
The Pathological Effects of Circulating Hydrophobic Bile Acids in Alzheimer’s Disease
Abstract
Recent clinical studies have revealed that the serum levels of toxic hydrophobic bile acids (deoxy cholic acid, lithocholic acid [LCA], and glycoursodeoxycholic acid) are significantly higher in patients with Alzheimer’s disease (AD) and amnestic mild cognitive impairment (aMCI) when compared to control subjects. The elevated serum bile acids may be the result of hepatic peroxisomal dysfunction. Circulating hydrophobic bile acids are able to disrupt the blood-brain barrier and promote the formation of amyloid-β plaques through enhancing the oxidation of docosahexaenoic acid. Hydrophobic bile acid may find their ways into the neurons via the apical sodium-dependent bile acid transporter. It has been shown that hydrophobic bile acids impose their pathological effects by activating farnesoid X receptor and suppressing bile acid synthesis in the brain, blocking NMDA receptors, lowering brain oxysterol levels, and interfering with 17β-estradiol actions such as LCA by binding to E2 receptors (molecular modelling data exclusive to this paper). Hydrophobic bile acids may interfere with the sonic hedgehog signaling through alteration of cell membrane rafts and reducing brain 24(S)-hydroxycholesterol. This article will 1) analyze the pathological roles of circulating hydrophobic bile acids in the brain, 2) propose therapeutic approaches, and 3) conclude that consideration be given to reducing/monitoring toxic bile acid levels in patients with AD or aMCI, prior/in combination with other treatments.
INTRODUCTION
As Alzheimer’s disease (AD) is becoming a major public health threat, there is an urgent unmet need to develop new therapies for AD [1]. As a result, several agents have been tested in clinical trials over the years with disease modifying therapies (DMTs) comprising more than 60% of the agents, and symptomatic cognitive enhancing agents forming about 30% of drugs [1–4]. DMTs refer to agents that either prevent, delay, or slow the progression of the disease [5]. DMTs themselves can be categorized into two groups: immunotherapy (mostly using antibodies) or small molecules [1]. There were 17 DMT agents in phase III of clinical trials in 2018, and eight agents were terminated in 2019 [4]. Preventing tau aggregation has been another approach in DMT. Leuco-methylthioninium bis(hydromethanesulfonate) (LMTM), a stable reduced form of the methylthioninium moiety, acts as a selective inhibitor of tau protein aggregation. LMTM did not show efficacy in a phase III clinical trial [6]. As another example, intranasal insulin showed significant improvements in learning, memory, and cognitive function in a pilot study of mild to moderate AD [7]. Then a clinical trial (NCT01767909) examined the effects of intranasally-administered insulin on cognition, entorhinal cortex, and hippocampal atrophy, and cerebrospinal fluid (CSF) biomarkers in amnestic mild cognitive impairment (aMCI) or mild AD. This trial completed, but no cognitive or functional benefits were observed with intranasal insulin treatment over a 12-month period [8]. Device malfunction was reported at the early stages of the trial [9].
With respect to symptomatic cognitive enhancing agents (such as donepezil, a cholinesterase inhibitor), these generally do not tackle the disease’s underlying mechanism. One of the drugs in this class, ITI-007, entered phase III clinical trial with the aim of improving neuropsychiatric symptoms (agitation) in AD patients. However, the trial was terminated in 2018, as interim data analysis revealed that the trial would not meet the primary outcome. On the other hand, suvorexant was tested in patients with AD for the treatment of insomnia [10]; and the drug was approved by the FDA for the indicated use.
The above observations show that there are other important disease underlying mechanisms such as oxidative stress [11] that should be considered in developing therapeutic agents for AD. This paper aims to expand on this point and present that altered bile acid metabolism could play major roles in developing AD.
LIVER DISFUNCTION CONTRIBUTING TO AD
A previous study found that the expression of peroxisomal D-bifunctional protein (D-BP) decreased significantly in the liver of patients with AD compared to control subjects [12]. This enzyme catalyzes conversion of tetracosahexaenoic acid into docosahexaenoic acid (DHA). Therefore, defective D-BP activity impairs DHA biosynthesis in the liver of patients with AD. It should be noted that DHA is found mainly as phospholipid species such as 38 : 6 and 40 : 6 in phosphatidylethanolamine (PE), phosphatidylinositol (PI), phosphatidylcholine (PC), phosphatidylserine (PS) [13], and lysophosphatidylcholine (LPC-DHA: LPC 22 : 6, CHEBI:73873, Mw: 567.7) [14]. Interestingly, association of blood lipids and AD has been shown [15, 16]. Therefore, reduced synthesis of DHA in the liver (due to defective D-BP activity) would lead to reduced serum lipids, such as PC 38 : 6 (CHEBI:74963, CAS: 59403-54-2), and PC ae C40 : 6 (CHEBI:86252) as shown in patients with AD [15]. Importantly, in vivo studies found that DHA in the form of LPC-DHA is transferred across the blood-brain barrier (BBB) by a member of major facilitator superfamily transporters (Mfsda2) [17]. Furthermore, dietary LPC-DHA significantly increased brain DHA content and improved brain function in adult mammals, but not DAH as free acid [18]. Therefore, reduction of DHA synthesis in the liver of patients with AD would reduce plasma levels of LPC-DHA (normally 21.5μM) [14]. Reduced plasma levels of LPC 18 : 2 have been reported in patients with AD [15] as well as LPC-DHA in these patients [19]. Now the important point is that D-BP is also involved in the biosynthesis of bile acids (shown schematically in Fig. 1) [20].
Fig. 1
The biosynthesis of bile acids from cholesterol by the neutral or acidic pathway A) in the cytoplasm and B) completion in peroxisomes (only for biosynthesis of conjugated cholic acid).
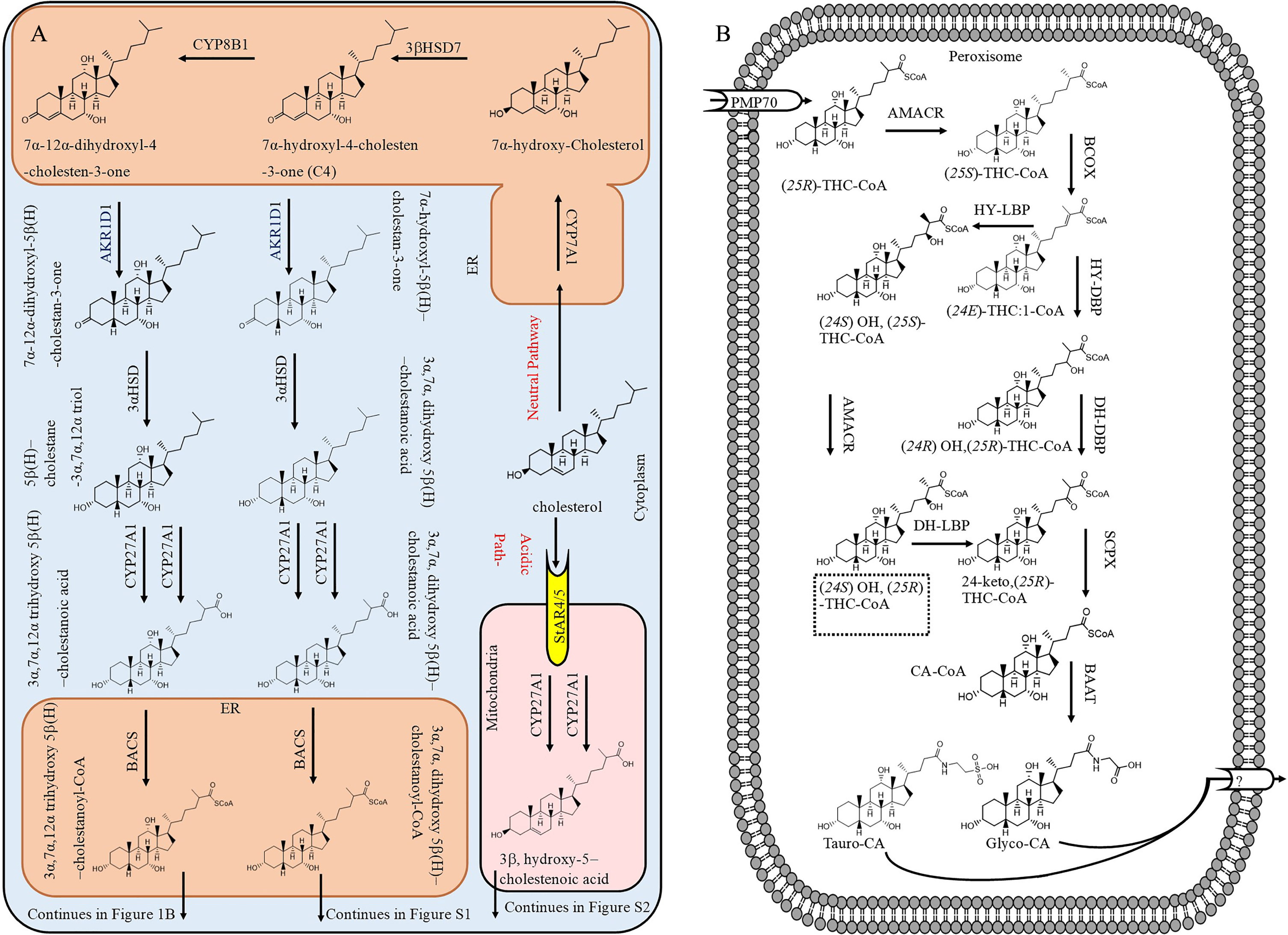
Figure 1A shows bile acid biosynthesis by classic and acidic pathways in the liver. In the classic pathway, cholesterol is transported into endoplasmic reticulum (ER) and oxidized to 7α-hydroxyl-4-cholesten-3-one (C4) by 3βHSD7. At this point, part of C4 is hydroxylated at position 12 by CYP8B1 to yield 7α, 12α, dihydroxyl-4-choleste-3-one. Both C4 and 7α, 12α, dihydroxyl-4-choleste-3-one are modified by several enzymes (AKR1D1, 3αHSD, CYP27A1, and BACS) to form 3α, 7α, dihydroxy-5β-cholestanoyl-CoA and 3α, 7α, 12α, trihydrohydroxy-cholestanoyl-CoA, respectively, which then are transported into peroxisome by the 70-kDa peroxisome membrane protein (PMP70). Figure 1B presents the side cleavage of 3α, 7α, 12α, trihydrohydroxy-cholestanoyl-CoA ((25 R)-THC-CoA). It can be seen that D-BP converts (24E) THC:1-CoA to 24-keto-(25 R)-THC-CoA at two steps. Figure 1B also depicts an alternative pathway for side chain oxidation employing L-bifunctional protein (L-BP) and AMACR enzymes. [21]. Therefore, when D-BP is not functioning, L-BP can step in and continue the formation of bile acids, but it appears that the activity of L-BP is much less than D-BP in bile acid biosynthesis. As in D-BP deficient patients, the hepatic ratio of C27/C24 bile acid increased to 1.45 compared to 0.04 in control subjects [20]. If L-BP was as active as D-BP, the C27/C24 bile acid would not have significantly increased in D-BP deficient patients. The side chain cleavage of 3α, dihydrohydroxy-cholestanoyl-CoA is presented in Supplementary Figure 1, which also shows the formation of conjugated CA and CDCA.
The acidic pathway (also known as alternative pathway) contributes about 10% of total bile acid production [22]. In addition, normally cholesterol is converted to other forms such as 27 hydroxy-cholesterol (3α, hydroxyl-5-cholestenoic acid) in extra-hepatic tissues or 24(S) hydroxycholesterol in the brain and it is transported into the liver by blood circulation for conversion mainly to CDCA via the acidic pathway [23]. This pathway can become the major bile acid production route when the classic pathway in the liver is not functioning normally due to deficiencies of enzymes such CYP7A1 [24, 25]. In one of the acidic pathways, which occurs in mitochondria, cholesterol is oxidized to 3β, hydroxy-5-cholestenoic acid by CYP27A1 at two steps and released to cytosol (Fig. 1A). Then, 3β, hydroxy-5-cholestenoic acid is uptaken into the ER and converted to 3β, 7α, dihydroxy-5-cholestenoic acid by CYP7B1. Afterwards, in one of metabolic pathways in the liver, 3β, 7α, dihydroxy-5-cholestenoic acid is modified to 3β, 7α, dihydroxy-5β-cholestanoyl-CoA (Supplementary Figure 2), which then is transported into peroxisome by PMP70 for side chain cleavage (Supplementary Figure 1).
Defective D-BP will affect levels of bile acids in the blood too [26], as these have been also observed in AD patients (increased plasma levels of GCA, GDCA, GCDCA) [27]. It is believed that defective D-BP results in the accumulation of C27 intermediate bile acids such as 3α,7α,12α trihydroxycholestanoic acid (THCA), 3α,7α dihydroxycholestanoic acid (DHCA), and OH-THCA in the liver, leading to cholestasis and consequent hepatic injury [20]. This hepatic injury may also result in further changes in the serum/plasma lipid levels. For example, six lipid metabolites were found to be cholesteryl esters (ChE 32 : 0, ChE 34 : 0, ChE 34 : 6, ChE; 33 : 6, ChE 40 : 4, ChE 32 : 4) that are reduced in plasma of patients with AD [28]. In another study, a combination of 24 blood metabolites classified AD patients with > 70% accuracy [29]. This study identified a blood lipid signature that predicts AD progression.
The neurological functions of bile acids have been reviewed recently [30–32], and neurological impairment has been reported with increased blood bile acid levels [33]. Air pollution [34] and genetic variations [35] may alter bile acid metabolism in the body and promote developing AD. In addition, elevated serum/plasma levels of toxic bile acids such as lithocholic acid (LCA) may originate from defective D-BP in the liver of patients with AD, which could play major roles in developing AD. This is further explained in the following sections.
HIGH BILE ACID SERUM/PLASMA LEVELS IN PATIENTS WITH AD
Clinical studies have indicated increased serum bile acid levels in patients with AD compared to control healthy subjects [15, 27, 36], as well [15] as increased urinary conjugated primary bile acid (taurochenodeoxycholic acid (TCDCA)) and conjugated secondary bile acid (taurodeoxycholic acid (TDCA)) levels [37]. It was originally reported by Greenberg et al. (2009) that the plasma levels of glycocholic acid (GCA), glycodeoxycholic acid (GDCA), and glycochenodeoxycholic acid (GCDCA) increased in patients with AD compared to control subjects [27]. Few years later, Mapstone et al. (2014) found that the serum levels of glycoursodeoxycholic acid were higher in patients with aMCI/AD compared to control subjects [15]. This was followed by the work of Olazaran et al. in 2015 that screened 495 plasma metabolites in patients with AD and normal control subjects. They found that plasma levels of deoxycholic acid (DCA) and LCA significantly increased in patients with AD compared to control subjects [38]. These observations were confirmed by Marksteiner et al. (2018), who reported that the plasma levels of LCA increased to 50±6 nM, which was significantly higher compared to control subjects (32±3 nM) [39] It should be noted that LCA is nontoxic to neurons at concentrations below 25μM [40]. Interestingly, the levels of LCA were higher in the brain of patients with AD compared to control subjects, suggesting that LCA may accumulate in the brain perhaps due to increased LCA serum levels, leading to declining of cognitive function in patients with AD [41]. MahmoudianDehkordi et al. (2019) found that the serum levels of secondary bile acids (DCA and its glycine and taurine conjugates) were significantly higher in patients with AD than control subjects, although serum levels of primary bile acids were significantly lower than control subjects [42]. While Shao et al. (2020) discovered that plasma levels of cholic acid were significantly higher than control subjects [36]. Organic anion transporter polypeptide 1 (OATP1) expressed in the choroid plexus and organic anion transporter polypeptide 2 (OATP2) expressed at the BBB both mediate the transport of bile acids from the systemic circulation into the brain [31]. Significant positive correlations have been observed between serum and brain bile acid concentrations for CA, CDCA, DCA, and taurocholic acid (TCA). LCA levels were below the limit of quantification in the brain [43].
On the other hand, Pan et al. (2017) did not find a significant difference in plasma levels of LCA between control subjects and AD patients, and in fact the levels of LCA in AD patients were lower (44±10 nM) than in control subjects (63±19 nM). Also, there was no significant difference in brain levels of LCA between AD patients (0.05±0.01 nmol/g) and control subjects (0.06±0.02 nmol/g) [44]. It should be noted that serum/plasma levels of LCA in young healthy subjects is 19.9±1.1 nM [45]. Therefore, these observations suggest that serum levels of LCA increase with age, which may have a role in gradual declining of cognitive function in non-cognitively impaired healthy elderly subjects [46]. Furthermore, these observations suggest that increased serum/plasma bile acid levels may happen at some stage of the disease and then may decrease due to other metabolic dysfunctions in the patients.
Total serum bile acids for general population is 3.8±0.2μM [47]. However, total serum bile acid levels less than 1.5μM are considered normal [48], but total serum bile acid level of 14μM is considered the normal upper limit [48]. The total serum bile acid levels can be as high as 100 times of normal levels [48]. Values of total serum bile acid ranged from 0.3 to 9.8μM in 216 pregnant women [49], while total serum bile acid levels increased from 4.2μM in control subjects to 6.5μM in patients with AD [39]. Therefore, total serum bile acid levels do not fall beyond normal upper limit in patients with AD, suggesting serious pathological effects are not expected, but chronic and steady declining effects on organs.
Patient and healthy subject data indicate that during aging the serum levels of LCA increase, however, the increase in AD patients is more noticeable [39, 47]. For example, serum LCA levels were 7.50±0.38 nM in healthy subjects aged between 19 and 65 years [47], while this increased to 32±3 nM in healthy subjects aged 77±1.2 (SEM), and 50±6 nM in AD patients aged 79±2.0 years [39]. Based on the available data in a clinical study, we plotted the serum LCA versus age for healthy subjects and AD patients (Fig. 2, raw data was obtained from Marksteiner et al. [39]). With age serum LCA level increases for AD patients. Also, Fig. 3 (raw data was obtained from Marksteiner et al. [39]) depicts Mini-Mental State Examination (MMSE) versus serum LCA showing a decrease in MMSE when serum LCA concentration increases.
Fig. 2
Serum lithocholic acid (LCA) levels versus the age for healthy subjects (\raisebox -2pt\epsfbox G:/Tex/IOSPRESS/ADR/220071/IF-01.eps) and AD patients (\raisebox -2pt\epsfbox G:/Tex/IOSPRESS/ADR/220071/IF-02.eps), demonstrating that LCA serum levels matches healthy subject before age 85. Raw data was obtained from Marksteiner et al. (2018) [39].
![Serum lithocholic acid (LCA) levels versus the age for healthy subjects (\raisebox -2pt\epsfbox G:/Tex/IOSPRESS/ADR/220071/IF-01.eps) and AD patients (\raisebox -2pt\epsfbox G:/Tex/IOSPRESS/ADR/220071/IF-02.eps), demonstrating that LCA serum levels matches healthy subject before age 85. Raw data was obtained from Marksteiner et al. (2018) [39].](https://content.iospress.com:443/media/adr/2023/7-1/adr-7-1-adr220071/adr-7-adr220071-g002.jpg)
Fig. 3
Serum lithocholic acid (LCA) levels versus MMSE score (n = 23), population includes healthy subjects, subjects with mild cognitive impairment and patients with AD, suggesting that generally MMSE levels tend to decrease by increasing serum LCA levels. Raw data was obtained from Marksteiner et al. (2018) [39]. Further studies are required to demonstrate this in larger populations.
![Serum lithocholic acid (LCA) levels versus MMSE score (n = 23), population includes healthy subjects, subjects with mild cognitive impairment and patients with AD, suggesting that generally MMSE levels tend to decrease by increasing serum LCA levels. Raw data was obtained from Marksteiner et al. (2018) [39]. Further studies are required to demonstrate this in larger populations.](https://content.iospress.com:443/media/adr/2023/7-1/adr-7-1-adr220071/adr-7-adr220071-g003.jpg)
HYDROPHOBIC BILE ACIDS DISRUPT THE BLOOD-BRAIN BARRIER
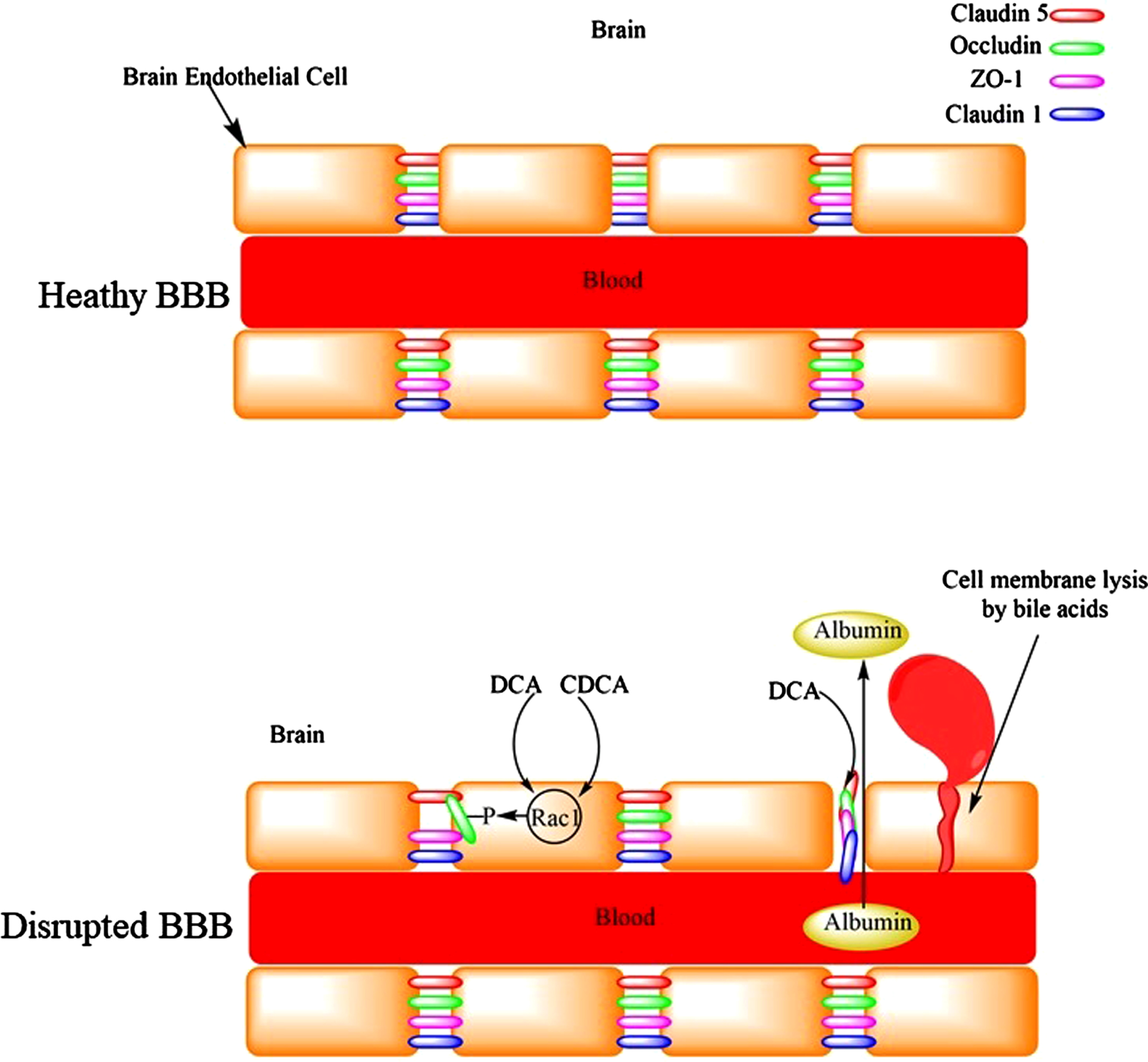
The BBB is a selective barrier that eclipses the brain and isolates it from the circulating blood. It is composed of the capillary basement membrane (BM), astrocyte end feet ensheathing the vessels, and pericytes embedded within the BM. It represents a major barrier for drug permeation especially those which are highly hydrophilic and have molecular mass greater than 400 Da. Hydrophobic bile acids such as DCA alter the permeability of the BBB to molecules that otherwise normally do not cross the BBB [50, 51]. Therefore, DCA potentially expose the brain neurons to harmful metabolites/chemicals in the blood (Fig. 4). Interestingly, DCA caused significant reduction in the brain mass of rats, [50] a phenomenon that is observed in patients with AD compared to cognitively normal subjects [52]. Peripapillary edema and alteration in tight junctions could be the reasons for increased BBB permeability by DCA [53]. In addition, bile acids are capable of damaging the BBB by their detergent and lytic effects on the cell membrane [53]. Cerebral endothelial cells form tight junctions with adjacent cells via interactions of the tight junction proteins: occluding, claudin-5, and intracellular docking proteins ZO-1and ZO-2. Bile acids (DCA and CDCA) increased permeability of the BBB by activation of Rac1 followed by phosphorylation of occludin, with CDCA being more potent [54]. The disruption of BBB significantly increased with hydrophobic bile acids (DCA and CDCA) than hydrophilic bile acids (ursodeoxycholic acid, GCDCA, TCDCA) [54]. Only doubling serum total bile acid concentration was sufficient to increase BBB permeability [54]. Seiffert et al. (2004) exposed the cerebral cortex of rats to bile salts (DCA), which resulted in long-lasting extravasation of serum albumin into the brain. Although this was associated with astrocyte activation, there was no inflammatory response or marked cell loss [55]. Higher CSF levels of albumin have been reported in patients with AD [56]. Increased BBB permeability was suggested for this observation. In another in vivo study, focal administration of deoxycholate resulted in BBB disruption (measured using in-vivo MRI brain imaging) leading to neuronal loss and deterioration of animals’ motor functions [57]. Previous studies have found increased permeability of the BBB in patients with AD compared to age-matched control subjects [58]. The disruption of BBB could lead to microhemorrhages with the release of neurotoxic hemoglobin-derived products [59]. The release of iron from hemoglobin can lead to lipid peroxidation and neuronal cell death [59]. It has been shown that only 1% oxidized DHA was sufficient to revert the protective effect of DHA and significantly increase Aβ production [60]. It has been proposed that the disruption of the BBB would facilitate the migration of inflammatory molecules into the brain leading to microglia activation and neuroinflammation [61]. Conjugated bile acids such as TCA activated Sphingosine 1 Phosphate Receptor 2 receptor, [62] which released CCL2 from neurons, leading to activated microglia [63], and neurological decline [64]. Furthermore, inflammation would increase plasmin system inhibitors, resulting in reduced plasmin activity, which leads to impaired Aβ degradation in the brain and accumulation of Aβ deposits in AD patients [65]. The plasmin system is a set of proteases and inhibitors in the brain expressed by neurons. This system contains urokinase-type plasminogen activator (uPA) and tissue-type plasminogen activator (tPA), which convert plasminogen to plasmin (active protease). In this system tPA and uPA are inhibited by both plasminogen inhibitor-1 (PAI-1) and plasminogen inhibitor-2 (PAI-2) [66, 67], with alpha-2-antiplasmin inhibiting plasmin [66]. Interestingly, PAI-1 levels increased (not significantly) in the CSF of patients with AD compared to control subjects [67]. Therefore, the accumulation of Aβ would be expected to increase in the brains of patients with AD (increased PAI-1 levels).
Fig. 4
The disruption of the blood-brain barrier by bile acids leading to the leakage of serum proteins like albumin into the brain or microhemorrhages.
Hydrophobic bile acids damage other epithelial barriers. DCA is a potent bile acid in decreasing the tight junctions of human bronchial epithelial cells at relatively mild acidic conditions (pH = 6) compared to TCA and glycocholic acid. This could be one of the reasons for pneumonia complications in AD patients [68].
POSSIBLE INCREASED C-27 BILE ACIDS LEVELS IN PLASMA OF PATIENTS WITH AD
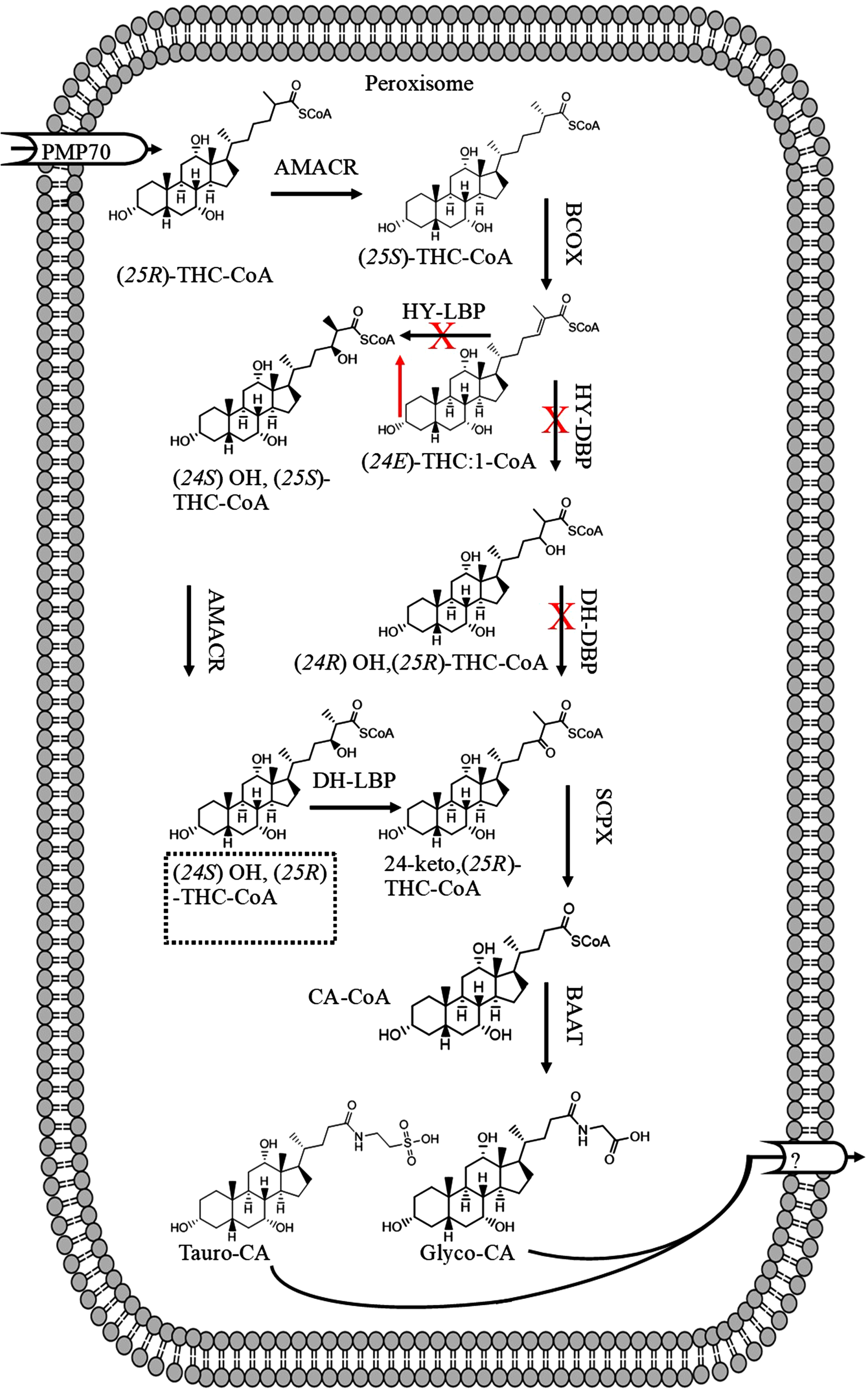
The downregulation of D-BP enzyme in the liver of patients with AD may suggest increased levels of C27 bile acids in the serum or liver (Fig. 5), as, C27 bile acids (derivatives of 3α, 7α, 12α, 24 tetrahydroxy-5β-cholestanoic acid) account for 74% of total bile acid in the serum of patients with D-BP deficiency [69]. C27 bile acids enhance mitochondrial ROS production by inhibiting respiratory chain [70]. However, the plasma levels of 3β-hydroxy-5-cholestenoic acid (C27 bile acid) ranged from 41 ng/ml to 134 ng/ml in 11 healthy subjects [71]. The total median levels of serum or plasma C27 bile acids (3β-hydroxy-5-cholestenoic acid, 3β,7α-dihydroxy-5-cholestenoic acid, 7α-hydroxy-3-oxo-4-cholestenoic acid), 172 ng/ml, was greater than median of C24 unconjugated bile acids (146 ng/ml) in healthy subjects [72]. Certain meals (oat bran) increased serum levels of C4 (7α-hydroxy-4-cholesten-3-one) [73]. C27 bile acids in human serum originate from 7α-hydroxy-cholesterol [74]. The serum levels of C4 also increased in patients with hypercholesterolemia and treated with cholestyramine from 12 ng/ml (heathy subjects) [72, 75] to 174 ng/ml. Gälman et al. (2003) showed that treatment with cholestyramine increased rat liver 7α-hyroxylase activity leading to increased serum levels of C4 [75], and potentially serum C27 bile acid levels [76]. Furthermore, increased liver 7α-hyroxylase activity would also increase serum 7α-hydroxycholesterol levels [76]. These observations indicate that serum levels of C27 bile acids may increase due to diet, treatment, GI or hepatic diseases, and not necessarily due to downregulation of D-BP enzyme in the liver.
Fig. 5
The suggested disruption of bile acid synthesis in patients with Alzheimer’s disease due to downregulation of HY-DBP and DH-DBP leading to accumulation of C-27 bile acids such as (24E) THC:1-CoA.
On the other hand, serum levels of C4 (and potentially C27 bile acid levels) decreased in patients with acute myelogenous leukemia or total parenteral nutrition [77, 78]. The levels of 7α-hydroxylase also significantly increased in patients treated with cholestyramine, but decreased in patients treated with chenodeoxycholic acid (both at the doses of 15 mg/kg) [79]. The mRNA of 7α-hydroxylase cholesterol increased in cholestatic patients, despite a reduction in 7α-cholesterol hydroxylation and bile acid synthesis [80]. These observations suggest that in certain liver diseases the serum levels of C27 bile acid and LCA may increase (derived from CDCA) due to increased serum levels of C4 [72, 81]. As CDCA is the precursor of LCA [82, 83] therefore, in AD patients when blood levels of CDCA are high, blood LCA levels tend to be high [39, 44]. As the prevalence of colorectal cancer decreases in patient with AD [84], then it may be suggested increased serum LCA levels (or potentially C4) would be transient.
On the other hand, when the serum levels of bile acids decrease in patients with AD [42], this could suggest reduction of bile acid synthesis by the liver. Then, in anyway, there would be a change in the existing balance within the intestinal microbiome of patients with AD, which is called dysbiosis. Reduced bile acid synthesis by the liver increases the small intestinal bacterial growth [85, 86], which leads to increased serum TNFα and systemic inflammation [87]. Due to proinflammatory mediators, the production of bile acids by the neutral pathway decreases and then the acid pathway produces more C4 and hence more CDCA [72]. Increased levels of CDCA increases intestinal Enterobacteriaceae, which are pathogenic [88]. The decreased levels of bile acids including CDCA in AD patients [42] would decrease secondary bile acids levels (including LCA) in the intestine produced by clostridum [89], which again leads to outgrowth of Enterobacteriaceae [89]. Then, the intestinal altered microbiome will lead to the formation of toxic bile acids such as DCA [90], which would damage the BBB and build up in the brain [42].
The levels of C4 are in the range of 0-15 ng/ml in healthy volunteers only for 49% of subjects. Although levels of C4 > 60 ng/ml are considered abnormal, 7% of healthy subjects fall in this range [91]. Therefore, the serum levels of C27 bile acids may be much higher in some healthy subjects compared to the others, yet with no obvious clinical symptoms.
In summary, although C27 bile acid levels may increase in the serum or liver of the patients with AD, the large variations in healthy subjects may prevent clearly seeing the increased C27 bile acids in these patients.
BILE ACIDS ENTER NEURONS VIA BILE ACID TRANSPORTERS
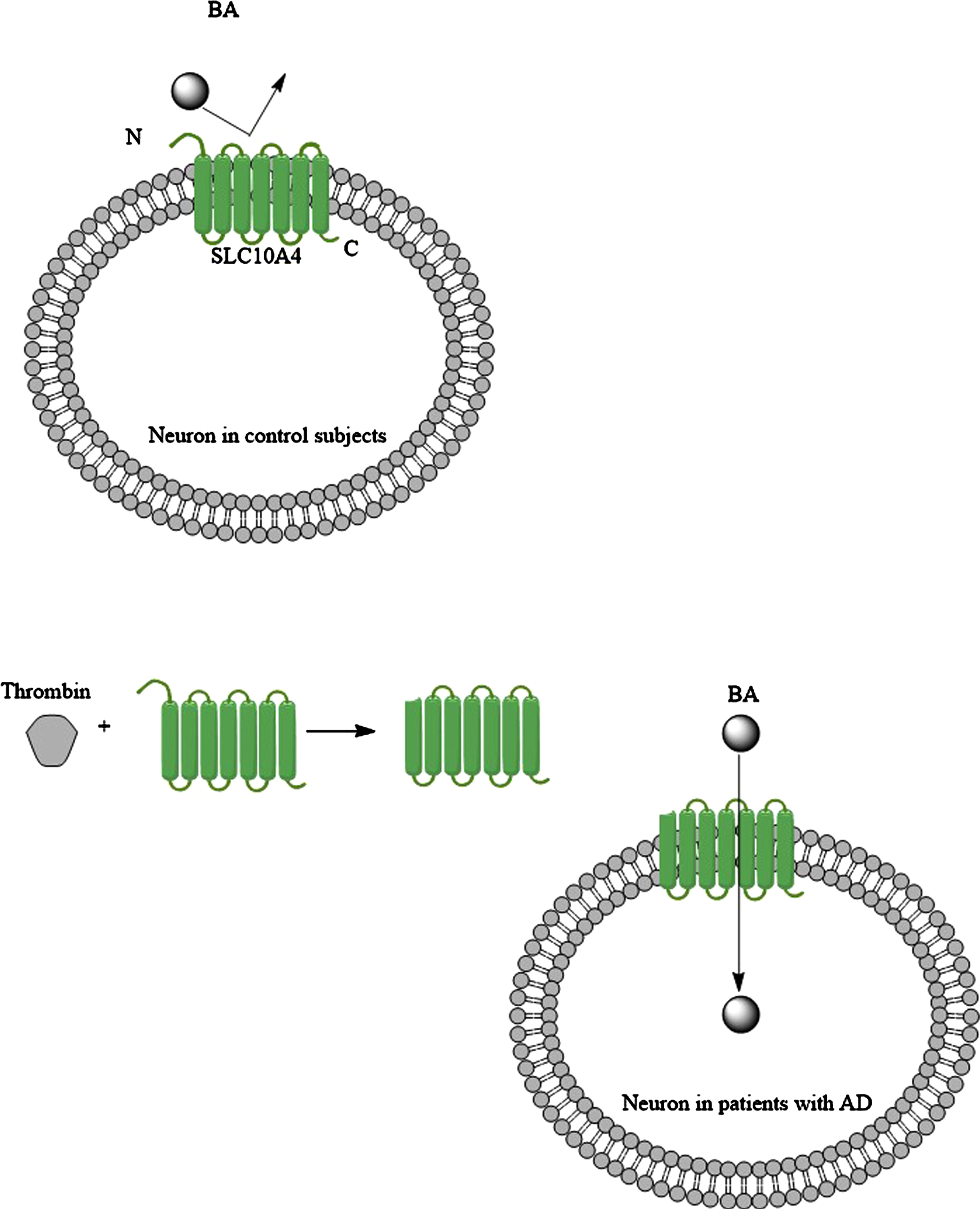
The solute carrier family 10 members 1 (SLC10A1; NTCP) and 2 (SLC10A2; ABST) transporter protein belong to the solute carrier super family with over 450 members [92, 93]. SLC10A1 and SLC10A2 carry bile acids. SLC10A4 is another member that is expressed in the brain [94–96]. It is located in the synaptic vesicles as either vesicular acetylcholinesterase or vesicular monoamine transporter 2 [92]. It has been proposed that SCL10A4 contributes to loading the vesicles in neurons with dopamine, which is released in synaptic clefts [92]. SCL10A4 normally has no bile acid transporter activity, but it can become an active bile acid transporter with the help of thrombin, which cleaves the N-terminus section of SLC10A4 protein (Fig. 6) [97]. Then SLC10A4 can transport LCA and DCA [97]. As thrombin levels are elevated in the brain of patients with AD [98], then this could be a mechanism that LCA finds its way into the neurons. When bile acids are in neurons, they exert their effects through a number of nuclear receptors, including farnesoid X receptor [99], pregnane X receptor [100], vitamin D receptor [101], and estrogen receptor α (discussed below). Furthermore, it has been shown that in cholestatic liver injury the serum bile acid levels increase. Bile acids both uncharged and negatively charged found their ways into neurons via SLC10A2 bile acid transporters, which is expressed in the rat hypothalamus; and suppressed the hypothalamic-pituitary-adrenal axis (HPA) [102]. It should be noted that SLC10A2 is not expressed in the human brain. HPA dysfunction has been reported in a substantial portion of patients with AD [103]. HPA axis dysfunction was correlated to the severity of dementia in patients with AD [103]. On the other hand, by increasing the severity of AD, depletion of SLC10A4 expression was noted in the transentorhinal cortex and hypothalamus of patients with AD [96]. Keitel et al. (2010) showed the expression of TGR5 (a membrane-bound bile acid receptor) on astrocytes and neurons [104], and the activation of TGR5 may promote the migration of bile acid transporter (ASBT) into the cell membrane [105]. However, the expression of TGR5 decreases in the brain by the elevation of serum bile acid levels [104]. This could be a further support for HPA dysfunction in AD patients in the short term, but not longitudinally [103].
Fig. 6
Under normal condition bile acids (BAs) cannot utilize bile acid transporter like SLC104A to enter neurons, but when brain levels of thrombin increase in the patients with AD (for example due to the BBB disruption), then the SLC104A is modified, and this time it carries BAs into the neurons.
THE INTERACTION OF BILE ACIDS WITH FARNESOID X RECEPTOR IN THE BRAIN
Farnesoid X receptor (FXR) protein expression was identified in the human brain abundantly [106]. In conditions when the liver is damaged, serum bile acids are known to increase, possibly due to the release of bile acid content from damaged hepatocytes [107]. When serum bile acids levels increase (for example in cholestasis), brain bile acid levels also increase [108] and toxic bile acids such as LCA tend to accumulate in the brain [108, 109]. This may also result in change of the brain bile acid profile, reduction of good bile acid levels such as CA and CDCA [109]. CDCA is the highest activator of FXR (346 folds), but LCA is the lowest FXR activator causing only 100 folds increase at 100μM [110]. Increased bile acid levels in the brain activated FXR signaling, which downregulated the expression of brain Cyp46A1 (catalyzes the synthesis of 24(S)-hydroxycholesterol) in mice (Fig. 7) [111].
Fig. 7
When bile acids find their ways into the neurons, then they activate FXR and possibly RXR, which collectively reduce the expression of Cyp46A1 leading to accumulation of cholesterol and reduced brain levels of 24(S)-hydroxycholesterol.
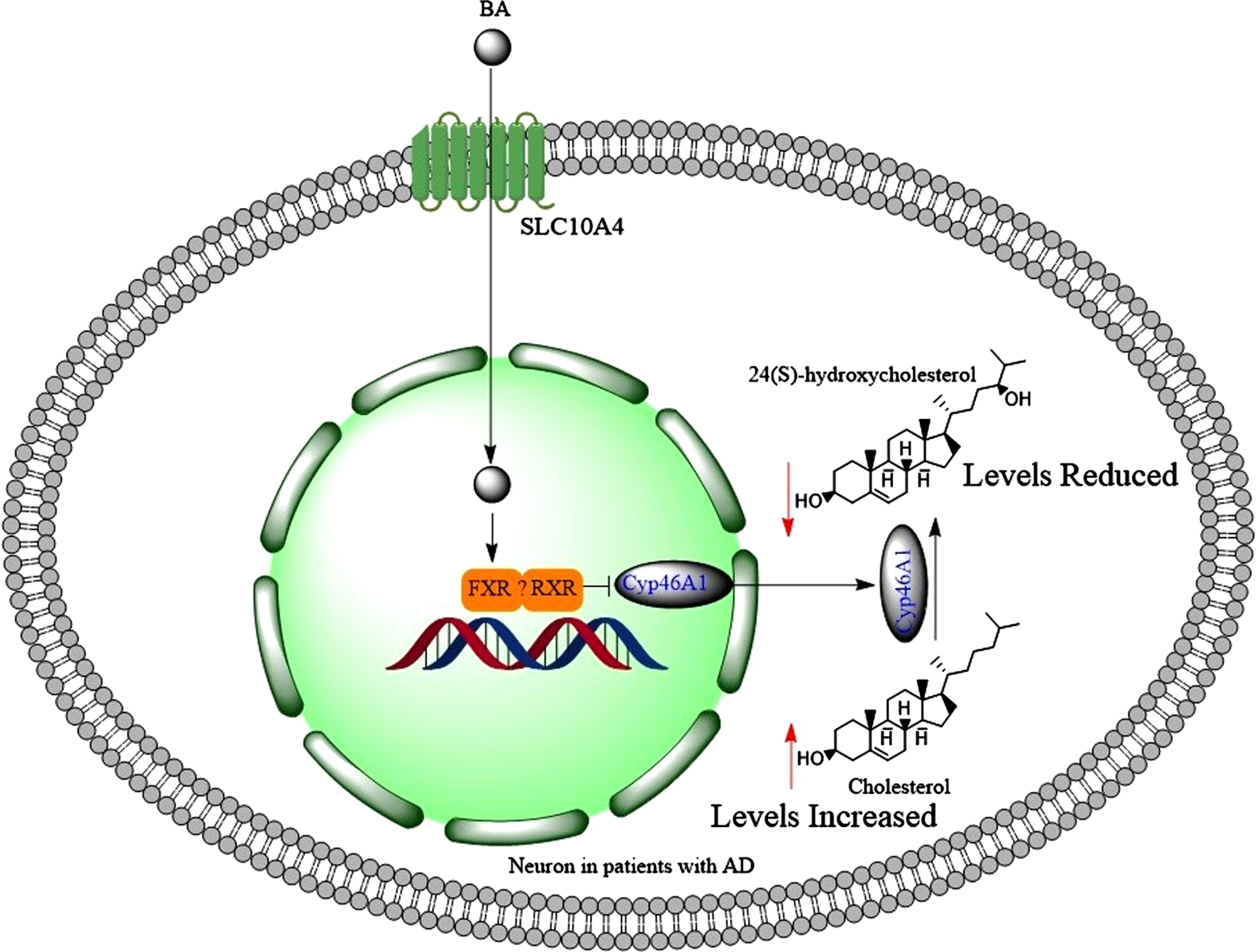
The expression of retinoid xenobiotic receptor (RXR) is reported in hippocampal neurons [112]. Therefore, RXR may heterodimerize with FXR in downregulation of Cyp46A1 (Fig. 7), as it does in the intestine [90]. This could be an explanation for the reduction of good bile acids in the brains of bile duct ligated rats [109]. In addition, the activation of FXR in the brain contributed to neurological decline [99]. Similar to the liver, activation of FXR by GW4064 increased the expression of small heterodimer partner (SHP) protein in brain primary cultured neurons [106]. More importantly, accumulation of bile acids in the brain led to neural cholesterol accumulation and neurological decline such as escape response and presence of server ataxia [111]. Bile acids bind to FXR and this prevents transactivation of LXRα, which is a positive regulator of cholesterol degradation, and LXRα is also expressed in the brain [113]. This means that activating FXR leads to accumulation of cholesterol [110]. In fact, increased total serum cholesterol levels have been observed in patients with non-cirrhotic, non-alcoholic steatohepatitis, when they were treated with 6α-ethyl CDCA (INT-747, [obeticholicacid]) [114]. This is a selective FXR agonist with 100 folds greater efficacy than CDCA [115]. Bile acids (CDCA, DCA, LCA) did not activate FXR significantly at concentrations less than 10μM [110]. The serum total bile acid levels can increase to 71μM in patients with liver disease compared to control subjects who have 9μM total serum bile acid level [116]. Therefore, significant activation of FXR can occur in the brain with serum bile acid levels under pathological conditions. These observations may explain increased levels of cholesteryl esters in the brains of patients with AD [117] through the activation of FXR signaling pathway in the brain. Reducing cortical bile acid levels alleviated accumulation of cholesterol in the brain [111]. Accumulation of cholesterol in the brain promotes the aggregation of Aβ1 - 42 [118], which increases the risk of AD.
There is an involvement of phospholipase A2 (PLA2) in AD (its levels either increase or decrease), and a recent study showed that activation of FXR by CDCA at 50μM concentration decreased phospholipase 2 G12B (PLA2G12B) expression in HepG2 (hepatoma cell line) cells [119]. Therefore, there is a possibility that part of alterations of PLA2 levels in the brains of patients with AD may originate from high toxic bile acid levels in the brain that activate FXR signaling pathway.
Activation of FXR reduced hepatic inflammation (through inhibition of NF-κB) [120]. NF-kβ inhibition also prevented the expression of proinflammatory cytokines from CNS-resident cells [121]. Furthermore, clinical aspects of experimental autoimmune encephalomyelitis (EAE) mice were significantly improved either by the inhibition of NF-kβ [122] or administration of obeticholic acid (a strong FXR agonist at the dose of 5 mg/kg) [123]. These suggest that activation of FXR would reduce neuroinflammation, a condition that has been observed in patients with AD [124]. However, toxic bile acids may contraindicate this benefit by promoting Aβ accumulation in the brain. On the other hand, NF-kβ can be activated by phosphatidylinositol 3-kinase through TNFR2 (TNF receptor 2), which promoted neuronal survival [125]. Therefore, inhibition of NF-kβ byFXR signaling could compromise neuronal survival in AD.
HYDROPHOBIC BILE ACIDS BLOCK NMDA RECEPTORS IN THE HIPPOCAMPAL NEURONS
N-methyl-D-aspartate receptors (NMDARs) play major roles in learning and memory [126–128]. NMDARs are capable of converting specific patterns of neuronal activity into long-term changes in synaptic structure (synaptic plasticity) leading to cognitive functions and learning [129]. NMDARs in the granule cells (GCs) of the dentate gyrus play a crucial role in the process of pattern separation [130]. Furthermore, new neurons are continuously generated in the hippocampus of the adult mammalian brain [131], and GluN2B containing NMDARs promote synaptic activation in adult born GCs of the olfactory bulb that integrate into circuits with high and correlated synaptic activity [132]. Two regions-the olfactory bulb (SVZ) and the dentate gyrus of the hippocampus-(SGZ)- receive and integrate new born neurons throughout the adult life [133]. NMDARs help new neurons to survive and integrate into circuits [131]. Work on rodent hippocampus has shown that NMDAR mediated plasticity (which is essential for the formation of the memory) [134] and NMDARs are crucial for the formation of temporal memory in the CA1 of the hippocampus [134]. Computational studies suggest that dentate gyrus and its connections to CA3 are responsible for pattern separation [135]. If the above pathway does not work, then the connections of CA3 to CA1 should complete the pattern [135]. Pattern completion and pattern separation complement each other. If one weakens, the other one strengthens [135]. There is a proper information encoding, normal pattern completion rate, and pattern recognition in patients with aMCI, but these are forgotten rapidly [135]. In contrast, there is a slow and even incomplete pattern recognition in patients with AD. These suggest that patients with AD have extensive hippocampal and parahippocamal damage, and therefore they cannot encode the information properly [135].
Both under-excitation (hypofunction) and excessive excitation of NMDARs are thought to play roles in AD [126, 127]. NMDAR antagonists impair spatial learning in rats [136] and nonhuman primates [137]. On the other hand, excessive activation of NMDARs (GluN2B containing NMDARs mediated by Aβ) results in deficit uptake of glutamate by excitatory amino acid transporters leading to impaired long term potentiation [126]. Although memantine is an NMDAR antagonist, memantine exerts its therapeutic efficacy by attenuating Aβ-induced tau-phosphorylation and associated signaling mechanisms [138], and it should be noted that memantine is a low-affinity NMDAR antagonist. Strong NMDAR antagonists such as phencyclidine can produce psychotomimetic effects in humans. Hydrophobic bile acids are able to inhibit NMDARs [139]. The affinity of bile acids to albumin determines the potency of a bile acid in blocking NMDARs [139] with LCA being the most potent molecule [140]. Blockade of NMDARs by bile acids would be expected, as pregnanolone sulfate is a non-competitive antagonist of NMDARs and structurally is similar to sulfated LCA. NMDARs are crucial for the formation of temporal memory in the CA1 [134], and the NR2A subunits of NMDARs are responsible for the retrieval of memory [141]. The blocking of NMDARs results in apoptosis of neuroblasts and this decreases the number of newly generated neurons [142] and hence formation of new memories [143]. The restoring of spine densities in the dentate gyrus rescued loss of long-term memories in mouse models of AD [144] and NMDARs are required for the formation of spines [145]. Furthermore, the blockade of NMDAR in retrosplenial cortex disrupts retrieval of remote and recent information [141], decreases synthesis of 17β estradiol in the brain [146], inactivates benefits of E2 via Rap/AF-6/ERK1/2 signaling pathway [147], reduces neural connectivity [148] (dendritic complexity, spine density and morphology), prevents persistent and maturation of newly formed synapses [149], and contributes to NMDAR hypofunction (which has been seen in AD) [150]. Hydrophobic bile acids reduce the levels of 24-OHC in the brain [151], which is a strong activator of NMDARs [152]. As presynaptic NMDARs (PreNMDARs) would enhance transmitter release in part via protein kinase C signaling [153] and subsequent glutamate release in CA3-CA1 circuit [154], then blockade of preNMDARs by toxic bile acids would further contribute to the dysfunction of remaining NMDARs in AD [155]. Blockade of NMDAR with toxic bile acids may lead to psychotic symptoms that has been observed in 50% of patients with AD [156].
Activation of NMDARs leads to the activation of mitogen-activated protein kinase (MAPK) and extracellular receptor kinase-1and 2 (ERK1/2), inducing phosphorylation of cAMP responsive element binding protein (CREB) and increasing expression of brain-derived neurotrophic factor (BDNF). This contributes to memory formation and cognitive function (Fig. 8) [157–159].
Fig. 8
Hydrophobic bile acids such as lithocholic acid block the activation of NMDARs by agonists such as glutamate (Glu); and prevent flux of Ca2 + into neurons leading to downregulation of BDNF, which contributes to the formation of memory and cognitive function. The flux of Ca2 + activates both cAMP-responsive element binding protein mitogen-activated protein kinase/extracellular regulated kinase MAPK/ERK and calcium/calmodulin-dependent protein kinases IIα (CamkIIα) [159] which results in phosphorylation of cAMP-responsive element binding protein (CREB) as well as activation of histone acetyl transferase (HAT). These lead to upregulation of BDNF.
![Hydrophobic bile acids such as lithocholic acid block the activation of NMDARs by agonists such as glutamate (Glu); and prevent flux of Ca2 + into neurons leading to downregulation of BDNF, which contributes to the formation of memory and cognitive function. The flux of Ca2 + activates both cAMP-responsive element binding protein mitogen-activated protein kinase/extracellular regulated kinase MAPK/ERK and calcium/calmodulin-dependent protein kinases IIα (CamkIIα) [159] which results in phosphorylation of cAMP-responsive element binding protein (CREB) as well as activation of histone acetyl transferase (HAT). These lead to upregulation of BDNF.](https://content.iospress.com:443/media/adr/2023/7-1/adr-7-1-adr220071/adr-7-adr220071-g008.jpg)
HYDROPHOBIC BILE ACIDS COULD IMPAIR THE ROLES OF E2 IN THE BRAIN
17β estradiol (E2) is a steroid hormone with molecular weight of 272 Da which is biosynthesized from cholesterol involving aromatase [160]. Hippocampal pyramidal neurons and granule neurons of adult male rats are equipped with a complete machinery for the synthesis of E2 from endogenous cholesterol [161]. Furthermore, brain has the capacity to convert circulatory testosterone into E2 through the enzymatic action of aromatase [162]. E2 plays major roles in the brain such as reducing neuronal loss following stroke, increasing neuronal connectivity, improving cognitive performance, inducing brain BDNF synthesis [163–166], improving learning in ovariectomized (OVX) mice [167], and contributing to leaning by increasing hippocampal dendritic spines [168-170]. E2 exerts its effects in the brain through estrogen receptor α (ERα), ERβ, and GPER1 [171]. ERα facilitated cognitive functions [172], contributed to the BBB functions [173], reduced Aβ accumulation in the brain [174], reduced tau phosphorylation by phosphorylating GSK3β [175], reduced BCL-associated death promoter (BAD) levels [176], and generated optimal signals of E2 through proline-, glutamic acid-, and leucine-rich protein-1 (PELP1) [177]. Furthermore, E2 interacts with GSK3β and β-catenin in the brain via ERα, which is crucial in ERα protein stabilization and turnover [178]. E2 mediates neuroprotection [179] via activation of plasma membrane ERα [180, 181], ERβ [182], GPER1 (GPR30) [183], extranuclear ER [184], via PELP1 interaction with GSK3β [185], and inhibition of NRLP3 inflammasome pathway activation [186]. E2 also improved episodic memory [187], LTP [188], and neurogenesis [189], through NMDARs. Spine density significantly increased with initial E2 treatment followed by NMDAR activation [147].
The estrogen receptor is unique in having a glutamate (Glu-353) to accept the hydrogen bond donated by the estrogenic 3-hydroxyl group [190]. The 17β-hydroxyl group of E2 makes a single hydrogen bond with the δ nitrogen of His-524 in hERα [191]. Most of ERα ligands contain 3-hydroxyl group [192], similar to LCA. However, LCA contains 19-methyl group, which makes LCA a weak partial agonist, and could displace more potent ligands from their binding sites [193]. Our molecular modelling studies suggests that LCA can bind to ERα receptors (Fig. 9), but possibly void of physiological actions (the molecular modelling investigations are exclusive to this review paper and were performed in Autodock vina version 1.1.2 [194, 195]). This is because upon binding of LCA with ERα, the helix 12 of ERα cannot cover as a lid over the ligand-binding pocket to secure the ligand in position. Hence, having ERα occupied by less potent ligands, this would reduce the efficacy of E2 to activate desired signaling pathways. Our results may be supported by previous observations about trans-repression of bile salt export pump (BSEP) mediated by E2 through ERα [196]. CDCA strongly transactivated BSEP expression and E2 repressed this activity. However, E2 efficacy was much lower in the presence of LCA [196]. Therefore, it may be interpreted that LCA reduced the efficacy of E2, through occupying ERα and preventing E2 to fully engage with ERα. Furthermore, LCA has been considered as a ligand which has similar lipophilicity to estrogen and possibly with the ability to interact with estrogen receptors such as ERα, but devoid of metabolic activity, which estrogen is able to exert [197].
Fig. 9
The molecular modelling of the interaction of estrogen receptor-α (ERα) with lithocholic acid. Lithocholic acid has a very similar shape to a known antagonist (compound 9 in reference [398], the molecular structure of the compound is given in Supplementary Figure 3) and down regulator of ERα which adopts a broadly L-shaped pose in the ERα structure with PDB code 5t92 and drapes a carboxylate at the exterior (solvent) of the protein.
![The molecular modelling of the interaction of estrogen receptor-α (ERα) with lithocholic acid. Lithocholic acid has a very similar shape to a known antagonist (compound 9 in reference [398], the molecular structure of the compound is given in Supplementary Figure 3) and down regulator of ERα which adopts a broadly L-shaped pose in the ERα structure with PDB code 5t92 and drapes a carboxylate at the exterior (solvent) of the protein.](https://content.iospress.com:443/media/adr/2023/7-1/adr-7-1-adr220071/adr-7-adr220071-g009.jpg)
Leu-387 in ER will act like a barrier from complete fitting of LCA to the ER ligand-binding pocket. E2 acts through ERα to acutely suppress GABA release [198]. The interference of LCA with E2 roles may explain the disturbed balance of excitatory and inhibitory signaling systems observed in AD brains [199]. LCA inhibited NLRP3 inflammasome activation [200], similar to E2. LCA does not activate ERβ [201, 202]. Furthermore, LCA reduced significantly the expression of ERα in MCF-7 cells [203], and the reduction of cytoplasmic ERα expression has been reported in the brains of patients with AD compared to control subjects [204].
The overall level of ERα does not significantly change in AD brains compared to control subjects, although there is a slight decrease compared to control subjects [205]. Therefore LCA could reduce the number of accessible ERα and diminish working memory performance [206]. Furthermore, the percentage of neurons expressing ER-mRNA decreases with age after 65 years [207], and there is a relationship between MMSE and levels of wild-type nuclear fraction ERα in superior frontal cortex of patients with AD [208].
The neuroprotection exerted by IGF-I also depends on estrogen receptors [209]. Although the levels of IGF-I increase in the CSF of patients with AD [210], the neuroprotective effects of IGF-1 are not seen. This might be explained by the blockade of ERα by LCA, and IGF-I requires involvement of ERα for its roles. Furthermore, BAD levels increase in the brains of patients with AD [211], along increased activity of GSK3β [212]. In conclusion, the above evidence suggests that LCA may interfere with the vast benefits of E2 in the brain via ERα receptor.
LITHOCHOLIC ACID AND THE FUNCTIONS OF ABCA1 AND P-GLYCOPROTEIN IN REDUCING BRAIN Aβ LEVELS
ATP-binding cassette (ABC) transporters utilize ATP to transport their substrates across membranes. ABCA1 (also known as CERP) is a member of ABC transporters that is expressed on the human BBB endothelial cells [213–215] (both luminal and abluminal sides in porcine [216]) and human astrocytes, pericytes and microglia [217]. There is a direct correlation between ABCA1 expression and lipidation of apoE in the CNS, indicating that ABCA1 transports cholesterol on to apoE [218] and interestingly, ABCA1 is required for normal levels of apoE in the brain [219]. The ABCA1 indirectly eliminates Aβ1 - 40 from the brain into the blood circulation through the BBB [220]. The ABCA1 of microglia cells is required for efflux of cholesterol to apoA-I, apoE2, and apoE3 [221], a major contributing pathways for regulating lipid homeostasis in the brain. As expected, the inhibition of ABCA1 resulted in the accumulation of cholesterol in the brain [222]. Furthermore, ABCA1 reduced neuroinflammation and neuronal death [223].
Lipidated apoE by ABCA1 promoted degradation of soluble Aβ both inside microglia and extracellularly [218, 224], and ABCA1 (even non-functional mutants) reduced Aβ production [225]. Astrocytes are the major source of extracellular apoE in the CNS and themselves express a high level of ABCA1, which plays a crucial role in extracellular lipidated apoE [226], facilitating degradation of soluble Aβ by insulin degrading enzyme [224], the main proteolytic degradation pathway of Aβ [227]. It should be noted that there are three major isoforms of apoE (apoE2, apoE3, apoE4), with lipidated apoE3 presenting 2-3 fold higher affinity for both Aβ1 - 40 and Aβ1 - 42 peptides than apoE4 [228]. On the other hand, delipidation of both apoE3 and apoE4 decreased their affinities for Aβ peptides by 5-10-fold and abolished the isoform-specificity [228]. In fact, overexpressing ABCA1 increased lipidation of apoE in the CNS and reduced amyloid burden [229]. Conversely, the absence of ABCA1 resulted in a significant increase in Aβ1 - 40 load in the brain of ABCA1-/- mice compared to the wild type [230]. It has been shown that increased brain ABCA1 levels by oral administration of rosiglitazone improved Aβ clearance from the brains of AD rat models [231]. ABCA1 is upregulated in AD hippocampal neurons two- to three-fold, potentially through Aβ mediated pathways [232]. The increased levels of ABCA1 in the brains of patients with AD may be a compensatory mechanism to reduce the brain Aβ load [233]. Overexpressing ABCA1 in APP/PS1 mice reduced brain Aβ burden [234]. It should be noted that environmental factors (such as dichlorodiphenyltrichloroethane) could increase brain Aβ levels by lowering ABCA1 expression and promoting Aβ synthesis [235].
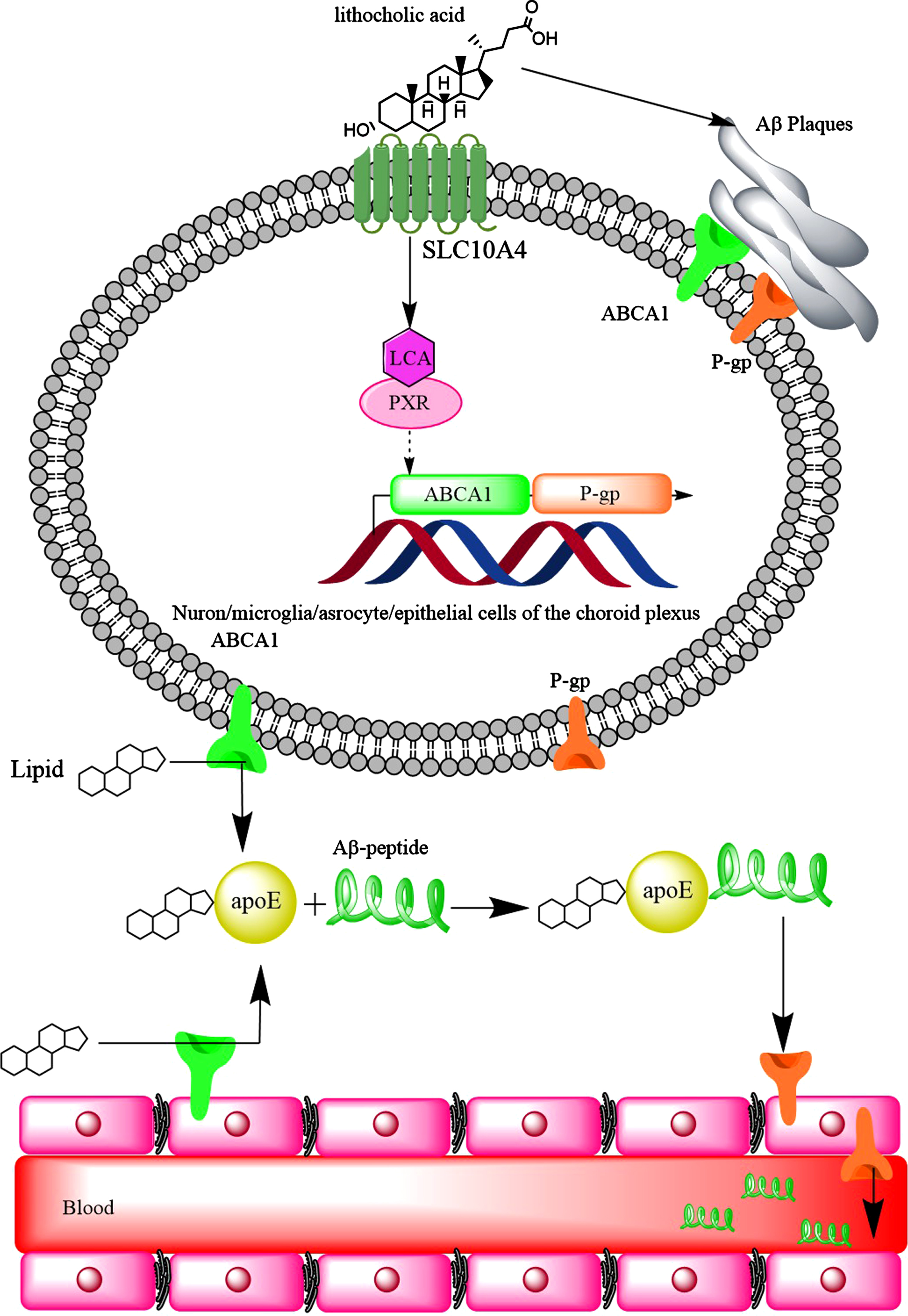
LCA reduced the expression of ABCA1 in HepG2 cells through the activation of nuclear pregnane X receptor (PXR) [100]. In fact, PXR serves as a physiological sensor of LCA [236]. Activation of PXR increased ABCA1 expression in kidneys [237]. Activation of PXR in the brain increased the expression of P-glycoprotein (P-gp) [238] and significantly reduced brain Aβ levels in a mouse model of AD. Therefore, it would be expected that P-gp expression is increased as the result of PXR activation by LCA. At the early stage of AD, P-gp expression is upregulated in the capillaries of the brain. However, the accumulation of Aβ completely disappeared the expression of P-gp on arterioles or reduced P-gp capillary expression [239, 240]. A compensatory mechanism may be suggested to increase Aβ clearance from the brain by upregulation of P-gp [239], high serum LCA levels may also contribute to increased P-gp expression via PXR activation. Furthermore, it was found that activation of PXR (by allopregnanolone or T0901317 [241]) led to increased expression of ABCA1 in mouse brain [242]. Therefore, the increased expression of ABCA1 in the brain of patients with AD could be due to Aβ-mediated pathway, as well as increased serum LCA levels. It appears that LCA may provide some neuroprotective effects against AD. However, this may lead to increased brain Aβ-levels, and contraindicates the neuroprotective effects (Fig. 10).
Fig. 10
The hydrophobic bile acids such as lithocholic acid increase the expression of ABCA1 transporter and P-gp. Although upregulation of these proteins should lead to better clearance of Aβ plaques from the brain of AD patients, the hydrophobic bile acids promote the formation of Aβ plaques which themselves make ABCA1 and P-gp out of function.
TOXIC BILE ACIDS AFFECT 24-OHC AND BILE ACID PROFILES IN THE BRAIN
Cholesterol 24-hydroxylase (CYP46A1) is highly expressed in the brain and catalyzes the synthesis of 24(S)-hydroxycholesterol (24-OHC) from cholesterol (Fig. 11) [243]. CYP46A1 is not uniformly distributed in the human brain, but it has been detected in the hippocampus [244]. The formation of 24-OHC is a major pathway for removal and excretion of cholesterol from the brain [245]. The net flux of 24-OHC is 6.4 mg/day to the blood circulation for human brain [246]. The brain contains about 80% of 24-OHC in the body [247], therefore, 24-OHC is also called cerebrosterol [248]. The net flux of 24-OHC remains age independent for adults older than 20 years [247]. As well as net flux of 24-OHC to the blood circulation and conversion to bile acids by the liver, brain has all the enzymes to biosynthesis for CDCA from 24-OHC [249].
Fig. 11
Hydrophobic bile acids (BAs) reduce brain levels of 24(S)-hydroxycholesterol, through downregulation of Cyp46A1. 24(S)-hydroxycholesterol increases the expression of ABCA1 transporter in astrocytes through activation of LXR-RXR signalling pathway, then reduction of 24(S)-hydroxycholesterol in the brain may contribute to downregulation of ABCA1 in astrocytes.
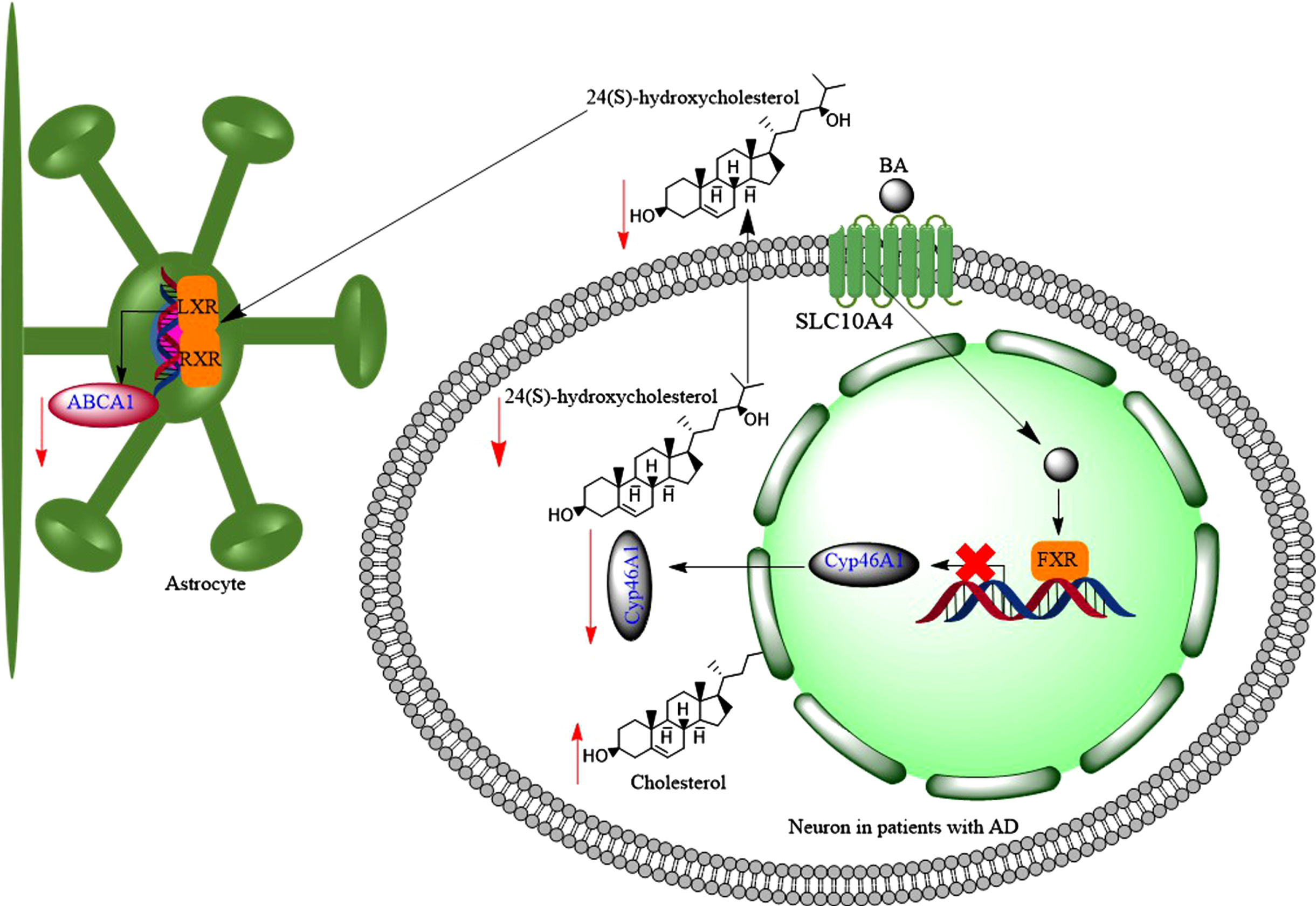
24-OHC induced transcription and protein synthesis in a dose dependent manner in astrocytes, but not in SH-SY5Y cells (neuronal origin) cells [250]. This led to increased efflux of cholesterol via apo-E and apoA-I [250]. Following a traumatic brain injury, there is an increased efflux of cholesterol from damaged cell membrane to the brain environment; and as a result the activity of CYP46A1 increases to convert excess insoluble cholesterol to soluble form of 24-OHC [251]. As well as trauma, inflammation and necrosis are associated with increased cholesterol production in the brain, which leads to increased amounts of 24-OHC in the brain [252]. 24-OHC is an LXR agonist, and consequently can increase the expression of ABCA1 [253], contributing to reduction of AβPP cleavage and Aβ production, i.e., potentially reducing the risk of developing AD [254]. Furthermore, 24-OHC has positive modulatory effects on NMDARs [152] promoting LTP and cognitive functions [152]. In fact, 24-OHC is a very potent, direct, and selective positive allosteric modulator of NMDARs with a mechanism that does not overlap with other allosteric modulators such cholesterol or 27-hydroxycholesterol [152]. Finally, under normal conditions, 24-OHC favors the processing of AβPP to the non-amyloidogenic pathway [255].
The serum levels of 24-OHC is a balance between the brain production of 24-OHC and its metabolism by the liver [256]. Then higher serum 24-OHC level would be expected in patients with liver disease compared to controlled subjects [257]. Serum levels of 24-OHC varied between 1.7-18μmol/L in patients with severe cholestatic liver disease (equivalent to 685 ng/mL-7,236 ng/mL, Mw = 402 g for 24-OHC) [258], while normal 24-OHC levels are in the range of 30-80 ng/mL [259]. Therefore, increased serum levels of 24-OHC in patients with liver disease could be due to the inability of the liver to convert it to bile acids such as CDCA [260]. On the other hand, the plasma or serum levels of 24-OHC were significantly lower in patients with AD (20-50 ng/mL) than matched control subjects (30-80 ng/mL) [259, 261]. The reduced plasma levels of 24-OHC could suggest the loss of CYP46A1 in the brain [262]. Then expectedly, variation in CYP46A1 gene may act as a risk factor for AD through influence on cholesterol metabolism in the brain [263]. It was found that the number of neurons and astrocytes expressing CYP46A1 decreased in the cortex of patients with AD compared to control subjects [264, 265]. Interestingly, the remaining CYP46A1 enzymes were accumulated around Aβ plaques [264].
Can elevated serum bile acid levels suppress CYP46A1 expression in the brain, and hence reduce 24-OHC levels in the brain? In vivo studies showed that mice with acute liver failure, which was induced by azoxymethane, developed increased serum bile acid levels [266]. Toxic bile acids such as LCA and DCA reduced the synthesis of 24-OHC and expression of CYP46A1 in the brain via activation of FXR and SHP [151] (Fig. 11). Therefore, decreased expression of CYP46A1 in the brains of patients with AD could be due to the increased serum bile acid levels. This is in line with reports that CSF [267] and brain levels of 24-OHC decrease as the severity of AD progresses [268]. Although, concentrations of 24-OHC in the CSF were either significantly higher in patients with AD (2.28±0.73 ng/mL at the early stages, or 2.03±0.36 ng/mL at the later stage) [269], or slightly higher than control subjects [270], higher early CSF 24-OHC levels in patients with AD could be due to the increased neurodegeneration [248]. Normal CSF 24-OHC concentration is 1.5 ng/mL [249]. Furthermore, it was confirmed that the reduction of 24-OHC was accompanied by the reduction in the expression of CYP46A1 in postmortem human AD brains [268]. Although, there has not been a report on the CSF levels of LCA in patients with AD and at the same time for 24-OHC levels, the CSF levels of GLCA and TLCA increased in patients with AD [271].
Therefore, it may be speculated that at the early stages of the AD, increased rate of neuron breakdown produces large amounts of 24-OHC in the brain and as a result CSF levels of 24-OHC increase. However, the negative feedback of serum high secondary bile acid levels such as LCA reduce the expression of CYP46A1 in the brain and hence CSF levels of 24-OHC decrease at the later stages of the AD.
THE INTERACTION OF HYDROPHOBIC BILE ACIDS WITH TGR5 RECEPTORS
Takeda G protein receptor 5 (TGR5) is a cell-surface G protein coupled receptor, which is responsive to bile acids [272]. TGR5 is expressed in gastrointestinal tract, on immune cell membranes, neurons and astrocytes [273]. TGR5 plasma membrane localization and responsiveness to extracellular ligands depends on a long (>9 residues) α-helical stretch at the C terminus [274]. Bile acids activate TGR5 with different potencies and in the rank order of LCA > DCA>CDCA>CA [275]. Among all bile acids, TLCA is the most potent agonist of TGR5 [276]. TLCA has a 3-hydroxyl group which forms hydrogen bond with Y240 of TGR5. This is essential for the high activity of TLCA [277]. Bile acids interact in different modes with TGR5 leading to different cellular responses [277]. For example, tauroursodeoxycholic acid (TUDCA) with a 7-hydroxyl (as well as 3-hydroxyl group) forms a hydrogen bond with N933.33 of TGR5 [277]. While LCA with 3-hydroxyl group (lack of 7-hydroxyl group) forms a hydrogen bond with Y893.29 or N933.33 (depending on the head-to-tail, or tail-to-head pose in the interaction site) of TGR5 [278]. Alternatively, toxic bile acids may transactivate other receptors, such as DC (deoxycholate) transactivating epidermal growth factor receptor (EGFR)-ERK1/2 signaling pathway via TGR5 [279], leading to different responses.
What are physiological roles of TGR5 in the brain?
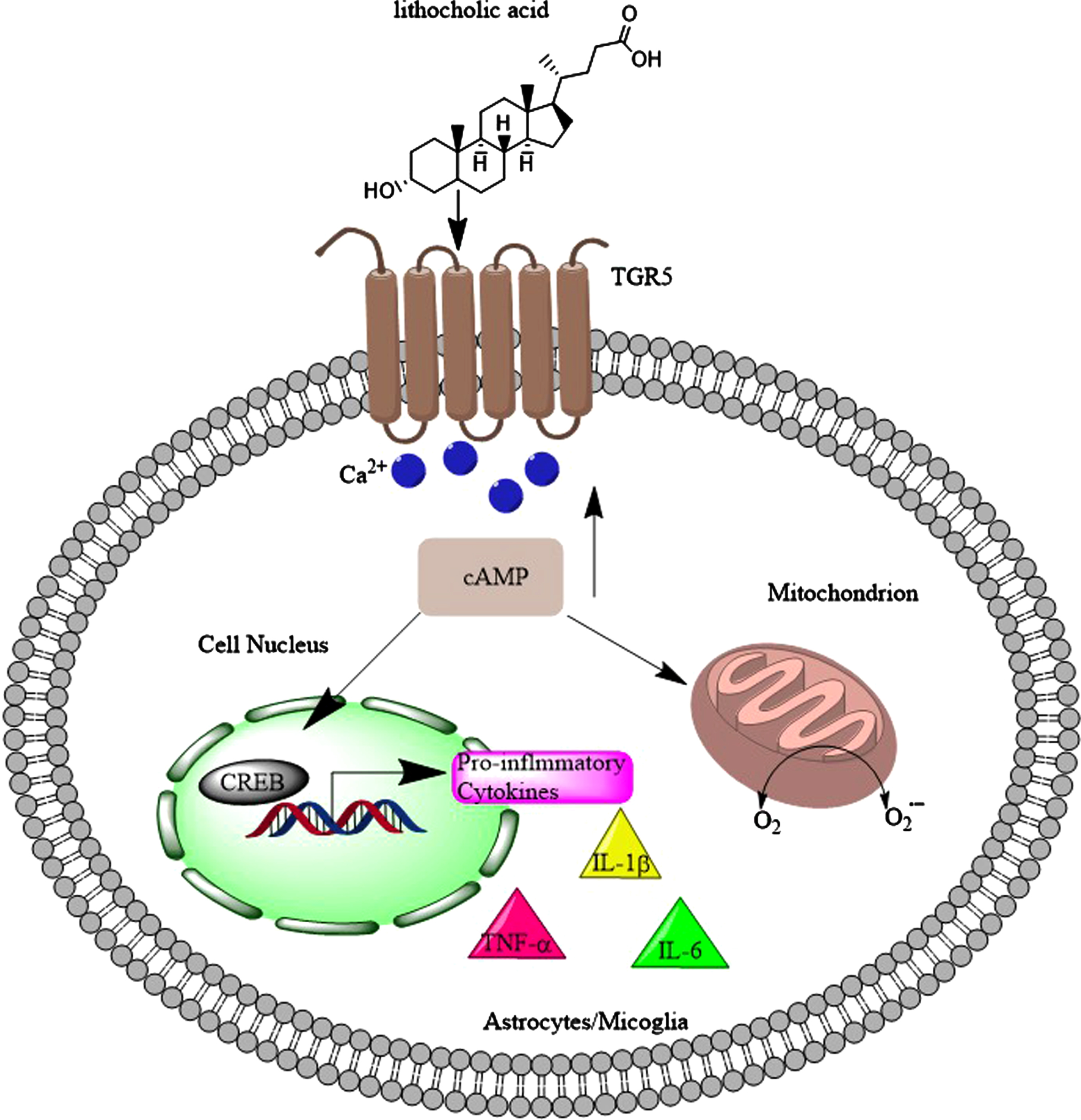
There is a body of evidence that activation of TGR5 by ligands (including bile acids) would reduce inflammatory, induce anti-inflammatory markers [280–286], reduced the progression of the disease in amyotrophic lateral sclerosis [287], and significantly decreased the levels of TNF-α, IL-1β and IL-6, following injection of Aβ1 - 42 into the brain [288]. On the other hand, TGR5 stimulation by 5β-pregnan-3α-ol-20-one and 5β-pregnan-3α-17α-21-triol-20-one (both structurally similar to LCA with 3-hydroxyl groups) are coupled to the elevation of intracellular Ca2 + and the generation of reactive oxygen species (Fig. 12) in astrocytes and neurons [104]. Furthermore, microglia cell lines showed slightly reduced inflammation following TGR5 activation by betulinic acid (a 3-hydroxyl group bile acid, but with two methyl groups at position 4, Supplementary Figure 3) in the range of 10μM to 1 mM [286].
Fig. 12
Toxic bile acids such as lithocholic acid activate TGR5 in microglia and astrocytes which leads to the flux of Ca2 + into these cells. As a result, the levels of cyclic adenosine monophosphate (cAMP) increase, which not only increases the reactive oxygen species (ROS) levels, but also leads to activation of CREB and production of pro-inflammatory cytokines (TNFα, IL-1β, and IL-6).
Bile acids may promote neurological functions through TGR5 activation. TUDCA improved neurological functions through TGR5/SIRT3 signaling pathway after subarachnoid hemorrhage (SAH) [289]. Also, INT-777 (Supplementary Figure 3) administration significantly decreased NLRP3-ASC inflammasome activation in microglia, reduced brain edema and neuroinflammation, leading to improved short-term neurobehavioral functions at 24 h after SAH [290], and significantly improved Aβ1–42-induced cognitive impairment (administered i.c.v.) [288]. Activating TGR5 by INT-777 reduced BBB permeability and improved neurological functions through the BRCA1/Sirt1 signaling pathway after middle cerebral artery occlusion [291]. The activation of TGR5 with INT-777 significantly improved the short-term and long-term neurological deficits, accompanied by reducing the oxidative stress and neuronal apoptosis at 24 h after SAH [292]. Oleanolic acid is a TGR5 agonist [293] and administration of oleanolic acid to EAE mice reduced the severity of the disease by reduction of the BBB leakage and lowering infiltration of inflammatory cells into the CNS [294]. TGR5 mRNA was downregulated in the cerebral cortex of cirrhotic patients dying with hepatic encephalopathy when compared with the brains from non-cirrhotic control subjects [104], perhaps due to increased serum bile acid levels in these patients [295]. Also, western diet increased brain levels of DCA, which was followed by reduced expression of TGR5 in the brain [296]. In addition, ammonia [104] downregulated the expression of TGR5 in the brain, and the brain levels of ammonia significantly increase in patients with AD [297]. Therefore, toxic metabolites such as bile acids may prevents the benefits of other bile acids by reducing TGR5 expression.
Does TGR5 play a role in AD?
Aβ1 - 42 in the brain downregulated the expression of TGR5 in the hippocampus [288]. Bile acids activate TGR5 on sensory nerves, stimulating the release of neuropeptides in the spinal cord that transmit itch and analgesia [298]. Patients with AD [299] or dementia [300] experience pruritus.
All the above observations lead to the conclusion that certain TGR5 signaling by bile acids such as TUDCA and INT-777 is neuroprotective and diminishes inflammation. However, TGR5 expression is downregulated in the brains of patients with AD, and toxic bile acids may contribution.
HYDROPHOBIC BILE ACIDS MAY IMPAIR SONIC HEDGEHOG SIGNALING PATHWAY IN THE BRAIN
Sonic hedgehog (SHH) is a mammalian glycoprotein involved in both during embryonic development and also post-embryonic tissue regeneration and repair [301]. The SHH expression increases by age in the cortex, hippocampus, and cerebellum regions of rat brains [302]. In addition, SHH signaling regulates the self-renewal of neuronal stems cell in the subventricular zone of adult brains [303] that continuously supply new neurons [304]. SHH is associated with cholesterol rich raft-like microdomains [305]. Lipid rafts represent discontinuous regions of the plasma membrane that form functional microdomains [306], which are enriched sterol, sphingolipids, and glycosylphosphatidylinositol-linked proteins (Fig. 13A) [307]. The SHH signaling pathway was reduced in 3xTgAD animals [306]. Inhibitors of γ-secretase restore SHH signaling, which are currently envisaged as tools for the cure of AD, because they lower Aβ levels [308]. The brains of AD patients show a specific down-regulation of seladin-1, a protein involved in cholesterol synthesis, and low membrane cholesterol was observed in hippocampal membranes of ApoE4 (apolipoprotein E4) AD cases [309]. As explained in the above, cholesterol is required for the activity of SHH protein. The expression of PTCH1 was almost missing in the brains of patients with AD, as well as decrease in Ptch2 [310]. Interestingly, Aβ1 - 42 decreased brain Ptch-Gli1 levels, while it elevated SHH expression [310]. Increased levels of SHH expression were found in the hippocampus of APP23 mice and AD patients [310, 311]. These observations indicate that although SHH levels increase in the brains of patients with AD, the other components for full SHH signaling are not available. The early and progressive cholinergic deficiency of basal forebrain cholinergic neurons (BFCNs), characterized by a reduction in acetylcholine synthesis, substantially contributes to the gradual cognitive decline of AD patients [312]. Work by Yue et al. (2015) showed the important role of SHH in derivation of embryonic stem cells to BFCNs [312]. This could highlight that lack of functional SHH in the brain of patients with AD, which may prevent derivation of endogenous stem cells in the brain to BFCNs.
Fig. 13
The possible interference of bile acids (BAs) with the sonic hedgehog (SHH) signaling. A) Under normal condition, SHH protein interacts with PTCH1, which inhibits smoothened protein (SMO). Upon the activation by the SHH, the SMO forms a complex with oxysterols like 24(S)-hydroxycholesterol and also is conjugated with cholesterol which leads to movement in the lipid raft and activation of Gli family proteins (Gli1, Gli2, Gli3). Initially the Gli proteins are in complex with SUFU protein, but upon interaction with SMO protein, SUFU is separated from the Gli family proteins. Then the released Gli family proteins are translocated to the nucleus, where over express SHH target genes. B) BAs may interfere with SHH signaling by preventing activation of SMO by oxysterols as well as modifications of lipid rafts which may prevent activation of Gli family proteins.
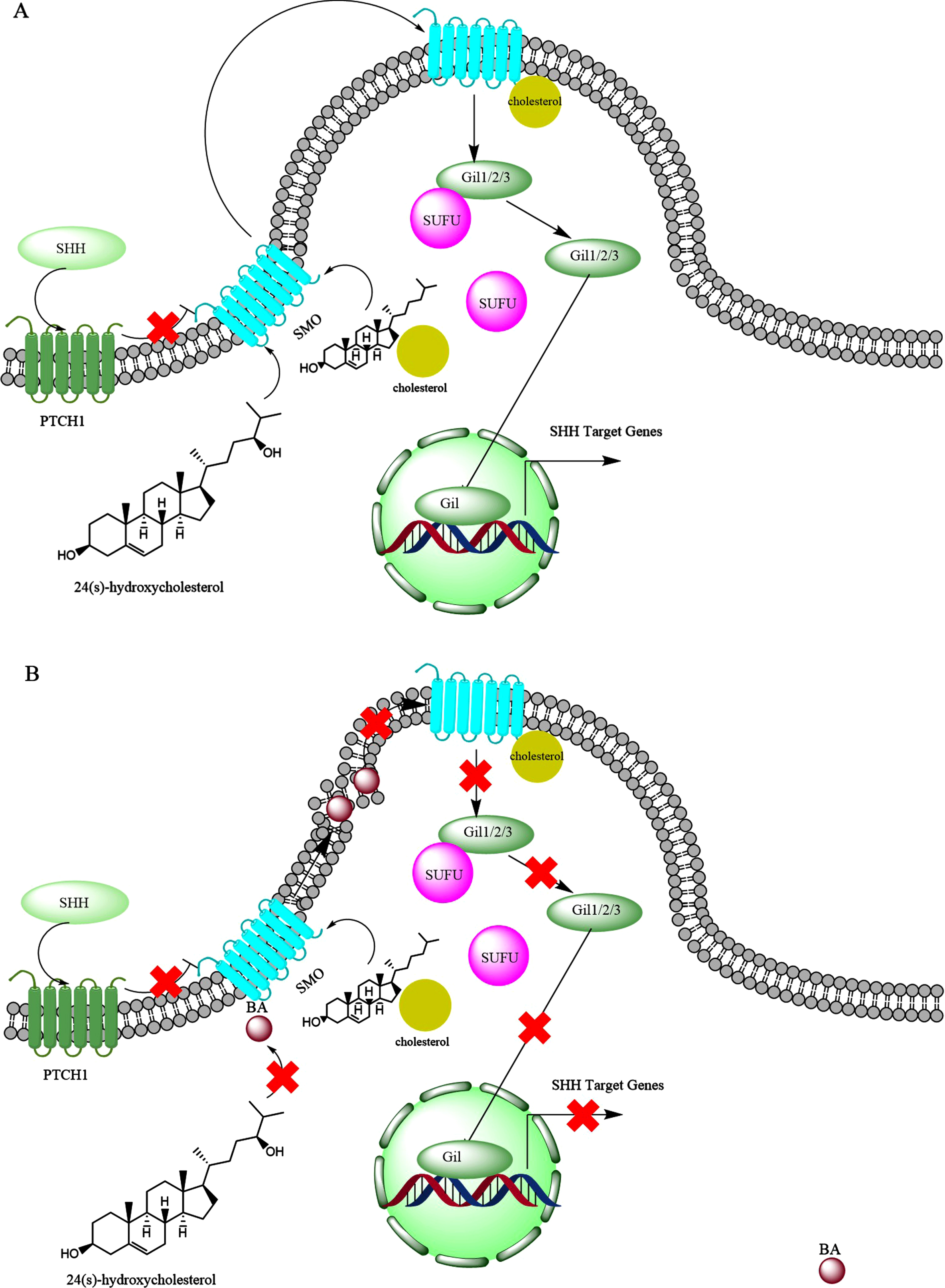
In the brain, the presence of ROS leads to autoxidation of cholesterol and production of 7-keto-cholesterol [313, 314]. CYP27A1(in the brain) [315] produces 7-keto-27-OHC from 7-keto-cholesterol, and 7-keto-27-OHC binds cysteine-rich domain of SMO activating SHH [316]. Interestingly, the levels of 7-keto-cholesterol significantly increase in the brain of patients with AD [268], suggesting activation of SHH signaling pathway in AD. However, it was shown that Aβ1 - 42 interrupted canonical SHH signal transduction, leading to distorted primary cilia structure, which are found in hippocampal neurons [317]. On the other hand, reducing SHH activity by protease nexin-1 helped improving memory functions of AD transgenic mice (APP/PA1) [318]. Meanwhile, facilitating the expression (over expressing) of SHH contributes to neurogenesis [319]. It has been suggested that the deregulation of PTCH1-Gli1 signaling in AD leads to the loss of NSCs and glial precursor cells and consequently decline in cognitive function [320].
The above controversial observations may be explained by the toxic bile acid interventions. Toxic bile acids like DCA preferentially interact with disordered membranes and makes them more disordered [321]. The hypothesis that changes in the lipid composition of rafts contribute to AD pathology has gained considerable support [306]. It was found that cortical lipid rafts are profoundly affected in the 3xTgAD mice [306]. Therefore, toxic bile acids might interfere with SHH signaling, by imposing perturbations into plasma membrane lipid rafts (Fig. 13B).
LITHOCHOLIC ACID MAY PROMOTE THE FORMATION OF Aβ PLAQUES
Amyloid formation by Aβ1 - 40, Aβ26 - 39, and Aβ26 - 40 can be nucleated-“seeded”-, by peptides which hold the critical C-terminal residues including Aβ1 - 42, Aβ26 - 42, Aβ26 - 43, and Aβ4 - 42 [322]. The key amino acids are isoleucine and alanine [322]. However, both Aβ1 - 42 and Aβ1 - 40 formed filaments [323]. Importantly, Aβ1 - 42 accelerated the nucleation of Aβ1 - 40 [324] with Aβ trimer sizes of 20 nm [325]. Aβ1 - 42 promoted Aβ deposition, but Aβ1 - 40 inhibited it [326–328]. Furthermore, Aβ aggregates induced formation of more Aβ aggregates [329]. Other metabolites or compounds also can affect the formation of Aβ aggregates such as retinola [330], vitamin D binding protein (vDBP) [331], vitamin D2 (V-D2) [332], and vitamin D (vitamin D3) [333]. Our molecular modelling studies also suggested that LCA forms a complex with Aβ1 - 40 similar to V-D2, hence, LCA may also promote the aggregation of Aβ1 - 40. Figure 14 represents the docking performed between Aβ1 - 40 taken from NMR structure with PDB code 1iyt and vitamin D2 or LCA. The docking was performed in Autodock vina version 1.1.2. and is analogous to that performed previously, the chain selected from docking was that labelled 2, which was found to resemble most closely that used previously [194, 195, 332].
Fig. 14
Molecular modelling simulation of docking between LCA (grey sticks), vitamin D2 (cyan lines) and Aβ1 - 40, demonstrating possible promotion of Aβ plaque formation by LCA.
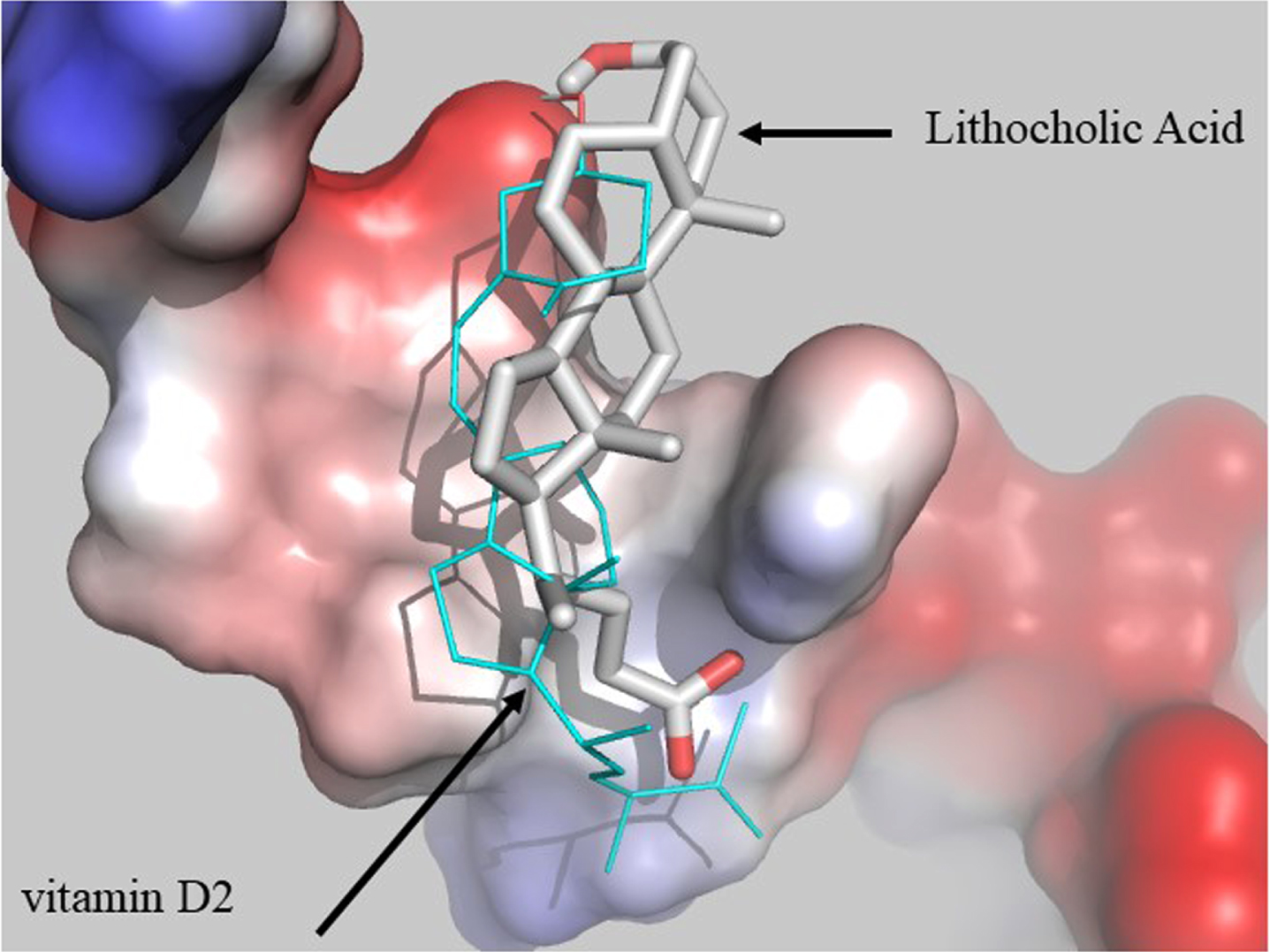
Both LCA and vitamin D3 (1,25 (OH)2D3) bind to vitamin D receptor, with LCA having less affinity [334]. Therefore, LCA may interfere with the biological benefits of vitamin D3 and could promote the aggregation of Aβ. This could have further consequences. In more details, two binding sites have been identified for vDBP, one for vitamin D and one for actin [335]. vDBP may also bind bile acids, because there is more ligand structure flexibility for vDBP compared to VDR [336]. Furthermore, vitamin D2 metabolites bind human vDBP with less affinity than vitamin D3 metabolites [337]. Hence, there may be other metabolites such as LCA that bind vDBP and reduce the efficiency of vDBP to perform its biological tasks in clearing Aβ plaques. Interestingly, perhaps as a compensatory mechanism, serum and CSF levels of vDBP increase in AD [338]. More precisely, the levels of vDBP increase from 0.6±0.0μg/ml in control patients to 1.2±0.1μg/ml in patients with AD [339]. However, this may not be high enough, then drug delivery systems such as PLGA nanoparticles may be employed to deliver vDBP to the brain to reduce accumulation of Aβ in the brain [340]. Therefore, if the functionality of vDBP is restored by reducing serum levels of LCA, then therapeutic benefits such as improvements may be observed in cognitive functions of patients with AD. It is important to start early treatment, as at the late stages, the brain is too compromised to take benefits from the drug [341].
THERAPEUTIC APPROACHES
Administration of TUDCA at the dose of 750 mg/day is safe in patients with liver cirrhosis [342]. This did not change serum levels of lithocholic acid, but decreased serum levels of CDCA and cholic acid at doses of 500 mg/day or higher [343]. Furthermore, TUDCA improved glucose homeostasis in streptozotocin-induced mouse AD model [344]. Certain bile acids have anti-inflammatory and anti-oxidative activities such as TUDCA and TCA [345]. However, it should be noted that administration of bile acids such as obeticholic acid (Supplementary Figure 3) comes with pruritus, which is common adverse event with FXR agonists [346]. Administering cholestyramine 16 g/day for three weeks decreased serum levels of lithocholic acid, but generally the change was not significant [347]. However, the limitations of the therapy are: unpleasant taste, diarrhea, and bloating. As 7α-hydroxylation by CYP7A is necessary for the conversion of cholesterol to bile acid, cholestyramine is an inducer of CYP7A, which is also active against 27-hydroxycholesterol. This is in addition to CYP7B [348]. Therefore, administration of cholestyramine will lead to more acidic pathway, and hence more CDCA, and potentially more LCA, and this could be an explanation why administration of cholestyramine does not significantly reduce serum LCA levels [81]. On the other hand, the probiotics cocktail VLS#3 could promote excretion of bile acids [349] and reduce LCA levels in AD patients (by increased bowel movement).
Bile acid therapy will be useful when serum C24 bile acid levels are low. Bile acid therapy will help absorption of lipids and vitamins [20]. Oral administration of non-absorbable antibiotics (vancomycin and polymyxin B) reduced amounts of LCA and DCA in the feces of mice after 5 days. This was due to the dysbiosis induced by the antibiotics. It should be noted that reduced levels of LCA and DCA changed the expression of hepatic enzymes such as CYP2b10, CYP3a25, and CYP51a1, which could affect metabolism of other drugs taken by the patient [350]. Can treatment with CDCA increase serum levels of LCA? There is a possibility, as treatment with DCA increases the amounts of DCA in the bile [351]. Therefore, treatment with CDCA may increase the content of this bile acids in the bile and consequently amounts of LCA in the blood. Sadeghi et al. (2020) also found that bile acid therapy by CA or sodium deoxycholate reduced acetylcholinesterase activity in the hippocampus of AD rat model, and this therapy also reduced the Aβ load in the brain of the animal models [352].
Table 1 presents a list of drugs that have been administered in different diseases such as intrahepatic cholestasis pregnancy and chronic idiopathic constipation to reduce serum levels of bile acids or where reduction of serum bile acids occurred unintentionally. Patients with AD may suffer from constipation, and this could be due to dysregulation of mAChr signaling pathways and ER stress response [353]. Therefore, treatment with drugs such as elobixibat (an inhibitor of ileal bile acid transporter [354]) may reduce the serum levels of LCA due to increased bowel movement, as it is suggested that constipation increases LCA serum levels due to greater LCA reabsorption [355]. High bile acid levels in feces are associated with diarrhea [356]. Importantly, delayed colon transit elevates serum C4 levels, indicating altered/increased bile acid synthesis and further elevation of serum LCA levels [357]. Interestingly, serum levels of LCA decreased below the detection level by applying transcutaneous neuromodulation [355]. In this method, electrical stimulation was delivered noninvasively via surface electrodes placed at both the acupoint ST36 and the posterior tibial nerve using an external watch-size stimulator. As an alternative method, total plasma exchange reduced serum bile acids by replacing the patient’s plasma volume with 5% albumin fluid [358]. This is known as therapeutic plasma exchange which is a procedure that the patient’s blood is passed through an apheresis machine. The filtered plasma is removed and discarded with reinfusion of red blood cells along with replacement fluid such as plasma or albumin in to the patient [359].
Table 1
List of drugs that can reduce serum bile acids including LCA
| Drug | Dose/day | Mechanism of action/ | Duration/ | Remarks | Limitations | Ref |
| Days-weeks | ||||||
| Cholestyramine | 16 g | Bile acid binding resin | 3 weeks | Decreases serum levels of lithocholic acid. The change is not large | Unpleasant taste, diarrhea, bloating, Vitamin K deficiency | [347, 361] |
| UDCA | 900 mg | Stimulation of biliary secretion of bile Acids | 15 days | Reduced serum LCA from 20μmol/L to 3μmol/L (normal level is 1μmol/L) | Not observed, rare mild diarrhea | [362–365] |
| Colestipol | 7.5 g | Bile acid sequestrant | 6 days | Reduced serum bile acids | Unpleasant taste | [366, 367] |
| Vancomycin | 1.5 g | Non-absorbable antibiotic affecting intestinal microbiota | 7 days | Serum secondary bile acids decreased | Diarrhea | [368] |
| Atrovastatin | 40 mg | Activation of pregnane X nuclear receptors | 12 weeks | Reduce serum GLCA from 42 ng/ml to 34 ng/ml | Increased serum alanine aminotransferase levels (liver damage) | [369, 370] |
| Dexamethasone | 12 mg | By reducing serum estriol and estradiol levels and their effects on the liver | 10 days (oral) | Reduced serum bile acids | Suppression of pituitary-adrenal axis | [371, 372] |
| S-adenosyl-L-methionine | 800 mg | By blocking the effects of estrogen on the liver | 10 days (i.v.) | Reduced total serum bile acids | No side effects | [373] |
| Elobixibat | 10 mg | SLC10A2 inhibitor | 14 days | Potential reduction of serum LCA due to shorter secondary bile acid time in the intestine | No serious adverse events, some or mild GI discomforts | [355, 357, 374, 375] |
| Activated charcoal | 150 g | Preventing bile acid enterohepatic recirculation | 8 days | Reduced serum bile acids | Preventing absorption of key nutrients such as vitamin K, and alarming black stool! | [376, 377] |
| Obeticholic acid | 5-10 mg | Selective farnesoid X receptor agonist | 12 months | Significantly reduced plasma bile acid levels including C4 | Pruritus | [346, 378] |
| Maralixibat | 20 mg | Apical sodium dependent bile acid competitive inhibitor | 13 weeks | Reduced serum bile acids from 52 ng/ml to 33 ng/ml | Gastrointestinal disorder (abdominal pain) | [379] |
| GSK2330672 | Starting with 90 mg then 180 mg | Ileal bile acid transporter inhibitor | 3 days at 90 mg then increase to 180 mg until day 14 | Reduced serum bile levels by 50% (from 30μM to 15μM) | Diarrhea | [380] |
| Colesevelam | 3.75 g | Anion-exchange resin with strong bile acid-binding capacity | 3 weeks | Reduced total serum bile acids from 155μmol/L to 140μmol/L. | Mild stool change | [381] |
| Rifampicin | 600 mg | Hydroxylation of hydrophobic bile acids and reducing their cytotoxicity | 1-2 weeks | Reduced total and conjugated bile acids | No adverse effects | [382, 383] |
| Guar gum | 15 g | Binds with bile acids in the intestinal lumen | At least 10 days | Prevented increase of serum bile acids levels | Mild abdominal distress | [384] |
| Cilofexor | 100 mg | Non-bile acid FXR agonist | 12 weeks | Reduced primary and secondary serum bile acids by 50% and 40%, respectively | Advert events (mainly pruritus) were similar to placebo | [385, 386] |
| NGM282 | 3 mg injection | FGF-19 mimetics | 12 weeks | Significantly reduced total serum bile acids (conjugated bile acids and DCA significantly reduced, LCA was also reduced but not significantly) | Gastrointestinal symptoms such as diarrhea and injection site reactions | [387] |
| Bezafibrate | 400 mg/day plus UDCA (600 mg/day) | PPAR agonist | 3 months | Reduced serum bile acids including LCA (did not reach statistical significance) | Adverse events were not reported when bezafibrate used with UCA, however, fibrates on their own may cause hepatotoxicity, increased risk of developing gallbladder disease and venothromboembolic disease (pulmonary embolism) | [388–390] |
| MBX-8025 | 200 mg | PPARσ agonist | 12 weeks | Reduced serum C4 and bile acid levels | Frequent adverse events were pruritus, diarrhea, and nausea | [391] |
| A4250 | 3 mg | Apical sodium dependent bile acid transporter inhibitor | 1 week | Reduced serum bile acids | Abdominal symptoms including increased bowel movements | [392] |
| Phenobarbitone | 3 mg/kg | Hepatic smooth endoplasmic reticulum enhancement, an increase in hepatic cytochrome P-450 content | 2 weeks | Reduced serum bile acids (did not reach statistical significance) | Not reported | [393] |
Elafibranor (GFT505) is agonist of PPARα/σ [360], and has been investigated in patients with primary biliary cholangitis in a phase II clinical trial (NCT03124108). Change in serum bile acids was a secondary outcome measure, but the report is due for publication. Elafibranor may reduce serum bile acid levels, as GENFIT reported reduction of serum C4 levels in the patients at the dose of 80 or 120 mg/day for 12 weeks [360].
CONCLUSIONS
Recent clinical studies have shown that serum bile acid levels increase in patients with AD including toxic bile acids such as LCA, perhaps due to the peroxisomal dysfunction. Hydrophobic bile acids such as DCA alter the permeability of the BBB to molecules that normally do not cross the BBB. Hydrophobic bile acids activate FXR signaling in the brain, which leads to recued brain levels of hydrophilic bile acids. Hydrophobic bile acids block NMDARs, with LCA being the most potent. NMDARs are crucial for the formation of temporal memory. Molecular modelling suggests that LCA has the ability to form a complex with E2 receptors, but without physiological actions, preventing estrogen from exerting its role in cognitive function and memory. LCA may reduce the expression of ABCA1, promote the formation of Aβ fibrils, leading to accumulation of Aβ plaques in the brain at the early stages of AD. The plasma or serum levels of 24-OHC were significantly lower in patients with AD and elevated serum bile acids levels have the ability to reduce synthesis of 24-OHC via activation of FXR. Bile acids have the ability of activating TGR5 and leading to neuroprotection and diminishing of inflammation. However, the expression of TGR5 decreases in the brains of patients with AD, with potential contributions from toxic bile acids. Therefore, the benefits from elevated serum bile acids may not be observed. Toxic bile acids may inhibit the activation of the SHH, by preventing the interaction of potent endogenous oxysterols with SMO protein. There are several approaches to reduce serum bile acids such as the use of transcutaneous neuromodulation, administration of cholestyramine (16 g/day), or obeticholic acid (5-10 mg/day).
FUTURE DIRECTIONS
Further studies should be conducted aiming to reduce serum levels of bile acids in patients with AD. These studies should include evaluation of the clinical outcomes, along the line of that undertaken by Marksteiner et al. (2018) [39], i.e., measuring the serum bile acids of patients with AD and recording basic cognitive scores (e.g., MMSE) at the beginning of trial. The intervention would be administering a therapeutic agent to reduce serum bile acids for a period of six months, followed by cognitive assessment scores, in parallel with serial measurements of serum bile acids. An alternative approach could be monitoring serum bile acid levels of individuals regularly, and establish association between serum bile acid levels and cognitive function, given link between secondary bile acids in the gut and brain [394]. Another direction would be to promote exercise in elderly subjects and monitoring serum bile acids and cognitive function. It has been shown that recreational physical activity is inversely associated with total fecal bile acid concentrations [395], and exercise is also linked to the reduction of total serum bile acid levels [396]. Optimizing the diet also could be another safe and inexpensive means of further investigation in patients with AD, as diet affects serum bile acid levels (vegans having higher serum bile acids compared to omnivores) [397].
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/ADR-220071.
REFERENCES
[1] | Cummings J , Lee G , Mortsdorf T , Ritter A , Zhong K ((2017) ) Alzheimer’s disease drug development pipeline: 2017. Alzheimers Dement (N Y) 3: , 367–384. |
[2] | Cummings J , Morstorf T , Lee G ((2016) ) Alzheimer’s drug-development pipeline: 2016. Alzheimers Dement (N Y) 2: , 222–232. |
[3] | Cummings J , Lee G , Ritter A , Zhong K ((2018) ) Alzheimer’s disease drug development pipeline: 2018. Alzheimers Dement (N Y) 4: , 195–214. |
[4] | Cummings J , Lee G , Ritter A , Sabbagh M , Zhong K ((2019) ) Alzheimer’s disease drug development pipeline: 2019. Alzheimers Dement (N Y) 5: , 272–293. |
[5] | Cummings JL , Zhong K ((2016) ) Symptomatic cognitive enhancing agents. In Developing Therapeutics for Alzheimer’s Disease, WolfeMS, ed. Academic Press, Boston, pp. 459–475. |
[6] | Gauthier S , Feldman HH , Schneider LS , Wilcock GK , Frisoni GB , Hardlund JH , Moebius HJ , Bentham P , Kook KA , Wischik DJ , Schelter BO , Davis CS , Staff RT , Bracoud L , Shamsi K , Storey JMD , Harrington CR , Wischik CM ((2016) ) Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: A randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet 388: , 2873–2884. |
[7] | de la Monte SM ((2012) ) Early intranasal insulin therapy halts progression of neurodegeneration: Progress in Alzheimer’s disease therapeutics. Aging Health 8: , 61–64. |
[8] | Craft S , Raman R , Chow TW , Rafii MS , Sun CK , Rissman RA , Donohue MC , Brewer JB , Jenkins C , Harless K , Gessert D , Aisen PS ((2020) ) Safety, efficacy, and feasibility of intranasal insulin for the treatment of mild cognitive impairment and Alzheimer disease dementia: A randomized clinical trial. JAMA Neurol 77: , 1099–1109. |
[9] | Craft S , Raman R , Chow T , Rafii M , Rissman R , Brewer J ((2018) ) Primary results from a phase II/III trial of intranasal insulin: A novel multi-target molecule and delivery mode for AD theraeutics. J Prev Alzheimers Dis 5: , S9. |
[10] | Hanazawa T , Kamijo Y ((2019) ) Effect of suvorexant on nocturnal delirium in elderly patients with Alzheimer’s disease: A case-series study. Clin Psychopharmacol Neurosci 17: , 547–550. |
[11] | Plascencia-Villa G , Perry G ((2021) ) Preventive and therapeutic strategies in Alzheimer’s disease: Focus on oxidative stress, redox metals, and ferroptosis. Antioxid Redox Signal 34: , 591–610. |
[12] | Astarita G , Jung KM , Berchtold NC , Nguyen VQ , Gillen DL , Head E , Cotman CW , Piomelli D ((2010) ) Deficient liver biosynthesis of docosahexaenoic acid correlates with cognitive impairment in Alzheimer’s disease. PLoS One 5: , e12538. |
[13] | Kim HY ((2007) ) Novel metabolism of docosahexaenoic acid in neural cells. J Biol Chem 282: , 18661–18665. |
[14] | Zhao Z , Xiao Y , Elson P , Tan H , Plummer SJ , Berk M , Aung PP , Lavery IC , Achkar JP , Li L , Casey G , Xu Y ((2007) ) Plasma lysophosphatidylcholine levels: Potential biomarkers for colorectal cancer. J Clin Oncol 25: , 2696–2701. |
[15] | Mapstone M , Cheema AK , Fiandaca MS , Zhong X , Mhyre TR , MacArthur LH , Hall WJ , Fisher SG , Peterson DR , Haley JM , Nazar MD , Rich SA , Berlau DJ , Peltz CB , Tan MT , Kawas CH , Federoff HJ ((2014) ) Plasma phospholipids identify antecedent memory impairment in older adults. Nat Med 20: , 415–418. |
[16] | Whiley L , Sen A , Heaton J , Proitsi P , García-Gómez D , Leung R , Smith N , Thambisetty M , Kloszewska I , Mecocci P , Soininen H , Tsolaki M , Vellas B , Lovestone S , Legido-Quigley C ((2014) ) Evidence of altered phosphatidylcholine metabolism in Alzheimer’s disease. Neurobiol Aging 35: , 271–278. |
[17] | Nguyen LN , Ma D , Shui G , Wong P , Cazenave-Gassiot A , Zhang X , Wenk MR , Goh EL , Silver DL ((2014) ) Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 509: , 503–506. |
[18] | Sugasini D , Thomas R , Yalagala PCR , Tai LM , Subbaiah PV ((2017) ) Dietary docosahexaenoic acid (DHA) as lysophosphatidylcholine, but not as free acid, enriches brain DHA and improves memory in adult mice. Sci Rep 7: , 11263–11263. |
[19] | González-Domínguez R , García-Barrera T , Gómez-Ariza JL ((2014) ) Combination of metabolomic and phospholipid-profiling approaches for the study of Alzheimer’s disease. J Proteomics 104: , 37–47. |
[20] | Ferdinandusse S , Denis S , Faust PL , Wanders RJ ((2009) ) Bile acids: The role of peroxisomes. J Lipid Res 50: , 2139–2147. |
[21] | Ferdinandusse S , Houten SM ((2006) ) Peroxisomes and bile acid biosynthesis. Biochim Biophys Acta 1763: , 1427–1440. |
[22] | Russell DW ((2003) ) The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem 72: , 137–174. |
[23] | Fuchs M ((2003) ) Bile acid regulation of hepatic physiology: III. Regulation of bile acid synthesis: Past progress and future challenges. Am J Physiol Gastrointest Liver Physiol 284: , G551–557. |
[24] | Vaz FM , Ferdinandusse S ((2017) ) Bile acid analysis in human disorders of bile acid biosynthesis. Mol Aspects Med 56: , 10–24. |
[25] | Pullinger CR , Eng C , Salen G , Shefer S , Batta AK , Erickson SK , Verhagen A , Rivera CR , Mulvihill SJ , Malloy MJ , Kane JP ((2002) ) Human cholesterol 7α-hydroxylase (CYP7A1) deficiency has a hypercholesterolemic phenotype. J Clin Invest 110: , 109–117. |
[26] | Lines MA , Jobling R , Brady L , Marshall CR , Scherer SW , Rodriguez AR , Lee L , Lang AE , Mestre TA , Wanders RJ , Ferdinandusse S , Tarnopolsky MA ((2014) ) Peroxisomal D-bifunctional protein deficiency: Three adults diagnosed by whole-exome sequencing. Neurology 82: , 963–968. |
[27] | Greenberg N , Grassano A , Thambisetty M , Lovestone S , Legido-Quigley C ((2009) ) A proposed metabolic strategy for monitoring disease progression in Alzheimer’s disease. Electrophoresis 30: , 1235–1239. |
[28] | Proitsi P , Kim M , Whiley L , Pritchard M , Leung R , Soininen H , Kloszewska I , Mecocci P , Tsolaki M , Vellas B , Sham P , Lovestone S , Powell JF , Dobson RJB , Legido-Quigley C ((2015) ) Plasma lipidomics analysis finds long chain cholesteryl esters to be associated with Alzheimer’s disease, Transl Psychiatry 5: , e494–e494. |
[29] | Proitsi P , Kim M , Whiley L , Simmons A , Sattlecker M , Velayudhan L , Lupton MK , Soininen H , Kloszewska I , Mecocci P , Tsolaki M , Vellas B , Lovestone S , Powell JF , Dobson RJ , Legido-Quigley C ((2017) ) Association of blood lipids with Alzheimer’s disease: A comprehensive lipidomics analysis. Alzheimers Dement 13: , 140–151. |
[30] | Kiriyama Y , Nochi H ((2019) ) The biosynthesis, signaling, and neurological functions of bile acids. Biomolecules 9,: , 232. |
[31] | Grant SM , DeMorrow S ((2020) ) Bile acid signaling in neurodegenerative and neurological disorders. Int J Mol Sci 21: , 5982. |
[32] | Mulak A ((2021) ) Bile acids as key modulators of the brain-gut-microbiota axis in Alzheimer’s disease. J Alzheimers Dis 84: , 461–477. |
[33] | Xie G , Wang X , Jiang R , Zhao A , Yan J , Zheng X , Huang F , Liu X , Panee J , Rajani C , Yao C , Yu H , Jia W , Sun B , Liu P , Jia W ((2018) ) Dysregulated bile acid signaling contributes to the neurological impairment in murine models of acute and chronic liver failure. EBioMedicine 37: , 294–306. |
[34] | Dutta M , Weigel KM , Patten KT , Valenzuela AE , Wallis C , Bein KJ , Wexler AS , Lein PJ , Cui JY ((2022) ) Chronic exposure to ambient traffic-related air pollution (TRAP) alters gut microbial abundance and bile acid metabolism in a transgenic rat model of Alzheimer’s disease. Toxicol Rep 9: , 432–444. |
[35] | Dumitrescu L , Mahoney ER , Mukherjee S , Lee ML , Bush WS , Engelman CD , Lu Q , Fardo DW , Trittschuh EH , Mez J , Kaczorowski C , HernandezSaucedo H , Widaman KF , Buckley R , Properzi M , Mormino E , Yang HS , Harrison T , Hedden T , Nho K , Andrews SJ , Tommet D , Hadad N , Sanders RE , Ruderfer DM , Gifford KA , Moore AM , Cambronero F , Zhong X , Raghavan NS , Vardarajan B , Pericak-Vance MA , Farrer LA , Wang LS , Cruchaga C , Schellenberg G , Cox NJ , Haines JL , Keene CD , Saykin AJ , Larson EB , Sperling RA , Mayeux R , Bennett DA , Schneider JA , Crane PK , Jefferson AL , Hohman TJ ((2020) ) Genetic variants and functional pathways associated with resilience to Alzheimer’s disease. Brain 143: , 2561–2575. |
[36] | Shao Y , Ouyang Y , Li T , Liu X , Xu X , Li S , Xu G , Le W ((2020) ) Alteration of metabolic profile and potential biomarkers in the plasma of Alzheimer’s disease. Aging Dis 11: , 1459–1470. |
[37] | Kurbatova N , Garg M , Whiley L , Chekmeneva E , Jiménez B , Gómez-Romero M , Pearce J , Kimhofer T , D’Hondt E , Soininen H , Kłoszewska I , Mecocci P , Tsolaki M , Vellas B , Aarsland D , Nevado-Holgado A , Liu B , Snowden S , Proitsi P , Ashton NJ , Hye A , Legido-Quigley C , Lewis MR , Nicholson JK , Holmes E , Brazma A , Lovestone S ((2020) ) Urinary metabolic phenotyping for Alzheimer’sdisease. Sci Rep 10: , 21745. |
[38] | Olazaran J , Gil-de-Gomez L , Rodriguez-Martin A , Valenti-Soler M , Frades-Payo B , Marin-Munoz J , Antunez C , Frank-Garcia A , Acedo-Jimenez C , Morlan-Gracia L , Petidier-Torregrossa R , Guisasola MC , Bermejo-Pareja F , Sanchez-Ferro A , Perez-Martinez DA , Manzano-Palomo S , Farquhar R , Rabano A , Calero M ((2015) ) A blood-based, 7-metabolite signature for the early diagnosis of Alzheimer’s disease. J Alzheimers Dis 45: , 1157–1173. |
[39] | Marksteiner J , Blasko I , Kemmler G , Koal T , Humpel C ((2018) ) Bile acid quantification of 20 plasma metabolites identifies lithocholic acid as a putative biomarker in Alzheimer’s disease. Metabolomics 14: , 1. |
[40] | Goldberg AA , Beach A , Davies GF , Harkness TA , Leblanc A , Titorenko VI ((2011) ) Lithocholic bile acid selectively kills neuroblastoma cells, while sparing normal neuronal cells. Oncotarget 2: , 761–782. |
[41] | Baloni P , Funk CC , Yan J , Yurkovich JT , Kueider-Paisley A , Nho K , Heinken A , Jia W , Mahmoudiandehkordi S , Louie G , Saykin AJ , Arnold M , Kastenmüller G , Griffiths WJ , Thiele I , Kaddurah-Daouk R , Price ND ((2020) ) Metabolic network analysis reveals altered bile acid synthesis and metabolism in Alzheimer’s disease. Cell Rep Med 1: , 100138. |
[42] | MahmoudianDehkordi S , Arnold M , Nho K , Ahmad S , Jia W , Xie G , Louie G , Kueider-Paisley A , Moseley MA , Thompson JW , St John Williams L , Tenenbaum JD , Blach C , Baillie R , Han X , Bhattacharyya S , Toledo JB , Schafferer S , Klein S , Koal T , Risacher SL , Kling MA , Motsinger-Reif A , Rotroff DM , Jack J , Hankemeier T , Bennett DA , De Jager PL , Trojanowski JQ , Shaw LM , Weiner MW , Doraiswamy PM , van Duijn CM , Saykin AJ , Kastenmüller G , Kaddurah-Daouk R ((2019) ) Altered bile acid profile associates with cognitive impairment in Alzheimer’s disease— An emerging role for gut microbiome. Alzheimers Dement 15: , 76–92. |
[43] | Koike S , Miyaji Y , Sano H , Aikawa N , Kai M , Kasahara S , Suzuki T , Nishimoto-Kusunose S , Ogasawara Y ((2021) ) Simultaneous determination of five bile acids as potential biomarkers for Alzheimer’s disease in mouse brain and plasma. Anal Sci 37: , 1165–1170. |
[44] | Pan X , Elliott CT , McGuinness B , Passmore P , Kehoe PG , Holscher C , McClean PL , Graham SF , Green BD ((2017) ) Metabolomic profiling of bile acids in clinical and experimental samples of Alzheimer’s disease. Metabolites 7: , 28. |
[45] | Bathena SP , Mukherjee S , Olivera M , Alnouti Y ((2013) ) The profile of bile acids and their sulfate metabolites in human urine and serum. J Chromatogr B Analyt Technol Biomed Life Sci 942-943: , 53–62. |
[46] | Pan X , Passmore P , Graham SF , Todd S , McGuinness B , Green BD ((2018) ) Significant age-related alterations in the blood plasma metabolome of noncognitively impaired healthy elderly subjects, Healthy Aging Res 7: , e16. |
[47] | Bathena SP , Thakare R , Gautam N , Mukherjee S , Olivera M , Meza J , Alnouti Y ((2015) ) Urinary bile acids as biomarkers for liver diseases I. Stability of the baseline profile in healthy subjects. Toxicol Sci 143: , 296–307. |
[48] | Geenes V , Williamson C ((2009) ) Intrahepatic cholestasis of pregnancy. World J Gastroenterol 15: , 2049–2066. |
[49] | Egan N , Bartels A , Khashan AS , Broadhurst DI , Joyce C , O’Mullane J , O’Donoghue K ((2012) ) Reference standard for serum bile acids in pregnancy. BJOG 119: , 493–498. |
[50] | Lalic-Popovic M , Vasovic V , Milijasevic B , Golocorbin-Kon S , Al-Salami H , Mikov M ((2013) ) Deoxycholic acid asa modifier of the permeation of gliclazide through the blood brainbarrier of a rat. J Diabetes Res 2013: , 598603. |
[51] | Mikov M , Kevresan S , Kuhajda K , Jakovljević V , Vasović V ((2004) ) 3Alpha,7alpha-dihydroxy-12-oxo-5beta-cholanate as blood-brainbarrier permeator. Pol J Pharmacol 56: , 367–371. |
[52] | Schuff N , Tosun D , Insel PS , Chiang GC , Truran D , Aisen PS , Jack CR Jr. , Weiner MW , Alzheimer’s Disease Neuroimaging Initiative ((2012) ) Nonlinear time course of brain volume loss in cognitively normal and impaired elders. Neurobiol Aging 33: , 845–855. |
[53] | Greenwood J , Adu J , Davey AJ , Abbott NJ , Bradbury MW ((1991) ) The effect of bile salts on the permeability and ultrastructure of the perfused, energy-depleted, rat blood-brain barrier. J Cereb Blood Flow Metab 11: , 644–654. |
[54] | Quinn M , McMillin M , Galindo C , Frampton G , Pae HY , DeMorrow S ((2014) ) Bile acids permeabilize the blood brain barrier after bile duct ligation in rats via Rac1-dependent mechanisms. Dig Liver Dis 46: , 527–534. |
[55] | Seiffert E , Dreier JP , Ivens S , Bechmann I , Tomkins O , Heinemann U , Friedman A ((2004) ) Lasting blood-brain barrier disruption induces epileptic focus in the rat somatosensory cortex. J Neurosci 24: , 7829–7836. |
[56] | Elovaara I , Icén A , Palo J , Erkinjuntti T ((1985) ) CSF inAlzheimer’s disease. Studies on blood-brain barrier function andintrathecal protein synthesis. J Neurol Sci 70: , 73–80. |
[57] | Tomkins O , Friedman O , Ivens S , Reiffurth C , Major S , Dreier JP , Heinemann U , Friedman A ((2007) ) Blood– brain barrier disruption results in delayed functional and structural alterations in the rat neocortex. Neurobiol Dis 25: , 367–377. |
[58] | Farrall AJ , Wardlaw JM ((2009) ) Blood-brain barrier: Ageing and microvascular disease–systematic review and meta-analysis. Neurobiol Aging 30: , 337–352. |
[59] | Zhong Z , Deane R , Ali Z , Parisi M , Shapovalov Y , O’Banion MK , Stojanovic K , Sagare A , Boillee S , Cleveland DW , Zlokovic BV ((2008) ) ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat Neurosci 11: , 420–422. |
[60] | Grimm MO , Haupenthal VJ , Mett J , Stahlmann CP , Blumel T , Mylonas NT , Endres K , Grimm HS , Hartmann T ((2016) ) Oxidized docosahexaenoic acid species and lipid peroxidation products increase amyloidogenic amyloid precursor protein processing. Neurodegener Dis 16: , 44–54. |
[61] | Patel JP , Frey BN ((2015) ) Disruption in the blood-brain barrier: The missing link between brain and body inflammation in bipolar disorder? Neural Plasticity 2015: , 708306. |
[62] | McMillin M , Frampton G , Grant S , Khan S , Diocares J , Petrescu A , Wyatt A , Kain J , Jefferson B , DeMorrow S ((2017) ) Bile acid-mediated sphingosine-1-phosphate receptor 2 signaling promotes neuroinflammation during hepatic encephalopathy in mice. Front Cell Neurosci 11: , 191. |
[63] | Matthew M , Sharon D ((2017) ) Neuroinflammatory signals during acute and chronic liver diseases. In Mechanisms of Neuroinflammation, Gonzalo Emiliano ArandaA, ed. IntechOpen, Rijeka, p. Ch. 11. |
[64] | McMillin M , Frampton G , Thompson M , Galindo C , Standeford H , Whittington E , Alpini G , DeMorrow S ((2014) ) Neuronal CCL2 is upregulated during hepatic encephalopathy and contributes to microglia activation and neurological decline. J Neuroinflammation 11: , 121. |
[65] | Gopalraj RK , Estus S ((2003) ) Plasmin system, Alzheimer’s disease and stroke. Proc Indian Natl Sci Acad B 69: , 239–250. |
[66] | Mahmood N , Mihalcioiu C , Rabbani SA ((2018) ) Multifaceted role of the urokinase-type plasminogen activator (uPA) and its receptor (uPAR): Diagnostic, prognostic, and therapeutic applications. Front Oncol 8,: , 24. |
[67] | Martorana A , Sancesario GM , Esposito Z , Nuccetelli M , Sorge R , Formosa A , Dinallo V , Bernardi G , Bernardini S , Sancesario G ((2012) ) Plasmin system of Alzheimer’s disease patients: CSF analysis. J Neural Transm (Vienna) 119: , 763–769. |
[68] | Manabe T , Mizukami K , Akatsu H , Teramoto S , Yamaoka K , Nakamura S , Ohkubo T , Kudo K , Hizawa N ((2016) ) Influence of pneumonia complications on the prognosis of patients with autopsy-confirmed Alzheimer’s disease, dementia with Lewy bodies, and vascular dementia. Psychogeriatrics 16: , 305–314. |
[69] | Une M , Konishi M , Suzuki Y , Akaboshi S , Yoshii M , Kuramoto T , Fujimura K ((1997) ) Bile acid profiles in a peroxisomal D-3-hydroxyacyl-CoA dehydratase/D-3-hydroxyacyl-CoA dehydrogenase bifunctional protein deficiency. J Biochem 122: , 655–658. |
[70] | Ferdinandusse S , Denis S , Dacremont G , Wanders RJ ((2009) ) Toxicity of peroxisomal C27-bile acid intermediates. Mol Genet Metab 96: , 121–128. |
[71] | Axelson M , Mork B , Sjovall J ((1988) ) Occurrence of 3 beta-hydroxy-5-cholestenoic acid, 3 beta,7 alpha-dihydroxy-5-cholestenoic acid, and 7 alpha-hydroxy-3-oxo-4-cholestenoic acid as normal constituents in human blood. J Lipid Res 29: , 629–641. |
[72] | Axelson M , Sjovall J ((1990) ) Potential bile acid precursors in plasma–possible indicators of biosynthetic pathways to cholic and chenodeoxycholic acids in man. J Steroid Biochem 36: , 631–640. |
[73] | Andersson M , Ellegard L , Andersson H ((2002) ) Oat bran stimulates bile acid synthesis within 8h as measured by 7alpha-hydroxy-4-cholesten-3-one. Am J Clin Nutr 76: , 1111–1116. |
[74] | Meaney S , Babiker A , Lutjohann D , Diczfalusy U , Axelson M , Bjorkhem I ((2003) ) On the origin of the cholestenoic acids in human circulation. Steroids 68: , 595–601. |
[75] | Galman C , Arvidsson I , Angelin B , Rudling M ((2003) ) Monitoring hepatic cholesterol 7alpha-hydroxylase activity by assay of the stable bile acid intermediate 7alpha-hydroxy-4-cholesten-3-one in peripheral blood. J Lipid Res 44: , 859–866. |
[76] | Hahn C , von Bergmann K ((1996) ) Relationship between the serum concentration of 7 alpha-hydroxycholesterol and fecal bile acid excretion in humans. Scand J Gastroenterol 31: , 804–808. |
[77] | Tatidis L , Vitols S , Gruber A , Paul C , Axelson M ((2001) ) Cholesterol catabolism in patients with acute myelogenous leukemia and hypocholesterolemia: Suppressed levels of a circulating marker for bile acid synthesis. Cancer Lett 170: , 169–175. |
[78] | Dawes LG , Laut HC , Woodruff M ((2007) ) Decreased bile acid synthesis with total parenteral nutrition. Am J Surg 194: , 623–627. |
[79] | Axelson M , Bjorkhem I , Reihner E , Einarsson K ((1991) ) The plasma level of 7 alpha-hydroxy-4-cholesten-3-one reflects the activity of hepatic cholesterol 7 alpha-hydroxylase in man. FEBS Lett 284: , 216–218. |
[80] | Bertolotti M , Carulli L , Concari M , Martella P , Loria P , Tagliafico E , Ferrari S , Del Puppo M , Amati B , De Fabiani E , Crestani M , Amorotti C , Manenti A , Carubbi F , Pinetti A , Carulli N ((2001) ) Suppression of bile acid synthesis, but not of hepatic cholesterol 7α-hydroxylase expression, by obstructive cholestasis in humans. Hepatology 34: , 234–242. |
[81] | Lorbek G , Lewinska M , Rozman D ((2012) ) Cytochrome P450s in the synthesis of cholesterol and bile acids–from mouse models to human diseases. FEBS J 279: , 1516–1533. |
[82] | Bazzoli F , Fromm H , Sarva RP , Sembrat RF , Ceryak S ((1982) ) Comparative formation of lithocholic acid from chenodeoxycholic and ursodeoxycholic acids in the colon. Gastroenterology 83: , 753–760. |
[83] | Fischer CD , Cooper NS , Rothschild MA , Mosbach EH ((1974) ) Effect of dietary chenodeoxycholic acid and lithocholic acid in the rabbit. Am J Dig Dis 19: , 877–886. |
[84] | Lee JE , Kim D , Lee JH ((2018) ) Association between Alzheimer’s disease and cancer risk in South Korea: An 11-year nationwide population-based study. Dement Neurocogn Disord 17: , 137–147. |
[85] | Bauer TM , Schwacha H , Steinbruckner B , Brinkmann FE , Ditzen AK , Aponte JJ , Pelz K , Berger D , Kist M , Blum HE ((2002) ) Small intestinal bacterial overgrowth in human cirrhosis is associated with systemic endotoxemia. Am J Gastroenterol 97: , 2364–2370. |
[86] | Qin N , Yang F , Li A , Prifti E , Chen Y , Shao L , Guo J , Le Chatelier E , Yao J , Wu L , Zhou J , Ni S , Liu L , Pons N , Batto JM , Kennedy SP , Leonard P , Yuan C , Ding W , Chen Y , Hu X , Zheng B , Qian G , Xu W , Ehrlich SD , Zheng S , Li L ((2014) ) Alterations of the human gut microbiome in liver cirrhosis. Nature 513: , 59–64. |
[87] | Wigg AJ , Roberts-Thomson IC , Dymock RB , McCarthy PJ , Grose RH , Cummins AG ((2001) ) The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia, and tumour necrosis factor alpha in the pathogenesis of non-alcoholic steatohepatitis. Gut 48: , 206–211. |
[88] | Kakiyama G , Pandak WM , Gillevet PM , Hylemon PB , Heuman DM , Daita K , Takei H , Muto A , Nittono H , Ridlon JM , White MB , Noble NA , Monteith P , Fuchs M , Thacker LR , Sikaroodi M , Bajaj JS ((2013) ) Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol 58: , 949–955. |
[89] | Acharya C , Bajaj JS ((2019) ) Altered microbiome in patients with cirrhosis and complications. Clin Gastroenterol Hepatol 17: , 307–321. |
[90] | Wahlström A , Sayin SI , Marschall HU , Bäckhed F ((2016) ) Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab 24: , 41–50. |
[91] | Camilleri M , Nadeau A , Tremaine WJ , Lamsam J , Burton D , Odunsi S , Sweetser S , Singh R ((2009) ) Measurement of serum 7alpha-hydroxy-4-cholesten-3-one (or 7alphaC4), a surrogate test for bile acid malabsorption in health, ileal disease and irritable bowel syndrome using liquid chromatography-tandem mass spectrometry, Neurogastroenterol Motil 21: , 734–e743. |
[92] | Larhammar M , Patra K , Blunder M , Emilsson L , Peuckert C , Arvidsson E , Ronnlund D , Preobraschenski J , Birgner C , Limbach C , Widengren J , Blom H , Jahn R , Wallen-Mackenzie A , Kullander K ((2015) ) SLC10A4 is a vesicular amine-associated transporter modulating dopamine homeostasis. Biol Psychiatry 77: , 526–536. |
[93] | Geyer J , Wilke T , Petzinger E ((2006) ) The solute carrier family SLC10: More than a family of bile acid transporters regarding function and phylogenetic relationships. Naunyn Schmiedebergs Arch Pharmacol 372: , 413–431. |
[94] | Schmidt S , Moncada M , Burger S , Geyer J ((2015) ) Expression, sorting and transport studies for the orphan carrier SLC10A4 in neuronal and non-neuronal cell lines and in Xenopus laevis oocytes. BMC Neurosci 16: , 35. |
[95] | Splinter P-L , Lazaridis K-N , Dawson P-A , LaRusso N-F ((2006) ) Cloning and expression of SLC10A4, a putative organic anion transport protein. World J Gastroenterol 12: , 6797–6805. |
[96] | Popova SN , Alafuzoff I ((2013) ) Distribution of SLC10A4, a synaptic vesicle protein in the human brain, and the association of this protein with Alzheimer’s disease-related neuronal degeneration. J Alzheimers Dis 37: , 603–610. |
[97] | Abe T , Kanemitu Y , Nakasone M , Kawahata I , Yamakuni T , Nakajima A , Suzuki N , Nishikawa M , Hishinuma T , Tomioka Y ((2013) ) SLC10A4 is a protease-activated transporter that transports bile acids. J Biochem 154: , 93–101. |
[98] | Grammas P , Samany PG , Thirumangalakudi L ((2006) ) Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: Implications for disease pathogenesis. J Alzheimers Dis 9: , 51–58. |
[99] | McMillin M , Frampton G , Quinn M , Ashfaq S , de los Santos M 3rd , Grant S , DeMorrow S ((2016) ) Bile acid signaling is involved in the neurological decline in a murine model of acute liver failure. Am J Pathol 186: , 312–323. |
[100] | Sporstol M , Tapia G , Malerod L , Mousavi SA , Berg T ((2005) ) Pregnane X receptor-agonists down-regulate hepatic ATP-binding cassette transporter A1 and scavenger receptor class B type I. Biochem Biophys Res Commun 331: , 1533–1541. |
[101] | Han S , Li T , Ellis E , Strom S , Chiang JY ((2010) ) A novel bile acid-activated vitamin D receptor signaling in human hepatocytes. Mol Endocrinol 24: , 1151–1164. |
[102] | McMillin M , Frampton G , Quinn M , Divan A , Grant S , Patel N , Newell-Rogers K , DeMorrow S ((2015) ) Suppression of the HPA axis during cholestasis can be attributed to hypothalamic bile acid signaling. Mol Endocrinol 29: , 1720–1730. |
[103] | Swanwick GR , Kirby M , Bruce I , Buggy F , Coen RF , Coakley D , Lawlor BA ((1998) ) Hypothalamic-pituitary-adrenal axis dysfunction in Alzheimer’s disease: Lack of association between longitudinal and cross-sectional findings. Am J Psychiatry 155: , 286–289. |
[104] | Keitel V , Gorg B , Bidmon HJ , Zemtsova I , Spomer L , Zilles K , Haussinger D ((2010) ) The bile acid receptor TGR5 (Gpbar-1) acts as a neurosteroid receptor in brain. Glia 58: , 1794–1805. |
[105] | Keitel V , Cupisti K , Ullmer C , Knoefel WT , Kubitz R , Häussinger D ((2009) ) The membrane-bound bile acid receptor TGR5 is localized in the epithelium of human gallbladders. Hepatology 50: , 861–870. |
[106] | Huang C , Wang J , Hu W , Wang C , Lu X , Tong L , Wu F , Zhang W ((2016) ) Identification of functional farnesoid X receptors in brain neurons. FEBS Lett 590: , 3233–3242. |
[107] | DeMorrow S ((2019) ) Bile acids in hepatic encephalopathy. J Clin Exp Hepatol 9: , 117–124. |
[108] | Higashi T , Watanabe S , Tomaru K , Yamazaki W , Yoshizawa K , Ogawa S , Nagao H , Minato K , Maekawa M , Mano N ((2017) ) Unconjugated bile acids in rat brain: Analytical method based on LC/ESI-MS/MS with chemical derivatization and estimation of their origin by comparison to serum levels. Steroids 125: , 107–113. |
[109] | Tripodi V , Contin M , Fernandez MA , Lemberg A ((2012) ) Bile acids content in brain of common duct ligated rats. Ann Hepatol 11: , 930–934. |
[110] | Wang H , Chen J , Hollister K , Sowers LC , Forman BM ((1999) ) Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell 3: , 543–553. |
[111] | McMillin M , Grant S , Frampton G , Petrescu AD , Kain J , Williams E , Haines R , Canady L , DeMorrow S ((2018) ) FXR-mediated cortical cholesterol accumulation contributes to the pathogenesis of type A hepatic encephalopathy. Cell Mol Gastroenterol Hepatol 6: , 47–63. |
[112] | Litwa E , Rzemieniec J , Wnuk A , Lason W , Krzeptowski W , Kajta M ((2016) ) RXRα, PXR and CAR xenobiotic receptors mediate the apoptotic and neurotoxic actions of nonylphenol in mouse hippocampal cells. J Steroid Biochem Mol Biol 156: , 43–52. |
[113] | Kruse MS , Rey M , Vega MC , Coirini H ((2012) ) Alterations of LXRα and LXRβ expression in the hypothalamus of glucose-intolerant rats. J Endocrinol 215: , 51–58. |
[114] | Neuschwander-Tetri BA , Loomba R , Sanyal AJ , Lavine JE , Van Natta ML , Abdelmalek MF , Chalasani N , Dasarathy S , Diehl AM , Hameed B , Kowdley KV , McCullough A , Terrault N , Clark JM , Tonascia J , Brunt EM , Kleiner DE , Doo E ((2015) ) Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 385: , 956–965. |
[115] | Ali AH , Carey EJ , Lindor KD ((2015) ) Recent advances in the development of farnesoid X receptor agonists. Ann Transl Med 3: , 5. |
[116] | Bathena SP , Thakare R , Gautam N , Mukherjee S , Olivera M , Meza J , Alnouti Y ((2015) ) Urinary bile acids as biomarkers for liver diseases II. Signature profiles in patients. Toxicol Sci 143: , 308–318. |
[117] | Astarita G , Jung K-M , Vasilevko V , DiPatrizio NV , Martin SK , Cribbs DH , Head E , Cotman CW , Piomelli D ((2011) ) Elevated stearoyl-CoA desaturase in brains of patients with Alzheimer’s disease, PLoS One 6: , e24777. |
[118] | Habchi J , Chia S , Galvagnion C , Michaels TCT , Bellaiche MMJ , Ruggeri FS , Sanguanini M , Idini I , Kumita JR , Sparr E , Linse S , Dobson CM , Knowles TPJ , Vendruscolo M ((2018) ) Cholesterol catalyses Abeta42 aggregation through a heterogeneous nucleation pathway in the presence of lipid membranes. Nat Chem 10: , 673–683. |
[119] | Liu Q , Yang M , Fu X , Liu R , Sun C , Pan H , Wong CW , Guan M ((2016) ) Activation of farnesoid X receptor promotes triglycerides lowering by suppressing phospholipase A2 G12B expression. Mol Cell Endocrinol 436: , 93–101. |
[120] | Liu H , Pathak P , Boehme S , Chiang JY ((2016) ) Cholesterol 7alpha-hydroxylase protects the liver from inflammation and fibrosis by maintaining cholesterol homeostasis. J Lipid Res 57: , 1831–1844. |
[121] | van Loo G , De Lorenzi R , Schmidt H , Huth M , Mildner A , Schmidt-Supprian M , Lassmann H , Prinz MR , Pasparakis M ((2006) ) Inhibition of transcription factor NF-kappaB in the central nervous system ameliorates autoimmune encephalomyelitis in mice. Nat Immunol 7: , 954–961. |
[122] | Brambilla R , Persaud T , Hu X , Karmally S , Shestopalov VI , Dvoriantchikova G , Ivanov D , Nathanson L , Barnum SR , Bethea JR ((2009) ) Transgenic inhibition of astroglial NF-kappa B improves functional outcome in experimental autoimmune encephalomyelitis by suppressing chronic central nervous system inflammation. J Immunol 182: , 2628–2640. |
[123] | Ho PP , Steinman L ((2016) ) Obeticholic acid, a synthetic bile acidagonist of the farnesoid X receptor, attenuatesexperimental autoimmune encephalomyelitis. Proc Natl Acad Sci US A 113: , 1600–1605. |
[124] | Heneka MT , Carson MJ , El Khoury J , Landreth GE , Brosseron F , Feinstein DL , Jacobs AH , Wyss-Coray T , Vitorica J , Ransohoff RM , Herrup K , Frautschy SA , Finsen B , Brown GC , Verkhratsky A , Yamanaka K , Koistinaho J , Latz E , Halle A , Petzold GC , Town T , Morgan D , Shinohara ML , Perry VH , Holmes C , Bazan NG , Brooks DJ , Hunot S , Joseph B , Deigendesch N , Garaschuk O , Boddeke E , Dinarello CA , Breitner JC , Cole GM , Golenbock DT , Kummer MP ((2015) ) Neuroinflammation in Alzheimer’s disease. Lancet. Neurol 14: , 388–405. |
[125] | Marchetti L , Klein M , Schlett K , Pfizenmaier K , Eisel ULM ((2004) ) Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-Methyl-D-aspartate receptor activation: Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-κB pathway. J Biol Chem 279: , 32869–32881. |
[126] | Paoletti P , Bellone C , Zhou Q ((2013) ) NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci 14: , 383–400. |
[127] | Newcomer JW , Farber NB , Olney JW ((2000) ) NMDA receptor function, memory, and brain aging. Dialogues Clin Neurosci 2: , 219–232. |
[128] | Traynelis SF , Wollmuth LP , McBain CJ , Menniti FS , Vance KM , Ogden KK , Hansen KB , Yuan H , Myers SJ , Dingledine R ((2010) ) Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol Rev 62: , 405–496. |
[129] | Bye CM , McDonald RJ ((2019) ) A specific role of hippocampal NMDA receptors and Arc protein in rapid encoding of novel environmental representations and a more general long-term consolidation function. Fron Behav Neurosci 13: , 8. |
[130] | McHugh TJ , Jones MW , Quinn JJ , Balthasar N , Coppari R , Elmquist JK , Lowell BB , Fanselow MS , Wilson MA , Tonegawa S ((2007) ) Dentate gyrus NMDA receptors mediate rapid pattern separation in the hippocampal network. Science 317: , 94–99. |
[131] | Mu Y , Zhao C , Toni N , Yao J , Gage FH ((2015) ) Distinct roles of NMDA receptors at different stages of granule cell development in the adult brain, Elife 4: , e07871. |
[132] | Platel JC , Kelsch W ((2013) ) Role of NMDA receptors in adult neurogenesis: An ontogenetic (re)view on activity-dependent development. Cell Mol Life Sci 70: , 3591–3601. |
[133] | Lledo PM , Alonso M , Grubb MS ((2006) ) Adult neurogenesis and functional plasticity in neuronal circuits. Nat Rev Neurosci 7: , 179–193. |
[134] | Huerta PT , Sun LD , Wilson MA , Tonegawa S ((2000) ) Formation of temporal memory requires NMDA receptors within CA1 pyramidal neurons. Neuron 25: , 473–480. |
[135] | Ally BA , Hussey EP , Ko PC , Molitor RJ ((2013) ) Pattern separation and pattern completion in Alzheimer’s disease: Evidence of rapid forgetting in amnestic mild cognitive impairment. Hippocampus 23: , 1246–1258. |
[136] | Morris RGM , Anderson E , Lynch GS , Baudry M ((1986) ) Selective impairment of learning and blockade of long-term potentiation by an N-methyl-D-aspartate receptor antagonist, AP5. Nature 319: , 774–776. |
[137] | Thompson DM , Winsauer PJ , Mastropaolo J ((1987) ) Effects of phencyclidine, ketamine and MDMA on complex operant behavior in monkeys. Pharmacol Biochem Behav 26: , 401–405. |
[138] | Song MS , Rauw G , Baker GB , Kar S ((2008) ) Memantine protects rat cortical cultured neurons against beta-amyloid-induced toxicity by attenuating tau phosphorylation. Eur J Neurosci 28: , 1989–2002. |
[139] | Schubring SR , Fleischer W , Lin JS , Haas HL , Sergeeva OA ((2012) ) The bile steroid chenodeoxycholate is a potent antagonist at NMDA and GABA(A) receptors. Neurosci Lett 506: , 322–326. |
[140] | Roda A , Cappelleri G , Aldini R , Roda E , Barbara L ((1982) ) Quantitative aspects of the interaction of bile acids with human serum albumin. J Lipid Res 23: , 490–495. |
[141] | Corcoran KA , Donnan MD , Tronson NC , Guzmán YF , Gao C , Jovasevic V , Guedea AL , Radulovic J ((2011) ) NMDA receptors in retrosplenialcortex are necessary for retrieval of recent and remote context fearmemory. J Neurosci 31: , 11655–11659. |
[142] | Platel JC , Dave KA , Gordon V , Lacar B , Rubio ME , Bordey A ((2010) ) NMDA receptors activated by subventricular zone astrocytic glutamate are critical for neuroblast survival prior to entering a synaptic network. Neuron 65: , 859–872. |
[143] | Deng W , Aimone JB , Gage FH ((2010) ) New neurons and new memories: How does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci 11: , 339–350. |
[144] | Roy DS , Arons A , Mitchell TI , Pignatelli M , Ryan TJ , Tonegawa S ((2016) ) Memory retrieval by activating engram cells in mouse models of early Alzheimer’s disease. Nature 531: , 508–512. |
[145] | Tian L , Stefanidakis M , Ning L , Van Lint P , Nyman-Huttunen H , Libert C , Itohara S , Mishina M , Rauvala H , Gahmberg CG ((2007) ) Activation of NMDA receptors promotes dendritic spine development through MMP-mediated ICAM-5 cleavage. J Cell Biol 178: , 687–700. |
[146] | Hojo Y , Hattori T-a , Enami T , Furukawa A , Suzuki K , Ishii H-t , Mukai H , Morrison JH , Janssen WGM , Kominami S , Harada N , Kimoto T , Kawato S ((2004) ) Adult male rat hippocampus synthesizes estradiol from pregnenolone by cytochromes P45017α and P450 aromatase localized in neurons. Proc Natl Acad Sci U S A 101: , 865–870. |
[147] | Srivastava DP , Woolfrey KM , Jones KA , Shum CY , Lash LL , Swanson GT , Penzes P ((2008) ) Rapid enhancement of two-step wiring plasticity by estrogen and NMDA receptor activity. Proc Natl Acad Sci U S A 105: , 14650–14655. |
[148] | Lambot L , Chaves Rodriguez E , Houtteman D , Li Y , Schiffmann SN , Gall D , de Kerchove d’Exaerde A ((2016) ) Striatopallidal neuron NMDA receptors control synaptic connectivity, locomotor, and goal-directed behaviors. J Neurosci 36: , 4976–4992. |
[149] | Chen SX , Tari PK , She K , Haas K ((2010) ) Neurexin-neuroligin cell adhesion complexes contribute to synaptotropic dendritogenesis via growth stabilization mechanisms in vivo. Neuron 67: , 967–983. |
[150] | Busse S , Brix B , Kunschmann R , Bogerts B , Stoecker W , Busse M ((2014) ) N-methyl-d-aspartate glutamate receptor (NMDA-R) antibodies in mild cognitive impairment and dementias. Neurosci Res 85: , 58–64. |
[151] | DeMorrow S , Grant S , Frampton G , McMillin M ((2017) ) Bile acid-mediated accumulation of brain cholesterol contributes to hepatic encephalopathy due to acute liver failure, FASEB 31: , 803.803. |
[152] | Paul SM , Doherty JJ , Robichaud AJ , Belfort GM , Chow BY , Hammond RS , Crawford DC , Linsenbardt AJ , Shu HJ , Izumi Y , Mennerick SJ , Zorumski CF ((2013) ) The major brain cholesterol metabolite24(S)-hydroxycholesterol is a potent allosteric modulator of N-methyl-D-aspartate receptors. J Neurosci 33: , 17290–17300. |
[153] | Kunz PA , Roberts AC , Philpot BD ((2013) ) Presynaptic NMDA receptor mechanisms for enhancing spontaneous neurotransmitter release. J Neurosci 33: , 7762–7769. |
[154] | McGuinness L , Taylor C , Taylor RD , Yau C , Langenhan T , Hart ML , Christian H , Tynan PW , Donnelly P , Emptage NJ ((2010) ) Presynaptic NMDARs in the hippocampus facilitate transmitter release at theta frequency. Neuron 68: , 1109–1127. |
[155] | Sze C , Bi H , Kleinschmidt-DeMasters BK , Filley CM , Martin LJ ((2001) ) N-Methyl-D-aspartate receptor subunit proteins and their phosphorylation status are altered selectively in Alzheimer’s disease. J Neurol Sci 182: , 151–159. |
[156] | Murray PS , Kumar S , Demichele-Sweet MAA , Sweet RA ((2014) ) Psychosis in Alzheimer’s disease. Biol Psychiatry 75: , 542–552. |
[157] | Chen YC , Hsu WL , Ma YL , Tai DJ , Lee EH ((2014) ) CREB SUMOylation by the E3 ligase PIAS1 enhances spatial memory. J Neurosci 34: , 9574–9589. |
[158] | Haddad JJ ((2005) ) N-methyl-D-aspartate (NMDA) and the regulation of mitogen-activated protein kinase (MAPK) signaling pathways: A revolving neurochemical axis for therapeutic intervention? Prog Neurobiol 77: , 252–282. |
[159] | Soleimanpour E , Bergado Acosta JR , Landgraf P , Mayer D , Dankert E , Dieterich DC , Fendt M ((2021) ) Regulation of CREB phosphorylation in nucleus accumbens after relief conditioning. Cells 10: , 238. |
[160] | Cignarella A , Kratz M , Bolego C ((2010) ) Emerging role of estrogen in the control of cardiometabolic disease. Trends Pharmacol Sci 31: , 183–189. |
[161] | Mukai H , Takata N , Ishii HT , Tanabe N , Hojo Y , Furukawa A , Kimoto T , Kawato S ((2006) ) Hippocampal synthesis of estrogens and androgens which are paracrine modulators of synaptic plasticity: Synaptocrinology. Neuroscience 138: , 757–764. |
[162] | Arevalo MA , Azcoitia I , Garcia-Segura LM ((2015) ) The neuroprotective actions of oestradiol and oestrogen receptors. Nat Rev Neurosci 16: , 17–29. |
[163] | Sohrabji F , Miranda RC , Toran-Allerand CD ((1995) ) Identification of a putative estrogen response element in the gene encoding brain-derived neurotrophic factor. Proc Natl Acad Sci U S A 92: , 11110–11114. |
[164] | Prossnitz ER , Barton M ((2011) ) The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol 7: , 715–726. |
[165] | Zhou J , Zhang H , Cohen RS , Pandey SC ((2005) ) Effects of estrogen treatment on expression of BDNF and CREB expression and phosphorylation in rat amygdaloid and hippocampal structures. Neuroendocrinology 81: , 294–310. |
[166] | Harte-Hargrove L , MacLusky NJ , Scharfman HE ((2013) ) BDNF-estrogen interactions in hippocampal mossy fiber pathway: Implications for normal brain function and disease. Neuroscience 239: , 46–66. |
[167] | Phan A , Suschkov S , Molinaro L , Reynolds K , Lymer JM , Bailey CD , Kow LM , MacLusky NJ , Pfaff DW , Choleris E ((2015) ) Rapid increases in immature synapses parallel estrogen-induced hippocampal learning enhancements. Proc Natl Acad Sci U S A 112: , 16018–16023. |
[168] | Khan MM , Dhandapani KM , Zhang QG , Brann DW ((2013) ) Estrogen regulation of spine density and excitatory synapses in rat prefrontal and somatosensory cerebral cortex. Steroids 78: , 614–623. |
[169] | Phan A , Gabor CS , Favaro KJ , Kaschack S , Armstrong JN , MacLusky NJ , Choleris E ((2012) ) Low doses of 17β-estradiol rapidly improve learning and increase hippocampal dendritic spines. Neuropsychopharmacology 37: , 2299–2309. |
[170] | Arevalo MA , Azcoitia I , Gonzalez-Burgos I , Garcia-Segura LM ((2015) ) Signaling mechanisms mediating the regulation of synaptic plasticity and memory by estradiol. Horm Behav 74: , 19–27. |
[171] | Almey A , Milner TA , Brake WG ((2015) ) Estrogen receptors in the central nervous system and their implication for dopamine-dependent cognition in females. Horm Behav 74: , 125–138. |
[172] | Bean LA , Kumar A , Rani A , Guidi M , Rosario AM , Cruz PE , Golde TE , Foster TC ((2015) ) Re-opening the critical window for estrogen therapy. J Neurosci 35: , 16077–16093. |
[173] | Han X , Aenlle KK , Bean LA , Rani A , Semple-Rowland SL , Kumar A , Foster TC ((2013) ) Role of estrogen receptor alpha and beta in preserving hippocampal function during aging. J Neurosci 33: , 2671–2683. |
[174] | Carroll JC , Pike CJ ((2008) ) Selective estrogen receptor modulators differentially regulate Alzheimer-like changes in female 3xTg-AD mice. Endocrinology 149: , 2607–2611. |
[175] | Cardona-Gomez P , Perez M , Avila J , Garcia-Segura LM , Wandosell F ((2004) ) Estradiol inhibits GSK3 and regulates interaction of estrogen receptors, GSK3, and beta-catenin in the hippocampus. Mol Cell Neurosci 25: , 363–373. |
[176] | Gollapudi L , Oblinger MM ((1999) ) Estrogen and NGF synergistically protect terminally differentiated, ERα-transfected PC12 cells from apoptosis. J Neurosci Res 56: , 471–481. |
[177] | Vadlamudi RK , Balasenthil S , Broaddus RR , Gustafsson JA , Kumar R ((2004) ) Deregulation of estrogen receptor coactivator proline-, glutamic acid-, and leucine-rich protein-1/modulator of nongenomic activity of estrogen receptor in human endometrial tumors. J Clin Endocrinol Metab 89: , 6130–6138. |
[178] | Grisouard J , Medunjanin S , Hermani A , Shukla A , Mayer D ((2007) ) Glycogen synthase kinase-3 protects estrogen receptor alpha from proteasomal degradation and is required for full transcriptional activity of the receptor. Mol Endocrinol 21: , 2427–2439. |
[179] | Ma Y , Liu M , Yang L , Zhang L , Guo H , Hou W , Qin P ((2020) ) Loss of estrogen efficacy against hippocampus damage in long-term OVX mice is related to the reduction of hippocampus local estrogen production and estrogen receptor degradation, Mol Neurobiol 57: , 3540–3551. |
[180] | Vegeto E , Belcredito S , Etteri S , Ghisletti S , Brusadelli A , Meda C , Krust A , Dupont S , Ciana P , Chambon P , Maggi A ((2003) ) Estrogen receptor-alpha mediates the brain antiinflammatory activity of estradiol. Proc Natl Acad Sci U S A 100: , 9614–9619. |
[181] | D’Astous M , Mendez P , Morissette M , Garcia-Segura LM , Di Paolo T ((2006) ) Implication of the phosphatidylinositol-3 kinase/protein kinase B signaling pathway in the neuroprotective effect of estradiol in the striatum of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mice. Mol Pharmacol 69: , 1492–1498. |
[182] | Zhao L , Brinton RD ((2007) ) Estrogen receptor α and β differentially regulate intracellular Ca2+dynamics leading to ERK phosphorylation and estrogen neuroprotection in hippocampal neurons. Brain Res 1172: , 48–59. |
[183] | Tang H , Zhang Q , Yang L , Dong Y , Khan M , Yang F , Brann DW , Wang R ((2014) ) GPR30 mediates estrogen rapid signalingand neuroprotection. Mol Cell Endocrinol 387: , 52–58. |
[184] | Yang LC , Zhang QG , Zhou CF , Yang F , Zhang YD , Wang RM , Brann DW ((2010) ) Extranuclear estrogen receptors mediate the neuroprotective effects of estrogen in the rat hippocampus, PLoS One 5: , e9851. |
[185] | Sareddy GR , Zhang Q , Wang R , Scott E , Zou Y , Connor JC , Chen Y , Dong Y , Vadlamudi RK , Brann D ((2015) ) Proline-, glutamic acid-, and leucine-rich protein 1 mediates estrogen rapid signaling and neuroprotection in the brain. Proc Natl Acad Sci U S A 112: , E6673. |
[186] | Thakkar R , Wang R , Sareddy G , Wang J , Thiruvaiyaru D , Vadlamudi R , Zhang Q , Brann D ((2016) ) NLRP3 inflammasome activation in the brain after global cerebral ischemia and regulation by 17beta-estradiol. Oxid Med Cell Longev 2016: , 8309031. |
[187] | Potier M , Georges F , Brayda-Bruno L , Ladepeche L , Lamothe V , Al Abed AS , Groc L , Marighetto A ((2016) ) Temporal memory and its enhancement by estradiol requires surface dynamics of hippocampal CA1 N-Methyl-D-Aspartate receptors. Biol Psychiatry 79: , 735–745. |
[188] | Smith CC , Smith LA , Bredemann TM , McMahon LL ((2016) ) 17beta estradiol recruits GluN2B-containing NMDARs and ERK during induction of long-term potentiation at temporoammonic-CA1 synapses. Hippocampus 26: , 110–117. |
[189] | Galea LA , Wainwright SR , Roes MM , Duarte-Guterman P , Chow C , Hamson DK ((2013) ) Sex, hormones and neurogenesis in the hippocampus: Hormonal modulation of neurogenesis and potential functional implications. J Neuroendocrinol 25: , 1039–1061. |
[190] | Tanenbaum DM , Wang Y , Williams SP , Sigler PB ((1998) ) Crystallographic comparison of the estrogen and progesterone receptor’s ligand binding domains. Proc Natl Acad Sci U S A 95: , 5998–6003. |
[191] | Brzozowski AM , Pike ACW , Dauter Z , Hubbard RE , Bonn T , Engström O , Öhman L , Greene GL , Gustafsson J-Å , Carlquist M ((1997) ) Molecular basis of agonism and antagonism in the oestrogen receptor. Nature 389: , 753–758. |
[192] | May FE ((2014) ) Novel drugs that target the estrogen-related receptor alpha: Their therapeutic potential in breast cancer. Cancer Manag Res 6: , 225–252. |
[193] | Fiorucci S , Clerici C , Antonelli E , Orlandi S , Goodwin B , Sadeghpour BM , Sabatino G , Russo G , Castellani D , Willson TM , Pruzanski M , Pellicciari R , Morelli A ((2005) ) Protective effects of 6-ethyl chenodeoxycholic acid, a farnesoid X receptor ligand, in estrogen-induced cholestasis. J Pharmacol Exp Ther 313: , 604–612. |
[194] | Eberhardt J , Santos-Martins D , Tillack AF , Forli S ((2021) ) AutoDock Vina 1.2.0: New docking methods, expanded force field, and python bindings. J Chem Inf Model 61: , 3891–3898. |
[195] | Trott O , Olson AJ ((2010) ) AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31: , 455–461. |
[196] | Chen Y , Vasilenko A , Song X , Valanejad L , Verma R , You S , Yan B , Shiffka S , Hargreaves L , Nadolny C , Deng R ((2015) ) Estrogen and estrogen receptor-alpha-mediated transrepression of bile salt export pump. Mol Endocrinol 29: , 613–626. |
[197] | Finan B , Yang B , Ottaway N , Stemmer K , Müller TD , Yi C-X , Habegger K , Schriever SC , García-Cáceres C , Kabra DG , Hembree J , Holland J , Raver C , Seeley RJ , Hans W , Irmler M , Beckers J , de Angelis MH , Tiano JP , Mauvais-Jarvis F , Perez-Tilve D , Pfluger P , Zhang L , Gelfanov V , DiMarchi RD , Tschöp MH ((2012) ) Targeted estrogen delivery reverses the metabolic syndrome. Nat Med 18: , 1847–1856. |
[198] | Huang GZ , Woolley CS ((2012) ) Estradiol acutely suppresses inhibition in the hippocampus through a sex-specific endocannabinoid and mGluR-dependent mechanism. Neuron 74: , 801–808. |
[199] | Kwakowsky A , Calvo-Flores Guzmán B , Govindpani K , Waldvogel HJ , Faull RL ((2018) ) Gamma-aminobutyric acid A receptors in Alzheimer’sdisease: Highly localized remodeling of a complex and diversesignaling pathway. Neural Regen Res 13: , 1362–1363. |
[200] | Guo C , Xie S , Chi Z , Zhang J , Liu Y , Zhang L , Zheng M , Zhang X , Xia D , Ke Y , Lu L , Wang D ((2016) ) Bile acids control inflammation and metabolic disorder through inhibition of NLRP3 inflammasome. Immunity 45: , 802–816. |
[201] | Makishima M , Lu TT , Xie W , Whitfield GK , Domoto H , Evans RM , Haussler MR , Mangelsdorf DJ ((2002) ) Vitamin D receptor as an intestinal bile acid sensor. Science 296: , 1313–1316. |
[202] | Tohgi H , Utsugisawa K , Yamagata M , Yoshimura M ((1995) ) Effects of age on messenger RNA expression of glucocorticoid, thyroid hormone, androgen, and estrogen receptors in postmortem human hippocampus. Brain Res 700: , 245–253. |
[203] | Luu TH , Bard JM , Carbonnelle D , Chaillou C , Huvelin JM , Bobin-Dubigeon C , Nazih H ((2018) ) Lithocholic bile acid inhibits lipogenesis and induces apoptosis in breast cancer cells. Cell Oncol (Dordr) 41: , 13–24. |
[204] | Ishunina TA , Swaab DF ((2001) ) Increased expression of estrogen receptor alpha and beta in the nucleus basalis of Meynert in Alzheimer’s disease. Neurobiol Aging 22: , 417–426. |
[205] | Wang C , Zhang F , Jiang S , Siedlak SL , Shen L , Perry G , Wang X , Tang B , Zhu X ((2016) ) Estrogen receptor-alpha is localized to neurofibrillary tangles in Alzheimer’s disease. Sci Rep 6: , 20352. |
[206] | Wang AC , Hara Y , Janssen WG , Rapp PR , Morrison JH ((2010) ) Synaptic estrogen receptor-alpha levels in prefrontal cortex in female rhesus monkeys and their correlation with cognitive performance. J Neurosci 30: , 12770–12776. |
[207] | Fortress AM , Frick KM ((2014) ) Epigenetic regulation of estrogen-dependent memory. Fron Neuroendocrinol 35: , 530–549. |
[208] | Kelly JF , Bienias JL , Shah A , Meeke KA , Schneider JA , Soriano E , Bennett DA ((2008) ) Levels of estrogen receptors α and β in frontal cortex of patients with Alzheimer’s disease: Relationship to Mini-Mental State Examination scores. Curr Alzheimer Res 5: , 45–51. |
[209] | Azcoitia I , Sierra A , Garcia-Segura LM ((1999) ) Neuroprotective effects of estradiol in the adult rat hippocampus: Interaction with insulin-like growth factor-I signalling. J Neurosci Res 58: , 815–822. |
[210] | Salehi Z , Mashayekhi F , Naji M ((2008) ) Insulin like growth factor-1 and insulin like growth factor binding proteins in the cerebrospinal fluid and serum from patients with Alzheimer’s disease. Biofactors 33: , 99–106. |
[211] | Kitamura Y , Shimohama S , Kamoshima W , Ota T , Matsuoka Y , Nomura Y , Smith MA , Perry G , Whitehouse PJ , Taniguchi T ((1998) ) Alteration ofproteins regulating apoptosis, Bcl-2, Bcl-x, Bax, Bak, Bad, ICH-1and CPP32, in Alzheimer’s disease., Brain Res 780: , 260–269. |
[212] | Leroy K , Yilmaz Z , Brion J-P ((2007) ) Increased level of active GSK-3β in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol Appl Neurobiol 33: , 43–55. |
[213] | Ohtsuki S , Watanabe Y , Hori S , Suzuki H , Bhongsatiern J , Fujiyoshi M , Kamoi M , Kamiya N , Takanaga H , Terasaki T ((2004) ) mRNA expression of the ATP-binding cassette transporter subfamily A (ABCA) in rat and human brain capillary endothelial cells. Biol Pharm Bull 27: , 1437–1440. |
[214] | Urich E , Lazic SE , Molnos J , Wells I , Freskgård PO ((2012) ) Transcriptional profiling of human brain endothelial cells reveals key properties crucial for predictive in vitroblood-brain barrier models. PLoS One 7: , e38149. |
[215] | Ahn SI , Sei YJ , Park H-J , Kim J , Ryu Y , Choi JJ , Sung H-J , MacDonald TJ , Levey AI , Kim Y ((2020) ) Microengineered human blood– brain barrier platform for understanding nanoparticle transport mechanisms. Nat Commun 11: , 175. |
[216] | Panzenboeck U , Balazs Z , Sovic A , Hrzenjak A , Levak-Frank S , Wintersperger A , Malle E , Sattler W ((2002) ) ABCA1 and scavenger receptor class B, type I, are modulators of reverse sterol transport at an in vitro blood-brain barrier constituted of porcine brain capillary endothelial cells. J Biol Chem 277: , 42781–42789. |
[217] | Fan J , Zhao RQ , Parro C , Zhao W , Chou H-Y , Robert J , Deeb TZ , Raynoschek C , Barichievy S , Engkvist O , Maresca M , Hicks R , Meuller J , Moss SJ , Brandon NJ , Wood MW , Kulic I , Wellington CL ((2018) ) Small molecule inducers of ABCA1 and apoE that act through indirect activation of the LXR pathway. J Lipid Res 59: , 830–842. |
[218] | Kim J , Yoon H , Horie T , Burchett JM , Restivo JL , Rotllan N , Ramírez CM , Verghese PB , Ihara M , Hoe H-S , Esau C , Fernández-Hernando C , Holtzman DM , Cirrito JR , Ono K , Kim J ((2015) ) microRNA-33 regulates ApoE lipidation and amyloid-β metabolism in the brain. J Neurosci 35: , 14717–14726. |
[219] | Wahrle SE , Jiang H , Parsadanian M , Legleiter J , Han X , Fryer JD , Kowalewski T , Holtzman DM ((2004) ) ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem 279: , 40987–40993. |
[220] | Akanuma S , Ohtsuki S , Doi Y , Tachikawa M , Ito S , Hori S , Asashima T , Hashimoto T , Yamada K , Ueda K , Iwatsubo T , Terasaki T ((2008) ) ATP-binding cassette transporter A1 (ABCA1) deficiency does not attenuate the brain-to-blood efflux transport of human amyloid-beta peptide (1-40) at the blood-brain barrier. Neurochem Int 52: , 956–961. |
[221] | Hirsch-Reinshagen V , Zhou S , Burgess BL , Bernier L , McIsaac SA , Chan JY , Tansley GH , Cohn JS , Hayden MR , Wellington CL ((2004) ) Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J Biol Chem 279: , 41197–41207. |
[222] | Do TM , Ouellet M , Calon F , Chimini G , Chacun H , Farinotti R , Bourasset F ((2011) ) Direct evidence of abca1-mediated efflux of cholesterol at the mouse blood– brain barrier. Mol Cell Biochem 357: , 397–404. |
[223] | Karasinska JM , de Haan W , Franciosi S , Ruddle P , Fan J , Kruit JK , Stukas S , Lutjohann D , Gutmann DH , Wellington CL , Hayden MR ((2013) ) ABCA1 influences neuroinflammation and neuronal death. Neurobiol Dis 54: , 445–455. |
[224] | Jiang Q , Lee CYD , Mandrekar S , Wilkinson B , Cramer P , Zelcer N , Mann K , Lamb B , Willson TM , Collins JL , Richardson JC , Smith JD , Comery TA , Riddell D , Holtzman DM , Tontonoz P , Landreth GE ((2008) ) ApoE promotes the proteolytic degradation of Abeta. Neuron 58: , 681–693. |
[225] | Kim WS , Hill AF , Fitzgerald ML , Freeman MW , Evin G , Garner B ((2011) ) Wild type and Tangier disease ABCA1 mutants modulate cellular amyloid-β production independent of cholesterol efflux activity. J Alzheimers Dis 27: , 441–452. |
[226] | Elali A , Rivest S ((2013) ) The role of ABCB1 and ABCA1 in beta-amyloid clearance at the neurovascular unit in Alzheimer’s disease. Front Physiol 4: , 45. |
[227] | Qiu WQ , Walsh DM , Ye Z , Vekrellis K , Zhang J , Podlisny MB , Rosner MR , Safavi A , Hersh LB , Selkoe DJ ((1998) ) Insulin-degrading enzyme regulates extracellular levels of amyloid beta-protein by degradation. J Biol Chem 273: , 32730–32738. |
[228] | Tokuda T , Calero M , Matsubara E , Vidal R , Kumar A , Permanne B , Zlokovic B , Smith JD , Ladu MJ , Rostagno A , Frangione B , Ghiso J ((2000) ) Lipidation of apolipoprotein E influences its isoform-specific interaction with Alzheimer’s amyloid beta peptides. Biochem J 348 Pt 2: , 359–365. |
[229] | Wahrle SE , Jiang H , Parsadanian M , Kim J , Li A , Knoten A , Jain S , Hirsch-Reinshagen V , Wellington CL , Bales KR , Paul SM , Holtzman DM ((2008) ) Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest 118: , 671–682. |
[230] | Wahrle SE , Jiang H , Parsadanian M , Hartman RE , Bales KR , Paul SM , Holtzman DM ((2005) ) Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J Biol Chem 280: , 43236–43242. |
[231] | Wang L , Liu W , Fan Y , Liu T , Yu C ((2017) ) Effect of rosiglitazone on amyloid precursor protein processing and Abeta clearance in streptozotocin-induced rat model of Alzheimer’s disease. Iran J Basic Med Sci 20: , 474–480. |
[232] | Kim WS , Bhatia S , Elliott DA , Agholme L , Kagedal K , McCann H , Halliday GM , Barnham KJ , Garner B ((2010) ) Increased ATP-binding cassette transporter A1 expression in Alzheimer’s disease hippocampal neurons. J Alzheimers Dis 21: , 193–205. |
[233] | Schreurs BG , Sparks DL ((2015) ) Dietary high cholesterol and trace metals in the drinking water increase levels of ABCA1 in the rabbit hippocampus and temporal cortex. J Alzheimers Dis 49: , 201–209. |
[234] | Donkin JJ , Stukas S , Hirsch-Reinshagen V , Namjoshi D , Wilkinson A , May S , Chan J , Fan J , Collins J , Wellington CL ((2010) ) ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J Biol Chem 285: , 34144–34154. |
[235] | Li G , Kim C , Kim J , Yoon H , Zhou H , Kim J ((2015) ) Common pesticide, dichlorodiphenyltrichloroethane (DDT), increases amyloid-β levels by impairing the function of ABCA1 and IDE: Implication for Alzheimer’s disease. J Alzheimers Dis 46: , 109–122. |
[236] | Staudinger JL , Goodwin B , Jones SA , Hawkins-Brown D , MacKenzie KI , LaTour A , Liu Y , Klaassen CD , Brown KK , Reinhard J , Willson TM , Koller BH , Kliewer SA ((2001) ) The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci U S A 98: , 3369–3374. |
[237] | Tovar-Palacio C , Torres N , Diaz-Villaseñor A , Tovar AR ((2012) ) The role of nuclear receptors in the kidney in obesity and metabolic syndrome. Genes Nutr 7: , 483–498. |
[238] | Hartz AM , Miller DS , Bauer B ((2010) ) Restoring blood-brain barrier P-glycoprotein reduces brain amyloid-beta in a mouse model of Alzheimer’s disease. Mol Pharmacol 77: , 715–723. |
[239] | Vogelgesang S , Warzok RW , Cascorbi I , Kunert-Keil C , Schroeder E , Kroemer HK , Siegmund W , Walker LC , Pahnke J ((2004) ) The role of P-glycoprotein in cerebral amyloid angiopathy; implications for theearly pathogenesis of Alzheimer’s disease. Curr Alzheimer Res 1: , 121–125. |
[240] | Jeynes B , Provias J ((2013) ) P-glycoprotein altered expression in Alzheimer’s disease: Regional anatomic variability. J Neurodegener Dis 2013: , 257953. |
[241] | Mitro N , Vargas L , Romeo R , Koder A , Saez E ((2007) ) T0901317 is a potent PXR ligand: Implications for the biology ascribed to LXR. FEBS Lett 581: , 1721–1726. |
[242] | Langmade SJ , Gale SE , Frolov A , Mohri I , Suzuki K , Mellon SH , Walkley SU , Covey DF , Schaffer JE , Ory DS ((2006) ) Pregnane X receptor (PXR) activation: A mechanism for neuroprotection in a mouse model of Niemann-Pick C disease. Proc Natl Acad Sci U S A 103: , 13807–13812. |
[243] | Lund EG , Xie C , Kotti T , Turley SD , Dietschy JM , Russell DW ((2003) ) Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J Biol Chem 278: , 22980–22988. |
[244] | Lund EG , Guileyardo JM , Russell DW ((1999) ) cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc Natl Acad Sci U S A 96: , 7238–7243. |
[245] | Dietschy JM , Turley SD ((2004) ) Thematic review series: Brain Lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res 45: , 1375–1397. |
[246] | Björkhem I , Lütjohann D , Diczfalusy U , Ståhle L , Ahlborg G , Wahren J ((1998) ) Cholesterol homeostasis in human brain: Turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J Lipid Res 39: , 1594–1600. |
[247] | Lütjohann D , Breuer O , Ahlborg G , Nennesmo I , Sidén A , Diczfalusy U , Björkhem I ((1996) ) Cholesterol homeostasis in human brain: Evidence for an age-deendent flux of 24S-hydroxycholesterolfrom the brain into the circulation. Proc Natl Acad Sci U S A 93: , 9799–9804 . |
[248] | Sun M-Y , Linsenbardt AJ , Emnett CM , Eisenman LN , Izumi Y , Zorumski CF , Mennerick S ((2016) ) 24(S)-hydroxycholesterol as a modulator of neuronal signaling and survival. Neuroscientist 22: , 132–144. |
[249] | Ogundare M , Theofilopoulos S , Lockhart A , Hall LJ , Arenas E , Sjovall J , Brenton AG , Wang Y , Griffiths WJ ((2010) ) Cerebrospinal fluid steroidomics: Are bioactive bile acids present in brain? J Biol Chem 285: , 4666–4679. |
[250] | Abildayeva K , Jansen PJ , Hirsch-Reinshagen V , Bloks VW , Bakker AH , Ramaekers FC , de Vente J , Groen AK , Wellington CL , Kuipers F , Mulder M ((2006) ) 24(S)-hydroxycholesterol participates in a liver X receptor-controlled pathway in astrocytes that regulates apolipoprotein E-mediated cholesterol efflux. J Biol Chem 281: , 12799–12808. |
[251] | Cartagena CM , Burns MP , Rebeck GW ((2010) ) 24S-hydroxycholesterol effects on lipid metabolism genes are modeled in traumatic brain injury. Brain Res 1319: , 1–12. |
[252] | Leoni V , Caccia C ((2013) ) 24S-hydroxycholesterol in plasma: A marker of cholesterol turnover in neurodegenerative diseases. Biochimie 95: , 595–612. |
[253] | Okabe A , Urano Y , Itoh S , Suda N , Kotani R , Nishimura Y , Saito Y , Noguchi N ((2014) ) Adaptive responses induced by 24S-hydroxycholesterol through liver X receptor pathway reduce 7-ketocholesterol-caused neuronal cell death. Redox Biol 2: , 28–35. |
[254] | Fukumoto H , Deng A , Irizarry MC , Fitzgerald ML , Rebeck GW ((2002) ) Induction of the cholesterol transporter ABCA1 in central nervous system cells by liver X receptor agonists increases secreted Abeta levels. J Biol Chem 277: , 48508–48513. |
[255] | Prasanthi JR , Huls A , Thomasson S , Thompson A , Schommer E , Ghribi O ((2009) ) Differential effects of 24-hydroxycholesterol and 27-hydroxycholesterol on beta-amyloid precursor protein levels and processing in human neuroblastoma SH-SY5Y cells. Mol Neurodegener 4: , 1. |
[256] | Bretillon L , Lütjohann D , Ståhle L , Widhe T , Bindl L , Eggertsen G , Diczfalusy U , Björkhem I ((2000) ) Plasma levels of 24S-hydroxycholesterol reflect the balance between cerebral production and hepatic metabolism and are inversely related to body surface. J Lipid Res 41: , 840–845. |
[257] | Takaki Y , Mizuochi T , Takei H , Eda K , Konishi K-i , Ishihara J , Kinoshita M , Hashizume N , Yamashita Y , Nittono H , Kimura A ((2020) ) Urinary and serum oxysterols in children: Developmental pattern and potential biomarker for pediatric liver disease. Sci Rep 10: , 6752. |
[258] | Meng LJ , Griffiths WJ , Nazer H , Yang Y , Sjövall J ((1997) ) High levels of (24S)-24-hydroxycholesterol 3-sulfate, 24-glucuronide in the serum and urine of children with severe cholestatic liver disease. J Lipid Res 38: , 926–934. |
[259] | Roy D , Chakrabarti SS , Banerjee A , Sharma P , Biswas A , Chakrabarti S ((2019) ) Serum 24-hydroxycholesterol in probable Alzheimer’s dementia: Reexploring the significance of a tentative Alzheimer’s disease biomarker. Aging Med (Milton) 2: , 74–81. |
[260] | Bjorkhem I , Andersson U , Ellis E , Alvelius G , Ellegard L , Diczfalusy U , Sjovall J , Einarsson C ((2001) ) From brain to bile. Evidence that conjugation and omega-hydroxylation are important for elimination of 24S-hydroxycholesterol (cerebrosterol) in humans. J Biol Chem 276: , 37004–37010. |
[261] | Bretillon L , Sidén Å , Wahlund L-O , Lütjohann D , Minthon L , Crisby M , Hillert J , Groth C-G , Diczfalusy U , Björkhem I ((2000) ) Plasma levels of 24S-hydroxycholesterol in patients withneurological diseases. Neurosci Lett 293: , 87–90. |
[262] | Papassotiropoulos A , Lütjohann D , Bagli M , Locatelli S , Jessen F , Rao ML , Maier W , Björkhem I , von Bergmann K , Heun R ((2000) ) Plasma 24S-hydroxycholesterol: A peripheral indicator of neuronal degeneration and potential state marker for Alzheimer’s disease. Neuroreport 11: , 1959–1962. |
[263] | Kölsch H , Lütjohann D , Jessen F , Popp J , Hentschel F , Kelemen P , Schmitz S , Maier W , Heun R ((2009) ) CYP46A1 variants influence Alzheimer’s disease risk and brain cholesterol metabolism. Eur Psychiatry 24: , 183–190. |
[264] | Brown J 3rd , Theisler C , Silberman S , Magnuson D , Gottardi-Littell N , Lee JM , Yager D , Crowley J , Sambamurti K , Rahman MM , Reiss AB , Eckman CB , Wolozin B ((2004) ) Differential expression of cholesterol hydroxylases in Alzheimer’s disease. J Biol Chem 279: , 34674–34681. |
[265] | Bogdanovic N , Bretillon L , Lund EG , Diczfalusy U , Lannfelt L , Winblad B , Russell DW , Björkhem I ((2001) ) On the turnover of brain cholesterol in patients with Alzheimer’s disease. Abnormal induction of the cholesterol-catabolic enzyme CYP46 in glial cells. Neurosci Lett 314: , 45–48. |
[266] | Grant S , McMillin M , Frampton G , Petrescu AD , Williams E , Jaeger V , Kain J , DeMorrow S ((2018) ) Direct comparison of the thioacetamide and azoxymethane models of type A hepatic encephalopathy in mice. Gene Expression 18: , 171–185. |
[267] | Simpson JE , Ince PG , Minett T , Matthews FE , Heath PR , Shaw PJ , Goodall E , Garwood CJ , Ratcliffe LE , Brayne C , Rattray M , Wharton SB ((2016) ) Neuronal DNA damage response-associated dysregulation of signalling pathways and cholesterol metabolism at the earliest stages of Alzheimer-type pathology. Neuropathol Appl Neurobiol 42: , 167–179. |
[268] | Testa G , Staurenghi E , Zerbinati C , Gargiulo S , Iuliano L , Giaccone G , Fanto F , Poli G , Leonarduzzi G , Gamba P ((2016) ) Changes in brain oxysterols at different stages of Alzheimer’s disease: Their involvement in neuroinflammation. Redox Biol 10: , 24–33. |
[269] | Papassotiropoulos A , Lütjohann D , Bagli M , Locatelli S , Jessen F , Buschfort R , Ptok U , Björkhem I , von Bergmann K , Heun R ((2002) ) 24S-hydroxycholesterol in cerebrospinal fluid is elevated in early stages of dementia. J Psychiatr Res 36: , 27–32. |
[270] | Griffiths WJ , Abdel-Khalik J , Yutuc E , Roman G , Warner M , Gustafsson J-Å , Wang Y ((2019) ) Concentrations of bile acid precursors in cerebrospinal fluid of Alzheimer’s disease patients. Free Radic Biol Med 134: , 42–52. |
[271] | Nho K , Kueider-Paisley A , MahmoudianDehkordi S , Arnold M , Risacher SL , Louie G , Blach C , Baillie R , Han X , Kastenmüller G , Jia W , Xie G , Ahmad S , Hankemeier T , van Duijn CM , Trojanowski JQ , Shaw LM , Weiner MW , Doraiswamy PM , Saykin AJ , Kaddurah-Daouk R , Alzheimer’s Disease Neuroimaging Initiative, the Alzheimer Disease Metabolomics Consortium ((2019) ) Altered bile acid profile in mild cognitive impairment and Alzheimer’s disease: Relationship to neuroimaging and CSF biomarkers. Alzheimers Dement 15: , 232–244. |
[272] | Kawamata Y , Fujii R , Hosoya M , Harada M , Yoshida H , Miwa M , Fukusumi S , Habata Y , Itoh T , Shintani Y , Hinuma S , Fujisawa Y , Fujino M ((2003) ) A G protein-coupled receptor responsive to bile acids. J Biol Chem 278: , 9435–9440. |
[273] | Eggink HM , Soeters MR , Pols TW ((2014) ) TGR5 ligands as potential therapeutics in inflammatory diseases. Int J Infereron Cytokine Mediator Res 6: , 27–38. |
[274] | Spomer L , Gertzen CG , Schmitz B , Haussinger D , Gohlke H , Keitel V ((2014) ) A membrane-proximal, C-terminal alpha-helix is required for plasma membrane localization and function of the G Protein-coupled receptor (GPCR) TGR5. J Biol Chem 289: , 3689–3702. |
[275] | Maruyama T , Miyamoto Y , Nakamura T , Tamai Y , Okada H , Sugiyama E , Nakamura T , Itadani H , Tanaka K ((2002) ) Identification of membrane-type receptor for bile acids (M-BAR). Biochem Biophys Res Commun 298: , 714–719. |
[276] | Li T , Chiang JYL ((2014) ) Bile acid signaling in metabolic disease and drug therapy. Pharmacol Rev 66: , 948–983. |
[277] | Gertzen CG , Spomer L , Smits SH , Haussinger D , Keitel V , Gohlke H ((2015) ) Mutational mapping of the transmembrane binding site of the G-protein coupled receptor TGR5 and binding mode prediction of TGR5 agonists. Eur J Med Chem 104: , 57–72. |
[278] | Macchiarulo A , Gioiello A , Thomas C , Pols TW , Nuti R , Ferrari C , Giacche N , De Franco F , Pruzanski M , Auwerx J , Schoonjans K , Pellicciari R ((2013) ) Probing the binding site of bile acids in TGR5. ACS Med Chem Lett 4: , 1158–1162. |
[279] | Yasuda H , Hirata S , Inoue K , Mashima H , Ohnishi H , Yoshiba M ((2007) ) Involvement of membrane-type bile acid receptor M-BAR/TGR5 in bile acid-induced activation of epidermal growth factor receptor and mitogen-activated protein kinases in gastric carcinoma cells. Biochem Biophys Res Commun 354: , 154–159. |
[280] | Yanguas-Casas N , Barreda-Manso MA , Nieto-Sampedro M , Romero-Ramirez L ((2017) ) TUDCA: An agonist of the bile acid receptor GPBAR1/TGR5with anti-inflammatory effects in microglial cells. J CellPhysiol 232: , 2231–2245. |
[281] | Karababa A , Groos-Sahr K , Albrecht U , Keitel V , Shafigullina A , Gorg B , Haussinger D ((2017) ) Ammonia attenuates LPS-induced upregulation of pro-inflammatory cytokine mRNA in co-cultured astrocytes and microglia. Neurochem Res 42: , 737–749. |
[282] | Hogenauer K , Arista L , Schmiedeberg N , Werner G , Jaksche H , Bouhelal R , Nguyen DG , Bhat BG , Raad L , Rauld C , Carballido JM ((2014) ) G-protein-coupled bile acid receptor 1 (GPBAR1, TGR5) agonists reduce the production of proinflammatory cytokines and stabilize the alternative macrophage phenotype. J Med Chem 57: , 10343–10354. |
[283] | Pols TW , Nomura M , Harach T , Lo Sasso G , Oosterveer MH , Thomas C , Rizzo G , Gioiello A , Adorini L , Pellicciari R , Auwerx J , Schoonjans K ((2011) ) TGR5 activation inhibits atherosclerosis by reducing macrophage inflammation and lipid loading. Cell Metab 14: , 747–757. |
[284] | Lewis ND , Patnaude LA , Pelletier J , Souza DJ , Lukas SM , King FJ , Hill JD , Stefanopoulos DE , Ryan K , Desai S , Skow D , Kauschke SG , Broermann A , Kuzmich D , Harcken C , Hickey ER , Modis LK ((2014) ) A GPBAR1 (TGR5) small molecule agonist shows specific inhibitory effects on myeloid cell activation in vitro and reduces experimental autoimmune encephalitis (EAE) in vivo. PLoS One 9: , e100883. |
[285] | Mobraten K , Haugbro T , Karlstrom E , Kleiveland CR , Lea T ((2015) ) Activation of the bile acid receptor TGR5 enhances LPS-induced inflammatory responses in a human monocytic cell line. J Recept Signal Transduct Res 35: , 402–409. |
[286] | McMillin M , Frampton G , Tobin R , Dusio G , Smith J , Shin H , Newell-Rogers K , Grant S , DeMorrow S ((2015) ) TGR5 signaling reduces neuroinflammation during hepatic encephalopathy. J Neurochem 135: , 565–576. |
[287] | Elia AE , Lalli S , Monsurrò MR , Sagnelli A , Taiello AC , Reggiori B , La Bella V , Tedeschi G , Albanese A ((2016) ) Tauroursodeoxycholic acid in the treatment of patients with amyotrophic lateral sclerosis. Eur J Neurol 23: , 45–52. |
[288] | Wu X , Lv YG , Du YF , Chen F , Reed MN , Hu M , Suppiramaniam V , Tang SS , Hong H ((2018) ) Neuroprotective effects of INT-777 against Abeta1-42-induced cognitive impairment, neuroinflammation, apoptosis, and synaptic dysfunction in mice. Brain Behav Immun 73: , 533–545. |
[289] | Wu H , Yu N , Wang X , Yang Y , Liang H ((2020) ) Tauroursodeoxycholic acid attenuates neuronal apoptosis via the TGR5/SIRT3 pathway after subarachnoid hemorrhage in rats. Biol Res 53: , 56–56. |
[290] | Hu X , Yan J , Huang L , Araujo C , Peng J , Gao L , Liu S , Tang J , Zuo G , Zhang JH ((2021) ) INT-777 attenuates NLRP3-ASC inflammasome-mediated neuroinflammation via TGR5/cAMP/PKA signaling pathway after subarachnoid hemorrhage in rats. Brain Behav Immun 91: , 587–600. |
[291] | Liang H , Matei N , McBride DW , Xu Y , Tang J , Luo B , Zhang JH ((2020) ) Activation of TGR5 protects blood brain barrier via the BRCA1/Sirt1pathway after middle cerebral artery occlusion in rats. J Biomed Sci 27: , 61–61. |
[292] | Zuo G , Zhang T , Huang L , Araujo C , Peng J , Travis Z , Okada T , Ocak U , Zhang G , Tang J , Lu X , Zhang JH ((2019) ) Activation of TGR5 with INT-777 attenuates oxidative stress and neuronal apoptosis via cAMP/PKCɛ/ALDH2 pathway after subarachnoid hemorrhage in rats. Free Radic Biol Med 143: , 441–453. |
[293] | Sato H , Genet C , Strehle A , Thomas C , Lobstein A , Wagner A , Mioskowski C , Auwerx J , Saladin R ((2007) ) Anti-hyperglycemic activity of a TGR5 agonist isolated from Olea europaea. Biochem Biophys Res Commun 362: , 793–798. |
[294] | Martin R , Carvalho-Tavares J , Hernandez M , Arnes M , Ruiz-Gutierrez V , Nieto ML ((2010) ) Beneficial actions of oleanolic acid in an experimental model of multiple sclerosis: A potential therapeutic role. Biochem Pharmacol 79: , 198–208. |
[295] | Liu N , Feng J , Lv Y , Liu Q , Deng J , Xia Y , Guo C , Zhou Y ((2019) ) Role of bile acids in the diagnosis and progression of liver cirrhosis: A prospective observational study. Exp Ther Med 18: , 4058–4066. |
[296] | Jena PK , Sheng L , Di Lucente J , Jin L-W , Maezawa I , Wan Y-JY ((2018) ) Dysregulated bile acid synthesis and dysbiosis are implicated in Western diet-induced systemic inflammation, microglial activation, and reduced neuroplasticity. FASEB 32: , 2866–2877. |
[297] | Adlimoghaddam A , Sabbir MG , Albensi BC ((2016) ) Ammonia as a potential neurotoxic factor in Alzheimer’s disease. Front Mol Neurosci 9: , 57–57. |
[298] | Alemi F , Kwon E , Poole DP , Lieu T , Lyo V , Cattaruzza F , Cevikbas F , Steinhoff M , Nassini R , Materazzi S , Guerrero-Alba R , Valdez-Morales E , Cottrell GS , Schoonjans K , Geppetti P , Vanner SJ , Bunnett NW , Corvera CU ((2013) ) The TGR5 receptor mediates bile acid-induced itch and analgesia. J Clin Invest 123: , 1513–1530. |
[299] | Steinhoff M , Bienenstock J , Schmelz M , Maurer M , Wei E , Biro T ((2006) ) Neurophysiological, neuroimmunological, and neuroendocrine basis of pruritus. J Invest Dermatol 126: , 1705–1718. |
[300] | Cohen KR , Frank J , Salbu RL , Israel I ((2012) ) Pruritus in the elderly: Clinical approaches to the improvement of quality of life. Pharm Ther 37: , 227–239. |
[301] | Choudhry Z , Rikani AA , Choudhry AM , Tariq S , Zakaria F , Asghar MW , Sarfraz MK , Haider K , Shafiq AA , Mobassarah NJ ((2014) ) Sonic hedgehog signalling pathway: A complex network. Ann Neurosci 21: , 28–31. |
[302] | Rivell A , Petralia RS , Wang Y-X , Clawson E , Moehl K , Mattson MP , Yao PJ ((2019) ) Sonic hedgehog expression in the postnatal brain. Biol Open 8: , bio040592. |
[303] | Palma V , Lim DA , Dahmane N , Sánchez P , Brionne TC , Herzberg CD , Gitton Y , Carleton A , Álvarez-Buylla A , Altaba ARi ((2005) ) Sonichedgehog controls stem cell behavior in the postnatal and adultbrain. Development 132: , 335–344. |
[304] | Briscoe J , Therond PP ((2013) ) The mechanisms of Hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol 14: , 416–429. |
[305] | Traiffort E , Moya KL , Faure H , Hassig R , Ruat M ((2001) ) High expression and anterograde axonal transport of aminoterminal sonic hedgehog in the adult hamster brain. Eur J Neurosci 14: , 839–850. |
[306] | Chadwick W , Brenneman R , Martin B , Maudsley S ((2010) ) Complex and multidimensional lipid raft alterations in a murine model of Alzheimer’s disease. Int J Alzheimers Dis 2010: , 604792. |
[307] | Rietveld A , Neutz S , Simons K , Eaton S ((1999) ) Association of sterol- and glycosylphosphatidylinositol-linked proteins with Drosophila raft lipid microdomains. J Biol Chem 274: , 12049–12054. |
[308] | Giacomini A , Stagni F , Trazzi S , Guidi S , Emili M , Brigham E , Ciani E , Bartesaghi R ((2015) ) Inhibition of APP gamma-secretase restores Sonic Hedgehog signaling and neurogenesis in the Ts65Dn mouse model of Down syndrome. Neurobiol Dis 82: , 385–396. |
[309] | Ledesma MD , Dotti CG ((2005) ) The conflicting role of brain cholesterol in Alzheimer’s disease: Lessons from the brain plasminogen system. Biochem Soc Symp 72: , 129–138. |
[310] | He P , Staufenbiel M , Li R , Shen Y ((2014) ) Deficiency of patched 1-induced Gli1 signal transduction results in astrogenesis in Swedish mutated APP transgenic mice. Hum Mol Genet 23: , 6512–6527. |
[311] | Hung YH , Chang SH , Huang CT , Yin JH , Hwang CS , Yang LY , Yang DI ((2016) ) Inhibitor of differentiation-1 and hypoxia-inducible factor-1 mediate sonic hedgehog induction by amyloid beta-peptide in rat cortical neurons. Mol Neurobiol 53: , 793–809. |
[312] | Yue W , Li Y , Zhang T , Jiang M , Qian Y , Zhang M , Sheng N , Feng S , Tang K , Yu X , Shu Y , Yue C , Jing N ((2015) ) ESC-derived basal forebrain cholinergic neurons ameliorate the cognitive symptoms associated with Alzheimer’s disease in mouse models. Stem Cell Rep 5: , 776–790. |
[313] | Vejux A , Abed-Vieillard D , Hajji K , Zarrouk A , Mackrill JJ , Ghosh S , Nury T , Yammine A , Zaibi M , Mihoubi W , Bouchab H , Nasser B , Grosjean Y , Lizard G ((2020) ) 7-Ketocholesterol and 7β-hydroxycholesterol: In vitro and animal models used to characterize their activities and to identify molecules preventing their toxicity. Biochem Pharmacol 173: , 113648. |
[314] | Zerbinati C , Iuliano L ((2017) ) Cholesterol and related sterols autoxidation. Free Radic Biol Med 111: , 151–155. |
[315] | Liao W-L , Heo G-Y , Dodder NG , Reem RE , Mast N , Huang S , DiPatre PL , Turko IV , Pikuleva IA ((2011) ) Quantification of cholesterol-metabolizing P450s CYP27A1 and CYP46A1 in neural tissues reveals a lack of enzyme-product correlations in human retina but not human brain. J Proteome Res 10: , 241–248. |
[316] | Myers BR , Sever N , Chong YC , Kim J , Belani JD , Rychnovsky S , Bazan JF , Beachy PA ((2013) ) Hedgehog pathway modulation by multiple lipid binding sites on the smoothened effector of signal response. Dev Cell 26: , 346–357. |
[317] | Vorobyeva AG , Saunders AJ ((2018) ) Amyloid-β interrupts canonical Sonic hedgehog signaling by distorting primary cilia structure. Cilia 7: , 5–5. |
[318] | Li X-L , Wang P , Xie Y ((2021) ) Protease nexin-1 protects against Alzheimer’s disease by regulating the sonic hedgehog signaling pathway. Int J Neurosci 131: , 1087–1096. |
[319] | Paganelli AR , Ocaña OH , Prat MaI , Franco PG , López SL , Morelli L , Adamo AM , Riccomagno MnM , Matsubara E , Shoji M , Affranchino JL , Castaño EM , Carrasco AE ((2001) ) TheAlzheimer-related gene presenilin-1 facilitates sonic hedgehogexpression in Xenopus primary neurogenesis. Mech Dev 107: , 119–131. |
[320] | Chen S-D , Yang J-L , Hwang W-C , Yang D-I ((2018) ) Emerging roles of sonic hedgehog in adult neurological diseases: Neurogenesis and beyond. Int J Mol Sci 19: , 2423. |
[321] | Zhou Y , Maxwell KN , Sezgin E , Lu M , Liang H , Hancock JF , Dial EJ , Lichtenberger LM , Levental I ((2013) ) Bile acids modulate signaling by functional perturbation of plasma membrane domains. J Biol Chem 288: , 35660–35670. |
[322] | Jarrett JT , Berger EP , Lansbury PT ((1993) ) The carboxy terminus of the.beta. amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry 32: , 4693–4697. |
[323] | Hasegawa K , Yamaguchi I , Omata S , Gejyo F , Naiki H ((1999) ) Interaction between A beta(1-42) and A beta(1-40) in Alzheimer’s beta-amyloid fibril formation in vitro . Biochemistry 38: , 15514–15521. |
[324] | Yoshiike Y , Chui DH , Akagi T , Tanaka N , Takashima A ((2003) ) Specific compositions of amyloid-beta peptides as the determinant of toxic beta-aggregation. J Biol Chem 278: , 23648–23655. |
[325] | Banerjee S , Sun Z , Hayden EY , Teplow DB , Lyubchenko YL ((2017) ) Nanoscale dynamics of amyloid beta-42 oligomers as revealed by high-speed atomic force microscopy. ACS Nano 11: , 12202–12209. |
[326] | Kim J , Onstead L , Randle S , Price R , Smithson L , Zwizinski C , Dickson DW , Golde T , McGowan E ((2007) ) Abeta40 inhibits amyloid deposition in vivo . J Neurosci 27: , 627–633. |
[327] | Murray MM , Bernstein SL , Nyugen V , Condron MM , Teplow DB , Bowers MT ((2009) ) Amyloid beta protein: Abeta40 inhibits Abeta42 oligomerization. J Am Chem Soc 131: , 6316–6317. |
[328] | Iljina M , Garcia GA , Dear AJ , Flint J , Narayan P , Michaels TCT , Dobson CM , Frenkel D , Knowles TPJ , Klenerman D ((2016) ) Quantitative analysis of co-oligomer formation by amyloid-beta peptide isoforms. Sci Rep 6: , 28658–28658. |
[329] | Ruiz-Riquelme A , Lau HHC , Stuart E , Goczi AN , Wang Z , Schmitt-Ulms G , Watts JC ((2018) ) Prion-like propagation of beta-amyloid aggregates in the absence of APP overexpression. Acta Neuropathol Commun 6: , 26. |
[330] | Ono K , Yoshiike Y , Takashima A , Hasegawa K , Naiki H , Yamada M ((2004) ) Vitamin A exhibits potent antiamyloidogenic and fibril-destabilizing effects. }, Exp Neurol 189: , 380–392. |
[331] | Moon M , Song H , Hong HJ , Nam DW , Cha MY , Oh MS , Yu J , Ryu H , Mook-Jung I ((2013) ) Vitamin D-binding protein interacts with Abeta and suppresses Abeta-mediated pathology. Cell Death Differ 20: , 630–638. |
[332] | Yoichi Matsunaga MS , Hironobu Takahashi , Akiko Furuta ((2016) ) Vitamin D affects neuronal peptides in neurodegenerative disease: Differences of V-D2 and V-D3 for affinity to amyloid-β and scrapie prion protein in vitro. In A Critical Evaluation of Vitamin D, Gowder SJT, ed., pp. 89–104. |
[333] | Grimm MOW , Thiel A , Lauer AA , Winkler J , Lehmann J , Regner L , Nelke C , Janitschke D , Benoist C , Streidenberger O , Stotzel H , Endres K , Herr C , Beisswenger C , Grimm HS , Bals R , Lammert F , Hartmann T ((2017) ) Vitamin D and its analogues decrease amyloid-beta (Abeta) formation and increase Abeta-degradation. Int J Mol Sci 18: , 2764. |
[334] | Masuno H , Ikura T , Morizono D , Orita I , Yamada S , Shimizu M , Ito N ((2013) ) Crystal structures of complexes of vitamin D receptor ligand-binding domain with lithocholic acid derivatives. J Lipid Res 54: , 2206–2213. |
[335] | Speeckaert M , Huang G , Delanghe JR , Taes YEC ((2006) ) Biological and clinical aspects of the vitamin D binding protein (Gc-globulin) and its polymorphism. Clin Chim Acta 372: , 33–42. |
[336] | Norman AW ((2008) ) Vitamin D nuclear receptor (VDR) and plasma vitamin D-binding protein (DBP) structures and ligand shape preferences for genomic and rapid biological responses. In Principles of Bone Biology (Third Edition) BilezikianJP, RaiszLG, MartinTJ, eds. Academic Press San Diego, pp. 749–778. |
[337] | Bouillon R , Pauwels S ((2018) ) The vitamin D-binding protein. In Vitamin D (Fourth Edition), FeldmanD, ed. Academic Press, pp. 97–115. |
[338] | Bishnoi RJ , Palmer RF , Royall DR ((2015) ) Vitamin D binding protein as a serum biomarker of Alzheimer’s disease. J Alzheimers Dis 43: , 37–45. |
[339] | Zhang J , Sokal I , Peskind ER , Quinn JF , Jankovic J , Kenney C , Chung KA , Millard SP , Nutt JG , Montine TJ ((2008) ) CSF multianalyte profiledistinguishes Alzheimer and Parkinson diseases. Am J ClinPathol 129: , 526–529. |
[340] | Jeon SG , Cha M-Y , Kim J-i , Hwang TW , Kim KA , Kim TH , Song KC , Kim J-J , Moon M ((2019) ) Vitamin D-binding protein-loaded PLGA nanoparticles suppress Alzheimer’s disease-related pathology in 5XFAD mice. Nanomedicine 17: , 297–307. |
[341] | Dhiman K , Blennow K , Zetterberg H , Martins RN , Gupta VB ((2019) ) Cerebrospinal fluid biomarkers for understanding multiple aspects of Alzheimer’s disease pathogenesis. Cell Mol Life Sci 76: , 1833–1863. |
[342] | Pan XL , Zhao L , Li L , Li AH , Ye J , Yang L , Xu KS , Hou XH ((2013) ) Efficacy and safety of tauroursodeoxycholic acid in the treatment of liver cirrhosis: A double-blind randomized controlled trial. J Huazhong Univ Sci Technolog Med Sci 33: , 189–194. |
[343] | Setchell KD , Rodrigues CM , Podda M , Crosignani A ((1996) ) Metabolism of orally administered tauroursodeoxycholic acid in patients with primary biliary cirrhosis. Gut 38: , 439–446. |
[344] | Zangerolamo L , Vettorazzi JF , Solon C , Bronczek GA , Engel DF , Kurauti MA , Soares GM , Rodrigues KS , Velloso LA , Boschero AC , Carneiro EM , Barbosa HCL ((2021) ) The bile acid TUDCA improves glucose metabolism in streptozotocin-induced Alzheimer’s disease mice model. Mol Cell Endocrinol 521: , 111116. |
[345] | Lirong W , Mingliang Z , Mengci L , Qihao G , Zhenxing R , Xiaojiao Z , Tianlu C ((2022) ) The clinical and mechanistic roles of bile acids in depression, Alzheimer’s disease, and stroke. Proteomics 22: , 2100324. |
[346] | Nevens F , Andreone P , Mazzella G , Strasser SI , Bowlus C , Invernizzi P , Drenth JPH , Pockros PJ , Regula J , Beuers U , Trauner M , Jones DE , Floreani A , Hohenester S , Luketic V , Shiffman M , van Erpecum KJ , Vargas V , Vincent C , Hirschfield GM , Shah H , Hansen B , Lindor KD , Marschall H-U , Kowdley KV , Hooshmand-Rad R , Marmon T , Sheeron S , Pencek R , MacConell L , Pruzanski M , Shapiro D ((2016) ) A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med 375: , 631–643. |
[347] | Einarsson K , Alvelius G , Hillebrant CG , Reihner E , Bjorkhem I ((1996) ) Concentration of unsulfated lithocholic acid in portal and systemic venous plasma: Evidence that lithocholic acid does not down regulate the hepatic cholesterol 7 alpha-hydroxylase activity in gallstone patients. Biochim Biophys Acta 1317: , 19–26. |
[348] | Norlin M , Andersson U , Bjorkhem I , Wikvall K ((2000) ) Oxysterol 7 alpha-hydroxylase activity by cholesterol 7 alpha-hydroxylase (CYP7A). J Biol Chem 275: , 34046–34053. |
[349] | Liu T , Song X , Khan S , Li Y , Guo Z , Li C , Wang S , Dong W , Liu W , Wang B , Cao H ((2020) ) The gut microbiota at the intersection of bile acids and intestinal carcinogenesis: An old story, yet mesmerizing. Int J Cancer 146: , 1780–1790. |
[350] | Kuno T , Hirayama-Kurogi M , Ito S , Ohtsuki S ((2018) ) Reduction in hepatic secondary bile acids caused by short-term antibiotic-induced dysbiosis decreases mouse serum glucose and triglyceride levels. Sci Rep 8: , 1253. |
[351] | Hillebrant C-G , Nyberg B , Angelin B , Axelson M , Björkhem I , Rudling M , Einarsson C ((1999) ) Deoxycholic acid treatment in patientswith cholesterol gallstones: Failure to detect a suppression ofcholesterol 7α-hydroxylase activity. J Int Med 246: , 399–407. |
[352] | Sadeghi L , Yekta R , Dehghan G ((2020) ) The inhibitory effects of bile acids on catalytic and non-catalytic functions of acetylcholinesterase as a therapeutic target in Alzheimer’s disease. Acta Neurobiol Exp (Wars) 80: , 108–116. |
[353] | Kim JE , Park JJ , Lee MR , Choi JY , Song BR , Park JW , Kang MJ , Son HJ , Hong JT , Hwang DY ((2019) ) Constipation in Tg2576 mice model for Alzheimer’s disease associated with dysregulation of mechanism involving the mAChR signaling pathway and ER stress response. PLoS One 14: , e0215205. |
[354] | Acosta A , Camilleri M ((2014) ) Elobixibat and its potential role in chronic idiopathic constipation. Therap Adv Gastroenterol 7: , 167–175. |
[355] | Ge Z , Duan Z , Yang H , Zhang S , Zhang S , Wang L , Yang D , Sun X , Zhang Z , Su L , Zhu H , Zhou D , Liu B , Shi H , Yu J , Yang H , Chang Q , Zhang N , Wu D , Chen JDZ ((2018) ) Home-based transcutaneous neuromodulation improved constipation via modulating gastrointestinal hormones and bile acids. Evid Based Complement Alternat Med 2018: , 2086163. |
[356] | Camilleri M , Busciglio I , Acosta A , Shin A , Carlson P , Burton D , Ryks M , Rhoten D , Lamsam J , Lueke A , Donato LJ , Zinsmeister AR ((2014) ) Effect of increased bile acid synthesis or fecal excretion in irritable bowel syndrome-diarrhea. Am J Gastroenterol 109: , 1621–1630. |
[357] | Abrahamsson H , Ostlund-Lindqvist AM , Nilsson R , Simren M , Gillberg PG ((2008) ) Altered bile acid metabolism in patients with constipation-predominant irritable bowel syndrome and functional constipation. Scand J Gastroenterol 43: , 1483–1488. |
[358] | Covach AJ , Rose WN ((2017) ) Intrahepatic cholestasis of pregnancy refractory to multiple medical therapies and plasmapheresis, AJP Rep 7: , e223–e225. |
[359] | Bobati SS , Naik KR ((2017) ) Therapeutic plasma exchange - an emerging treatment modality in patients with neurologic and non-neurologic diseases, J Clin Diagn Res 11: , EC35–EC37. |
[360] | Cruz-Ramón V , Chinchilla-López P , Ramírez-Pérez O , Méndez-Sánchez N ((2017) ) Bile acids in nonalcoholic fattyliver disease: New concepts and therapeutic advances, AnnHepatol 16: , S58–S67. |
[361] | Lammert F , Marschall HU , Glantz A , Matern S ((2000) ) Intrahepatic cholestasis of pregnancy: Molecular pathogenesis, diagnosis and management. J Hepatol 33: , 1012–1021. |
[362] | Lucangioli SE , Castano G , Contin MD , Tripodi VP ((2009) ) Lithocholic acid as a biomarker of intrahepatic cholestasis of pregnancy during ursodeoxycholic acid treatment. Ann Clin Biochem 46: , 44–49. |
[363] | Ambros-Rudolph CM , Glatz M , Trauner M , Kerl H , Mullegger RR ((2007) ) The importance of serum bile acid level analysis and treatment with ursodeoxycholic acid in intrahepatic cholestasis of pregnancy: A case series from central Europe. Arch Dermatol 143: , 757–762. |
[364] | Geenes V , Lovgren-Sandblom A , Benthin L , Lawrance D , Chambers J , Gurung V , Thornton J , Chappell L , Khan E , Dixon P , Marschall HU , Williamson C ((2014) ) The reversed feto-maternal bile acid gradient in intrahepatic cholestasis of pregnancy is corrected by ursodeoxycholic acid. PLoS One 9: , e83828. |
[365] | Ikegami T , Matsuzaki Y ((2008) ) Ursodeoxycholic acid: Mechanism of action and novel clinical applications. Hepatol Res 38: , 123–131. |
[366] | Fleishaker JC , Rossi SS , Smith RB , Welshman IR , Daei F , Angellotti MA , Hofmann AF ((1990) ) The effect of colestipol dose on postprandial serum bile acid concentration: Assessment by an enzymic bioluminescence procedure. Aliment Pharmacol Ther 4: , 623–633. |
[367] | Vuorio A , Kuoppala J , Kovanen PT , Humphries SE , Tonstad S , Wiegman A , Drogari E , Ramaswami U ((2019) ) Statins for children with familial hypercholesterolemia, Cochrane Database Syst Rev 2019: , CD006401. |
[368] | Vrieze A , Out C , Fuentes S , Jonker L , Reuling I , Kootte RS , van Nood E , Holleman F , Knaapen M , Romijn JA , Soeters MR , Blaak EE , Dallinga-Thie GM , Reijnders D , Ackermans MT , Serlie MJ , Knop FK , Holst JJ , van der Ley C , Kema IP , Zoetendal EG , de Vos WM , Hoekstra JBL , Stroes ES , Groen AK , Nieuwdorp M ((2014) ) Impact of oral vancomycin on gut microbiota, bile acid metabolism, and insulin sensitivity. J Hepatol 60: , 824–831. |
[369] | Stojakovic T , Putz-Bankuti C , Fauler G , Scharnagl H , Wagner M , Stadlbauer V , Gurakuqi G , Stauber RE , Marz W , Trauner M ((2007) ) Atorvastatin in patients with primary biliary cirrhosis and incomplete biochemical response to ursodeoxycholic acid. Hepatology 46: , 776–784. |
[370] | Stanley FM , Linder KM , Cardozo TJ ((2015) ) Statins increase plasminogen activator inhibitor type 1 gene transcription through a pregnane X receptor regulated element. PloS One 10: , e0138097–e0138097. |
[371] | Hirvioja ML , Tuimala R , Vuori J ((1992) ) The treatment of intrahepatic cholestasis of pregnancy by dexamethasone. Br J Obstet Gynaecol 99: , 109–111. |
[372] | Kauppila A , Jouppila P , Karvonen P , Tuimala R , Ylikorkala O ((1976) ) Effect of dexamethasone on blood levels of ACTH, cortisol,progesterone, estradiol and estriol during late pregnancy. IntJ Gynaecol Obstet 14: , 177–181. |
[373] | Frezza M , Pozzato G , Chiesa L , Stramentinoli G , Padova CD ((1984) ) Reversal of intrahepatic cholestasis of pregnancy in women after high dose S-adenosyl-L-methionine administration. Hepatology 4: , 274–278. |
[374] | Simren M , Bajor A , Gillberg PG , Rudling M , Abrahamsson H ((2011) ) Randomised clinical trial: The ileal bile acid transporter inhibitor A3309 vs. placebo in patients with chronic idiopathic constipation–a double-blind study. Aliment Pharmacol Ther 34: , 41–50. |
[375] | Nakajima A , Seki M , Taniguchi S , Ohta A , Gillberg P-G , Mattsson JP , Camilleri M ((2018) ) Safety and efficacy of elobixibat for chronic constipation: Results from a randomised, double-blind, placebo-controlled, phase 3 trial and an open-label, single-arm, phase 3 trial. Lancet Gastroenterol Hepatol 3: , 537–547. |
[376] | Kaaja RJ , Kontula KK , Raiha A , Laatikainen T ((1994) ) Treatment of cholestasis of pregnancy with peroral activated charcoal. A preliminary study. Scand J Gastroenterol 29: , 178–181. |
[377] | Gurung V , Middleton P , Milan SJ , Hague W , Thornton JG ((2013) ) Interventions for treating cholestasis in pregnancy. Cochrane Database Sys Rev 2013: , CD000493. |
[378] | Hirschfield GM , Mason A , Luketic V , Lindor K , Gordon SC , Mayo M , Kowdley KV , Vincent C , Bodhenheimer HC , Parés A , Trauner M , Marschall H-U , Adorini L , Sciacca C , Beecher-Jones T , Castelloe E , Böhm O , Shapiro D ((2015) ) Efficacy of obeticholic acid inpatients with primary biliary cirrhosis and inadequate response toursodeoxycholic acid. Gastroenterology 148: , 751–761.e758. |
[379] | Mayo MJ , Pockros PJ , Jones D , Bowlus CL , Levy C , Patanwala I , Bacon B , Luketic V , Vuppalanchi R , Medendorp S , Dorenbaum A , Kennedy C , Novak P , Gu J , Apostol G , Hirschfield GM ((2019) ) A randomized, controlled, phase 2 study of Maralixibat in the treatment of itching associated with primary biliary cholangitis. Hepatol Commun 3: , 365–381. |
[380] | Hegade VS , Kendrick SFW , Dobbins RL , Miller SR , Thompson D , Richards D , Storey J , Dukes GE , Corrigan M , Oude Elferink RPJ , Beuers U , Hirschfield GM , Jones DE ((2017) ) Effect of ileal bile acid transporter inhibitor GSK2330672 on pruritus in primary biliary cholangitis: A double-blind, randomised, placebo-controlled, crossover, phase 2a study. Lancet 389: , 1114–1123. |
[381] | Kuiper EM , van Erpecum KJ , Beuers U , Hansen BE , Thio HB , de Man RA , Janssen HL , van Buuren HR ((2010) ) The potent bile acid sequestrant colesevelam is not effective in cholestatic pruritus: Results of a double-blind, randomized, placebo-controlled trial. Hepatology 52: , 1334–1340. |
[382] | Marschall HU , Wagner M , Zollner G , Fickert P , Diczfalusy U , Gumhold J , Silbert D , Fuchsbichler A , Benthin L , Grundstrom R , Gustafsson U , Sahlin S , Einarsson C , Trauner M ((2005) ) Complementary stimulation of hepatobiliary transport and detoxification systems by rifampicin and ursodeoxycholic acid in humans. Gastroenterology 129: , 476–485. |
[383] | Hoensch HP , Balzer K , Dylewizc P , Kirch W , Goebell H , Ohnhaus EE ((1985) ) Effect of rifampicin treatment on hepatic drug metabolism and serum bile acids in patients with primary biliary cirrhosis. Eur J Clin Pharmacol 28: , 475–477. |
[384] | Riikonen S , Savonius H , Gylling H , Nikkila K , Tuomi AM , Miettinen TA ((2000) ) Oral guar gum, a gel-forming dietary fiber relieves pruritus in intrahepatic cholestasis of pregnancy. Acta Obstet Gynecol Scand 79: , 260–264. |
[385] | Trauner M , Gulamhusein A , Hameed B , Caldwell S , Shiffman ML , Landis C , Eksteen B , Agarwal K , Muir A , Rushbrook S , Lu X , Xu J , Chuang JC , Billin AN , Li G , Chung C , Subramanian GM , Myers RP , Bowlus CL , Kowdley KV ((2019) ) The nonsteroidal farnesoid X receptor agonist cilofexor (GS-9674) improves markers of cholestasis and liver injury in patients with primary sclerosing cholangitis. Hepatology 70: , 788–801. |
[386] | Samant H , Manatsathit W , Dies D , Shokouh-Amiri H , Zibari G , Boktor M , Alexander JS ((2019) ) Cholestatic liver diseases: An era of emerging therapies. World J Clin Cases 7: , 1571–1581. |
[387] | Hirschfield GM , Chazouilleres O , Drenth JP , Thorburn D , Harrison SA , Landis CS , Mayo MJ , Muir AJ , Trotter JF , Leeming DJ , Karsdal MA , Jaros MJ , Ling L , Kim KH , Rossi SJ , Somaratne RM , DePaoli AM , Beuers U ((2019) ) Effect of NGM282, an FGF19analogue, in primary sclerosing cholangitis: A multicenter,randomized, double-blind, placebo-controlled phase II trial. JHepatol 70: , 483–493. |
[388] | Honda A , Ikegami T , Nakamuta M , Miyazaki T , Iwamoto J , Hirayama T , Saito Y , Takikawa H , Imawari M , Matsuzaki Y ((2013) ) Anticholestatic effects of bezafibrate in patients with primary biliary cirrhosis treated with ursodeoxycholic acid. Hepatology 57: , 1931–1941. |
[389] | Davidson MH , Armani A , McKenney JM , Jacobson TA ((2007) ) Safety considerations with fibrate therapy. Am J Cardiol 99: , 3c–18c. |
[390] | Bahar R , Wong KA , Liu CH , Bowlus CL ((2018) ) Update on new drugs and those in development for the treatment of primary biliary cholangitis. Gastroenterol Hepatol (N Y) 14: , 154–163. |
[391] | Jones D , Boudes PF , Swain MG , Bowlus CL , Galambos MR , Bacon BR , Doerffel Y , Gitlin N , Gordon SC , Odin JA , Sheridan D , Worns MA , Clark V , Corless L , Hartmann H , Jonas ME , Kremer AE , Mells GF , Buggisch P , Freilich BL , Levy C , Vierling JM , Bernstein DE , Hartleb M , Janczewska E , Rochling F , Shah H , Shiffman ML , Smith JH , Choi YJ , Steinberg A , Varga M , Chera H , Martin R , McWherter CA , Hirschfield GM ((2017) ) Seladelpar (MBX-8025), a selective PPAR-delta agonist, in patients with primary biliary cholangitis with an inadequate response to ursodeoxycholic acid: A double-blind, randomised, placebo-controlled, phase 2, proof-of-concept study. Lancet Gastroenterol Hepatol 2: , 716–726. |
[392] | Graffner H , Gillberg PG , Rikner L , Marschall HU ((2016) ) The ileal bile acid transporter inhibitor A4250 decreases serum bile acids by interrupting the enterohepatic circulation. Aliment Pharmacol Ther 43: , 303–310. |
[393] | Bachs L , Pares A , Elena M , Piera C , Rodes J ((1989) ) Comparison of rifampicin with phenobarbitone for treatment of pruritus in biliary cirrhosis. Lancet 1: , 574–576. |
[394] | Monteiro-Cardoso VF , Corlianò M , Singaraja RR ((2021) ) Bile acids: A communication channel in the gut-brain axis. Neuro Molecular Med 23: , 99–117. |
[395] | Wertheim BC , Martínez ME , Ashbeck EL , Roe DJ , Jacobs ET , Alberts DS , Thompson PA ((2009) ) Physical activity as a determinant of fecal bile acid levels. Cancer Epidemiol Biomarkers Prev 18: , 1591–1598. |
[396] | Danese E , Salvagno GL , Tarperi C , Negrini D , Montagnana M , Festa L , Sanchis-Gomar F , Schena F , Lippi G ((2017) ) Middle-distance running acutely influences the concentration and composition of serum bile acids: Potential implications for cancer risk? Oncotarget 8: , 52775–52782. |
[397] | Trefflich I , Marschall HU , Giuseppe RD , Ståhlman M , Michalsen A , Lampen A , Abraham K , Weikert C ((2019) ) Associations between dietary patterns and bile acids-results from a cross-sectional study in vegans and omnivores. Nutrients 12: , 47. |
[398] | Burks HE , Abrams T , Kirby CA , Baird J , Fekete A , Hamann LG , Kim S , Lombardo F , Loo A , Lubicka D , Macchi K , McDonnell DP , Mishina Y , Norris JD , Nunez J , Saran C , Sun Y , Thomsen NM , Wang C , Wang J , Peukert S ((2017) ) Discovery of an acrylic acid based tetrahydroisoquinoline as an orally bioavailable selective estrogen receptor degrader for ERα+breast cancer. J Med Chem 60: , 2790–2818. |


