
Building a Network of Adverse Outcome Pathways (AOPs) Incorporating the Tau-Driven AOP Toward Memory Loss (AOP429)
Abstract
The adverse outcome pathway (AOP) concept was first proposed as a tool for chemical hazard assessment facilitating the regulatory decision-making in toxicology and was more recently recommended during the BioMed21 workshops as a tool for the characterization of crucial endpoints in the human disease development. This AOP framework represents mechanistically based approaches using existing data, more realistic and relevant to human biological systems. In principle, AOPs are described by molecular initiating events (MIEs) which induce key events (KEs) leading to adverse outcomes (AOs). In addition to the individual AOPs, the network of AOPs has been also suggested to beneficially support the understanding and prediction of adverse effects in risk assessment. The AOP-based networks can capture the complexity of biological systems described by different AOPs, in which multiple AOs diverge from a single MIE or multiple MIEs trigger a cascade of KEs that converge to a single AO. Here, an AOP network incorporating a recently proposed tau-driven AOP toward memory loss (AOP429) related to sporadic (late-onset) Alzheimer’s disease is constructed. This proposed AOP network is an attempt to extract useful information for better comprehending the interactions among existing mechanistic data linked to memory loss as an early phase of sporadic Alzheimer’s disease pathology.
INTRODUCTION
During the last decade, new approaches for as-sessing chemical toxicity have been developed by regulatory and academic bodies following the recent technological advances. These new developments represent more mechanistically based approaches, shifting to more realistic and efficient strategies, relevant to human biological systems. Considering the technological advances, within the field of chemical risk assessment, applying more predictive approaches can facilitate the determination of more accurate endpoints. Subsequently, more cost-effective, accurate, and human relevant testing strategies can be applied [1, 2]. The concept of adverse outcome pathway (AOP) has been proposed to improve the human relevance of chemical toxicity testing based on available mechanistically relevant approaches using existing human relevant data, resulting in a better understanding of the adverse effects of exposure to various chemicals [3].
During the BioMed21 workshop, the application of AOPs has been suggested for mapping the perturbation of normal human physiology during disease development [4, 5]. Notably, the AOP concept has been already proposed for the characterization of crucial endpoints in the human pathogenesis of COVID-19 (https://ec.europa.eu/jrc/en/event/webinar/intro-webinar-ciao-project). In compliance with the Organisation for Economic Co-operation and Development (OECD), guidance on the development of the AOP concept is available, providing assistance on defining, constructing, and assessing the development of AOPs [6]. An AOP is characterized by a linear series of events, including a molecular initiating event (MIE) triggered by a stressor, which directly interacts with a biological target at molecular level, followed by intermediate key events (KEs) at cellular level, and ultimately leading to an adverse outcome (AO) at an organism or population level [3]. These KEs are linked to each other by key event relationships (KERs) which describe the causal links between a downstream event and an upstream event based on existing biological knowledge. Biological plausibility, essentiality and supporting evidence, among the reduced modified set of Bradford Hill criteria, are required for weighing the available evidence [6, 7].
In addition, the development of AOP networks has been proposed not only to improve understanding but also in predicting adverse effects used in risk assessment, research, and regulatory decision-making [8, 9]. The AOP networks are derived from existing individual AOPs, available from the AOP knowledgebase (AOP-wiki [6]), sharing common events. Hence, the AOP network concept is believed to improve the mechanistic understanding by providing insights into possible interactions among individual AOPs. The AOP-based networks can capture the complexity of biological systems described by different AOPs, in which multiple AOs diverge from a single MIE or multiple MIEs trigger a cascade of KEs that converge to a single AO [8].
Recently, a tau-driven AOP for the memory loss in sporadic (late-onset) Alzheimer’s disease (sAD) has been proposed [10]. In this AOP blueprint, several environmental neurotoxicants were also suggested as plausible MIE plug-ins, portraying possible chemical interference with early sAD pathology. In more detail, 27 tentative MIEs triggered by different stressors, including environmental chemicals and drugs, were plugged into the proposed processes of the tau-driven AOP under development (AOP429). It was hypothesized that neurotoxicity and sAD pathology may share common pathological processes [10]. Next, this hypothesis was further confirmed by identifying microRNA (miR)-gene interactions commonly regulated by the processes described in this tau-driven AOP [11].
In this present manuscript, individual AOPs listed in the AOP-wiki and sharing common processes/events with the plausible tau-driven AOP are assembled into networks. This AOP network concept is an attempt to better understand the interactions among existing mechanistic data linked to memory loss as an early phase of sAD pathology. In addition, this AOP network-based approach can also provide an effective tool for using the already available knowledge in order to predict same adverse effects of other stressors which could trigger common key events leading to memory loss.
METHODS
Selection of AOPs
We selected publicly available AOPs, endorsed by OECD, from the AOP-wiki [6], with commonly shared events, as those described by the proposed tau-driven AOP toward memory loss. An exemplified map of the tau-driven AOP was used for defining the different events of interest at molecular, cellular, and organism level (Fig. 1). Thus, seven known AOPs, namely AOP3 (inhibition of the mitochondrial complex I of nigro-striatal neurons leads to parkinsonian motor deficits), AOP10 (binding to the picrotoxin site of ionotropic gamma-aminobutyric acid (GABA) receptors leading to epileptic seizures in adult brain), AOP12 (chronic binding of antagonist to N-methyl-D-aspartate receptors (NMDARs) during brain development leads to neurodegeneration with impairment in learning and memory in aging), AOP13 (chronic binding of antagonist to N-methyl-D-aspartate receptors (NMDARs) during brain development induces impairment of learning and memory abilities), AOP42 (inhibition of thyroperoxidase and subsequent adverse neurodevelopmental outcomes in mammals), AOP48 (binding of agonists to ionotropic glutamate receptors in adult brain causes excitotoxicity that mediates neuronal cell death, contributing to learning and memory impairment), and AOP54 (inhibition of Na+/I-symporter (NIS) decreases thyroid hormone (TH) synthesis leading to learning and memory impairment), were included. In Table 1, the detailed data of the triggered MIEs, which can induce the KEs and ultimately lead to the AOs of these selected AOPs, are provided.
Fig. 1
Schematic representation of the proposed tau-driven AOP for memory loss, presenting a starting point (bidirectional relationship between glucose and cholesterol metabolism) which can trigger a cascade of key events (KEs) (mitochondrial dysfunction, oxidative stress, hyperphosphorylated tau, dysfunctional autophagy, toxic tau oligomers, dysfunctional axonal transport, dysfunctional synapses, neuroinflammation, and neuronal dysfunction), which eventually can lead to the adverse outcome (AO), memory loss (modified from [10]).
![Schematic representation of the proposed tau-driven AOP for memory loss, presenting a starting point (bidirectional relationship between glucose and cholesterol metabolism) which can trigger a cascade of key events (KEs) (mitochondrial dysfunction, oxidative stress, hyperphosphorylated tau, dysfunctional autophagy, toxic tau oligomers, dysfunctional axonal transport, dysfunctional synapses, neuroinflammation, and neuronal dysfunction), which eventually can lead to the adverse outcome (AO), memory loss (modified from [10]).](https://content.iospress.com:443/media/adr/2022/6-1/adr-6-1-adr220015/adr-6-adr220015-g001.jpg)
Table 1
Selected AOPs from AOP-wiki based on their shared common events with those of the tau-driven AOP. The AOP ID, AOP description, including potential stressors, molecular initiating events (MIEs), key events (KEs), and adverse outcome (AO), are provided
| AOP ID: Description | Stressor | MIEs | KE1 | KE2 | KE3 | KE4 | KE5 | KE6 | KE7 | KE8 | AO |
| AOP3: Inhibition of the mitochondrial complex I of nigro-striatal neurons leads to parkinsonian motor deficits | Binding of inhibitor, NADH-ubiquinone oxidoreductase (complex I) (MIE:888) | Inhibition, NADH-ubiquinone oxidoreductase (complex I) (KE:887) | N/A, Mitochondrial dysfunction 1 (KE:177) | Impaired, Proteostasis (KE:889) | Neuroinflamma-tion (KE:188) | Degeneration of dopaminergic neurons of the nigrostriatal pathway (KE:890) | Parkinsonian motor deficits (AO:896) | ||||
| AOP10: Binding to the picrotoxin site of ionotropic GABA receptors leading to epileptic seizures in adult brain | Picrotoxin, Lindane, Dieldrin, Heptachlor, Endosulfan, RDX, Fipronil | Binding at picrotoxin site, iGABAR chloride channel (MIE:667) | Reduction, Ionotropic GABA receptor chloride channel conductance (KE:64) | Reduction, Neuronal synaptic inhibition (KE:669) | Generation, Amplified excitatory postsynaptic potential (EPSP) (KE:682) | Occurrence, A paroxysmal depolarizing shift (KE:616) | Occurrence, Epileptic seizure (AO:613) | ||||
| AOP12: Chronic binding of antagonist to N-methyl-D-aspartate receptors (NMDARs) during brain development leads to neurodegeneration with impairment in learning and memory in aging | Lead (Pb) | Binding of antagonist, NMDA receptors (MIE:201) | Inhibition, NMDARs (KE:195) | Decreased, Calcium influx (KE:52) | Reduced levels of BDNF (KE:381) | Cell injury/death (KE:55) | Neuroinflammation (KE:188) | N/A, Neurodegeneration (AO:352), Impairment, Learning and memory (AO:341) | |||
| AOP13: Chronic binding of antagonist to N-methyl-D-aspartate receptors (NMDARs) during brain development induces impairment of learning and memory abilities | Binding of antagonist, NMDA receptors (MIE:201) | Decreased, Calcium influx (KE:52) | Inhibition, NMDARs (KE:195) | Reduced levels of BDNF (KE:381) | Aberrant, Dendritic morphology (KE:382) | Decrease of synaptogenesis (KE:385) | Decrease of neuronal network function (KE:386) | Reduced, Presynaptic release of glutamate (KE:383) | Cell injury/death (KE:55) | Impairment, Learning and memory (AO:341) | |
| AOP42: Inhibition of Thyroperoxidase and Subsequent Adverse Neurodevelopmental Outcomes in Mammals | Thyroperoxidase, Inhibition (MIE:279) | Thyroid hormone synthesis, Decreased (KE:277) | Thyroxine (T4) in serum, Decreased (KE:281) | Thyroxine (T4) in neuronal tissue, Decreased (KE:280) | Hippocampal gene expression, Altered (KE:756) | Hippocampal anatomy, Altered (KE:757) | Hippocampal Physiology, Altered (KE:758) | Cognitive Function, Decreased (AO:402) | |||
| AOP48: Binding of agonists to ionotropic glutamate receptors in adult brain causes excitotoxicity that mediates neuronal cell death, contributing to learning and memory impairment | Glufosinate, Domoic acid | Binding of agonist, Ionotropic glutamate receptors (MIE:875) | N/A, Mitochondrial dysfunction (KE:177) | Cell injury/death (KE:55) | N/A, Neurodegeneration (KE:352) | Overactivation, NMDARs (KE:388) | Increased, Intracellular Calcium overload (KE:389) | Decreased, Neuronal network function in adult brain (KE:618) | Neuroinflammation (KE:188) | Impairment, Learning and memory (AO:341) | |
| AOP54: Inhibition of Na+/I-symporter (NIS) decreases thyroid hormone (TH) synthesis leading to learning and memory impairment | Perchlorate, Nitrate, Thiocynate, Dysidenin, Aryltrifluoroborates | Inhibition, Na+/I- symporter (NIS) (MIE:424) | Decrease of Thyroidal iodide (KE:425) | Thyroid hormone synthesis, Decreased (KE:277) | Thyroxine (T4) in serum, Decreased (KE:281) | Thyroxine (T4) in neuronal tissue, Decreased (KE:280) | Reduced levels of BDNF (KE:381) | Decrease of GABAergic interneurons (KE:851) | Decrease of synaptogenesis (KE:385) | Decrease of neuronal network function (KE:386) | Impairment, Learning and memory (AO:341) |
Besides the approved AOPs by OECD guidelines, in AOP-wiki, relevant AOPs, still under development, were also carefully looked up and found to commonly share some of the affected events represented by the tau-driven AOP: AOP16 (acetylcholinesterase inhibition leading to acute mortality), AOP17 (binding of electrophilic chemicals to SH (thiol)-group of proteins and /or to seleno-proteins involved in protection against oxidative stress during brain development leads to impairment of learning and memory), AOP26 (calcium-mediated neuronal ROS production and energy imbalance), AOP134 (Sodium iodide symporter (NIS) inhibition and subsequent adverse neurodevelopmental outcomes in mammals), AOP152 (interference with thyroid serum binding protein transthyretin and subsequent adverse human neurodevelopmental toxicity), AOP279 (microtubule interacting drugs lead to peripheral neuropathy), AOP281 (acetylcholinesterase inhibition leading to neurodegeneration), and AOP405 (organo-phosphate chemicals induced inhibition of acetylcholinesterase (AChE) leading to impaired cognitive function).
RESULTS AND DISCUSSION
Building the AOP network for memory loss
According to the proposed tau-driven AOP, the hypothetical starting point, glucose and cholesterol dysmetabolism, can induce a series of intermediate KEs such as mitochondrial dysfunction (KE1), oxidative stress (KE2), hyperphosphorylation of tau (KE3), or dysfunctional autophagy (KE4), converging to formation of toxic tau oligomers (KE5). Next, dysfunctional of axonal transport (KE6), synaptic dysfunction (KE7), or neuroinflammation, can be induced, resulting in neuronal dysfunction, which eventually leads to memory loss (AO). Several neurotoxicants/stressors [6, 10] can trigger MIEs, which may induce a cascade of KEs, leading to AOs, including memory loss or neurodegenerative-related disorders, as illustrated in Fig. 2. Many neurotoxicants can potentially bind to molecular targets (triggering MIEs), which in turn can induce cellular responses (intermediate KEs), and subsequently be linked to the presented AOs, which are part of different AOPs (either approved by OECD or under development). In Table 2, several stressors with their potentially targeted molecular events, induced effects, experimental evidence, involved in different events and/or AOPs, are given. By linking these potential stressors to MIEs which in turn can induce intermediate KEs and/or can be linked to the target AO, memory loss, possible interactions among the commonly affected events driven by different AOPs can be identified.
Fig. 2
Potential stressors linked to plausible molecular initiating events (MIEs), which induce a cascade of key events (KEs), leading to adverse outcomes (AOs), including memory loss or neurodegenerative-related disorders, are provided. Stressors are indicated in yellow, molecular targets of the MIEs in orange, KEs in blue, and AO in red color. The hypothetical starting point, glucose and cholesterol dysmetabolism is shown in light blue color. The IDs of AOPs under development are shown in italics.
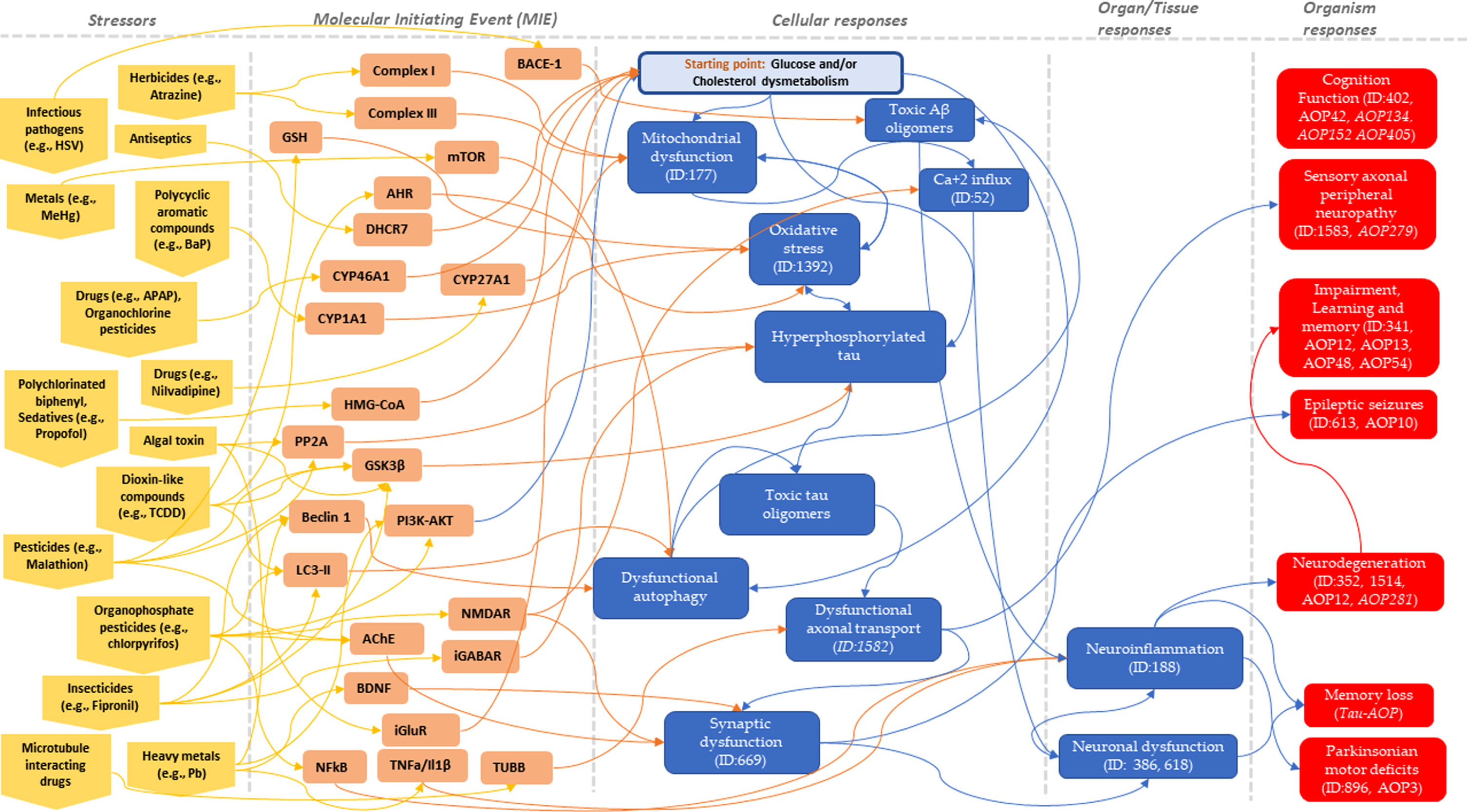
Table 2
Included environmental neurotoxicants as stressors for triggering the plausible molecular initiating events (MIEs), linked to the proposed tau-driven AOP for memory loss
| Stressors | Category | Molecular Target | Effect | Empirical Support | Involved KEs | Involved AOPs | Link to tau-driven AOP |
| 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) | Drug | Complex I | Inhibition of ETC, increased Ca2+ levels and reduced AKT phosphorylation, Mitochondrial complex I inhibitor | Mouse brain concentrations of MPP+ (approximately 12–47μM) triggered approximately a 50% inhibition of Complex I. Even small (20μM) amount of MPP+ exhibited pronounced deleterious effect on both the Ca2+-induced changes in the membrane potential and the Ca2+-accumulating capacity of mitochondria [12]. | Event: 177 Mitochondrial dysfunction, Event: 887 NADH-ubiquinone oxidoreductase (complex I), Inhibition | AOP3: Mitochondrial dysfunction and Neurotoxicity | Mitochondrial dysfunction |
| Acetaminophen | Drug | CYP46A1 | Increased 24S-OHC levels driving increased cholesterol turn-over and hypocholesterolemia | A CYP46A1 activator activated the enzyme in mouse brains at low concentrations (up to 30μM) but inhibited the P450 at higher concentrations. It increased by > 30% the formation of cholesterol 24-hydroxylation in isolated mouse brain microsomes [13]. | Cholesterol dysmetabolism | ||
| Alcohol | Solvent | NMDAR, NR2B expression, NR1 expression, PSD-95 | Blocking NMDAR, NMDAR modulation resulting in excitotoxicity and neuronal damage reflect reduction in synaptic activity | 1. After a 7-day ethanol (50 mM) exposure of human embryonic stem cell (hESC)-derived cortical neurons followed by a 24-hour ethanol withdrawal treatment, four NMDA receptor subunit genes, including GRIN1, GRIN2A, GRIN2B, GRIN2D were highly upregulated [14]. | MIE: Event: 201 NMDA receptors, Binding of antagonist | AOP13: Binding of antagonist to NMDARs impairs cognition, AOP12: Binding of antagonist to NMDARs can lead to neuroinflammation and neurodegeneration | Synaptic dysfunction, Neuroinflammation, Neurodegeneration, Impaired cognition |
| 2. Inhibition of NMDA receptor function by ethanol (and interactions between ethanol and the noncompetitive NMDA receptor antagonist ifenprodil) in neocortical neurons from rat and human embryonic kidney (HEK) 293 cells expressing recombinant NMDA receptors [15]. | |||||||
| 3. A significant association between high life time drinking and high daily alcohol intake with lower DNA methylation of NR2B in alcohol-dependent patients undergoing alcohol withdrawal [16]. | |||||||
| 4. NR1 isoforms co-expressed in various combinations with one of the four NR2 subtypes in human embryonic kidney 293 cells, the sensitivity depended on the combination of NR1-3b/NR2C, NR1-3b/NR2D, and NR1-4b/NR2C pairs, and the NR1-2b/NR2C pair were inhibited by ethanol [17]. | |||||||
| 5. In rat primary hippocampal cultures, chronic exposure to ethanol induced increase in PSD-95 expression and dendritic spine size [18]. | |||||||
| BDNF | Presynaptic effects mediated by disruption of NMDAR-activity dependent BDNF signaling by inhibiting BDNF activity | An association between BDNF serum levels and the history of alcohol consumption in humans[19]. | |||||
| Atrazine (ATZ) | Herbicides | Complex I and III | Inhibition of ETC, increased Ca2+ levels and reduced AKT phosphorylation | Sprague-Dawley rats (n = 48) were treated for 5 months with low concentrations (30 or 300μg/ kg /day) of ATZ provided in drinking water. ATZ blocked the activities of oxidative phosphorylation complexes I and III, resulting in decreased oxygen consumption. It also suppressed the insulin-mediated phosphorylation of Akt [20]. | Glucose dysmetabolism via mitochondrial dysfunction | ||
| Benzalkonium chlorides | Antiseptics | DHCR7 | Impairment of cholesterol biosynthesis | Exposure to benzalkonium chlorides exhibited high potency in inhibiting DHCR7 when tested in mouse and human neuroblastoma cells by analyzing cholesterol and its precursor using gas chromatography-mass spectrometry [21]. | Cholesterol dysmetabolism | ||
| Benzo[a]pyrene (B[a]P) | Polycyclic aromatic compounds (PACs) | CYPA1 | Oxidation of macromolecules before conjugation with glutathione, decreased cellular ATP production, MMP, oxidative phosphorylation and mitochondrial protein complexes I, II, and IV activity, and eventually apoptosis | 1. Intraperitoneal injection of B[a]P from embryonic day 7 at a dose of 250 mg kg-1 induced NTDs (13.3% frequency) in mice. BaP exposure significantly increased expression of genes associated with oxidative stress, Cyp1a1 [22]. | Mitochondrial dysfunction, Oxidative stress | ||
| 2. B[a]P-induced neurotoxicity occurred through mitochondria-mediated apoptosis (rat cerebral neurons). B[a]P-induced apoptosis was accompanied by loss of mitochondrial membrane potential, release of cytochrome c from mitochondria to the cytosol [23]. | |||||||
| 3. Exposure of human neuroblastoma SK-N-SH cells to B[a]P at 0.5–40μM for 24 h, increased in levels of ROS, significantly decreased mitochondrial membrane potential (MMP), up-regulated pro-apoptotic genes and down-regulated antioxidative genes expression, elevated cytochrome c protein levels and lipid peroxidation (LPO) in a concentration-related manner [24]. | |||||||
| Carbofuran | Pesticides | PP2A, GSK3β | Disturbed mitochondrial ETC, low ATP levels, increased oxidative stress, intracellular Ca2+ levels and tau hyperphosphorylation. Reduced dephosphorylation supporting tau hyperphosphorylation | Carbofuran exposure (rats) led to tau hyperphosphorylation at multiple AD-related phosphorylation sites with activation of GSK-3β and inhibition of PP2A [25]. | Mitochondrial dysfunction, Oxidative stress, tau hyperphos-phorylation | ||
| NMDAR | Carbofuran was administered respectively into the rats once a day for 28 days by gavage. Pesticide exposure induced spatial learning and memory deficits with a simultaneous decrease of NMDAR1, synaptophysin, and synapsin I, all of which are memory-related synaptic proteins [25]. | Synaptic dysfunction | |||||
| Cadmium (Cd) | Heavy metals | PI3K/Beclin/BCL2 signaling | p-tau induction and PI3K/Beclin/BCL2 signaling followed by excessive autophagy and apoptosis | 1. Exposure of human SH-SY5Y cells to Cd increased intracellular ROS levels [26]. | Oxidative stress, Dysfunctional autophagy | ||
| 2. Cd exposure to rat cerebral cortical neurons induced cytoprotective autophagy by activating the class III PI3K/beclin-1/Bcl-2 signaling pathway [27]. | |||||||
| Chlorophenotane | Pesticides | CYP51, DHCR7? | Impairment of cholesterol biosynthesis | Acute sublethal and chronic administration of chlorophenotane (DDT) decreased brain lipid metabolism of rhesus monkeys [28]. | Cholesterol dysmetabolism | ||
| Chlorpyrifos | Organophos-phate pesticides | AChE | Irreversible binding between AChE and OP pesticides due to phosphorylation of enzyme. AChE inhibitors (pesticides) bind to the enzyme and interfere with the breakdown of AChE, leading to deposition of AChE in nerve synapses, causing disrupted neurotransmission. | 1. Estuarine fish (Oreochromis mossambicus) to a 24 h LC50 concentration of chlorpyrifos, after 6 h reached > 40% AChE inhibition while after 24 h reached 90% AChE inhibition [29]. | MIE: Event: 12 Acetylcholinesterase (AChE), Inhibition | AOP16: Acetylcholinesterase inhibition leading to acute mortality, AOP405: Organo-Phosphate Chemicals induced inhibition of AChE leading to impaired cognitive function | Impaired cognitive function |
| 2. An acute sublethal exposure of chlorpyrifos to Sprague-Dawley rats increased inhibition of AChE, increased levels of acetylcholine, and significantly impacted to motor activity [30]. | |||||||
| PI3K-AKT | Chronic cholinergic activity linked to defective PI3K-AKT pathway activation | Chlorpyrifos exposure of human neural precursor cells (hNPCs) derived from human embryonic stem cells (hESCs) reduced the expression of AKT and ERK proteins involved in intracellular survival pathways [31]. | Glucose metabolism | ||||
| NMDAR | Glutamate excitotoxicity leading to neuronal damage | Chlorpyrifos -induced neurotoxicity after CPF exposure with and without Ifenprodil (IFN) on 4-week differentiated human neural progenitor stem cell culture model (ReNcell CX) [32]. | Synaptic dysfunction | ||||
| NFkB | Release IL1β and TNFα? | Chlorpyrifos exposure of human neural precursor cells (hNPCs) derived from human embryonic stem cells (hESCs) induced nuclear accumulation of NFκB manner via ROS generation in a concentration-dependent [31]. | Neuroinflammation | ||||
| LC3-II expression | Oxidative stress induced increased in numbers of autophagic vacuoles, autophagy, and apoptosis | 1. Chlorpyrifos-induced cytotoxicity in human SH-SY5Y cells, and induced autophagic cell death by upregulating the expression of LC3-II and p62 [33]. | Oxidative stress, Dysfunctional autophagy | ||||
| 2. Chlorpyrifos generated oxidative stress and lipid peroxidation in different rat cell types causing neuronal damage by elevating the production of ROS, DNA damage, and lipid peroxidation in the CNS [34, 35]. | |||||||
| Conazole | Fungicides | CYP51, DHCR7 | Impairment of cholesterol biosynthesis | Inhibits CYP51 by coordinating with the heme group, which halts substrate binding with a resulting increase of lanosterol in mouse Neuro2a cells [36]. | Cholesterol dysmetabolism | ||
| Copper (Cu) | Metals | LC3-II expression | Oxidative stress induced increased in numbers of autophagic vacuoles, autophagy, and apoptosis | CuCl2 induced a dose-dependent accumulation of the autophagosome marker, LC3-II, in human neuroblastoma cell line SK-N-SH [37]. | Oxidative stress, Dysfunctional autophagy | ||
| Deltamethrin | Pesticides | PP2A, GSK3β | Disturbed mitochondrial ETC, low ATP levels, increased oxidative stress, intracellular Ca2+ levels and tau hyperphosphorylation. Reduced dephosphorylation supporting tau hyperphosphorylation. | Exposure of deltamethrin (30μM), PP2A/PP2B inhibitors, induced tau aggregation in human SH-SY5Y tau-BiFC cells and in HEK293 tau-BiFC cells. | Mitochondrial dysfunction, Oxidative stress, tau hyperphos-phorylation | ||
| Dibenzothiopene | Alkyl-polycyclic aromatic compounds (PACs) | CYP1A1, CYP1B1 | Oxidation of macromolecules before conjugation with glutathione, decreased cellular ATP production, MMP, oxidative phosphorylation and mitochondrial protein complexes I, II, and IV activity, and eventually apoptosis | Exposure of human neuroblastoma SK-N-SH cells to dibenzothiophene (at 0.5–40μM for 24 h) increased in levels of ROS, significantly decreased mitochondrial membrane potential, upregulated pro-apoptotic genes and down-regulated antioxidative genes expression, elevated cytochrome c protein levels and lipid peroxidation in a concentration-related manner [24]. | Mitochondrial dysfunction, Oxidative stress | ||
| Dichlorvos | Organophos-phate pesticides | AChE | Irreversible binding between AChE and OP pesticides is due to phosphorylation of enzyme. The AChE inhibitors (pesticides) bind to the enzyme and interfere with the breakdown of ACh, leading to the deposition of ACh in the nerve synapses and causing disrupted neurotransmission | Rats injected with a sublethal concentration of dichlorvos found a significant decrease in AChE activity, increased ACh concentrations, and enhanced contractile responses in jejunum muscle. At sublethal concentrations (56% of the LD50), researchers found a significant (18%) increase in the amount of ACh in brain tissue of rats exposed to disulfoton for 3 days and resulted in AChE inhibition of 68% with respect to controls [38]. | MIE: Event: 12 Acetylcholinesterase (AChE), Inhibition | AOP405: Organo-Phosphate Chemicals induced inhibition of AChE leading to impaired cognitive function | Impaired cognitive function |
| Dieldrin | Organochloride insecticides | iGABARs | Directly blocks chloride conductance through the ion channel | Long-term exposure of mouse primary cerebellar granule cell cultures to 3μM dieldrin reduced the GABAA receptor function to 55% of control, as measured by the GABA-induced 36Cl- uptake [39]. | MIE: Event: 667 iGABAR chloride channel, Binding at picrotoxin site | AOP10: Blocking iGABA receptor ion channel leading to seizures | Seizures |
| Domoic acid (DomA) | Kainic acid-type neurotoxin (ASP) | COX2 | 1. DomA (4 mg/kg at 30, 60 and 240 min post-injection) promoted the expression of early inflammatory genes in mouse brain, such as COX2 and the development of neurodegeneration [40]. | AOP48: Binding of agonists to ionotropic glutamate receptors in adult brain causes excitotoxicity that mediates neuronal cell death, contributing to learning and memory impairment. | Neuroinflammation | ||
| 2. DomA treatment (2 mg/kg per day for 3 weeks) in mice significantly stimulated the expression of inflammatory mediators, including IL-1β (1.7 fold increase), TNF-α (2 fold increase), GFAP (1.4 fold increase), Cox-2 (3 fold increase), and iNOS (1.6 fold increase) compared to controls [41]. | |||||||
| 3. DomA (0.75 mg/kg body weight) when administered intravenously in adult rats induced neuronal degeneration followed by glial activation [42]. | |||||||
| iGlu, NMDRAs, KA &ARs | DomA induces excitotoxicity by an integrative action on ionotropic glutamate receptors at pre- and post-synaptic sides. DomA directly activates KA/AMPARs receptors followed by indirect activation of the NMDARs | 1. In rat hippocampal neurons at 5 days after DomA administration, NMDAR1 immunoreactivity increased, and glutamate receptor genes were induced in response to DomA-induced neurotoxicity [43]. | MIE: Event: 875 Ionotropic glutamate receptors, Binding of agonist | Synaptic dysfunction | |||
| 2. DomA exposure of immature and mature primary cultures of neurons and glial cells from rat cerebellum induced neurotoxicity mediated by the AMPA/KA receptors [44]. | |||||||
| Efavirenz | Drug | CYP46A1 | Increased 24S-OHC levels driving increased cholesterol turn-over and hypocholesterolemia | Efavirenz a CYP46A1 activator activated the enzyme in mouse brains at low concentrations (up to 20μM) but inhibited the P450 at higher concentrations. It increased the formation of cholesterol 24-hydroxylation in isolated mouse brain microsomes [13]. | Cholesterol dysmetabolism | ||
| Endosulfan | Organochloride insecticide | iGABARs | Non-competitive ion channel blocker | Poisoning with endosulfan caused seizure, status epilepticus, or refractory status epilepticus in humans [45, 46], and eventually led to the death of a farmer [45] and a toddler [47]. | MIE: Event: 667 iGABAR chloride channel, Binding at picrotoxin site | AOP10: Blocking iGABA receptor ion channel leading to seizures | Seizures |
| CYP51, DHCR7 | Impairment of cholesterol biosynthesis C | Exposure of mouse Neuro-2a cells to endosulfan (1.1μM) induced elevation of lanosterol by inhibiting either CYP51 or DHCR7 [48]. | Cholesterol dysmetabolism | ||||
| Felodipine | Drugs | CYP27A1 | Reduced 27-OHC production, elevated cholesterol biosynthesis and reduced steroidal acid production | Felodipine administration to mice at a 1 mg/kg of body weight/day for 7 days induced reduction in 27-hydroxycholesterol levels in plasma, brain and liver, whereas tissue levels of total cholesterol were unchanged [49]. | Cholesterol dysmetabolism | ||
| Fenobucarb, Propoxur | Organophosphate pesticides | AChE | Irreversible binding between AChE and OP pesticides is due to phosphorylation of enzyme. The AChE inhibitors (pesticides) bind to the enzyme and interfere with the breakdown of ACh, leading to the deposition of ACh in the nerve synapses and causing disrupted neurotransmission. | Female mice exposed to either fenobucarb or propoxur, reported a major increase in AChE in brain tissue 10 minutes after injection, simultaneously major elevation in AChE inhibition [50]. | MIE: Event: 12 Acetylcholinesterase (AChE), Inhibition | AOP405: Organo-Phosphate Chemicals induced inhibition of AChE leading to impaired cognitive function (AOP under development) | Impaired cognitive function |
| Fenpropimorph | Fungicides | CYP51, DHCR7 | Impairment of cholesterol biosynthesis | Exposure of human cell lines and human neuroprogenitor model derived from hiPSCs to fenpropimorph caused hypocholesterolemia, decreased absolute cholesterol levels, inhibited CYP51 and elevated 7-DHC levels [48]. | Cholesterol dysmetabolism | ||
| Fipronil | Insecticides | iGABARs, Cl- channels regulated by GABA receptors | Directly blocks chloride conductance through the ion channel. | Acute human intoxication with fipronil revealed symptoms associated with the GABA transmission within the central nervous system, including seizure, agitation, and headache. In two patients, plasma fipronil concentrations were measured at 1600 and 3744μg/L. In another patient, a peak measured plasma concentration at 1040μg/L [51]. | MIE: Event: 667 iGABAR chloride channel, Binding at picrotoxin site), Event: 64 iGABAR chloride conductance, Reduction | AOP10: Blocking iGABA receptor ion channel leading to seizures | Seizures |
| GSK3β | Alteration of AKT/GSK3β phosphorylation | In human dopaminergic SH-SY5Y cells, fipronil exposure altered the level of Akt/GSK3β phosphorylation, reduced the Akt phosphorylation on Ser473, and in parallel with the inactivation of Akt, phosphorylation of GSK3β on Ser9 which inactivated GSK3β, decreased after treatment [52]. | Tau hyperphos-phorylation | ||||
| LC3-II &Beclin-1 expression | Oxidative stress induced increased in numbers of autophagic vacuoles, autophagy, and apoptosis | 1. Exposure to fipronil in human neuroblastoma SH-SY5Y cells induced autophagic death by monitoring LC3-Ii and Beclin-1 expression [53]. | Oxidative stress, Dysfunctional autophagy | ||||
| 2. Fipronil exposure to human dopaminergic SH-SY5Y cells induced dopaminergic cell death involved in increase of ROS generation [52]. | |||||||
| Galantamine | Drugs | CYP46A1 | Increased 24S-OHC levels driving increased cholesterol turn-over and hypocholesterolemia | Galantine treatment in isolated bovine brain microsomes stimulated CYP46A1 activity by 6–7-fold [13]. | Cholesterol dysmetabolism | ||
| Glufosinate | Phosphorous herbicides, Fungicides | NMDARs | Agonist action at the NMDAR is expected to occur through direct interaction with the glutamate binding site and requires binding of the glycine co-agonist as well as release of the magnesium block from the channel pore. | 1. In one human poisoning case (267 mg/kg d,l-glufosinate, oral ingestion), the concentrations respectively of d- and l-GLF 1 h after exposure were 1050 and 1070μM in plasma and after 27 h were 7.95 and 1.93μM in plasma and 2.6 and 0.66μM in cerebrospinal fluid, respectively. d,l-Glufosinate concentrations in excess of 100μM are needed to affect the NMDAR [54]. | MIE: Event: 875 Ionotropic glutamate receptors, Binding of agonist | AOP48: Binding of agonists to ionotropic glutamate receptors in adult brain causes excitotoxicity that mediates neuronal cell death, contributing to learning and memory | Synaptic dysfunction |
| 2. Chronic exposure to glufosinate induced structural changes in the NMDAR rich hippocampal region of the mouse brain, disrupting activation of NMDARs [55]. | |||||||
| Iron (Fe) | Metals | LC3-II &Beclin expression | Oxidative stress induced increased in numbers of autophagic vacuoles, autophagy, and apoptosis | Fe given in the neonatal period in rats (single daily oral dose of vehicle or iron carbonyl (30 mg/kg) at postnatal days 12–14) impaired inhibitory avoidance memory and induced a decrease in proteins critically involved in the autophagy pathway, Beclin-1 and LC3, in hippocampus [56]. | Oxidative stress, Dysfunctional autophagy | ||
| Ketamine | Anaesthetics | NMDARs | Blocking NMDA receptor channel activity | In humans, ketamine exposure produced profound decrements in both attention and memory [57, 58] | Synaptic dysfunction | ||
| Lindane | Insecticides | iGABARs | Non-competitive ion channel blocker | Toxic doses of lindane produced neuronal hyperexcitability in humans and other vertebrates by inhibiting both glycine and GABAA receptors [59]. | Synaptic dysfunction | ||
| Lead (Pb) | Heavy metals | BDNF | Presynaptic effects mediated by disruption of NMDAR-activity dependent BDNF signaling by inhibiting BDNF activity | In primary rat hippocampal neurons exposed to 1μM Pb2+ for 5 days during the period of synaptogenesis (DIV7–DIV12), decreased cellular pro-BDNF protein (40% compared to control) and extracellular levels of mBDNF (25% compared to control [60]. | Event: 389 (Release of BDNF, Reduced) | AOP13: Binding of antagonist to NMDARs impairs cognition, AOP54: NIS inhibition and DNT effects, AOP12: Binding of antagonist to NMDARs can lead to neuroinflammation and neurodegeneration | Synaptic dysfunction |
| NMDAR | Glutamate excitotoxicity leading to neuronal damage | 1. In rat hippocampal neurons, Pb2+ (2.5–50μM) inhibited NMDA-induced whole-cell and single-channel currents in a concentration-dependent manner, suggesting that Pb2+ can decrease the frequency of NMDA-induced channel activation. Pb2+ also affected the binding of [3H]MK-801 to the rat brain hippocampal membranes [61]. | Event:195 NMDARs, Inhibition, Event: 201 NMDA receptors, Binding of antagonist | AOP12: Binding of antagonist to NMDARs can lead to neuroinflammation and neurodegeneration | Synaptic dysfunction, Neuroinflammation, Neurodegeneration | ||
| 2. Studies in animal models of developmental Pb2+ exposure exhibit altered NMDAR subunit ontogeny and disruption of NMDAR-dependent intracellular signaling [60]. | |||||||
| Zn2+ regulatory site of the NMDAR | Potent, non-competitive antagonist of the NMDAR binding at the Zn2+ regulatory site of the NMDAR in a voltage-independent manner causing inhibition of Ca2+ channels, presynaptic neurotransmission and NMDARs signaling | Pb2+ exposure decreased Ca2+ ion concentration and increased Ca2+ efflux by a calmodulin-dependent mechanism in embryonic rat hippocampal neurons [62]. | Event:52 Ca2+ influx, decreased | Synaptic dysfunction? | |||
| IL-1b, TNF-a | Proinflammatory cytokines | In vivo and in vitro models (rats), Pb exposure caused microglial activation, which upregulated the levels of pro-inflammatory cytokines IL-1β, TNF-α and of iNOS and caused neuronal injury and neuronal death in hippocampus [63]. | Event:188 Neuroinflammation | Neuroinflammation | |||
| Tau | Phosphorylation of tau | 1. Monkeys exposed to Pb 1.5 mg/kg/day from birth to 400 days at 23 years of age tau accumulation, Overexpression of amyloid-beta protein precursor and of amyloid-beta enhanced pathologic neurodegeneration [64]. | Tau hyperphospho-rylation? | ||||
| 2. Mice exposed toPb 0.2% in drinking water from PND 1–20 or from PND 1–20 and from 3–7 months at 700 days of age, elevated protein and mRNA for tau and aberrant site-specific tau [65]. | |||||||
| 3. Perinatal exposure to Pb leading to a blood concentration of 10μg/dl promoted tau phosphorylation in rat forebrain, cerebellum and hippocampus [66]. | |||||||
| 4. Chronic exposure of rats to Pb via drinking water induced hyperphosphorylation of tau and excessive increase in autophagy, which might induce cell-programmed death and increase neurotoxicity [67]. | |||||||
| Beclin-1/ LC3-II signaling, Akt/mTOR pathway | Beclin-1/LC3-II signaling followed by excessive autophagy | Pb exposure induced autophagy in astrocytes, by increased LC3II and Beclin 1 protein levels in both the rat hippocampus and astrocytes through blocking the downstream Akt/mTOR pathway in astrocytes [68]. | Oxidative stress, Dysfunctional autophagy | ||||
| PI3K-Akt signaling | Pb exposure induced a decrease in rat hippocampal glucose metabolism by reducing GLUT4 levels in the cell membrane through the PI3K-Akt pathway. In vivo and in vitro GLUT4 over-expression increased the membrane translocation of GLUT4 and glucose uptake, and reversed the Pb-induced impairment to synaptic plasticity and cognition [69]. | Glucose dysmetabolism? | |||||
| Malathion | Organophosphate pesticides | AChE | Irreversible binding between AChE and OP pesticides is due to phosphorylation of enzyme. The AChE inhibitors (pesticides) bind to the enzyme and interfere with the breakdown of AChE, leading to the deposition of AChE in the nerve synapses and causing disrupted neurotransmission | 1. In rats, administration of malathion induced reduction of plasma acetylcholinesterase activity [70]. | MIE: Event: 12 Acetylcholinesterase (AChE), Inhibition | AOP405: Organo-Phosphate Chemicals induced inhibition of AChE leading to impaired cognitive function (AOP under development) | Impaired cognitive function |
| 2. In silkworm exposed to malathion for 5 days increased mortality, decreased AChE, and increased in AChE as compared to controls [71]. | |||||||
| GSK3β, PP2A | Reduced dephosphorylation supporting tau hyperphosphorylation | In rats, administration of malathion induced spatial learning and memory deficits with a simultaneous decrease of PSD93 and tau hyperphosphorylation at multiple AD-related phosphorylation sites with activation of GSK-3β and inhibition of PP2A [70]. | Tau hyperphospho-rylation | ||||
| SH containing proteins | Depletion of glutathione buffer, oxidation of macromolecules, and disturbed cellular redox homeostasis | 1. Subchronic exposure of rats to malathion increased malondialdehyde and 8-OHdG levels, whereas it decreased glutathione levels, also acetylcholinesterase, superoxide dismutase, and catalase activities in the blood and brain tissues [72]. | Mitochondrial dysfunction, Oxidative stress | ||||
| 2. In rats, administration of malathion induced amelioration of reduced GSH in hippocampus [70]. | |||||||
| TNF-a, IL-6 | Proinflammatory cytokines | In rats, administration of malathion induced elevation of TNF α and IL-6 levels in the hippocampus [70]. | Neuroinflammation | ||||
| Methylmercury (MeHg) | Metals | Beclin-1 expression | Oxidative stress induced increased in numbers of autophagic vacuoles, autophagy, and apoptosis | 1. Exposure of human neural stem cells to MeHg upregulated autophagy by inhibiting mTOR levels and by enhancing Atg6/Beclin-1 protein levels [73]. | Oxidative stress, Dysfunctional autophagy | ||
| 2. Oxidative stress has been suggested to enhance autophagy signaling pathway, as the underlying mechanism of MeHg-induced neurotoxicity [74]. | |||||||
| m-TOR expression | |||||||
| LC3-II expression | In rat astrocytes, MeHg decreased cell viability in a concentration- and time-dependent manner, with induction of both apoptosis and autophagy via increasing the LC3-II–LC3-I ratio, and of Beclin-1 protein [75]. | ||||||
| Methyl parathion | Organophosphate pesticides | AChE | Irreversible binding between AChE and OP pesticides is due to phosphorylation of enzyme. The AChE inhibitors (pesticides) bind to the enzyme and interfere with the breakdown of ACh, leading to the deposition of ACh in the nerve synapses and causing disrupted neurotransmission | Sublethal exposure (12–48 h) to methyl parathion highly inhibited AChE levels in brain tissue in fish, (Oreochromis mossambicus) with inhibition increasing from 36–62% as in comparison to controls over the time elapse [76]. | MIE: Event: 12 Acetylcholinesterase (AChE), Inhibition | AOP405: Organo-Phosphate Chemicals induced inhibition of AChE leading to impaired cognitive function (AOP under development) | Impaired cognitive function |
| Mirtazapine | Drugs | CYP46A1 | Increased 24S-OHC levels driving increased cholesterol turn-over and hypocholesterolemia | Exposure of bovine isolated brain microsomes to mirtazapine increased CYP46A1 activity [13]. | Cholesterol dysmetabolism | ||
| Manganese (Mn) | Metals | SH containing proteins | Depletion of glutathione buffer, oxidation of macromolecules, and disturbed cellular redox homeostasis | 1. Exposure of rat-derived mesencephalic dopaminergic neuronal (N27) cells to Mn induced ROS formation [77]. | Mitochondrial dysfunction, Oxidative stress, tau hyperphos-phorylation | ||
| 2. Exposure of rat primary striatal neurons to Mn induced a dose-dependent decrease in MMP and complex II activity [78]. | |||||||
| LC3-II &Beclin-1 expression | Oxidative stress induced increased in numbers of autophagic vacuoles, autophagy, and apoptosis | After a single intrastriatal injection of Mn, the short- (4–12 h) and long-term (1–28 days) effect of Mn on rat dopaminergic neurons, increased number of abnormal lysosomes, decreased protein expression of Beclin-1, and decreased ratio of LC3 II over LC3 I, concomitant with activated mTOR/p70s6k signaling [79]. | Oxidative stress, Dysfunctional autophagy | ||||
| Nilvadipine | Drugs | CYP27A1 | Reduced 27-OHC production, elevated cholesterol biosynthesis and reduced steroidal acid production | Administration of nilvadipine to mice at a 1 mg/kg of body weight per day, for 7 days, reduced 27-hydroxycholesterol levels in the plasma, brain, and liver, whereas tissue levels of total cholesterol were unchanged [49]. | Cholesterol dysmetabolism | ||
| Organochlorine pesticides (OCPs) | Pesticides | CYP46A1 | Increased 24S-OHC levels driving increased cholesterol turn-over and hypocholesterolemia | In human studies, exposure of OCPs induced elevated levels of the total cholesterol in blood [80]. | Cholesterol dysmetabolism | ||
| Okadaic acid (OKA) | Algal toxins | GSK3β, PP2A | Tau hyperphosphorylation | Administration of OKA in rats increased tau gene and protein expression, Ca2+/ CAMKII, calpain and GSK3β in the hippocampus and cerebral cortex, while decreased the PP2A gene and protein expression in these brain regions [81]. | Tau hyperphos-phorylation | ||
| Disturbed mitochondrial ETC, low ATP levels, increased oxidative stress, intracellular Ca2+ levels | Intracerebroventricular administration of OKA increased intracellular Ca2+, impairing the mitochondrial ETC and generating intracellular ROS and RNS (reactive nitrogen species) in rat brain areas [82]. | Mitochondrial dysfunction, Oxidative stress | |||||
| NMDARs | Involvement of NMDA receptor in OKA intracerebroventricular -induced tau hyperphosphorylation in rat brain areas [81]. | Synaptic dysfunction | |||||
| Paraoxon | Organophosphate pesticides | AChE | Irreversible binding between AChE and OP pesticides is due to phosphorylation of enzyme. The AChE inhibitors (pesticides) bind to the enzyme and interfere with the breakdown of AChE, leading to the deposition of AChE in the nerve synapses and causing disrupted neurotransmission | Inhibition of striatal AChE activity and decreased extracellular AChE levels in rats intracerebrally perfused after exposure to paraoxon (0, 0.03, 0.1, 1, 10 or 100μM, 1.5μl/min for 45 min) [83]. | MIE: Event: 12 Acetylcholinesterase (AChE), Inhibition | AOP405: Organo-Phosphate Chemicals induced inhibition of AChE leading to impaired cognitive function (AOP under development) | Impaired cognitive function |
| Paraquat | Pesticides | NFkB | Release IL1β, IL6 and TNFα | 1. Upon paraquat exposure, HMGB1 increased, translocated into cytosol and then released to the extracellular milieu of human neuroblastoma SH-SY5Y cells, via activation of RAGE-P38-NF-κB signaling pathway and the expression of inflammatory cytokines such as TNF-α and IL-6 [84]. | Neuroinflammation | ||
| 2. Paraquat-induced ROS inhibited human blood neutrophil apoptosis via a p38 MAPK/NF-κB–IL-6/TNF-α [85]. | |||||||
| Parathion | Organophosphate insecticides and acaricides | AChE | Irreversible binding between AChE and OP pesticides is due to phosphorylation of enzyme. The AChE inhibitors (pesticides) bind to the enzyme and interfere with the breakdown of AChE, leading to the deposition of AChE in the nerve synapses and causing disrupted neurotransmission. | An acute (48 h) sublethal exposure to methyl parathion found that AChE levels in brain tissue in fish (Oreochromis mossambicus) were significantly inhibited at all measured durations ranging from 12–48 h with inhibition increasing from 36–62% as compared to controls over the time span [76]. | MIE: Event: 12 Acetylcholinesterase (AChE), Inhibition | AOP16: Acetylcholinesterase inhibition leading to acute mortality | |
| Polychlorinated Biphenyls (PCBs) | Persistent organic pollutants (POPs) | CYP450 | Oxidation of macromolecules before conjugation with GSH, decreased cellular ATP production, MMP, oxidative phosphorylation and mitochondrial protein complexes I, II, and IV activity, and eventually apoptosis. | Chronic exposure of human SH-SY5Y cells to PCBs decreased cellular ATP production, MMP, oxidative phosphorylation and the activity of mitochondrial protein complexes I, II, and IV, resulting in mitochondrial dysfunction [86]. | Mitochondrial dysfunction, Oxidative stress | ||
| HMG-CoA reductase | Increased cholesterol serum levels result in impaired cholesterol biosynthesis | In human studies, exposure of PCBs induced elevated levels of the total cholesterol in blood [80]. | Cholesterol dysmetabolism | ||||
| Particle matter (PM) &Sulfur dioxide (SO2) | Air pollutants | PSD-95, NR2B, BDNF, Synp, tau, expression | Reduction in these synaptic markers reflect reduction in synaptic activity | 1. PM2.5 and SO2 co-exposure led to neurodegeneration at low doses, reduced synaptic structural protein PSD-95 and NMDA receptor subunits (NR2B), and elevated tau phosphorylation, in vitro (mouse primary cortical neuron culture) and in vivo (mice) [87]. | Synaptic dysfunction, tau hyperphos-phorylation | ||
| 2. Human cocultured neurons and astrocytes with PM2.5 treatment exhibited reduction in the number of synapsin I by ≈49.6% compared to the nontreated cocultured model [88]. | |||||||
| 3. Human in utero exposure to PM2.5 was inversely associated with placental BDNF expression [89]. | |||||||
| IL-1β | Proinflammatory cytokines | Human astrocytes cocultured with neurons were activated by PM2.5 treatment produced significant levels of proinflammatory chemokines (CCL1 and CCL2) and cytokines (IL-1β and IFN-γ), which can recruit and activate microglia [88]. | Neuroinflammation | ||||
| Profenofos (PFF) | Organophosphate insecticides | AChE | Irreversible binding between AChE and OP pesticides is due to phosphorylation of enzyme. The AChE inhibitors (pesticides) bind to the enzyme and interfere with the breakdown of AChE, leading to the deposition of AChE in the nerve synapses and causing disrupted neurotransmission. | A time course study of earthworms (Eisenis foetida) exposed to the 4.56±0.14 and 3.55±0.10μg cm–2 for 24 and 48 h (LC50), respectively, of PPF found a significant relationship between increases in percent inhibition of AChE and increase in time of exposure from 8–48 h [90]. | MIE: Event: 12 Acetylcholinesterase (AChE), Inhibition | AOP16: Acetylcholinesterase inhibition leading to acute mortality | |
| Propofol | Sedative drugs | HMG-CoA reductase | Increased cholesterol serum levels result in impaired cholesterol biosynthesis | Treatment of rats with propofol induced increased levels of serum total cholesterol, possibly due to the increase in HMG-CoA reductase activity, a rate limiting step in cholesterol biosynthesis. Propofol at 2 and 4 mg/kg increased the levels of serum total cholesterol by 74% and 55%, triglycerides by 97% and 115%, and LDL-C by 45% and 73%, respectively, while HDL-C decreased by 41% and 54%, respectively [91]. | Cholesterol dysmetabolism | ||
| RDX | iGABARs | Directly blocks chloride conductance through the ion channel. | 1. Human exposure to high doses of RDX causes headache, dizziness, vomiting, and confusion, followed by tonic-clonic seizures [92]. 2. RDX binds to GABAA) receptor causing reduction of GABAA receptor–mediated synaptic transmission and induction of epileptiform activity, in the amygdala, a seizure-prone structure of the limbic system, in rats [92]. | Event: 667 iGABAR chloride channel, Binding at picrotoxin site | AOP10: Blocking iGABA receptor ion channel leading to seizures | Seizures | |
| Retene | Alkyl-polycyclic aromatic compounds (PACs) | CYP1A1, CYP1B1 | Oxidation of macromolecules before conjugation with GSH, decreased cellular ATP production, MMP, oxidative phosphorylation and mitochondrial protein complexes I, II, and IV activity | Exposure of human neuroblastoma SK-N-SH cells to retene at 0.5–40μM for 24 h, increased in levels of ROS, significantly decreased MMP, up-regulated pro-apoptotic genes and down-regulated antioxidative genes expression, elevated cytochrome c protein levels and lipid peroxidation in a concentration-related manner [24]. | Mitochondrial dysfunction, Oxidative stress | ||
| Rotenone | Isoflavone pesticides, insecticides, piscicides | LC3-II expression, Beclin-1 expression, mTOR | Oxidative stress induced increased in numbers of autophagic vacuoles, autophagy, and apoptosis | 1. Robust increase in steady state expression of LC3 (LC3I and LC3II), upon rotenone treatment compared to untreated cells, and a significant decrease in phosphorylation of Akt and beclin1, in human SHSY-5Y cells [93]. 2. Exposure to rotenone at a higher dose (10μM) decreased mTORC1 activity, in human SHSY-5Y cells. Rotenone-treated cells showed ∼2 fold increase in ROS generation compared to untreated cells [93]. | Oxidative stress, Dysfunctional autophagy | ||
| Complex I | Inhibition of ETC, increased Ca2+ levels and reduced AKT phosphorylation, Mitochondrial complex I inhibitor | Mouse brain concentrations of rotenone (20–30 nM) triggered approximately a 50% inhibition of Complex I [12]. | Event: 177 Mitochondrial dysfunction, Event: 887 NADH-ubiquinone oxidoreductase (complex I), Inhibition | AOP48: Binding of agonists to ionotropic glutamate receptors in adult brain causes excitotoxicity that mediates neuronal cell death, contributing to learning and memory impairment. AOP3: Mitochondrial dysfunction and Neurotoxicity | Glucose dysmetabolism via mitochondrial dysfunction | ||
| Silica (SiO2-NPs), Silver (AgNPs) nanoparticles | Nanoparticles | PSD-95, NR2B expression | Reduction in these synaptic markers reflect reduction in synaptic activity | 1. In brains of immature rats subjected to a low dose of AgNPs, decreased expression of several NMDA receptor complex-related proteins, such as GluN1 and GluN2B subunits, scaffolding proteins PSD95 and SynGAP, as well as neuronal nitric oxide synthase (nNOS) [94]. | Synaptic dysfunction | ||
| 2. AgNPs oral exposure significantly decreased levels of the presynaptic proteins synapsin I and synaptophysin, as well as PSD-95 protein which is an indicator of postsynaptic densities, in hippocampal region of rat brain [95]. | |||||||
| GSK3β | Induction of tau hyperphosphorylation | Exposure of human SK-N-SH to SiO2-NPs significantly increased the intracellular levels of ROS and the hyperphosphorylation of tau at Ser262 and Ser396 accompanied by increased GSK3β activity [96] | Tau hyperphospho-rylation | ||||
| APP | Immunofluorescent staining indicated a significantly increased number of in cells containing intracellular Aβ1-42 positive deposit and upregulated APP and downregulated Aβ-degrading enzyme neprilysin. in SiNP-treated human SK-N-SH and mouse N2a cells in comparison to the control or micro-sized SiO2-treated cells [96]. | Intracellular Aβ | |||||
| Sodium azide (NaN3) | Gas-forming inorganic compound | Complex IV | Inhibition of the mitochondrial ETC, oxidative stress | Exposure of in rat PC12 cells to NaN3 inhibited cytochrome oxidase in the mitochondrial ETC and accumulation of mitochondrial ROS. NaN3-induced apoptosis was associated with the expression levels of Pgc-1α family proteins and Cox IV in mitochondria-mediated signaling pathway [97]. | Mitochondrial dysfunction, Oxidative stress | ||
| Spiroxamine | Fungicides | CYP51, DHCR7 | Impairment of cholesterol biosynthesis | Exposure of human hiPSCs and mouse neuroblastoma Neuro-2a cells to spiroxamine induced decreased cholesterol biosynthesis either by inhibiting CYP51 or DHCR7 [48]. | Cholesterol dysmetabolism | ||
| Taxol &epothilones | Microtubule interacting drugs (MSAs) | Tubulin (TUBB) | MSAs bind to polymerized tubulin. Impairment in axonal transport leads to an inadequate supply of the neuronal periphery. | 1. Disruption of microtubule dynamic instability decreased transport of horseradish peroxidase in dorsal root ganglia neurons resulting in less microtubule crosslinks. Intact axonal transport regained after taxol wash-out (1 day treatment, 2 days wash-out) [98]. | AOP279: Microtubule interacting drugs lead to peripheral neuropathy | Impaired axonial transport | |
| 2. Taxol inhibited anterograde fast (and retrograde) axonal transport in rat sciatic nerves [99] and anterograde transport in SK-N-SH human neuroblastoma cells and mice sciatic nerves [100]. | |||||||
| 3. Suppressed microtubule dynamic instability had inhibitory effects on anterograde fast axonal transport in isolated squid axoplasm [101]. | |||||||
| 2,3,7,8-tetrachloordi-benzo-p-dioxine (TCDD) | Dioxin-like compounds | LC3-II expression | Oxidative stress induced increased in numbers of autophagic vacuoles, autophagy, and apoptosis | In human neuronal cell line SHSY5Y, expression of LC3II was significantly increased in cells exposed to 200 nM TCDD, compared with control cells [102]. | Oxidative stress, Dysfunctional autophagy | ||
| AHR | Potent aryl hydrocarbon receptor (AHR) ligands | TCDD toxicity in human SHSY5Y neuroblastoma cells depended on dioxin concentration and time of incubation, with a main role of aryl hydrocarbon receptor at low nanomolar TCDD concentrations (3 nM at 24 h). Induced apoptosis by the disruption of calcium homeostasis, affecting membrane structural integrity [103]. | MIE: Event:165 Activation, Long term AHR receptor driven direct and indirect gene expression changes, Event: 18 Activation, AhR | AOP41: Sustained AhR Activation leading to Rodent Liver Tumours, AOP150: Aryl hydrocarbon receptor activation leading to early life stage mortality, via reduced VEGF, AOP21: Aryl hydrocarbon receptor activation leading to early life stage mortality, via increased COX-2, AOP131: Aryl hydrocarbon receptor activation leading to uroporphyria | Oxidative stress | ||
| GSK3β | tau hyperphosphorylation | Acute exposure to TCDD (25μg/kg body weight) induced neuronal toxicity in the cortex of female Sprague-Dawley rats. Exposure of rat PC12 cells to TCDD induced activation of GSK3β and decreased β-catenin in neural cells [104]. | Tau hyperphospho-rylation | ||||
| Tri-ortho-cresyl phosphate (TOCP) | Organophos-phorus based compound | LC3-II &Beclin expression | Oxidative stress induced increased in numbers of autophagic vacuoles, autophagy, and apoptosis | Low concentrations of TOCP induced autophagy and inhibited neurite outgrowth in a dose-dependent manner in human neuroblastoma SH-SY5Y cells. After treatment with TOCP, both LC3-II and the ratio of LC3-II/LC3-I, and autophagy proteins Atg5 and Beclin 1 were increased [105]. | Oxidative stress, Dysfunctional autophagy | ||
| Ultra-Fine Particles (UFP) | Air pollutants | SH containing proteins | Oxidation of intracellular GSH, resulting in the formation of GSSG which alters the redox state of the cell | Exposure to 50μg/mL UFP for 6 hours led to a decrease in GSH levels (from 17.1±1.8μM to 12.0±2.4μM) and an increase in GSSG (from 0.62±0.26μM to 1.60±0.2μM) in human aortic endothelial cells [106]. | AOP149: Oxidative Stress Leading to Hypertension | Oxidative stress | |
| Viruses (HSV-1/2/3, HIV) | Infectious pathogens | BACE-1 and nicastrin | A large proportion of Aβ plaques contain viral or bacterial DNA. Infections activate the amyloidogenic pathway of APP processing while inhibiting Aβ degradation, leading to intracellular Aβ accumulation. Infected cells also showed a disruption of Aβ autophagy, as evidenced by an accumulation of A-containing autophagic compartments that failed to fuse with lysosomes | 1. HSV-1 infection of human and rat neuronal cultures activated amyloidogenic pathway of APP processing while inhibiting Aβ degradation, leading to intracellular Aβ accumulation [107]. | Aβ oligomers | ||
| 2. Cell culture experiments revealed that intracellular concentrations of Aβ were significantly increased as early as 24-h post-infection and that infected cells increased their expression of both BACE-1 and nicastrin, a component of γ-secretase [108]. | |||||||
| 3. Antiviral treatments greatly reduced Aβ accumulation in HSV-1 infected cells. Treatment with the antiviral agent Acyclovir reduced the intensity of intracellular Aβ staining to 28% of that in untreated infected cells, while also reducing the levels of BACE-1 and nicastrin [109]. | |||||||
| 4. In cell culture aggregates from human AD brains, treatment with the HIV factor Tat led to increased concentrations of soluble A and a reduced activity of the Aβ-degrading enzyme neprilysin [110]. | |||||||
| Voriconazole | Antifungal drug | CYP46A1 | Decreased 24S-OHC levels, reduction of HMG-CoA reductase levels, downregulation of cholesterol synthesis and hypocholesterolemia | Intraperitoneal injections of voriconazole in mice reduced the levels of 24S- hydroxycholesterol in the brain, inhibiting the CYP46A1 [111]. | Cholesterol dysmetabolism | ||
| Zink (Zn) | Metals | Binding to Aβ oligomers, Aβ1-40-Zn2+ and Aβ1-42-Zn2+ | Neurotoxic soluble Aβ oligomers, affecting the calcium ion channel activity in synapsis, through disrupting nerve signal transmission and damage mitochondrial causes to increase free radial lead to cell death. | Zn2+ binding decreased the solvation energy (increase hydrophobicity) of Aβ oligomer, which enhanced the aggregation propensity, and that a higher concentration of Zn2+ could reduce aggregation kinetics. Aβ peptide can reduce Cu2+ to Cu+, and Fe3+ to Fe2+, facilitating the generation of reactive oxygen species H202 and OH• radical [112]. | Aβ oligomers, oxidative stress |
Further, a potential network of AOPs, including the seven approved AOPs (AOP3, AOP10, AOP12, AOP13, AOP42, AOP48, and AOP54) and the seven (still developing) AOPs (AOP17*, AOP26*, AOP134*, AOP152*, AOP279*, AOP281*, and AOP405*) is built, displaying the events commonly shared by these AOPs and the tau-driven AOP (Fig. 3). In this network, different MIEs, KEs, or AOs are shown to be shared by individual AOPs. The AOs of these included under development AOPs, namely AOP134*, AOP152*, AOP281*, and AOP405*, are described by decreased cognitive function or neurodegeneration, which are indirectly linked to the AO of the tau-driven AOP429. Apparently, all shown individual AOPs seem to share a common event, either directly or indirectly.
Fig. 3
Network of AOPs linked to the proposed tau-driven AOP for memory loss (ID:429, under development). This network is assembled of individual AOPs, available in AOP-wiki, sharing one or more events at molecular, cellular or organism level. Plausible molecular initiating events (MIEs) plugged into this tau-driven AOP are depicted by possible molecular targets of the environmental neurotoxicants [10]. The dotted lines indicate indirect links between the source and the target.
![Network of AOPs linked to the proposed tau-driven AOP for memory loss (ID:429, under development). This network is assembled of individual AOPs, available in AOP-wiki, sharing one or more events at molecular, cellular or organism level. Plausible molecular initiating events (MIEs) plugged into this tau-driven AOP are depicted by possible molecular targets of the environmental neurotoxicants [10]. The dotted lines indicate indirect links between the source and the target.](https://content.iospress.com:443/media/adr/2022/6-1/adr-6-1-adr220015/adr-6-adr220015-g003.jpg)
In this proposed AOP network for neurodegene-rative-related disorders or memory impairment, we integrated plausible MIEs which are potentially triggered by environmental neurotoxicants. Notably, only few of these stressors, including fipronil, 1,3,5-trinitro-1,3,5-triazine (RDX), endosulfan, and lead (Pb) have been elsewhere reported to trigger MIEs of the selected AOPs, confirming their potential involvement in neurodegenerative disorders. In more detail, fipronil, RDX, and endosulfan have been described as potential stressors for binding at picrotoxin site, iGABAR chloride channel (ID:667) leading to occurrence of epileptic seizures (ID:613) in the AOP10, while lead Pb for binding of antagonist, NMDA receptors (ID:201) leading to neurodegeneration (ID:352) and learning and memory impairment (ID:341) in the AOP12.
Among the presented AOPs, the AOP3 (AO: parkinsonian motor deficits (ID:896)) shares mitochondrial dysfunction 1 (KE:177) and neuroinflammation (KE:188), the AOP10 (AO: occurrence of epileptic seizure (ID:613)) shares the neuronal synaptic inhibition (KE:669), the AOP12 (AO: neurodegeneration (ID:352), AO: learning and memory impairment (ID:341)) shares neuroinflammation (KE:188), both AOP13 and AOP54 (AO: learning and memory impairment (ID:341)) share the decreased neuronal function (KE:386), the AOP48 (AO: learning and memory impairment (ID:341)) mitochondrial dysfunction 1 (KE:177), decreased neuronal network function (KE:618) and neuroinflammation (KE:188), with tau-driven AOP. Although the AOP42 does not seem to share any event with the tau-driven AOP, its AO (decreased cognitive function (ID:402)) is highly relevant to the memory loss, therefore, it is included in our network.
It is important to mention that some events of the tau-driven AOP, such as glucose and cholesterol dysmetabolism, dysfunctional autophagy, tau toxic oligomers, and hyperphosphorylation of tau, have not earlier been described in any of the existing AOPs in AOP-wiki. In this network of AOPs, we presented possible molecular targets for few of these not well-defined events in the context of the AOPs. Such an example, in the AOP3, its MIE, binding of inhibitor, NADH-ubiquinone oxidoreductase (complex I), has been also linked to glucose dysmetabolism via mitochondrial dysfunction. Other cases, in the AOP48, its MIE, binding of agonist, ionotropic glutamate receptors, such as NMDARs, and in the AOP10, its MIE, binding at picrotoxin site, iGABAR chloride channel, have been also linked to synaptic dysfunction and hyperphosphorylation of tau, respectively. These connections among events of different AOPs may support the usefulness of these networks for filling the gaps in understanding how same stressors-MIE interactions can lead to different adverse outcomes or different stressors-MIE interactions can lead to same adverse outcomes.
Despite the fact that some of the selected AOPs in this proposed AOP network have not approved by OECD yet, they can still provide useful information relevant to the adverse outcome of concern, memory loss. The available data for these AOPs still under development may need reassessment for confirming their potential involvement in the tau-driven AOP after endorsement by OECD.
Moreover, this presented network of AOPs for memory loss is consisting of individual AOPs which have earlier been linked to memory or cognitive related disorders, based on existing knowledge. These AOPs are available in AOP-wiki, providing all relevant information and allowing scientists to comment or update them. Of course, the developed AOPs are endorsed by OECD requiring extensive review of the provided data by experts.
It is worth noticing that human studies were prioritized for the empirical support used for weighing the links between the presented stressors and the potential MIEs, in this attempt to create this proposed AOP network. However, most of the available studies for the exposures of neurotoxicants have been performed on animals. This limitation may lead to potential weaknesses of our AOP network to adverse outcomes such as memory loss in humans. More human data are needed to further support the evidence for implicating these neurotoxicants or plausible MIEs in memory loss in humans.
CONCLUSIONS
The application of AOP network, using existing data, can serve a useful tool for a better understanding of the complexity of biological systems and for predicting the adverse effects. A proposed AOP network for the tau-driven AOP may help to contribute into unravelling of the interactions among existing mechanistic data linked to memory loss as an early phase of sAD pathology.
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This work was supported by ToxGenSolutions BV (https://toxgensolutions.eu) and 3Rs Management and Consulting ApS (https://3rsmc-aps.com).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Krewski D , Acosta D , Andersen M , Anderson H , Bailar JC , Boekelheide K , Brent R , Charnley G , Cheung VG , Green S , Kelsey KT , Kerkvliet NI , Li AA , McCray L , Meyer O , Patterson RD , Pennie W , Scala RA , Solomon GM , Stephens M , Yager J , Zeise L ((2010) ) Toxicity testing in the 21st century: A vision and a strategy. J Toxicol Environ Health, Part B 13: , 51–138. |
[2] | Bal-Price A , Meek ME ((2017) ) Adverse outcome pathways: Application to enhance mechanistic understanding of neurotoxicity. Pharmacol Ther 179: , 84–95. |
[3] | Ankley GT BR , Erickson RJ , Hoff DJ , Hornung MW , Johnson RD , Mount DR , Nichols JW , Russom CL , Schmieder PK , Serrrano JA , Tietge JE , Villeneuve DL ((2010) ) Adverse outcome pathways: A conceptual framework to support ecotoxicology research and risk assessment. Environ Toxicol Chem. 29: , 730–741. |
[4] | Langley GR , Adcock IM , Busquet F , Crofton KM , Csernok E , Giese C , Heinonen T , Herrmann K , Hofmann-Apitius M , Landesmann B , Marshall LJ , McIvor E , Muotri AR , Noor F , Schutte K , Seidle T , van de Stolpe A , Van Esch H , Willett C , Woszczek G ((2017) ) Towards a 21st-century roadmap for biomedical research and drug discovery: Consensus report and recommendations. Drug Discov Today 22: , 327–339. |
[5] | Marshall LJ , Austin CP , Casey W , Fitzpatrick SC , Willett C ((2018) ) Recommendations toward a human pathway-based approach to disease research. Drug Discov Today 23: , 1824–1832. |
[6] | AOP-Wiki ((2018) ) Adverse Outcome PathwayWiki (AOP-Wiki), https://aopwiki.org/. |
[7] | Becker RA , Ankley GT , Edwards SW , Kennedy SW , Linkov I , Meek B , Sachana M , Segner H , Van Der Burg B , Villeneuve DL , Watanabe H , Barton-Maclaren TS ((2015) ) Increasing scientific confidence in adverse outcome pathways: Application of tailored Bradford-Hill considerations for evaluating weight of evidence. Regul Toxicol Pharmacol 72: , 514–537. |
[8] | Knapen D , Angrish MM , Fortin MC , Katsiadaki I , Leonard M , Margiotta-Casaluci L , Munn S , O’Brien JM , Pollesch N , Smith LC , Zhang X , Villeneuve DL ((2018) ) Adverse outcome pathway networks I: Development and applications. Environ Toxicol Chem 37: , 1723–1733. |
[9] | Villeneuve DL , Angrish MM , Fortin MC , Katsiadaki I , Leonard M , Margiotta-Casaluci L , Munn S , O’Brien JM , Pollesch NL , Smith LC , Zhang X , Knapen D ((2018) ) Adverse outcome pathway networks II: Network analytics. Environ Toxicol Chem 37: , 1734–1748. |
[10] | Tsamou M , Pistollato F , Roggen EL ((2021) ) A tau-driven adverse outcome pathway blueprint toward memory loss in sporadic (late-onset) Alzheimer’s disease with plausible molecular initiating event plug-ins for environmental neurotoxicants. J Alzheimers Dis 81: , 459–485. |
[11] | Tsamou M , Carpi D , Pistollato F , Roggen EL ((2022) ) Sporadic Alzheimer’s disease- and neurotoxicity-related microRNAs affecting key events of tau-driven adverse outcome pathway toward memory loss. J Alzheimers Dis 86: , 1427–1457. |
[12] | Thomas B , Banerjee R , Starkova NN , Zhang SF , Calingasan NY , Yang L , Wille E , Lorenzo BJ , Ho DJ , Beal MF , Starkov A ((2012) ) Mitochondrial permeability transition pore component cyclophilin D distinguishes nigrostriatal dopaminergic death paradigms in the MPTP mouse model of Parkinson’s disease. Antioxid Redox Signaling 16: , 855–868. |
[13] | Mast N , Li Y , Linger M , Clark M , Wiseman J , Pikuleva IA ((2014) ) Pharmacologic stimulation of cytochrome P450 46A1 and cerebral cholesterol turnover in mice. J Biol Chem 289: , 3529–3538. |
[14] | Xiang Y , Kim KY , Gelernter J , Park IH , Zhang H ((2015) ) Ethanol upregulates NMDA receptor subunit gene expression in human embryonic stem cell-derived cortical neurons. PLoS One 10: , e0134907. |
[15] | Lovinger DM ((1995) ) Developmental decrease in ethanol inhibition of N-methyl-D-aspartate receptors in rat neocortical neurons: Relation to the actions of ifenprodil. J Pharmacol Exp Ther 274: , 164–172. |
[16] | Biermann T , Reulbach U , Lenz B , Frieling H , Muschler M , Hillemacher T , Kornhuber J , Bleich S ((2009) ) N-methyl-d-aspartate 2b receptor subtype (NR2B) promoter methylation in patients during alcohol withdrawal. J Neural Transm 116: , 615–622. |
[17] | Chandrasekar R ((2013) ) Alcohol and NMDA receptor: Current research and future direction. Front Mol Neurosci 6: , 14. |
[18] | Carpenter-Hyland EP , Chandler LJ ((2006) ) Homeostatic plasticity during alcohol exposure promotes enlargement of dendritic spines. Eur J Neurosci 24: , 3496–3506. |
[19] | Carito V , Ceccanti M , Ferraguti G , Coccurello R , Ciafrè S , Tirassa P , Fiore M ((2019) ) NGF and BDNF alterations by prenatal alcohol exposure. Curr Neuropharmacol 17: , 308–317. |
[20] | Lim S , Ahn SY , Song IC , Chung MH , Jang HC , Park KS , Lee K-U , Pak YK , Lee HK ((2009) ) Chronic exposure to the herbicide, atrazine, causes mitochondrial dysfunction and insulin resistance. PloS One 4: , e5186–e5186. |
[21] | Herron J , Reese RC , Tallman KA , Narayanaswamy R , Porter NA , Xu L ((2016) ) Identification of environmental quaternary ammonium compounds as direct inhibitors of cholesterol biosynthesis. Toxicol Sci 151: , 261–270. |
[22] | Lin S , Ren A , Wang L , Huang Y , Wang Y , Wang C , Greene ND ((2018) ) Oxidative stress and apoptosis in benzo[a]pyrene-induced neural tube defects. Free Radic Biol Med 116: , 149–158. |
[23] | Nie JS , Zhang HM , Zhao J , Liu HJ , Niu Q ((2013) ) Involvement of mitochondrial pathway in benzo[a]pyrene-induced neuron apoptosis. Hum Exp Toxicol 33: , 240–250. |
[24] | Sarma SN , Blais JM , Chan HM ((2017) ) Neurotoxicity of alkylated polycyclic aromatic compounds in human neuroblastoma cells. J Toxicol Environ Health, Part A 80: , 285–300. |
[25] | Chen NN , Luo DJ , Yao XQ , Yu C , Wang Y , Wang Q , Wang JZ , Liu GP ((2012) ) Pesticides induce spatial memory deficits with synaptic impairments and an imbalanced tau phosphorylation in rats. J Alzheimers Dis 30: , 585–594. |
[26] | Kim S , Cheon HS , Kim SY , Juhnn YS , Kim YY ((2013) ) Cadmium induces neuronal cell death through reactive oxygen species activated by GADD153. BMC Cell Biol 14: , 4. |
[27] | Wang QW , Wang Y , Wang T , Zhang KB , Jiang CY , Hu FF , Yuan Y , Bian JC , Liu XZ , Gu JH , Liu ZP ((2015) ) Cadmium-induced autophagy promotes survival of rat cerebral cortical neurons by activating class III phosphoinositide 3-kinase/beclin-1/B-cell lymphoma 2 signaling pathways. Mol Med Rep 12: , 2912–2918. |
[28] | Sanyal S , Agarwal N , Subrahmanyam D ((1986) ) Effect of acute sublethal and chronic administration of ddt (chlorophenotane) on brain lipid metabolism of rhesus monkeys. Toxicol Lett 34: , 47–54. |
[29] | Venkateswara Rao J ((2008) ) Brain acetylcholinesterase activity as a potential biomarker for the rapid assessment of chlorpyrifos toxicity in a euryhaline fish, Oreochromis mossambicus. Environ Bioindic 3: , 11–22. |
[30] | Karanth S , Liu J , Mirajkar N , Pope C ((2006) ) Effects of acute chlorpyrifos exposure on in vivo acetylcholine accumulation in rat striatum. Toxicol Appl Pharmacol 216: , 150–156. |
[31] | Lee JE , Lim MS , Park JH , Park CH , Koh HC ((2014) ) Nuclear NF-κB contributes to chlorpyrifos-induced apoptosis through p53 signaling in human neural precursor cells. Neurotoxicology 42: , 58–70. |
[32] | Sherif M ((2020) ) Chlorpyrifos-induced Developmental Neurotoxicity Using Human Neural Progenitor Stem Cells: Clue for Involvement of N-methyl-D-aspartate (NMDA) Receptors, University of Nottingham, Notting-ham eTheses. |
[33] | Park JH , Lee JE , Shin IC , Koh HC ((2013) ) Autophagy regulates chlorpyrifos-induced apoptosis in SH-SY5Y cells. Toxicol Appl Pharmacol 268: , 55–67. |
[34] | Lee JE , Park JH , Shin IC , Koh HC ((2012) ) Reactive oxygen species regulated mitochondria-mediated apoptosis in PC12 cells exposed to chlorpyrifos. Toxicol Appl Pharmacol 263: , 148–162. |
[35] | Saulsbury MD , Heyliger SO , Wang K , Johnson DJ ((2009) ) Chlorpyrifos induces oxidative stress in oligodendrocyte progenitor cells. Toxicology 259: , 1–9. |
[36] | Korade Z , Kim H-YH , Tallman KA , Liu W , Koczok K , Balogh I , Xu L , Mirnics K , Porter NA ((2016) ) The effect of small molecules on sterol homeostasis: Measuring 7-dehydrocholesterol in Dhcr7-deficient Neuro2a cells and human fibroblasts. J Med Chem 59: , 1102–1115. |
[37] | Anandhan A , Rodriguez-Rocha H , Bohovych I , Griggs AM , Zavala-Flores L , Reyes-Reyes EM , Seravalli J , Stanciu LA , Lee J , Rochet J-C , Khalimonchuk O , Franco R ((2015) ) Overexpression of alpha-synuclein at non-toxic levels increases dopaminergic cell death induced by copper exposure via modulation of protein degradation pathways. Neurobiol Dis 81: , 76–92. |
[38] | Stavinoha WB , Ryan LC , Smith PW ((1969) ) Biochemical effects of an organophosphorus cholinesterase inhibitor on the rat brain. Ann NY Acad Sci 160: , 378–382. |
[39] | Babot Z , Vilaró MT , Suñol C ((2007) ) Long-term exposure to dieldrin reduces γ-aminobutyric acid type A and N-methyl-D-aspartate receptor function in primary cultures of mouse cerebellar granule cells. J Neurosci Res 85: , 3687–3695. |
[40] | Ryan JC , Morey JS , Ramsdell JS , van Dolah FM ((2005) ) Acute phase gene expression in mice exposed to the marine neurotoxin domoic acid. Neuroscience 136: , 1121–1132. |
[41] | Lu J , Wu D , Zheng Y , Hu B , Cheng W , Zhang Z , Li M ((2013) ) Troxerutin counteracts domoic acid–induced memory deficits in mice by inhibiting CCAAT/enhancer binding protein β–mediated inflammatory response and oxidative stress. J Immunol 190: , 3466–3479. |
[42] | Ananth C , Thameem Dheen S , Gopalakrishnakone P , Kaur C ((2001) ) Domoic acid-induced neuronal damage in the rat hippocampus: Changes in apoptosis related genes (Bcl-2, Bax, caspase-3) and microglial response. J Neurosci Res 66: , 177–190. |
[43] | Ananth C , Dheen ST , Gopalakrishnakone P , Kaur C ((2003) ) Distribution of NADPH-diaphorase and expression of nNOS, N-methyl-D-aspartate receptor (NMDAR1) and non-NMDA glutamate receptor (GlutR2) genes in the neurons of the hippocampus after domoic acid-induced lesions in adult rats. Hippocampus 13: , 260–272. |
[44] | Hogberg HT , Bal-Price AK ((2011) ) Domoic acid-induced neurotoxicity is mainly mediated by the AMPA/KA receptor: Comparison between immature and mature primary cultures of neurons and glial cells from rat cerebellum. J Toxicol 2011: , 543512. |
[45] | Roberts DM , Dissanayake W , Rezvi Sheriff MH , Eddleston M ((2004) ) Refractory status epilepticus following self-poisoning with the organochlorine pesticide endosulfan. J Clin Neurosci 11: , 760–762. |
[46] | Jeong Mi M , Byeong Jo C ((2009) ) Acute endosulfan poisoning: A retrospective study. Hum Exp Toxicol 28: , 309–316. |
[47] | Parbhu B , Rodgers G , Sullivan JE ((2009) ) Death in a toddler following endosulfan ingestion. Clin Toxicol 47: , 899–901. |
[48] | Wages PA , Joshi P , Tallman KA , Kim H-YH , Bowman AB , Porter NA ((2020) ) Screening ToxCast™ for chemicals that affect cholesterol biosynthesis: Studies in cell culture and human induced pluripotent stem cell-derived neuroprogenitors. Environ Health Perspect 128: , 17014–17014. |
[49] | Lam M , Mast N , Pikuleva IA ((2018) ) Drugs and scaffold that inhibit cytochrome P450 27A1 in vitro and in vivo. Mol Neurobiol 93: , 101–108. |
[50] | Haruo K , Akira Y , Toshihiko K , Keiko S , Tetsuji O , Kumi S ((1985) ) Effects of insecticidal carbamates on brain acetylcholine content, acetylcholinesterase activity and behavior in mice. Toxicol Lett 29: , 153–159. |
[51] | Mohamed F , Senarathna L , Percy A , Abeyewardene M , Eaglesham G , Cheng R , Azher S , Hittarage A , Dissanayake W , Sheriff MHR , Davies W , Buckley NA , Eddleston M ((2004) ) Acute human self-poisoning with the N-phenylpyrazole insecticide fipronil–a GABAA-gated chloride channel blocker. J Toxicol Clin Toxicol 42: , 955–963. |
[52] | Lee JE , Kang JS , Ki YW , Lee SH , Lee SJ , Lee KS , Koh HC ((2011) ) Akt/GSK3β signaling is involved in fipronil-induced apoptotic cell death of human neuroblastoma SH-SY5Y cells. Toxicol Lett 202: , 133–141. |
[53] | Park JH , Lee JE , Lee S-J , Park SJ , Park KH , Jeong M , Koh HC ((2013) ) Potential autophagy enhancers protect against fipronil-induced apoptosis in SH-SY5Y cells. Toxicol Lett 223: , 25–34. |
[54] | Hori Y , Tanaka T , Fujisawa M , Shimada K ((2003) ) Toxicokinetics of DL-glufosinate enantiomer in human BASTA poisoning. Biol Pharm Bull 26: , 540–543. |
[55] | Calas AG , Richard O , MÊme S , Beloeil JC , Doan BT , Gefflaut T , MÊme W , Crusio WE , Pichon J , Montécot C ((2008) ) Chronic exposure to glufosinate-ammonium induces spatial memory impairments, hippocampal MRI modifications and glutamine synthetase activation in mice. Neurotoxicology 29: , 740–747. |
[56] | Uberti VH , de Freitas BS , Molz P , Bromberg E , Schröder N ((2020) ) Iron overload impairs autophagy: Effects of rapamycin in ameliorating iron-related memory deficits. Mol Neurobiol 57: , 1044–1054. |
[57] | Malhotra AK , Pinals DA , Weingartner H , Sirocco K , Missar CD , Pickar D , Breier A ((1996) ) NMDA receptor function and human cognition: The effects of ketamine in healthy volunteers. Neuropsychopharmacology 14: , 301–307. |
[58] | Krystal JH , Karper LP , Seibyl JP , Freeman GK , Delaney R , Bremner JD , Heninger GR , Bowers MB Jr , Charney DS ((1994) ) Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry 51: , 199–214. |
[59] | Islam R , Lynch JW ((2012) ) Mechanism of action of the insecticides, lindane and fipronil, on glycine receptor chloride channels. Br J Pharmacol 165: , 2707–2720. |
[60] | Neal AP , Guilarte TR ((2010) ) Molecular neurobiology of lead (Pb(2+)): Effects on synaptic function. Mol Neurobiol 42: , 151–160. |
[61] | Alkondon M , Costa ACS , Radhakrishnan V , Aronstam RS , Albuquerque EX ((1990) ) Selective blockade of NMDA-activated channel currents may be implicated in learning deficits caused by lead. FEBS Lett 261: , 124–130. |
[62] | Ferguson C KM , Audesirk G ((2000) ) Nanomolar concentrations of inorganic lead increase Ca2+ efflux and decrease intracellular free Ca2+ ion concentrations in cultured rat hippocampal neurons by a calmodulin-dependent mechanism. Neurotoxicology 21: , 365–378. |
[63] | Liu MC , Liu XQ , Wang W , Shen XF , Che HL , Guo YY , Zhao MG , Chen JY , Luo WJ ((2012) ) Involvement of microglia activation in the lead induced long-term potentiation impairment. PloS One 7: , e43924–e43924. |
[64] | Bihaqi SW , Zawia NH ((2013) ) Enhanced taupathy and AD-like pathology in aged primate brains decades after infantile exposure to lead (Pb). Neurotoxicology 39: , 95–101. |
[65] | Bihaqi SW , Bahmani A , Adem A , Zawia NH ((2014) ) Infantile postnatal exposure to lead (Pb) enhances tau expression in the cerebral cortex of aged mice: Relevance to AD. Neurotoxicology 44: , 114–120. |
[66] | Gąssowska M , Baranowska-Bosiacka I , Moczydłowska J , Tarnowski M , Pilutin A , Gutowska I , Strużyńska L , Chlubek D , Adamczyk A ((2016) ) Perinatal exposure to lead (Pb) promotes tau phosphorylation in the rat brain in a GSK-3β and CDK5 dependent manner: Relevance to neurological disorders. Toxicology 347-349: , 17–28. |
[67] | Zhang J , Cai T , Zhao F , Yao T , Chen Y , Liu X , Luo W , Chen J ((2012) ) The role of α-synuclein and tau hyperphosphorylation-mediated autophagy and apoptosis in lead-induced learning and memory injury. Int J Biol Sci 8: , 935–944. |
[68] | Huang Y , Liao Y , Zhang H , Li S ((2020) ) Lead exposure induces cell autophagy via blocking the Akt/mTOR signaling in rat astrocytes. J Toxicol Sci 45: , 559–567. |
[69] | Zhao Z-H , Du K-J , Wang T , Wang J-Y , Cao Z-P , Chen X-M , Song H , Zheng G , Shen X-F ((2021) ) Maternal lead exposure impairs offspring learning and memory via decreased GLUT4 membrane translocation. Front Cell Dev Biol 9: , 648261. |
[70] | Mohammadzadeh L , Abnous K , Razavi BM , Hosseinzadeh H ((2020) ) Crocin-protected malathion-induced spatial memory deficits by inhibiting TAU protein hyperphosphorylation and antiapoptotic effects. Nutr Neurosci 23: , 221–236. |
[71] | Pant R , Katiyar SK ((1983) ) Effect of malathion and acetylcholine on the developing larvae ofPhilosamia ricini (Lepidoptera: Saturniidae). J Biosci 5: , 89–95. |
[72] | Akbel E , Arslan-Acaroz D , Demirel HH , Kucukkurt I , Ince S ((2018) ) The subchronic exposure to malathion, an organophosphate pesticide, causes lipid peroxidation, oxidative stress, and tissue damage in rats: The protective role of resveratrol. Toxicol Res 7: , 503–512. |
[73] | Chang SH , Lee HJ , Kang B , Yu KN , Minai-Tehrani A , Lee S , Kim SU , Cho M-H ((2013) ) Methylmercury induces caspase-dependent apoptosis and autophagy in human neural stem cells. J Toxicol Sci 38: , 823–831. |
[74] | Farina M , Rocha JBT , Aschner M ((2011) ) Mechanisms of methylmercury-induced neurotoxicity: Evidence from experimental studies. Life Sci 89: , 555–563. |
[75] | Yuntao F , Chenjia G , Panpan Z , Wenjun Z , Suhua W , Guangwei X , Haifeng S , Jian L , Wanxin P , Yun F , Cai J , Aschner M , Rongzhu L ((2016) ) Role of autophagy in methylmercury-induced neurotoxicity in rat primary astrocytes. Arch Toxicol 90: , 333–345. |
[76] | Rao KSP , Rao KVR ((1984) ) Impact of methyl parathion toxicity and eserine inhibition on acetylcholinesterase activity in tissues of the teleost (Tilapia mossambica) —a correlative study. Toxicol Lett 22: , 351–356. |
[77] | Harischandra DS , Jin H , Anantharam V , Kanthasamy A , Kanthasamy AG ((2015) ) α-Synuclein protects against manganese neurotoxic insult during the early stages of exposure in a dopaminergic cell model of Parkinson’s disease. Toxicol Sci 143: , 454–468. |
[78] | Malecki EA ((2001) ) Manganese toxicity is associated with mitochondrial dysfunction and DNA fragmentation in rat primary striatal neurons. Brain Res Bull 55: , 225–228. |
[79] | Zhang J , Cao R , Cai T , Aschner M , Zhao F , Yao T , Chen Y , Cao Z , Luo W , Chen J ((2013) ) The role of autophagy dysregulation in manganese-induced dopaminergic neurodegeneration. Neurotoxic Res 24: , 478–490. |
[80] | Aminov Z , Haase RF , Pavuk M , Carpenter DO , Anniston Environmental Health Research C ((2013) ) Analysis of the effects of exposure to polychlorinated biphenyls and chlorinated pesticides on serum lipid levels in residents of Anniston, Alabama. Environ Health 12: , 108–108. |
[81] | Kamat PK , Rai S , Swarnkar S , Shukla R , Ali S , Najmi AK , Nath C ((2013) ) Okadaic acid-induced tau phosphorylation in rat brain: Role of NMDA receptor. Neuroscience 238: , 97–113. |
[82] | Kamat PK , Nath C ((2015) ) Okadaic acid: A tool to study regulatory mechanisms for neurodegeneration and regeneration in Alzheimer’s disease. Neural Regener Res 10: , 365–367. |
[83] | Ray A , Liu J , Karanth S , Gao Y , Brimijoin S , Pope C ((2009) ) Cholinesterase inhibition and acetylcholine accumulation following intracerebral administration of paraoxon in rats. Toxicol Appl Pharmacol 236: , 341–347. |
[84] | Huang M , Guo M , Wang K , Wu K , Li Y , Tian T , Wang Y , Yan W , Zhou Z , Yang H ((2020) ) HMGB1 mediates paraquat-induced neuroinflammatory responses via activating RAGE signaling pathway. Neurotoxic Res 37: , 913–925. |
[85] | Wang X , Luo F , Zhao H ((2014) ) Paraquat-induced reactive oxygen species inhibit neutrophil apoptosis via a p38 MAPK/NF-κB–IL-6/TNF-α positive-feedback circuit. PLoS One 9: , e93837. |
[86] | Cocco S , Secondo A , Del Viscovo A , Procaccini C , Formisano L , Franco C , Esposito A , Scorziello A , Matarese G , Di Renzo G , Canzoniero LMT ((2015) ) Polychlorinated biphenyls induce mitochondrial dysfunction in SH-SY5Y neuroblastoma cells. PloS One 10: , e0129481. |
[87] | Ku T , Chen M , Li B , Yun Y , Li G , Sang N ((2017) ) Synergistic effects of particulate matter (PM2.5) and sulfur dioxide (SO2) on neurodegeneration via the microRNA-mediated regulation of tau phosphorylation. Toxicol Res 6: , 7–16. |
[88] | Kang YJ , Tan H-Y , Lee CY , Cho H ((2021) ) An air particulate pollutant induces neuroinflammation and neurodegeneration in human brain models. Adv Sci 8: , 2101251. |
[89] | Saenen ND , Plusquin M , Bijnens E , Janssen BG , Gyselaers W , Cox B , Fierens F , Molenberghs G , Penders J , Vrijens K , De Boever P , Nawrot TS ((2015) ) In utero fine particle air pollution and placental expression of genes in the brain-derived neurotrophic factor signaling pathway: An ENVIRONAGE Birth Cohort Study. Environ Health Perspect 123: , 834–840. |
[90] | Chakra Reddy N , Venkateswara Rao J ((2008) ) Biological response of earthworm, Eisenia foetida (Savigny) to an organophosphorous pesticide, profenofos. Ecotoxicol Environ Saf 71: , 574–582. |
[91] | Adaramoye OA , Akinwonmi O , Akanni O ((2013) ) Effects of propofol, a sedative-hypnotic drug, on the lipid profile, antioxidant indices, and cardiovascular marker enzymes in wistar rats. ISRN Pharmacol 2013: , 230261. |
[92] | Williams LR , Aroniadou-Anderjaska V , Qashu F , Finne H , Pidoplichko V , Bannon DI , Braga MFM ((2011) ) RDX binds to the GABA(A) receptor-convulsant site and blocks GABA(A) receptor-mediated currents in the amygdala: A mechanism for RDX-induced seizures. Environ Health Perspect 119: , 357–363. |
[93] | Pal R , Bajaj L , Sharma J , Palmieri M , Di Ronza A , Lotfi P , Chaudhury A , Neilson J , Sardiello M , Rodney GG ((2016) ) NADPH oxidase promotes Parkinsonian phenotypes by impairing autophagic flux in an mTORC1-independent fashion in a cellular model of Parkinson’s disease. Sci Rep 6: , 22866. |
[94] | Dąbrowska-Bouta B , Sulkowski G , Sałek M , Frontczak-Baniewicz M , Strużyńska L ((2021) ) Early and delayed impact of nanosilver on the glutamatergic NMDA receptor complex in immature rat brain. Int J Mol Sci 22: , 3067. |
[95] | Skalska J , Frontczak-Baniewicz M , Strużyńska L ((2015) ) Synaptic degeneration in rat brain after prolonged oral exposure to silver nanoparticles. Neurotoxicology 46: , 145–154. |
[96] | Yang X , He Ce , Li J , Chen H , Ma Q , Sui X , Tian S , Ying M , Zhang Q , Luo Y , Zhuang Z , Liu J ((2014) ) Uptake of silica nanoparticles: Neurotoxicity and Alzheimer-like pathology in human SK-N-SH and mouse neuro2a neuroblastoma cells. Toxicol Lett 229: , 240–249. |
[97] | Zuo Y , Hu J , Xu X , Gao X , Wang Y , Zhu S ((2019) ) Sodium azide induces mitochondria-mediated apoptosis in PC12 cells through Pgc-1α-associated signaling pathway. Mol Med Rep 19: , 2211–2219. |
[98] | Theiss C , Meller K ((2000) ) Taxol impairs anterograde axonal transport of microinjected horseradish peroxidase in dorsal root ganglia neurons in vitro. Cell Tissue Res 299: , 213–224. |
[99] | Nakata T , Yorifuji H ((1999) ) Morphological evidence of the inhibitory effect of taxol on the fast axonal transport. Neurosci Res 35: , 113–122. |
[100] | Smith JA , Slusher BS , Wozniak KM , Farah MH , Smiyun G , Wilson L , Feinstein S , Jordan MA ((2016) ) Structural basis for induction of peripheral neuropathy by microtubule-targeting cancer drugs. Cancer Res 76: , 5115–5123. |
[101] | LaPointe NE , Morfini G , Brady ST , Feinstein SC , Wilson L , Jordan MA ((2013) ) Effects of eribulin, vincristine, paclitaxel and ixabepilone on fast axonal transport and kinesin-1 driven microtubule gliding: Implications for chemotherapy-induced peripheral neuropathy. Neurotoxicology 37: , 231–239. |
[102] | Zhao J , Tang C , Nie X , Xi H , Jiang S , Jiang J , Liu S , Liu X , Liang L , Wan C , Yang J ((2016) ) Autophagy potentially protects against 2,3,7,8-tetrachlorodibenzo-p-Dioxin induced apoptosis in SH-SY5Y cells. Environ Toxicol 31: , 1068–1079. |
[103] | Morales-Hernández A , Corrales-Redondo M , Marcos-Merino JM , González-Rico FJ , Sánchez-Martín FJ , Merino JM ((2016) ) AhR-dependent 2,3,7,8-tetrachloro-dibenzo-p-dioxin toxicity in human neuronal cell line SHSY5Y. Neurotoxicology 56: , 55–63. |
[104] | Xu G , Zhou Q , Wan C , Wang Y , Liu J , Li Y , Nie X , Cheng C , Chen G ((2013) ) 2,3,7,8-TCDD induces neurotoxicity and neuronal apoptosis in the rat brain cortex and PC12 cell line through the down-regulation of the Wnt/β-catenin signaling pathway. Neurotoxicology 37: , 63–73. |
[105] | Chen JX , Sun YJ , Wang P , Long DX , Li W , Li L , Wu YJ ((2013) ) Induction of autophagy by TOCP in differentiated human neuroblastoma cells lead to degradation of cytoskeletal components and inhibition of neurite outgrowth. Toxicology 310: , 92–97. |
[106] | Du Y , Navab M , Shen M , Hill J , Pakbin P , Sioutas C , Hsiai TK , Li R ((2013) ) Ambient ultrafine particles reduce endothelial nitric oxide production via S-glutathionylation of eNOS. Biochem Biophys Res Commun 436: , 462–466. |
[107] | De Chiara G , Marcocci ME , Civitelli L , Argnani R , Piacentini R , Ripoli C , Manservigi R , Grassi C , Garaci E , Palamara AT ((2010) ) APP processing induced by herpes simplex virus type 1 (HSV-1) yields several APP fragments in human and rat neuronal cells. PloS One 5: , e13989–e13989. |
[108] | Wozniak MA , Itzhaki RF , Shipley SJ , Dobson CB ((2007) ) Herpes simplex virus infection causes cellular β-amyloid accumulation and secretase upregulation. Neurosci Lett 429: , 95–100. |
[109] | Wozniak MA , Frost AL , Preston CM , Itzhaki RF ((2011) ) Antivirals reduce the formation of key Alzheimer’s disease molecules in cell cultures acutely infected with herpes simplex virus type 1. PloS One 6: , e25152–e25152. |
[110] | Rempel HC , Pulliam L ((2005) ) HIV-1 Tat inhibits neprilysin and elevates amyloid β. AIDS 19: , 127–135. |
[111] | Shafaati M , Mast N , Beck O , Nayef R , Heo GY , Björkhem-Bergman L , Lütjohann D , Björkhem I , Pikuleva IA ((2010) ) The antifungal drug voriconazole is an efficient inhibitor of brain cholesterol 24S-hydroxylase in vitro and in vivo. J Lipid Res 51: , 318–323. |
[112] | Boopathi S , Poma AB , Garduño-Juárez R ((2021) ) An overview of several inhibitors for Alzheimer’s disease: Characterization and failure. Int J Mol Sci 22: , 10798. |


