
The Amyloid Cascade Hypothesis 2.0: On the Possibility of Once-in-a-Lifetime-Only Treatment for Prevention of Alzheimer’s Disease and for Its Potential Cure at Symptomatic Stages
Abstract
We posit that Alzheimer’s disease (AD) is driven by amyloid-β (Aβ) generated in the amyloid-β protein precursor (AβPP) independent pathway activated by AβPP-derived Aβ accumulated intraneuronally in a life-long process. This interpretation constitutes the Amyloid Cascade Hypothesis 2.0 (ACH2.0). It defines a tandem intraneuronal-Aβ (iAβ)-anchored cascade occurrence: intraneuronally-accumulated, AβPP-derived iAβ triggers relatively benign cascade that activates the AβPP-independent iAβ-generating pathway, which, in turn, initiates the second, devastating cascade that includes tau pathology and leads to neuronal loss. The entire output of the AβPP-independent iAβ-generating pathway is retained intraneuronally and perpetuates the pathway’s operation. This process constitutes a self-propagating, autonomous engine that drives AD and ultimately kills its host cells. Once activated, the AD Engine is self-reliant and independent from Aβ production in the AβPP proteolytic pathway; operation of the former renders the latter irrelevant to the progression of AD by relegating its iAβ contribution to insignificant, and brands its manipulation for therapeutic purposes, such as BACE (beta-site AβPP-cleaving enzyme) inhibition, as futile. In the proposed AD paradigm, the only valid direct therapeutic strategy is targeting the engine’s components, and the most effective feasible approach appears to be the activation of BACE1 and/or of its homolog BACE2, with the aim of exploiting their Aβ-cleaving activities. Such treatment would collapse the iAβ population and ‘reset’ its levels below those required for the operation of the AD Engine. Any sufficiently selective iAβ-depleting treatment would be equally effective. Remarkably, this approach opens the possibility of a short-duration, once-in-a-lifetime-only or very infrequent, preventive or curative therapy for AD; this therapy would be also effective for prevention and treatment of the ‘common’ pervasive aging-associated cognitive decline. The ACH2.0 clarifies all ACH-unresolved inconsistencies, explains the widespread ‘resilience to AD’ phenomenon, predicts occurrences of a category of AD-afflicted individuals without excessive Aβ plaque load and of a novel type of familial insusceptibility to AD; it also predicts the lifespan-dependent inevitability of AD in humans if untreated preventively. The article details strategy and methodology to generate an adequate AD model and validate the hypothesis; the proposed AD model may also serve as a research and drug development platform.
INTRODUCTION
Two recently concluded large stage III clinical trials, one for mild-to-moderate and another for prodromal Alzheimer’s disease (AD), of verubecestat, a Merck-developed candidate AD drug, were both terminated prematurely due to the complete lack of efficacy and the absence of any indication that symptomatic relief, or clinical improvement, or at least symptomatic arrest or any other benefit to the patients could be expected if trials were to continue [1, 2]. These major failures came as a surprise.
Several lines of evidence strongly indicate, and this hypothesis takes it as its only assumption, that AD is caused by amyloid-β (Aβ), a peptide encoded by a segment of the amyloid-β protein precursor (AβPP) gene embedded within its coding region. In the AβPP proteolytic pathway, therefore, Aβ is produced by two cleavages. First cleavage, by β-secretase (β-site AβPP cleaving enzyme, BACE) between residues 671 and 672 (numbering according to the AβPP770 isoform), generates the C-terminal fragment of AβPP (C99, reflecting the number of its amino acid residues) and forms the N-terminus of Aβ. Subsequent second cleavage of C99 by γ-secretase (γ-site AβPP cleaving enzyme), mostly on the plasma membrane, forms the C-terminus of Aβ and completes its production; simultaneously, the resulting Aβ peptide is secreted from the cell. According to the currently prevailing interpretation of AD, the Amyloid Cascade Hypothesis (ACH) formulated by Hardy and Higgins in 1992 [147], Aβ, produced and exported in the AβPP proteolytic/secretory pathway, accumulates in the extracellular space; this triggers a cascade of molecular and cellular events, which lead to neurodegeneration that eventually manifests as AD symptoms. Within the framework of this interpretation, suppression of β-secretase activity should be very effective as AD therapy. Inhibition of BACE1 by different means in animal models overexpressing Aβ apparently provided evidence that this indeed could be the case: BACE inhibition by NB360 facilitated repair of Aβ overproduction-caused pathophysiology and rescued neuronal hyperactivity, impaired long-range circuit function, and memory defects [3], whereas BACE1 deletion in the adult mouse overexpressing Aβ reversed preformed amyloid deposition and improved cognitive functions [4]. Verubecestat exhibited the same traits and was shown to be very effective in animal studies and preclinical trials [5]; its success in stage III clinical trials, therefore, was highly anticipated (for reasons why it worked so well in mouse AD models but not at all in human AD patients, see Discussion section).
The most puzzling aspect of the failures of stage III clinical trials of verubecestat was that they were not due to the drug inefficiency in AD patients. To the contrary, it proved to be very effective within constrains of its design and purpose. Verubecestat was designed to penetrate the brain and then the neurons, to inhibit the β-site cleavage and thus Aβ production and secretion in the AβPP proteolytic pathway, and, consequently, to reduce levels of extracellular Aβ. And this is precisely what it did, and did very well, in AD patients [1, 2], yet without any positive impact whatsoever on the progression of the disease. Provided that Aβ is the primary cause of the disease, a notion that is taken here as an assumption, a ‘given’, and considering all of the above, the failures of stage III clinical trials of verubecestat suggest, and could be best explained by, the following conclusions: (a) In AD, A β is produced in the A βPP-independent pathway; this pathway operates only in AD-afflicted humans in addition to and in parallel with the AβPP proteolytic/secretory process of Aβ generation. (b) The entire output or at least the bulk of A β produced in the A βPP-independent pathway is retained within affected neurons. (c) It is this pool of intraneuronal A β that drives and sustains AD. It also anchors a cascade that includes tau pathology and ultimately leads to the neuronal loss, hence ‘Amyloid Cascade Hypothesis 2.0’ (ACH2.0). In this AD paradigm, the operation of the AβPP-independent Aβ generation pathway is compulsory for the elicitation of AD; without it, the disease-triggering levels of intraneuronal Aβ cannot be reached and maintained.
How the AβPP-independent pathway of generation of intraneuronally retained Aβ is regulated, what activates it, and in what manner it is sustained are, in this context, the ultimate questions in the quest for understanding and controlling AD. The present study proposes that the activation of this pathway is mediated by AβPP-derived Aβ accumulated, in a life-long process, intracellularly to the critical above-the-threshold levels. Aβ produced in the AβPP-independent pathway is retained within the cell and its levels drastically increase. This sustains and promotes further operation of the pathway; in this context, the contribution of the AβPP proteolytic pathway to the intraneuronal Aβ pool is rendered insignificant. Such intraneuronal Aβ-driven AβPP-independent generation of intraneuronally retained Aβ constitutes the autonomous AD Engine that initiates and powers a cascade, which includes tau pathology and ultimately kills its host cells. In this AD paradigm, the only direct and effective AD therapy is to interfere with its cause, i.e., the components of the AD Engine, with the goal of ceasing its operation. Likewise, precluding the activation of the AD Engine would prevent the disease. Remarkably, both aims, the therapy at the symptomatic stages of AD and its prevention prior to the manifestation of AD symptoms, appear to be achievable by a treatment of a limited duration administered only once-in-a-lifetime or very infrequently. Moreover, it is possible that at least some of the ubiquitous milder instances of ‘common’ aging-related cognitive deterioration are, in fact, a low-grade AD, i.e., neurons are dying via operation of the AD Engine, but in much lower numbers or, possibly, only at particular locations associated with the retention of cognitive functionality in aging. If this were the case, a one-time-only preventive AD treatment would also protect from the pervasive aging-associated cognitive decline, a notion supported by the existing data. The ACH2.0 clarifies all principal inconsistencies unresolved by the ACH, such as why ACH-based candidate AD drugs worked so well in mouse AD models and not at all in human AD patients and why current mouse AD models cannot display the full spectrum of AD pathology. It explains the widespread ‘resilience to AD’ phenomenon, i.e., amyloid-PET brain-positive individuals not exhibiting any cognitive dysfunction, and predicts occurrences of a category of AD-afflicted individuals with normal, non-excessive amyloid plaque load and of a novel type of familial insusceptibility to AD; it also predicts the lifespan-dependent inevitability of AD if not treated preventively (on the exclusivity of AD to humans, see [6]). Most importantly, it predicts that any intraneuronal Aβ-depleting activity would constitute an effective curative and preventive AD drug, and names two plausible agents: BACE1 and BACE2 activators. The strategy and methodology to generate an adequate AD model and validate the hypothesis are detailed below.
THE ENGINE THAT DRIVES AD
Previously, we proposed a succession of molecular events that leads to neurodegeneration and eventually results in neuronal death, and thus fosters and advances AD [6]. Akin to an automobile’s propulsion system, it comprises two components: a ‘starter motor’, which ignites an autonomous self-perpetuating ‘engine’ that sustains the disease. The AD Starter Motor entails intraneuronal accumulation of AβPP-derived Aβ to critical threshold levels sufficient to activate the AβPP-independent AD Engine that generates intraneuronally retained Aβ, which, in turn, promotes further operation of the engine. The activation of this self-sustaining engine, described in more detail below, leads to increased neurotoxicity, hyperphosphorylation and aggregation of tau protein, and ultimately results in neuronal loss; the disease commences and its symptoms manifest only subsequently to the activation of the AβPP-independent Aβ generation pathway and, consequently of the AD Engine. The susceptibility to the disease, its sporadic nature, is defined by intraneuronal Aβ threshold levels required for the activation of the engine, which probably vary individually, and by the rates of accumulation of AβPP-derived Aβ within neuronal cells.
Intraneuronal accumulation of AβPP-derived Aβ, whereas apparently occurring at higher rates in the build-up to the disease, is not specific for AD. It occurs universally in both healthy and AD-afflicted individuals. Moreover, in the framework of proposed interpretation of AD, it is of significance only prior to the commencement of the disease, which occurs subsequently to the activation of the AβPP-independent Aβ generation pathway and, consequently, of the AD Engine: the AD Engine, when ignited, is self-sustaining and completely independent from the input of AβPP-derived Aβ, i.e., the Starter Motor (which is not needed after fulfilling its purpose). As discussed below, the AβPP proteolytic pathway is rendered irrelevant for the progression of the disease when the AD Engine is operational; this explains why targeting it at this stage would be, and in fact was proven to be, futile.
Conversion of AβPP-derived extracellular amyloid-β into iAβ: Cellular uptake of secreted Aβ
Numerous investigations of intraneuronal Aβ (iAβ) accumulation have shown that it is neurotoxic in vivo, correlates with synaptic impairment and neuronal loss, and constitutes the principal pathological trigger in AD [7–18]. Moreover, these studies suggested that iAβ accumulation, rather than extracellular plaque pathology, correlates with neuronal loss [19]. There are two potential sources of iAβ produced in the AβPP proteolytic pathway. One is the cellular uptake of secreted extracellular amyloid-β, effectively converting it into iAβ. Data obtained conclusively show that soluble extracellular Aβ42 and Aβ40 use endocytosis [20] to enter the cell and that Aβ42 is taken up two times more efficiently than Aβ40 [21]. The latter feature, combined with the increased toxicity of Aβ42, potentially contributes to the early onset of the disease in familial AD (FAD) mutants associated with the increased Aβ42/Aβ40 ratio [22]. Beta-sheet-rich Aβ42 aggregates were observed to enter cells at low nanomolar concentrations [23]. In contrast, monomers were shown to bind to plasma membrane and to form aggregates there before cellular uptake and accumulation in endocytic vesicles [24], thus indicating that formation of Aβ aggregates may be a prerequisite for its cellular uptake [23–25]. Moreover, it was suggested that oligomer-specific Aβ toxicity in cell models is mediated by its selective uptake [20]. Cellular uptake of Aβ was also shown to be APOE isoform-dependent and mediated by lipoprotein receptor LR11/SorLA [24]. APOE4, a major genetic risk factor for AD, was shown to be much more efficient in mediating Aβ uptake than APOE3 and APOE2 [12, 24]. LRP, another member of the lipoprotein receptor family, binds to Aβ directly or through ligands, such as APOE, and undergoes endocytosis, thus facilitating cellular uptake of Aβ [26]. The internalization of extracellular Aβ can also be mediated by α7 nicotinic acetylcholine receptor [27–29], the scavenger receptor for advanced glycation, RAGE [30–32], the formyl peptide receptor-like1, FPRL1 [33], and N-methyl-d-aspartate, NMDA, receptors [34]. Internalization of Aβ was observed in neurons and other cell types; it occurs in cells of normal subjects as well as in cells of AD-affected individuals [35].
Generation of iAβ by AβPP proteolysis: Intraneuronal retention of a fraction of AβPP-derived Aβ
Another source of AβPP-derived iAβ is its retention within the neuronal cell. Whether Aβ is retained intracellularly or is secreted into the extracellular pool is defined by the location at which the immediate amyloid-β precursor, C99, is cleaved by the γ-secretase complex. The vast majority of Aβ produced in the AβPP proteolytic pathway is generated by cleavage at the plasma membrane and is secreted. However, γ-cleavage can also occur in the endoplasmic reticulum (ER) [36], Golgi and trans Golgi network (TGN) [37], and at endosomal [36], lysosomal [36], and mitochondrial [38] membranes; such cleavages generate intracellularly retained Aβ. It has been shown that different isoforms of intracellular Aβ can be generated at different locations. For example, cleavage within the ER produces predominantly Aβ42 [39–43] whereas cleavage within the TGN mostly generates Aβ40 [44]; interestingly, these locations of intracellular Aβ generation are limited to neurons [39, 40]. It appears that a certain familial AD-associated AβPP mutation, namely the Swedish mutation, facilitates the generation of AβPP-derived iAβ by promoting the AβPP γ-cleavage on intracellular membranes [45], i.e., Swedish mutants are characterized by the increased production of AβPP-derived iAβ; this, apparently, contributes to and possibly underlies the early onset of AD associated with this mutation. In addition, subcellular localization of presenilins (PSENs) is known to direct the assembly of γ-secretase complex to specific cellular compartments and thus to contribute to the balance between intracellular accumulation and secretion of Aβ [21, 46]; FAD-associated PSENs mutations were shown to augment the intracellular pool of Aβ by determining subcellular localization of γ-secretase [46].
Elicitation of the integrated stress response by the iAβ-activated PKR kinase
When, as a result of life-long accumulation, AβPP-derived iAβ reaches critical levels, it initiates the operation of the AβPP-independent pathway of iAβ generation, and thus triggers the activation of the self-sufficient AD Engine. The first step in this proposed process is the iAβ-mediated elicitation of the integrated stress response (ISR). This can occur in at least two different ways. One is through the activation of the PKR kinase. In vitro studies with human neuroblastoma cell lines as well as with primary neuronal cultures showed that in neuronal cells PKR is activated by Aβ peptide toxicity [47]. Studies with animal AD models have confirmed the outcomes of the in vitro studies [48, 49]. In AD patients, a link was established between PKR and the disease by a demonstration that degenerating neurons in the hippocampus and the frontal cortex displayed marked immunohistochemical positivity for phosphorylated PKR and alpha subunit of eukaryotic translation initiation factor 2 (eIF2α) [50]. It was concluded that the PKR-eIF2α pro-apoptotic pathway could be involved in neuronal degeneration [50, 51]. The molecular mechanism of neuronal Aβ-mediated PKR activation in AD is not well understood. One possibility is that it occurs via TNFα, as was suggested by animal studies [52]. Another possibility is that the PKR activator protein PACT is involved in this process. Regarding the latter, it was shown that PACT and phosphorylated PKR are co-localized in degenerating neurons in human AD brains [53]. Moreover, PACT shRNA treatment of human neuroblastoma cells decreased PKR activation produced by the Aβ exposure [53], consistent with the notion that the Aβ exposure could induce PKR activation via the increase in PACT levels.
Postulated role of the ISR in the activation of the AβPP-independent iAβ production pathway
The integrated stress response is a complex signaling pathway operating in eukaryotic cells, which is activated in response to a wide range of cellular stresses [54–63]. The “integrating” feature of this pathway is the convergence of all stimuli that activate the ISR to the one common event: phosphorylation of eIF2α at serine 51. In mammalian cells, this phosphorylation is catalyzed by the family of eIF2α kinases. This family is comprised of four members: PKR kinase; PKR-like ER kinase (PERK); general control none-derepressible-2 kinase (GCN2); and heme-regulated kinase (HRI). Phosphorylation of eIF2α at serine 51 elicits the ISR, which manifests as a severe reduction in global cellular protein synthesis, primarily through the inhibition of the cap-dependent initiation of translation, and, simultaneously, the facilitation of cap-independent translation of selected mRNAs, including those encoding specific transcription factors. It was proposed previously [6] that among these transcriptions factors, or among products of genes activated by them, are crucial components required for the activation of the AβPP-independent iAβ production pathway in AD. The ability of iAβ to induce neuronal PKR activation and eIF2α phosphorylation, and to elicit the ISR was indeed confirmed in human neuroblastoma cells and in primary neuronal cultures [47].
Elicitation of the ISR via iAβ-mediated activation of the HRI kinase
Another mode in which AβPP-derived iAβ can trigger the elicitation of the ISR in neuronal cells and mediate the activation of the AβPP-independent iAβ production pathway in AD is via the OMA1-DELE1-HRI signaling pathway. AD has been associated with mitochondrial dysfunction, and the overproduction of Aβ was shown to be sufficient to activate mitochondrial dysfunction and to elicit cellular stress response in model systems [64–80] (reviewed in [6]). The question of how the stress signal is transmitted from mitochondria to the cytosol was always of significant interest; it was answered in two recent studies [81, 82]. In these investigations, it was shown that depolarization, a change in the electrical charge, which occurs during mitochondrial dysfunction, activates the protease OMA1, which is located on the inner of the two mitochondrial membranes surrounding the organelle. In turn, the activated OMA1 cleaves or facilitates the cleavage of another mitochondrial protein, DELE1, which resides in the space between the mitochondrial membranes and is associated with the inner membrane, the locality of OMA1. Following the cleavage, a cleaved-off fragment of DELE1 is released to the cytosol where it binds to the HRI kinase and activates it. Activated HRI phosphorylates eIF2α; this leads to the integrated stress response. This pathway was shown to operate in different cell types including neuronal cells [82].
Cellular mechanisms capable of enabling the operation of the AβPP-independent iAβ generation pathway
AβPP-derived iAβ-mediated elicitation of the ISR (or some other AβPP-derived iAβ-mediated event) activates the AβPP-independent iAβ generation pathway, which appears to be exclusive to humans [6]. The molecular mechanism underlying this pathway drives and is at the core of AD. Four distinct mechanisms, each capable of generating Aβ independently of AβPP, have been previously proposed [6, 146]. The common principal feature of all these mechanisms is that translation initiates at the AUG codon normally encoding Met671 of AβPP and preceding contiguously the Aβ-encoding nucleotide sequence. Accordingly, regardless of a mechanism employed, the primary translation product would be C100, i.e., C99 containing the translation-initiating N-terminal Met, which is removed post- rather than co-translationally because it is followed by an aspartate (Asp1 of Aβ) (reviewed in [6], also see Discussion section below). γ-secretase cleavage of C99 (or C100) would produce Aβ (or Met-Aβ eventually processed into Aβ). Unlike AβPP, C100 does not contain N-terminal signal peptide that would guide if into a secretory pipeline, and for Aβ produced from it to be retained intracellularly, it is sufficient that γ-secretase cleavage would take place on an internal cellular membrane, a process known to occur in neuronal cells [39, 40].
In three of the four proposed mechanisms, C100 is translated from severely 5’-truncated AβPP mRNAs produced in distinct pathways; in all, the first in-frame AUG is the one contiguously preceding the Aβ-encoding segment. One of those three mechanisms is RNA-dependent amplification of AβPP mRNA. The mammalian RNA-dependent mRNA amplification pathway has been implicated in physiological differentiation-specific overproduction of certain polypeptides in different cell types [83–86]. RNA-dependent amplification of human AβPP mRNA was proposed early on [87–89, 90–94, 146] due to the observed self-priming of human AβPP antisense strand. The activation of this pathway requires the inducible expression, presumably mediated by the ISR, of one or more components of the RNA-dependent RNA polymerase complex [6, 90–94, 146]. Another mechanism utilizing 5’-truncated AβPP mRNA and capable of AβPP-independent generation of Aβ in AD is the internal initiation of transcription within the coding region of the AβPP gene and upstream from the AUG normally encoding Met671 of AβPP (reviewed in [6]). The activation of the internal initiation of transcription within the coding region of the AβPP gene would require induction of a suitable transcription factor (or a co-factor) presumably under conditions of the ISR [6]. The third mechanism potentially responsible for AβPP-independent generation of iAβ in AD is cleavage of the intact AβPP mRNA at a suitable position within its coding region (reviewed in [6]). Its activation would require the ISR-enabled expression of an appropriate enzyme (or an enzyme’s component). The mRNA products of these three mechanisms would be conceptually similar; in each, the first in-frame AUG would be the one normally encoding Met671 and each would be conventionally translated into the C100 fragment of AβPP. The fourth potential mechanism of iAβ production in the AβPP-independent manner is the internal initiation of translation within coding region of intact AβPP mRNA, at the AUG normally encoding Met671 of AβPP. This, in fact, was chronologically (1987) the first suggestion of a possibility of the AβPP-independent generation of Aβ in AD [95]. It was presumably “ruled out”, but not in the AD context and only in non-neuronal cells [131, 132]; it certainly was not ruled out in AD-affected human neuronal cells where it remains a valid possibility (reviewed in [6, 146]). The basis for this proposal, which was posited shortly after the elucidation of the nucleotide sequence of human AβPP gene [96–98], was the exceptional position of the Met671-encoding AUG: of twenty methionine-encoding AUGs in human AβPP mRNA, only this AUG is situated within an optimal translation initiation context (not even the AUG encoding the translation-initiating Met of AβPP is) [95]. The activation of the internal initiation of translation would require the ISR-induced expression of a translation factor or of its co-factor [6, 146].
It should be emphasized that the operation of the AβPP-independent iAβ generation pathway in AD, by whatever mechanism, is mandated by the results of the clinical trials discussed above. It is highly probable that this pathway is enabled by one of the four mechanisms described above. It cannot be excluded, however, that yet another, unforeseen, mechanism underlies this pathway; regardless, the end result in every case would be the same: the retained iAβ produced independently of AβPP. Likewise, the role of iAβ-mediated elicitation of the ISR in the activation and maintenance of AβPP-independent production of iAβ is only a plausible option, it can be any other suitable process; the only requirement for this step is that it is initiated by AβPP-derived iAβ and sustained by iAβ produced in the AβPP-independent pathway.
The AD Engine: iAβ-driven AβPP-independent generation of iAβ
To summarize, it has been proposed that the life-long accumulation of AβPP-derived, i.e., conventionally produced in the AβPP proteolytic pathway, iAβ to levels sufficient to trigger, via PKR activation and/or OMA1-DELE1-HRI signaling or through some other, yet undetermined, pathway, the elicitation of the ISR and, consequently, activation of the AβPP-independent iAβ generation pathway, plays the role of a starter motor. Once switched on, the “Starter Motor” ignites the “Engine” that drives AD. Indeed, over-the-threshold levels of AβPP-derived iAβ lead to the activation of PKR and/or HRI, phosphorylation of eIF2α, and elicitation of the ISR. Within the framework of the ISR (or in a yet undefined context), it also leads to the activation of the AβPP-independent iAβ generation pathway. The bulk, if not the entire output of this pathway is retained intraneuronally. Drastically increased levels of iAβ promote, in turn, further activity of eIF2α kinases; eIF2α phosphorylation is maintained, and the ISR and, consequently, the AβPP-independent iAβ production pathway are sustained, thus generating self-perpetuating (intraneuronal Aβ-driven AβPP-independent generation of intraneuronally retained Aβ) mutual feedback cycles, which constitute the autonomous, independent from AβPP-derived Aβ, AD Engine; activation of the engine precedes and signifies the commencement of the disease. The bottom line is that the AβPP-independent mode of iAβ production requires certain defined levels of iAβ for its operation; this is the essence of the AD Engine. These relationships are presented diagrammatically in Fig. 1. It depicts the mutual feedback cycles as a two-stroke engine, the engine that drives AD; ultimately, it instigates neuronal host cells’ death. To develop sporadic AD (SAD), it takes, apparently, a lifetime of accumulation of AβPP-derived iAβ to levels sufficient to switch on the AD Engine. In FAD, because of abnormal AβPP proteolysis and/or other factors, critical levels of conventionally produced (AβPP-derived) iAβ40, or of its more toxic isoforms, are reached sooner and the disease occurs earlier in life. In this context, the rate of retention of AβPP-derived iAβ, combined with that of its cellular uptake from the secreted Aβ pool, appear to play the key role in defining the susceptibility to AD. The occurrence of the AβPP-independent iAβ generation and the consequent operation of the AD Engine appear to be limited to humans; this explains why not even long-lived mammals, e.g., elephants, suffer from AD (reviewed in [6, 146]). Potentially (but not necessarily) this process may be feasible in primates, but their ability to sufficiently accumulate AβPP-derived iAβ is, because of their limited longevity, apparently inadequate for this process to occur and for AD to manifest.
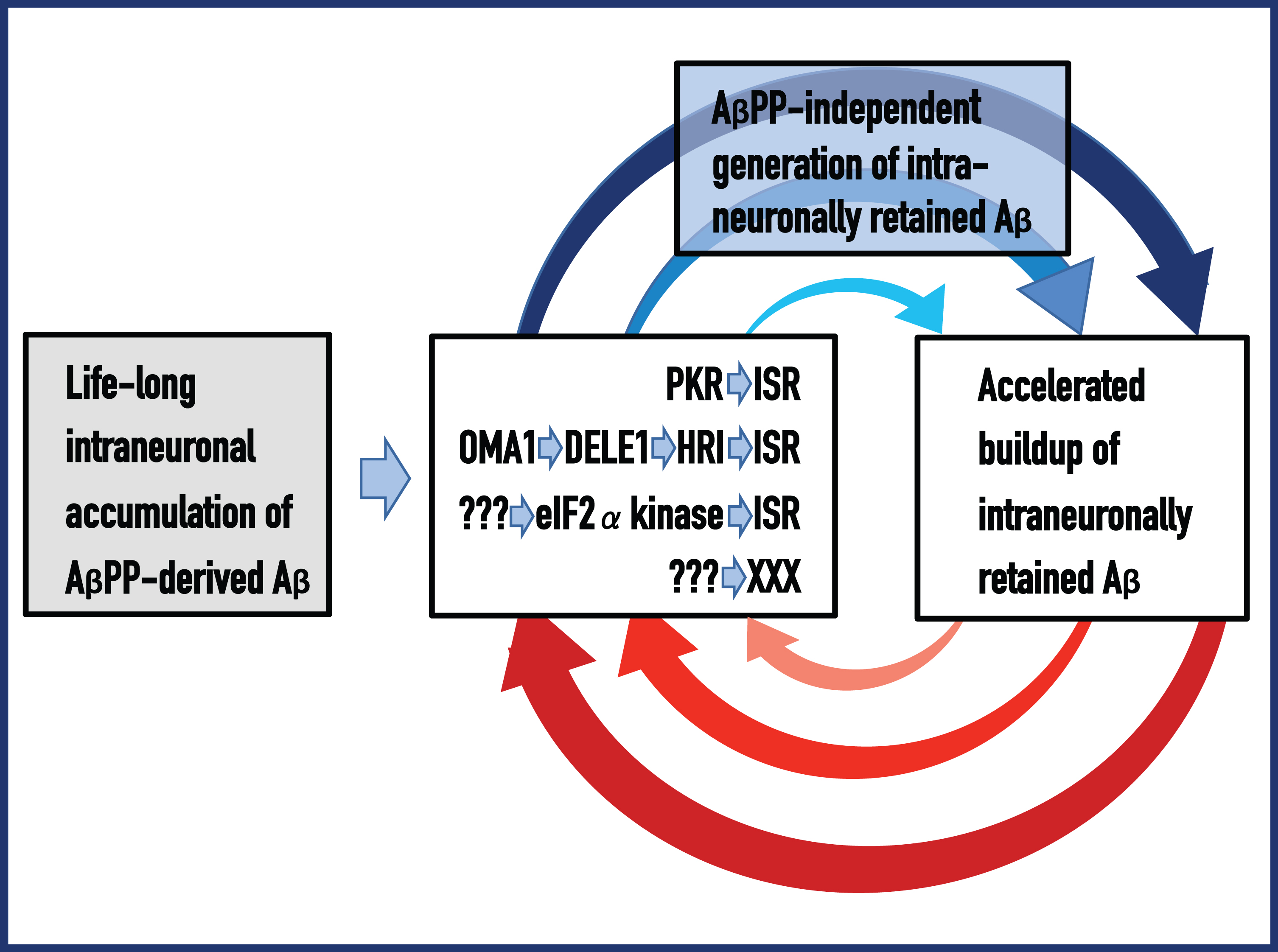
Fig. 1
Amyloid Cascade Hypothesis 2.0: The engine that drives AD; Etiology and primum mobile of the disease in the proposed AD paradigm. ACH2.0 defines a tandem, Aβ-anchored cascade occurrence: intraneuronally-accumulated, AβPP-derived Aβ triggers relatively benign cascade (left to middle boxes) that activates the AβPP-independent pathway generating intraneuronally-retained Aβ, which, in turn, initiates the second, devastating cascade that is driven by the AD Engine, includes tau pathology, and ultimately leads to the neuronal loss. Left Box (highlighted in grey): The “Starter Motor” – life-long accumulation of AβPP-derived iAβ, through the cellular uptake of secreted peptide and intracellular retention of a fraction of AβPP-derived Aβ, to levels sufficient to ignite the AD Engine (rest of the figure). Middle Box: Several pathways, both actual (top two) and hypothetical (each line represents a pathway), of the Aβ-mediated elicitation of the integrated stress response or of yet unknown process capable of activating the AβPP-independent iAβ production pathway. Top Box (highlighted in blue): AβPP-independent generation of iAβ via one of the following mechanisms— RNA-dependent AβPP mRNA amplification; the internal initiation of transcription within the AβPP gene; cleavage within AβPP mRNA; the internal initiation of translation within intact AβPP mRNA. It cannot be excluded that yet another, unforeseen, mechanism underlies the AβPP-independent iAβ generation pathway. Regardless of the mechanism employed, translation initiates at the AUG normally encoding Met671 of AβPP (which contiguously precedes Asp1 of C99 and of Aβ) and results in C100 (N-terminal Met-containing C99, converted post-translationally into C99), which is processed by γ-secretase cleavage into Aβ (or Met-Aβ eventually converted to Aβ) that is retained intraneuronally. Right Box: The entire output, or at least the bulk of Aβ generated in the AβPP-independent pathway is retained within the cell, and the iAβ levels rapidly increase. This sustains the operation of one or more of the iAβ-mediated pathways shown in the Middle Box, which, in turn, support the AβPP-independent generation of iAβ. Blue and red arched arrows: Mutually perpetuating feedback cycles. They constitute an autonomous, self-sustained, two-stroke engine, the engine that drives AD. The disease commences and manifests symptomatically only following the activation of the AD Engine.
DYNAMICS OF Aβ ACCUMULATION AND OF THE DISEASE IN AD-AFFLICTED HUMAN POPULATION: TWO PARADIGMS
In both the currently prevailing interpretation of AD and the one presented above, the disease is caused by Aβ, and in both paradigms the rate of its accumulation defines the susceptibility to the disorder. The two interpretations, however, are drastically different: In one the disease is caused by AβPP-derived secreted Aβ accumulating extracellularly, whereas in another the culprit is intraneuronally accumulated amyloid-β (iAβ). Moreover, dynamics of Aβ accumulation and, correspondingly, of the disease are also distinctly diverse in two paradigms. These dynamics are illustrated in Fig. 2, where every continuous line represents an affected individual in a population of AD patients. Figure 2A and 2B (SAD and FAD respectively) depict the prevailing interpretation of the disease. The dynamics of extracellular Aβ accumulation (blue lines) and that of neurodegeneration and the disease (red lines) are single-phased. The latter can be divided into two stages: asymptomatic (red lines) and symptomatic (red blocks); the symptomatic stage commences after extracellular Aβ levels and corresponding cell damage cross the threshold T. Aβ, it is assumed, is overproduced (and secreted) solely in the AβPP proteolytic/secretory pathway. As its extracellular levels increase, it triggers neurodegeneration (red lines) starting early in life; its severity corresponds to Aβ levels. Damages accumulate and manifest symptomatically (red blocks) late in life in sporadic cases (Fig. 2A). In familial AD cases, where mutations in the AβPP gene or in presenilins increase production of either common Aβ isoform or of its more toxic isoforms, neurodegeneration reaches critical threshold sooner and AD symptoms occur earlier in life, mostly in the late forties and fifties (Fig. 2B). In this paradigm, the disease is considered untreatable in the symptomatic phase and there are currently no preventive AD therapies, but if they were available, according to this viewpoint, it would be largely futile to intervene late in life in sporadic AD cases or at mid-age in cases of FAD because, although AD symptoms have not yet manifested, the irreversible damage has already occurred; to be effective, any preventive therapy should commence years, if not decades, prior to the symptomatic stage and continue for life.
Fig. 2
Dynamics of Aβ accumulation and the disease in AD-affected patients: Two paradigms. Images on the left: Dynamics of Aβ accumulation (extracellular in A and B, intraneuronal in C-E); Images on the right: Dynamics of neurodegeneration. Blue lines: Levels of Aβ; Red lines: Extent of neurodegeneration; Black lines: Indicator lines, no noticeable neurodegeneration; Red blocks: Symptomatic manifestation of AD. T: Threshold of extracellular Aβ levels and the corresponding extent of cell damage triggering AD symptoms; T1: Threshold of AβPP-derived iAβ levels required for the activation of the AβPP-independent iAβ production pathway (genetic aspects and epigenetic factors influence the timing of the T and T1 crossings, hence the fanning lines); T2: Threshold of iAβ levels and the corresponding extent of neuronal damage triggering AD symptoms (T, T1, and T2 are patient-specific). A (SAD ), B (FAD): Dynamics of AD in the old paradigm. Levels of extracellular Aβ increase and so does the extent of neurodegeneration; when the T is reached, AD symptoms manifest. C (SAD), D (FAD): Dynamics of AD in the new paradigm. As AβPP-derived iAβ cross the T1, AβPP-independent production of iAβ is activated and its levels rapidly increase. After a lag period during which iAβ further accumulates, neurodegeneration commences; when the T2 is reached, AD symptoms manifest. Red and blue lines over the T1 threshold are shown arbitrarily as parallel (i.e., identical rates of the accumulation of iAβ produced independently of AβPP and of the corresponding extent of cellular damage) and as of uniform heights over the T2 threshold (i.e., equal extents of damage); in reality, both the rates and the extents and, accordingly, lines’ angles and their heights over the T2 are likely different and define the duration of the disease in individual AD patients. E: Presumed dynamics of iAβ accumulation in subjects with an inoperative AD Engine. AβPP-derived iAβ levels cross the T1 threshold but the AβPP-independent iAβ production pathway is not activated. Neither neurodegeneration-triggering iAβ levels nor the T2 threshold are reached; there is no noticeable neurodegeneration, no AD symptoms manifest, no disease occurs.
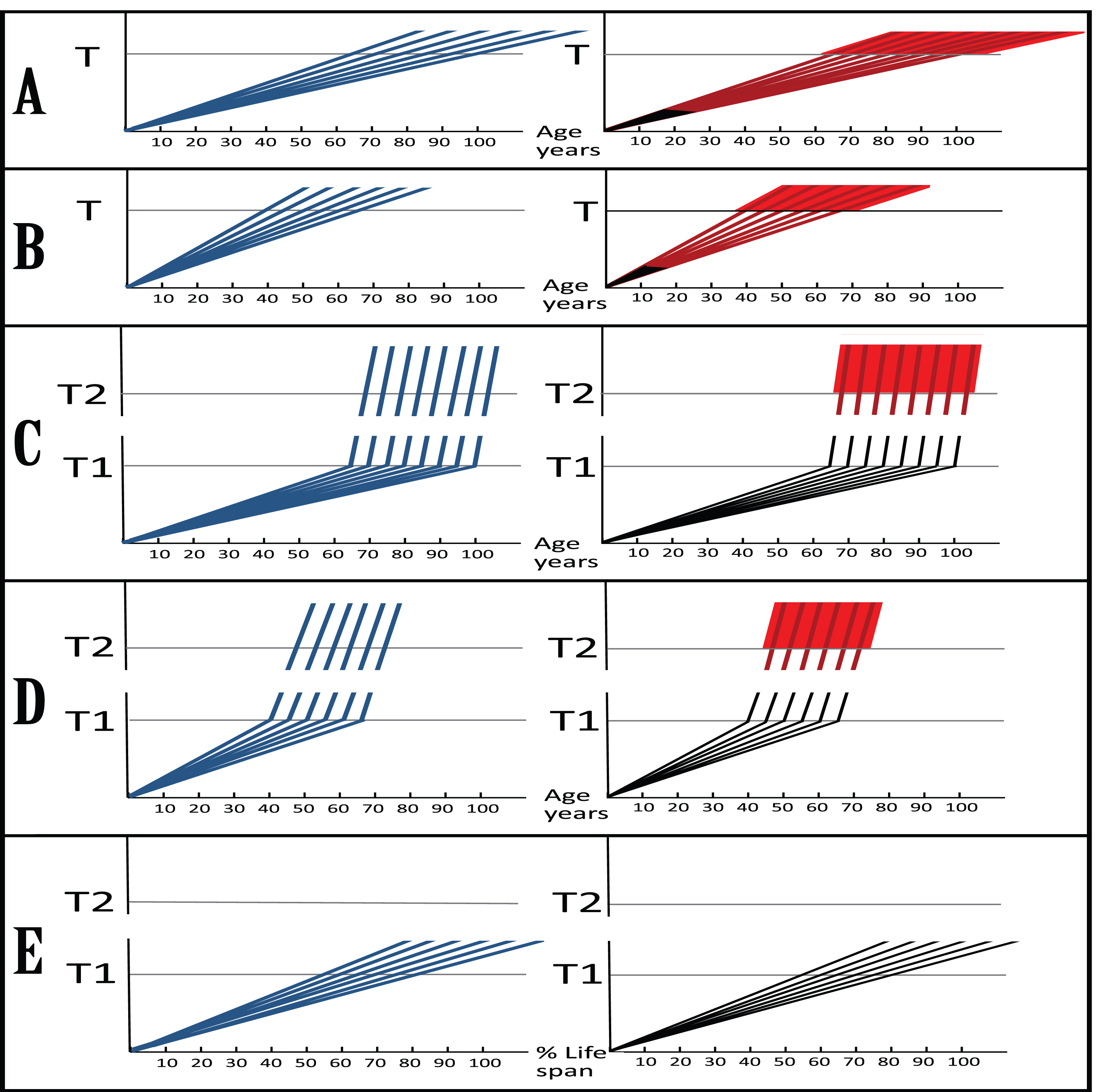
Dynamics of the disease in the proposed paradigm, illustrated in Fig. 2C (SAD) and Fig. 2D (FAD), are radically different. This dynamic is biphasic. In the first phase, only the AβPP proteolytic pathway of Aβ production is in operation. This phase is a slow, life-long process of the AβPP-derived iAβ accumulation (blue lines). It occurs via the cellular uptake of secreted Aβ and the intracellular retention of a fraction of AβPP-derived Aβ. These processes are common to Homo sapiens, including healthy humans, and to non-human mammals, and result neither in noticeable damage (black indicator lines), nor in any manifestation of the disease; there is, in fact, no disease in this phase. The second phase occurs exclusively in AD-affected human patients [6] and commences when levels of AβPP-derived iAβ cross the T1 threshold. This leads to the activation of PKR and/or HRI kinases, phosphorylation of eIF2α and elicitation of the ISR, which, in turn, mediates the activation of the AβPP-independent pathway of iAβ generation and, eventually, symptomatic onset of the disease. In this, second, phase, the rate of production and the extent of accumulation of iAβ (blue lines) sharply accelerate, causing, after a lag period during which iAβ further accumulates (black indicator lines), significant neurodegeneration (red lines). When iAβ levels cross the T2 threshold, so does the corresponding extent of neurodegeneration; neuronal cells start losing the functionality and committing apoptosis, and AD symptoms (red blocks) manifest. In this paradigm, a preventive therapy for AD would be effective when initiated at any time prior to the commencement of the second phase, i.e., of the activation of the AβPP-independent iAβ production pathway and, consequently, of the AD Engine; if this is prevented, neither neurodegeneration-triggering iAβ levels nor T2 threshold are reached, no AD symptoms manifest, no disease occurs (Fig. 2E). If the AD Engine is disabled after being operational, even after AD symptoms manifest, the disease can still be potentially cured or contained, as described in the following sections below.
DYNAMICS OF iAβ ACCUMULATION IN THE AFFECTED NEURONAL POPULATION OF AND PROGRESSION OF THE DISEASE IN AN AD PATIENT
The present section attempts to analyze the dynamics of iAβ accumulation in affected neuronal population and, accordingly, the progression of the disease in an individual AD patient within the framework of the proposed interpretation of AD. The understanding of this dynamics is of importance because it could help to define the types of therapeutic options available in the proposed paradigm. Two possible basic scenarios (variants of both are discussed in this section below) can be envisioned in this regard. In one, depicted in Fig. 3A and 3B and showing levels of intracellular iAβ in affected neurons of an AD patient, after the life-long accumulation of AβPP-derived iAβ, neurons reach and cross the T1 threshold within a wide temporal window in a stochastic manner characteristic for the majority of biological processes. In this scenario, the duration of the disease is defined primarily by the distribution of neuronal crossings of the T1 threshold as a function of time. Following the T1 crossing, the AD Engine described above is switched on, and levels of iAβ rapidly increase. Upon crossing the T2 level, neurons commit to the apoptotic pathway and eventually die. When enough cells become non-functional or die, AD symptoms manifest (Fig. 3A, commencement of symptomatic manifestation of AD is denoted by vertical arrows). As AD progresses, more and more affected neurons cross the T1 and, subsequently, the T2, and the disease reaches its end-stage (Fig. 3B). This scenario, however, can be ruled out. Indeed, as shown in Fig. 3A, when sufficient fraction of neurons crosses the T2 threshold and symptoms manifest, a substantial proportion of neuronal cells do not yet reach the T1 threshold. If, at this point, the production of AβPP-derived Aβ were suppressed by BACE1 inhibition, neuronal crossings of the T1 threshold should be stopped or delayed and so would be the progression of the disease. This, however, was not the case when an effective BACE1 inhibitor was used in mild-to-moderate AD cohort [1] or even with prodromal AD patients [2]; neither improvements nor delay in progression were seen.
Fig. 3
Dynamics of iAβ accumulation and the disease in the affected neuronal population of an AD patient. iA β: Intraneuronal Aβ levels; T1: The level of iAβ that triggers elicitation of the integrated stress response, initiation of AβPP-independent generation of iAβ, and activation of the AD Engine; T2: The level of iAβ that triggers cell’s commitment to the apoptotic pathway; Red blocks: Fraction of affected neurons either committed to apoptosis or dead; Vertical arrows: Indicate minimal fraction of neurons over the T2 threshold that causes symptomatic manifestation of AD. A: The initial symptomatic manifestation of the disease. The affected neurons cross the T1 threshold stochastically in a wide temporal window; at the time when the initial symptoms manifest, a substantial fraction of affected neurons did not yet cross the T1 threshold. B: The end-stage of the disease. A’: The initial symptomatic manifestation of the disease. The affected neurons reach and cross the T1 threshold within a narrow temporal window; when the initial symptoms manifest, the T1 threshold has been crossed by and the AD Engine has been activated in all or the bulk of affected neuronal cells. B’: The end-stage of the disease. Of the two, only the scenario depicted in A’ and B’ is viable since it conforms (unlike the scenario shown in A and B) to experimental data.
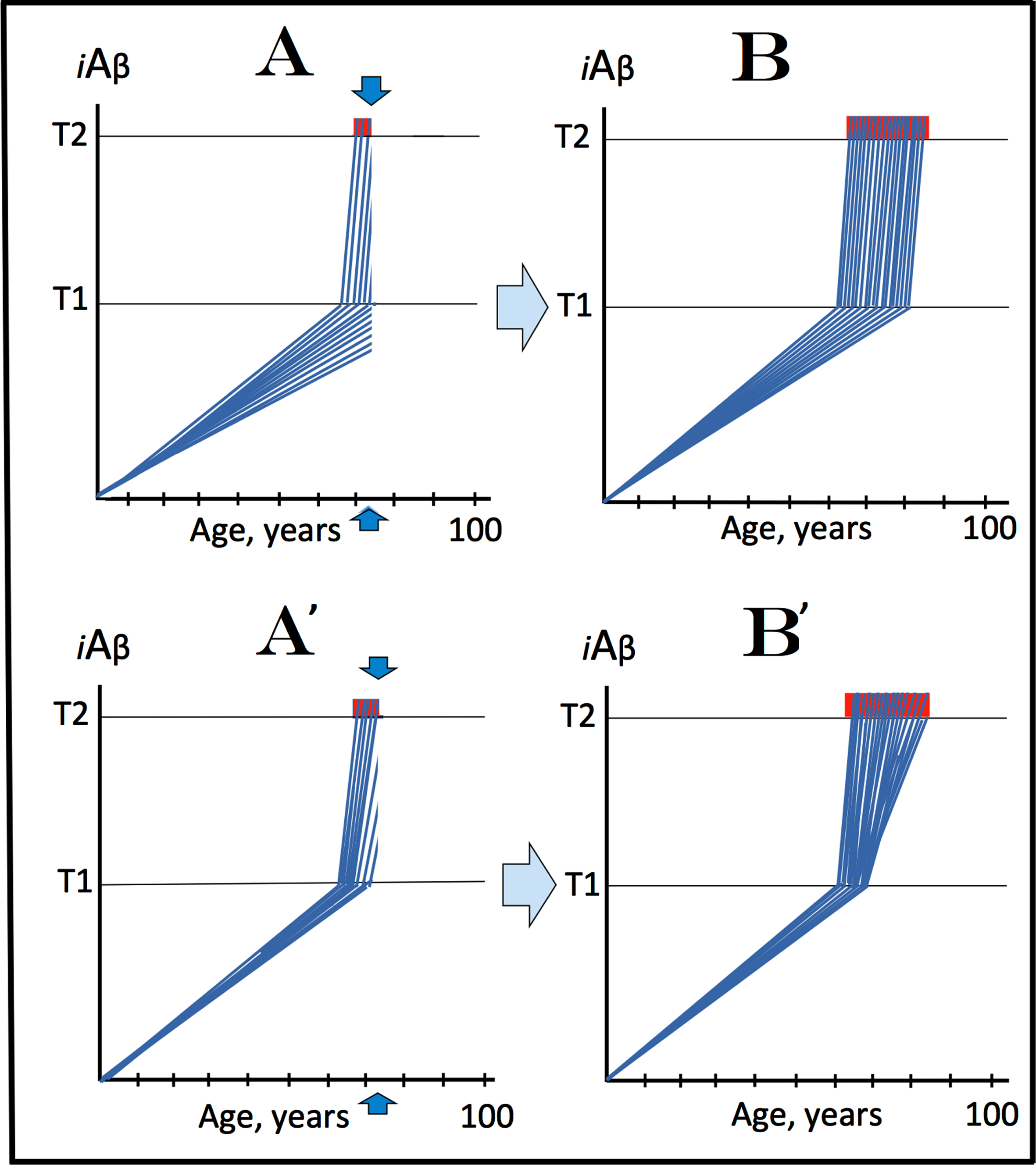
The alternative scenario, conforming to experimental data, is depicted in Fig. 3A’ and 3B’. In it, all affected neurons cross the T1 threshold within a narrow temporal window. Following the crossing of the T1, they reach and cross the T2 threshold in a wide stochastic manner. In this scenario, the duration of the disease is defined primarily by the temporal distribution of neuronal crossings of the T2 threshold. By the time a sufficient fraction of affected cells cross the T2 for AD symptoms to manifest (Fig. 3A’, commencement of AD symptoms is indicated by vertical arrows), all, or the bulk of affected neurons have crossed the T1 (activating the AD Engine and rendering BACE inhibition ineffective). With the progression of AD, additional neurons cross the T2 until the disease reaches the end-stage (Fig. 3B’). Thus, the principal distinguishing feature of this scenario is that by the time AD symptoms manifest, the T1 threshold has been crossed by and, consequently, the AD Engine has been switched on in all, or the bulk of affected neurons. Intermediate variants combining features of both scenarios (e.g., temporally relatively wide crossings of T1 followed by stochastically distributed crossings of the T2) are also possible. In any such variant, however, the experimentally supported bottom line is that by the time of symptomatic manifestation of AD, AβPP-derived iAβ levels have crossed the T1 threshold, and AβPP-independent iAβ production as well as the AD Engine have been activated in all or the bulk of all affected neurons.
THERAPEUTIC OPTIONS FOR THE SYMPTOMATIC STAGES OF AD AND FOR THE PREVENTION OF THE DISEASE IN THE PROPOSED AD PARADIGM
Why some approaches did not or could not work
The consequences of the viable scenario (or of its variants), supported by experimental data and described in the preceding section, are clear: when symptoms of AD manifest, it is futile to administer BACE inhibitors or to employ extracellular Aβ-targeting antibodies. This is because by this time, AβPP-derived iAβ levels crossed the T1 threshold and AβPP-independent production of iAβ, as well as the AD Engine, have been activated in all, or at least the decisive bulk of affected neurons. The AD Engine is autonomous and completely self-sufficient; the operation of the AβPP-independent iAβ generation pathway provides continuous influx of iAβ thus sustaining its own activity. At this stage, the AβPP proteolytic pathway becomes irrelevant to the progression of AD because its contribution to the iAβ pool is insignificant in comparison with that of the AβPP-independent pathway; suppressing its operation with BACE inhibitors or depleting extracellular Aβ with antibodies (thus limiting its cellular uptake) would be fruitless.
It follows that at symptomatic stages of AD, the only effective therapeutic targets are the components of the AD Engine. Suppressing processes leading to the activation of the AβPP-independent iAβ generation pathway would be challenging, if even possible. If it occurs within the framework of the integrated stress response, as suggested above, it involves redundancies, such as PKR and/or HRI-mediated phosphorylation of eIF2α. Moreover, inhibition of one or both of these kinases is likely to induce a compensatory activation of other eIF2α kinases [54]. The interference with eIF2α phosphorylation is also not an option because of its role in basic physiology. Moreover, there is no sufficient certainty regarding the exclusive involvement of the ISR in activation of the AβPP-independent iAβ production pathway, in the first place, to entertain its manipulation; a redundancy can be involved at this stage too. Moreover, as reflected in Fig. 1, a pathway that activates the AβPP-independent iAβ production could be distinct from the ISR.
The next potential therapeutic target is the AβPP-independent iAβ production pathway. Whereas the results of verubecestat’s clinical trials impart sufficient confidence in the operation of this pathway in AD, there is no certainty regarding the nature of the mechanism underlying this process. Moreover, such a mechanism could play a vital physiological function and, therefore, be ‘untouchable’. For example, the principal candidate mechanism for this role, mammalian RNA-dependent mRNA amplification, was shown to be crucial in both erythropoiesis [83–85] and the deposition of extracellular matrix [86]. It is probably involved in numerous other processes of cellular differentiation and, therefore, cannot be interfered with without potential deleterious consequences, at least not at the current state of its understanding.
The above considerations leave the only viable option: depletion of iAβ, by any means, to levels below those required for the operation of the AD Engine. This could be attempted in several ways, some potentially more effective than others. For example, it was proposed previously [6] that since the final step in iAβ generation in the AβPP-independent pathway is γ-cleavage of C99 (or C100) on an intracellular membrane, it could be potentially shifted to the plasma membrane; this would result in secretion of Aβ and prevent its intraneuronal accumulation. However, even if practically possible, this would be a partial remedy at best because the existing intracellular pool of iAβ would not be depleted and would continue to be resupplied by the cellular intake of secreted Aβ. Another obvious approach is the inhibition of γ-secretase; this would affect production of Aβ in both AβPP-dependent and -independent pathways. This approach was attempted more than once [99–102], every time with deleterious outcomes, and was eventually abandoned. γ-secretase is a prominent member of the Notch signaling pathway, with numerous AβPP-unrelated substrates in many cell types; this is why its suppression has proved to be detrimental. Modulators of γ-secretase, compounds that shift the spectrum of Aβ toward shorter, less ‘toxic’ peptides by altering its C-termini, are more promising as therapeutic agents but their trials were disappointing [101, 102]. This approach continues being developed, with some hopeful candidates appearing in recent studies [103, 104]. However, whereas, potentially, γ-secretase modulators could reduce the detrimental effect of iAβ, it would not eliminate it. This is because it would not reduce total levels of iAβ and its more harmful isoforms would still remain in the iAβ population, albeit in a decreased proportion.
An approach that potentially could work: Activation of α-secretase
Another approach to reduce iAβ levels is to activate α-secretase, a protease that cleaves at Lys16 of Aβ segment of both AβPP and C99 [105–107]. This would eliminate intracellular Aβ produced not only by AβPP proteolysis but also, importantly, in the AβPP-independent pathway. Regulation of α-secretase and its activation were subjects of numerous studies [108–110], and its therapeutic potential is well documented in model systems [110–113]. On the other hand, α-secretase is a member of the ADAM family of metalloproteases. It has over thirty known AβPP-unrelated substrates, and it was shown in genetic studies that overproduction of α-secretase affects the expression of over 300 genes [114]; importantly, it was also shown to control some Notch-dependent pathways [115]. With the reminder of γ-secretase inhibitors trials fiasco, there is a certain caution in the field regarding the development of α-secretase activators as possible AD drugs. If, however, the potential detrimental effects of α-secretase activators could be reduced to acceptable levels, for example by minimizing the duration of treatment, as discussed in the following sections, they could be effective in AD therapy provided the subcellular distribution of their target enzyme is sufficiently broad.
An approach that should work: Activation (yes, activation) of BACE1 and/or BACE2
The depletion of iAβ appears to be the ideal therapeutic strategy for AD. α-secretase activators may or may not provide such option. There is, however, an alternative to α-secretase that also cleaves Aβ amyloidolytically but does not carry the accompanying ‘baggage’. This alternative is a familiar one: β-secretase. Of the three secretases that cleave either at the termini or within Aβ, β-secretase appears to be the most amenable to manipulations, as evidenced by the extensive and prolonged use of BACE1 inhibitor in human clinical trials with no significant deleterious effects [1, 2]. β-secretase is instrumental in producing AβPP-derived Aβ, and thus is a major contributor to the processes leading to AD. The same β-secretase, on the other hand, appears to also hold a key to the treatment, cure, and prevention of the disease. Indeed, consider the following [116–119].
1) BACE1 was shown to cleave not only (albeit predominantly) at the β-site, prior to residue 1 of Aβ (between residues 671 and 672 of AβPP) but also at the β’-site between residues 10 and 11 of Aβ. Whereas cleavage at the β-site generates C99 and, after γ-secretase cleavage, Aβ1-XX (XX stands for any number between 37 and 43), cleavage at the β’-site generates C89 and, after γ-secretase cleavage, Aβ11-XX. 2) BACE1 cleaves with equal efficiency at the β’-site of AβPP, C99, and Aβ. 3) Overproduction of human BACE1 in mice increases both the rate of cleavage at the β’-site and the ratio of Aβ11-XX/Aβ1-XX. 4) Overproduction of human BACE1 strongly decreases deposition of extracellular Aβ in mouse brain 5) Protective effect of the Icelandic AβPP mutation A673T is associated with and appears to be due to the increased rate of BACE1-mediated cleavage at the β’-site [133]. Since C100 (eventually converted to C99) generated in the AβPP-independent iAβ production pathway and the retained iAβ (or Met-Aβ eventually converted to Aβ), produced from it, are, along with AβPP-derived iAβ, all valid substrates for BACE1 cleavage at the β’-site, increasing the activity of BACE1 appears to be an effective therapeutic strategy in the proposed AD paradigm.
Moreover, numerous studies showed that BACE1 is also capable of cleavage between residues 34 and 35 of human Aβ (yet unnamed BACE1 cleavage site) [120–123]. This BACE1 cleavage occurs within fully formed Aβ following γ-secretase cleavage of C99 [120, 121]. The rate of this cleavage correlates with the main activity of BACE1 and substantially increases when BACE1 is overexpressed; this cleavage generates an intermediate (Aβ34) in Aβ degradation/clearing process [120–123]. Taken together, the above observations strongly suggest that a sufficient increase in the activity of BACE1 in AD, possibly accompanied by its differential shift toward cleavages at the β’ and/or 34/35 sites of Aβ, could initiate amyloidolytic processing of C100/C99 and Aβ/Met-Aβ, and thus deplete the affected neurons of iAβ.
Furthermore, as if the above is insufficient, the activation of β-secretase holds even more therapeutic potential for AD. BACE2, a homolog of BACE1, was shown to cleave AβPP at the β-site, but its predominant activity is the internal amyloidolytic cleavage of Aβ at the Phe19 and Phe20 residues; these cleavages can occur within the fully formed Aβ [124]. Physiologically, BACE2 appears to reduce the production of Aβ; when its activity is suppressed in cell models, more Aβ is generated [125]. The protective potential of the Aβ-cleaving activity of BACE2 is indicated by a naturally occurring mutant. Indeed, it appears that the Flemish FAD mutation at the residue 21 of Aβ interferes with the Aβ-cleaving activity of BACE2 and thus increases not only the production of Aβ but also the iAβ content in affected individuals, consequently causing the early onset of AD [124]. Sufficient activation of BACE2 would deplete iAβ and have therapeutic effect in its own right; the depletion would be most effective, however, in combination with BACE1 activation. The former would not only supplement the latter by targeting additional iAβ cleavage sites, but also complement it because of their distinctly different subcellular localization [126]. It should be emphasized that this therapeutic option can be pursued, either at symptomatic stages or preventively (as described below), independently from and in parallel with the investigation of the mechanism underlying the AβPP-independent iAβ production pathway. Whereas this mechanism is of substantial interest and remains to be elucidated, the understanding of its nature is not requisite for the prevention of its activation or for its suppression by the depletion of iAβ levels below those required for its operation.
It should be commented that the activation of BACE1 (but not of BACE2, where cleavages at the 19/20 and 20/21 sites of Aβ dominate that at the β-site of AβPP) appears capable of increasing Aβ production in the AβPP proteolytic pathway. However, the expected increase in the cleavage at the AβPP β-site would be balanced (possibly outbalanced) by the increased cleavage within Aβ and may not result to the elevated production of AβPP-derived Aβ; even if it does, in the framework of the proposed paradigm this might not be detrimental because this AβPP-derived Aβ would be secreted. Moreover, as described in the following sections, remarkably, this type of therapeutic intervention could be administered, both at the symptomatic stage and preventively, for a limited duration only, thus minimizing possible deleterious effects, and at infrequent intervals of years or decades, and, possibly, even as once-in-a-lifetime treatment.
TREATMENT OF AD: EFFECT OF iAβ DEPLETION AT THE SYMPTOMATIC STAGES OF THE DISEASE
Within the framework of the proposed interpretation of AD, the goal of the treatment at symptomatic stages of the disease is to reduce levels of iAβ below those required for the activation and operation of the AD Engine. Considering that a mechanism, presumably the ISR but potentially any other iAβ-dependent process, responsible for the initiation and functioning of the AβPP-independent iAβ generation pathway depends on sufficient levels of iAβ, the reduction of the latter would terminate the mechanism’s operation. This would deprive the AβPP-independent iAβ production pathway of its crucial component(s) produced in the framework of the ISR, and it would cease to operate. Consequently, the continuous influx of iAβ will end, and the AD Engine would be interrupted and will come to a stop; this would allow the viable affected neurons to recover and reconnect. The efficiency of the treatment would depend on the extent of the depletion of iAβ and on the viability of the affected neurons. Provided that the treatment is of limited duration, its frequency would be defined by how soon the depleted iAβ levels could be restored, presumably by the accumulation of AβPP-derived iAβ, to those (T1) required for the activation and operation of the AβPP-independent iAβ production pathway and of the AD Engine.
In Fig. 4, which depicts effects of the iAβ depletion treatment at different symptomatic stages of AD, it is assumed that the activation of BACE1 and/or BACE2 (or the utilization of any suitable iAβ-depleting agent) is sufficient to completely, or nearly completely, deplete intracellular iAβ, and that the rate of accumulation of AβPP-derived iAβ to the T1 level remains constant and linear pre- and post-treatment (both assumptions are further discussed below). It is also assumed that a drug was administered for a limited, potentially short, duration and then withdrawn. In every case illustrated in Fig. 4, the effect of the treatment would be a ‘RESET’ of intracellular iAβ levels in surviving neurons. Figure 4A shows the effect of iAβ depletion treatment at the very early symptomatic stage (timing of the administration of the treatment is denoted by vertical arrows). At this time, a fraction of neurons have already crossed the T2 threshold and committed to the apoptotic pathway; they are either lost or irredeemable. On the other hand, the reset of iAβ levels in the remaining, viable, neurons, which at this stage of AD comprise the bulk of neuronal cells, would switch off the AD Engine and enable cells to recover and reconnect. At this point, the de novo accumulation of AβPP-derived iAβ to the T1 threshold and the consequent activation of the AβPP-independent iAβ production pathway and of the AD Engine would require a substantial time, possibly decades, prior to the recurrence of the disease. As shown in the Fig. 4, this may not happen in the remaining lifespan of an individual, at least in cases of sporadic AD.
Fig. 4
Effect of iAβ depletion treatment administered for limited duration at different symptomatic stages of AD. iA β: Intraneuronal Aβ levels; T1: iAβ level that triggers elicitation of the integrated stress response, initiation of AβPP-independent generation of iAβ, and activation of the AD Engine; T2: iAβ level that triggers cell’s commitment to the apoptotic pathway; Red blocks: Fraction of affected neurons committed to apoptosis or dead; Vertical arrows: Timing of the administration of a treatment; the drug is withdrawn after the complete or nearly complete depletion of iAβ is achieved. A: The drug is administered at the early symptomatic stage of the disease. Levels of iAβ in surviving cells have been ‘reset’ and the AD Engine switched off. The bulk of affected neurons, which did not yet reach the T2 threshold, recover and reconnect. At this point Aβ is produced only in the AβPP proteolytic pathway. The de novo accumulation of AβPP-derived iAβ resumes via cellular uptake of secreted peptide and intracellular retention of a fraction of its output in the AβPP proteolytic pathway. If the rate of accumulation remains constant and linear pre- and post-treatment (other options are considered below), the levels of AβPP-derived iAβ are unlikely to reach the T1 threshold and the disease to recur within the remaining lifespan of an individual (at least in SAD cases). B-D: The drug is administered at increasingly advanced stages of the disease. Outcomes are similar to that shown in A, except, as the disease progresses, there are less and less viable affected neurons that can be “reset” and thus redeemed.
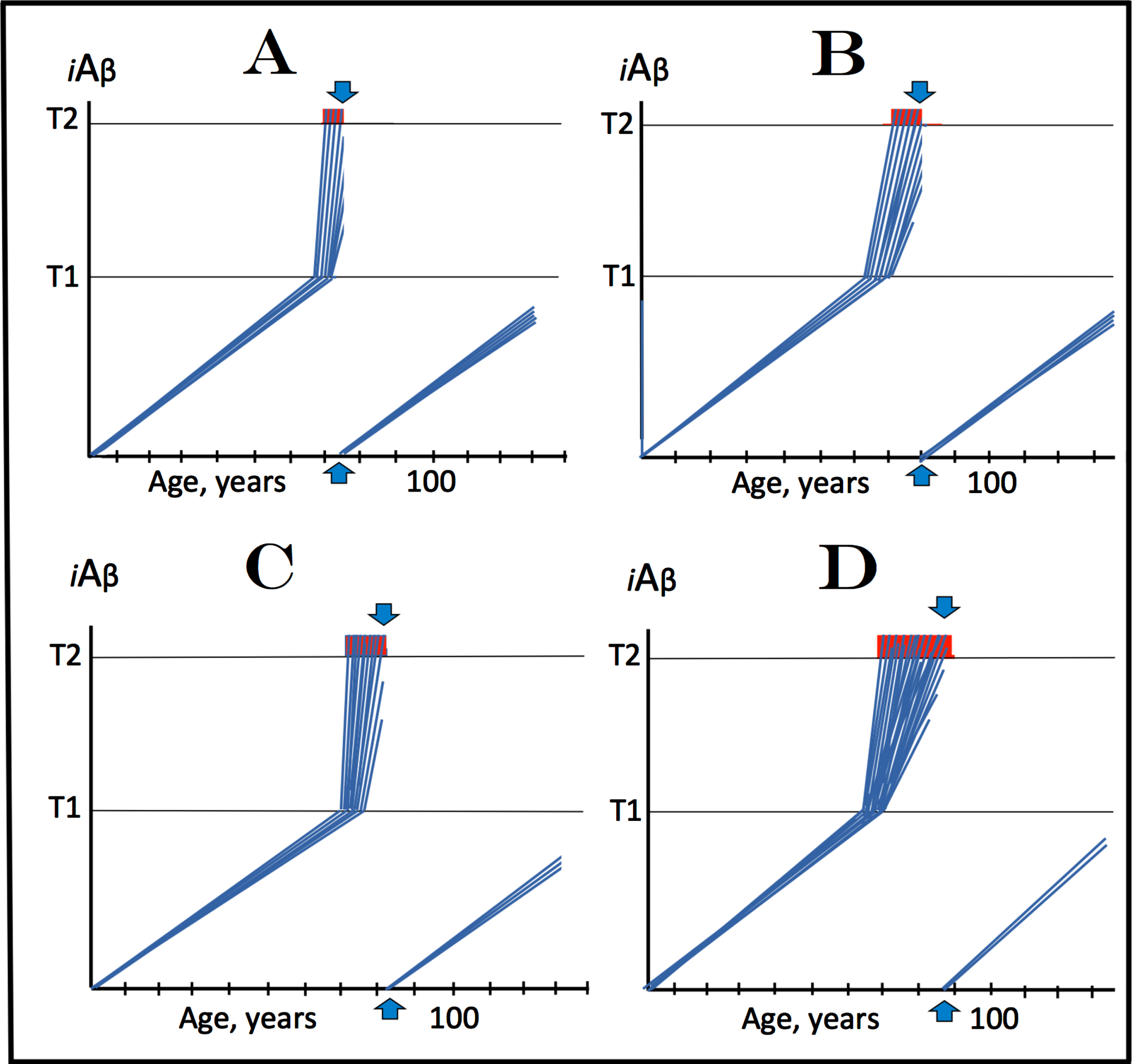
As the disease progresses, more and more neurons reach the T2 threshold and commit to the apoptotic pathway. Accordingly, this leaves less and less viable, still redeemable, neurons. This process is illustrated in Fig. 4B through 4D. Administration of the treatment (indicated by vertical arrows) and the resulting depletion and reset of iAβ levels would enable at least some neurons (probably depending on the extent of iAβ-inflicted cell damage at the time of treatment’s administration) to recuperate and reconnect. At the advanced symptomatic stages, however, while the progression of the disease would, probably, be stopped, the lost brain functionality is unlikely to be fully restored. If the ‘reset’ is complete and if the post-treatment rate of accumulation of AβPP-derived iAβ to the T1 level remains constant and is comparable to that operating linearly prior to symptomatic stages, the progression of the disease is not expected to resume within the remaining lifespan of an individual.
PREVENTION OF AD: EFFECT OF iAβ DEPLETION PRIOR TO SYMPTOMATIC MANIFESTATION OF THE DISEASE
The projected outcomes of the treatment of AD at its symptomatic stages by iAβ depletion via activation of BACE1 and/or BACE2 (or by other suitable iA β-depleting agent), described above, contain some uncertainties regarding the ability of neurons that crossed the T1, but have not yet reach the T2 threshold, to recover and reconnect following the disabling of the AD Engine; such ability, probably, would be inversely proportional to levels of iAβ and the accompanying cellular damage at the time of the treatment’s administration. There are no such uncertainties when this treatment is employed for the prevention of the disease. In this approach, the treatment is administered prior to the manifestation of AD symptoms, i.e., before iAβ levels have reached the T1 threshold and the AD Engine has been activated in any neurons. With the complete “reset” of the iAβ levels and with the rate of intracellular accumulation of AβPP-derived iAβ to the T1 level remaining constant and linear both pre- and post-treatment (see further discussion below), one such treatment in a lifetime could be, if properly timed, i.e., close to but below the statistical age of the onset of symptomatic AD, sufficient to prevent the occurrence of the disease within the remaining lifespan of an individual. This scenario is depicted in Fig. 5.
Fig. 5
Effect of iAβ depletion by a treatment of limited duration prior to symptomatic manifestation of AD. iA β: Intraneuronal Aβ levels; T1: iAβ level that triggers elicitation of the integrated stress response, initiation of AβPP-independent generation of iAβ, and activation of the AD Engine; T2: iAβ level that triggers cell’s commitment to the apoptotic pathway; Vertical arrows: timing of the administration of a treatment; the drug is withdrawn after the complete or nearly complete depletion of iAβ is achieved. A: Prevention of SAD. The drug is administered in the early sixties; statistically prior to the late onset of the disease and before levels of intracellular AβPP-derived iAβ reach the T1 threshold in any neuronal cells. Levels of iAβ have been ‘reset’ and the de novo accumulation of AβPP-derived iAβ resumes via the cellular uptake of secreted peptide and the intracellular retention of a fraction of its output in the AβPP proteolytic pathway. If the rate of this accumulation remains constant and linear through the lifespan of a neuron (other options are described in the Discussion section below), the levels of AβPP-derived iAβ are unlikely to reach the T1 threshold and the disease to occur within the remaining lifespan of an individual. B: Prevention of FAD. The drug is administered in the early forties, statistically prior to the early onset of the disease and before any neurons reach the T1 threshold. Levels of AβPP-derived iAβ are reset and their restoration would take decades. This could occur still within the lifespan of an individual; in such a case, a repeated administration of the drug could be required for the prevention of AD.
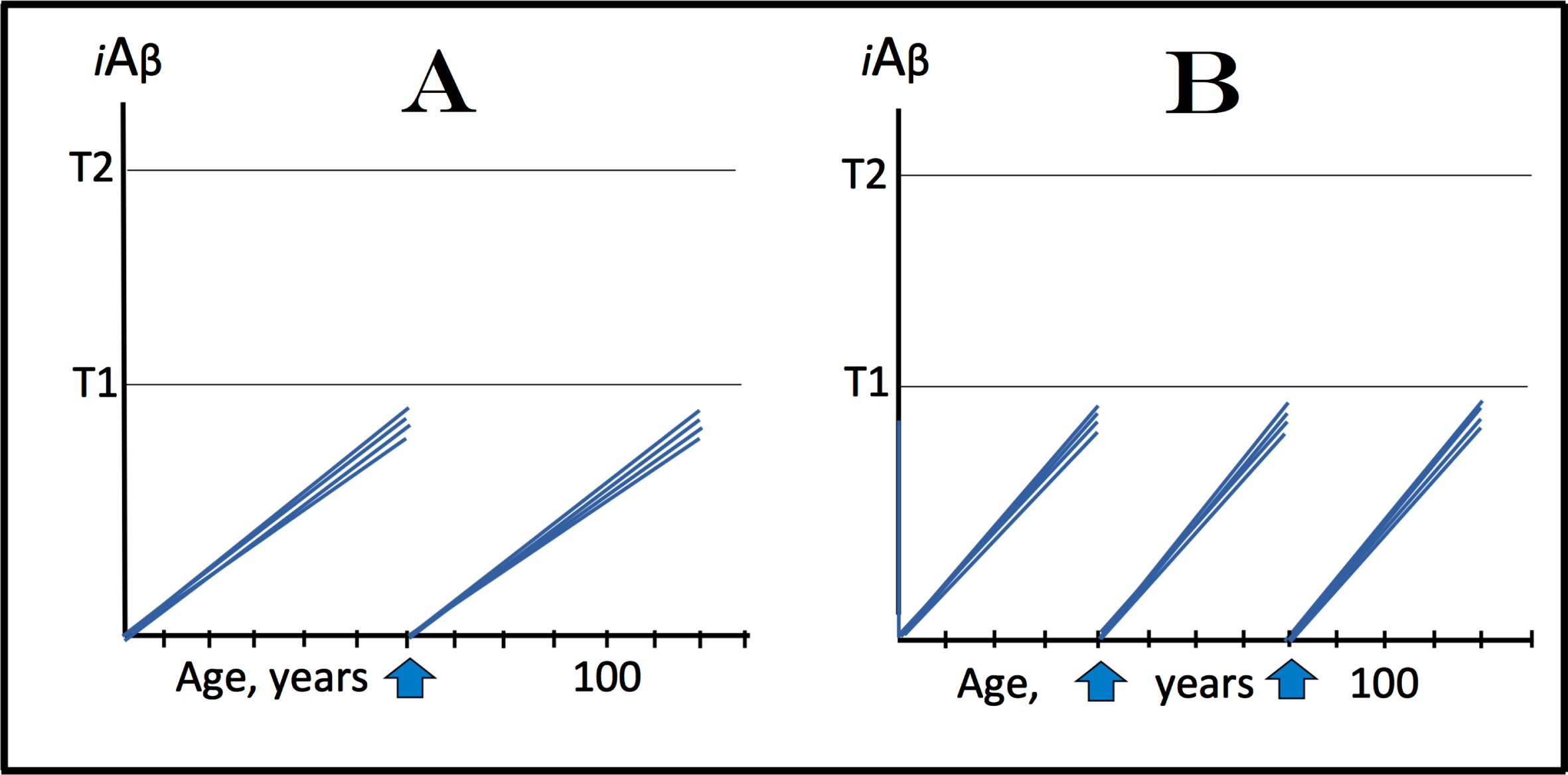
For prevention of sporadic AD, the treatment is shown in Fig. 5A to be administered at early sixties. At this age, in none of the neurons has AβPP-derived iAβ yet reached the level sufficient for the activation of the AβPP-independent iAβ production pathway and, consequently, of the AD Engine. Whatever level AβPP-derived iAβ has attained is reset by the iAβ depletion treatment, and the duration of its restoration, more precisely of its build-up to the T1 threshold, could exceed the remaining lifespan of an individual. For prevention of familial AD, the treatment is shown in Fig. 5B to be administered in early forties. At this stage, in none of the neurons have the AβPP-independent pathway of iAβ generation and the AD Engine been activated. The level of AβPP-derived iAβ accumulated within neurons is reset by the treatment, and its build-up to the T1 threshold could potentially take decades. Since in prospective FAD cases this build-up may plausibly occur within the remaining lifespan of an individual, the iAβ depletion treatment could have to be administered more than once.
DISCUSSION
AβPP-derived iAβ defines susceptibility to AD and triggers the disease
The notion of the causative role of Aβ in AD has strong experimental support and serves as a single assumption in the proposed interpretation of the disease. Taking this notion as a given, the results of multiple stage III clinical trials of verubecestat [1, 2], a potent BACE1 inhibitor, combined with those of its preclinical studies [5], mandate the occurrence of intraneuronally retained Aβ produced in the AβPP-independent pathway in AD, and assign to this intraneuronal pool of amyloid-β the causal function in the disease. In the herein proposed interpretation of AD, the activation of the AβPP-independent iAβ production pathway in the disease is preceded and mediated by a life-long accumulation of conventionally generated, AβPP-derived, iAβ to the critical over-the-threshold level. In this scenario, it is the rate of the AβPP-derived iAβ accumulation, which is primarily accountable for the susceptibility to and determines the timing of the onset of AD by causing the activation of the AβPP-independent iAβ production pathway. Two mechanisms are known to be responsible for the accumulation of AβPP-derived iAβ. One is the cellular uptake of a portion of secreted Aβ, in effect its conversion into iAβ. Another mechanism is the intraneuronal retention of a fraction of Aβ produced in the AβPP proteolytic pathway, which is generated as iAβ. Upon reaching critical levels, the accumulated AβPP-derived iAβ triggers the elicitation of the integrated stress response, presumed to mediate the activation of the AβPP-independent iAβ generation pathway (other candidates for this role cannot be excluded, as discussed above and shown in Fig. 1). This occurs via iAβ-mediated activation of PKR and/or HRI kinase; the latter through the mitochondrial distress-facilitated OMA1-DELE1-HRI signaling pathway. In both cases, eIF2α (target of both, activated PKR and HRI kinases) gets phosphorylated and the ISR ensues.
Aβ generated in the AβPP-independent pathway can be distinguished from Aβ produced by AβPP proteolysis
The activation of the AβPP-independent pathway of iAβ production occurs not necessarily via the ISR or only via the ISR (discussed above and shown in Fig. 1) and its details remain to be elucidated, but it can be plausibly suggested that among new proteins expressed within the framework of the ISR are components crucial for the operation of the AβPP-independent iAβ production pathway. Four mechanisms, each capable of underlying this pathway, have been proposed and described above. Which one of them operates in the disease remains to be ascertained, but they all share a key common feature, namely that in any such mechanism, translation of either intact or severely 5’-truncated AβPP mRNA initiates at the AUG normally encoding Met671 and preceding immediately and contiguously the Aβ-encoding sequence. The rationale for this notion, initially proposed for and limited to the internal initiation of translation within coding region of AβPP mRNA, is the exceptional position (literally exceptional in human AβPP mRNA) of the Met671-encoding AUG: this is the only Met-encoding AUG within human AβPP mRNA that is situated within an optimal translation initiation context; not even the AUG encoding the translation-initiating methionine of AβPP is. In each of the proposed mechanisms, the primary translation product is C100, i.e., C99 containing translation-initiating N-terminal Met (possibly acetylated), which is eventually removed post- rather than co-translationally. This is because it is followed by an aspartate (Asp1 of Aβ) and, therefore, cannot be cleaved-off by N-terminal methionine aminopeptidases (MAP) 1 and/or 2, which operate co-translationally provided that the translation-initiating Met is followed by Gly, Ala, Ser, Cys, Thr, Pro, or Val (listed in order of increasing size) [128]. Residues larger than Val do not fit, together with N-terminal Met, into the MAP active site, and in such cases the translation-initiating Met is removed post-translationally by one of multiple aminopeptidases with broad specificity (reviewed in [6]).
It follows that when the AβPP-independent iAβ production pathway is operational, steady-state pools of C100 (the equilibrium of C100 influx versus the removal of its N-terminal Met, i.e., its conversion to C99, and its cleavage by γ-secretase) and, possibly, of Met-Aβ (if it is sufficiently abundant, which depends on relative rates of the removal of N-terminal Met from versus γ-secretase cleavage of C100) should be detectable in affected live neuronal cells. These pools would not be present in postmortem samples because in dying neurons, C100 influx would cease while aminopeptidases are still operational; the equilibrium is thus shifted to the exclusion of both C100 and Met-Aβ. The detection of either or both pools in a model system would serve as a confirmation of the activity of the AβPP-independent iAβ generation pathway. On the other hand, when this pathway is disabled, and no influx of C100 occurs, both pools will dissipate; assaying for the occurrence of C100 and Met-Aβ would, therefore, provide means of assessing the efficiency of iAβ depletion therapy in model systems. Unlike AβPP, C100 does not contain N-terminal signal peptide that would guide it into a secretory pipeline. For Aβ produced in the AβPP-independent pathway to be retained intraneuronally, it is sufficient, therefore, that γ-secretase cleavage of C100 (Met-C99) or, after the N-terminal Met removal, of C99 would occur on an internal membrane, a phenomenon documented in neuronal cells [39, 40].
Peculiar case of the Swedish FAD mutant
It should be noted that in Swedish FAD mutants, where the AUG normally encoding Met671 of AβPP is replaced by CUG, the same scenario for the AβPP-independent production of C100 and eventually of iAβ, i.e., translation initiation from a codon for the residue 671 of AβPP, also plays out (discussed as a case study in [6]). This is because a leucine-encoding CUG, when situated within an optimal translation initiation context (and in Swedish FAD mutants it is), is capable of initiating translation with 82% efficiency of an AUG within the same context [130]. In Swedish FAD mutants’ case, the C100 fragment does not necessarily starts with Leu (translated from the initiating CUG codon) at the N-end, it could still be Met; which one depends on the translation initiation factor in play. If it were eIF2A (not to confuse with eIF2α), which is likely to operate under the ISR, C100 would begin with the translation-initiating leucine residue (i.e., forming Leu-C99). However, if it were eIF2, C100 would start with Met, which is always utilized for translation initiation, regardless of a start codon, when eIF2 is involved [130] (thus producing, in this case, Met-C99 despite initiating translation from leucine-encoding CUG). If C100 translation were initiated with Leu, it would be removed post-translationally by the leucyl aminopeptidase.
Mutation(s) disrupting translation initiation from the AUG encoding Met671 of AβPP would convey to its carriers insusceptibility to AD
Within the proposed AD paradigm, because of the pivotal role of the AUG encoding AβPP’s Met671 or, more generally, because of the role of the competency to efficiently initiate translation from a codon in this position (e.g., Swedish FAD mutant) in the AβPP-independent production of iAβ, it can be predicted with a considerable certainty that a mutation that replaces the AUG in question with a codon that is incapable of initiating translation (or of initiating it with sufficient efficiency; even a single-nucleotide substitution might suffice), or changes the surrounding nucleotide context so that the AUG in question can no longer initiate translation (or initiate it with sufficient efficiency), would disable the AβPP-independent iAβ generation pathway and render its bearers insusceptible to AD. In this context, it is not unreasonable to search for such mutation(s) in families of centenarians resistant to AD. Such induced (edited in) mutations of the AβPP gene can also be exploited for research and drug-development purposes in human neuronal cells AD model described below.
Activation of the AβPP-independent iAβ generation pathway renders the AβPP proteolytic pathway irrelevant for the progression of AD and brands its targeting futile
The production of intraneuronally retained Aβ in the de novo activated AβPP-independent pathway significantly increases its cellular levels. As a result, PKR and/or HRI remain in the activated state, eIF2α phosphorylation is maintained, the integrated stress response is perpetually elicited, and the AβPP-independent iAβ generation pathway is sustained. These self-propagating feedback cycles, where newly produced iAβ stimulates its own generation in the AβPP-independent pathway, constitute the engine that drives AD. In this context, the life-long accumulation of AβPP-derived iAβ to levels sufficient to activate eIF2α kinases and elicit the ISR, constitute the Starter Motor. Once switched on, the Starter Motor ignites the autonomous, self-sufficient, AβPP-independent AD Engine, which provides the incessant influx of iAβ. Thus, in cells with the AβPP-independent source of iAβ operational, the continuous production of Aβ in the AβPP proteolytic pathway becomes irrelevant to the progression of the disease, and its targeting is rendered futile because its contribution of iAβ turns into insignificant in comparison with that of the AβPP-independent iAβ generation pathway.
Not all components of the AD Engine can be targeted for therapeutic purposes
Moreover, it appears that by the time AD symptoms manifest, intracellular levels of AβPP-derived iAβ have reached critical threshold, the AβPP-independent production of iAβ has been activated, and the AD Engine been switched on in all, or the bulk of all affected neurons of an AD patient. It follows that at this stage, therapeutic approaches targeting solely the AβPP proteolytic pathway of Aβ production, such as BACE1 inhibition, or pursuing the reduction of levels of extracellular Aβ (and thus of its cellular uptake) with antibodies are unproductive and the only viable direct therapeutic strategy is pursuing components of the AD Engine with the goal of ceasing its activity. Targeting the activation of the AβPP-independent iAβ production pathway, however, is challenging because of redundancies involved and the lack of the absolute certainty that it is indeed mediated by the ISR or solely by the ISR. Pursuing the AβPP-independent iAβ generation pathway could be no less demanding prior to identifying the underlying mechanism, which can turn out to be physiologically vitally important and therefore “untouchable”.
The only remaining target, therefore, is iAβ; a sufficient depletion of its levels by any means would stop the operation of the AD Engine. The obvious way to do this is via the inhibition of γ-secretase, which would prevent the processing of C99 or C100 into Aβ (or Met-Aβ), or through the activation of α-secretase, which potentially would be even more efficient because it could not only prevent the de novo generation of Aβ but also eliminate pre-formed Aβ. Inhibition of γ-secretase, however, is not feasible; it is a prominent member of the Notch signaling pathway and has multiple AβPP-unrelated substrates. Its administration in clinical trials elicited deleterious effects in AD patients. A milder related approach, the modulation of γ-secretase toward production of shorter, less toxic Aβ isoforms, has so far been proven unsuccessful in clinical trials. The activation of α-secretase, although highly promising, could also be problematic. This protease is a member of the ADAM family of metalloproteases and has dozens of known AβPP-unrelated substrates in multiple cell types; its overproduction was shown to affect expression of over three hundred genes.
Any iAβ-depleting activity would constitute an effective curative and preventive AD drug: BACE1 and BACE2 activators as feasible iAβ depletion agents
On the other hand, an alternative approach proposed in the present study, namely the activation of one or, preferably, both homologs of β-secretase, BACE1 and BACE2, has all the advantages of α-secretase activation (and more) without its drawbacks. BACE1 cleaves predominantly at the β-site of AβPP but also between residues 10/11 and 34/35 of Aβ. Its homolog, BACE2, is also capable of cleaving at the β-site of AβPP but its predominant cleaving sites are between residues 19/20 and 20/21 of Aβ. Two naturally occurring human AβPP mutations provide illustration of the protective potential of Aβ-cleaving activities of BACE1 and BACE2. Icelandic mutation at the second residue of Aβ confers protection from AD to its carriers. This protective effect is associated with and appears to be due to the increased rate of BACE1-mediated cleavage at the 10/11 site of Aβ. This is precisely the anticipated effect of the proposed BACE1 activation. On the other hand, Flemish mutation at the residue 21 of Aβ interferes with the Aβ-cleaving activity of BACE2 and thus increases levels of iAβ in affected individuals, causing early onset of the disease. By implication, increasing the activity of BACE2 would deplete iAβ and protect from the disease. A treatment combining the activation of both, BACE1 and BACE2, would be the most effective therapeutic approach for AD: the two are not only mutually supplementary by virtue of targeting different sites within Aβ, but also complementary, due to their distinctly different subcellular localization. The administration of BACE1 and/or BACE2 activator(s), possibly only for a limited duration, would result in the depletion of iAβ. Astonishingly, such therapy, either at the symptomatic stage or preventively, prior to the manifestation of AD symptoms, could open up the possibility of a once-in-a-lifetime (or at least very infrequent) curative or preventive treatment for the disease.
Indeed, as described above and illustrated in Figs. 4 and 5, the activation of BACE1 and/or BACE2 has the potential to deplete iAβ and to “reset” its levels. In such a case, the operation of the AD Engine would be stopped (or prevented) and, provided that the rate of accumulation of AβPP-derived iAβ remains linear and constant, the restoration of iAβ levels to those sufficient to activate the AβPP-independent pathway of its production, and thus to re-ignite (or ignite) the AD Engine, would take decades, probably exceeding the remaining lifespan of an individual. Since, as the disease progresses, the increasing proportion of affected neurons die or commit to the apoptotic pathway and become irredeemable, the outcome of BACE activation at the symptomatic stage of AD would depend on its timing relative to the symptomatic onset of the disease; the sooner it is administered, the more affected viable neurons would be enabled to recover and reconnect. Preventively, the BACE activation treatment could be administered at the age that is statistically close to but below that of the onset of symptomatic AD: early sixties for preempting sporadic AD and early forties for prospective familial AD. Following the reset of iAβ levels, it could be anticipated that no additional treatment would be required within the remaining lifespan of an individual, at least in cases of sporadic AD treatment or prevention.
Once-in-a-lifetime-only iAβ depletion treatment by BACE1 and/or BACE2 activation could protect from the ‘common’ pervasive aging-related cognitive decline
Moreover, it is possible that at least some of the ubiquitous (and milder than AD) instances of aging-related cognitive deterioration are, in fact, a low-grade AD, i.e., neurons are dying by the same mechanism as in AD (via operation of the AD Engine) but in much lower numbers and/or only at particular sites, e.g., the anterior midcingulate cortex, a location associated with the retention of cognitive functionality in aging. Alternatively, it is possible that in these cases AβPP-derived iAβ accumulates sufficiently within neurons to cause cognitive dysfunction symptoms but insufficiently to activate the AβPP-independent iAβ production pathway and trigger AD. If either of these alternatives were the case, a potentially one-time-only AD-preventive or curative iAβ depletion treatment, described above, would also afford the protection from the occurrence or the cure of the pervasive aging-associated cognitive decline. The feasibility of this notion is strongly supported by studies of the Icelandic AβPP mutation A673T. This mutation protects its bearers from AD. The protective effect is apparently due to the increased rate of BACE1 cleavage at residues 10/11 of Aβ (β’-site) and the consequent iAβ depletion within neuronal cells. The same mutation also protects its carriers from aging-associated cognitive decline [133–135]. The proposed activation of BACE1 (and of BACE2) would cause conceptually similar iAβ depletion and is, therefore, expected to have similar protective or curative effect from both AD and aging-related cognitive decline. In fact, the iAβ depletion by any suitable agent can be expected to be as protective.
Administration of iAβ-depleting drugs: Practical aspects
The outcomes depicted in Figs. 4 and 5 and discussed above are contingent upon the following two presumptions: 1) The completeness of BACE1 and/or BACE2 activation-driven (in fact, any iAβ depleting agent-driven) depletion of iAβ and 2) The constancy and linearity of the rate of accumulation of AβPP-derived iAβ. These presumptions may not necessarily be operative, and the projected outcomes could be not attainable. It is important, therefore, to consider possible results of BACE activation under less than optimal parameters. First, it is necessary to define what extent of depletion of iAβ would be therapeutically effective. The answer is clear: in symptomatic AD cases, for the treatment to be effective, i.e., therapeutically meaningful (in cases of the preventive therapy, any reduction of iAβ levels would be therapeutically meaningful in the proposed AD paradigm), iAβ should be depleted to levels below those required for the activation and operation of the AβPP-independent iAβ production pathway and, consequently, of the AD Engine; this can be referred to as the “minimal extent of iAβ depletion”. Such definition of the required extent of iAβ depletion determines the “minimal duration of the treatment”: long enough to achieve the minimal extent of iAβ depletion. The actual duration of the treatment, aiming for the complete or nearly complete iAβ depletion, can potentially be very short, days akin to an antibiotics treatment, but even much longer term does not constitute a hindrance because, unlike α- and γ-secretases, BACE was shown, in human clinical trials, to be amenable to prolonged and extensive manipulations without deleterious consequences.
In practical terms, however, the therapeutically effective duration of treatment at symptomatic stages of AD could be longer than that needed for a sufficient iAβ depletion. This is because, whereas the ISR would no longer be driven by iAβ, it could be transiently sustained by the persistent activity of eIF2α kinases and/or the residual stress-generating cellular damage (caused by iAβ and requiring an additional time to be healed) and, in turn, continue to support operation of the AβPP-independent iAβ production pathway. Therefore, the duration of treatment at symptomatic stages of AD should be long enough to achieve (a) a sufficient depletion of iAβ and (b) the cessation of AβPP-independent generation of iAβ, if only the (a) were satisfied and the drug withdrawn, iAβ would be rapidly replenished via its continuous AβPP-independent production (these considerations do not apply to cases of the preventive treatment where the AβPP-independent iAβ production pathway is not operative). To ascertain its cessation, the activity of the AβPP-independent iAβ production pathway could, as discussed above, be assessed in model systems by the occurrence of C100 (and/or Met-Aβ) or the lack thereof, with the presence of a methionine at the N-termini of the C99 fragment or of intraneuronal Aβ reporting on the operation of the pathway. A practical, minimally invasive, method for such an assessment in AD patients would need to be developed.
The last parameter affecting the outcomes of the proposed AD therapy and defining the manner of its application is the duration of the therapeutically meaningful effect of the treatment, i.e., the time period needed for the build-up of depleted iAβ levels to those (T1) sufficient to activate and maintain the operation of the AD Engine, an attribute that would determine the required frequency of the administration of the treatment. This parameter depends on two factors: the extent of the iAβ depletion and the subsequent rate of intracellular accumulation of AβPP-derived iAβ. The first of these factors could be regulated by the duration of the treatment, and, because BACE is evidently amenable to extensive manipulations, it can be assumed, for practical considerations, that either a complete or nearly complete, or at least a very substantial depletion could be achieved. As for the rate of the accumulation of AβPP-derived iAβ to the T1 level, it is the only remaining uncertainty. In Figs. 4 and 5 above, it is shown, both pre- and post-depletion treatment, as identical and linear. This, possibly, is correct, but potentially could be an oversimplification; in the run-up to the disease, the rate in question could be increasing in affected neurons with time in, say, exponential or semi-parabolic manner rather than maintaining linearity. In such a case, if intracellular accumulation of AβPP-derived iAβ would resume, post-treatment, at the maximal rate it had exhibited pre-treatment, and proceed in the same manner, the critical, AβPP-independent iAβ production-activating, AD Engine-igniting, iAβ levels could be re-established (or established, in preventive treatment cases) sooner than in decades; it is likely, however that even in such a case it would take years rather than months, and the frequency of treatments would have to be adjusted accordingly.
Amyloid plaques as physiological protective measure, the ‘resilience to AD’ phenomenon, AD without excessive Aβ plaques, and the lifespan-dependent inevitability of AD
As for secreted Aβ, whereas a contributory role of extracellular Aβ plaques in AD cannot be excluded, they occur physiologically and their increased presence in AD patients could be only a correlation. Indeed, the increased production of AβPP-derived Aβ in general or of its 1–42 variant (with increased amyloidogenicity and uptake efficiency) in particular would result in increases in Aβ retention, secretion, and cellular uptake, i.e., factors instrumental in triggering the disease in the proposed interpretation of AD. It would also, coincidentally, result in increased plaque formation. In fact, in the proposed AD paradigm it is conceivable that Aβ sequestration in plaques is, at least in part, a protective measure to hold up the cellular uptake of secreted Aβ, i.e., its conversion into iAβ. The observed association of iAβ, rather than amyloid plaques, with neuronal loss [19], as well as the occurrence of a substantial cohort of AD-free (in fact, any cognitive dysfunction-free) aged individuals who die of old age or from unrelated causes yet whose brains exhibit, upon amyloid-PET scan or postmortem analysis, levels of extracellular amyloid plaques equal to or exceeding those typically seen in the disease [35, 135–141], a phenomenon referred to as the ‘resilience to AD’ [141], suggest that, anyway, just Aβ plaques are insufficient for the occurrence of AD, are neither the cause nor necessarily a consequence of the disease, and provide additional support to the centrality of iAβ produced in the AβPP-independent pathway in AD. As to why some amyloid-PET brain positive individuals do not show cognitive dysfunction, the answer, within the proposed AD paradigm, is simple: a high, even a normal, rate of Aβ secretion, combined with a low rate of cellular uptake of secreted Aβ and/or of intracellular retention of AβPP-derived iAβ would result in excessive Aβ plaque formation yet in slow accumulation of AβPP-derived iAβ, insufficient to reach the T1 threshold and trigger the activation of the AβPP-independent iAβ production pathway and the disease within the lifespan of an individual. As a prediction, a diametrically opposite scenario could be envisioned: no excessive Aβ plaque formation yet the occurrence of AD. This phenomenon, yet to be observed (or recognized), could be explained by a low, or even a normal, rate of Aβ secretion combined with a high rate of the retention of AβPP-derived iAβ and/or of cellular uptake of secreted Aβ; in either case, levels of AβPP-derived iAβ can reach the T1 threshold, activate the AβPP-independent iAβ production pathway and trigger AD in the absence of Aβ plaque pathology. In this AD paradigm, any individual (except carriers of certain protective mutations or factors), who hypothetically lives sufficiently long (120 or 150 years for some few; much less for others), and was not treated preventively, would inevitably develop AD because, given enough time, the levels of AβPP-derived iAβ would reach the T1 threshold and trigger the disease. In the light of the steadily increasing human lifespan, a preventive AD (and, possibly, the ‘common’ aging-associated cognitive decline) treatment, when available, would be essential.
Why current mouse AD models cannot display a complete spectrum of AD pathology
Following the above lines of reasoning, it could be also argued that the immense non-physiological overproduction of AβPP-derived Aβ in mouse AD models causes neurodegeneration, potentially, by a sheer volume of cell-‘suffocating’ Aβ plaques. If this is the case, such models have little relevance to AD. Another, more likely possibility supported by the observation that the levels of iAβ, rather than amyloid plaques, correlate with neuronal loss in a mouse AD model [19] is that because of great quantity of produced and secreted AβPP-derived Aβ, its combined iAβ fraction, both retained within and taken up by neuronal cells, is sufficient to cause partial AD symptoms in mouse models. It is, however, patently insufficient to elicit the complete spectrum of AD pathology in mouse models, including, notably, the formation of neurofibrillary tangles (NFTs), a basic AD hallmark. This is because the AβPP-independent generation of the retained iAβ is inoperative in either wild or transgenic mice [6, 90–94] and without it, levels of iAβ required for the elicitation of AD cannot be reached; in any adequate mouse AD model (which remains to be constructed), this pathway would have to be built in as a mandatory requirement. On the other hand, this pathway is operative in human AD patients, and this provides an explanation as to why, experimentally, the formation of NFTs was seen only in human neuronal cells AD model [129] where the overproduction of exogenous Aβ apparently activated the endogenous AβPP-independent iAβ production pathway (further discussed below).
Why candidate AD drugs worked so well in mouse AD models and not at all in AD patients
The above notion, namely the lack of the AβPP-independent generation of the retained iAβ in mouse AD models and its occurrence in human AD patients, central to the proposed AD paradigm, explains why suppression of AβPP proteolysis by BACE inhibitors or depletion of extracellular Aβ by monoclonal antibodies (operating extracellularly) worked so well in the former by either eliminating the only Aβ production source with BACE inhibitors, or removing extracellular Aβ thus suppressing its cellular uptake with antibodies, yet had no positive effect whatsoever on the progression of the disease in the latter because the operation of the autonomous, AβPP-independent, AD Engine was not and could not be affected in any way by either BACE inhibitors or Aβ antibodies.
VALIDATION OF THE ACH2.0 AND OF ACH2.0-BASED THERAPEUTIC STRATEGIES FOR AD
Advantages of human neuronal cells-based AD model system
The present study considers the results of stage III clinical trials of verubecestat an attestation of the production of iAβ in the AβPP-independent pathway in AD. In this and following sections, we describe further experimental designs devised to validate the occurrence of this pathway, to establish its causative effect in the disease, and to evaluate the proposed iAβ depletion therapy for AD. If successful, the experimental approaches outlined below could potentially form a basis for research and therapeutic strategies in the proposed AD paradigm. Arguably, the best conceivable model system for both research purposes and the initial testing of potential AD drugs is the one based on human neuronal cells. Beside utilizing the actual cell type affected by the disease, the most important attribute of such AD model system is that it carries human AβPP gene capable of supporting, via mRNA expressed from it, the endogenous AβPP-independent iAβ production, a process apparently operating in AD-affected human neuronal cells within the framework of the exclusivity of AD to humans (reviewed in [6, 146]; briefly, in non-human mammals Aβ cannot be endogenously produced in the AβPP-independent pathway). As described below, in such model system, processes could be relatively easily initiated that potentially lead to the endogenous AβPP-independent production of C100 (i.e., Met-C99) and, subsequently, of Met-Aβ, which are readily distinguishable from their AβPP-derived counterparts by the presence of the translation-initiating methionine residue at their N-termini.
Design of an adequate human neuronal cells-based AD model
Within the framework of the proposed interpretation of AD, the most “physiological” (i.e., emulating processes presumably occurring in the disease) method for the activation of the AβPP-independent production of iAβ from the endogenous AβPP gene in human neuronal cells (thus making the model “adequate”) is to accelerate the operation of the Starter Motor, i.e., the intracellular accumulation of iAβ to levels (T1) sufficient to trigger the activation of the AβPP-independent iAβ generation pathway and, consequently, of the autonomous AD Engine. This can be achieved by transiently supplying cells with exogenous iAβ42 either by importing the peptide or by expressing it from appropriate DNA constructs or from transfected mRNA. The DNA constructs or mRNA should encode solely Aβ42; translation would be initiated from the AUG codon preceding the Aβ nucleotide sequence and the resulting peptide would remain within the cell, i.e., it would be iAβ. However, for purposes of the proposed experiments, the translation-initiating AUG in a DNA construct or in transfected mRNA cannot precede the Aβ-coding sequence contiguously. The reason for this is that the principal detection assay for the activity of the endogenous AβPP-independent iAβ production pathway would be for the occurrence of N-terminal Met-containing Aβ (along with C100, i.e., Met-C99). If, in an iAβ expression construct, the translation-initiating AUG precedes the Aβ-coding sequence contiguously, the primary translation product would be, as described above, Met-Aβ, and its presence would interfere with the detection of the anticipated N-terminal Met-containing iAβ produced endogenously in the AβPP-independent pathway.
Therefore, the design of an iAβ-expressing DNA construct or of transfected mRNA should be such that the initiating N-terminal Met would be removed co-translationally, by MAP1 or MAP2. To achieve this, one of the seven residues compatible with the co-translational removal of N-terminal Met by MAP1/MAP2 (listed above) should be inserted between the translation-initiating Met and the first aspartate (Asp1) of Aβ. The guide for which residue out of the seven should be employed is provided by the “N-end rule” that defines the extent of stability (half-life) afforded to a polypeptide by its N-terminal residue [127]. According to this rule, the best choice would be valine. This, however, is not an option because, when the translation-initiating Met is followed by Val or Thr (position 1, P1), the removal of the N-terminal Met depends on the identity of a residue in the P2. If it is Asp, Glu, or Pro, the N-terminal Met cannot be removed co-translationally [128]. Therefore, if the position following the translation-initiating Met (P1) is occupied by Val and the P2 contains aspartate (Asp1 of Aβ), the primary translation product would be Met-Val-Aβ. The second best choice, according to the N-end rule, is glycine. If Gly occupies the P1, the N-terminal Met would be readily removed co-translationally by MAP1/MAP2 [128], and the resulting primary product would be Gly-Aβ, easily distinguishable from the anticipated Met-Aβ generated in the endogenous AβPP-independent pathway. Another possible approach, which takes a page out of the Swedish FAD mutant book, would be to substitute, in an iAβ-expressing DNA construct or in transfected mRNA, the AUG preceding the Aβ-coding sequence with CUG. The expression from such a construct or from transfected mRNA would be less efficient [130] but the resulting Leu-Aβ would be readily distinguishable from the anticipated endogenous Met-Aβ produced in the AβPP-independent pathway. This approach is, however, conditional: for the reasons discussed above, it requires that, for translation initiation with Leu rather than with Met, eIF2A, rather than eIF2, is engaged [130], an aspect that remains to be ascertained in human neuronal cells AD models. It can be anticipated that at the stage of the activation of the endogenous AβPP-independent iAβ generation pathway, when cells are presumed to be under the ISR conditions, eIF2A would be operational [130]; this might not be the case at the early stages of exogenous iAβ overproduction.
On the other hand, the detection of C100, i.e., Met-C99, is completely sufficient for the validation purposes. If this approach is taken, exogenous Met-Aβ can be freely expressed in human neuronal cells, in order to activate the endogenous AβPP-independent iAβ generation pathway, without interfering with the detection procedure. Several additional, albeit less physiological, methods of activation of the endogenous AβPP-independent iAβ production pathway in human neuronal cells, which bypass the stage of initial intracellular accumulation of exogenous iAβ, are discussed in [6]. They include the activation of OMA1, thus triggering the OMA1-DELE1-HRI-ISR cascade, and stressor-specific activation of one of eIF2α kinases, also resulting in the elicitation of the integrated stress response; in both cases, provided that the elicitation of the ISR alone is sufficient, the initiation of the endogenous AβPP-independent production of iAβ and the consequent activation of the AD Engine would, potentially, follow.
Testing the proposed human neuronal cells-based AD model: Validation of the ACH2.0 and of ACH2.0-based therapeutic strategies for AD
In any procedure for the activation of the endogenous AβPP-independent iAβ production pathway in human neuronal cells, the detection of the pathway’s activity is based principally on the identification of its distinctly unique primary translation product, C100 (i.e., N-terminal Met-containing C99), and, possibly, of N-terminal Met-containing Aβ (if sufficiently abundant; see above). The detection of either or both of these signature molecules within neuronal cells would constitute a proof for the operation of the endogenous AβPP-independent pathway of generation of the retained iAβ. Additional assays, described in [6], could pinpoint the mechanism which underlies and enables this pathway. Among those, of special interest would be testing for the presence of chimeric RNA molecules containing, in a defined and predicted manner [6, 84, 85, 87], both sense and antisense segments of AβPP mRNA; its detection would indicate the occurrence of the RNA-dependent AβPP mRNA amplification process resulting in C100-encoding mRNA [6, 87]. Confirmation of the activity of the AβPP-independent pathway generating the retained iAβ in human neurons would allow to assess the potential of iAβ depletion to suppress or prevent this activity. This could be done by exogenously overexpressing BACE1, BACE 2, or both from a constitutive or an inducible promoter and either prior or subsequent to the activation of the AβPP-independent iAβ generation pathway. The evaluation of the proposed human neuronal cells-based AD model system and of the efficacy, as well as of the consequences, of the iAβ depletion would be rendered not only upon the activity (or lack thereof) of the AβPP-independent iAβ generation pathway and the levels of the intact iAβ, but also upon the occurrence (or its prevention) of the formation of neurofibrillary tangles (see below). The ability to regulate the activity of the AβPP-independent iAβ generation pathway via BACE-mediated iAβ depletion would constitute a proof of principle for the usage of BACE activators (or other iAβ-depleting agents) as potential AD drugs and justify a major effort to develop such agents. Moreover, the tau hyperphosphorylation and NFTs formation can be utilized as the only AD biomarkers in the proposed AD model system; in such a case, the suppression or prevention of their occurrence by BACE1 and/or BACE2 activation (or exogenous overexpression) would provide the proof of principle without the necessity to analyze the activity of the AβPP-independent iAβ generation pathway, thus circumventing potential technical challenges of the detection of N-terminal methionine-containing C99 and iAβ, due to their possibly low abundance.
Human neuronal cells-based AD model can potentially display a full spectrum of cellular AD pathology and serve as a research and drug development platform
Even if the experiments proposed above are successful and prove the occurrence of the endogenous AβPP-independent generation of the retained iAβ in human neuronal cells under certain conditions, a connection with AD will have to be ascertained in order to establish the cause-and-effect relationship. In animal model systems, the observation and evaluation of symptoms of the disease could provide such connection. Could “symptoms” of AD be observed in cultured neuronal cells? The answer is affirmative. Indeed, it was shown [129] that in cultured human neuronal cells, overproduction of Aβ could trigger the formation of NFTs, the major hallmark (i.e., a cellular “symptom”) of AD. In that study, its authors interpreted the results within the framework of the ACH and attributed the observed NFTs formation to the effects of secreted and extracellularly accumulated AβPP-derived Aβ. In our interpretation of the results presented in [129], the investigators inadvertently, as an unintended consequence, activated the AD Engine (analyzed as a case study in [6]). Indeed, in the framework of the proposed AD paradigm it is highly plausible that the formation of NFTs, observed in that study, occurred within a cascade initiated by increased levels of iAβ produced in the AβPP-independent pathway, i.e., by the operation of the AD Engine. Briefly, in the study under discussion, human AβPP harboring both the Swedish K670N/M671L and the London V717I FAD mutations, as well as PSEN1 with ΔE9 FAD mutation, were acutely overexpressed in human neural progenitor cells from polycistronic lentiviral constructs, and cells were differentiated in three-dimensional matrigel cultures. Swedish FAD mutation facilitates the increased retention of AβPP-derived iAβ (described above, [45]), whereas other two FAD mutations facilitate generation of Aβ42 isoform. This model, thus, is characterized by the augmented accumulation of the retained AβPP-derived iAβ and by very efficient cellular uptake of secreted Aβ. The latter occurs because secreted Aβ does not diffuse in matrigel and accumulates in the vicinity of the cell, and also because Aβ42 is taken up preferentially, with the efficiency twice that of other Aβ species. The AβPP-derived ‘toxic’ iAβ42 isoform rapidly accumulates to critical levels and triggers activation of the endogenous AβPP-independent generation of the retained iAβ; this drastically increases the iAβ levels and eventually leads to the formation of NFTs. Indeed, sufficiently high levels of iAβ, such as those anticipated to result from the sustained activity of the AβPP-independent iAβ generation pathway are, apparently, requisite for the AD-associated tauopathy. As was shown experimentally, they could lead to the inhibition of the ubiquitin-proteosome system, facilitation of the build-up and phosphorylation of tau protein, and the formation of NFTs [142–145]. Therefore, it can be anticipated that targeted activation of the AβPP-independent iAβ production and, consequently, of the AD Engine in human neuronal cells by methods described above (including the procedure utilized in [129]) or via alternative procedures would lead to the formation of NFTs. Observation of NFTs in human neuronal cells with the activated AβPP-independent iAβ production pathway would provide direct connection with the disease and be consistent with a notion that activation of the AβPP-independent generation of iAβ is essential for the development and progression of AD. It would also establish this AD model as a legitimate experimental platform for both research purposes and the development of AD therapies.
CONCLUSION
In conclusion, the proposed interpretation of AD postulates that the disease is caused and sustained by iAβ generated in the AβPP-independent pathway and retained within neuronal cells, a notion that is mandated by the results of multiple stage III clinical trials of a BACE inhibitor. This interpretation, in fact a reformulation of the ACH, constitutes the Amyloid Cascade Hypothesis 2.0, ACH2.0 (‘The iAβ Cascade Hypothesis’ and, accordingly, ‘iACH’ is more precise but lacks a reference to its origins). The 2.0 denotes the second generation rendering of the ACH; coincidentally it also signifies a tandem, iAβ-anchored cascade occurrence: sufficiently accumulated AβPP-derived iAβ triggers a relatively benign cascade capable of activating the AβPP-independent iAβ-generating pathway, which initiates the second, devastating cascade that is driven by the AD Engine, includes tau pathology, and ultimately leads to the neuronal loss. The ACH2.0 retains the centrality of Aβ in AD, the main premise of the ACH. Moreover, in both ACH versions Aβ is postulated to initiate cascades, albeit of different mechanistic nature, eventually leading to neuronal loss. What differ are the location of the ‘problematic’ Aβ (iAβ in the ACH2.0, rather than extracellular Aβ in the ACH), and the principal modes of its generation in the disease: the AβPP-independent pathway in the ACH2.0, rather than and in addition to AβPP proteolysis, the presumed sole Aβ source in the ACH. The activation of the AβPP-independent iAβ generation pathway is mediated by a life-long intracellular accumulation of AβPP-derived iAβ, accomplished, in part, by conversion of some of extracellular Aβ into iAβ, to over-the-threshold levels, a process which serves as the Starter Motor that elicits the integrated stress response and triggers the activation of AβPP-independent production of iAβ (a proposition directly testable in human neuronal cells AD model system as described above). The bulk of, if not the entire Aβ output produced in the AβPP-independent pathway is retained intraneuronally, i.e., is created as iAβ, and perpetuates the operation of this pathway. These mutual feedback cycles constitute the self-propagating Engine that drives AD and ultimately kills its host neuronal cells; its operation renders the AβPP proteolytic pathway of Aβ production irrelevant to the progression of the disease (because in this context its contribution to the iAβ pool becomes insignificant) and marks its manipulation for therapeutic purposes, such as BACE inhibition, as futile. In the proposed AD paradigm, the only valid direct therapeutic strategy is pursuing the components of the AD Engine, and the most effective approach appears to be the activation of BACE1 and/or BACE2 with the aim of exploiting their Aβ-cleaving activities (“and”, rather than “or”, would be the most efficient tactic: the activities in question are mutually supplementary due to diverse cleavage sites and also complementary because of their discrete subcellular localization). This would collapse the iAβ population and reset its levels below those required for the operation of the AD Engine. In fact, any sufficiently specific iAβ-depleting treatment would be equally effective in achieving this objective. Such treatment could be efficiently employed curatively, at symptomatic stages of the disease, or preventively, prior to the manifestation of AD symptoms. Astonishingly, this approach opens up the possibility of a short-duration, once-in-a-lifetime only or at least very infrequent, curative or preventive therapy for AD; this therapy would be, potentially, also effective for the prevention or treatment of the ‘common’ pervasive aging-associated cognitive decline. If the infrequent nature of such treatment is indeed the case, the Big Pharma’s enthusiasm to commit necessary investments could be limited; it would fall, then, mostly to academic researchers to develop effective iAβ-depleting drugs.
ACKNOWLEDGMENTS
The authors are grateful to Dr. Bjorn R. Olsen (Harvard Medical School) for his support, for critical reading of the manuscript, and for help with the preparation of the figures.
FUNDING
This work was supported by grant from the National Institutes of Health (R21 GM056179; R01 AR036819).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Egan MF , Kost J , Tariot PN , Aisen PS , Cummings JL , Vellas B , Sur C , Mukai Y , Voss T , Furtek C , Mahoney E , Harper Mozley L , Vandenberghe R , Mo Y , Michelson D ((2018) ) Randomized trials of verubecestat for mild-to-moderate Alzheimer’s disease, N Engl J Med 378: , 1691–1703. |
[2] | Egan MF , Kost J , Voss T , Mukai Y , Aisen PS , Cummings JL , Tariot PN , Vellas B , van Dyck CH , Boada M , Zhang Y , Li W , Furtek C , Mahoney E , Harper Mozley L , Mo Y , Sur C , Michelson D ((2019) ) Randomized trial of verubecestat for prodromal Alzheimer’s disease, N Engl J Med 380: , 1408–1420. |
[3] | Keskin AD , Kekus M , Adelsberger H , Neumann U , Shimshek DR , Song B , Zott B , Peng T , Förstl H , Staufenbiel M , Nelken I , Sakmann B , Konnerth A , Busche MA ((2017) ) BACE inhibition-dependent repair of Alzheimer’s pathophysiology, Proc Natl Acad Sci U S A 114: , 8631–8636. |
[4] | Hu X , Das B , Hou H , He W , Yan R ((2018) ) BACE1 deletion in the adult mouse reverses preformed amyloid deposition and improves cognitive functions, J Exp Med 215: , 927–940. |
[5] | Kennedy ME , Stamford AW , Chen X , Cox K , Cumming JN , Dockendorf MF , Egan M , Ereshefsky L , Hodgson RA , Hyde LA , Jhee S , Kleijn HJ , Kuvelkar R , Li W , Mattson BA , Mei H , Palcza J , Scott JD , Tanen M , Troyer MD , Tseng JL , Stone JA , Parker EM , Forman MS ((2016) ) The BACE1 inhibitor verubecestat (MK-8931) reduces CNS β-amyloid in animal models and in Alzheimer’s disease patients, Sci Transl Med 8: , 363ra150. |
[6] | Volloch V , Rits-Volloch S ((2021) ) Alzheimer’s disease is driven by beta-amyloid generated in the amyloid precursor protein-independent pathway and retained intraneuronally: Research and therapeutic strategies in a new AD paradigm, Ann Integr Mol Med 2: , 1010. |
[7] | Casas C , Sergeant N , Itier JM , Blanchard V , Wirths O , van der Kolk N , Vingtdeux V , van de Steeg E , Ret G , Canton T , Drobecq H , Clark A , Bonici B , Delacourte A , Benavides J , Schmitz C , Tremp G , Bayer TA , Benoit P , Pradier L ((2004) ) Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Abeta42 accumulation in a novel Alzheimer transgenic model, Am J Pathol 165: , 1289–1300. |
[8] | Bayer TA , Wirths O ((2008) ) Review on the APP/PS1KI mouse model: Intraneuronal Abeta accumulation triggers axonopathy, neuron loss and working memory impairment. (Suppl), Genes Brain Behav 1: , 6–11. |
[9] | Bayer TA , Breyhan H , Duan K , Rettig J , Wirths O ((2008) ) Intraneuronal beta-amyloid is a major risk factor–novel evidence from the APP/PS1KI mouse model, Neurodegener Dis 5: , 140–142. |
[10] | Wirths O , Breyhan H , Cynis H , Schilling S , Demuth HU , Bayer TA ((2009) ) Intraneuronal pyroglutamate-Abeta 3-42 triggers neurodegeneration and lethal neurological deficits in a transgenic mouse model, Acta Neuropathol 118: , 487–496. |
[11] | Christensen DZ , Bayer TA , Wirths O ((2010) ) Intracellular Aβ triggers neuron loss in the cholinergic system of the APP/PS1KI mouse model of Alzheimer’s disease, Neurobiol Aging 31: , 1153–1163. |
[12] | Christensen DZ , Schneider-Axmann T , Lucassen PJ , Bayer TA , Wirths O ((2010) ) Accumulation of intraneuronal Abeta correlates with ApoE4 genotype, Acta Neuropathol 119: , 555–566. |
[13] | Bayer TA , Wirths O ((2010) ) Intracellular accumulation of amyloid-Beta - a predictor for synaptic dysfunction and neuron loss in Alzheimer’s disease, Front Aging Neurosci 2: , 8. |
[14] | Bayer TA , Wirths O ((2011) ) Intraneuronal Aβ as a trigger for neuron loss: Can this be translated into human pathology? , Biochem Soc Trans 39: , 857–861. |
[15] | Wirths O , Bayer TA ((2012) ) Intraneuronal Aβ accumulation and neurodegeneration: Lessons from transgenic models, Life Sci 91: , 1148–1152. |
[16] | Kumar S , Wirths O , Theil S , Gerth J , Bayer TA , Walter J ((2013) ) Early intraneuronal accumulation and increased aggregation of phosphorylated Abeta in a mouse model of Alzheimer’s disease, Acta Neuropathol 125: , 699–709. |
[17] | Ripoli C , Cocco S , Li Puma DD , Piacentini R , Mastrodonato A , Scala F , Puzzo D , D’Ascenzo M , Grassi C ((2014) ) Intracellular accumulation of amyloid-β (Aβ) protein plays a major role in Aβ-induced alterations of glutamatergic synaptic transmission and plasticity, J Neurosci 34: , 12893–12903. |
[18] | Scala F , Fusco S , Ripoli C , Piacentini R , Li Puma DD , Spinelli M , Laezza F , Grassi C , D’Ascenzo M ((2015) ) Intraneuronal Aβ accumulation induces hippocampal neuron hyperexcitability through A-type K(+) current inhibition mediated by activation of caspases and GSK-3, Neurobiol Aging 36: , 886–900. |
[19] | Christensen DZ , Kraus SL , Flohr A , Cotel MC , Wirths O , Bayer TA ((2008) ) Transient intraneuronal Abeta rather than extracellular plaque pathology correlates with neuron loss in the frontal cortex of APP/PS1KI mice, Acta Neuropathol 116: , 647–655. |
[20] | Chafekar S , Baas F , Scheper W ((2008) ) Oligomer-specific amyloid-beta toxicity in cell models is mediated by selective uptake, Biochem Biophys Acta 9: , 523–531. |
[21] | Wesen E , Jeffries G , Dzebo M , Esbjorner M ((2017) ) Endocytic uptake of monomeric amyloid-β peptides is clathrin- and dynamin-independent and results in selective accumulation of Aβ(1–42) compared to Aβ(1–40), Sci Rep 7: , 2021. |
[22] | Kumar-Singh S , Theuns J , Van Broeck B , Pirici D , Vennekens K , Corsmit E , Cruts M , Dermaut B , Wang R , Van Broeckhoven C ((2006) ) Mean age-of-onset of familial Alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40, Hum Mutat 27: , 686–695. |
[23] | Hu X , Crick SL , Bu G , Frieden C , Pappu RV , Lee JM ((2009) ) Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide, Proc Natl Acad Sci U S A 106: , 20324–20329. |
[24] | Yajima R , Tokutake T , Koyama A , Kasuga K , Tezuka T , Nishizawa M , Ikeuchi T ((2015) ) ApoE-isoform-dependent cellular uptake of amyloid-β is mediated by lipoprotein receptor LR11/SorLA, Biochem Biophys Res Commun 456: , 482–488. |
[25] | Omtri RS , Davidson MW , Arumugam B , Poduslo JF , Kandimalla KK ((2012) ) Differences in the cellular uptake and intracellular itineraries of amyloid beta proteins 40 and 42: Ramifications for the Alzheimer’s drug discovery, Mol Pharmaceutics 9: , 1887. |
[26] | Bu G , Cam J , Zerbinatti C ((2006) ) LRP in amyloid-β production and metabolism, Ann NY Acad Sci 1086: , 35–53. |
[27] | Wang HY , Lee DH , D’Andrea MR , Peterson PA , Shank RP , Reitz AB ((2000) ) beta-Amyloid(1-42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer’s disease pathology, J Biol Chem 275: , 5626–5632. |
[28] | Nagele R , D’Andrea M , Anderson W , Wang H ((2002) ) Intracellular accumulation of Aβ42 in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease, Neuroscience 110: , 199–211. |
[29] | Oddo S , Caccamo A , Green KN , Liang K , Tran L , Chen Y , Leslie FM , LaFerla FM ((2005) ) Chronic nicotine administration exacerbates tau pathology in a transgenic model of Alzheimer’s disease, Proc Natl Acad Sci U S A 102: , 3046–3051. |
[30] | Yan SD , Chen X , Fu J , Chen M , Zhu H , Roher A , Slattery T , Zhao L , Nagashima M , Morser J , Migheli A , Nawroth P , Stern D , Schmidt AM ((1996) ) RAGE and amyloid-β peptide neurotoxicity in Alzheimer’s disease, Nature 382: , 685–691. |
[31] | Sasaki N , Toki S , Chowei H , Saito T , Nakano N , Hayashi Y , Takeuchi M , Makita Z ((2001) ) Immunohistochemical distribution of the receptor for advanced glycation end products in neurons and astrocytes in Alzheimer’s disease, Brain Res 888: , 256–262. |
[32] | Deane R , Du Yan S , Submamaryan RK , LaRue B , Jovanovic S , Hogg E , Welch D , Manness L , Lin C , Yu J , Zhu H , Ghiso J , Frangione B , Stern A , Schmidt AM , Armstrong DL , Arnold B , Liliensiek B , Nawroth P , Hofman F , Kindy M , Stern D , Zlokovic B ((2003) ) RAGE mediates amyloid-β peptide transport across the blood–brain barrier and accumulation in brain, Nat Med 9: , 907–913. |
[33] | Iribarren P , Zhou Y , Hu J , Le Y , Wang J , ((2005) ) Role of formyl peptide receptor-like 1 (FPRL1/FPR2) in mononuclear phagocyte responses in Alzheimer disease, Immunol Res 31: , 165–176. |
[34] | Snyder EM , Nong Y , Almeida CG , Paul S , Moran T , Choi EY , Nairn AC , Salter MW , Lombroso PJ , Gouras GK , Greengard P ((2005) ) Regulation of NMDA receptor trafficking by amyloid-β, Nat Neurosci 8: , 1051–1058. |
[35] | LaFerla F , Green K , Oddo S ((2007) ) Intracellular amyloid-beta in Alzheimer’s disease, Nat Rev Neurosci 8: , 499–509. |
[36] | Kinoshita A , Fukumoto H , Shah T , Whelan CM , Irizarry MC , Hyman BT ((2003) ) Demonstration by FRET of BACE interaction with the amyloidprecursor protein at the cell surface and in early endosomes, JCell Sci 116: , 3339–3346. |
[37] | Xu H , Greengard P , Gandy S ((1995) ) Regulated formation of Golgi secretory vesicles containing Alzheimer β-amyloid precursor protein, J Biol Chem 270: , 23243–23245. |
[38] | Mizuguchi M , Ikeda K , Kim S ((1992) ) Differential distribution of cellular forms of β-amyloid precursor protein in murine glial cell cultures, Brain Res 584: , 219–225. |
[39] | Cook DG , Forman MS , Sung JC , Leight S , Kolson DL , Iwatsubo T , Lee VM , Doms RW ((1997) ) Alzheimer’s Aβ42 is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells, Nat Med 3: , 1021–1023. |
[40] | Hartmann T , Bieger SC , Brühl B , Tienari PJ , Ida N , Allsop D , Roberts GW , Masters CL , Dotti CG , Unsicker K , Beyreuther K ((1997) ) Distinct sites of intracellular production for Alzheimer’s disease Aβ40/42 amyloid peptides, Nat Med 3: , 1016–1020. |
[41] | Wild-Bode C , Yamazaki T , Capell A , Leimer U , Steiner H , Ihara Y , Haass C ((1997) ) Intracellular generation and accumulation of amyloid beta-peptide terminating at amino acid 42, J Biol Chem 272: , 16085–16088. |
[42] | Lee SJ , Liyanage U , Bickel PE , Xia W , Lansbury PT Jr , Kosik KS ((1998) ) A detergent-insoluble membrane compartment contains Aβ, Nat Med 4: , 730–734. |
[43] | Skovronsky D , Doms R , Lee V ((1998) ) Detection of a novel intraneuronal pool of insoluble amyloid β protein that accumulates with time in culture, J Cell Biol 141: , 1031–1039. |
[44] | Manczak M , Anekonda TS , Henson E , Park BS , Quinn J , Reddy PH ((2006) ) Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: Implications for free radical generation and oxidative damage in disease progression, Hum Mol Genet 15: , 1437–1449. |
[45] | Martin B , Schrader-Fischer G , Busciglio J , Duke M , Paganetti P , Yankner B ((1995) ) Intracellular accumulation of beta-amyloid in cells expressing the Swedish mutant amyloid precursor protein, J Biol Chem 270: , 26727–26730. |
[46] | Sannerud R , Esselens C , Ejsmont P , Mattera R , Rochin L , Tharkeshwar AK , De Baets G , De Wever V , Habets R , Baert V , Vermeire W , Michiels C , Groot AJ , Wouters R , Dillen K , Vints K , Baatsen P , Munck S , Derua R , Waelkens E , Basi GS , Mercken M , Vooijs M , Bollen M , Schymkowitz J , Rousseau F , Bonifacino JS , Van Niel G , De Strooper B , Annaert W ((2016) ) Restricted location of PSEN2/gamma-secretase determines substrate specificity and generates an intracellular Abeta pool, Cell 166: , 193–208. |
[47] | Chang RC , Suen KC , Ma CH , Elyaman W , Ng HK , Hugon J ((2002) ) Involvement of double-stranded RNA-dependent protein kinase and phosphorylation of eukaryotic initiation factor-2alpha in neuronal degeneration, J Neurochem 83: , 1215–1225. |
[48] | Peel AL ((2004) ) PKR activation in neurodegenerative disease, J Neuropathol Exp Neurol 63: , 97–105. |
[49] | Peel AL , Bredesen DE ((2003) ) Activation of the cell stress kinase PKR in Alzheimer’s disease and human amyloid precursor protein transgenic mice, Neurobiol Dis 14: , 52–62. |
[50] | Chang RC , Wong AK , Ng HK , Hugon J ((2002) ) Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer’s disease, Neuroreport 13: , 2429–2432. |
[51] | Onuki R , Bando Y , Suyama E , Katayama T , Kawasaki H , Baba T , TohyamaM , Taira K ((2004) ) An RNA-dependent protein kinase is involved intunicamycin-induced apoptosis and Alzheimer’s disease., EMBO J 23: , 959–968. |
[52] | Lourenco MV , Clarke JR , Frozza RL , Bomfim TR , Forny-Germano L , Batista AF , Sathler LB , Brito-Moreira J , Amaral OB , Silva CA , Freitas-Correa L , Espírito-Santo S , Campello-Costa P , Houzel JC , Klein WL , Holscher C , Carvalheira JB , Silva AM , Velloso LA , Munoz DP , Ferreira ST , De Felice FG ((2013) ) TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys, Cell Metab 18: , 831–843. |
[53] | Paquet C , Mouton-Liger F , Meurs EF , Mazot P , Bouras C , Pradier L , Gray F , Hugon J ((2012) ) The PKR activator PACT is induced by Abeta: Involvement in Alzheimer’s disease, Brain Pathol 22: , 219–229. |
[54] | Pakos-Zebrucka K , Koryga I , Mnich K , Ljujic M , Samali A , Gorman AM ((2016) ) The integrated stress response, EMBO Rep 17: , 1374–1395. |
[55] | Ron D ((2002) ) Translational control in the endoplasmic reticulum stress response, J Clin Invest 110: , 1383–1388. |
[56] | Harding HP , Zhang Y , Zeng H , Novoa I , Lu PD , Calfon M , Sadri N , Yun C , Popko B , Paules R , Stojdl DF , Bell JC , Hettmann T , Leiden JM , Ron D ((2003) ) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress, Mol Cell 11: , 619–633. |
[57] | Brostrom CO , Prostko CR , Kaufman RJ , Brostrom MA ((1996) ) Inhibition of translational initiation by activators of the glucose-regulated stress protein and heat shock protein stress response systems. Role of the interferon-inducible double-stranded RNA-activated eukaryotic initiation factor 2alpha kinase, J Biol Chem 271: , 24995–25002. |
[58] | Dever TE , Feng L , Wek RC , Hinnebusch AG ((1992) ) Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast, Cell 68: , 585–596. |
[59] | Wek RC , Jiang HY , Anthony TG ((2006) ) Coping with stress: eIF2 kinases and translational control, Biochem Soc Trans 34: , 7–11. |
[60] | Rzymski T , Milani M , Pike L , Buffa F , Mellor HR , Winchester L , Pires I , Hammond E , Ragoussis I , Harris AL ((2010) ) Regulation of autophagy by ATF4 in response to severe hypoxia, Oncogene 29: , 4424–4435. |
[61] | Ye J , Kumanova M , Hart LS , Sloane K , Zhang H , De Panis DN , Bobrovnikova-Marjon E , Diehl JA , Ron D , Koumenis C ((2010) ) The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation, EMBO J 29: , 2082–2096. |
[62] | Garcia MA , Meurs EF , Esteban M ((2007) ) The dsRNA protein kinase PKR: Virus and cell control, Biochimie 89: , 799–811. |
[63] | Harding HP , Zhang Y , Ron D ((1999) ) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase, Nature 397: , 271–274. |
[64] | Zhu X , Perry G , Moreira PI , Aliev G , Cash AD , Hirai K , Smith MA ((2006) ) Mitochondrial abnormalities and oxidative imbalance in Alzheimer disease, J Alzheimers Dis 9: , 147–153. |
[65] | Blass JP ((2000) ) The mitochondrial spiral. An adequate cause of dementia in the Alzheimer’s syndrome, Ann N Y Acad Sci 92: , 170–183. |
[66] | Manczak M , Park BS , Jung Y , Reddy PH ((2004) ) Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: Implications for early mitochondrial dysfunction and oxidative damage, Neuromolecular Med 5: , 147–162. |
[67] | Qin W , Haroutunian V , Katsel P , Cardozo CP , Ho L , Buxbaum JD , Pasinetti GM ((2009) ) PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia, Arch Neurol 66: , 352–361. |
[68] | Du H , Guo L , Yan S , Sosunov AA , McKhann GM , Yan SS ((2010) ) Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model, Proc Natl Acad Sci U S A 107: , 18670–18675. |
[69] | Lin MT , Simon DK , Ahn CH , Kim LM , Beal MF ((2002) ) High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer’s disease brain, Hum Mol Genet 11: , 133–145. |
[70] | Calkins M , Manczak M , Mao P , Shirendeb U , Reddy PH ((2011) ) Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease, Hum Mol Genet 20: , 4515–4529. |
[71] | Anandatheerthavarada HK , Biswas G , Robin MA , Avadhani NG ((2003) ) Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells, J Cell Biol 161: , 41–54. |
[72] | Caspersen C , Wang N , Yao J , Sosunov A , Chen X , Lustbader JW , Xu HW , Stern D , McKhann G , Yan SD ((2005) ) Mitochondrial Abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease, FASEB J 19: , 2040–2041. |
[73] | Chen JX , Yan SS ((2010) ) Role of mitochondrial amyloid-beta in Alzheimer’s disease, J Alzheimers Dis 20: (Suppl 2), 569–578. |
[74] | Hansson Petersen CA , Alikhani N , Behbahani H , Wiehager B , Pavlov PF , Alafuzoff I , Leinonen V , Ito A , Winblad B , Glaser E , Ankarcrona M ((2008) ) The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae, Proc Natl Acad Sci U S A 105: , 13145–13150. |
[75] | de la Monte SM , Luong T , Neely TR , Robinson D , Wands JR ((2000) ) Mitochondrial DNA damage as a mechanism of cell loss in Alzheimer’s disease, Lab Invest 80: , 1323–1335. |
[76] | Brooks WM , Lynch PJ , Ingle CC , Hatton A , Emson PC , Faull RL , Starkey MP ((2007) ) Gene expression profiles of metabolic enzyme transcripts in Alzheimer’s disease, Brain Res 1127: , 127–135. |
[77] | Wang X , Perry G , Smith MA , Zhu X ((2007) ) Amyloid-beta-derived diffusible ligands cause impaired axonal transport of mitochondria in neurons, Neurodegener Dis 7: , 56–59. |
[78] | Wang X , Su B , Fujioka H , Zhu X ((2008) ) Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients, J Pathol 173: , 470–482. |
[79] | Wang X , Su B , Lee HG , Li X , Perry G , Smith MA , Zhu X ((2009) ) Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease, J Neurosci 29: , 9090–9103. |
[80] | Wang X , Su B , Siedlak SL , Moreira PI , Fujioka H , Wang Y , Casadesus G , Zhu X ((2008) ) Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins, Proc Natl Acad Sci U S A 105: , 19318–19323. |
[81] | Guo X , Aviles G , Liu Y , Tian R , Unger BA , Lin YT , Wiita AP , Xu K , Correia MA , Kampmann M ((2020) ) Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway, Nature 579: , 427–432. |
[82] | Fessler E , Eckl EM , Schmitt S , Mancilla IA , Meyer-Bender MF , Hanf M , Philippou-Massier J , Krebs S , Zischka H , Jae LT ((2020) ) A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol, Nature 579: , 433–437. |
[83] | Volloch V , Schweitzer B , Rits S ((1996) ) Antisense globin RNA in murine erythroid tissues: Structure, origin and possible function, Proc Natl Acad Sci U S A 93: , 2476–2481. |
[84] | Volloch V ((2019) ) Protein-encoding RNA-to-RNA information transfer in mammalian cells: Principles of RNA-dependent mRNA amplification, Ann Integr Mol Med 1: , 1002. |
[85] | Rits S , Olsen B , Volloch V ((2019) ) Protein-encoding RNA to RNA information transfer in mammalian cells: RNA-dependent mRNA amplification. Identification of chimeric RNA intermediates and putative RNA end products, Ann Integr Mol Med 1: , 1003. |
[86] | Volloch V , Rits S , Olsen B ((2019) ) RNA-dependent amplification of mammalian mRNA encoding extracellullar matrix proteins: Identification of chimeric RNA intermediates for α1, β1, and γ1 chains of laminin, Ann Integr Mol Med 1: , 1004. |
[87] | Volloch V ((1996) ) A mechanism for β-amyloid overproduction in Alzheimer’s disease: Precursor-independent generation of β-amyloid via antisense RNA-primed mRNA synthesis, FEBS Lett 390: , 124–128. |
[88] | Volloch V (1997) Mechanism for β-amyloid overproduction in sporadic Alzheimer’s Disease: Possible antisense RNA-mediated generation of a 5’-truncated ßAPP mRNA encoding 12 kDa C-terminal fragment of βAPP, the immediate precursor of Aβ. In, Wasco W, Tanzi R, eds. New York: Springer Science+Business Media, pp, 45 –56Molecular Mechanisms of Dementia. |
[89] | Volloch V ((1997) ) Possible mechanism for resistance to Alzheimer’s disease (AD) in mice suggests new approach to generate a mouse model for sporadic AD and may explain familial resistance to AD in man, Exp Neurobiol 144: , 214–218. |
[90] | Volloch V , Rits S ((2018) ) Results of beta-secretase-inhibitor clinical trials support amyloid precursor protein-independent generation of beta amyloid in sporadic Alzheimer’s disease, (Basel) 6: , 45. |
[91] | Volloch V , Olsen B , Rits S ((2019) ) Precursor-independent overproduction of beta-amyloid in AD: Mitochondrial dysfunction as possible initiator of asymmetric RNA-dependent βAPP mRNA amplification. An engine that drives Alzheimer’s disease, Ann Integr Mol Med 1: , 1005. |
[92] | Volloch V , Olsen B , Rits S ((2020) ) AD “Statin”: Alzheimer’s disorder is a “fast” disease preventable by therapeutic intervention initiated even late in life and reversible at the early stages, Ann Integr Mol Med 2: , 1006. |
[93] | Volloch V , Olsen B , Rits S ((2020) ) Alzheimer’s disease is driven by intraneuronally retained beta-amyloid produced in the AD-specific, βAPP-independent pathway: Current perspective and experimental models for tomorrow, Ann Integr Mol Med 2: , 1007. |
[94] | Volloch V , Olsen B , Rits S ((2020) ) Alzheimer’s disease prevention and treatment: Case for optimism, Ann Integr Mol Med 2: , 1008. |
[95] | Breimer L , Denny P ((1987) ) Alzheimer amyloid aspects, Nature 326: , 749–750. |
[96] | Kang J , Lemaire HG , Unterbeck A , Salbaum JM , Masters CL , Grzeschik KH , Multhaup G , Beyreuther K , Müller-Hill B ((1987) ) The precursor of Alzheimer’s disease amyloid A4 protein resembles a cell-surface receptor, Nature 325: , 733–736. |
[97] | Robakis NK , Ramakrishna N , Wolfe G , Wisniewski HM ((1987) ) Molecular cloning and characterization of a cDNA encoding the cerebrovascular and the neuritic plaque amyloid peptides, Proc Natl Acad Sci U S A 84: , 4190–4194. |
[98] | Tanzi RE , Gusella JF , Watkins PC , Bruns GA , St George-Hyslop P , Van Keuren ML , Patterson D , Pagan S , Kurnit DM , Neve RL ((1987) ) Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus, Science 235: , 880–884. |
[99] | Henley DB , Sundell KL , Sethuraman G , Dowsett SA , May PC ((2014) ) Safety profile of semagacestat, a gamma-secretase inhibitor: IDENTITY trial findings, Curr Med Res 30: , 2021–2032. |
[100] | Tagami S , Yanagida K , Kodama TS , Takami M , Mizuta N , Oyama H , Nishitomi K , Chiu YW , Okamoto T , Ikeuchi T , Sakaguchi G , Kudo T , Matsuura Y , Fukumori A , Takeda M , Ihara Y , Okochi M ((2017) ) Semagacestat is a pseudo-inhibitor of γ-secretase, Rep 21: , 259–273. |
[101] | Imbimbo B , Giardina G ((2011) ) γ-secretase inhibitors and modulators for the treatment of Alzheimer’s disease: Disappointments and hopes, Curr Top Med Chem 11: , 1555–1570. |
[102] | Zhao J , Liu X , Xia W ((2020) ) Targeting amyloidogenic processing of APP in Alzheimer’s disease, Front Mol Neurosci 13: , 137. |
[103] | Mekala S , Nelson G , Li Y ((2020) ) Recent developments of small molecule γ-secretase modulators for Alzheimer’s disease, RSC Med Chem 11: , 1003–1022. |
[104] | Rynearson KD , Ponnusamy M , Prikhodko O , Xie Y , Zhang C , Nguyen P , Hug B , Sawa M , Becker A , Spencer B , Florio J , Mante M , Salehi B , Arias C , Galasko D , Head BP , Johnson G , Lin JH , Duddy SK , Rissman RA , Mobley WC , Thinakaran G , Tanzi RE , Wagner SL ((2021) ) Preclinical validation of a potent γ-secretase modulator for Alzheimer’s disease prevention, J Exp Med 218: , e20202560. |
[105] | Jäger S , Leuchtenberger S , Martin A , Czirr E , Wesselowski J , Dieckmann M , Waldron E , Korth C , Koo EH , Heneka M , Weggen S , Pietrzik CU ((2009) ) Alpha-secretase mediated conversion of the amyloid precursor protein derived membrane stub C99 to C83 limits Aβ generation, J Neurochem 6: , 1369–1382. |
[106] | Kuhn PH , Wang H , Dislich B , Colombo A , Zeitschel U , Ellwart JW , Kremmer E , Rossner S , Lichtenthaler SF ((2010) ) ADAM10 is the physiologically relevant, constitutive α-secretase of the amyloid precursor protein in primary neurons, EMBO J 29: , 3020–3032. |
[107] | Lichtenthaler S ((2011) ) Alpha-secretase in Alzheimer’s disease, J Neurochem 116: , 10–21. |
[108] | Postina R ((2012) ) Activation of alpha secretase cleavage, J Neurochem 120: , 46–54. |
[109] | Endres K , Deller T ((2017) ) Regulation of alpha-secretase ADAM10 and: Genetic, epigenetic, and protein-based mechanisms, Front Mol Neurosci 10: , 56. |
[110] | Ray B , Maloney B , Sambamurti K , Karnati HK , Nelson PT , Greig NH , Lahiri DK ((2020) ) Rivastigmine modifies the α-secretase pathway and potentially early Alzheimer’s disease, Transl Psychiatry 10: , 47. |
[111] | Fahrenholz F ((2007) ) Alpha-secretase as a therapeutic target, Curr Alzheimer Res 4: , 412–417. |
[112] | Postina R , Schroeder A , Dewachter I , Bohl J , Schmitt U , Kojro E , Prinzen C , Endres K , Hiemke C , Blessing M , Flamez P , Dequenne A , Godaux E , van Leuven F , Fahrenholz F ((2004) ) A disintegrin- metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model, J Clin Invest 113: , 1456–1464. |
[113] | MacLeod R , Hillert E , Cameron R , Baillie G ((2015) ) The role and therapeutic targeting of α-, β- and γ-secretase in Alzheimer’s disease., Future Sci OA 1: , FSO11. |
[114] | Prinzen C , Trumbach D , Wurst W , Endres K , Postina R , Fahrenholz F ((2009) ) Differential gene expression in ADAM10 and mutant ADAM10 transgenic mice, BMC Genomics 10: , 66. |
[115] | Jorissen E , Prox J , Bernreuther C , Weber S , Schwanbeck R , Serneels L , Snellinx A , Craessaerts K , Thathiah A , Tesseur I , Bartsch U , Weskamp G , Blobel CP , Glatzel M , De Strooper B , Saftig P ((2010) ) The disintegrin/metal-loproteinase ADAM10 is essential for the establishment of the brain cortex, J Neurosci 30: , 4833–4844. |
[116] | Huse J , Liu K , Pijak D , Carlin D , Lee V , Doms R ((2002) ) Beta-secretase processing in the trans-Golgi network preferentially generates truncated amyloid species that accumulate in Alzheimer’s disease brain, J Biol Chem 277: , 16278–16284. |
[117] | Liu K , Doms R , Lee M ((2002) ) Glu11 site cleavage and N-terminally truncated A beta production upon BACE overexpression, Biochemistry 41: , 3128–3136. |
[118] | Lee EB , Zhang B , Liu K , Greenbaum EA , Doms RW , Trojanowski JQ , Lee VM ((2005) ) BACE overexpression alters the subcellular processing of APP and inhibits Aβ deposition, J Cell Biol 168: , 291–302. |
[119] | Kimura A , Hata S , Suzuki T ((2016) ) Alternative selection of beta-site APP-cleaving enzyme 1 (BACE1) cleavage sites in amyloid beta-protein precursor (APP) harboring protective and pathogenic mutations within the Abeta sequence, J Biol Chem 291: , 24041–24053. |
[120] | Fluhrer R , Multhaup G , Schlicksupp A , Okochi M , Takeda M , Lammich S , Willem M , Westmeyer G , Bode W , Walter J , Haass ((2003) ) Identification of a beta-secretase activity, which truncates amyloid beta-peptide after its presenilin-dependent generation, J Biol Chem 278: , 5531–5538. |
[121] | Shi XP , Tugusheva K , Bruce JE , Lucka A , Wu GX , Chen-Dodson E , Price E , Li Y , Xu M , Huang Q , Sardana MK , Hazuda DJ ((2003) ) Beta-secretase cleavage at amino acid residue 34 in the amyloid beta peptide is dependent upon gamma-secretase activity, J Biol Chem 278: , 21286–21294. |
[122] | Liebsch F , Kulic L , Teunissen C , Shobo A , Ulku I , Engelschalt V , Hancock MA , van der Flier WM , Kunach P , Rosa-Neto P , Scheltens P , Poirier J , Saftig P , Bateman RJ , Breitner J , Hock C , Multhaup G ((2019) ) Abeta34 is a BACE1-derived degradation intermediate associated with amyloid clearance and Alzheimer’s disease progression, Nat Commun 10: , 2240. |
[123] | Garcia-Gonzales L , Pilat D , Rivera S ((2019) ) Emerging alternative proteinases in APP metabolism and Alzheimer’s disease pathogenesis, Front Aging Neurosci 11: , 244. |
[124] | Farzan M , Schnitzler CE , Vasilieva N , Leung D , Choe H ((2000) ) BACE2,a β-secretase homolog, cleaves at the β site andwithin the amyloid-β region of the amyloid-βprecursor protein, Proc Natl Acad Sci U S A 97: , 9712–9717. |
[125] | Yan R , Munzner J , Shuck M , Bienkowski M ((2001) ) BACE2 functions as an alternative alpha secretase in cells, J Biol Chem 276: , 34019–34027. |
[126] | Basi G , Frigon N , Barbour R , Doan T , Gordon G , McConlogue L , Sinha S , Zeller M ((2003) ) Antagonistic effects of beta-site amyloid precursor protein-cleaving enzymes 1 and 2 on beta-amyloid peptide production in cells, J Biol Chem 278: , 31512–31520. |
[127] | Varshavsky A ((2011) ) The N-end rule pathway and regulation by proteolysis, Protein Sci 20: , 1298–1345. |
[128] | Wingfield P ((2017) ) N-terminal methionine processing, Curr Protoc Protein Sci 88: , 6.14.1–6.14.3. |
[129] | Choi SH , Kim YH , Hebisch M , Sliwinski C , Lee S , D’Avanzo C , Chen H , Hooli B , Asselin C , Muffat J , Klee JB , Zhang C , Wainger BJ , Peitz M , Kovacs DM , Woolf CJ , Wagner SL , Tanzi RE , Kim DY ((2014) ) A three-dimensional human neural cell culture model of Alzheimer’s disease, Nature 515: , 274–278. |
[130] | Kearse M , Wilusz J ((2017) ) Non-AUG translation: A new start for protein synthesis in eukaryotes, Genes Dev 31: , 1717–1731. |
[131] | Citron M , Haass C , Selkoe D ((1993) ) Production of amyloid beta peptide by cultured cells: No evidence for internal initiation of translation at Met596, Neurobiol Aging 14: , 571–573. |
[132] | Macq A , Philippe B , Octave N ((1994) ) The amyloid peptide of Alzheimer’s disease is not produced by internal initiation of translation generating C-terminal amyloidogenic fragments of its precursor, Neurosci Lett 182: , 227–230. |
[133] | Jonsson T , Atwal JK , Steinberg S , Snaedal J , Jonsson PV , Bjornsson S , Stefansson H , Sulem P , Gudbjartsson D , Maloney J , Hoyte K , Gustafson A , Liu Y , Lu Y , Bhangale T , Graham RR , Huttenlocher J , Bjornsdottir G , Andreassen OA , Jönsson EG , Palotie A , Behrens TW , Magnusson OT , Kong A , Thorsteinsdottir U , Watts RJ , Stefansson K ((2012) ) A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline, Nature 488: , 96–99. |
[134] | Harper AR , Nayee S , Topol EJ ((2015) ) Protective alleles and modifier variants in human health and disease, Nat Rev Genet 16: , 689–701. |
[135] | Katzman R , Terry R , DeTeresa R , Brown T , Davies P , Fuld P , Rebing X , Peck A ((1988) ) Clinical, pathological, and neurochemical changes in dementia: A subgroup with preserved mental status and numerous neocortical plaques, Ann Neurol 23: , 138–144. |
[136] | Delaère P , Duyckaerts C , Masters C , Beyreuther K , Piette F , Hauw J ((1990) ) Large amounts of neocortical beta A4 deposits without neuritic plaques nor tangles in a psychometrically assessed, non-demented person, Neurosci Lett 116: , 87–93. |
[137] | Dickson D , Crystal H , Mattiace L , Masur D , Blau A , Davies P , Yen S , Aronson M ((1992) ) Identification of normal and pathological aging in prospectively studied nondemented elderly humans, Neurobiol Aging 13: , 178–189. |
[138] | Aizenstein H , Nebes R , Saxton J , Price J , Mathis C , Tsopelas N , Ziolko S , James J , Snitz B , Houck P , Bi W , Cohen A , Lopresti B , DeKosky S , Halligan E , Klunk W ((2008) ) Frequent amyloid deposition without significant cognitive impairment among the elderly, Arch Neurol 65: , 1509–1517. |
[139] | Klunk W , Mathis C , Price J , DeKosky S , Lopresti B , Tsopelas N , Saxton J , Nebes R ((2009) ) Amyloid imaging with PET in Alzheimer’s disease, mild cognitive impairment, and clinically unimpaired subjects. In PET in the Evaluation of Alzheimer’s Disease and Related Disorders, Silverman D, ed. Springer, New York, pp. 119–147. |
[140] | Villemagne V , Pike K , Chételat G , Ellis K , Mulligan R , Bourgeat P , Ackermann U , Jones G , Szoeke C , Salvado O , Martins R , O’Keefe G , Mathis C.A , Klunk W , Ames D , Masters C , Rowe C ((2011) ) Longitudinalassessment of Aβ and cognition in aging and Alzheimerdisease, Ann Neurol 69: , 181–192. |
[141] | Seto M , Weiner RL , Dumitrescu L , Hohman TJ ((2021) ) Protective genes and pathways in Alzheimer’s disease: Moving towards precision interventions, Mol Neurodegen 16: , 29. |
[142] | Almeida C , Takahashi R , Gouras G ((2006) ) β-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin–proteasome system, J Neurosci 26: , 4277–4288. |
[143] | Gregori L , Fuchs C , Figueiredo-Pereira M , Van Nostrand W , Goldgaber D ((1995) ) Amyloid β-protein inhibits ubiquitin-dependent protein degradation, J Biol Chem 270: , 19702–19708. |
[144] | Oh S , Hong HS , Hwang E , Sim HJ , Lee W , Shin SJ , Mook-Jung I ((2005) ) Amyloid peptide attenuates the proteasome activity in neuronal cells, Mech Ageing Dev 126: , 1292–1299. |
[145] | Tseng B , Green K , Chan J , Blurton-Jones M , LaFerla F ((2008) ) Aβ inhibits the proteasome and enhances amyloid and tau accumulation, Neurobiol Aging 29: , 1607–1618. |
[146] | Volloch V , Rits-Volloch S ((2021) ) News from Mars: Two-tier paradox, intracellular PCR, chimeric junction shift, dark matter mRNA and other remarkable features of mammalian RNA-dependent mRNA amplification. Implications for Alzheimer’s disease, RNA-based vaccines and mRNA therapeutics, Ann Integr Mol Med 2: , 131–173. |
[147] | Hardy JA , Higgins GA ((1992) ) Alzheimer’s disease: The amyloid cascade hypothesis, Science 256: , 184–185. |


