
Discovery of a Missense Mutation (Q222K) of the APOE Gene from the Australian Imaging, Biomarker and Lifestyle Study
Abstract
After age, polymorphisms of the Apolipoprotein E (APOE) gene are the biggest risk factor for the development of Alzheimer’s disease (AD). During our investigation to discovery biomarkers in plasma, using 2D gel electrophoresis, we found an individual with and unusual apoE isoelectric point compared to APOE ɛ2, ɛ3, and ɛ4 carriers. Whole exome sequencing of APOE from the donor confirmed a single nucleotide polymorphism (SNP) in exon 4, translating to a rare Q222K missense mutation. The apoE ɛ4 (Q222K) mutation did not form dimers or complexes observed for apoE ɛ2 & ɛ3 proteins.
INTRODUCTION
Sporadic (non-familial) Alzheimer’s disease (AD) is a condition with progressively grave consequences for cognitive function, resulting from complex and incompletely understood etiology. The biggest risk factor, after age, for developing the disease are allelic variations within the gene encoding the cholesterol and lipid transporter, apolipoprotein E (APOE) [1]. There are three common APOE alleles in the general population defined as: APOE ɛ2, APOE ɛ3, and APOE ɛ4 [2]. Each allele encodes for an apoE proteoform where two cysteine residues are present in apoE ɛ2, or mutated to arginine C158R in apoE ɛ3, or C112R/C158R in apoE ɛ4 [3]. The mutation to arginine produces one or two additional positive charges relative to apoE ɛ2, at biological pH. The introductionof the additional basic residues in apoE ɛ3 and apoE ɛ4 results in a shift in the isoelectric point of the proteins and was the basis of their nomenclature [4, 5]. The importance of apoE in AD is demonstrated by the significantly different effects that APOE’s three main alleles, ɛ2, ɛ3, and ɛ4, have on the prevalence and age of inception AD in their carriers. APOE ɛ4 is the most jeopardizing allele, conferring a dose-dependent increase to the risk for AD and reducing the mean age of onset about ten years in homozygous ɛ4 persons, compared to persons without ɛ4 allele [6, 7].
Amyloid plaques are one of the major pathological hallmarks in AD. The peptide amyloid-β (Aβ) is a major component of amyloid plaques [8]. The buildup and eventual deposition of Aβ is due to a 2–5% decrease in the clearance of Aβ from the brain [9, 10]. ApoE ɛ2 and ɛ3 bind to Aβ; however, this is lost in the apoE ɛ4 variant [11, 12]. The loss in binding is thought to contribute to a decrease in the clearance of Aβ and thus is part of the mechanism for the increased risk of apoE ɛ4 allele carriers to develop AD [13–15]. And in mouse models blocking the apoE:Aβ interaction can have significant effects on amyloid and tangle formation [16]. The formation of homodimers of apoE and the ability to form complexes with Aβ and other proteins is only one functional difference between the apoE variants. This includes the important discoveries about rare APOE missense variants that impact protein function and modify disease risk. The discovery of the protective roles of Christchurch (R136S) in autosomal dominant PSEN1 familial AD and the Jacksonville (V236E), & R251G mutations that are protective against sporadic AD [17–20]. Similarly, the ɛ7 (E244K,E245K) mutant can increase the risk for AD. Other mutations like R145C mutant can results in altered function including mild triglyceride dyslipidemia [21]. As genome sequencing continues to expand new potential disease modifying variants for AD are being discovered [22]. The combination of rare variants and sufficiently large studies to determine their disease impact have potential to develop new avenues of drug development that target APOE [23].
The Australian Imaging, Biomarker and Lifestyle (AIBL) study of aging is a longitudinal study of over 1,117 volunteers and includes amyloid positron emission tomography (PET) imagining, as well as several repeated cognitive and behavioral tests [24]. The findings reported in this paper were made while we were conducting a discovery plasma proteomic project, searching for blood-based biomarkers, that could aid in the early diagnosis of AD [25]. The technique we used for our proteomic approach was large format (24 cm) 2D gel electrophoresis, where proteins are differentially labelled with high sensitivity fluorescent Zdyes™ [26]. The use of 2D gels was fortuitous as it allowed us to discover the APOE mutation we report here, because it caused a clearly detectable shift in the isoelectric point of the mutant protein. This mutation would have been missed via standard bottom-up mass spectrometry, because of the requirement that the database contain the mutant protein sequence to provide a match. Here we report Q222K mutation in an individual within the ABIL cohort. The three main alleles, ɛ2, ɛ3, and ɛ4, of APOE modify disease risk; however, it is not clear if it is due to a gain of loss in function. Several functions are prescribed to apoE protein including lipid transport [27], innate and adaptive immume responses [28], inhibitor of ferroptosis [29], and in the context of AD is implicated in the clearance of Aβ [15, 30]. Because one hypothesis for loss of Aβ clearance is due to the loss of a direct interaction with apoE, we investigated if the Q222K mutation modulated the ability of apoE to form complexes with itself or other proteins in vivo.
MATERIALS AND METHODS
Ethics approval
The AIBL study was approved by the following institutional human research ethics committees of Austin Health, St Vincent’s Health, Hollywood Private Hospital and Edith Cowan University.
APOE genotyping
DNA extraction was carried out using either QIAamp DNA blood Midi or Maxi kits (Qiagen), according to the manufacturer’s protocol. APOE genotype was determined through TaqMan® genotyping assays (Life Technologies) for rs7412 (Assay ID: C_904973_10) and rs429358 (Assay ID: C_3084793_20) on a QuantStudio 12K-Flex™ real-time PCR system (Applied Biosystems) using the TaqMan® GTXpress™ Master Mix (Life Technologies) as per manufacturer’s instructions.
Whole exome sequencing
The sequencing of the four coding exons of APOE was undertaken using a five amplicon design, with Exons 1 through 3 amplified as a single amplicon and Exon 4 split in two overlapping amplicons. Exons were amplified via PCR using GoTaq DNA polymerase, 200μM of each dNTP, 1.5 mM MgCl2 and primers specific for each exon (Table 1). Thermocycler conditions included an initial denaturation step at 95°C for 2 min, followed by 35 cycles of: 94°C for 30 s, annealing temperature (Table 1) for 30 s and 72°C for 30 s. Finally, at the end of the 35 cycles a 7-min adenylation step was undertaken. To remove impurities, unpurified PCR sample templates were cleaned by Solid Phase Reversible Immobilization (SPRI), which utilized paramagnetic beads. The second part of the Exon 4 required the band to be gel purified using the Qiagen gel purification kit. Samples were quantified by gel electrophoresis against a DNA ladder of known concentration. Purified DNA fragments were then sequenced using the Big Dye version 3.1 chemistry (Applied Biosystems) and post-cleaned using SPRI. Fragments were separated on a 3730xI DNA Analyzer, using a 96-capillary array (Applied Biosystems) at the Australian Genome Research Facility (Perth, Australia).
Table 1
Primers for exome sequencing
| Primer | Annealing Temp (°C) | Primer Sequence |
| Apoe_EX1_F | 56 | TGTGGGGGGTGGTCAAAAGA |
| Apoe_EX1_R | 56 | CCCCGAGTAGCTCTCCTGAGAC |
| Apoe_EX2_F | 56 | GCTAACCTGGGGTGAGGCC |
| Apoe_EX2_R | 56 | GCCCAGGGTCTGCCTGAAT |
| Apoe_EX3_F | 56 | CCACCATGGCTCCAAAGAAG |
| Apoe_EX3_R | 56 | ACTTAGCGACAGGGGCAGAA |
| Apoe_EX4_part1_F | 58 | TGGGTCTCTCTGGCTCATCC |
| Apoe_EX4_part1_R | 58 | TGCCCATCTCCTCCATCCG |
| Apoe_EX4_part2_F | 58 | GCCGCCACTGTGGGCTCC |
| Apoe_EX4_part2_R | 58 | AGGAGGGGGCTGAGAATTGTG |
EX#, Exon; F/R, Forward/Reverse.
Validation and genotyping of Q222K (Gln222Lys)
The novel non-synonymous SNP 45412217C > A (variant ID 19-44908960-C-A, Q222K; Gln222Lys); was validated using a custom TaqMan® genotyping assay (Life Technologies), designed via the Life Technologies’ online Custom TaqMan® Assay Design Tool (entered sequence and designed primer/probe sets available upon request). The nomenclature for the residue numbering used here includes the first 18 amino acids which are a signal peptide that is removed from the endogenous apoE protein. The numbering without the signal peptide yields C112 and C158 as the location of C⟶R mutation for the different APOE 2, 3, and 4 alleles types. The location of this mutation without the signal sequence is Q204K.
Plasma preparation
Plasma was collected as previously described by [31]. A 250μL aliquot plasma (using EDTA anti-coagulant) was thawed on ice for 1 h. An aliquot (45μL) was diluted (1:4) with 50 mM TRIS pH 8.0 before affinity enrichment of proteins, using a prepacked 1 mL heparin sepharose column (GE Lifescience Sweden). The equivalent of 40μL of neat plasma was injected on column at a flow rate of 0.5 mL*min–1 and washed with buffer A (20 mM NaCl 50 mM TRIS pH 8.0) for 4.5 column volumes (CV). The absorbance at 280 nM was monitored and when the absorbance returned to baseline the bound proteins were eluted in a single isocratic step using 100% buffer B (1.5 M NaCl 50 mM TRIS pH 8.0). The eluted proteins were collected in a 15 mL falcon tube (VWR Australia). The 2.5 mL volume of the collected material was reduced and alkylated with 10 mM Tris(2-carboxyethyl) phosphine (Pierce Bond Breaker) and 20 mM 4-vinylpyridine (Sigma) for 60 min at room temperature, while rotating the sample slowly on a mechanical wheel. The reduced and alkylated proteins were then precipitated with the addition 7.5 mL of chilled acetone (–20°C, HPLC grade, Sigma). After several inversions of the tubes the samples were incubated overnight at –20°C. The next day the tubes were centrifuged at 5,000×g for 20 min at room temperature, the acetone was carefully decanted, and the pellet was left to dry for 10 min at room temperature in a ventilated hood. The pellet was then dissolved with 50μL of 8 M urea 4% CHAPS. The protein concentration was determined by BCA protein assay and typically yielded 5–8 mg*mL–1. An aliquot of 40μg of protein was diluted with labeling buffer (45 mM bicine, 8M urea, 4% CHAPS pH 8.0) to a final total volume of 10μL (i.e., 7μL of 6 mg*mL–1 with 3μL labeling buffer). The samples from different individuals were labeled with one of two spectrally resolved NHS-fluorescent Zdyes dyes (JAV-I-187, blue emitting or BDR-I227, green emitting) as previously described by Laffoon et al. [25]. The different colored samples were mixed before isoelectric focusing (IEF) and 2D gel separation.
2D-page
24 cm pH 4.9–5.7 narrow range IEF strips (BioRad) were passively rehydrated with a mixture of the labelled sample and 472μL of rehydration buffer (8 M urea, 4% CHAPS, 1% Destreak™ (GE Healthcare), 1% pH 3–11 IPG buffer (GE Healthcare), and a trace of bromophenol blue (Sigma-Aldrich) under biotechnology grade mineral oil (Bio-Rad), for nine hours at 4°C, prior to isoelectric focusing. Isoelectric focusing was performed on an Ettan™ IPGphor II™ ceramic manifold, according to the follow protocol: Step 500 V for 1000 VHrs; linear gradient to 1000 V over 1500 VHrs hours; linear gradient to 10,000 V over 16,500 VHrs hours; step at 10,000 V for 90,000 VHrs and held at 50 V to prevent defocusing. After focusing, the strips were gently rocked in the dark for 15 min in 10 ml of 1% DTT (Bio-Rad) in equilibration buffer (6 M urea, 2% SDS, 20% glycerol, 50 mM Tris-HCl pH 8.8), followed by 15 min in 10 ml of 2.5% iodoacetamide (Sigma-Aldrich) in equilibration buffer. For the second dimension PAGE, the focused strips were sealed in agarose with a trace of bromophenol blue onto 24 cm ×1.5 mm, 11% acrylamide gels. PAGE was run on a water-cooled DALT6 tank for 1 h at 2 W per gel, followed by approximately 5 h at 15 W per gel, until the blue dye front migrated to the bottom of the gels. Gels were scanned, at 100μm resolution, for fluorescence using a Typhoon™ Trio scanner (GE Healthcare). Photomultiplier tube voltages were selected to assure sub-saturating intensities of protein spots. Scans of the blue emitting JAV-I-187 labeled proteins used the 488 nm excitation laser and an HQ 510 nm, 30 nm bandpass emission filter (Chroma Technology). For the green emitting BDR-I-227 labeled proteins, the 532 nm excitation laser was used with a 580 nm, 30 nm bandpass emission filter (GE Healthcare).
Spot matching and quantitation
Gel image files were imported into Progenesis SameSpots software (Nonlinear Dynamics V3.1.3030.23662). Artefacts were removed from the analysis, and misalignments were corrected by manual warping and by adding landmarks when appropriate.
Western blot analysis
EDTA plasma was diluted 1:20 in distilled water, briefly vortexed and then 10μL of the diluted plasma was mixed with 3μL of 4x Laemmli SDS loading buffer (non-reducing conditions). To reduce samples 4x Laemmli SDS loading buffer contained 83 mM Tris(2-carboxyethyl)phosphine (TCEP, ThermoFisher), final concentration in the sample of 20 mM TCEP. Samples were heated for 10 min at 90°C before being loaded on to 4–20% gradient acrylamide gels (Criterion, Bio-rad). Proteins were transferred to PVDF membranes blocked with 4% skim milk in tris buffer saline with 0.1% tween-20. The primary antibody was a rabbit monoclonal anti-apolipoprotein E (EP1373Y, Novus biologicals), secondary was a polyclonal Goat, anti-rabbit-HRP (Dako). Images were captured on ChemiDoc imaging system (Bio-Rad).
RESULTS AND DISCUSSION
We compared the relative peptide spot positions of plasma apolipoprotein E (ApoE) isoforms in the isoelectric dimension of two-dimensional gels (2D-PAGE), to verify the APOE genotypes of 153 samples. The genotypes: 6 ɛ2/ɛ3, 6 ɛ2/ɛ4, 44 ɛ3/ɛ3, 78 ɛ3/ɛ4, and 19 ɛ4/ɛ4, as determined by DNA genotyping, were correctly indicated by gel spot positions in all but one sample donor. That anomalous sample, nominally ɛ4/ɛ4 by DNA genotyping, showed an apoE spot position with an unexpected basic shift, predicting a peptide with an additional positive charge to that occupied by standard apoE ɛ4 (labelled μ in Fig. 1). The donor of the novel APOE variant was an Australian Caucasian male, 82 years of age at the time of testing, with no cognitive impairment, as measured by the Mini-Mental State Exam (baselineMMSE = 28, T1 = 30, T2 = 28, T3 = 30, T4 = 27, T5 = 28, T6 = 26, T7 = 28, T8 = 26, T9 = 28, T10 = 28, bold indicates the timepoint for the blood used in this study, each timepoint is approximately 18 months apart). The AIBL study is a longitudinal study, and this individual has undergone eight amyloid PET scans over the past 14 years. The amyloid PET centiloid (CL) values have increased over time as follows despite lack of clinically observed cognitive decline (BaselinePIB = 22.4, T1PIB = 29.5, T2PIB = 28.5, T3PIB = 37.6, T4PIB = 31.1, T5NAV = 27.5, T7NAV =47.9, T10NAV = 71.2; subscript indicates amyloid tracer used). The advanced age, stability of cognition over time and only recent increase in brain amyloid accumulation on an apoE ɛ4 background could suggest the mutation is protective, or at least not deleterious. However, the observation of a greater number of cases or corroborating biochemical evidence would be required before any conclusions could be made.
Fig. 1
Large format 2D gel (pH range 4.7–5.9) comparing the ApoE proteins from different genotypes. ApoE protein has three main groups of protein isoforms (circled, A). The white arrows indicate representative non-apoE “landmark” spots, showing that there is no appreciable pI shift between the gels. Comparison of and ɛ3 and ɛ4 carriers demonstrates the well know shift in isoelectric point between different apoE forms as well as the shift introduced by the missense mutation of cys to arginine. The additional mutation of Q222K (μ) induces a basic shift of similar magnitude as the cys to arg mutation. ApoE ɛ2 and ɛ3 protein variants are known to form a 45 kDa complex with apoAII:Aβ. This complex is present in the ɛ3 carriers but not in the Q222K (ɛ4/μ)carrier (B).
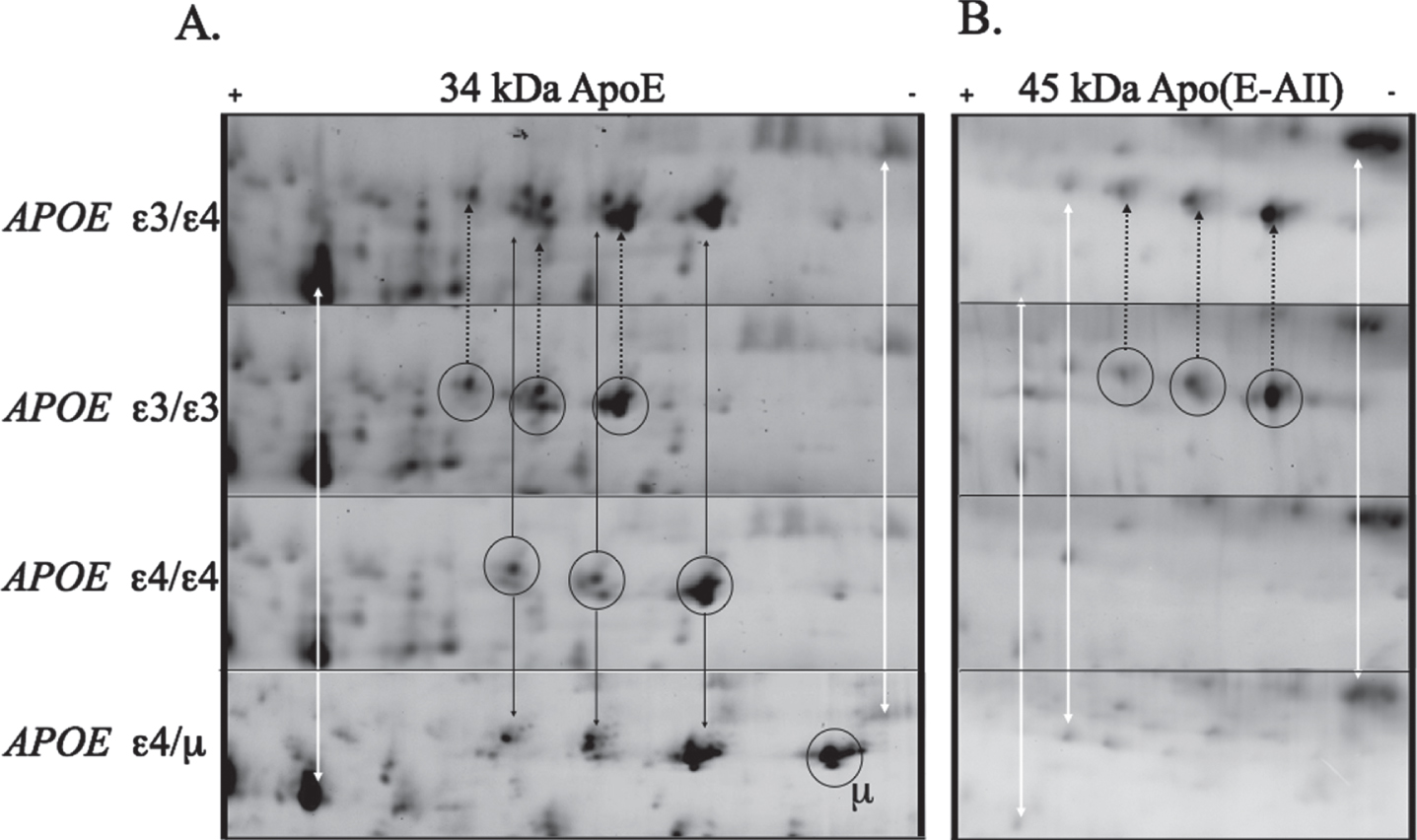
Based on the anomalous apoE protein pattern we conducted a complete APOE sequencing of the donor’s APOE coding exons (Table 1). The exon sequencing revealed a novel, non-synonymous, single nucleotide polymorphism (SNP) in exon 4 (Supplementary Fig. 1), translating to Q222K, and validating the shift in the protein isoelectric point. To investigate the novelty of this mutation we looked at frequency that it has been observed in gnomAD [32]. This misssense mutation has been observed four previous times, twice in an African ancestry and once in European. The Q222K rare missense mutation has a mean allele frequency (MAF) of 2.6 e-5. Structurally, position 222 is at a flexible loop region of apoE, corresponding to the shoulder of a domain (residues 244–272) hypothesized to bind Aβ [15] and involved in lipid binding [33]. Due to the proximity of the mutation to a hypothesized Aβ binding domain, we undertook further investigation to see if the Q222K mutation resulted in restoration of the ability of apoE to form complexes with Aβ or other proteins. We first investigated the heterodimer apoE-apoAII complex that were measurable in the 2D gels (Fig. 1B). The apoE-apoAII complex in plasma was discovered in the late 1970s [34] and this complex has been shown to occur in ɛ2 and ɛ3 carriers but not ɛ4/ ɛ4 homozygotes [35]. We found that the Q222K mutant protein did not have detectable levels of the 45 kDa apoE-apoAII complex (Fig. 1B). This contrasted with ɛ3 carriers that display heteromeric and homomeric protein complexes.
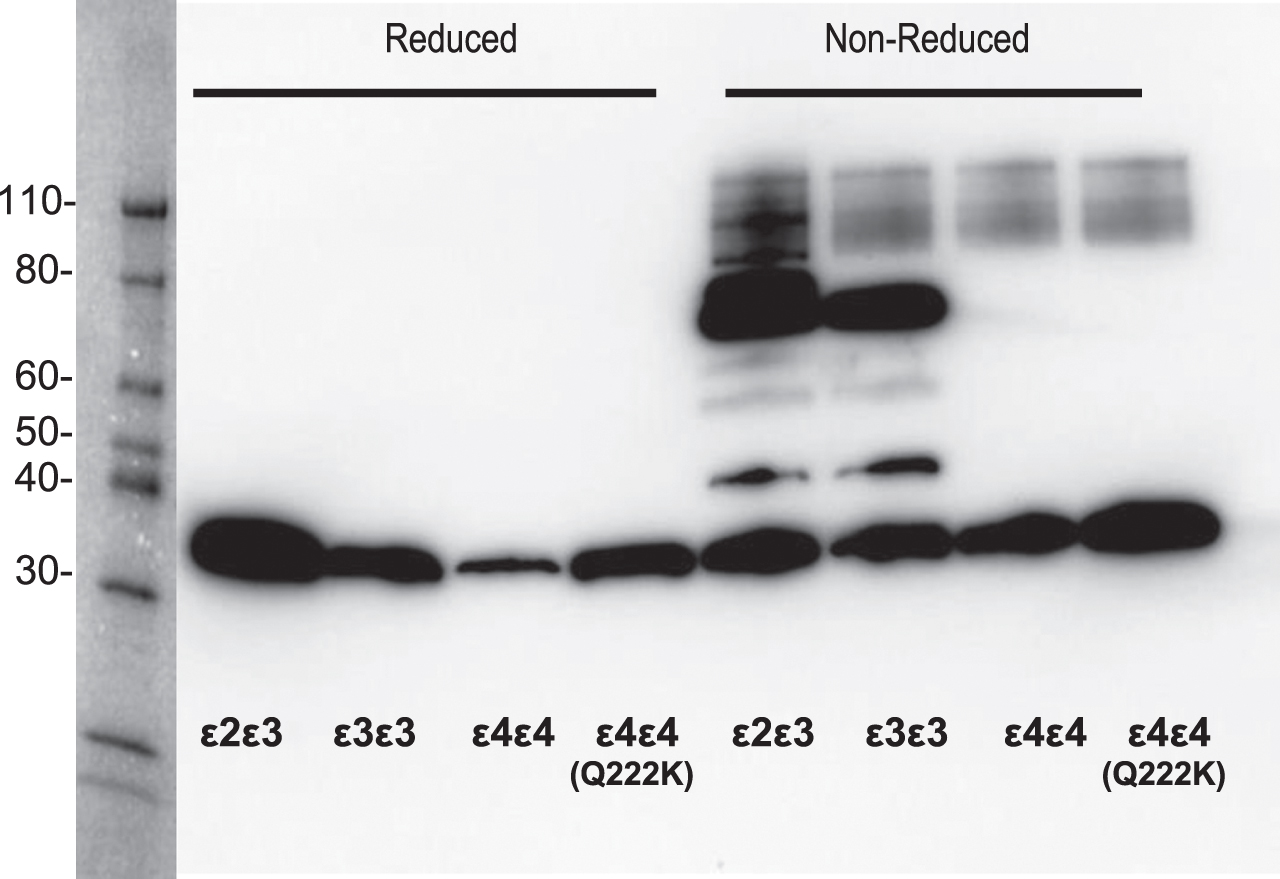
To determine if other heterodimer complexes form due to the Q222K mutation, we conducted a western blot for apoE under disulfide reducing or non-reducing conditions (Fig. 2). We did not observe any novel complexes with apoE Q222K under these conditions. As expected, apoE ɛ2 or ɛ3 under non-reducing conditions contained homodimer and heterodimer complexes between apoE (∼70 kDa band) and apoE-AII (34 kDa band), respectively. When exposed to denaturing and reducing conditions, complexes between apoE ɛ2 or ɛ3 collapse to a single 34 kDa band corresponding to apoE. The observation of the apoE-apoAII heterodimer in the 2D-gels (Fig. 1B) was surprising to us, as reduction and alkylation were part of the sample processing. In contrast, the western under reducing conditions show a complete breakdown of the apoE homodimers and heterodimers (Fig. 2), consistent with previous literature [35]. We attribute this discrepancy to differences in the reduction and alkylation conditions used for the 2D gels. The reduction and alkylation of proteins prior to 2D gel analysis occurred under native conditions versus the sample preparation of the western that uses heat and the denaturing detergent sodium dodecyl sulfate. The combination of detergent and heat denature the proteins and expose disulfides to the reductant (TECP) that is otherwise excluded due to the disulfides being buried within the protein-protein interface. In conclusion, this mutation does not appear to significantly impact the ability to bind other proteins and form complexes that are resistant stable in SDS-PAGE analysis.
Fig. 2
Comparison of ApoE protein complexes between the Q222K mutation and APOE ɛ2, ɛ3, and ɛ4 carriers. Western blot analysis for apoE forms on neat plasma in the presence or absence of reducing agent (10 mM Tris(2-carboxyethyl) phosphine (TCEP)). The full length apoE protein is observed at 34 kDa under both conditions. However, under non-reducing conditions the presence of apoE proteins complexes were detected, as apoE:apoAII (45 kDa, arrow), apoE dimer (75 kDa).
ACKNOWLEDGMENTS
We would like to acknowledge the Australian Imaging and Biomarker, Lifestyle (AIBL) research team (a complete list of researchers can be found at www.aibl.csiro.au) as well as the volunteer subjects and their families.
FUNDING
We would also like to acknowledge funding from The Cooperative Research Centre for Mental Health, Alzheimer’s Drug Discovery Foundation grant (BRR, PI), Knott Family Equipment Grant (BRR, PI), and the Pierce Armstrong Trust (BRR, PI), The Victorian Government’s Operational Infrastructure Support Program and the Wicking Trust. And support from The Florey Neuroproteomics facility. National Institutes of Health- Small Business Technology Transfer (NIH STTR grant 5R42RR021790, EAD PI), Center for Biological Research Excellence (NIH CoBRE grants 1P20RR024237 and 2P20GM 104935, EAD PI), Montana Board of Research and Commercialization Technology Grants: (MBRCT: 05–14, 06–46, and 08–17, EAD PI) and the Murdock Charitable Trust (EAD, PI).
CONFLICT OF INTEREST
EAD is a founder and shareholder in Zdye LLC, which licenses patents issued to Montana StateUniversity and reagents used in the present study. All others have no conflicts to declare.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/ADR-220075.
REFERENCES
[1] | Heffernan AL , Chidgey C , Peng P , Masters CL , Roberts BR ((2016) ) The neurobiology and age-related prevalence of the epsilon4 allele of Apolipoprotein E in Alzheimer’s disease cohorts, J Mol Neurosci 60: , 316–324. |
[2] | Zannis VI , Breslow JL , Utermann G , Mahley RW , Weisgraber KH , Havel RJ , Goldstein JL , Brown MS , Schonfeld G , Hazzard WR , Blum C ((1982) ) Proposed nomenclature of apoE isoproteins, apoE genotypes, and phenotypes, J Lipid Res 23: , 911–914. |
[3] | Mahley RW ((1988) ) Apolipoprotein E: cholesterol transport protein with expanding role in cell biology, Science 240: , 622–630. |
[4] | Utermann G , Steinmetz A , Weber W ((1982) ) Genetic control of human apolipoprotein E polymorphism: comparison of one- and two-dimensional techniques of isoprotein analysis, Hum Genet 60: , 344–351. |
[5] | Zannis VI , Breslow JL ((1981) ) Human very low density lipoprotein apolipoprotein E isoprotein polymorphism is explained by genetic variation and posttranslational modification, Biochemistry 20: , 1033–1041. |
[6] | Corder EH , Saunders AM , Strittmatter WJ , Schmechel DE , Gaskell PC , Small GW , Roses AD , Haines JL , Pericak-Vance MA ((1993) ) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families, Science 261: , 921–923. |
[7] | Smirnov DS , Galasko D , Hiniker A , Edland SD , Salmon DP ((2021) ) Age-at-onset and APOE-related heterogeneity in pathologically confirmed sporadic Alzheimer disease, Neurology 96: , e2272–e2283. |
[8] | Masters CL , Simms G , Weinman NA , Multhaup G , McDonald BL , Beyreuther K ((1985) ) Amyloid plaque core protein in Alzheimer disease and Down syndrome, Proc Natl Acad Sci U S A 82: , 4245–4249. |
[9] | Mawuenyega KG , Sigurdson W , Ovod V , Munsell L , Kasten T , Morris JC , Yarasheski KE , Bateman RJ ((2010) ) Decreased clearance of CNS beta-amyloid in Alzheimer’s disease, Science 330: , 1774. |
[10] | Roberts BR , Lind M , Wagen AZ , Rembach A , Frugier T , Li QX , Ryan TM , McLean CA , Doecke JD , Rowe CC , Villemagne VL , Masters CL ((2017) ) Biochemically-defined pools of amyloid-beta in sporadic Alzheimer’s disease: correlation with amyloid PET, Brain 140: , 1486–1498. |
[11] | LaDu MJ , Falduto MT , Manelli AM , Reardon CA , Getz GS , Frail DE ((1994) ) Isoform-specific binding of apolipoprotein E to beta-amyloid, J Biol Chem 269: , 23403–23406. |
[12] | Strittmatter WJ , Saunders AM , Schmechel D , Pericak-Vance M , Enghild J , Salvesen GS , Roses AD ((1993) ) Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease, Proc Natl Acad Sci U S A 90: , 1977–1981. |
[13] | Fagan AM , Watson M , Parsadanian M , Bales KR , Paul SM , Holtzman DM ((2002) ) Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of Alzheimer’s disease, Neurobiol Dis 9: , 305–318. |
[14] | Holtzman DM , Fagan AM , Mackey B , Tenkova T , Sartorius L , Paul SM , Bales K , Ashe KH , Irizarry MC , Hyman BT ((2000) ) Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer’s disease model, Ann Neurol 47: , 739–747. |
[15] | Verghese PB , Castellano JM , Garai K , Wang Y , Jiang H , Shah A , Bu G , Frieden C , Holtzman DM ((2013) ) ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions, Proc Natl Acad Sci U S A 110: , E1807–1816. |
[16] | Liu S , Breitbart A , Sun Y , Mehta PD , Boutajangout A , Scholtzova H , Wisniewski T ((2014) ) Blocking the apolipoprotein E/amyloid beta interaction in triple transgenic mice ameliorates Alzheimer’s disease related amyloid beta and tau pathology, J Neurochem 128: , 577–591. |
[17] | Arboleda-Velasquez JF , Lopera F , O’Hare M , Delgado-Tirado S , Marino C , Chmielewska N , Saez-Torres KL , Amarnani D , Schultz AP , Sperling RA , Leyton-Cifuentes D , Chen K , Baena A , Aguillon D , Rios-Romenets S , Giraldo M , Guzman-Velez E , Norton DJ , Pardilla-Delgado E , Artola A , Sanchez JS , Acosta-Uribe J , Lalli M , Kosik KS , Huentelman MJ , Zetterberg H , Blennow K , Reiman RA , Luo J , Chen Y , Thiyyagura P , Su Y , Jun GR , Naymik M , Gai X , Bootwalla M , Ji J , Shen L , Miller JB , Kim LA , Tariot PN , Johnson KA , Reiman EM , Quiroz YT ((2019) ) Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: a case report, Nat Med 25: , 1680–1683. |
[18] | Medway CW , Abdul-Hay S , Mims T , Ma L , Bisceglio G , Zou F , Pankratz S , Sando SB , Aasly JO , Barcikowska M , Siuda J , Wszolek ZK , Ross OA , Carrasquillo M , Dickson DW , Graff-Radford N , Petersen RC , Ertekin-Taner N , Morgan K , Bu G , Younkin SG ((2014) ) ApoE variant p.V236E is associated with markedly reduced risk of Alzheimer’s disease, Mol Neurodegener 9: , 11. |
[19] | Liu CC , Murray ME , Li X , Zhao N , Wang N , Heckman MG , Shue F , Martens Y , Li Y , Raulin AC , Rosenberg CL , Doss SV , Zhao J , Wren MC , Jia L , Ren Y , Ikezu TC , Lu W , Fu Y , Caulfield T , Trottier ZA , Knight J , Chen Y , Linares C , Wang X , Kurti A , Asmann YW , Wszolek ZK , Smith GE , Vemuri P , Kantarci K , Knopman DS , Lowe VJ , Jack CR Jr. , Parisi JE , Ferman TJ , Boeve BF , Graff-Radford NR , Petersen RC , Younkin SG , Fryer JD , Wang H , Han X , Frieden C , Dickson DW , Ross OA , Bu G ((2021) ) APOE3-Jacksonville (V236E) variant reduces self-aggregation and risk of dementia, Sci Transl Med 13: , eabc9375. |
[20] | Le Guen Y , Belloy ME , Grenier-Boley B , de Rojas I , Castillo-Morales A , Jansen I , Nicolas A , Bellenguez C , Dalmasso C , Küçükali F , et al ((2022) ) Association of rare APOE missense variants V236E and R251G with risk of Alzheimer disease, JAMA Neurol 79: , 652–663. |
[21] | Abou Ziki MD , Strulovici-Barel Y , Hackett NR , Rodriguez-Flores JL , Mezey JG , Salit J , Radisch S , Hollmann C , Chouchane L , Malek J , Zirie MA , Jayyuosi A , Gotto AM Jr. , Crystal RG ((2014) ) Prevalence of the apolipoprotein E Arg145Cys dyslipidemia at-risk polymorphism in African-derived populations, Am J Cardiol 113: , 302–308. |
[22] | Bagaria J , Moon Y , Bagyinszky E , Shim KH , An SSA , Kim S , Han SH ((2022) ) Whole exome sequencing reveals a novel APOE mutation in a patient with sporadic early-onset Alzheimer’s disease, Front Neurol 13: , 899644. |
[23] | Bu G ((2022) ) APOE targeting strategy in Alzheimer’s disease: lessons learned from protective variants, Mol Neurodegener 17: , 51. |
[24] | Fowler C , Rainey-Smith SR , Bird S , Bomke J , Bourgeat P , Brown BM , Burnham SC , Bush AI , Chadunow C , Collins S , et al. ((2021) ) Fifteen tears of the Australian Imaging, Biomarkers and Lifestyle (AIBL) Study: Progress and observations from 2,359 older adults spanning the spectrum from cognitive normality to Alzheimer’s disease, J Alzheimers Dis Rep 5: , 443–468. |
[25] | Laffoon SB , Doecke JD , Roberts AM , Vance JA , Reeves BD , Pertile KK , Rumble RL , Fowler CJ , Trounson B , Ames D , Martins R , Bush AI , Masters CL , Grieco PA , Dratz EA , Roberts BR ((2022) ) Analysis of plasma proteins using 2D gels and novel fluorescent probes: in search of blood based biomarkers for Alzheimer’s disease, Proteome Sci 20: , 2. |
[26] | Epstein MG , Reeves BD , Maaty WS , Fouchard D , Dratz EA , Bothner B , Grieco PA ((2013) ) Enhanced sensitivity employing zwitterionic and pI balancing dyes (Z-CyDyes) optimized for 2D-gel electrophoresis based on side chain modifications of CyDye fluorophores. New tools for use in proteomics and diagnostics, Bioconjug Chem 24: , 1552–1561. |
[27] | Hauser PS , Narayanaswami V , Ryan RO ((2011) ) Apoli-poprotein E: from lipid transport to neurobiology, Prog Lipid Res 50: , 62–74. |
[28] | Kloske CM , Wilcock DM ((2020) ) The important interface between apolipoprotein E and neuroinflammation in Alzheimer’s disease, Front Immunol 11: , 754. |
[29] | Belaidi AA , Masaldan S , Southon A , Kalinowski P , Acevedo K , Appukuttan AT , Portbury S , Lei P , Agarwal P , Leurgans SE , Schneider J , Conrad M , Bush AI , Ayton S ((2022) ) Apolipoprotein E potently inhibits ferroptosis by blocking ferritinophagy. Mol Psychiatry, doi: 10.1038/s41380-022-01568-w. |
[30] | Tai LM , Mehra S , Shete V , Estus S , Rebeck GW , Bu G , LaDu MJ ((2014) ) Soluble apoE/Abeta complex: mechanism and therapeutic target for APOE4-induced AD risk, Mol Neurodegener 9: , 2. |
[31] | Ellis KA , Bush AI , Darby D , De Fazio D , Foster J , Hudson P , Lautenschlager NT , Lenzo N , Martins RN , Maruff P , Masters C , Milner A , Pike K , Rowe C , Savage G , Szoeke C , Taddei K , Villemagne V , Woodward M , Ames D ((2009) ) The Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging: methodology and baseline characteristics of 1112 individuals recruited for a longitudinal study of Alzheimer’s disease, Int Psychogeriatr 21: , 672–687. |
[32] | Karczewski KJ , Francioli LC , Tiao G , Cummings BB , Alfoldi J , Wang Q , Collins RL , Laricchia KM , Ganna A , Birnbaum DP , Gauthier LD , Brand H , Solomonson M , Watts NA , Rhodes D , Singer-Berk M , England EM , Seaby EG , Kosmicki JA , Walters RK , Tashman K , Farjoun Y , Banks E , Poterba T , Wang A , Seed C , Whiffin N , Chong JX , Samocha KE , Pierce-Hoffman E , Zappala Z , O’Donnell-Luria AH , Minikel EV , Weisburd B , Lek M , Ware JS , Vittal C , Armean IM , Bergelson L , Cibulskis K , Connolly KM , Covarrubias M , Donnelly S , Ferriera S , Gabriel S , Gentry J , Gupta N , Jeandet T , Kaplan D , Llanwarne C , Munshi R , Novod S , Petrillo N , Roazen D , Ruano-Rubio V , Saltzman A , Schleicher M , Soto J , Tibbetts K , Tolonen C , Wade G , Talkowski ME , Genome Aggregation Database C Neale BM , Daly MJ , MacArthur DG ((2020) ) The mutational constraint spectrum quantified from variation in 141,456 humans, Nature 581: , 434–443. |
[33] | Dong LM , Wilson C , Wardell MR , Simmons T , Mahley RW , Weisgraber KH , Agard DA ((1994) ) Human apolipoprotein E. Role of arginine 61 in mediating the lipoprotein preferences of the E3 and E4 isoforms, J Biol Chem 269: , 22358–22365. |
[34] | Innerarity TL , Mahley RW , Weisgraber KH , Bersot TP ((1978) ) Apoprotein (E–A-II) complex of human plasma lipoproteins. II. Receptor binding activity of a high density lipoprotein subfraction modulated by the apo(E–A-II) complex, J Biol Chem 253: , 6289–6295. |
[35] | Weisgraber KH , Shinto LH ((1991) ) Identification of the disulfide-linked homodimer of apolipoprotein E3 in plasma. Impact on receptor binding activity, J Biol Chem 266: , 12029–12034. |


