: , 333–345. |
[91] | Kumar A , Gupta V , Sharma S (2023) Donepezil. In Stat-Pearls Publishing, https://www.ncbi.nlm.nih.gov/books/NBK513257/, Accessed on August 17. 2023. |
[92] | Kalola UK , Nguyen H (2023) Galantamine. In Stat-Pearls Publishing, https://www.ncbi.nlm.nih.gov/books/NBK574546/, Accessed on August 17. 2023. |
[93] | Kandiah N , Pai M-C , Senanarong V , Looi I , Ampil E , Park KW , Karanam AK , Christopher S ((2017) ) Rivastigmine: The advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clin Interv Aging 12: , 697–707. |
[94] | Patel PH , Gupta V (2023) Rivastigmine. In Stat-Pearls Publishing, https://www.ncbi.nlm.nih.gov/books/NBK557438/, Accessed on August 17. 2023. |
[95] | Lo D , Grossberg GT ((2011) ) Use of memantine for the treatment of dementia. Expert Rev Neurother 11: , 1359–1370. |
[96] | Health Canada acepta la presentacion de un nuevo farmaco para lecanemab como tratamiento para la enfermedad de Alzheimer temprana | Biogen, https://investors.biogen.com/news-releases/news-release-details/healthcanada-accepts-new-drug-submission-lecanemabtreatment, Accessed on June 7, 2023. |
[97] | Lecanemab Receives Priority Review Status in Japan | Biogen. https://investors.biogen.com/news-releases/newsrelease-details/lecanemab-receives-priority-review-statusjapan, Accessed on June 7, 2023. |


