
Creutzfeldt-Jakob Disease in a Patient with Previous COVID-19 Infection: “The Virus Caused the Derangement in My Brain”
Abstract
Recent studies have speculated a link between Creutzfeldt-Jakob disease (CJD) and COVID-19, following the description of CJD cases after COVID-19 infection. We report the case of a 71-year-old female patient who developed neuropsychiatric and neurological symptoms after COVID-19 infection and was later diagnosed with CJD. Cerebrospinal fluid (CSF) total tau levels were slightly increased. She resulted prion protein gene (PRNP) M129V heterozygous. We aim to emphasize the role of the polymorphism at codon 129 of PRNP gene on the clinical phenotype and duration of CJD, and the CSF total tau levels that likely correlate with the rate of disease progression.
INTRODUCTION
Creutzfeldt-Jakob disease (CJD) is a rare neurodegenerative disorder caused by the accumulation of abnormally shaped prion protein Scrapie (PrPSc) in neurons of the central nervous system. It is characterized by different clinical phenotypes and different ages of onset and disease duration, determinedin part by prion protein gene (PRNP) codon 129 polymorphism and prion conformation. It typically presents as a rapidly progressive dementia (RPD) with myoclonus, but prominent visual signs, dyskinetic, thalamic, and cerebellar symptoms may be present in the different clinical forms of the pathology [1]. The clinical manifestations, the presence of a typical electroencephalogram (EEG) pattern, the occurrence of distinctive brain magnetic resonance imaging (MRI) changes and a panel of cerebrospinal fluid (CSF) biomarkers including real-time quaking-induced conversion (RT-QuIC) assay are valuable for CJD possible or probable diagnosis in vivo, especially in cases with clinically limited sporadic CJD (sCJD) and prolonged survival [2].
Recent reports have proposed a putative link between CJD and SARS-CoV-2, the causative agent of coronavirus disease 2019 (COVID-19), following the description of CJD cases after COVID-19 infection. The hypothesis is that COVID-19 may accelerate the pathogenesis of CJD [3–8]. Furthermore, COVID-19 is responsible for other neurological manifestations like anosmia, headache, dizziness, encephalitis, stroke, and seizures. The underlying pathophysiological mechanism of these neurological conditions has been attributed to viral neurotropism and/or “cytokine storm” [9].
In this context, this report presents the case of a 71-year-old woman who developed neuropsychiatric and neurological symptoms subsequently to SARS-CoV-2 infection and was diagnosed with sCJD after a long period from the infection.
CASE PRESENTATION
A 71-year-old woman with a medical history of hemicrania without aura had SARS-CoV-2 nasopharyngeal swab PCR positivity in November 2020 that resulted in mild, uncomplicated flu-like symptoms. Since then she referred feeling different with a slight depression, social withdrawal, reduced appetite, and sleep disturbances. During the following months, family members reported episodes of spatial disorientation, episodic memory impairment, and a single episode of uncontrolled buying. More than a year after the COVID-19 infection and the persistence of this symptomatology, she underwent a psychiatrist consult who diagnosed her depressive and hypochondriacal disorders, and a brain MRI that showed a symmetrical frontal, fronto-parietal, temporal and insular cortical signal enhancement in diffusion-weighted imaging (DWI) scans. Thus, she was visited by a neurology specialist who suggested hospitalization to study the case.
At the admission to the hospital, 15 months after SARS-CoV-2 infection, the patient appeared disoriented in time, anxious with perseverant references to her previous infection with COVID-19 which she considered responsible for her symptomatology (“The virus caused the derangement in my brain”). A mild clinical picture of attention deficit, ideo-motor apraxia, lateral left pulsion during walking, and neglect syndrome was detected. EEG showed diffuse slowing anomalies with sharp waves with some triphasic complex. Blood biochemistry tests, serum electrolytes, and hepatic, renal and thyroid function were normal. CFS analysis was normal and, waiting for the other results, empiric therapy for infectious and autoimmune encephalitis was administrated with intravenous acyclovir, ceftriaxone, and methylprednisolone.
Afterward, the patient manifested a major episode of psychomotor agitation, which required olanzapine therapy, language disturbances, more prominent ideo-motor apraxia and developed a clear left neglect syndrome. Neuropsychological tests revealed severe cognitive deficits featured by marked language impairment, and attention and executive deficits with motor, ideo-motor, and constructive apraxia (Table 1). A full-body CT scan was performed in the hypothesis of a paraneoplastic disease, and onconeural and neuronal surface antibodies in serum and CFS were negative, using commercial kits (Euroimmun) (Table 2). Microbiological analysis and PCR for common viruses, including SARS-CoV-2 RNA, on CFS resulted in negative so the empiric therapy was discontinued. CSF amyloid-β peptide 1–42 and 1–42/1–40, phosphorylated-tau (p-tau) were normal, while total tau (t-tau) level was mildly increased with a value of 782 pg/mL (normal value < 400 pg/mL) (Table 3). The brain MRI was repeated and showed frontal, parietal, temporal, and insular cortical ribboning in DWI and corresponding hyperintensity in fluid-attenuated inversion recovery (FLAIR) sequences (Fig. 1). Brain 18F-fluorodeoxyglucose (FDG) positron emission tomography (PET) showed severe cortical hypometabolism involving right frontal lobe, temporo-parietal lobe bilaterally, precuneus, and posterior cingulate cortex (Fig. 2). EEG was performed serially until rare quasi-periodic right frontotemporal sharp wave complexes were detected (Fig. 3). CSF RT-QuIC assay was positive for pathologic prion protein. Analysis of the PRNP gene did not detect pathogenic mutations and showed the Methionine/Valine (M/V) polymorphism at codon 129. According to the Revised International CJD Surveillance Network diagnostic criteria for sCJD [2], the diagnosis of probable sCJD was made. Currently, the patient is getting worse due to generalized epilepsy onset.
Table 1
Neuropsychological battery
| Test | Score | Cut Off | E.S. | Pathological (*) | |
| Normal (+) | |||||
| Raw | Adjusted | ||||
| Mini-Mental State Examination | 3 | 2.3 | < 23.8 | * | |
| Visuo-Spatial Analysis and Attention | |||||
| Bells test: total omissions | 29 | ≥5 | * | ||
| Bells test: right/left difference | 5 | ≥5 | * | ||
| Total false positives | 18 | > 0 | * | ||
| Bells test: execution time | 300 | 216,49 | * | ||
| Apraxia | |||||
| Gesture imitation test (right) | 9 | < 53 | * | ||
| Gesture imitation test (left) | 20 | < 53 | * | ||
| Object’s pantomime of use | 6 | ≤18 | * | ||
| Copy of drawings | 4 | 4.60 | ≤7,18 | 0 | * |
| Clock drawing test | 4 | > 2 | * | ||
| Language | |||||
| S.A.N.D. – Screening for Aphasia in NeuroDegeneration | |||||
| Picture naming | 9 | 9.273 | ≤9,69 | * | |
| Picture naming – Living | 3 | 3.242 | ≤3,839 | * | |
| Picture naming – Non living | 6 | ≤5 | + | ||
| Auditory sentence comprehension | 0 | – | ≤6,157 | * | |
Score is adjusted for age and education. E.S., equivalent score.
Table 2
Blood and CSF antibodies
| Normal values | Blood | CSF | |
| Anti-onconeural | Negative | Negative | Negative |
| Anti-NMDA-R IgG | Negative | Negative | Negative |
| Anti-LGI1 IgG | Negative | Negative | Negative |
| Anti-CASPR2 IgG | Negative | Negative | Negative |
| Anti-GABA-B1/2-R IgG | Negative | Negative | Negative |
| Anti-GluR1 IgG | Negative | Negative | Negative |
| Anti-GluR2 IgG | Negative | Negative | Negative |
NMDA-R, N-methyl-D-aspartate receptor; LGI1, leucine rich glioma inactivated 1; CASPR2, Contactin-associated protein-like 2; GABA-B1/2, Gamma-aminobutyric acid B receptor, 1/2; GluR1,2, subunit glutamate receptor 1,2.
Table 3
CSF analysis
| Parameter | Reference range | Value |
| Aspect | – | Clear |
| Color | – | Colorless |
| Proteins (g/L) | < 0,45 | 0,25 |
| Chlorides (mEq/L) | 118–132 | 126 |
| LDH (U/L) | – | 25 |
| Lactate (mmol/L) | 0,2–2,2 | 2,0 |
| White blood cells (/microL) | 0,00–5,00 | 3,00 |
| Erythrocytes (/microL) | < 50 | < 50 |
| Glucose (mg/dL) | – | 81 |
| GluCSF/GluBlood (%) | > 45% | 75 |
| Albumin (g/L) | – | 0,138 |
| IgG (g/L) | – | 0.016 |
| Link index | < 0,7 | 0,054 |
| Albumin quotient | < 7,0 | 4,1 |
| Oligoclonal bands | Absents | Absents |
| Amyloid-β 1–42 (pg/mL) | > 670 | 907 |
| Amyloid- β 1–42/1–40 | > 0,062 | 0,097 |
| p-tau (pg/mL) | < 60,0 | 49,4 |
| t-tau (pg/mL) | < 400 | 782* |
| SARS-CoV-2 RNA | – | Not detected |
Fig.1
Brain MRI DWI showed cortical ribboning of the frontal, parietal, temporal, and insular regions (A-D) that corresponded to hyperintensity in FLAIR sequences (E-H).
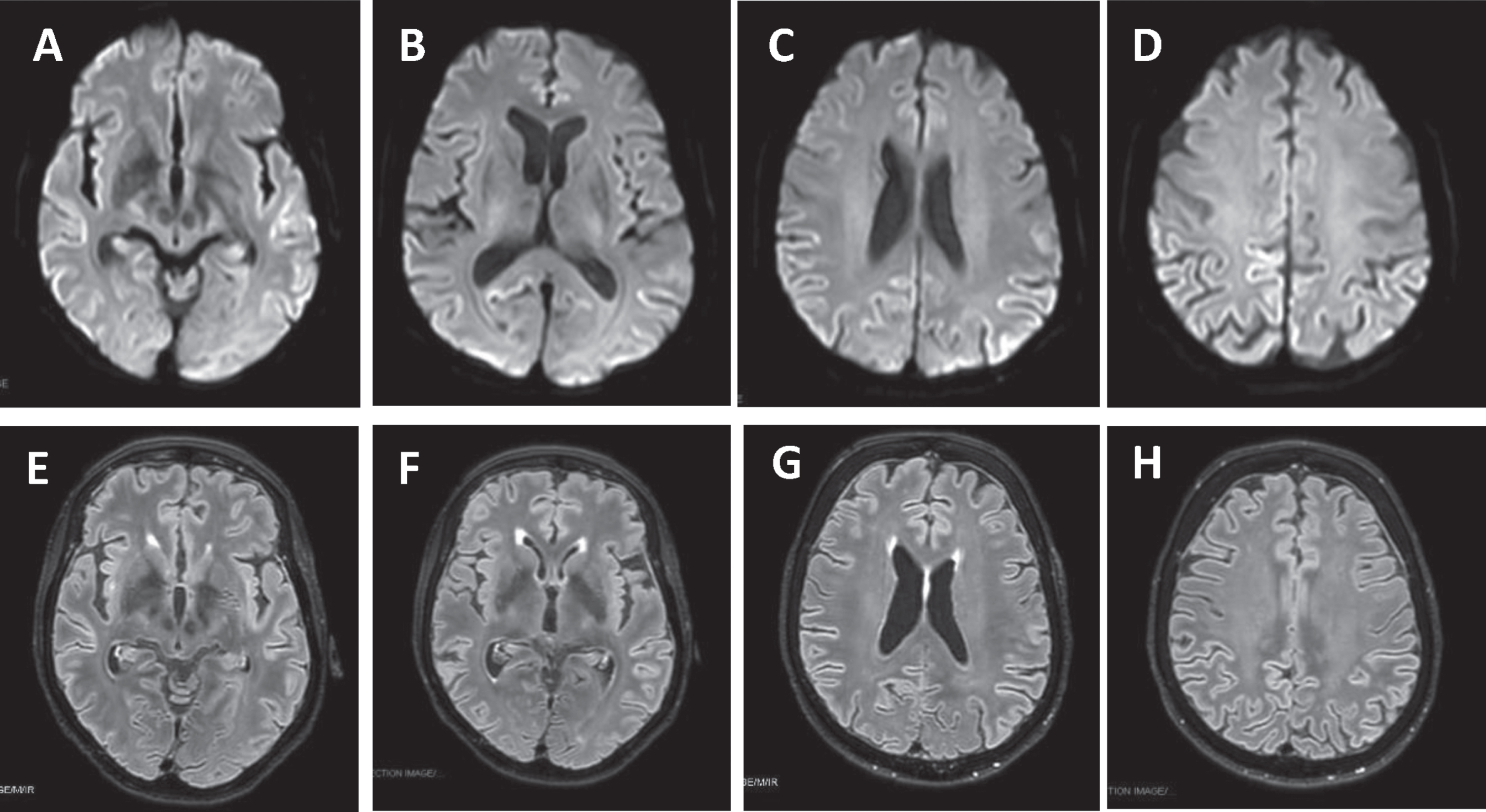
Fig.2
Brain 18F-FDG PET showed a severe cortical hypometabolism involving right frontal lobe, temporo-parietal lobe bilaterally, precuneus, and posterior cingulate cortex.
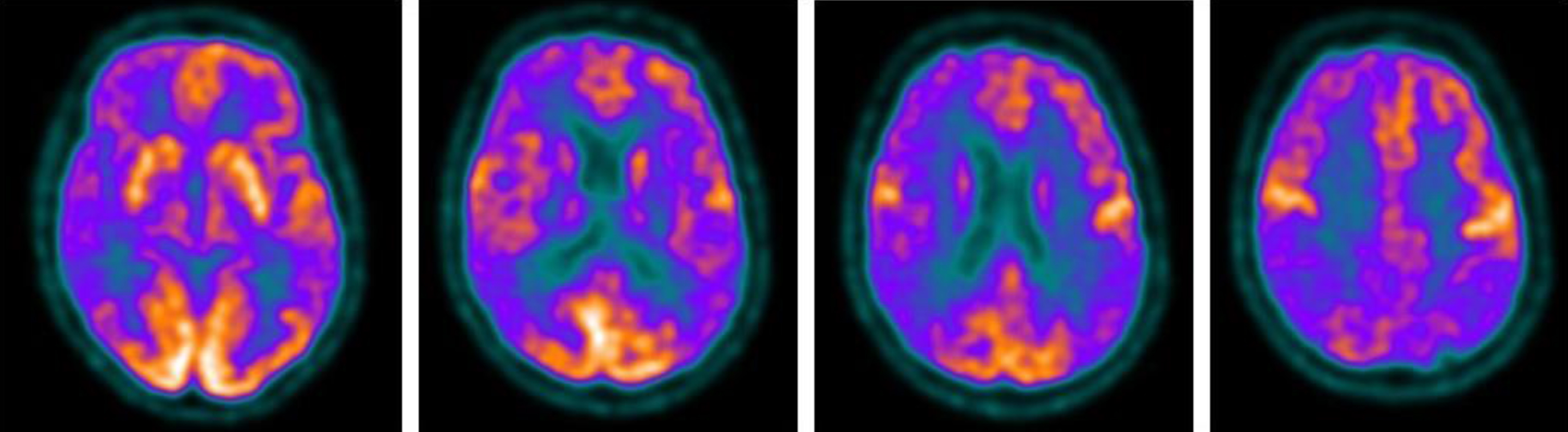
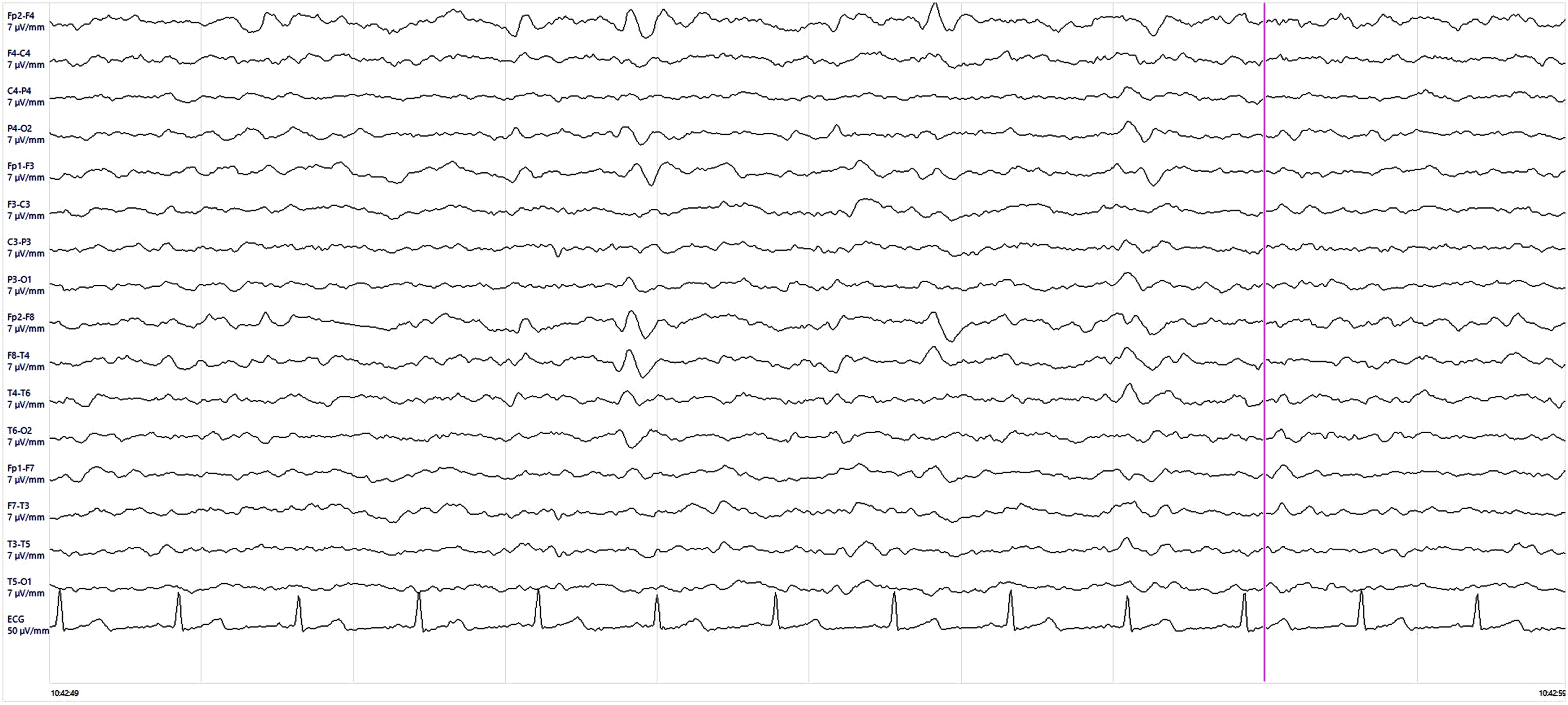
Fig.3
Electroencephalogram (EEG) findings. EEG showed rare quasi-periodic right frontotemporal sharp wave complexes.
DISCUSSION
Here we present the case of a 71-year-old female patient who developed neuropsychiatric and neurological symptoms subsequently to SARS-CoV-2 infection and was diagnosed with sCJD after a long period from the infection.
SARS-CoV-2 has been hypothesized to accelerate neurodegeneration and CJD pathogenesis by facilitating the loss of astrocyte homeostasis and promoting a neuroinflammatory transcriptional signature [3–5]. Indeed, the virus has been shown to possess neurotropic and neuroinvasive properties [9]. A recent case report described the association between SARS-CoV-2 and a higher probability of developing or accelerating neurodegenerative diseases such as CJD in addition to rapidly progressive Alzheimer’s disease and frontotemporal dementia [6]. Other reports showed an RPD condition occurring directly after COVID-19 infection and patients died a few months later [3, 6] or 15 days after hospitalization with an atypical clinical phenotype and fulminant evolution [8]. Our patient did not experience rapidly progressive deterioration and was hospitalized 15 months after the onset of symptoms, but it is worth mentioning that not all cases of sCJD are the same in terms of clinical phenotype and disease duration. These remarkable heterogeneities are influenced by the polymorphism M/V at codon 129 of PRNP gene and by prion types (based on PrPSc molecular mass). This assumption led to a classification of sCJD on six subtypes: MM, MV, and VV, each of which could be associated with PrPSc glicotype 1 or 2, in addition to differences in the resistances of the PrPSc to proteinase K digestion [1]. Traditionally, disease duration averages 5 months from clinical onset to death, but ranges from weeks to several years [9]. Our patient was PRNP M129V heterozygous and, in the literature, this polymorphism correlates with long disease duration (mean 17.1 months) when associated with PrPSc type 2 [1].
Moreover, sCJD heterogeneity also concerns CSF biomarker profile. Several proteins act as surrogate CFS biomarkers such as 14–3–3 proteins, t-tau, neurofilaments, S100b, and alpha-synuclein, indicativeof neuronal degeneration. However, none of these proteins are formally accepted as part of the pathology-defining criteria. There is no consensus regarding the t-tau cut-off that should be linked to sCJD; several studies have proposed either > 1,072 pg/mL, > 1,250 pg/mL, > 1,300 pg/mL, or > 1,400 pg/mL [10]. However, in our analysis, t-tau level was only slightly increased with a value of 782 pg/mL. It is noteworthy that recent studies proved that t-tau concentration, measured in either blood or CSF, was strongly associated with survival time and correlated with the rate of disease progression in sCJD [11, 12]. Therefore, our CSF t-tau levels could reflect the slow clinical course of sCJD in our patient.
Conclusions
It remains intricate to assert a link between COVID-19 and CJD. We aim to emphasize the extreme heterogeneity of clinical presentation and duration of sCJD, mainly determined by the polymorphism at codon 129 of the PRNP gene, and the role of surrogate CSF biomarkers like t-tau which can only be slightly increased and probably correlate with the rate of progression of sCJD.
ACKNOWLEDGMENTS
The authors are grateful to Francesca Piattellini, Edoardo Fronzoni, Giulio Pastorelli, Valentina Moschini and Carmen Barbarato, part of the patient’s treatment team.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
REFERENCES
[1] | Parchi P , Giese A , Capellari S , Brown P , Schulz-Schaeffer W , Windl O , Zerr I , Budka H , Kopp N , Piccardo P , Poser S , Rojiani A , Streichemberger N , Julien J , Vital C , Ghetti B , Gambetti P , Kretzschmar H ((1999) ) Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46: , 224–233. |
[2] | Watson N , Hermann P , Ladogana A , Denouel A , Baiardi S , Colaizzo E , Giaccone G , Glatzel M , Green AJE , Haïk S , Imperiale D , MacKenzie J , Moda F , Smith C , Summers D , Tiple D , Vaianella L , Zanusso G , Pocchiari M , Zerr I , Parchi P , Brandel J-P , Pal S ((2022) ) Validation of Revised International Creutzfeldt-Jakob Disease Surveillance Network Diagnostic Criteria for Sporadic Creutzfeldt-Jakob Disease. JAMA Netw Open 5: , e2146319. |
[3] | Young MJ , O’Hare M , Matiello M , Schmahmann JD ((2020) ) Creutzfeldt-Jakob disease in a man with COVID-19: SARS-CoV-2-accelerated neurodegeneration? Brain Behav Immun 89: , 601–603. |
[4] | Nasiri E , Naseri A , Yazdchi M , Talebi M ((2021) ) Is there a link between COVID-19 and Creutzfeldt-Jakob disease? A case report. J Res Clin Med 9: , 26. |
[5] | Ciolac D , Racila R , Duarte C , Vasilieva M , Manea D , Gorincioi N , Condrea A , Crivorucica I , Zota E , Efremova D , Crivorucica V , Ciocanu M , Movila A , Groppa SA ((2021) ) Clinical and radiological deterioration in a case of Creutzfeldt-Jakob disease following SARS-CoV-2 infection: Hints to accelerated age-dependent neurodegeneration. Biomedicines 9: , 1730. |
[6] | Pimentel GA , Guimarães TG , Silva GD , Scaff M ((2022) ) Case Report: Neurodegenerative diseases after severe acute respiratory syndrome coronavirus 2 infection, a report of three cases: Creutzfeldt–Jakob disease, rapidly progressive Alzheimer’s disease, and frontotemporal dementia. Front Neurol 13: , 731369. |
[7] | Bernardini A , Gigli GL , Janes F , Pellitteri G , Ciardi C , Fabris M , Valente M Creutzfeldt-Jakob disease after COVID-19: Infection-induced prion protein misfolding? A case report. Prion 16: , 78–83. |
[8] | Olivo S , Furlanis G , Buoite Stella A , Fabris M , Milanic R , Zanusso G , Manganotti P ((2022) ) Rapidly evolving Creutzfeldt-Jakob disease in COVID-19: From early status epilepticus to fatal outcome. Acta Neurol Belg, doi: 10.1007/s13760-022-02023-x. |
[9] | Ferini-Strambi L , Salsone M ((2021) ) COVID-19 and neuro-logical disorders: Are neurodegenerative or neuroimmunological diseases more vulnerable? J Neurol 268: , 409–419. |
[10] | Hermann P , Appleby B , Brandel J-P , Caughey B , Collins S , Geschwind MD , Green A , Haïk S , Kovacs GG , Ladogana A , Llorens F , Mead S , Nishida N , Pal S , Parchi P , Pocchiari M , Satoh K , Zanusso G , Zerr I ((2021) ) Biomarkers and diagnostic guidelines for sporadic Creutzfeldt-Jakob disease. Lancet Neurol 20: , 235–246. |
[11] | Thompson AGB , Luk C , Heslegrave AJ , Zetterberg H , Mead SH , Collinge J , Jackson GS ((2018) ) Neurofilament light chain and tau concentrations are markedly increased in the serum of patients with sporadic Creutzfeldt-Jakob disease, and tau correlates with rate of disease progression. J Neurol Neurosurg Psychiatry 89: , 955–961. |
[12] | Staffaroni AM , Kramer AO , Casey M , Kang H , Rojas JC , Orrú CD , Caughey B , Allen IE , Kramer JH , Rosen HJ , Blennow K , Zetterberg H , Geschwind MD ((2019) ) Association of blood and cerebrospinal fluid tau level and other biomarkers with survival time in sporadic Creutzfeldt-Jakob disease. JAMA Neurol 76: , 969–977. |


