
Virus-Like Cytosolic and Cell-Free Oxidatively Damaged Nucleic Acids Likely Drive Inflammation, Synapse Degeneration, and Neuron Death in Alzheimer’s Disease
Abstract
Oxidative stress, inflammation, and amyloid-β are Alzheimer’s disease (AD) hallmarks that cause each other and other AD hallmarks. Most amyloid-β-lowering, antioxidant, anti-inflammatory, and antimicrobial AD clinical trials failed; none stopped or reversed AD. Although signs suggest an infectious etiology, no pathogen accumulated consistently in AD patients. Neuropathology, neuronal cell culture, rodent, genome-wide association, epidemiological, biomarker, and clinical studies, plus analysis using Hill causality criteria and revised Koch’s postulates, indicate that the virus-like oxidative damage-associated molecular-pattern (DAMP) cytosolic and cell-free nucleic acids accumulated in AD patients’ brains likely drive neuroinflammation, synaptotoxicity, and neurotoxicity. Cytosolic oxidatively-damaged mitochondrial DNA accumulated outside mitochondria dose-dependently in preclinical AD and AD patients’ hippocampal neurons, and in AD patients’ neocortical neurons but not cerebellar neurons or glia. In oxidatively-stressed neural cells and rodents’ brains, cytosolic oxidatively-damaged mitochondrial DNA accumulated and increased antiviral and inflammatory proteins, including cleaved caspase-1, interleukin-1β, and interferon-β. Cytosolic double-stranded RNA and DNA are DAMPs that induce antiviral interferons and/or inflammatory proteins by oligomerizing with various innate-immune pattern-recognition receptors, e.g., cyclic GMP-AMP synthase and the nucleotide-binding-oligomerization-domain-like-receptor-pyrin-domain-containing-3 inflammasome. In oxidatively-stressed neural cells, cytosolic oxidatively-damaged mitochondrial DNA caused synaptotoxicity and neurotoxicity. Depleting mitochondrial DNA prevented these effects. Additionally, cell-free nucleic acids accumulated in AD patients’ blood, extracellular vesicles, and senile plaques. Injecting cell-free nucleic acids bound to albumin oligomers into wild-type mice’s hippocampi triggered antiviral interferon-β secretion; interferon-β injection caused synapse degeneration. Deoxyribonuclease-I treatment appeared to improve a severe-AD patient’s Mini-Mental Status Exam by 15 points. Preclinical and clinical studies of deoxyribonuclease-I and a ribonuclease for AD should be prioritized.
INTRODUCTION
Oxidative stress, inflammation, and amyloid-β (Aβ) signaling are core Alzheimer’s disease (AD) hallmarks that can cause each other and other AD hallmarks experimentally [1, 2]. Paradoxically though, most clinical trials for AD patients of Aβ-lowering, antioxidant, anti-inflammatory, and antimicrobial agents failed, and even the few that met their primary endpoints or even slowed early-AD progression failed to stop or reverse AD progression [2–7]. Furthermore, although many signs have pointed to an infectious viral or bacterial etiology of AD spanning from Oskar Fischer’s seminal neuropathology work to that of contemporary researchers, no specific pathogen has ever been shown to accumulate consistently in the brains of different AD patient cohorts [8–17].
Based on the currently available directly relevant neuropathology [12, 15, 18–25], neuronal cell culture [21, 26], rodent [21, 26], genome-wide association [27, 28], epidemiological [1, 2, 26, 29–32], and biomarker studies [33–37], plus the only currently available relevant clinical experiment [38], it appears likely that the virus-like, damage-associated molecular pattern (DAMP) cytosolic and cell-free oxidatively damaged nucleic acids accumulated in AD patients’ brains, and the innate immune system reacting to them as if they were viruses, contribute non-trivially to multiple aspects of AD pathophysiology, including neuroinflammation, synaptotoxicity, and neurotoxicity.
CYTOSOLIC OXIDATIVELY DAMAGED MITOCHONDRIAL DNA ACCUMULATED DOSE-DEPENDENTLY IN PCAD AND AD PATIENTS’ VULNERABLE NEURONS (I.E., OPPORTUNITY)
Some of the crucial facts in this case include that, postmortem, oxidatively damaged mitochondrial DNA accumulated dose-dependently outside mitochondria in the cytosol in preclinical AD (PCAD) and AD patients’ hippocampal neurons, and in AD patients’ neocortical neurons, but not in their relatively spared cerebellar neurons or glia [18–20, 39]. This is a meta-claim formed from the following individual findings. Oxidatively damaged mitochondrial DNA has been found to accumulate in mild cognitive impairment (MCI) and AD patients’ diseased brain regions [39–42]. Hirai et al. [18] found elevated levels of mitochondrial DNA free in the cytosol in AD patients’ hippocampal and neocortical neurons, but not in their cerebellar neurons or glia [18]. They also found significantly reduced mitochondria via morphometric analysis, and that markers of oxidative damage and accumulated mitochondrial DNA colocalized to the same neurons [18]. Lovell et al. and Majd and Power, respectively, found significantly elevated levels of oxidatively damaged DNA in the cytosols of PCAD and AD patients’ hippocampus and parahippocampal gyrus or hippocampal neurons, and both groups interpreted this as oxidatively damaged mitochondrial DNA since it was cytosolic (compare nuclear DAPI and cytosolic 8-OHdG staining in Fig. 6 of Majd and Power (2018) [20]; see colocalization of neuronal marker Tuj-1 and 8-OHG RNA in Fig. 1 of Lovell et al. [19]; see DNA-associated 8-OHG surrounding a nucleus-shaped mostly non-fluorescent spherical space near the center of the cells in Fig. 2 of Lovell et al. [19, 20]).
Fig. 1
Reprinted from Current Alzheimer Research, Vol. 15, Shohreh Majd and John H.T. Power, Oxidative Stress and Decreased Mitochondrial Superoxide Dismutase 2 and Peroxiredoxins 1 and 4 Based Mechanism of Concurrent Activation of AMPK and mTOR in Alzheimer’s Disease, 1-13, Copyright 2018, with permission from Bentham Science.
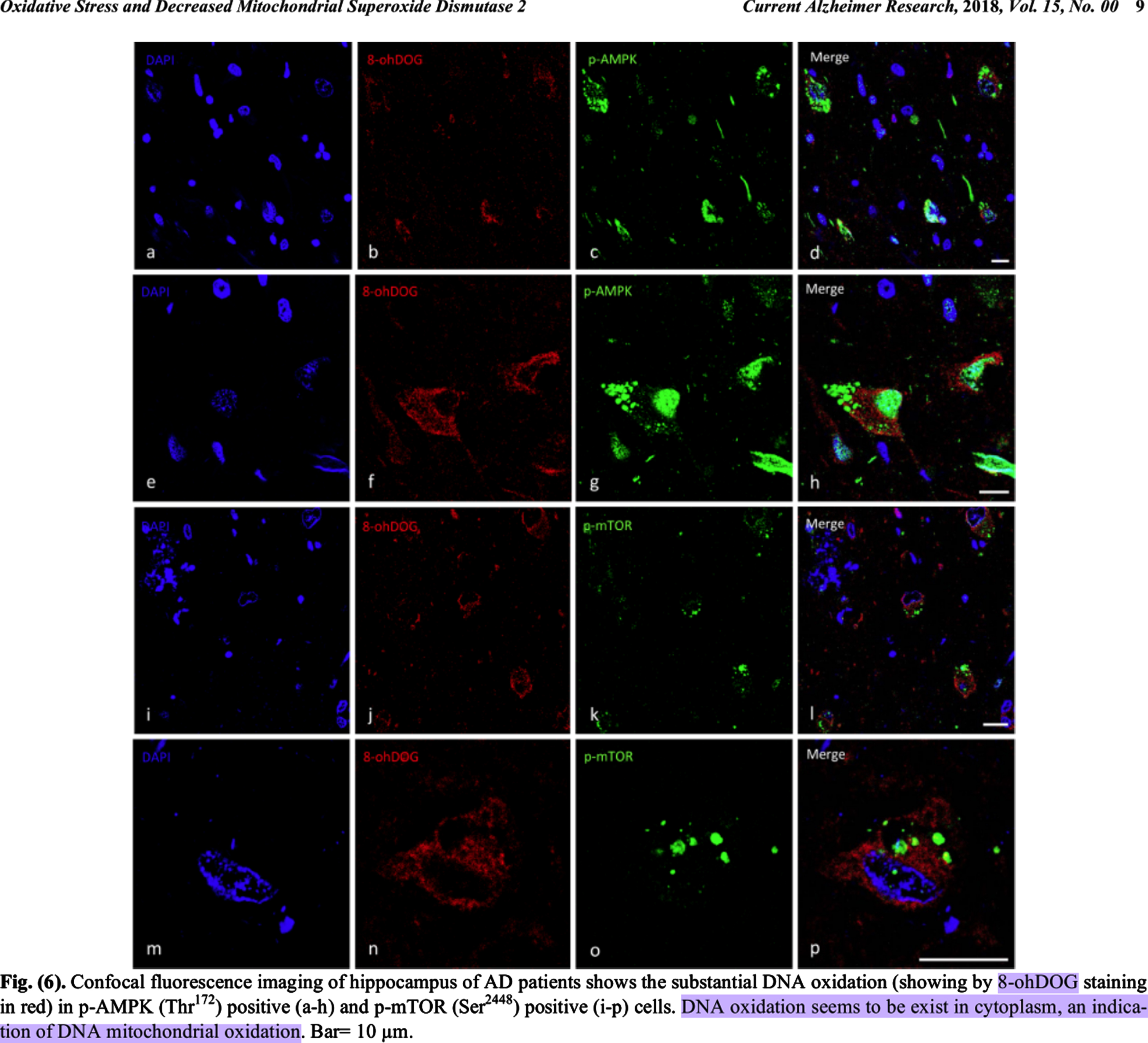
Fig. 2
Reprinted from The Journal of Neuroscience, Vol. 21, Keisuke Hirai, Gjumrakch Aliev, Akihiko Nunomura, Hisashi Fujioka, Robert L. Russell, Craig S. Atwood, Anne B. Johnson, Yvonne Kress, Harry V. Vinters, Massimo Tabaton, Shun Shimohama, Adam D. Cash, Sandra L. Siedlak, Peggy L. R. Harris, Paul K. Jones, Robert B. Petersen, George Perry, Mark A. Smith, Mitochondrial Abnormalities in Alzheimer's Disease, 3017-23, Copyright 2001, with permission from Society for Neuroscience.
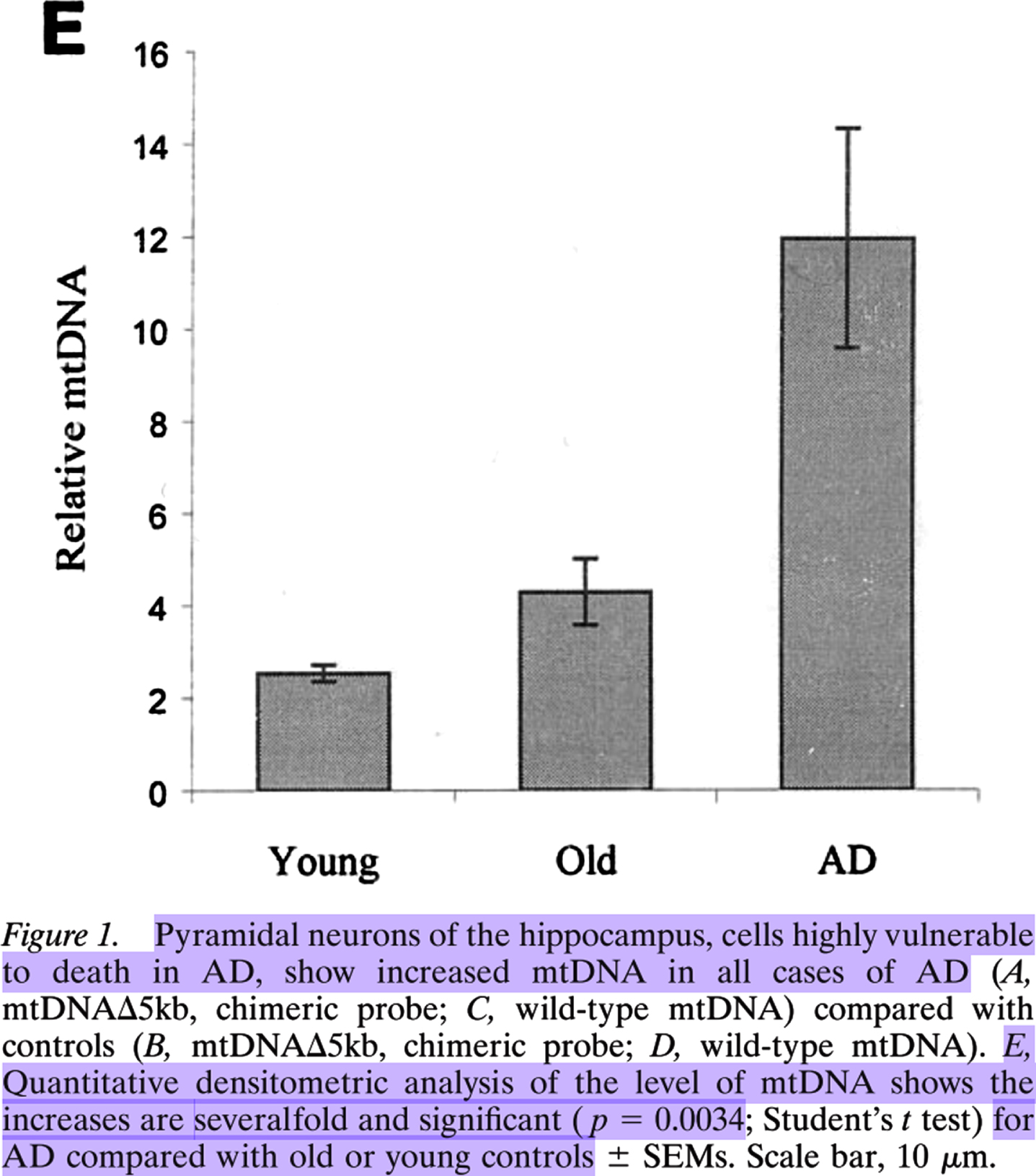
Lovell et al. found, compared to in those of elderly controls, 37% more oxidatively damaged DNA immunofluorescence in PCAD patients’ hippocampal and parahippocampal gyrus neurons1, the majority of which was cytosolic (see the images in the right column in Fig. 2 of Lovell et al. [19]). Hirai et al. [18] found significantly and severalfold more mitochondrial DNA in AD patients’ pyramidal hippocampal neurons compared to those in elderly controls via quantitative densitometric analysis (see Fig. 1E from Hirai et al. [18], Copyright 2001 Society for Neuroscience [18]).
Fig. 3
Reprinted from Acta Neuropathologica, Vol. 19, Christensen DZ, Schneider-Axmann T, Lucassen PJ, Bayer TA, Wirths O (2010) Accumulation of intraneuronal Aβ correlates with ApoE4 genotype, 555-566, 2010.
Estimating from Fig. 1E of Hirai et al., there was approximately 3 times as much mitochondrial DNA in AD patients’ pyramidal hippocampal neurons as in those of elderly-control individuals, i.e., ∼300% as much. ∼300% - ∼137% =∼163%. This indicates that AD patients’ pyramidal hippocampal neurons contained ∼1.6-fold as much largely cytosolic mitochondrial DNA as PCAD individuals’ hippocampal and parahippocampal gyrus neurons contained mostly cytosolic oxidatively damaged DNA presumed to be mitochondrial. If these two cytosolic nucleic acid species are mostly the same population of hippocampal neural cytosolic oxidatively damaged mitochondrial DNA, as they appear to be, then this suggests that there is a dose-response relationship between hippocampal neuron cytosolic oxidatively damaged mitochondrial DNA and the progression of non-demented low-pathology aging to PCAD (a 37% increase), and from asymptomatic PCAD to AD dementia (a 163% increase). Of course, future studies should repeat this comparison directly in a new, single cohort using a standardized and updated set of methods. Until then though, this is the most reasonable retrospective interpretation of the evidence. Therefore, it appears that hippocampal neural cytosolic oxidatively damaged mitochondrial DNA accumulated dose-dependently in PCAD and AD patients’ hippocampal neurons[18–20, 39].
CYTOSOLIC OXIDATIVELY DAMAGED MITOCHONDRIAL DNA IN NEURAL CELLS CAUSES NEUROINFLAMMATION, SYNAPSE DEGENERATION, AND NEURAL DEATH BY OLIGOMERIZING WITH PATTERN-RECOGNITION RECEPTORS (i.e., MEANS)
In vitro in chronically oxidatively stressed neural cells, cytosolic oxidatively damaged mitochondrial DNA accumulated, caused significant synaptotoxicity and neurotoxicity, and upregulated inflammatory proteins, including cyclic GMP-AMP (cGAMP) synthase (cGAS), Stimulator of interferon genes (STING), Interferon regulatory factor 3 (IRF3), antiviral type-I interferons interferon-β and interferon-α, phosphorylated Nuclear-factor κB (NF-κB), cleaved caspase-1, interleukin-1β, interleukin-6, and interleukin-18 [26].
Mechanistically linking cytosolic and cell-free nucleic acids and innate inflammation, ectopic DAMP and pathogen-associated molecular pattern (PAMP) nucleic acids provoke antiviral IRF3/type-I interferon and/or inflammatory NF-κB signaling in part by oligomerizing with several cytosolic innate immune pattern-recognition receptors (PRR) that were upregulated in AD patients’ brains, including the Nucleotide-binding oligomerization domain-Like Receptor Pyrin domain-containing 3 (NLRP3) inflammasome [24, 25, 43–50], Protein Kinase double-stranded RNA-activated interferon-induced (PKR) [51–55], STING downstream of cGAS [56, 57], the Absent In Melanoma 2 (AIM2) inflammasome [1, 57, 58], and the Interferon-Inducible Protein 16 (IFI16) inflammasome [21, 57, 58].
A complex question this raises is whether any of these pattern recognition receptor proteins upregulated in AD patients’ brains were expressed by neurons, and therefore whether they were at the right place to sense the cytosolic mitochondrial DNA accumulated there. The double-stranded RNA sensor PKR was upregulated in AD patients’ hippocampal and neocortical neurons [55] and in their cerebrospinal fluid (CSF) [54], but PKR would not sense mitochondrial DNA. AIM2 and IFI16 were significantly upregulated at the mRNA level in AD patients’ brains (search the AlzData.org database for these findings) [58], making their cellular localization unknown. Elevated STING and phosphorylated IRF3 proteins were found in AD patients’ cortices according to a poster abstract [56], making their cellular localization also unknown. NLRP3 proteins were increased in AD patients’ temporal cortices and colocalized with glial maturation factor (GMF) proteins near and at the edges of amyloid plaques and neurofibrillary tangles [25]. Autophagic vesicle markers p62, LC3, and LAMP1 were also increased and also colocalized with GMF [25]. However, the cell type-specific localization of the NLPR3 proteins was not determined in this study, making it unclear whether they and the neural cytosolic oxidatively damaged mitochondrial DNA colocalized and could have interacted [18, 25]. Both neurons and glia express GMF [59, 60]. Accumulation of autolysosomal vesicle markers p62 and LC3-II was observed in AD patients’ CA1 hippocampal neurons [61]. Neurofibrillary tangles occur intracellularly in neurons. NLRP3 proteins were upregulated in mesencephalic neurons in Parkinson’s disease patients, indicating that neurons can express NLRP3 proteins [24]. Therefore, at least some of these NLRP3 inflammasomes upregulated in AD patients’ temporal cortices were probably expressed by neurons and could have colocalized with the neural cytosolic mitochondrial DNA in AD patients’ temporal cortices [18, 25]. Furthermore, in other cell types, oxidatively damaged mitochondrial DNA fragments activate the NLRP3 inflammasome [50]. This indicates that NRLP3 inflammasome proteins likely had AD-specific, temporal cortex-specific, and neuron-specific opportunity and mechanistic means to be activated by the cytosolic oxidatively damaged mitochondrial DNA in AD patients’ temporal cortex neurons.
Oligomerized inflammasomes, such as the NLRP3 and AIM2 inflammasomes, catalyze the proteolytic activation of inflammatory proteins, such as caspase-1, interleukin-1β, and interleukin-18, which accumulated in oxidative stressed neural cells [62–64]. Consistent with the notion that this may also occur to some extent in AD patients, cleaved caspase-1, interleukin-1β, and interleukin-18 proteins accumulated in AD patients’ brains and systemically too [25, 43, 65, 66].
Caspase-1 cleaves Gasdermin-D, proteins of which accumulated in AD patients [65]. Gasdermin-D proteins oligomerize and form pores in cellular membranes, triggering inflammatory cell death via pyroptosis [67–70]. This suggests that cytosolic DAMP mitochondrial DNA may promote neuronal pyroptosis in AD.
Other PRRs, such as cGAS, oligomerize with cytosolic double-stranded DNA or other nucleic species and then induce an antiviral type I interferon response [26]. Consistent with the notion that this may occur in AD patients’ brains, interferon-stimulated genes were progressively upregulated in AD patients’ brains in a Braak-stage-correlating manner [21].
In vivo, this cytosolic oxidatively damaged mitochondrial DNA-associated inflammatory phenotype was also observed in oxidatively stressed mice’s brains [26]. In neural cells, depleting mitochondrial DNA rescued the neuroinflammation, synapse degeneration, and neural cell death [26]. Clearing ectopic damaged DNA with deoxyribonuclease-I (DNase-I) protein transfection rescued the inflammatory phenotype, although its effects on synapse degeneration and neural cell death were not tested [26].
That is arguably the most direct experimental evidence currently available, since it was carried out in oxidatively-stressed neural cells [26], which seems to be a reasonable preclinical model of sporadic AD because it simulates the cytosolic oxidatively damaged mitochondrial DNA accumulated in AD patients’ vulnerable neurons [18–20]. However, an honorable mention is arguably due to Matsui et al., who showed that, in neuroblastoma cells and other cell types with Parkinson’s disease-related mutations in the autophagy-related genes PINK1, GBA, and/or ATP13A2, cytosolic mitochondrial DNA accumulated and induced type I interferons and cell death, effects which were rescued by either IFI16 depletion or lysosomal DNase-II overexpression [71]. DNase-II overexpression also rescued dopaminergic cell loss and movement disorders in Parkinson’s disease modeling zebrafish [71]. This study may not be as directly relevant to this discussion as the previous study, since the authors did not mention that they differentiated the neuroblastoma cells into neural cells, and the trigger of the mitochondrial DNA accumulation was Parkinson’s disease-related mutations associated with autophagic dysfunction, not oxidative stress, which is hypothesized to be a root cause of AD [2, 30, 31]. That being said, AD patients’ CA1 hippocampal neurons appear to suffer severe autophagic flux block as shown by the accumulation there of colocalized LC3-II and p62 in fused but uncleared autolysosomes [61], and mitochondrial DNA accumulated not only in the cytosol but also in uncleared autophagic vacuoles in AD patients’ neurons [18, 72], so autophagic flux block may contribute to the cytosolic mitochondrial DNA accumulation in AD patients’ hippocampal neurons too [18].
Another honorable mention is arguably due to Song et al., who found that Ataxia Telangiectasia Mutated kinase (ATM) deficiency caused cytosolic single-stranded and double-stranded DNA accumulation in microglia, cerebellar Purkinje cell neurons, and other cell types, and this induced robust inflammatory mediator production in microglia that was toxic to neurons [73]. This is intriguing in part because ATM proteins have been found to be depleted in AD patients’ brains [74]. However, this is arguably somewhat indirect evidence because only the effects of microglial cytosolic DNA were tested [74], and Hirai et al. noted that cytosolic mitochondrial DNA did not accumulate in glia [18]. Still, this does not exclude the possibility that cytosolic DNA may transiently appear in AD patients’ microglia and provoke inflammatory responses but then be rapidly degraded so that it does not accumulate.
NEURAL CYTOSOLIC OXIDATIVELY DAMAGED MITOCHONDRIAL DNA HAD MEANS AND OPPORTUNITY TO DRIVE NEUROINFLAMMATION, SYNAPSE DEGENERATION, AND NEURON DEATH IN AD PATIENTS
This demonstrates that cytosolic oxidatively damaged mitochondrial DNA had dose-dependent, disease stage-specific, brain region-specific, and cell type-specific opportunity and experimentally-confirmed mechanistic means to drive neuroinflammation, synaptotoxicity, and neurotoxicity in PCAD and AD patients’ degenerating (but not relatively spared) neurons, whereas transfection of ectopic-DNA-degrading DNase-I protein largely rescued at least some of these effects in neural cells.
INTRANEURONAL CYTOSOLIC Aβ PROTOFIBRILS AGGREGATED WITH NUCLEIC ACIDS MAY ACCUMULATE IN AD PATIENTS’ HIPPOCAMPI AND FUNCTION ANALOGOUSLY TO PRRs
Furthermore, intraneuronal Aβ has been identified in many though not all studies of AD patients’ brains (see Table 1 in Aho et al. [75–77]), and Christensen et al. found that immunoreactivity to the OC antibody— which binds to Aβ protofibrils, fibrillar oligomers, and other fibrillar amyloids [78–80]— accumulated in AD patients’ hippocampal neurons in a staining pattern that was noted to be similar to that of N-terminal Aβ peptides, which was “a granular staining pattern and concentration around the nucleus (Fig. 1b)” [81]. (See Fig. 1b from Christensen et al. reproduced with permission).
Several amyloid-like antimicrobial peptides have been shown to form protofibrils with ectopic nucleic acids that function as innate immune signalosomes to cause inflammation by activating Toll-like receptors [82]. On the other hand, Aβ functions as an antimicrobial peptide [83]. DNA binds to and aggregates with the positively-charged N-termini of both Aβ and cGAS proteins, especially with facilitation by Zn2+ ions [84, 85]. In vitro, nucleic acids accelerate and increase Aβ aggregation, appearing to bind to monomers, dimers, oligomers, protofibrils, and mature fibrils [2, 84, 86–90]. Interestingly, the Aβ protofibril-targeting antibody BAN2401/Lecanemab significantly slowed early-AD progression in a phase II clinical trial [4], suggesting that Aβ protofibrils may be important therapeutic targets.
Therefore, intraneuronal cytosolic and/or endolysosomal Aβ may bind to and form protofibrils [81] with ectopic and damaged nucleic acids, such as the cytosolic and autophagic vesicle oxidatively damaged mitochondrial DNA [18], in AD patients’ hippocampal neurons [2, 18, 81, 82, 84–90], potentially increasing inflammation by acting somewhat analogously to a PRR [82, 84, 85].
CELL-FREE NUCLEIC ACIDS ACCUMULATED IN AD PATIENTS’ SENILE AMYLOID PLAQUES AND MAY DRIVE NEUROINFLAMMATION
Cell-free RNA and/or DNA of unidentified origin and unknown oxidation accumulated in all of the neuritic/senile-amyloid plaques that were checked for this in AD patients’ brains in one study [21]. Specifically, neuronal messenger RNA [22, 91], herpes-simplex virus-1 DNA [15], bacterial-biofilm DNA [12], and oxidatively damaged neutrophil DNA forming neutrophil-extracellular traps [23] accumulated in AD patients’ senile/neuritic-amyloid plaques.
It is unclear to what extent these extracellular amyloid plaque-associated nucleic acids may drive neuroinflammation in AD. Supporting the possibility that they may contribute non-trivially, however, it was found that microglia with upregulated interferon-stimulated gene expression surrounded AD patients’ senile/neuritic-amyloid plaques [21]. Another possibility though is that nucleic acid-Aβ complexes may do most of their damage while cytosolic and/or soluble in the brain interstitium, before depositing as neuritic plaques.
NUCLEIC ACIDS IN MCI AND AD PATIENTS’ EXTRACELLULAR VESICLES MAY DRIVE NEUROINFLAMMATION IN AD
It remains to be determined whether nucleic acids in PCAD, MCI, or AD patients’ extracellular vesicles drive neuroinflammation. Favoring this possibility, mitochondrial RNA accumulated in MCI and AD patients’ circulating extracellular vesicles [34]. Several studies have shown that DNA accumulates in extracellular vesicles such as exosomes in various other pathological states and induces inflammatory responses in immune cells [92–97]. However, only mitochondrial RNA (not DNA) has been identified in MCI or AD patients’ extracellular vesicles so far [34], and there does not appear to be any clear evidence about whether extracellular vesicle RNA induces inflammation. To clarify this issue, future studies should investigate whether extracellular vesicle mitochondrial RNA can induce inflammatory responses, and whether DNA (particularly oxidized mitochondrial DNA) accumulates in PCAD, MCI, and AD patients’ extracellular vesicles.
CELL-FREE NUCLEIC ACIDS ACCUMULATED IN MCI AND AD PATIENTS’ BLOOD
Peripherally, cell-free nucleic acids accumulated in MCI and AD patients’ blood plasma [33]. CSF Aβ and mitochondrial DNA was lower in AD patients [35–37], suggesting decreased brain clearance of both. These findings plus the experimental results summarized below suggest that cell-free nucleic acids may contribute non-trivially to neuroinflammation and synapse degeneration in AD patients.
CELL-FREE NUCLEIC ACIDS UPREGULATED INTERFERONS AND THE COMPLEMENT CASCADE, WHICH CAUSED SYNAPSE DEGENERATION
Mirroring results in cells and AD-modeling transgenic animals, in vivo injection of amyloid fibrils composed of cell-free RNA and albumin into the hippocampi of wild-type animals induced inflammatory genes, type-I interferon-stimulated gene expression, and synaptotoxic-complement C3 [21]. Injection of recombinant interferon-β into wild-type animals’ hippocampi did the same and induced C3-dependent synapse degeneration [21].
Does an analogous process of protein-bound cell-free nucleic acid-induced neuroinflammation and synapse degeneration occur in AD patients’ brains? Whether there are soluble cell-free nucleic acids bound to albumin, Aβ, or other proteins inside AD patients’ brains specifically has not yet been determined, although their possible presence is suggested by previously mentioned findings such as the accumulation of nucleic acids in AD patients’ neuritic plaques [12, 15, 21–23, 91], neural cytosols [18–20], blood [33], and extracellular vesicles [34], the depletion of mitochondrial DNA and Aβ from AD patients’ CSF [35–37], and the aggregation of Aβ with DNA in vitro [2, 84, 86–90]. Also consistent with this possibility are the findings that interferon-stimulated gene expression was upregulated in AD patients’ brains in a Braak-stage correlating manner [21], that C1q, C3, and other classical complement pathway proteins accumulated in AD patients’ hippocampal neurons and neuritic/senile-amyloid plaques [98–102], and that C3 proteins were elevated in AD patients’ CSF [102].
The preceding discussion suggests that at least some of the ectopic cell-free nucleic acid species accumulated in AD patients may have had opportunity and mechanistic means, even independently of Aβ, to drive inflammation and synaptotoxicity in AD patients’ brains.
DNASE-I APPEARED TO UNPRECEDENTLY IMPROVE A SEVERE AD PATIENTS’ MMSE BY 15 POINTS
No clinical trials of nuclease enzymes for AD patients have yet been conducted. However, in a case report, a severe AD patient was treated with high-dose oral DNase-I for two months, and his cognition on the Mini-Mental Status Exam (MMSE) improved by 15 points [38]. This raises several questions. Firstly, did the oral DNase-I enter the brain and engage the target? Target engagement was not assessed in this case report, and proteins typically do not enter the bloodstream or brain from the gut. However, it is possible that the oral DNase-I could have entered the brain because AD patients have been shown to have intestinal [103] and blood-brain barrier permeability [104–106], which would allow small proteins such as DNase-I to pass from the gut into the bloodstream, and from there into the brain. Another possibility is that the DNase-I proteins could have exerted an indirect effect via the gut microbiota, the enteric nervous system, or the vagus nerve by degrading cell-free DNA in the gut, an intriguing possibility given the recent research into the role of the gut microbiota in AD [107–109].
Secondly, even if any DNase-I proteins did degrade DNA in the gut, bloodstream, or brain, did they only degrade cell-free DNA in the extracellular space, leaving the neuronal cytosolic DNA untouched? This seems more plausible, since although cells can take up extracellular proteins, these proteins are typically degraded rapidly in lysosomes [110, 111]. On the other hand though, lysosomes appear to be dysfunctional and overburdened with mitochondrial DNA in AD patients’ neurons [61, 72], so yet another possibility is that some of the patient’s neurons could have taken up a small number of the DNase-I proteins that made it into the brain and used them to degrade some of that lysosomal mitochondrial DNA, perhaps thereby ameliorating inflammation or autophagic flux to some extent.
Thirdly, in the extracellular space, would the DNase-I proteins have only degraded cell-free DNA diffusing freely or bound to proteins, or could they have degraded some DNA associated with extracellular vesicles too? Surprisingly, DNase-I and other DNases have been shown to reduce the DNA content of exosomes by degrading the portion of their DNA cargo that is embedded in the extracellular vesicle membrane and external [112, 113]. Therefore, the DNase-I treatment could have reduced the amount of cell-free DNA associated with and on the outside of extracellular vesicles.
Fourthly, even if the oral DNase-I did degrade any of the ectopic DNA species in the patient’s gut, bloodstream, or brain, was this the cause of the patients’ 15-point improvement on the MMSE? Or was it a coincidence? One possibility is that the patient could have been temporarily delirious, or demented and temporarily delirious in addition, and so would have gotten better anyway without the DNase-I. Arguing against this possibility though, one way to distinguish between dementia and delirium is that dementia symptoms emerge chronically, whereas delirium symptoms emerge acutely, and G. Tetz reported that this patient’s symptoms emerged chronically (G. Tetz, personal communication, May 18, 2022), indicating he had dementia and not delirium. Although this leaves several important questions unanswered, if this preliminary clinical experiment is corroborated by future rigorous randomized clinical trials, it might constitute the first example of partial AD reversal.
THE RS141639275 DNASE1 POLYMORPHISM IS ASSOCIATED WITH AD
Genetically, a genome-wide association study of 5,740 late-onset AD patients and 5,096 cognitively normal controls found that the rs141639275 G:A transition single nucleotide polymorphism upstream of the DNASE1 gene is strongly associated with AD, with p values on the order of 10–81 to 10–63 (search https://www.niagads.org/genomics for DNASE1 to see this data) [27, 28]. Since this single nucleotide polymorphism was associated with AD and could alter ectopic DNA degradation, this also appears to be consistent with the notion that cytosolic and/or cell-free DNA could drive AD pathogenesis.
AD CAUSAL MUTATIONS AND RISK FACTORS ARE ASSOCIATED WITH ECTOPIC NUCLEIC ACID-GENERATING OXIDATIVE STRESS
Epidemiologically, if oxidatively damaged cytosolic and cell-free nucleic acids drive AD pathogenesis, then we would predict that oxidative stress should be associated with AD etiology. This is because, unlike self-replicating PAMP nucleic acids, cytosolic and cell-free DAMP oxidatively damaged nucleic acids accumulated in neuronal and other cell types due to chronic oxidative stress and injury [26, 29]. AD is caused rarely by specific mutations, and commonly by the combination of numerous diverse risk factors, chiefly aging and being heterozygous or homozygous for the APOE4 allele. Indeed, many if not all AD causal mutations and risk factors— including aging and APOE4 genotype— have been associated with increased oxidative stress/damage/injury [1, 2, 30–32]. Furthermore, oxidatively damaged lipids and nucleic acids have been shown to accumulate in PCAD, MCI, and AD patients’ brains [18–20, 39–41, 114–116], and oxidatively damaged proteins have been shown to accumulate in MCI and AD patients’ brains [117–119].
ANALYSIS FROM HILL CAUSALITY CRITERIA
Analysis with Hill causality criteria and Fredricks-Relman molecular revised Koch’s postulates of this preliminary but wide set of data with supportive evidence from testing in different experiments under different conditions indicates that ectopic nucleic acids should be considered probable etiological agents in AD [120].
The first Hill causality criterion is “Strength of association \dots What is the relative risk?” [120]. The epidemiological concept of relative risk does not apply to this situation since oxidatively damaged cytosolic and/or cell-free nucleic acids are not an environmental agent that one is either exposed or not exposed to. However, as discussed, various oxidative damage-promoting risk factors have been associated with AD [1, 2, 30–32], and elevated levels of cytosolic and cell-free nucleic acids have been associated with AD [12, 15, 18–23, 33, 34, 39, 91].
The second Hill causality criterion is “Consistency of association \dots Is there agreement among repeated observations in different places, at different times, using different methodology, by different researchers, under different circumstances?” [120]. In short, yes. Multiple groups have found oxidatively damaged cytosolic DNA presumed to be mitochondrial, or cytosolic mitochondrial DNA, in PCAD or AD patients’ hippocampal neurons [18–20]. Additionally, multiple groups have found different cell-free nucleic acid species in AD patients’ neuritic plaques [12, 15, 21–23, 91].
The third Hill causality criterion is “Specificity of association \dots Is the outcome unique to the exposure?” [120]. Admittedly, oxidatively damaged nucleic acids and cytosolic mitochondrial DNA are not unique to AD; however, it appears likely based on the evidence so far that the brain region-specific accumulation of these nucleic acid species is specific to AD. Specifically, in AD patients, cytosolic mitochondrial DNA has been shown to accumulate in neurons of the hippocampus and neocortex [18], two of the most severely affected regions in AD. By contrast, in Parkinson’s disease patients, accumulations have been demonstrated of cytosolic oxidatively damaged RNA and DNA in substantia nigra neurons [121] and of cytosolic mitochondrial double-stranded DNA together with the IFI16 inflammasome and Lewy bodies in the medulla oblongata [71], with the substantia nigra and medulla oblongata being two severely affected brain regions in Parkinson’s disease [122]. This suggests that, although harmful cytosolic nucleic acid species accumulate in both AD and Parkinson’s disease patients, their accumulation in hippocampal and neocortical neurons may be specific to AD, whereas their accumulation in substantia nigra and medulla oblongata neurons may be specific to Parkinson’s disease.
The fourth Hill causality criterion is “Temporality \dots Does exposure precede the outcome variable?” [120]. If the outcome variable is dementia or cognitive impairment, then “exposure” does indeed precede it, since PCAD is a stage of moderate AD neurodegeneration that may precede cognitive impairment, and cytosolic oxidatively damaged DNA presumed to be of mitochondrial origin accumulated in PCAD individuals’ hippocampal neurons [19].
The fourth Hill causality criterion is “Biological gradient \dots Is there evidence of a dose-response relationship?” [120]. There is. As discussed in the section of this manuscript titled “Cytosolic oxidatively damaged mitochondrial DNA accumulated dose-dependently in PCAD and AD patients’ vulnerable neurons (i.e., opportunity),” it appears that cytosolic DNA accumulated in a dose-dependent fashion in PCAD individuals’ hippocampal and parahippocampal gyrus neurons compared to elderly controls’ [19], and in AD patients’ hippocampal neurons compared to elderly controls’ and PCAD individuals’ [18, 19].
The fifth Hill causality criterion is “Plausibility \dots Does the causal relationship make biological sense?” [120]. The causal relationship between cytosolic and cell-free nucleic acids and neuroinflammation, synapse, degeneration, and neuron death does make sense. As reviewed in previous sections of this manuscript, in brief, cytosolic mitochondrial DNA has been shown to accumulate in PCAD and AD patients’ vulnerable neurons [18, 19], and to be capable in preclinical models of driving inflammation, synapse degeneration, and neuron death [26, 71]. Furthermore, various cell-free nucleic acid species have been shown to accumulate in AD patients [12, 15, 21–23, 33, 34, 91], and relatively indirect evidence suggests that at least some of these cell-free nucleic acid species may also promote neuroinflammation and synaptotoxicity in AD [21, 92–97].
The sixth Hill causality criterion is “Coherence \dots Is the causal association compatible with present knowledge of the disease?” [120]. Yes, it is. There does not appear to be any inconsistencies between this causal association and present knowledge of the disease.
The seventh Hill causality criterion is “Experimentation \dots Does controlled manipulation of the exposure variable change the outcome?” Yes, it has. As reviewed in the sections “Cytosolic oxidatively damaged mitochondrial DNA in neural cells causes neuroinflammation, synapse degeneration, and neural death by oligomerizing with pattern-recognition receptors (i.e., means)” and “Cell-free nucleic acids upregulated interferons and the complement cascade, which caused synapse degeneration,” clearing cytosolic and/or mitochondrial DNA in different experiments was shown to improve inflammatory gene expression, synapse degeneration, neurotoxicity, or functional outcomes [26, 71]. For a discussion of the only currently available clinical experiment, see “DNase-I appeared to unprecedently improve a severe AD patients’ MMSE by 15 points,” although it must be noted that this was not a controlled experiment.
The eighth Hill causality criterion is “Analogy \dots Does the causal relationship conform to a previously described relationship?” [120]. Yes, it does. First of all, that DAMP nucleic acids induce innate inflammation by oligomerizing with innate immune pattern recognition receptors is settled science in basic immunology, so no analogy is needed. That being said, it is analogous to the idea of PAMP nucleic acids causing inflammation in infectious diseases.
Therefore, at least cytosolic DNA appears to provisionally meet the Hill causality criteria to be considered a probable etiological agent in AD.
ANALYSIS FROM FREDRICKS-RELMAN REVISED KOCH’S POSTULATES
The association between ectopic nucleic acids and AD can also be assessed using Koch’s postulates. Although Koch’s postulates were one of the first and most well-known attempts to construct an instrument to estimate the probability of causality, there were problems with the original version that led to the proposal of various revised Koch’s postulates. Here Fredricks and Relman’s revision of Koch’s postulates is used because it takes into account advances made possible by molecular biology.
The first Fredricks-Relman revised Koch’s postulate is that “(i) A nucleic acid sequence belonging to a putative pathogen should be present in most cases of an infectious disease. Microbial nucleic acids should be found preferentially in those organs or gross anatomic sites known to be diseased (i.e., with anatomic, histologic, chemical, or clinical evidence of pathology) and not in those organs that lack pathology” [120]. Indeed, cytosolic mitochondrial DNA accumulated primarily in hippocampal and neocortical neurons, which are two of the most affected regions and cell populations in AD, but not in cerebellar neurons, which are relatively spared [18–20]. Oxidatively damaged nucleic acids and cytosolic mitochondrial DNA appear to be present in most if not all cases of the diseases including in different cohorts of patients [18–20, 39, 40].
The second Fredricks-Relman revised Koch’s postulate is that “(ii) Fewer, or no, copy numbers of pathogen-associated nucleic acid sequences should occur in hosts or tissues without disease” [120]. Indeed, there was less mitochondrial DNA, oxidatively damaged DNA, and cytosolic oxidatively damaged DNA in elderly controls than in PCAD or AD patients [18–20, 39–41], and cytosolic mitochondrial DNA was not found in relatively spared cerebellar neurons in AD patients [18].
The third Fredricks-Relman revised Koch’s postulate is that “(iii) With resolution of disease (for example, with clinically effective treatment), the copy number of pathogen-associated nucleic acid sequences should decrease or become undetectable. With clinical relapse, the opposite should occur” [120]. This has yet to be tested.
The fourth Fredricks-Relman revised Koch’s postulate is that “(iv) When sequence detection predates disease, or sequence copy number correlates with severity of disease or pathology, the sequence-disease association is more likely to be a causal relationship” [120]. As discussed in the answers above to “Temporality \dots Does exposure precede the outcome variable?” and “Biological gradient \dots Is there evidence of a dose-response relationship?”, this does appear to be the case, as cytosolic DNA accumulates in PCAD patients’ hippocampal and parahippocampal gyrus neurons, predating disease (i.e., dementia) emergence, and neural cytosolic DNA appears to accumulate dose-dependently in AD patients more than in PCAD patients [18, 19].
The fifth Fredricks-Relman revised Koch’s postulate is that “(v) The nature of the microorganism inferred from the available sequence should be consistent with the known biological characteristics of that group of organisms. When phenotypes (e.g., pathology, microbial morphology, and clinical features) are predicted by sequence-based phylogenetic relationships, the meaningfulness of the sequence is enhanced” [120]. Indeed, mitochondrial DNA is known to enter the cytosol of neural cells and various other cell types in response to different stressors, including oxidative stress and autophagy disruption, and induce inflammatory protein expression [26, 71]. In terms of predicting phenotypes, cytosolic mitochondrial DNA would be predicted based on in vitro evidence in neural cells to cause inflammatory protein expression, synapse degeneration, and neuron death [26], which are core features of AD dementia. Another phenotype that this cytosolic mitochondrial DNA predicts is arguably that the disease should be treatable with nucleases to varying extents depending on the administration method, but not with antimicrobials, which appears to potentially be the case with AD based on the evidence so far [5, 6, 38].
The sixth Fredricks-Relman revised Koch’s postulate is that “(vi) Tissue-sequence correlates should be sought at the cellular level: efforts should be made to demonstrate specific in situ hybridization of microbial sequence to areas of tissue pathology and to visible microorganisms or to areas where microorganisms are presumed to be located” [120]. Indeed, mitochondrial DNA was found specifically in the cytosols of AD patients’ severely affected hippocampal and neocortical neurons, but not relatively spared cerebellar neurons or glia [18].
The seventh Fredricks-Relman revised Koch’s postulate is that “(vii) These sequence-based forms of evidence for microbial causation should be reproducible.” As discussed above in “Consistency of association \dots Is there agreement among repeated observations in different places, at different times, using different methodology, by different researchers, under different circumstances?”, at least three independent groups have found accumulations of cytosolic DNA in PCAD or AD patients’ hippocampal neurons [18–20], and various groups have found different cell-free nucleic acid species in AD patients’ neuritic plaques [12, 15, 21–23, 91].
Therefore, the evidence appears to conform to 6/7 of Fredricks-Relman revised Koch’s postulates, suggesting again that ectopic nucleic acids such as cytosolic mitochondrial DNA may be considered potential etiological agents in AD.
POPPER’S FALSIFICATION-BASED THEORY OF SCIENCE ARGUABLY FAVORS THIS HYPOTHESIS OVER THE AMYLOID HYPOTHESIS
There is much more evidence confirming the incumbent amyloid hypothesis than the virus-mimetic oxidatively damaged ectopic nucleic acid hypothesis. However, that may be because the amyloid hypothesis has dominated the field for decades, whereas this hypothesis has been studied relatively less (analogously to incumbency advantage). Moreover, there are numerous inconsistencies between the empirical evidence and the amyloid hypothesis [2, 7, 23, 123–130], but none that this author could find between the empirical evidence and this hypothesis as of mid-2022, despite looking and making every reasonable attempt to not cherry pick evidence. Therefore, although the two hypotheses are essentially consistent with each other, according to Karl Popper’s exceptionally rigorous falsification-based theory of science [131] and based on this preliminary but wide body evidence so far, the virus-mimetic oxidatively damaged ectopic nucleic acid hypothesis is arguably currently more likely to be correct than previous and current amyloid-hypothesis variants.
Here are a few examples of inconsistencies between the amyloid hypothesis and the empirical evidence. In vitro, synthetic Aβ oligomers and fibrils alone induced little to no inflammatory gene expression in optimally-human-microglia-modeling human induced-pluripotent stem-cell-derived microglia and several other human microglia, astrocyte, and macrophage cell lines [124]. Synthetic Aβ oligomers and protofibrils alone did not induce formation of neutrophil-extracellular traps composed of oxidatively-damaged citrullinated histone-3-bound genomic DNA and myeloperoxidase like those that accumulated in AD patients’ brain vasculature and senile/neuritic amyloid plaques [23]. The Aβ in PCAD and AD patients’ hippocampi was found to be in high-molecular weight assemblies that are non-inflammatory and non-synaptotoxic, and no synaptotoxic low-molecular weight oligomers or protofibrils were found [127, 128]. In terms of staging, whereas neurofibrillary tangles start in and remain concentrated in subcortical nuclei and medial-temporal lobe entorhinal cortex and hippocampus, neuritic plaques start in and remain concentrated in neocortical regions [129, 132, 133]. The extent of amyloid deposition is not significantly different between relatively spared and severely affected neocortical regions, and it does not correlate consistently with the severity of alterations such as glucose hypo-metabolism, hypo-perfusion, or neuronal atrophy [130, 134, 135]. Glucose hypo-metabolism precedes amyloid deposition in APOE4 carriers by decades [136, 137]. Oxidatively-damaged RNA 8-hydroxyguanosine accumulated significantly in Down syndrome patients’ cerebral neurons starting in their teens and twenties, whereas Aβ deposition only increased after age 30 [138]. PS1, PS2, APP, and trisomy 21 mutations appear capable of driving AD independently of Aβ too [31, 139–141], and only cause 5% of early-onset AD cases [142].
This is not an exhaustive list of inconsistencies with different variants of the amyloid hypothesis. The point is though that, whereas I could not find any inconsistencies between this hypothesis and the empirical evidence, I found numerous inconsistencies between the amyloid hypothesis and the empirical evidence. Therefore, Popper’s falsification-based theory of science rejects the amyloid hypothesis but does not reject this hypothesis.
LINKAGES TO MAJOR THEORIES
This DAMP/virus-mimetic oxidatively damaged ectopic nucleic acid hypothesis is arguably more consistent than previous amyloid, oxidative-stress, inflammation, infection, and antimicrobial-protection hypothesis variants not only with the empirical evidence so far, but also simultaneously with the up-to-date prion-cofactor model (proponents of the “protein only” prion hypothesis updated to a prion-cofactor model because recombinant prion proteins alone do not cause disease in wild-type animals, but prion proteins bound to nucleic acids or other cofactors do [143]), contemporary basic immunology, germ theory (in the sense that accumulating inflammatory nucleic acids cause diseases), the central dogma (in the sense that nucleic acids are generally more important than proteins), and theories of aging, aging-associated degenerative diseases, and AD centered on free radicals, oxidative stress, damage, injury, entropy, inflammation, inflammaging, and immunosenescence.
IMPLICATIONS ANALOGOUS TO IN INFECTIOUS DISEASES
The relative impact of ectopic nucleic acids on AD pathophysiology has yet to be determined. Critically though, it is worth noting that infectious diseases cannot be cured without reducing the levels of the causative pathogen’s nucleic acids. Therefore, an important implication of this hypothesis is that, if ectopic oxidatively damaged nucleic acids centrally drive AD pathophysiology, analogously to specific pathogen nucleic acids in infectious diseases (as predicted by basic immunology [57] and supported by currently available relevant neuropathology [12, 15, 18–23], cell [21, 26], genome-wide association [27, 28], epidemiological [1, 2, 26, 29–32], biomarker [33–37], animal, and patient studies [21, 26, 38]), then AD may be impossible to stop or reverse without clearing the ectopic nucleic acids accumulated in patients’ brains. If so, this may have hindered clinical trials of other therapeutic approaches, analogously to how giving patients with infectious diseases conventional anti-inflammatories and antioxidants but not the appropriate antimicrobial(s) leads to suboptimal results. A corollary of this is that clearing ectopic nucleic acids with nucleases (especially if targeted to the cytosols of neurons) might synergize with and unlock the full potential of other rational treatment modalities, such as the Aβ protofibril-targeting antibody BAN2401/Lecanemab, anti-inflammatories, and antioxidants such as intranasal N-acetylcysteine and melatonin in combination-treatment clinical trials.
CONCLUSION
These experiments should be replicated and improved upon in additional cell and animal models, including in cultured neurons and sporadic AD-modeling rodents such as those generated with chronic aluminum exposure [144]. As an example of a future preclinical study, future research questions could include, does transfection of DNase-I proteins decrease inflammatory protein levels, synapse degeneration, or neuron death in cultured neurons or mice with AD induced by chronic aluminum, iron, and/or H2O2 exposure?
As discussed, it would fill an important gap in the literature to test the hypothesis that DNA, specifically mitochondrial and oxidatively damaged DNA, accumulate in PCAD, MCI, and AD patients’ neuron-derived plasma exosomes to a greater extent than in those of controls.
In addition, future studies could determine the following: whether the rs141639275 G:A DNASE1 polymorphism decreases ectopic DNA degradation and promotes its accumulation [27]; whether this mutation occurs somatically in any of PCAD or AD patients’ neurons; and whether the reactive oxygen species hydrogen peroxide (H2O2) in the absence of iron can cause this mutation in vitro, since H2O2 treatment experimentally caused predominantly transition mutations in multiple eukaryotic cell types [145–149] (not predominantly transversion mutations as previously predicted and erroneously believed by some based on preliminary indirect evidence [150–153]), predominantly transition mutations accumulated in elderly individuals’ brain mitochondrial DNA compared to in that of young individuals [152], and predominantly transition mutations accumulated in PCAD patients’ brain synaptosomal mitochondrial DNA more than in that of cognitively-intact elderly-control individuals[150].
Clinicals trials for AD patients comparing standard-of-care plus placebo to standard-of-care plus DNase-I combined with its cofactor Mg2+ and an RNase are warranted and should be given top priority. It would also be useful to test in a clinical trial the hypothesis that DNase-I, Mg2+, and RNase delay progression to AD in PCAD patients compared to placebo, since cytosolic oxidatively damaged DNA accumulates in PCAD patients’ vulnerable neurons [19]. The overall hypothesis and the evidence are based upon are what suggest these courses of action, not the clinical evidence per se because the clinical evidence in these areas is quite sparse so far. That being said, there is the case report of DNase-I treatment correlating with a severe AD patient improving 15 points on the MMSE [38]. In addition, DNase-I is safe and FDA-approved for the treatment of cystic fibrosis [154]. A phase II randomized, double-blind, placebo-controlled clinical trial for 30 patients with primary Sjögren’s syndrome found that an RNase Fc fusion protein significantly improved patients’ cognition on the Digit Symbol Substitution Test, as well as the severity of fatigue on the Profile of Fatigue and Functional Assessment of Chronic Illness Therapy–Fatigue, and disease severity on the European League Against Rheumatism Sjögren’s Syndrome Patient Reported Index [155]. This very preliminary clinical evidence is nonetheless still suggestive that DNase-I and an RNase should be at least safe, and possibly effective at improving cognition, in AD patients [38, 154, 155].
I propose that, ideally, these agents should be administered intranasally both in freely diffusible/soluble forms and transfected into neurons and selectively targeted to the cytosol using extracellular vesicles or virus-like particles, techniques discussed elsewhere [156, 157]. I suggest administering them intranasally because intranasal administration has been successfully used to deliver peptides into the brain, including in clinical trials for AD patients [158]. I suggest administering some of the nuclease proteins in the freely diffusible/soluble form since doing so correlated with a 15-point improvement on the MMSE in one AD patient [38], and as discussed, this approach should target cell-free nucleic acids well. I suggest transfecting some of the nuclease proteins into neurons using some form of extracellular vesicles or virus-like particles targeted to the cytosol because, as discussed, it appears that some of the most harmful nucleic acid species that need to be degraded are the accumulated free cytosolic mitochondrial DNA [18, 72]. These innovative nuclease delivery methods should be tested for safety and efficacy first in cultured neurons and rodents.
ACKNOWLEDGMENTS
The author has no acknowledgments to report.
FUNDING
The author has no funding to report.
CONFLICT OF INTEREST
The author has no conflict of interest to report.
REFERENCES
[1] | Sanders OD , Rajagopal L , Rajagopal JA ((2021) ) Does oxidatively damaged DNA drive amyloid-β generation in Alzheimer’s disease? A hypothesis. J Neurogenet 35: , 351–357. |
[2] | Sanders OD , Rajagopal L , Rajagopal JA ((2022) ) The oxidatively damaged DNA and amyloid-β oligomer hypothesis of Alzheimer’s disease. Free Radic Biol Med 179: , 403–412. |
[3] | Budd Haeberlein S , Aisen PS , Barkhof F , Chalkias S , Chen T , Cohen S , Dent G , Hansson O , Harrison K , von Hehn C , Iwatsubo T , Mallinckrodt C , Mummery CJ , Muralidharan KK , Nestorov I , Nisenbaum L , Rajagovindan R , Skordos L , Tian Y , van Dyck CH , Vellas B , Wu S , Zhu Y , Sandrock A ((2022) ) Two randomized phase 3 studies of Aducanumab in early Alzheimer’s disease. J Prev Alzheimers Dis 9: , 197–210. |
[4] | Swanson CJ , Zhang Y , Dhadda S , Wang J , Kaplow J , Lai RYK , Lannfelt L , Bradley H , Rabe M , Koyama A , Reyderman L , Berry DA , Berry S , Gordon R , Kramer LD , Cummings JL ((2021) ) A randomized, double-blind, phase 2b proof-of-concept clinical trial in early Alzheimer’s disease with lecanemab, an anti-Aβ protofibril antibody. Alzheimers Res Ther 13: , 80. |
[5] | Molloy DW , Standish TI , Zhou Q , Guyatt G ((2013) ) A multicenter, blinded, randomized, factorial controlled trial of doxycycline and rifampin for treatment of Alzheimer’s disease: The DARAD trial. Int J Geriatr Psychiatry 28: , 463–470. |
[6] | Lindblom N , Lindquist L , Westman J , Aström M , Bullock R , Hendrix S , Wahlund L-O ((2021) ) Potential virus involvement in Alzheimer’sdisease: Results from a phase IIa trial evaluating Apovir, anantiviral drug combination. J Alzheimers Dis Rep 5: , 413. |
[7] | Plascencia-Villa G , Perry G ((2020) ) Status and future directions of clinical trials in Alzheimer’s disease. Int Rev Neurobiol 154: , 3–50. |
[8] | Broxmeyer L ((2017) ) Are the infectious roots of Alzheimers buried deep in the past? J Mol Path Epidemiol 3: , 2. |
[9] | Moir RD , Lathe R , Tanzi RE ((2018) ) The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement 14: , 1602–1614. |
[10] | Alonso R , Pisa D , Fernández-Fernández AM , Carrasco L ((2018) ) Infection of fungi and bacteria in brain tissue from elderly personsand patients with Alzheimer’s disease. Front Aging Neurosci 10: , 159. |
[11] | Wormser GP , Marques A , Pavia CS , Schwartz I , Feder HM , Pachner AR ((2021) ) Lack of convincing evidence that Borrelia burgdorferi infection causes either Alzheimer’s disease or Lewy body dementia. Clin Infect Dis 75: , 342–346. |
[12] | Miklossy J ((2016) ) Bacterial amyloid and DNA are important constituents of senile plaques: Further evidence of the spirochetal and biofilm nature of senile plaques. J Alzheimers Dis 53: , 1459–1473. |
[13] | Fülöp T , Itzhaki RF , Balin BJ , Miklossy J , Barron AE ((2018) ) Role of microbes in the development of Alzheimer’s disease: State of the Art - An International Symposium Presented at the 2017 IAGG Congress in San Francisco. Front Genet 9: , 362. |
[14] | Readhead B , Funk CC , Ehrlich ME , Gandy S , Dudley JT , Readhead B , Funk CC , Richards MA , Shannon P ((2018) ) Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human Herpesvirus. Neuron 99: , 64–82. |
[15] | Wozniak M , Mee A , Itzhaki R ((2009) ) Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J Pathol 217: , 131–138. |
[16] | Chorlton SD ((2020) ) Reanalysis of Alzheimer’s brain sequencing data reveals absence of purported HHV6A and HHV7. J Bioinform Comput Biol 18: , 2050012. |
[17] | Itzhaki RF ((2021) ) Overwhelming evidence for a major role for herpes simplex virus type 1 (HSV1) in Alzheimer’s disease (AD); Underwhelming evidence against. Vaccines 9: , 679. |
[18] | Hirai K , Aliev G , Nunomura A , Fujioka H , Russell RL , Atwood CS , Johnson AB , Kress Y , Vinters H V , Tabaton M , Shimohama S , Cash AD , Siedlak SL , Harris PL , Jones PK , Petersen RB , Perry G , Smith MA ((2001) ) Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci 21: , 3017–3023. |
[19] | Lovell MA , Soman S , Bradley MA ((2011) ) Oxidatively modified nucleic acids in preclinical Alzheimer’s disease (PCAD) brain. Mech Ageing Dev 132: , 443–448. |
[20] | Majd S , Power JHT ((2018) ) Oxidative stress and decreased mitochondrial superoxide dismutase 2 and peroxiredoxins 1 and 4 based mechanism of concurrent activation of AMPK and mTOR in Alzheimer’s disease. Curr Alzheimer Res 15: , 1–13. |
[21] | Roy ER , Wang B , Wan YW , Chiu G , Cole A , Yin Z , Propson NE , Xu Y , Jankowsky JL , Liu Z , Lee VM-YMY , Trojanowski JQ , Ginsberg SD , Butovsky O , Zheng H , Cao W ((2020) ) Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest 130: , 1912–1930. |
[22] | Ginsberg SD , Crino PB , Hemby SE , Weingarten JA , Lee VM-Y , Eberwine JH , Trojanowski JQ ((1999) ) Predominance of neuronal mRNAs in individual Alzheimer’s disease senile plaques. Ann Neurol 45: , 174–181. |
[23] | Smyth LCD , Murray HC , Hill M , van Leeuwen E , Highet B , Magon NJ , Osanlouy M , Mathiesen SN , Mockett B , Singh-Bains MK , Morris VK , Clarkson AN , Curtis MA , Abraham WC , Hughes SM , Faull RLM , Kettle AJ , Dragunow M , Hampton MB ((2022) ) Neutrophil-vascular interactions drive myeloperoxidase accumulation in the brain in Alzheimer’s disease. Acta Neuropathol Commun 10: , 38. |
[24] | von Herrmann KM , Salas LA , Martinez EM , Young AL , Howard JM , Feldman MS , Christensen BC , Wilkins OM , Lee SL , Hickey WF , Havrda MC ((2018) ) NLRP3 expression in mesencephalic neurons and characterization of a rare NLRP3 polymorphism associated with decreased risk of Parkinson’s disease. NPJ Parkinsons Dis 4: , 24. |
[25] | Ahmed ME , Iyer S , Thangavel R , Kempuraj D , Selvakumar GP , Raikwar SP , Zaheer S , Zaheer A ((2017) ) Co-localization of glia maturation factor with NLRP3 inflammasome and autophagosome markers in human Alzheimer’s disease brain. J Alzheimers Dis 60: , 1143–1160. |
[26] | Jauhari A , Baranov S V. , Suofu Y , Kim J , Singh T , Yablonska S , Li F , Wang X , Oberly P , Minnigh MB , Poloyac SM , Carlisle DL , Friedlander RM ((2020) ) Melatonin inhibits cytosolic mitochondrial DNA-induced neuroinflammatory signaling in accelerated aging and neurodegeneration. J Clin Invest 130: , 3124–3136. |
[27] | Kuzma A , Valladares O , Cweibel R , Greenfest-Allen E , Childress DM , Malamon J , Gangadharan P , Zhao Y , Qu L , Leung YY , Naj AC , Stoeckert CJ , Schellenberg GD , Wang L-S ((2016) ) NIAGADS: The NIA Genetics of Alzheimer’s Disease Data Storage Site. Alzheimers Dement 12: , 1200–1203. |
[28] | Bis JC , Jian X , Kunkle BW , Chen Y , Hamilton-Nelson KL , Bush WS , Salerno WJ , Lancour D , Ma Y , Renton AE , Marcora E , Farrell JJ , Zhao Y , Qu L , Ahmad S , Amin N , Amouyel P , Beecham GW , Below JE , Campion D , Cantwell L , Charbonnier C , Chung J , Crane PK , Cruchaga C , Cupples LA , Dartigues JF , Debette S , Deleuze JF , Fulton L , Gabriel SB , Genin E , Gibbs RA , Goate A , Grenier-Boley B , Gupta N , Haines JL , Havulinna AS , Helisalmi S , Hiltunen M , Howrigan DP , Ikram MA , Kaprio J , Konrad J , Kuzma A , Lander ES , Lathrop M , Lehtimäki T , Lin H , Mattila K , Mayeux R , Muzny DM , Nasser W , Neale B , Nho K , Nicolas G , Patel D , Pericak-Vance MA , Perola M , Psaty BM , Quenez O , Rajabli F , Redon R , Reitz C , Remes AM , Salomaa V , Sarnowski C , Schmidt H , Schmidt M , Schmidt R , Soininen H , Thornton TA , Tosto G , Tzourio C , van der Lee SJ , van Duijn CM , Valladares O , Vardarajan B , Wang LS , Wang W , Wijsman E , Wilson RK , Witten D , Worley KC , Zhang X , Bellenguez C , Lambert JC , Kurki MI , Palotie A , Daly M , Boerwinkle E , Lunetta KL , Destefano AL , Dupuis J , Martin ER , Schellenberg GD , Seshadri S , Naj AC , Fornage M , Farrer LA ((2020) ) Whole exome sequencing studyidentifies novel rare and common Alzheimer’s-Associated variantsinvolved in immune response and transcriptional regulation. MolPsychiatry 25: , 1859–1875. |
[29] | Takahashi A , Okada R , Nagao K , Kawamata Y , Hanyu A , Yoshimoto S , Takasugi M , Watanabe S , Kanemaki MT , Obuse C , Hara E ((2017) ) Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat Commun 8: , 15287. |
[30] | Guglielmotto M , Giliberto L , Tamagno E , Tabaton M ((2010) ) Oxidative stress mediates the pathogenic effect of different Alzheimer’s disease risk factors. Front Aging Neurosci 2: , 3. |
[31] | Meraz-Ríos MA , Franco-Bocanegra D , Toral Rios D , Campos-Peña V ((2014) ) Early onset Alzheimer’s disease and oxidative stress. Oxid Med Cell Longev 2014: , 375968. |
[32] | Turpin C , Catan A , Guerin-Dubourg A , Debussche X , Bravo SB , Álvarez E , van Den Elsen J , Meilhac O , Rondeau P , Bourdon E ((2020) ) Enhanced oxidative stress and damage in glycatederythrocytes, PLoS One 15: , e0235335. |
[33] | Pai M , Kuo Y , Wang I , Chiang P , Tsai K ((2019) ) The role of methylated circulating nucleic acids as a potential biomarker in Alzheimer’s disease. Mol Neurobiol 56: , 2440–2449. |
[34] | Kim KM , Meng Q , Perez de Acha O , Mustapic M , Cheng A , Eren E , Kundu G , Piao Y , Munk R , Wood WH , De S , Noh JH , Delannoy M , Cheng L , Abdelmohsen K , Kapogiannis D , Gorospe M ((2020) ) Mitochondrial RNA in Alzheimer’s disease circulating extracellular vesicles. Front Cell Dev Biol 8: , 1049. |
[35] | Podlesniy P , Llorens F , Puigròs M , Serra N , Sepúlveda-Falla D , Schmidt C , Hermann P , Zerr I , Trullas R ((2020) ) Cerebrospinal fluid mitochondrial DNA in rapid and slow progressive forms of Alzheimer’s disease. Int J Mol Sci 21: , 6298. |
[36] | Podlesniy P , Figueiro-Silva J , Llado A , Antonell A , Sanchez-Valle R , Alcolea D , Lleo A , Molinuevo JL , Serra N , Trullas R ((2013) ) Low cerebrospinal fluid concentration of mitochondrial DNA in preclinical Alzheimer disease. Ann Neurol 74: , 655–668. |
[37] | Podlesniy P , Llorens F , Golanska E , Sikorska B , Liberski P , Zerr I , Trullas R ((2016) ) Mitochondrial DNA differentiates Alzheimer’s disease from Creutzfeldt-Jakob disease. Alzheimers Dement 12: , 546–555. |
[38] | Tetz V , Tetz G ((2016) ) Effect of deoxyribonuclease I treatment for dementia in end-stage Alzheimer’s disease: A case report. J Med Case Rep 10: , 131. |
[39] | Mecocci P , MacGarvey U , Beal MF ((1994) ) Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol 36: , 747–751. |
[40] | Wang J , Markesbery WR , Lovell MA ((2006) ) Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J Neurochem 96: , 825–832. |
[41] | Wang J , Xiong S , Xie C , Markesbery WR , Lovell MA ((2005) ) Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J Neurochem 93: , 953–962. |
[42] | Davis Sanders O , Rajagopal L , Chase Barton C , Archa Rajagopal J , Lopez O , Lopez K , Malik F ((2022) ) Does oxidative DNA damage trigger histotoxic hypoxia via PARP1/AMP-driven mitochondrial ADP depletion-induced ATP synthase inhibition in Alzheimer’s disease? Mitochondrion 67: , 59–64. |
[43] | Heneka MT , Kummer MP , Stutz A , Delekate A , Schwartz S , Vieira-saecker A , Griep A , Axt D , Remus A , Tzeng T , Gelpi E , Halle A , Korte M , Latz E , Golenbock DT ((2013) ) NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493: , 674–678. |
[44] | Van Zeller M , Dias D , Sebastião AM , Valente CA ((2021) ) NLRP3 inflammasome: A starring role in amyloid-β- and tau-driven pathological events in Alzheimer’s disease. J Alzheimers Dis 83: , 939–961. |
[45] | Haseeb M , Javaid N , Yasmeen F , Jeong U , Han JH , Yoon J , Seo JY , Heo JK , Shin HC , Kim MS , Kim W , Choi S ((2022) ) Novel small-molecule inhibitor of NLRP3 inflammasome reverses cognitive impairment in an Alzheimer’s disease model. ACS Chem Neurosci 13: , 818–833. |
[46] | Feng YS , Tan ZX , Wu LY , Dong F , Zhang F ((2020) ) The involvement of NLRP3 inflammasome in the treatment of Alzheimer’s disease. Ageing Res Rev 64: , 101192. |
[47] | Bai H , Zhang Q ((2021) ) Activation of NLRP3 inflammasome and onset of Alzheimer’s disease. Front Immunol 12: , 2998. |
[48] | Choudhury SM , Ma X , Abdullah SW , Zheng H ((2021) ) Activation and inhibition of the NLRP3 inflammasome by RNA viruses. J Inflamm Res 14: , 1145. |
[49] | da Costa LS , Outlioua A , Anginot A , Akarid K , Arnoult D ((2019) ) RNA viruses promote activation of the NLRP3 inflammasome through cytopathogenic effect-induced potassium efflux. Cell Death Dis 10: , 346. |
[50] | Zhong Z , Liang S , Sanchez-Lopez E , He F , Shalapour S , Lin X-J , Wong J , Ding S , Seki E , Schnabl B , Hevener AL , Greenberg HB , Kisseleva T , Karin M ((2018) ) New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560: , 198–203. |
[51] | Gal-Ben-Ari S , Barrera I , Ehrlich M , Rosenblum K ((2019) ) PKR: A kinase to remember. Front Mol Neurosci 11: , 480. |
[52] | Hugon J , Mouton-Liger F , Dumurgier J , Paquet C ((2017) ) PKR involvement in Alzheimer’s disease. Alzheimers Res Ther 9: , 83. |
[53] | Ohno M ((2014) ) Roles of eIF2α kinases in the pathogenesis of Alzheimer’s disease. Front Mol Neurosci 7: , 22. |
[54] | Mouton-Liger F , Paquet C , Dumurgier J , Lapalus P , Gray F , Laplanche J-L , Hugon J , Groupe d’Investigation du Liquide Céphalorachidien Study Network ((2012) ) Increased cerebrospinal fluid levels of double-stranded RNA-dependant protein kinase in Alzheimer’s disease. Biol Psychiatry 71: , 829–835. |
[55] | Peel AL , Bredesen DE ((2003) ) Activation of the cell stress kinase PKR in Alzheimer’s disease and human amyloid precursor protein transgenic mice. Neurobiol Dis 14: , 52–62. |
[56] | Martorell P , Brouwer V , Schwarz S , Heneka M ((2020) ) cGAS-STING activation in Alzheimer’s disease. In BonnBrain3 Meeting, Bonn. |
[57] | Kumar V ((2021) ) The trinity of cGAS, TLR9, and ALRs guardians of the cellular galaxy against host-derived self-DNA. Front Immunol 11: , 1. |
[58] | Xu M , Zhang DF , Luo R , Wu Y , Zhou H , Kong LL , Bi R , Yao YG ((2018) ) A systematic integrated analysis of brain expression profiles reveals YAP1 and other prioritized hub genes as important upstream regulators in Alzheimer’s disease. Alzheimers Dement 14: , 215–229. |
[59] | Wang BR , Zaheer A , Lim R ((1992) ) Polyclonal antibody localizes glia maturation factor beta-like immunoreactivity in neurons and glia. Brain Res 591: , 1–7. |
[60] | Fan J , Fong T , Chen X , Chen C , Luo P , Xie H ((2018) ) Glia maturation factor-β: A potential therapeutic target in neurodegeneration and neuroinflammation. Neuropsychiatr Dis Treat 14: , 495. |
[61] | Bordi M , Berg MJ , Mohan PS , Peterhoff CM , Alldred MJ , Che S , Ginsberg SD , Nixon RA ((2016) ) Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 12: , 2467–2483. |
[62] | Choubey D ((2019) ) Type i interferon (IFN)-inducible Absent in Melanoma 2 proteins in neuroinflammation: Implications for Alzheimer’s disease. J Neuroinflammation 16: , 236. |
[63] | van de Veerdonk FL , Netea MG , Dinarello CA , Joosten LAB ((2011) ) Inflammasome activation and IL-1β and IL-18 processing during infection. Trends Immunol 32: , 110–116. |
[64] | Elliott EI , Sutterwala FS ((2015) ) Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev 265: , 35–52. |
[65] | Rui W , Xiao H , Fan Y , Ma Z , Xiao M , Li S , Shi J ((2021) ) Systemic inflammasome activation and pyroptosis associate with the progression of amnestic mild cognitive impairment and Alzheimer’s disease. J Neuroinflammation 18: , 280. |
[66] | Malaguarnera L , Motta M , Di Rosa M , Anzaldi M , Malaguarnera M ((2006) ) Interleukin-18 and transforming growth factor-beta 1 plasma levelsin Alzheimer’s disease and vascular dementia. Neuropathology 26: , 307–312. |
[67] | Russo HM , Rathkey J , Boyd-Tressler A , Katsnelson MA , Abbott DW , Dubyak GR ((2016) ) Active caspase-1 induces plasma membrane pores that precede pyroptotic lysis and are blocked by lanthanides. J Immunol 197: , 1353–1367. |
[68] | Chen X , He W , Hu L , Li J , Fang Y , Wang X , Xu X , Wang Z , Huang K , Han J ((2016) ) Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res 26: , 1007–1020. |
[69] | He W , Wan H , Hu L , Chen P , Wang X , Huang Z , Yang Z-H , Zhong C-Q , Han J ((2015) ) Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res 25: , 1285–1298. |
[70] | Cypryk W , Nyman TA , Matikainen S ((2018) ) From inflammasome to exosome-does extracellular vesicle secretion constitute an inflammasome-dependent immune response? Front Immunol 9: , 2188. |
[71] | Matsui H , Ito J , Matsui N , Uechi T , Onodera O , Kakita A ((2021) ) Cytosolic dsDNA of mitochondrial origin induces cytotoxicity and neurodegeneration in cellular and zebrafish models of Parkinson’s disease. Nat Commun 12: , 3101. |
[72] | Moreira PI , Siedlak SL , Wang X , Santos MS , Oliveira CR , Tabaton M , Nunomura A , Szweda LI , Aliev G , Smith MA , Zhu X , Perry G ((2007) ) Autophagocytosis of mitochondria is prominent in Alzheimer disease. J Neuropathol Exp Neurol 66: , 525–532. |
[73] | Song X , Ma F , Herrup K ((2019) ) Accumulation of cytoplasmic DNA due to ATM deficiency activates the microglial viral response system with neurotoxic consequences. J Neurosci 39: , 6378–6394. |
[74] | Shen X , Chen J , Li J , Kofler J , Herrup K ((2016) ) Neurons in vulnerable regions of the Alzheimer’s disease brain display reduced ATM signaling. eNeuro 3: , ENEURO.0124–15.2016. |
[75] | Aho L , Pikkarainen M , Hiltunen M , Leinonen V , Alafuzoff I ((2010) ) Immunohistochemical visualization of amyloid-beta protein precursor and amyloid-beta in extra- and intracellular compartments in the human brain. J Alzheimers Dis 20: , 1015–1028. |
[76] | Blair J , Siedlak S , Wolfram J , Nunomura A , Castellani R , Ferreira S , Klein W , Wang Y , Casadesus G , Smith M , Perry G , Zhu X , Lee H ((2014) ) Accumulation of intraneuronal amyloid-β is common in normal brain. Curr Alzheimer Res 11: , 317–324. |
[77] | Welikovitch LA , Do Carmo S , Maglóczky Z , Szocsics P , Lőke J , Freund T , Cuello AC ((2018) ) Evidence of intraneuronal Aβ accumulation preceding tau pathology in the entorhinal cortex. Acta Neuropathol 136: , 901–917. |
[78] | Wahlberg E , Rahman MM , Lindberg H , Gunneriusson E , Schmuck B , Lendel C , Sandgren M , Löfblom J , Ståhl S , Härd T ((2017) ) Identification of proteins that specifically recognize and bind protofibrillar aggregates of amyloid-β. Sci Rep 7: , 5949. |
[79] | Tomic JL , Pensalfini A , Head E , Glabe CG ((2009) ) Soluble fibrillar oligomer levels are elevated in Alzheimer’s disease brain and correlate with cognitive dysfunction. Neurobiol Dis 35: , 352–358. |
[80] | Kayed R , Head E , Sarsoza F , Saing T , Cotman CW , Necula M , Margol L , Wu J , Breydo L , Thompson JL , Rasool S , Gurlo T , Butler P , Glabe CG ((2007) ) Fibril specific, conformation dependent antibodies recognize a generic epitope common to amyloid fibrils and fibrillar oligomers that is absent in prefibrillar oligomers. Mol Neurodegener 2: , 18. |
[81] | Christensen DZ , Schneider-Axmann T , Lucassen PJ , Bayer TA , Wirths O ((2010) ) Accumulation of intraneuronal Aβ correlates with ApoE4 genotype. Acta Neuropathol 119: , 555–566. |
[82] | Lee EY , Srinivasan Y , de Anda J , Nicastro LK , Tükel Ç , Wong GCL ((2020) ) Functional reciprocity of amyloids and antimicrobial peptides: Rethinking the role of supramolecular assembly in host defense, immune activation, and inflammation. Front Immunol 11: , 1629. |
[83] | Soscia SJ , Kirby JE , Washicosky KJ , Tucker SM , Ingelsson M , Hyman B , Burton MA , Goldstein LE , Duong S , Tanzi RE , Moir RD ((2010) ) The Alzheimer’s disease-associated amyloid β-protein is an antimicrobial peptide. PLoS One 5: , e9505. |
[84] | Khmeleva SA , Radko SP , Kozin SA , Kiseleva YY , Mezentsev Y V. , Mitkevich VA , Kurbatov LK , Ivanov AS , Makarov AA ((2016) ) Zinc-mediated binding of nucleic acids to amyloid-β aggregates: Role of histidine residues. J Alzheimers Dis 54: , 809–819. |
[85] | Du M , Chen ZJ ((2018) ) DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361: , 704–709. |
[86] | Tetz G , Tetz V ((2021) ) Bacterial extracellular DNA promotes β-amyloid aggregation. Microorganisms 9: , 1301. |
[87] | Barrantes A , Rejas MT , Benítez MJ , Jiménez JS ((2007) ) Interaction between Alzheimer’s Aβ1-42 peptide and DNAdetected by surface plasmon resonance. J Alzheimers Dis 12: , 345–355. |
[88] | Ahn BW , Song DU , Jung Y Do , Chay KO , Chung MA , Yang SY , Shin BA ((2000) ) Detection of β-amyloid peptide aggregation using DNA electrophoresis. Anal Biochem 284: , 401–405. |
[89] | Yu H , Ren J , Qu X ((2007) ) Time-dependent DNA condensation induced by amyloid β-peptide. Biophys J 92: , 185–191. |
[90] | Camero S , Ayuso JM , Barrantes A , Benítez MJ , Jiménez JS ((2013) ) Specific binding of DNA to aggregated forms of Alzheimer’s disease amyloid peptides. Int J Biol Macromol 55: , 201–206. |
[91] | Ginsberg SD , Crino PB , Lee VMY , Eberwine JH , Trojanowski JQ ((1997) ) Sequestration of RNA in Alzheimer’s disease neurofibrillary tangles and senile plaques. Ann Neurol 41: , 200–209. |
[92] | Ye W , Tang X , Yang Z , Liu C , Zhang X , Jin J , Lyu J ((2017) ) Plasma-derived exosomes contribute to inflammation via the TLR9-NF-κB pathway in chronic heart failure patients. Mol Immunol 87: , 114–121. |
[93] | Tsilioni I , Theoharides TC ((2018) ) Extracellular vesicles are increased in the serum of children with autism spectrum disorder, contain mitochondrial DNA, and stimulate human microglia to secrete IL-1β. J Neuroinflammation 15: , 239. |
[94] | Torralba D , Baixauli F , Villarroya-Beltri C , Fernández-Delga I , Latorre-Pellicer A , Acín-Pérez R , Martín-Cófreces NB , Jaso-Tamame ÁL , Iborra S , Jorge I , González-Aseguinolaza G , Garaude J , Vicente-Manzanares M , Enríquez JA , Mittelbrunn M , Sánchez-Madrid F ((2018) ) Primingof dendritic cells by DNA-containing extracellular vesicles fromactivated T cells through antigen-driven contacts. Nat Commun 9: , 2658. |
[95] | Sisquella X , Ofir-Birin Y , Pimentel MA , Cheng L , Abou Karam P , Sampaio NG , Penington JS , Connolly D , Giladi T , Scicluna BJ , Sharples RA , Waltmann A , Avni D , Schwartz E , Schofield L , Porat Z , Hansen DS , Papenfuss AT , Eriksson EM , Gerlic M , Hill AF , Bowie AG , Regev-Rudzki N ((2017) ) Malaria parasite DNA-harbouring vesicles activate cytosolic immune sensors. Nat Commun 8: , 1985. |
[96] | Kitai Y , Kawasaki T , Sueyoshi T , Kobiyama K , Ishii KJ , Zou J , Akira S , Matsuda T , Kawai T ((2017) ) DNA-containing exosomes derived from cancer cells treated with topotecan activate a STING-dependent pathway and reinforce antitumor immunity. J Immunol 198: , 1649–1659. |
[97] | Lian Q , Xu J , Yan S , Huang M , Ding H , Sun X , Bi A , Ding J , Sun B , Geng M ((2017) ) Chemotherapy-induced intestinal inflammatory responses are mediated by exosome secretion of double-strand DNA via AIM2 inflammasome activation. Cell Res 27: , 784–800. |
[98] | McGeer PL , Akiyama H , Itagaki S , McGeer EG ((1989) ) Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci Lett 107: , 341–346. |
[99] | Terai K , Walker DG , McGeer EG , McGeer PL ((1997) ) Neurons express proteins of the classical complement pathway in Alzheimer disease. Brain Res 769: , 385–390. |
[100] | Eikelenboom P , Stam FC ((1982) ) Immunoglobulins and complement factors in senile plaques. An immunoperoxidase study. Acta Neuropathol 57: , 239–242. |
[101] | Ishii T , Haga S ((1984) ) Immuno-electron-microscopic localization of complements in amyloid fibrils of senile plaques. Acta Neuropathol 63: , 296–300. |
[102] | Krance SH , Wu CY , Zou Y , Mao H , Toufighi S , He X , Pakosh M , Swardfager W ((2021) ) The complement cascade in Alzheimer’s disease: A systematic review and meta-analysis. Mol Psychiatry 26: , 5532–5541. |
[103] | Stadlbauer V , Engertsberger L , Komarova I , Feldbacher N , Leber B , Pichler G , Fink N , Scarpatetti M , Schippinger W , Schmidt R , Horvath A ((2020) ) Dysbiosis, gut barrier dysfunction and inflammation in dementia: A pilot study. BMC Geriatr 20: , 248. |
[104] | Starr JM , Farrall AJ , Armitage P , McGurn B , Wardlaw J ((2009) ) Blood– brain barrier permeability in Alzheimer’s disease: A case– control MRI study. Psychiatry Res Neuroimaging 171: , 232–241. |
[105] | Van De Haar HJ , Burgmans S , Jansen JFA , Van Osch MJP , Van Buchem MA , Muller M , Hofman PAM , Verhey FRJ , Backes WH ((2016) ) Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology 281: , 527–535. |
[106] | van de Haar HJ , Jansen JFA , van Osch MJP , van Buchem MA , Muller M , Wong SM , Hofman PAM , Burgmans S , Verhey FRJ , Backes WH ((2016) ) Neurovascular unit impairment in early Alzheimer’s disease measured with magnetic resonance imaging. Neurobiol Aging 45: , 190–196. |
[107] | Bairamian D , Sha S , Rolhion N , Sokol H , Dorothée G , Lemere CA , Krantic S ((2022) ) Microbiota in neuroinflammation and synaptic dysfunction: A focus on Alzheimer’s disease. Mol Neurodegener 17: , 1–23. |
[108] | Vogt NM , Kerby RL , Dill-McFarland KA , Harding SJ , Merluzzi AP , Johnson SC , Carlsson CM , Asthana S , Zetterberg H , Blennow K , Bendlin BB , Rey FE ((2017) ) Gut microbiome alterations in Alzheimer’s disease. Sci Rep 7: , 13537. |
[109] | Jiang C , Li G , Huang P , Liu Z , Zhao B ((2017) ) The gut microbiota and Alzheimer’s disease. J Alzheimers Dis 58: , 1–15. |
[110] | Ryser HJP ((1968) ) Uptake of protein by mammalian cells: An underdeveloped area. Science 159: , 390–396. |
[111] | Wall DA , Maack T ((1985) ) Endocytic uptake, transport, and catabolism of proteins by epithelial cells. Am J Physiol 248: , C12–20. |
[112] | Thakur BK , Zhang H , Becker A , Matei I , Huang Y , Costa-Silva B , Zheng Y , Hoshino A , Brazier H , Xiang J , Williams C , Rodriguez-Barrueco R , Silva JM , Zhang W , Hearn S , Elemento O , Paknejad N , Manova-Todorova K , Welte K , Bromberg J , Peinado H , Lyden D ((2014) ) Double-stranded DNA in exosomes: A novel biomarker in cancer detection. Cell Res 24: , 766–769. |
[113] | Liu H , Tian Y , Xue C , Niu Q , Chen C , Yan X ((2022) ) Analysis of extracellular vesicle DNA at the single-vesicle level by nano-flow cytometry. J Extracell Vesicles 11: , e12206. |
[114] | Bradley MAA , Markesbery WRR , Lovell MAA ((2010) ) Increased levels of 4-hydroxynonenal and acrolein in the brain in preclinical Alzheimer disease. Free Radic Biol Med 48: , 1570–1576. |
[115] | Bradley MA , Xiong-Fister S , Markesbery WR , Lovell MA ((2012) ) Elevated 4-hydroxyhexenal in Alzheimer’s disease (AD) progression. Neurobiol Aging 33: , 1034–1044. |
[116] | Hofer T , Perry G ((2016) ) Nucleic acid oxidative damage in Alzheimer’s disease— explained by the hepcidin-ferroportin neuronal iron overload hypothesis? J Trace Elem Med Biol 38: , 1–9. |
[117] | Butterfield DA , Sultana R ((2007) ) Redox proteomics identification of oxidatively modified brain proteins in Alzheimer’s disease and mild cognitive impairment: Insights into the progression of this dementing disorder. J Alzheimers Dis 12: , 61–72. |
[118] | Aluise CD , Robinson RAS , Cai J , Pierce WM , Markesbery WR , Butterfield DA ((2011) ) Redox proteomics analysis of brains from subjects with amnestic mild cognitive impairment compared to brains from subjects with preclinical alzheimer’s disease: Insights into memory loss in MCI. J Alzheimers Dis 23: , 257–269. |
[119] | Butterfield DA , Poon HF , St. Clair D , Keller JN , Pierce WM , Klein JB , Markesbery WR ((2006) ) Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: Insights into the development of Alzheimer’s disease. Neurobiol Dis 22: , 223–232. |
[120] | Fredricks DN , Relman DA ((1996) ) Sequence-based identification of microbial pathogens: A reconsideration of Koch’s postulates. Clin Microbiol Rev 9: , 18–33. |
[121] | Zhang J , Perry G , Smith MA , Robertson D , Olson SJ , Graham DG , Montine TJ ((1999) ) Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol 154: , 1423–1429. |
[122] | Seidel K , Mahlke J , Siswanto S , Krüger R , Heinsen H , Auburger G , Bouzrou M , Grinberg LT , Wicht H , Korf HW , Den Dunnen W , Rüb U ((2015) ) The brainstem pathologies of Parkinson’s disease and dementia with Lewy bodies. Brain Pathol 25: , 121. |
[123] | Scheff SW , Ansari MA , Mufson EJ ((2016) ) Oxidative stress and hippocampal synaptic protein levels in elderly cognitively intact individuals with Alzheimer’s disease pathology. Neurobiol Aging 42: , 1–12. |
[124] | Quiroga IY , Cruikshank AE , Bond ML , Reed KSM , Evangelista BA , Tseng JH , Ragusa JV , Meeker RB , Won H , Cohen S , Cohen TJ , Phanstiel DH ((2022) ) Synthetic amyloid beta does not induce a robust transcriptional response in innate immune cell culture systems. J Neuroinflammation 19: , 99. |
[125] | Morris GP , Clark IA , Vissel B ((2014) ) Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun 2: , 135. |
[126] | Castellani RJ , Lee H-G , Zhu X , Nunomura A , Perry G , Smith MA ((2006) ) Neuropathology of Alzheimer disease: Pathognomonic but not pathogenic. Acta Neuropathol 111: , 503–509. |
[127] | Jimenez S , Navarro V , Moyano J , Sanchez-Mico M , Torres M , Davila JC , Vizuete M , Gutierrez A , Vitorica J ((2014) ) Disruption of amyloid plaques integrity affects the soluble oligomers content from Alzheimer disease brains. PLoS One 9: , e114041. |
[128] | Yang T , Li S , Xu H , Walsh DM , Selkoe DJ ((2017) ) Large soluble oligomers of amyloid β-protein from Alzheimer Brain are far less neuroactive than the smaller oligomers to which they dissociate. J Neurosci 37: , 152–163. |
[129] | Braak H , Braak E , Braak Braak ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[130] | Altmann A , Ng B , Landau SM , Jagust WJ , Greicius MD ((2015) ) Regional brain hypometabolism is unrelated to regional amyloid plaque burden. Brain 138: , 3734–3746. |
[131] | Karl Raimund Popper ((2002) ) The Logic of Scientific Discovery, Psychology Press. |
[132] | Thal DR , Rüb U , Orantes M , Braak H ((2002) ) Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology 58: , 1791–1800. |
[133] | Braak H , Thal DR , Ghebremedhin E , Tredici K Del ((2011) ) Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J Neuropathol Exp Neurol 70: , 960–969. |
[134] | Nelson PT , Abner EL , Scheff SW , Schmitt FA , Kryscio RJ , Jicha GA , Smith CD , Patel E , Markesbery WR ((2009) ) Alzheimer’s-type neuropathology in the precuneus is not increased relative to other areas of neocortex across a range of cognitive impairment. Neurosci Lett 450: , 336–339. |
[135] | Iacono D , O’Brien R , Resnick SM , Zonderman AB , Pletnikova O , Rudow G , An Y , West MJ , Crain B , Troncoso JC ((2008) ) Neuronal hypertrophy in asymptomatic Alzheimer disease. J Neuropathol Exp Neurol 67: , 578–589. |
[136] | Butterfield DA , Mattson MP ((2020) ) Apolipoprotein E and oxidative stress in brain with relevance to Alzheimer’s disease. Neurobiol Dis 138: , 104795. |
[137] | Reiman EM , Chen K , Alexander GE , Caselli RJ , Bandy D , Osborne D , Saunders AM , Hardy J ((2004) ) Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc Natl Acad Sci U S A 101: , 284–289. |
[138] | Nunomura A , Perry G , Pappolla MA , Friedland RP , Hirai K , Chiba S , Smith MA ((2000) ) Neuronal oxidative stress precedes amyloid-β deposition in Down syndrome. J Neuropathol Exp Neurol 59: , 1011–1017. |
[139] | Kepp KP ((2016) ) Alzheimer’s disease due to loss of function: A new synthesis of the available data. Prog Neurobiol 143: , 36–60. |
[140] | Chan SL , Culmsee C , Haughey N , Klapper W , Mattson MP ((2002) ) Presenilin-1 mutations sensitize neurons to DNA damage-induced death by a mechanism involving perturbed calcium homeostasis and activation of calpains and caspase-12. Neurobiol Dis 11: , 2–19. |
[141] | Soto-Mercado V , Mendivil-Perez M , Velez-Pardo C , Lopera F , Jimenez-Del-Rio M ((2020) ) Cholinergic-like neurons carrying PSEN1 E280A mutation from familial Alzheimer’s disease reveal intraneuronal sAPPβ fragments accumulation, hyperphosphorylation of tau, oxidative stress, apoptosis and Ca2+ dysregulation: Therapeutic implications, PLoS One 15: , e0221669. |
[142] | Cacace R , Sleegers K , Van Broeckhoven C ((2016) ) Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement 12: , 733–748. |
[143] | Ma J , Wang F ((2014) ) Prion disease and the “protein-only hypothesis”. Essays Biochem 56: , 181–191. |
[144] | Singh NA , Bhardwaj V , Ravi C , Ramesh N , Mandal AKA , Khan ZA ((2018) ) EGCG nanoparticles attenuate aluminum chloride induced neurobehavioral deficits, beta amyloid and tau pathology in a rat model of Alzheimer’s disease. Front Aging Neurosci 10: , 244. |
[145] | Degtyareva NP , Saini N , Sterling JF , Placentra VC , Klimczak LJ , Gordenin DA , Doetsch PW ((2019) ) Mutational signatures of redox stress in yeast single-strand DNA and of aging in human mitochondrial DNA share a common feature, PLOS Biol 17: , e3000263. |
[146] | Moraes EC , Keyse SM , Tyrrell RM ((1990) ) Mutagenesis by hydrogen peroxide treatment of mammalian cels: A molecular analysis. Carcinogenesis 11: , 283–293. |
[147] | Felício DL , Almeida CEB , Silva AB , Leitão AC ((2009) ) Hydrogen peroxide induces a specific DNA base change profile in thepresence of the iron chelator 2,2’ dipyridyl in Escherichia coli. Braz J Med Biol Res 42: , 1015–1019. |
[148] | Degtyareva NP , Heyburn L , Sterling J , Resnick MA , Gordenin DA , Doetsch PW ((2013) ) Oxidative stress-induced mutagenesis in single-strand DNA occurs primarily at cytosines and is DNA polymerase zeta-dependent only for adenines and guanines. Nucleic Acids Res 41: , 8995–9005. |
[149] | Ruiz-Laguna J , Pueyo C ((1999) ) Hydrogen peroxide and coffee induce G:C⟶T:A transversions in the lacI gene of catalase-defective Escherichia coli. Mutagenesis 14: , 95–102. |
[150] | Hoekstra JG , Hipp MJ , Montine TJ , Kennedy SR ((2016) ) Mitochondrial DNA mutations increase in early stage Alzheimer disease and are inconsistent with oxidative damage. Ann Neurol 80: , 301–306. |
[151] | Itsara LS , Kennedy SR , Fox EJ , Yu S , Hewitt JJ , Sanchez-Contreras M , Cardozo-Pelaez F , Pallanck LJ ((2014) ) Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. PLoS Genet 10: , e1003974. |
[152] | Kennedy SR , Salk JJ , Schmitt MW , Loeb LA ((2013) ) Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet 9: , e1003794. |
[153] | Cheng KC , Cahill DS , Kasai H , Nishimura S , Loeb LA ((1992) ) 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G—-T and A—-C substitutions. J Biol Chem 267: , 166–172. |
[154] | Lauková L , Konečcná B , Janovičcová L , Vlková B , Celec P ((2020) ) Deoxyribonucleases and their applications inbiomedicine. Biomolecules 10: , 1036. |
[155] | Posada J , Valadkhan S , Burge D , Davies K , Tarn J , Casement J , Jobling K , Gallagher P , Wilson D , Barone F , Fisher BA , Ng WF ((2021) ) Improvement of severe fatigue following nuclease therapy in patientswith primary Sjögren’s syndrome: A randomized clinical trial. Arthritis Rheumatol 73: , 143–150. |
[156] | van der Koog L , Gandek TB , Nagelkerke A ((2022) ) Liposomes and extracellular vesicles as drug delivery systems: A comparison of composition, pharmacokinetics, and functionalization, Adv Healthc Mater 11: , e2100639. |
[157] | Kaczmarczyk SJ , Sitaraman K , Young HA , Hughes SH , Chatterjee DK ((2011) ) Protein delivery using engineered virus-like particles. Proc Natl Acad Sci U S A 108: , 16998–17003. |
[158] | Craft S , Raman R , Chow TW , Rafii MS , Sun CK , Rissman RA , Donohue MC , Brewer JB , Jenkins C , Harless K , Gessert D , Aisen PS ((2020) ) Safety, efficacy, and feasibility of intranasal insulin for the treatment of mild cognitive impairment and Alzheimer disease dementia: A randomized clinical trial. JAMA Neurol 77: , 1099–1109. |


