
Common Signaling Pathways Involved in Alzheimer’s Disease and Stroke: Two Faces of the Same Coin
Abstract
Alzheimer’s disease (AD) and stroke are two interrelated neurodegenerative disorders which are the leading cause of death and affect the neurons in the brain and central nervous system. Although amyloid-β aggregation, tau hyperphosphorylation, and inflammation are the hallmarks of AD, the exact cause and origin of AD are still undefined. Recent enormous fundamental discoveries suggest that the amyloid hypothesis of AD has not been proven and anti-amyloid therapies that remove amyloid deposition have not yet slowed cognitive decline. However, stroke, mainly ischemic stroke (IS), is caused by an interruption in the cerebral blood flow. Significant features of both disorders are the disruption of neuronal circuitry at different levels of cellular signaling, leading to the death of neurons and glial cells in the brain. Therefore, it is necessary to find out the common molecular mechanisms of these two diseases to understand their etiological connections. Here, we summarized the most common signaling cascades including autotoxicity, ApoE4, insulin signaling, inflammation, mTOR-autophagy, notch signaling, and microbiota-gut-brain axis, present in both AD and IS. These targeted signaling pathways reveal a better understanding of AD and IS and could provide a distinguished platform to develop improved therapeutics for these diseases.
INTRODUCTION
Alzheimer’s disease (AD) is the most prevalent form of dementia causing higher death rates worldwide. Extracellular amyloid-β (Aβ) aggregation, intracellular tau hyperphosphorylation, and inflammation are the main pathophysiological hallmarks of AD [1, 2]. Accumulating evidence suggests that the amyloid hypothesis of AD has not been proven and anti-amyloid therapies that remove amyloid deposition have not yet slowed cognitive decline [3–5]. Around 7.1 million people will be diagnosed with AD in 2025 and it will increase to 13.8 million by 2060 [6]. In addition, cerebral amyloid angiopathy (CAA) a major contributor to AD pathogenesis is defined by the deposition of Aβ in the walls of small leptomeningeal, cortical arteries, and cortical capillaries. The chief clinical appearances of CAA are lobar intracerebral hemorrhage (ICH) and cognitive impairment. CAA is the predominant vascular lesion in AD and more prevalent in strokes especially in ischemic stroke (IS) which is caused by cerebral blood flow interruption that induces severe neural injuries leading to death. Every year globally around 15 million elderly people are affected by stroke [7, 8]. The impaired Aβ clearance could accelerate IS and this might make the brain more susceptible to AD pathology. The association between AD and IS seems to be strong in the presence of common vascular risk factors. In addition, common risk factors such as diabetes, hypertension, and hyperlipidemia may contribute to both AD and IS.
Epidemiological studies indicate that AD patients have a higher incidence of stroke than normal people [9]. Also the occurrence of dementia is fifty times higher after a major stroke but the widespread dementia risk is substantially reduced after a transient ischemic attack or minor stroke [10]. Cross-sectional epidemiological studies have reported that within three months after an IS incidence, one-fourth of elderly patients meet the criteria for dementia [11]. Another population-based study indicated that the increased risk of dementia might be started five years before the onset of stroke and would reach the maximum in one year after the stroke, also the risk continued for more than eleven years [12]. Therefore, it is intricate to disclose the occurrence of dementia as pre- and post-stroke or vice-versa.
In AD and IS, loss of neuronal cell bodies and axons cause pathological changes in the neuronal circuitry. Microglia and astrocytes are also drastically changed after IS and AD. Ramified microglia can be converted to archaeocytes or amoebocytes [13–15] and accelerate the production of reactive oxygen species (ROS) and pro-inflammatory cytokines [16]. These substances disrupt the blood-brain barrier (BBB) permeability and also release a variety of neurotoxic elements all over the brain [17]. AD and IS cause cerebral neuropathological changes such as cellular excitotoxicity, mitochondrial dysfunctions, neuroinflammation, BBB impairment, neuronal cell death, and eventually severe neurological deficits. Several signaling pathways become stimulated or deactivated during these pathophysiological shifts [18]. Therefore, it is essential to better understand the pathophysiological association between AD and IS. Hence the present review provides the common targeted signaling pathways between AD and IS, also proposing the therapeutic strategies for these diseases (Fig. 1).
Fig. 1
A brief summary of the contributing factors present in both Alzheimer’s disease (AD) and ischemic stroke (IS). Excessive glutamate secretion from astrocytes cause extrasynaptic NMDA activation leading to AD [19, 20] and IS [21–23] pathologies. Cell signaling systems mainly PI3K/Akt, mTOR-autophagy, and Notch signaling pathway are involved in AD and IS; oxidative stress due to mitochondrial dysfunction and excessive ROS production; NFkB and NLRP1/3 inflammasome are involved in neuroinflammation; and microbiota-gut-brain-axis is dysregulated by blood-brain-barrier (BBB) break down and gut dysbiosis due to the reduction of small chain fatty acids (SCFAs) producing bacteria. NMDA receptor, N-methyl-D-aspartate receptors; PI3K, phosphatidylinositol 3-kinase; Akt, protein kinase B; mTOR, mammalian target of rapamycin; ROS, reactive oxygenated species; NFkB, Nuclear factor-κB; NLRP, nucleotide-binding oligomerization domain like receptor pyrin domain-containing protein.
![A brief summary of the contributing factors present in both Alzheimer’s disease (AD) and ischemic stroke (IS). Excessive glutamate secretion from astrocytes cause extrasynaptic NMDA activation leading to AD [19, 20] and IS [21–23] pathologies. Cell signaling systems mainly PI3K/Akt, mTOR-autophagy, and Notch signaling pathway are involved in AD and IS; oxidative stress due to mitochondrial dysfunction and excessive ROS production; NFkB and NLRP1/3 inflammasome are involved in neuroinflammation; and microbiota-gut-brain-axis is dysregulated by blood-brain-barrier (BBB) break down and gut dysbiosis due to the reduction of small chain fatty acids (SCFAs) producing bacteria. NMDA receptor, N-methyl-D-aspartate receptors; PI3K, phosphatidylinositol 3-kinase; Akt, protein kinase B; mTOR, mammalian target of rapamycin; ROS, reactive oxygenated species; NFkB, Nuclear factor-κB; NLRP, nucleotide-binding oligomerization domain like receptor pyrin domain-containing protein.](https://content.iospress.com:443/media/adr/2023/7-1/adr-7-1-adr220108/adr-7-adr220108-g001.jpg)
EXCITOTOXICITY IN AD AND STROKE
Excitotoxicity is known as neuronal cell death by the deleterious effects of excitatory amino acids, deficiency of glucose and oxygen, disruption of oxidative phosphorylation, and deprivation of energy in the brain [21]. It creates an ionic imbalance, calcium (Ca2 +) overload, and finally cell membrane depolarization which triggers the excessive secretion of glutamate (Fig. 2). Excitotoxicity creates harmful events on neuronal cells such as dysregulation of calcium homeostasis, free radical formation, increasing oxidative stress, mitochondrial dysfunction, and stimulation of several transcription factors and gene expression [17]. Therefore, excitotoxicity is recommended as a common main contributing aspect to the initial stage of AD and IS.
Fig. 2
Schematic representation of excitotoxicity pathway in Alzheimer’s disease (AD) and ischemic stroke. In AD, Aβ deposition reduces the blood flow in the brain and causes excitotoxicity by the excess secretion of glutamate. In addition, activation of extrasynaptic N-methyl-D-aspartate receptors (NMDA) accelerates the production of Aβ and tau protein in AD [24, 25]. Similarly, in ischemic stroke reduced blood flow in the brain causes neuronal cell death.
![Schematic representation of excitotoxicity pathway in Alzheimer’s disease (AD) and ischemic stroke. In AD, Aβ deposition reduces the blood flow in the brain and causes excitotoxicity by the excess secretion of glutamate. In addition, activation of extrasynaptic N-methyl-D-aspartate receptors (NMDA) accelerates the production of Aβ and tau protein in AD [24, 25]. Similarly, in ischemic stroke reduced blood flow in the brain causes neuronal cell death.](https://content.iospress.com:443/media/adr/2023/7-1/adr-7-1-adr220108/adr-7-adr220108-g002.jpg)
Association between excitotoxicity and AD
Glutamate is the key excitatory neurotransmitter involved in perception and cognition. The excess glutamate and glutamatergic receptor activation can cause neuronal dysfunction and cell death. Glutamate-mediated neurotoxicity is associated with AD pathology. Continuous activation of N-methyl-D-aspartate (NMDA) receptors causes an enormous influx of Ca+2 and boost in synaptic “noise,” which diminishes the long-term potentiation process, neuronal plasticity, and neuronal damage [19, 26]. Disturbance of glutamate homeostasis can be provoked by energy deficits, free radical formation, and dysregulation of other signaling pathways [26]. Literature also proposed that there is a relationship between Aβ production and NMDA receptors activation, Aβ activates NMDARs, whereas activation of NMDARs enhances Aβ and tau protein production in AD [24, 25] (Fig. 2). Glutamatergic dysfunction was observed in postmortem AD brains [27] and in epidemiological studies [28]. Therefore, the most favorable action should be essential to restore the concentration of glutamate for the maintenance of synaptic plasticity and ongoing neurodegeneration.
In recent years, AD pathology has spread in multiple dimensions including the pathogenic role of the nonneuronal cells such as astrocytes, and microglia. In normal physiological conditions, astrocyte plays important role in several cerebral functions such as the development and maintenance of the BBB [23], promotion of neurovascular coupling [29], the attraction of cells through the release of chemokines [23], K+ buffering [30], uptake of glutamate [31], and GABA by specific transporters [32], control of pH in the brain [33]; and their dysfunction promote neurodegeneration in AD [34]. By the deposition of Aβ in the AD brain, astrocytes become activated and cause detrimental consequences including the reduction of the glutamate uptake due to low expression of re uptake transporters, altered ions homeostasis (K+ and Ca+), increased the release of cytokines and inflammatory mediators and reduced levels of aquaporins [35].
Over-accumulation of extracellular glutamate during Aβ aggregation causes neurotoxicity in AD pathogenesis [25]. In AD, morphological alteration of astrocytes causes modifications in K+ neurovascular regulation by downregulation of rectifying K+ channels (Kir) 4.1 which causes irregular cerebral blood flow. Disruption of gliotransmission by enhancing calcium signaling in astrocytes is altered by Aβ accumulation [23]. This calcium/gliotransmission alteration could underlie an important role of astrocytes in AD pathology. These alterations cause the over-expression and overstimulation of α7 subunit-containing nicotinic acetylcholine receptors (7αnAChR), purinergic receptor P2Y1, metabotropic glutamate receptor 5 (mGluR5), and changes in Ca2 + homeostasis which increases the levels of glutaminase and glutamine in the astrocyte of the hippocampal regions. It increases internal Ca2 + levels and finally stimulates hemichannels to release more glutamate and led to the downregulation of glutamate transporter (GLT1) in astrocytes causing glutamate excitotoxicity in AD [36]. In addition, long-term exposure to Aβ1–42 can cause GLT-1 loss in astrocytes that are mediated by numerous inflammation-related transcriptional processes [37].
Furthermore, aquaporins 4 (AQP4) controls various functions of astrocytes. AQP4 plays an important role in the clearance of Aβ [38]. About 55–65% of Aβ clearance was decreased in AQP4 knockout mice [39]. Increasing Aβ accumulation and memory deficits were found in the AQP4 deleted 12-month-old APP/PS1 mice [40]. From the postmortem AD brain, it was found that loss of perivascular AQP4 localization and its low expression level might be reduced Aβ disaggregation and Aβ clearance in AD [41]. Literature indicated that deficiency of AQP4 leads to the downregulation of GLT1 which affects synaptic plasticity and memory [42]. Using GLT1 expression regulators such as ceftriaxone rescues hippocampal memory deficit in AQP4 knockout mice via GLT-1 activation [43]. Therefore, AQP4 can be a molecular target for AD treatment.
Another important innate immune cell of the central nervous system (CNS), microglia contribute to the Aβ clearance through phagocytosis and degradation of Aβ. M2 microglia secretes anti-inflammatory cytokines and growth factors which have a neuroprotective effect. In AD progression, Aβ triggers microglial activation (M1 microglia) which secretes pro-inflammatory cytokines and reduces the efficacy of Aβ clearance, causing inflammation in AD [44].
Association between excitotoxicity and stroke
Cerebral blood flow is substantially diminished immediately after IS. As a result, the availability of glucose and oxygen in the neuronal tissues are reduced too. Deprivation of energy creates mitochondrial dysfunction, induces oxidative stress, and eventually triggers ROS production. Insufficiency of energy simultaneously contributes to the ionic imbalance of Na+, K+, and Ca2 + concentration, causing cellular depolarization and glutamate secretion. Excessive glutamate activates NMDA receptors initiating cell toxicity and serious damage to the CNS [45] (Fig. 2). Enhanced Ca2 + influx disturbs ionic homeostasis, resulting Ca2 + overload in mitochondria and cytosol [46]. These events trigger the production of free radicals, lipases, kinases, phosphatases, endonucleases [47] and stimulate calpain [48], mitochondrial dysfunction and oxidative stress, and finally neuronal cell death [49].
Glutamate has an important role in ischemia-induced excitotoxicity. Astrocytes dynamically control synaptic transmission and trigger excitotoxicity by the secretion of glutamate in stroke [22]. The mechanism of astrocytic glutamate secretion has been mysterious. Some researchers believe that glutamate transporters such as EAAT1 (also known as GLAST) and GLT-1 (known as EAAT2) are downregulated which increases glutamate concentration, and leads to cell death via excitotoxicity in ischemia or reperfusion injury conditions [50]. During post-cerebral injury, more excitotoxicity was found in mutated GLT-1 gene knockout mice than in their wild-type counterparts [51]. Other researchers suggested that astrocytic Swell1 subunit containing volume-regulated anion channel release glutamate, regulate synaptic transmission, and contribute to the excitotoxicity in stroke [52]. Furthermore, another glutamate transporter, EAAT4, is highly expressed following brain injury [53]. Under conditions of ischemia, the excessive influx of Na+ into astrocytes triggers cell death by the overload of Ca2 + through a reversed activity of the cell membrane Na+ -Ca2 + exchanger [54].
Like AD, AQP4 also plays a significant in IS. In the early stage of a stroke, APQP4 prevents the swelling of astrocytes, leads to decrease water inflation in the brain, and decreases BBB destruction, inflammation, and neuronal death [55]. After 1-hour post-stroke, the expression of AQP4 is rapidly increased in perivascular endfeet of astrocytes. Upregulated AQP4 promotes water uptake, emerges cellular damage, disrupts BBB, and finally, shifts vasogenic edema to hemorrhagic conversion [56]. Brain edema was reduced after IS in AQP4-deficient mice [57]. Therefore, inhibition of AQP4 may contribute to a novel therapeutic option for stroke treatment.
Additionally, microglia have a ramified morphology in homeostatic conditions. During all stages of IS, microglia are activated and can produce both detrimental and neuroprotective mediators. After instant IS, microglia (M1) immediately shift towards the lesion site and aggravate tissue injury by producing pro-inflammatory cytokines and cytotoxic substances. On the other hand, microglia (M2) also engulf debris and produce anti-inflammatory cytokines and growth factors, maintaining a suitable microenvironment for new neural circuity [58].
In addition, there is a dynamic interaction between microglia and other cells like astrocytes, neurons, oligodendrocytes, and endothelial cells in the neurovascular unit. This dynamic interaction plays a significant role in continuing acute injury and post-stroke recovery. After IS, microglia are activated earlier and promote the activation of astrocytes. Astrocyte activation accelerates pro-inflammatory cytokines and damages oligodendrocytes in ischemic white matter injury. It also disrupts BBB integrity and damage blood vessels. M2 microglia normalize endothelial cell proliferation and help angiogenesis in a biphasic manner in the brain [59, 60]. Therefore, M1/M2 microglial activation has significant translational value to boost the defensive role and /or diminish the adverse effect of stroke.
APOLIPOPROTEIN E IN AD AND STROKE
Apolipoprotein E (ApoE) is a main lipid transporter that has a fundamental role in the development, maintenance, and repair of CNS. It also controls various crucial signaling pathways in the brain. ApoE is mostly expressed by astrocytes and microglia, and it plays dynamic roles in AD and stroke pathogenesis [61] (Fig. 3). Three major isoforms namely ApoE2, ApoE3, and Apo4 are encoded by three different alleles namely APOE ɛ2, APOE ɛ3, and APOE ɛ4, located on chromosome 19. Three isoforms are structurally and functionally different due to the substitution of a single amino acid residue. ApoE4 is involved in the neurodegeneration in AD and IS while ApoE2 and ApoE3 are protective in nature [62].
Fig. 3
Schematic representation of APOE isoform-dependent effects on the common features of Alzheimer’s disease (AD) and stroke. Specifically, APOE4 carriers are more susceptible to produce more plaques and tangles, and to have advanced insulin resistance, mitochondrial dysfunction, neuroinflammation, and synaptic deficits. The APOE4 isoform is also responsible for decrease metabolism of glucose, lipid, cholesterol, autophagy, Aβ clearance across the blood-brain barrier (BBB), and a gradual loss of Aβ uptake and clearance by microglia causing neurodegenerative diseases such as AD and ischemic stroke.
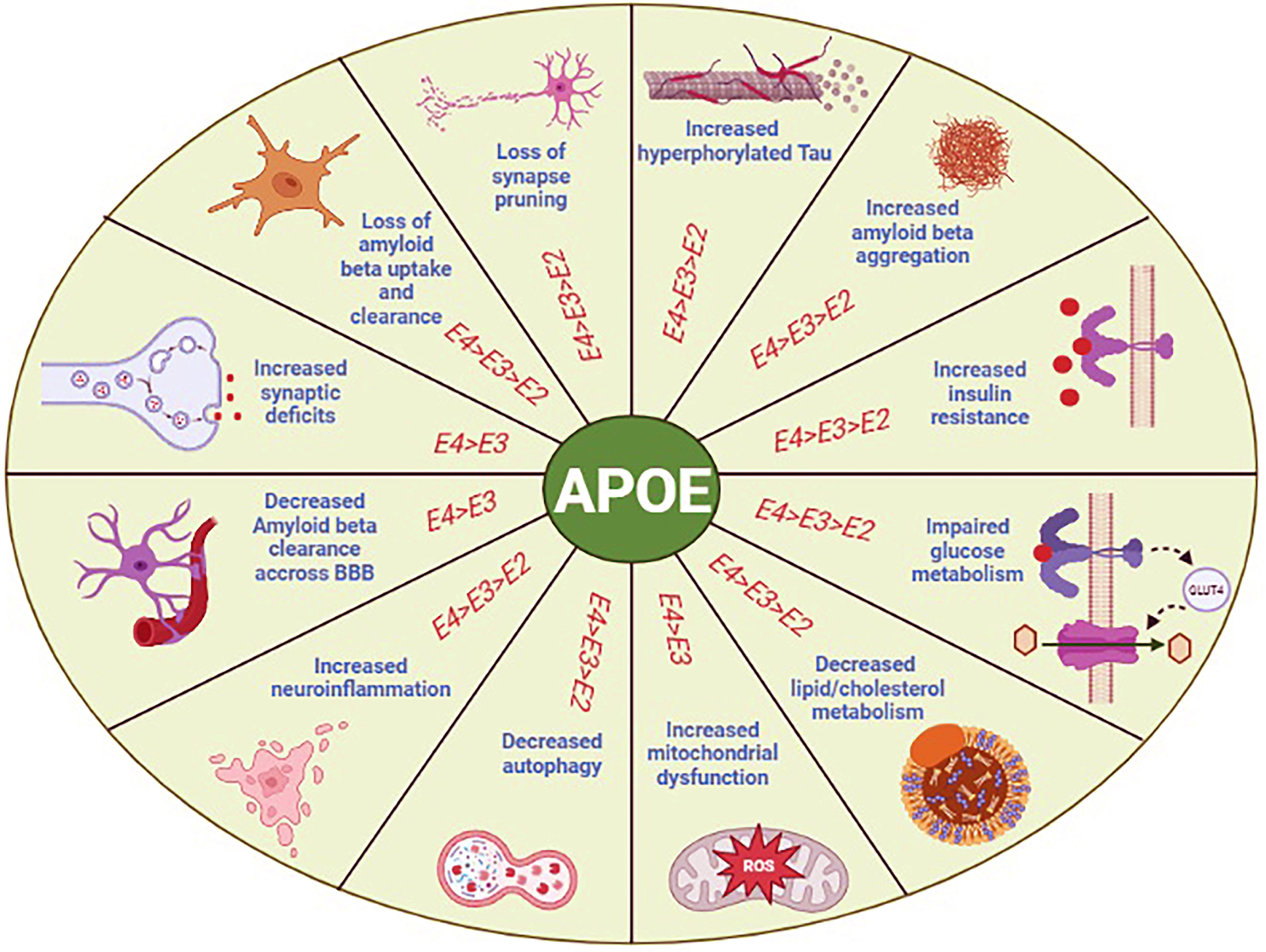
Association between ApoE4 and AD
APOE ɛ4 is a major threat for late-onset AD and increases the risk of AD by 3-fold [63]. Although the exact mechanism of APOE ɛ4 in the onset of AD progression is not fully revealed, numerous evidence suggests that ApoE4 influences Aβ aggregation and deposition in AD. The isoform ApoE4 impairs synaptic function, causes neurotoxicity, tau hyperphosphorylation, and neuroinflammation which are the contributing factors in the onset of AD [64] (Fig. 3). The molecular mechanism of ApoE4 remains unclear. Recently, it was found that ApoE4 quickens the BBB disruption by the activation of cyclophilin A-matrix metalloproteinase-9 (CypA-MMP-9) in aged APOE knock-in mice crossed with 5xFAD. The reduced level of cerebral blood flow deficits the behavioral activity and causes neuronal loss in this mouse model [65], suggesting more research work will be needed to understand the role of ApoE4 in AD.
Association between APOE ɛ4 and stroke
Several studies revealed the connection between APOE gene polymorphisms and the possibility of developing a stroke. APOE ɛ4 allele increases cholesterol levels, amyloid deposition in blood vessels, development of hypertension, hypometabolism of glucose, and insulin resistance which are underlying triggers for ischemic cerebrovascular disorders [66] (Fig. 3). It is well documented that carriers of the ɛ4 allele show an increased risk of ICH but ɛ2 allele carriers are not at risk of having ICH in the future. Recent research indicated that the occurrence of the APOE ɛ4 allele significantly increases the risk of IS in the populations from rural Eastern India [67], China [68], and Korea [69], and deteriorates cognitive function [70]. However, the influence of ApoE4 in pre-and post-stroke is not clear. Some researchers believe that ApoE4 quickens dementia progression after stroke [65]. Though the interaction between ApoE4, Aβ, and tau pathways after transient IS in the cerebrovascular system, and their contribution to cognitive impairment in the development of dementia remains uncertain [65].
PHOSPHATIDYLINOSITOL 3-KINASE/AKT (PI3K/AKT) SIGNALING IN AD AND STROKE
Hypometabolism of glucose is one of the remotest factors responsible for the onset of AD and IS and is linked with PI3K/Akt signaling pathway. Dysregulated PI3K/Akt signaling may have a vital role in impaired cognition such as in AD [71] and IS [17]. In aged people with no dementia, PI3K/Akt signaling maintains both the volume and function of the brain and protects the brain from atrophy and slow the progression of associated complications [72]. Therefore, an impaired PI3K/Akt signaling pathway may cause neurodegenerative diseases such as stroke and AD (Fig. 4).
Fig. 4
Regulation of PI3K/Akt signaling in Alzheimer’s disease (AD) and ischemic stroke (IS). In both of the diseases, PI3K/Akt signaling is dysregulated by the reduced protein expression levels of insulin signaling related proteins such as IR, IRS, PI3K, PIP2, and Akt. The downstream signaling proteins such as GSK3 is over activated and Cdk5/p25 pathway leads to tau hyperphosphorylation in AD. In AD and IS, insulin resistance in the brain diminishes the glucose receptor GLUT4, and due to impaired PI3k/Akt signaling pathway, the glucose receptors such as GLUT 1, 2, and 3 may reduce in the brain, causing the decrease of glucose metabolism. In addition, lower levels of glucose reduce UDP-GlcNAc concentration via the hexosamine biosynthetic pathway (HBP) and subsequently diminished tau O-GlcNAcylation. Reduced O-GlcNAcylation helps in tau hyperphosphorylation, tau oligomerization and ultimately neurofibrillary tangles are produced in AD [73]. Akt, protein kinase B; CdK 5, cyclin-dependent kinase 5; Fructose-6-p, fructose-6-phosphate; GlcNAc, β-N-acetylglucosamine; Glucosamine-6-P, glucosamine-6-Phosphate; GLUT, glucose transporter; GSK3, glycogen synthetase kinases 3; HBP, hexosamine biosynthetic pathway; HIF-1, hypoxia inducible factor-1; IGF, insulin growth factor; IR, insulin receptor; IRS, insulin receptor substrate; m-TOR, mammalian target of rapamycin; NFTs, neurofibrillary tangles; OGA, O-GlcNAcase; OGT, O-GlcNAc transferase; P, phosphate group; p70S6K, ribosomal protein S6 kinase with a molecular weight of 70 kD; PI3K, phosphatidylinositol 3-kinase; PIP2, phosphatidylinositol (3,4)-bisphosphate; TCA, tricarboxylic acid cycle; UDP, uridine diphosphate.
![Regulation of PI3K/Akt signaling in Alzheimer’s disease (AD) and ischemic stroke (IS). In both of the diseases, PI3K/Akt signaling is dysregulated by the reduced protein expression levels of insulin signaling related proteins such as IR, IRS, PI3K, PIP2, and Akt. The downstream signaling proteins such as GSK3 is over activated and Cdk5/p25 pathway leads to tau hyperphosphorylation in AD. In AD and IS, insulin resistance in the brain diminishes the glucose receptor GLUT4, and due to impaired PI3k/Akt signaling pathway, the glucose receptors such as GLUT 1, 2, and 3 may reduce in the brain, causing the decrease of glucose metabolism. In addition, lower levels of glucose reduce UDP-GlcNAc concentration via the hexosamine biosynthetic pathway (HBP) and subsequently diminished tau O-GlcNAcylation. Reduced O-GlcNAcylation helps in tau hyperphosphorylation, tau oligomerization and ultimately neurofibrillary tangles are produced in AD [73]. Akt, protein kinase B; CdK 5, cyclin-dependent kinase 5; Fructose-6-p, fructose-6-phosphate; GlcNAc, β-N-acetylglucosamine; Glucosamine-6-P, glucosamine-6-Phosphate; GLUT, glucose transporter; GSK3, glycogen synthetase kinases 3; HBP, hexosamine biosynthetic pathway; HIF-1, hypoxia inducible factor-1; IGF, insulin growth factor; IR, insulin receptor; IRS, insulin receptor substrate; m-TOR, mammalian target of rapamycin; NFTs, neurofibrillary tangles; OGA, O-GlcNAcase; OGT, O-GlcNAc transferase; P, phosphate group; p70S6K, ribosomal protein S6 kinase with a molecular weight of 70 kD; PI3K, phosphatidylinositol 3-kinase; PIP2, phosphatidylinositol (3,4)-bisphosphate; TCA, tricarboxylic acid cycle; UDP, uridine diphosphate.](https://content.iospress.com:443/media/adr/2023/7-1/adr-7-1-adr220108/adr-7-adr220108-g004.jpg)
Association between PI3K/Akt signaling and AD
Abundant evidence suggests that impaired brain insulin signaling and dysregulation of brain glucose metabolism may lead to cognitive deficits in AD [71]. Recently, we found the insulin signaling proteins IRβ, IGF-1, IRS-1, IRS-2, p-Akt (Ser473), and Akt were noticeably decreased in AD rats. The downregulation of PI3K/Akt signaling was conveyed by the activation of glycogen synthetase kinases 3 (GSK3), phospho-GSK3β (Tyr216), total GSK3β and cyclin-dependent kinase 5 (Cdk5) has been identified as important candidates for irregular tau hyperphosphorylation. The levels of phospho-GSK3 at Ser9 and phospho-GSK3 at Ser21 were significantly decreased in AD. The PI3K-Akt signaling molecules are negatively connected with the tau hyperphosphorylation at Ser396, Ser202/Thr205, and Cdk5 and to its cofactors p35 and p25 in the brain. The downregulation of Akt decreases the expression of main glucose transporters such as GLUT3 and GLUT4. The overactivation of GSK3β increases the tau hyperphosphorylation and decreases the microtubule-binding activity of tau of a sporadic AD rat model [71, 73, 74] (Fig. 4). It has also been recommended that impaired brain insulin signaling may lead to the reduction of IRS-1 and PI3K phosphorylation, downregulation of O-GlcNAcylation, overactivation of GSK3β, and acceleration of tau hyperphosphorylation in 3xTg-AD mice before the onset of peripheral insulin resistance, whereas Tg2576 mice show dysregulation of these signaling proteins after the onset of peripheral insulin resistance. These inconsistencies may be specific to the tau pathology that develops in 3xTg-AD mice [75]. Reducing CNS insulin signaling dysfunction in patients with mild cognitive impairment or early-stage AD may support a decrease in peripheral insulin resistance [76]. Analysis of postmortem human AD brain revealed that the protein expression of IRS, IGFs, and glucose receptors are significantly diminished [77]. Therefore, it is suggested that impairment of glucose metabolism is due to both pre-and post-peripheral insulin resistance, causing Aβ and tau pathologies.
Association between PI3K/Akt signaling and stroke
PI3K/Akt signaling pathway is directly involved in the neuroprotection against ischemic brain damage and displays a critical role in promoting neuronal survival after IS [17]. Decreased level of phosphorylation of GSK3β on Ser9 was found in the penumbra cortex in IS rat brain [78]. The expression levels of p70S6K on Thr389 reduced at 1, 4, and 24 h of reperfusion, while the levels of total p70S6K remained the same at 1 and 4 h of reperfusion and then declined at 24 h of reperfusion [79]. The consequences suggest that cell survival pathways by Akt1 and p70S6K signaling are suppressed after ischemia, which may donate to the progression of neuronal cell death after ischemic stroke (Fig. 4). Protein expression of p-Akt and total Akt were also reduced after 24 hours of cerebral ischemia in male rat brains [80]. Therefore, PI3K/Akt signaling pathways is broadly considered to play a critical role in the neuroprotective effect against ischemic brain injury.
Hyperglycemia is an indicator of IS, contributing to a significant increase in brain ischemia [81, 82]. Considerable evidence demonstrates that glucose transporters mainly GLUT1 and GLUT3 are important to maintain energy homeostasis, especially in IS patients [83]. In the post-ischemic condition after day-1, the expression of GLUT1 mRNA increases at 175% and returns to the normal level by day 3 [84]. During ischemia, the cerebral glucose level is lower than normal [85]. Three days after ischemic stroke, the expression of mRNA of GLUT3 protein starts improving but is still below the normal level, causing neuronal death [85]. Therefore, increasing the expression of GLUT3 and regulating the brain glucose metabolism during pre and post-ischemic cerebral injury is urgently needed.
INFLAMMATORY SIGNALING PATHWAY IN AD AND STROKE
Inflammation is one of the most important pathophysiological conditions in AD and stroke (Fig. 5). It is mainly mediated by microglia [86, 87], oligodendrocytes [88, 89], and astrocytes [90, 91] in the brain.
Fig. 5
Schematic representation of inflammasome in Alzheimer’s disease (AD) and ischemic stroke (IS). Inflammation is triggered by the activation of the NFkB pathway and NLRP1/3 inflammasome pathway in AD and IS. Finally, IL-18 and IL-1β are produced as proinflammatory cytokines. IL, interleukin; IL-1B, interleukin 1-beta; NF-KB, nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3, nucleotide-binding oligomerization domain like receptor pyrin domain-containing protein 3; TLR, toll-like receptor.
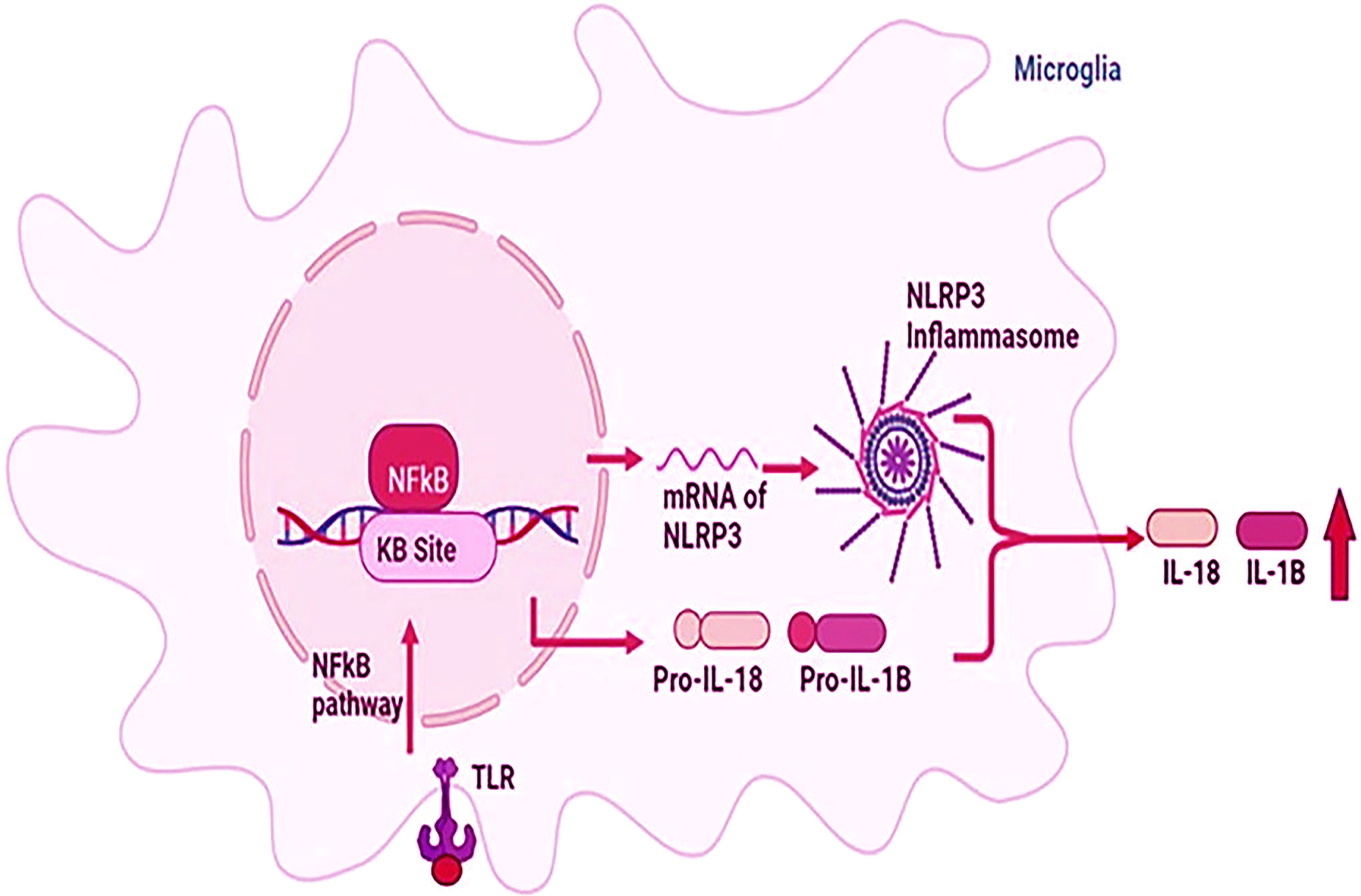
Association between inflammation and AD
Extracellular Aβ deposition activates microglia and triggers pro-inflammatory cytokines such as interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α), and interleukin-1β (IL-1β) in the AD brain [92]. Microglia and astrocytes have an important role in Aβ clearance and degradation [93], but in case of overproduction of Aβ and pro-inflammatory cytokines, microglia fail to activate the effective phagocytic response to clear the Aβ from the brain, causing the deposition of more pro-inflammatory cytokines in the AD brain [93].
Recent findings showed significant elevated TLR2 activation in Tg2576 AD mice [94, 95] which may also contribute to Aβ accumulation in the brain [96]. However, TLR9 can decrease in both Aβ and tau pathologies in various AD transgenic mouse models and rescue their cognitive deficits [97]. In AD pathogenesis, NF-kB stimulates NLRP3 inflammasome, and proinflammatory cytokines such as IL-1β and IL-18 are produced, finally getting mature IL-1β and IL-18 and released from microglia to the extracellular environment [98] (Fig. 5).
Association between inflammation and stroke
Oxidative stress and inflammation are mainly involved in the pathological development of late post-ischemic conditions [99]. NF-kB signaling cascade is activated within 2 hours after brain injury in a middle cerebral artery occlusion (MCAO) rat model [100]. Stimulated NF-kB controls the gene expression of proinflammatory cytokines such as TNF-α and IL-1β. In addition, toll-like receptors (TLR) mediated signaling pathway via NF-kB is also stimulated and upregulated in ischemic brain injury by MCAO in C57BL/6 mice [101]. TLRs play a vital role in cerebral brain injury. The protein expression of TLRs is augmented which is linked with the inflammatory cascade in IS [102]. In addition, TLR4 also rises brain ischemic lesions. The protein expression of TLR4, TNF-α, caspase-1, NLRP3, and IL-8 significantly increased in the cerebral ischemia-reperfusion mice model [103]. Oxidative stress markers such as nitric oxide and malondialdehyde are increased whereas superoxide dismutase is reduced in these mice brains. The levels of NLRP1, NLRP3, IL-1β, and IL-18 were elevated in the animal model and human brain with IS [104] (Fig. 5). Therefore, a thorough understanding of the widespread mechanisms of post-ischemic neuroinflammatory signaling pathways is needed for the new treatment strategies for IS.
AUTOPHAGY PATHWAY IN AD AND STROKE
Autophagy is a cellular degradation process and is essential for the maintenance of cellular homeostasis through the synthesis, deprivation, and turnover of different cellular components. The mechanism of autophagy is carried out through the formation of vesicle nucleation, vesicle elongation, and autophagosome. Then autophagosome is fused with the lysosome, forming an autolysosome in which engulfed material and inner membrane is degraded [105]. Dysfunction of autophagy accelerates AD and stroke progression and pathogenesis [106, 107] (Fig. 6).
Fig. 6
Schematic representation of mTOR-autophagy pathway in Alzheimer’s disease (AD) and ischemic stroke (IS). In AD, Aβ participate in autophagy via the endosome. The downregulation of mTOR-autophagy pathway is found in AD and IS. AICD, amyloid precursor protein intracellular domain; Akt, protein kinase B; AMP, adenosine 5’-monophosphate; AMPK, adenosine monophosphate-activated protein kinase; APP, amyloid precursor protein; ATP, adenosine tri-phosphate; Aβ, amyloid-β; C99, 99-residue C-terminal fragment; mTOR, mammalian target of rapamycin; PI3k, phosphatidylinositol 3-kinase; TFEB, transcription factor EB; ULK1, Unc-51 like autophagy activating kinase 1.
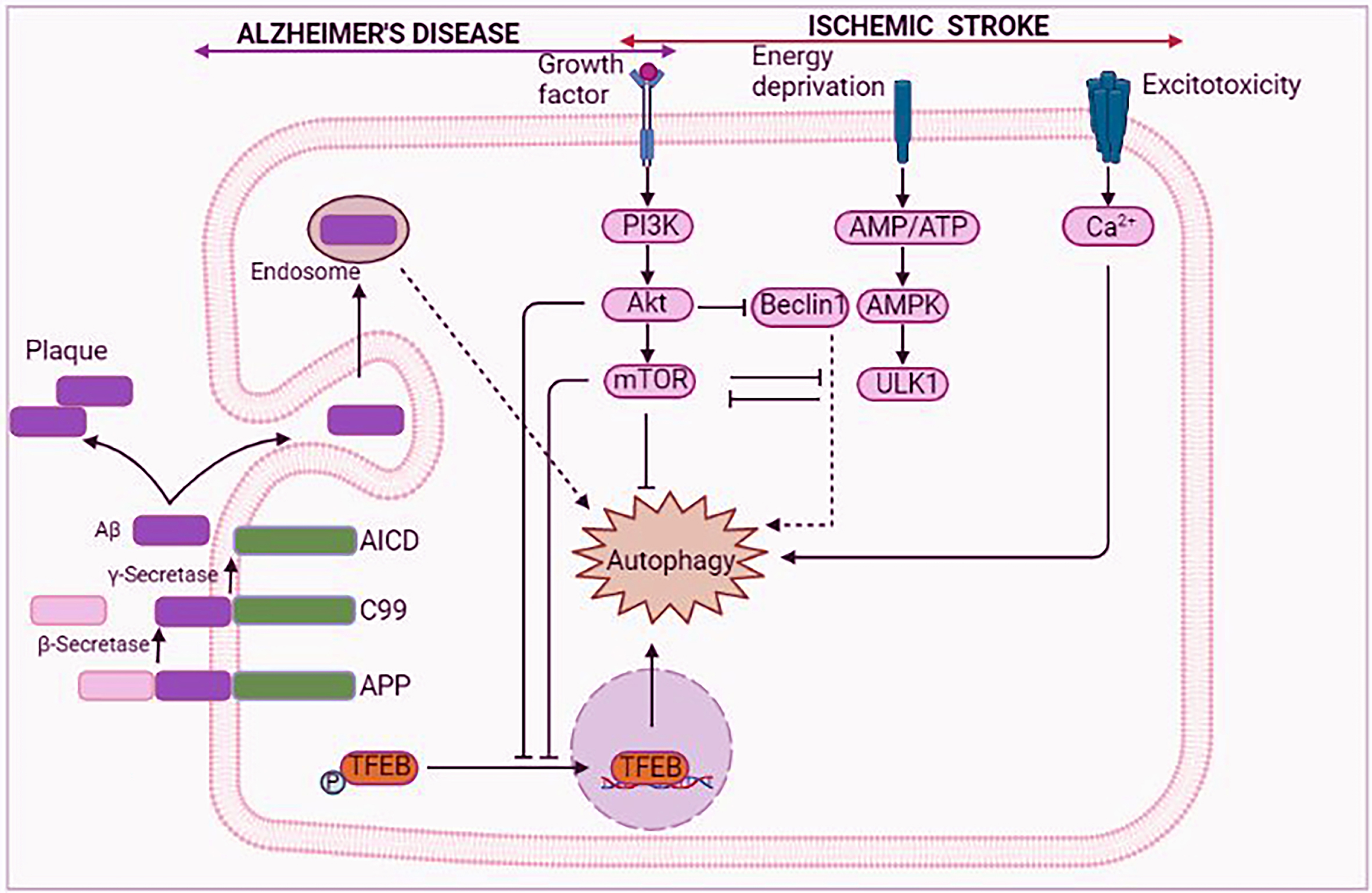
Association between autophagy and AD
Dysregulation of autophagy is associated with AD pathogenesis. The precise role of autophagy in AD through the endosomal-lysosomal system is still debatable. Some researchers suggested that abnormal autophagy induction helps to create the necessary compartment for the accumulation of autophagic vacuoles holding Aβ and finally forming Aβ plaques [108, 109]. Other evidence indicated that either impaired autophagic clearance or deteriorating autophagic activity may play in AD pathogenesis and progression [110]. Therefore, autophagy is a key regulator of Aβ generation and clearance. Aβ is released in an autophagy-dependent manner from neurons in AD pathology [111]. In the early stage of AD, amyloid-β protein precursor (AβPP) processing and Aβ accumulation-mediated autophagy facilitate the removal of toxic protein aggregates via mechanistic target of the rapamycin (mTOR)-dependent and -independent pathways [112, 113]. In addition, autophagy-related genes and AβPP are believed to influence AD development and offer a bidirectional link between autophagy and AD progression. Fewer extracellular Aβ plaques were found in the autophagy-deficient AβPP transgenic mice by the inability of cells with disrupted autophagy to secrete Aβ in the brain [114]. In AD the mechanism of autophagy modulation in AβPP processing and Aβ metabolism and its pathogenesis are not well known. The mTOR signaling pathway controls autophagy in AD (Fig. 6).
Numerous studies suggest that the levels of phospho-mTOR and its downstream signaling molecules such as ribosomal protein S6 kinase beta-1 (p70S6K) and the eukaryotic translation factor 4E (eIF4E) are elevated in postmortem human AD brains, suggesting increased mTOR activity in the AD brains [115]. The increased mTOR activity was connected with enhanced tau hyperphosphorylation [112]. In short, the downregulation of mTOR signaling proteins may reduce autophagy which accelerates the Aβ aggregation and tau hyperphosphorylation in AD pathogenesis.
Association between autophagy and stroke
Numerous evidence indicates that autophagy is triggered in neurons, glial cells, and brain microvascular cells upon IS [116]. But the exact molecular mechanism of autophagy in IS is not well known. However, recent accumulating evidence in IS showed that autophagy inhibits inflammasome activation by triggering mTOR and AMPK pathways in cellular homeostasis. The autophagy-forming markers such as LC3-II and Beclin-1 are upregulated in IS. Autophagy can retrieve the neuronal tissue after IS by the phenotypic alteration of microglia through the NF-κB pathway [107]. Additionally, the autophagy-mediated death-promoting mechanism promotes numerous relevant stimuli in post-ischemic neuronal damage through the downregulation of autophagy signaling molecules such as autophagy-related protein 7, Beclin-1, and LC3-II [117]. In addition, mTOR signaling molecules are reduced in autophagy via phosphorylation of its downstream signaling factors (p70S6K) and 4E binding protein 1 (4EBP1) [117, 118] (Fig. 6). Hence, activation of the mTOR signaling pathway could be an effective strategy for the reduction of infarct size after post-cerebral ischemia.
NOTCH SIGNALING PATHWAY IN AD AND STROKE
Notch signaling is intermediated by the communication of one the ligand of Delta/Serrate/LAG2 (i.e., JAG1, JAG2, DLL3, and DLL4) in one cell with one of the Notch receptors (i.e., Notch1, Notch2, Notch3, and Notch4) in the neighboring cell during development and aging processes. The notch intercellular domain (NICD) is the most contributing factor in Notch signaling. It is produced by the proteolytic cleavage of the receptor-ligand interaction cascade and then NICD is translocated into the nucleus and induces the gene expression which is involved in proliferation, differential, and development (Fig. 7). Notch signaling is activated in AD [119] and IS [120].
Fig. 7
Schematic representation of the Notch signaling. Notch signaling is started by three sequential proteolytic cleavages. First, Notch is cut by Furin at the trans-Golgi and it is known as S1 cleavage. Glycosylation occurs in this step. Then the extracellular domain of Notch, EGF repeats, interacts with several ligands such as Jagged-1, -3, and -4 and Delta 1 and 2 on signal sending cells is cleaved (S2 cleavage) by metalloproteases of the ADAM family. Then Notch intercellular domain (NICD) is cut by a γ-secretase activity of presenilin (PS) (S3 cleavage). γ-secretase activity is improved in AD and ischemic stroke conditions. Activated Notch increases apoptotic and inflammation in the brain. DLL, Delta-like; JAG, jagged protein; NICD, Notch intracellular domain.
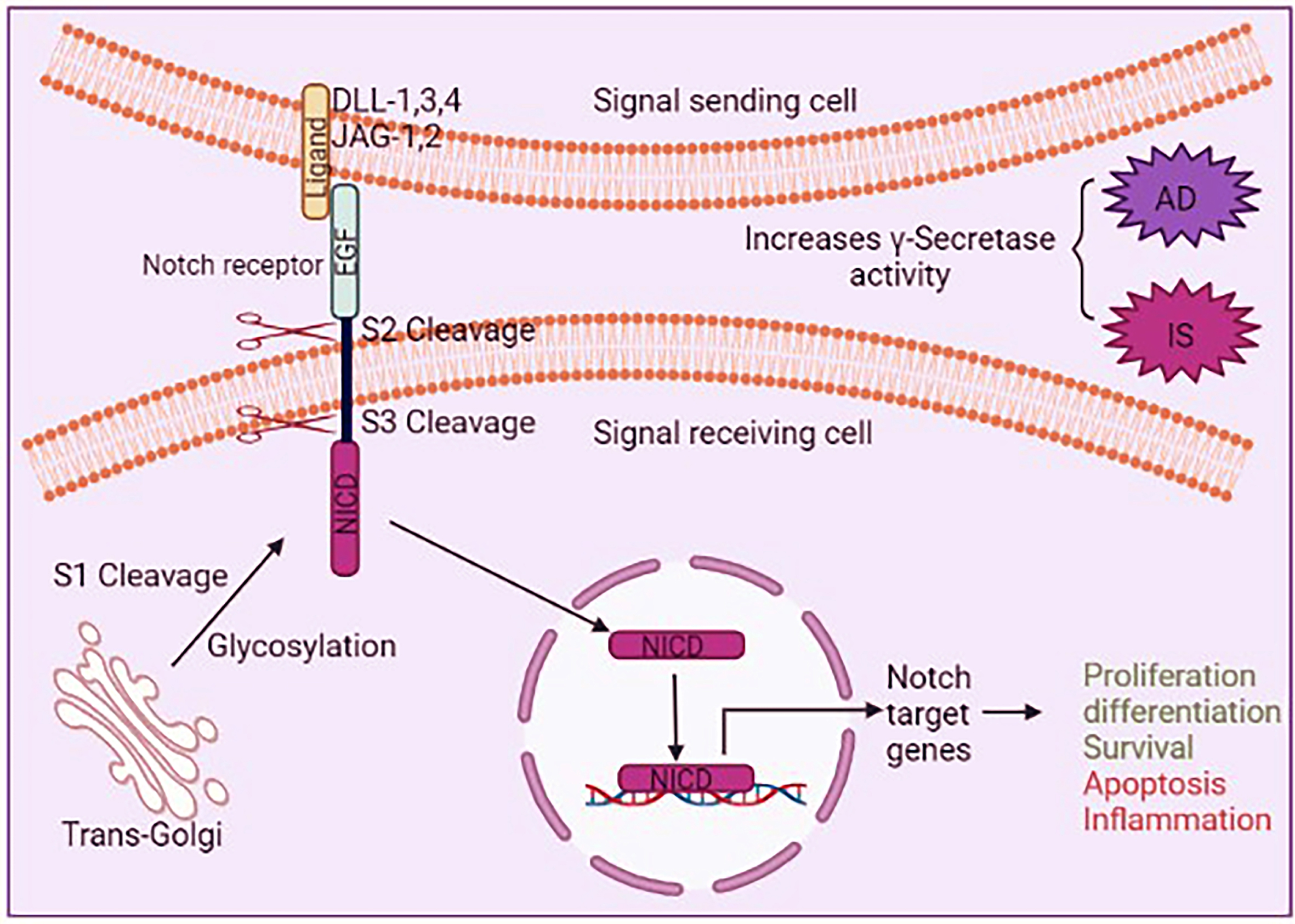
Association between Notch signaling and AD
Presenilin (PS), the catalytic member of the γ-secretase proteolytic complex, plays an important role in the cleavage of AβPP and Aβ production through Notch signaling in the early-onset familial AD pathogenesis. Alterations in proteolysis of the Notch by γ-secretase may participate in the pathogenesis of AD [121]. Notch proteins also interact with PSs and with AβPP in postmitotic neurons in AD. Mutations in the genes encoding the AβPP and PS1, and PS2 are occasionally responsible for early-onset AD. Loss of specific memory and learning and behavioral and cognitive impairment is associated with the Notch signaling pathway in AD [119, 121] (Fig. 7). High expression of Notch1 was observed in the AD cortex, indicating the enhancement of Aβ production and alteration of Notch signaling in the AD brain [122]. Two-fold Notch expression was higher in the AD brain than in the age-matched control human brain [123]. It suggested that higher Notch signaling and expression could be a detrimental effect on AD. Therefore, altered Notch signaling in AD is needed to be further investigated.
Association between Notch signaling and stroke
In vivo and in vitro studies indicated that Notch signaling contributes to IS. The levels of γ-secretase and NICD were augmented in post-ischemia-transgenic mice [124]. Increased levels of NICD were also found in primary neuron culture upon ischemia-like conditions. Furthermore, it has been conveyed that Notch is upregulated instantly after initiation of ischemia in animal models [124, 125]. The contribution of the Notch pathway has been recognized in the growth and proliferation of blood vessels in IS pathogenesis. The levels of NICD and Jag1 positive endothelial cells amplified by activation of downstream signals including Bim, P65, and NFκB donated to the development and deterioration of IS in stroke brains of human and mouse models [120, 125]. The γ-secretase inhibitor might recover the stroke signs through the downregulation of NICD and downstream targets (Fig. 7). Therefore, Notch signaling has a vital role in brain damage in IS.
GUT-BRAIN AXIS IN AD AND STROKE
Gut microbiota has a proven role in regulating multiple neurochemical pathways through the highly interconnected “gut-brain axis (GBA)”. “Gut-brain axis” states a bidirectional communication network between the CNS and the gastrointestinal tract involving multiple overlapping pathways such as the autonomic, neuroendocrine, and immune systems and; directly affecting the vagus nerve to the brain. The gut microbiome controls BBB integrity, neural development, aging, and CNS immune activation. Alteration of gut-microbiota diversity breaks the gut barrier integrity and activates systemic neuroinflammation, promotes neuronal injury, and finally neurodegenerative diseases such as AD and stroke. Dysbiosis accelerates or causes impaired GBA signaling in AD [126] and in stroke [127]. Therefore, the GBA is an important pathway of communication between the gut and the brain, and dynamically contributes to AD and IS pathogenesis (Fig. 8).
Fig. 8
Schematic representation of microbiome-gut-brain axis in (A) Alzheimer’s disease (AD) and (B) stroke. The bidirectional gut-brain interaction communicates by mainly four pathways such as metabolic, immune, neural and endocrine signaling pathways. Gut dysbiosis changes gut-microbiota diversity and gut microbiota enter to blood circulation by leaky gut in both AD and stroke. In addition, the ischemic brain triggers the gut-microbiota diversity by the hypothalamic-pituitary-adrenal (HPA) pathway which plays an important role in stroke outcomes. BBB, blood-brain barrier; BDNF, brain derived neurotrophic factor; GABA, gamma-aminobutyric acid; GLP-1, glucagon-like peptide 1; HPA, hypothalamus-pituitary-adrenal; LPS, lipopolysaccharide; NFTs, neurofibrillary tangles; SCFAs, short-chain fatty acids; TMAO, trimethylamine N-oxide.
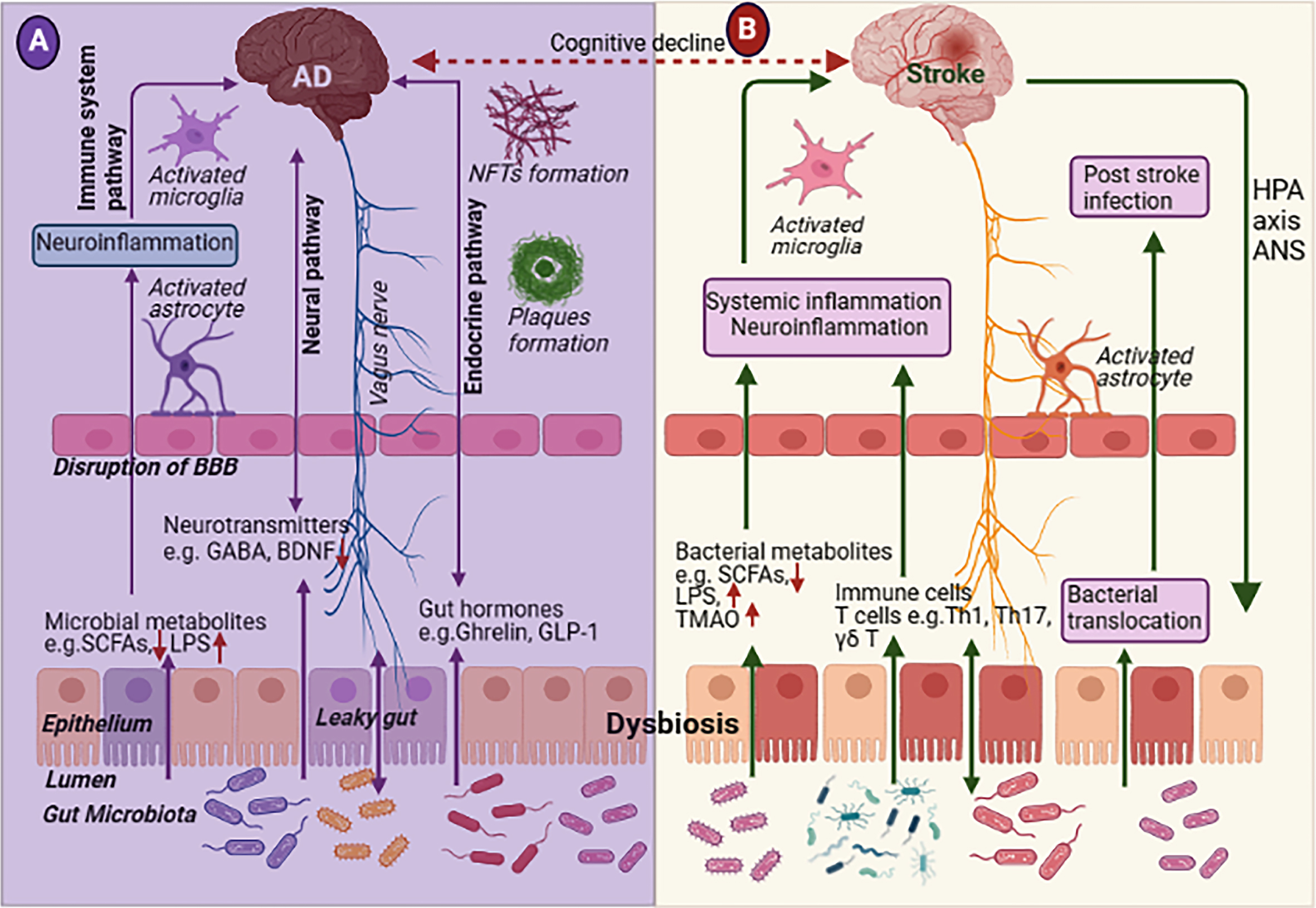
Association between gut-brain axis and AD
Gut dysbiosis may worsen BBB permeability and cause AD pathogenesis. Numerous evidence suggests that gut microbiota can control brain and behavioral function and produce bacterial amyloid (also known as curli). Curli may trigger the immune cells in the gut and/or brain and activates neuroinflammation [128]. Bacteria containing lipopolysaccharides may upregulate pro-inflammatory cytokines through GBA [129]. Also, reduced beneficial bacteria may impact the levels of short-chain fatty acid (SCFAs) bioavailability which may subsequently change the metabolic pathways and further contribute Aβ deposition in AD brain [130, 131]. Elevated levels of Helicobacteraceae Desulfovibrionaceae and Odoribacter were found in APP/PSI mice [132]. Reduced levels of Acetobacter and Lactobacilli were found in an AD Drosophila model, and it was associated with reduced SCFAs levels [133]. Higher quantities of Bacteroids, Ruminococci, Actinobacteriae, Lachnospiraceae, and Selenomonadales levels were observed in the fecal samples of AD patients [134]. Low loads of Firmicutes and Bifidobacterium and high loads of Bacteroidetes were found in the fecal samples of AD patients, suggesting the presence of gut dysbiosis in AD patients [135]. Gut microbiota may impact AD pathogenesis via numerous pathways such as neuroinflammation, oxidative stress, pathogenic translocation, and neurotransmitter dysregulation. The gut microbiome needs more attention and understanding the interaction in terms of amyloid pathology may help with beneficially modifying the bacterial consortium to improve the quality of life in pre-symptomatic AD patients.
Association between gut-brain axis and stroke
Emerging clues propose the important role of gut microbiota in the pathology of stroke. Dysbiosis increases the risk factors of strokes such as systemic inflammation, atherosclerosis, and cardiometabolic disorders. Dysbiosis induces systemic inflammation, neuroinflammation, and infection in acute cerebral ischemia which has a detrimental effect on stroke outcomes. Several pathways such as bacterial components, gut metabolites, immune cells, and bacterial translocation are involved in stroke. The ischemia brain also stimulates the gut microbial diversity through the neural or hypothalamic-pituitary-adrenal pathways, which have a harmful effect on stroke outcomes [136]. In addition, many gut microbiota such as Bacteroides , Prevotella, and Faecali bacterium are significantly reduced in IS patients compared with their healthy counterparts [137]. Increased levels of Lactobacillus ruminis and decreased Lactobacillus sakei were identified in IS patients, which induce the proinflammatory cytokine IL-8 secretion [137, 138]. Though the research on the gut inflammatory and immune response after stroke is still in its initial stages, recent investigations between clinical trials [138–142] and animal experiments [143–145] have provided some interesting clues regarding the role of gut microbiota in IS outcomes. Hence, the gut microbiome plays a pivotal role in gut inflammation and the immune response to pre/post-ischemic brain injury and stroke.
CONCLUSION
In a nutshell, we concluded that both diseases share several common abnormalities including excitotoxicity, ApoE4, impaired PI3K/Akt pathway, increased neuroinflammation, decreased mTOR-autophagy signaling, decreased Notch signaling, and gut dysbiosis. It has been recommended that AD and stroke disrupt common cellular and molecular pathways and each disease reinforces the progression of the other. This review mainly focused on the biochemical pathways shared by AD and IS. Understanding the key mechanisms underlying the toxic interaction between these diseases may provide opportunities for designing effective therapeutic strategies. Whether stroke is directly involved in AD or acts indirectly as a contributing factor to AD pathogenesis needs to be established. Hence, the prevention of stroke and its treatment may have important implications for the alleviation of AD and warrant further investigations.
ACKNOWLEDGMENTS
All figures were made by using Bio Render App.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Das TK , Wati MR , Fatima-Shad K ((2014) ) Oxidative stress gated by Fenton and Haber Weiss reactions and its association with Alzheimer’s disease. Arch Neurosci 2: , 1–8. |
[2] | Zhang H , Wei W , Zhao M , Ma L , Jiang X , Pei H , Cao Y , Li H ((2021) ) Interaction between Aβ and tau in the pathogenesis of Alzheimer’s disease. Int J Biol Sci 17: , 2181–2192. |
[3] | Morris GP , Clark IA , Vissel B ((2014) ) Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Communs 2: , 135. |
[4] | Long JM , Holtzman DM ((2019) ) Alzheimer disease: An update on pathobiology and treatment strategies. Cell 179: , 312–339. |
[5] | Haass C , Selkoe D ((2022) ) If amyloid drives Alzheimer disease, why have anti-amyloid therapies not yet slowed cognitive decline? PLoS Biol 20: , e3001694. |
[6] | ((2022) ) 2022 Alzheimer’s disease facts and figures. Alzheimers Dementia 18: , 700–789. |
[7] | Thayabaranathan T , Kim J , Cadilhac DA , Thrift AG , Donnan GA , Howard G , Howard VJ , Rothwell PM , Feigin V , Norrving B , Owolabi M , Pandian J , Liu L , Olaiya MT ((2022) ) Global stroke statistics 2022. Int J Stroke 17: , 946–956. |
[8] | Greenberg SM , Bacskai BJ , Hernandez-Guillamon M , Pruzin J , Sperling R , van Veluw SJ ((2020) ) Cerebral amyloid angiopathy and Alzheimer disease — one peptide, two pathways. Nat Rev Neurol 16: , 30–42. |
[9] | Waziry R , Chibnik LB , Bos D , Ikram MK , Hofman A ((2020) ) Risk of hemorrhagic and ischemic stroke in patients with Alzheimer disease. Neurology 94: , 265–272. |
[10] | Pendlebury ST , Rothwell PM ((2019) ) Incidence and prevalence of dementia associated with transient ischemic attack and stroke: Analysis of the population-based Oxford Vascular Study. Lancet Neurol 18: , 248–258. |
[11] | Desmond DW , Moroney JT , Sano M , Stern Y ((2002) ) Incidence of dementia after ischemic stroke: Results of a longitudinal study. Stroke 33: , 2254–2262. |
[12] | Guo X , Östling S , Kern S , Johansson L , Skoog I ((2018) ) Increased risk for dementia both before and after stroke: A population-based study in women followed over 44 years. Alzheimers Dement 14: , 1253–1260. |
[13] | Nguyen TV , Frye JB , Zbesko JC , Stepanovic K , Hayes M , Urzua A , Serrano G , Beach TG , Doyle KP ((2016) ) Multiplex immunoassay characterization and species comparison of inflammation in acute and non-acute ischemic infarcts in human and mouse brain tissue. Acta Neuropathol Commun 4: , 100. |
[14] | Park JY , Byeon JH , Park SW , Eun SH , Chae KY , Eun BL ((2013) ) Neuroprotective effect of human placental extract on hypoxic– ischemic brain injury in neonatal rats. Brain Dev 35: , 68–74. |
[15] | Barthels D , Das H ((2020) ) Current advances in ischemic stroke research and therapies. Biochim Biophys Acta Mol Basis Dis 1866: , 165260. |
[16] | Yenari MA , Kauppinen TM , Swanson RA ((2010) ) Microglial activation in stroke: Therapeutic targets. Neurotherapeutics 7: , 378–391. |
[17] | Qin C , Yang S , Chu YH , Zhang H , Pang XW , Chen L , Zhou LQ , Chen M , Tian DS , Wang W ((2022) ) Signaling pathways involved in ischemic stroke: Molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther 7: , 215. |
[18] | Eskandari S , Sajadimajd S , Alaei L , Soheilikhah Z , Derakhshankhah H , Bahrami G ((2021) ) Targeting common signaling pathways for the treatment of stroke and Alzheimer’s: A comprehensive review. Neurotox Res 39: , 1589–1612. |
[19] | Wang R , Reddy PH ((2017) ) Role of glutamate and NMDA receptors in Alzheimer’s disease. J Alzheimers Dis 57: , 1041–1048. |
[20] | Talantova M , Sanz-Blasco S , Zhang X , Xia P , Akhtar MW , Okamoto S , Dziewczapolski G , Nakamura T , Cao G , Pratt AE , Kang YJ , Tu S , Molokanova E , McKercher SR , Hires SA , Sason H , Stouffer DG , Buczynski MW , Solomon JP , Michael S , Powers ET , Kelly JW , Roberts A , Tong G , Newmeyer TF , Parker J , Holland EA , Zhang D , Nakanishi N , Chen HSV , Wolosker H , Wang Y , Parsons LH , Ambasudhan R , Masliah E , Heinemann SF , Crespo JCP , Stuart A . Lipton SA ((2013) ) Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc Natl Acad Sci U S A 110: , E2518–E2527. |
[21] | Kirdajova DB , Kriska J , Tureckova J , Anderova M ((2020) ) Ischemia-triggered glutamate excitotoxicity from the perspective of glial cells. Front Cell Neurosci 19: , 51. |
[22] | Wang F , Xie X , Xing X , Sun X ((2022) ) Excitatory synaptic transmission in ischemic stroke: A new outlet for classical neuroprotective strategies. Int J Mol Sci 23: , 9381. |
[23] | Siracusa R , Fusco R , Cuzzocrea S ((2019) ) Astrocytes: Role and functions in brain pathologies. Front Pharmacol 10: , 1114. |
[24] | Danysz W , Parsons CG ((2012) ) Alzheimer’s disease, β-amyloid, glutamate, NMDA receptors and memantine - searching for the connections. Br J Pharmacol 167: , 324–352. |
[25] | Conway ME ((2020) ) Alzheimer’s disease: Targeting the glutamatergic system. Biogerontology 21: , 257–274. |
[26] | Dong XX , Wang Y , Qin ZH ((2009) ) Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin 30: , 379–387. |
[27] | Yeung JHY , Walby JL , Palpagama TH , Turner C , Waldvogel HJ , Faull RLM , Kwakowsky A ((2021) ) Glutamatergic receptor expression changes in the Alzheimer’s disease hippocampus and entorhinal cortex. Brain Pathol 31: , e13005. |
[28] | Madeira C , Vargas-Lopes C , Brandão CO , Reis T , Laks J , Panizzutti R , Ferreira ST ((2018) ) Elevated glutamate and glutamine levels in the cerebrospinal fluid of patients with probable Alzheimer’s disease and depression. Front Psychiatry 9: , 561. |
[29] | MacVicar BA , Newman EA ((2015) ) Astrocyte regulation of blood flow in the brain. Cold Spring Harb Perspect Biol 7: , a020388. |
[30] | Kim Y , Park J , Choi YK ((2019) ) The role of astrocytes in the central nervous system focused on BK channel and heme oxygenase metabolites: A review. Antioxidants 8: , 121. |
[31] | Mahmoud S , Gharagozloo M , Simard C , Gris D ((2019) ) Astrocytes maintain glutamate homeostasis in the CNS by controlling the balance between glutamate uptake and release. Cells 8: , 184. |
[32] | Ishibashi M , Egawa K , Fukuda A ((2019) ) Diverse actions of astrocytes in GABAergic signaling. Int J Mol Sci 20: , 2964. |
[33] | Theparambil SM , Hosford PS , Ruminot I , Kopach O , Reynolds JR , Sandoval PY , Rusakov DA Barros LF , Gourine AV ((2020) ) Astrocytes regulate brain extracellular pH via a neuronal activity-dependent bicarbonate shuttle. Nat Commun 11: , 5073. |
[34] | Phatnani H , Maniatis T ((2015) ) Astrocytes in neurodegenerative disease. Cold Spring Harb Perspect Biol 7: , a020628. |
[35] | Assefa BT , Gebre AK , Altaye BM ((2018) ) Reactive astrocytes as drug target in Alzheimer’s disease. Biomed Res Int 2018: , 4160247. |
[36] | Satarker S , Bojja SL , Gurram PC , Mudgal J , Arora D , Nampoothiri M ((2022) ) Astrocytic glutamatergic transmission and its implications in neurodegenerative disorders. Cells 11: , 1139. |
[37] | Scimemi A , Meabon JS , Woltjer RL , Sullivan JM , Diamond JS , Cook DG ((2013) ) Amyloid-β 1–42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. J Neurosci 33: , 5312–5318. |
[38] | Simon M , Wang MX , Ismail O , Braun M , Schindler AG , Reemmer J , Jesica Reemmer J , Wang Z , Haveliwala MA , O’Boyle RP , Han WY , Roese N , Grafe M , Woltjer R , Boison D , Jeffrey J . Iliff JJ ((2022) ) Loss of perivascular aquaporin-4 localization impairs glymphatic exchange and promotes amyloid β plaque formation in mice. Alzheimers Res Ther 14: , 59. |
[39] | Iliff JJ , Wang M , Liao Y , Plogg BA , Peng W , Gundersen GA , Benveniste H , Vates GE , Deane R , Goldman SA , Nagelhus EA , Nedergaard M ((2012) ) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med 4: , 147ra111. |
[40] | Xu Z , Xiao N , Chen Y , Huang H , Marshall C , Gao J , Cai Z , Wu T , Hu G , Xiao M ((2015) ) Deletion of aquaporin-4 in APP/PS1 mice exacerbates brain Aβ accumulation and memory deficits. Mol Neurodegener 10: , 58. |
[41] | Burfeind KG , Murchison CF , Westaway SK , Simon MJ , Erten-Lyons D , Kaye JA , Quinn JF , Iliff JJ ((2017) ) The effects of noncoding aquaporin-4 single-nucleotide polymorphisms on cognition and functional progression of Alzheimer’s disease. Alzheimers Dement 3: , 348–359. |
[42] | Li YK , Wang F , Wang W , Luo Y , Wu PF , Xiao JL , Hu ZL , Jin Y , Hu G , Chen JG ((2012) ) Aquaporin-4 deficiency impairs synaptic plasticity and associative fear memory in the lateral amygdala: Involvement of downregulation of glutamate transporter-1 expression. Neuropsychopharmacology 37: , 1867–1878. |
[43] | Yang J , Li MX , Luo Y , Chen T , Liu J , Fang P , Jiang B , Hu ZL , Jin Y , Chen JG , Wang F ((2013) ) Chronic ceftriaxone treatment rescues hippocampal memory deficit in AQP4 knockout mice via activation of GLT-1. Neuropharmacology 75: , 213–222. |
[44] | Cai Y , Liu J , Wang B , Sun M , Yang H ((2022) ) Microglia in the neuroinflammatory pathogenesis of Alzheimer’s disease and related therapeutic targets. Front Immunol 13: , 856376. |
[45] | Willard SS , Koochekpour S ((2013) ) Glutamate, glutamate receptors, and downstream signaling pathways. Int J Biol Sci 9: , 948–959. |
[46] | George PM , Steinberg GK ((2015) ) Novel stroke therapeutics: Unraveling stroke pathophysiology and its impact on clinical treatments. Neuron 87: , 297–309. |
[47] | Weber JT ((2012) ) Altered calcium signaling following traumatic brain injury. Front Pharmacol 3: , 60. |
[48] | Xu J , Kurup P , Zhang Y , Goebel-Goody SM , Wu PH , Hawasli AH , Baum ML , Bibb JA , Lombroso PJ ((2009) ) Extrasynaptic NMDA receptors couple preferentially to excitotoxicity via calpain-mediated cleavage of STEP. J Neurosci 29: , 9330–9343. |
[49] | Dirnagl U , Iadecola C , Moskowitz MA ((1999) ) Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci 22: , 391–397. |
[50] | Neves JD , Vizuete AF , Nicola F , da Ré C , Rodrigues AF , Schmitz F , Mestriner RG , Aristimunha D , Wyse ATS , Netto CA ((2018) ) Glial glutamate transporters expression, glutamate uptake, and oxidative stress in an experimental rat model of intracerebral hemorrhage. Neurochem Int 116: , 13–21. |
[51] | Rimmele TS , Li S , Andersen JV , Westi EW , Rotenberg A , Wang J , Aldana BI , Selkoe DJ , Aoki CJ , Dulla CG , Rosenberg PA ((2021) ) Neuronal loss of the glutamate transporter GLT-1 promotes excitotoxic injury in the hippocampus. Front Cell Neurosci 15: , 788262. |
[52] | Yang J , Vitery M del C , Chen J , Osei-Owusu J , Chu J , Qiu Z ((2019) ) Glutamate-releasing SWELL1 channel in astrocytes modulates synaptic transmission and promotes brain damage in stroke. Neuron 102: , 813–827. |
[53] | Yi JH , Herrero R , Chen G , Hazell AS ((2007) ) Glutamate transporter EAAT4 is increased in hippocampal astrocytes following lateral fluid-percussion injury in the rat. Brain Res 1154: , 200–205. |
[54] | Uria-Avellanal C , Robertson NJ ((2014) ) Na+/H+ exchangers and intracellular pH in perinatal brain injury. Transl Stroke Res 5: , 79–98. |
[55] | Sun L , Zhang Y , Liu E , Ma Q , Anatol M , Han H , Yan J ((2019) ) The roles of astrocyte in the brain pathologies following ischemic stroke. Brain Inj 33: , 712–716. |
[56] | Clément T , Rodriguez-Grande B , Badaut J ((2020) ) Aquaporins in brain edema. J Neurosci Res 98: , 9–18. |
[57] | Manley GT , Fujimura M , Ma T , Noshita N , Filiz F , Bollen AW , Chan P , Verkman AS ((2000) ) Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med 6: , 159–163. |
[58] | Qin C , Zhou LQ , Ma XT , Hu ZW , Yang S , Chen M , Dale B Bosco DB , Wu LJ , Tian DS ((2019) ) Dual functions of microglia in ischemic stroke. Neurosci Bull 35: , 921–933. |
[59] | Liu LR , Liu JC , Bao JS , Bai QQ , Wang GQ ((2020) ) Interaction of microglia and astrocytes in the neurovascular unit. Front Immunol 11: , 1024. |
[60] | Barakat R , Redzic Z ((2016) ) The role of activated microglia and resident macrophages in the neurovascular unit during cerebral ischemia: Is the jury still out? Med Princ Pract 25: (Suppl 1), 3–14. |
[61] | Calle RF , Konings SC , Rubio JF , Revilla JG , Ferrer LC , Svensson M , Martinson I , Serrano B , Venero JL , Nielsen HM , Gouras GK , Deierborg T ((2022) ) APOE in the bullseye of neurodegenerative diseases: Impact of the APOE genotype in Alzheimer’s disease pathology and brain diseases. Mol Neurodegener 17: , 62. |
[62] | Liu CC , Kanekiyo T , Xu H , Bu G ((2013) ) Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat Rev Neurol 9: , 106–118. |
[63] | Husain MA , Laurent B , Plourde M ((2021) ) APOE and Alzheimer’s disease: From lipid transport to physiopathology and therapeutics. Front Neurosci 15: , 630502. |
[64] | Shi Y , Yamada K , Liddelow SA , Smith ST , Zhao L , Luo W , Tsai RM , Spina S , Lea T Grinberg LT , Rojas JC , Gallardo G , Wang K , Roh J , Robinson G , Finn MB , Jiang H , Sullivan PM , Baufeld C , Wood MW , Sutphen C , McCue L , Xiong C , Aguila JLD , Morris JC , Cruchaga C , Fagan AM , Miller BL , Boxer AL , Seeley WW , Butovsky O , Barres BA , Paul SM , Holtzman DM ((2017) ) ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature 549: , 523–527. |
[65] | Montagne A , Nation DA , Zlokovic BV ((2020) ) APOE4 accelerates development of dementia after stroke. Stroke 51: , 699–700. |
[66] | Johnson LA , Torres ER , Weber Boutros S , Patel E , Akinyeke T , Alkayed NJ , Raber J ((2019) ) Apolipoprotein E4 mediates insulin resistance-associated cerebrovascular dysfunction and the post-prandial response. J Cereb Blood Flow Metab 39: , 770–781. |
[67] | Ganaie H , Biswas A , Bhattacharya A , Pal S , Ray J , Das S ((2020) ) Association of APOE gene polymorphism with stroke patients from rural Eastern India. Ann Indian Acad Neurol 23: , 504–509. |
[68] | Gan C , Zhang Y , Liang F , Guo X , Zhong Z ((2022) ) Effects of APOE gene ɛ4 allele on serum lipid profiles and risk of cardiovascular disease and tumorigenesis in southern Chinese population. World J Surg Oncol 20: , 280. |
[69] | Lee JS , Ko KH , Oh JH , Kim JG , Kang CH , Song SK , Kang SY , Kang jH , Park JH , Koh MJ , Lee HK , Choi JC ((2020) ) Apolipoprotein E ɛ4 is associated with the development of incident dementia in cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy patients with p.Arg544Cys mutation. Front Aging Neurosci 12: , 591879. |
[70] | Rajan KB , Aggarwal NT , Schneider JA , Wilson RS , Everson-Rose SA , Evans DA ((2016) ) Role of APOE ɛ4 allele and incident stroke on cognitive decline and mortality. Alzheimer Dis Assoc Disord 30: , 318–323. |
[71] | Das TK , Chakrabarti SK , Zulkipli IN , Abdul Hamid MRW ((2019) ) Curcumin ameliorates the impaired insulin signaling involved in the pathogenesis of Alzheimer’s disease in rats. J Alzheimers Dis Rep 3: , 59–70. |
[72] | Chen Y , Li Y , Hsieh T , Wang C , Cheng K , Wang L , Lin TY , Cheung CHA , Wu CL , Chiang HC ((2019) ) Aging-induced Akt activation involves in aging-related pathologies and Aβ-induced toxicity. Aging Cell 18: , e12989. |
[73] | Deng Y , Li B , Liu Y , Iqbal K , Grundke-Iqbal I , Gong CX ((2009) ) Dysregulation of insulin signaling, glucose transporters, O-GlcNAcylation, and phosphorylation of tau and neurofilaments in the brain. Am J Pathol 175: , 2089–2098. |
[74] | Das TK , Jana P , Chakrabarti SK , Abdul Hamid MRW ((2019) ) Curcumin downregulates GSK3 and Cdk5 in scopolamine-induced Alzheimer’s disease rats abrogating Aβ40/42 and tau hyperphosphorylation. J Alzheimers Dis Rep 3: , 257–267. |
[75] | Velazquez R , Tran A , Ishimwe E , Denner L , Dave N , Oddo S , Dineley KT ((2017) ) Central insulin dysregulation and energy dyshomeostasis in two mouse models of Alzheimer’s disease. Neurobiol Aging 58: , 1–13. |
[76] | Talbot K , Wang HY , Kazi H , Han LY , Bakshi KP , Stucky A , Fuino RL , Kawaguchi KR , Samoyedny AJ , Wilson RS , Arvanitakis Z , Schneider JA , Wolf BA , Bennett DA , Trojanowski JQ , Arnold SE ((2012) ) Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest 122: , 1316–1338. |
[77] | de La Monte SM ((2009) ) Insulin resistance and Alzheimer’s disease. BMB Rep 42: , 475–481. |
[78] | Lan R , Xiang J , Zhang Y , Wang GH , Bao J , Li WW , Zhang W , Xu LL ((2013) ) PI3K/Akt pathway contributes to neurovascular unit protection of Xiao-Xu-Ming decoction against focal cerebral ischemia and reperfusion injury in rats. Evid Based Complement Alternat Med 2013: , 459467. |
[79] | Wang Z , Han Y , Tian S , Bao J , Wang Y , Jiao J ((2020) ) Lupeol alleviates cerebral ischemia–reperfusion injury in correlation with modulation of PI3K/Akt pathway. Neuropsychiatr Dis Treat 16: , 1381–1390. |
[80] | Gao X , Zhang H , Steinberg G , Zhao H ((2010) ) The Akt pathway is involved in rapid ischemic tolerance in focal ischemia in rats. Transl Stroke Res 1: , 202–209. |
[81] | Martín A , Rojas S , Chamorro Á , Falcón C , Bargalló N , Planas AM ((2006) ) Why does acute hyperglycemia worsen the outcome of transient focal cerebral ischemia? Role of corticosteroids, inflammation, and protein O-glycosylation. Stroke 37: , 1288–1295. |
[82] | Robbins NM , Swanson RA ((2014) ) Opposing effects of glucose on stroke and reperfusion injury. Stroke 45: , 1881–1886. |
[83] | Koepsell H ((2020) ) Glucose transporters in brain in health and disease. Pflugers Arch 472: , 1299–1343. |
[84] | McCall AL , van Bueren AM , Nipper V , Moholt-Siebert M , Downes H , Lessov N ((1996) ) Forebrain ischemia increases Glut1 protein in brain microvessels and parenchyma. J Cereb Blood Flow Metab 16: , 69–76. |
[85] | Alquisiras-Burgos I , Aguilera P ((2022) ) Involvement of glucose transporter overexpression in the protection or damage after ischemic stroke. Neural Regen Res 17: , 783–784. |
[86] | Zhao SC , Ma LS , Chu ZH , Xu H , Wu WQ , Liu F ((2017) ) Regulation of microglial activation in stroke. Acta Pharmacol Sin 38: , 445–458. |
[87] | Hansen DV , Hanson JE , Sheng M ((2018) ) Microglia in Alzheimer’s disease. J Cell Biol 217: , 459–472. |
[88] | Roth AD , Ramírez G , Alarcón R , Bernhardi RV ((2005) ) Oligodendrocytes damage in Alzheimer’s disease: Beta amyloid toxicity and inflammation. Biol Res 38: , 381–387. |
[89] | Sozmen EG , Rosenzweig S , Llorente IL , DiTullio DJ , Machnicki M , Vinters HV , Havton LA , Giger RJ , Hinman JD , Carmichael ST ((2016) ) Nogo receptor blockade overcomes remyelination failure after white matter stroke and stimulates functional recovery in aged mice, Proc Natl Acad Sci U S A 113: , E8453–E8462. |
[90] | Roy-O’Reilly M , McCullough LD ((2017) ) Astrocytes fuel the fire of lymphocyte toxicity after stroke. Proc Natl Acad Sci U S A 114: , 425–427. |
[91] | Phillips EC , Croft CL , Kurbatskaya K , O’Neill MJ , Hutton ML , Hanger DP , Garwood CJ , Nobel W ((2014) ) Astrocytes and neuroinflammation in Alzheimer’s disease. Biochem Soc Trans 42: , 1321–1325. |
[92] | Wang WY , Tan MS , Yu JT , Tan L ((2015) ) Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med 3: , 136. |
[93] | Ries M , Sastre M ((2016) ) Mechanisms of Aβ clearance and degradation by glial cells. Front Aging Neurosci 8: , 160. |
[94] | Honarpisheh P , Reynolds CR , Blasco Conesa MP , Moruno Manchon JF , Putluri N , Bhattacharjee MB , Urayama A , McCullough LD , Ganesh BP ((2020) ) Dysregulated gut homeostasis observed prior to the accumulation of the brain amyloid-β in Tg2576 Mice. Int J Mol Sci 21: , 1711. |
[95] | Das TK , Blasco-Conesa MP , Korf J , Honarpisheh P , Chapman MR , Ganesh BP ((2022) ) Bacterial amyloid Curli associated gut epithelial neuroendocrine activation predominantly observed in Alzheimer’s disease mice with central amyloid-β pathology. J Alzheimers Dis 88: , 191–205. |
[96] | Calvo-Rodriguez M , García-Rodríguez C , Villalobos C , Núñez L ((2020) ) Role of toll like receptor 4 in Alzheimer’s disease. Front Immunol 11: , 1588. |
[97] | Scholtzova H , Do E , Dhakal S , Sun Y , Liu S , Mehta PD , Wisniewski T ((2017) ) Innate immunity stimulation via toll-like receptor 9 ameliorates vascular amyloid pathology in Tg-SwDI mice with associated cognitive benefits. J Neurosci 37: , 936–959. |
[98] | Bai H , Zhang Q ((2021) ) Activation of NLRP3 inflammasome and onset of Alzheimer’s disease. Front Immunol 12: , 701282. |
[99] | Wu L , Xiong X , Wu X , Ye Y , Jian Z , Zhi Z , Gu L ((2020) ) Targeting oxidative stress and inflammation to prevent ischemia-reperfusion injury. Front Mol Neurosci 13: , 28. |
[100] | Sun BZ , Chen L , Wu Q , Wang HL , Wei XB , Xiang YX , Zhang XM ((2014) ) Suppression of inflammatory response by flurbiprofen following focal cerebral ischemia involves the NF-κB signaling pathway. Int J Clin Exp Med 7: , 3087–3095. |
[101] | Shen F , Jiang L , Han F , Degos V , Chen S , Su H ((2019) ) Increased inflammatory response in old mice is associated with more severe neuronal injury at the acute stage of ischemic stroke. Aging Dis 10: , 12–22. |
[102] | Wang YC , Lin S , Yang QW ((2011) ) Toll-like receptors in cerebral ischemic inflammatory injury. J Neuroinflammation 8: , 134. |
[103] | Fard SG , Shoorei H , Poornajaf Y , Hussen BM , Hajiesmaeili Y , Abak A , Taheri M , Eghbali A ((2022) ) NLRP3: Role in ischemia/reperfusion injuries. Front Immunol 13: , 926895. |
[104] | Saresella M , la Rosa F , Piancone F , Zoppis M , Marventano I , Calabrese E , Rainone V , Nemni R , Mancuso R , Clerici M ((2016) ) The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol Neurodegener 11: , 23. |
[105] | Parzych KR , Klionsky DJ ((2014) ) An overview of autophagy: Morphology, mechanism, and regulation. Antioxid Redox Signal 20: , 460–473. |
[106] | Bar-Yosef T , Damri O , Agam G ((2019) ) Dual role of autophagy in diseases of the central nervous system. Front Cell Neurosci 13: , 196. |
[107] | Mo Y , Sun YY , Liu KY ((2020) ) Autophagy and inflammation in ischemic stroke. Neural Regen Res 15: , 1388–1396. |
[108] | Orr ME , Oddo S ((2013) ) Autophagic/lysosomal dysfunction in Alzheimer’s disease. Alzheimers Res Ther 5: , 53. |
[109] | Varo RS , Estrada LT , Mejias ES , Torres M , Vargas DB , Gonzalez IM , Castro VD , Jimenez S , Ruano D , Vizuete M , Davila JC , Verdugo JMG , Jimenez AJ , Vitorica J , Gutierrez A ((2012) ) Abnormal accumulation of autophagic vesicles correlates with axonal and synaptic pathology in young Alzheimer’s mice hippocampus. Acta Neuropathol 123: , 53–70. |
[110] | Zhang W , Xu C , Sun J , Shen HM , Wang J , Yang C ((2022) ) Impairment of the autophagy– lysosomal pathway in Alzheimer’s diseases: Pathogenic mechanisms and therapeutic potential. Acta Pharm Sin B 12: , 1019–1040. |
[111] | Zhang Z , Yang X , Song YQ , Tu J ((2021) ) Autophagy in Alzheimer’s disease pathogenesis: Therapeutic potential and future perspectives. Ageing Res Rev 72: , 101464. |
[112] | Mueed Z , Tandon P , Maurya SK , Deval R , Kamal MA , Poddar NK ((2019) ) Tau and mTOR: The hotspots for multifarious diseases in Alzheimer’s development. Front Neurosci 12: , 1017. |
[113] | Zuroff L , Daley D , Black KL , Hamaoui MK ((2017) ) Clearance of cerebral Aβ in Alzheimer’s disease: Reassessing the role of microglia and monocytes. Cell Mol Life Sci 74: , 2167–2201. |
[114] | Zhao Y , Zhang Y , Zhang J , Zhang X , Yang G ((2020) ) Molecular mechanism of autophagy: Its role in the therapy of Alzheimer’s disease. Curr Neuropharmacol 18: , 720–739. |
[115] | Oddo S ((2012) ) The role of mTOR signaling in Alzheimer disease. Front Biosci 4: , 941–952. |
[116] | Wang P , Shao BZ , Deng Z , Chen S , Yue Z , Miao CY ((2018) ) Autophagy in ischemic stroke. Prog Neurobiol 163: , 98–117. |
[117] | Shi R , Weng J , Zhao L , Li X , Gao T , Kong J ((2012) ) Excessive autophagy contributes to neuron death in cerebral ischemia. CNS Neurosci Ther 18: , 250–260. |
[118] | Chen W , Sun Y , Liu K , Sun X ((2014) ) Autophagy: A double-edged sword for neuronal survival after cerebral ischemia. Neural Regen Res 9: , 1210–1216. |
[119] | Woo HN , Park JS , Gwon AR , Arumugam TV , Jo DG ((2009) ) Alzheimer’s disease and Notch signaling. Biochem Biophys Res Commun 390: , 1093–1097. |
[120] | Arumugam TV , Baik SH , Balaganapathy P , Sobey CG , Mattson MP , Jo DG ((2018) ) Notch signaling and neuronal death in stroke. Prog Neurobiol 165: , 103–116. |
[121] | Strooper BD , Iwatsubo T , Wolfe MS ((2012) ) Presenilins and γ-secretase: Structure, function, and role in Alzheimer disease, Cold Spring Harb Perspect Med 2: , a006304. |
[122] | Cho SJ , Yun SM , Jo C , Jeong J , Park MH , Han C , Koh YH ((2019) ) Altered expression of Notch1 in Alzheimer’s disease, PLoS One 14: , e0224941. |
[123] | Berezovska O , Xia MQ , Hyman BT ((1998) ) Notch is expressed in adult brain, is coexpressed with presenilin-1, and is altered in Alzheimer disease. J Neuropathol Exp Neurol 57: , 738–745. |
[124] | Gridley T , Groves AK ((2014) ) Overview of genetic tools and techniques to study notch signaling in mice. Methods Mol Biol 1187: , 47–61. |
[125] | Liu J , Dong F , Jeong J , Masuda T , Lobe CG ((2014) ) Constitutively active Notch1 signaling promotes endothelial-mesenchymal transition in a conditional transgenic mouse model. Int J Mol Med 34: , 669–676. |
[126] | doifode T , Giridharan VV , Generoso JS , Bhatti G , Collodel A , Schulz PE , Orestes V , Forlenza OV , Barichello T ((2021) ) The impact of the microbiota-gut-brain axis on Alzheimer’s disease pathophysiology. Pharmacol Res 164: , 105314. |
[127] | Durgan DJ , Lee J , McCullough LD , Bryan RM ((2019) ) Examining the role of the microbiota-gut-brain axis in stroke. Stroke 50: , 2270–2277. |
[128] | Megur A , Baltriukienė D , Bukelskienė V , Burokas A ((2020) ) The microbiota– gut– brain axis and Alzheimer’s disease: Neuroinflammation is to blame? Nutrients 13: , 37. |
[129] | Tang W , Zhu H , Feng Y , Guo R , Wan D ((2020) ) The impact of gut microbiota disorders on the blood– brain barrier. Infect Drug Resist 13: , 3351–3363. |
[130] | Colombo AV , Sadler RK , Llovera G , Singh V , Roth S , Heindl S , Monasor LS , Verhoeven A , Peters F , Parhizkar S , Kamp F , de Aguero MG , MacPherson AJ , Winkler E , Herms J , Benakis C , Dichgans M , Steiner H , Giera M , Haass C , Tahirovic S , Liesz A ((2021) ) Microbiota-derived short chain fatty acids modulate microglia and promote Aβ plaque deposition. Elife 10: , e59826. |
[131] | Qian XH , Xie RY , Liu XL , Chen SD , Tang HD ((2022) ) Mechanisms of short-chain fatty acids derived from gut microbiota in Alzheimer’s disease. Aging Dis 13: , 1252–1266. |
[132] | Shen L , Liu L , Ji HF ((2019) ) Alzheimer’s disease histological and behavioral manifestations in transgenic mice correlate with specific gut microbiome state. J Alzheimers Dis 56: , 385–390. |
[133] | Kong Y , Jiang B , Luo X ((2018) ) Gut microbiota influences Alzheimer’s disease pathogenesis by regulating acetate in Drosophila model. Future Microbiol 13: , 1117–1128. |
[134] | Zhuang ZQ , Shen LL , Li WW , Fu X , Zeng F , Gui L , Lü Y , Cai M , Zhu C , Tan YL , Zheng P , Li HY , Zhu J , Zhou HD , Bu XL , Wang YJ ((2018) ) Gut microbiota is altered in patients with Alzheimer’s disease. J Alzheimers Dis 63: , 1337–1346. |
[135] | Vogt NM , Kerby RL , Dill-McFarland KA , Harding SJ , Merluzzi AP , Johnson SC , Carlsson CM , Asthana S , Zetterberg H , Blennow K , Bendlin BB , Federico E Rey FE ((2017) ) Gut microbiome alterations in Alzheimer’s disease. Sci Rep 7: , 13537. |
[136] | Yamashiro K , Kurita N , Urabe T , Hattori N ((2021) ) Role of the gut microbiota in stroke pathogenesis and potential therapeutic implications. Ann Nutr Metab 77: , 36–44. |
[137] | Chang Y , Woo HG , Jeong JH , Kim GH , Park KD , Song TJ ((2021) ) Microbiota dysbiosis and functional outcome in acute ischemic stroke patients. Sci Rep 11: , 10977. |
[138] | Li N , Wang X , Sun C , Wu X , Lu M , Si Y , Ye X , Wang T , Yu X , Zhao X , Wei N , Wang X ((2019) ) Change of intestinal microbiota in cerebral ischemic stroke patients. BMC Microbiol 19: , 191. |
[139] | Tan C , Wu Q , Wang H , Gao X , Xu R , Cui Z , Zhu j , Zeng X , Zhou H , He Y , Yin J ((2021) ) Dysbiosis of gut microbiota and short-chain fatty acids in acute ischemic stroke and the subsequent risk for poor functional outcomes. JPEN J Parenter Enteral Nutr 45: , 518–529. |
[140] | Yin J , Liao SX , He Y , Wang S , Xia GH , Liu FT , Zhu JJ , You C , Chen Q , Zhou L , Pan SY , Zhou HW ((2015) ) Dysbiosis of gut microbiota with reduced trimethylamine-N-oxide level in patients with large-artery atherosclerotic stroke or transient ischemic attack. J Am Heart Assoc 4: , e002699. |
[141] | Xia GH , You C , Gao XX , Zeng XL , Zhu JJ , Xu KY , Tan CH , Xu RT , Wu QH , Zhou HW , He Y , Yin J ((2019) ) Stroke Dysbiosis Index (SDI) in gut microbiome are associated with brain injury and prognosis of stroke. Front Neurol 10: , 397. |
[142] | Yamashiro K , Tanaka R , Urabe T , Ueno Y , Yamashiro Y , Nomoto K , Takahashi T , Tsuji H , Asahara T , Hattori N ((2017) ) Gut dysbiosis is associated with metabolism and systemic inflammation in patients with ischemic stroke. PLoS One 12: , e0171521. |
[143] | Chen YL , Bai L , Dilimulati D , Shao S , Qiu C , Liu T , Xu S , Bai XB , Du LJ , Lu-Jun Zhou LJ , Lin WZ , Meng XQ , Jin YC , Liu Y , Zhang XH , Duan SZ , Jia F ((2022) ) Periodontitis salivary microbiota aggravates ischemic stroke through IL-17A. Front Neurosci 16: , 876582. |
[144] | Singh V , Roth S , Llovera G , Sadler R , Garzetti D , Stecher B , Garzetti D , Stecher B , Dichgans M , Liesz A ((2016) ) Microbiota dysbiosis controls the neuroinflammatory response after stroke. J Neurosci 36: , 7428–7440. |
[145] | Houlden A , Goldrick M , Brough D , Vizi ES , Lénárt N , Martinecz B , Roberts IS , Denes A ((2016) ) Brain injury induces specific changes in the caecal microbiota of mice via altered autonomic activity and mucoprotein production. Brain Behav Immun 57: , 10–20. |


