
A New Hypothesis for Alzheimer’s Disease: The Lipid Invasion Model
Abstract
This paper proposes a new hypothesis for Alzheimer’s disease (AD)—the lipid invasion model. It argues that AD results from external influx of free fatty acids (FFAs) and lipid-rich lipoproteins into the brain, following disruption of the blood-brain barrier (BBB). The lipid invasion model explains how the influx of albumin-bound FFAs via a disrupted BBB induces bioenergetic changes and oxidative stress, stimulates microglia-driven neuroinflammation, and causes anterograde amnesia. It also explains how the influx of external lipoproteins, which are much larger and more lipid-rich, especially more cholesterol-rich, than those normally present in the brain, causes endosomal-lysosomal abnormalities and overproduction of the peptide amyloid-β (Aβ). This leads to the formation of amyloid plaques and neurofibrillary tangles, the most well-known hallmarks of AD. The lipid invasion model argues that a key role of the BBB is protecting the brain from external lipid access. It shows how the BBB can be damaged by excess Aβ, as well as by most other known risk factors for AD, including aging, apolipoprotein E4 (APOE4), and lifestyle factors such as hypertension, smoking, obesity, diabetes, chronic sleep deprivation, stress, and head injury. The lipid invasion model gives a new rationale for what we already know about AD, explaining its many associated risk factors and neuropathologies, including some that are less well-accounted for in other explanations of AD. It offers new insights and suggests new ways to prevent, detect, and treat this destructive disease and potentially other neurodegenerative diseases.
INTRODUCTION
This introduction sets out the basic evidence for the lipid invasion model of Alzheimer’s disease (AD).
AD is a neurodegenerative disorder first described by the German physician Alois Alzheimer in 1907 [1]. It is a cause of dementia characterized by the extensive death of brain cells and associated with two types of strongly-staining deposits within the brain, called amyloid plaques and neurofibrillary tangles (NFTs). While both plaques and NFTs are individually seen in other forms of neurodegeneration [2–5], their occurrence together is largely unique to AD.
AD has emerged as the most common dementia, accounting for over half of all such disorders, with an especially high prevalence among over-85 year-olds in the developed world [6]. Yet, in the century or so since AD’s discovery, relatively limited progress has been made in understanding its etiology or in developing effective treatments to stop its progression [7–10].
There have been two major hypotheses so far as to what drives AD progression: the cholinergic hypothesis and the amyloid cascade hypothesis.
The cholinergic hypothesis
The cholinergic hypothesis, which emerged in the 1980s, sought to explain the disease in terms of reduced synthesis of acetylcholine within the brain [11]. However, while substantial evidence points to AD-associated deficits in the cholinergic projection system of the brain [11], treatments based on this model do not greatly slow disease progression [11, 12].
The amyloid cascade hypothesis
Since the 1990s the dominant model for explaining AD has been the amyloid cascade hypothesis. This postulates that amyloid-β (Aβ), a proteolytic product of amyloid-β protein precursor (AβPP), is the fundamental cause of the disease [13]. It is backed by a substantial body of evidence, not least the fact that Aβ is the main component of amyloid plaques [13].
Key evidence of the important role of AβPP and Aβ in AD includes:
1. Patients with inherited forms of the disease, collectively referred to as familial AD (FAD), have a number of genes related to AβPP processing that are abnormal, leading to excess production of Aβ [14].
2. People with Down’s syndrome (DS) possess an extra copy of chromosome 21, on which AβPP resides. It is common for them to develop a form of dementia largely indistinguishable from AD [15], if they survive into their 40s.
3. There is substantial evidence, in postmortem human brains and animal and in vitro studies, that amyloid plaque formation in AD is associated with failure of mechanisms that normally clear Aβ from the brain [16], including reduced levels of expression and activity of a number of key proteins. These include low-density lipoprotein receptor-related protein-1 (LRP1), P-glycoprotein, and phosphatidylinositol binding clathrin assembly protein (PICALM) [17–20].
4. Postmortem studies show that receptor for advanced glycation end products (RAGE), a receptor that transports Aβ into the brain (i.e., in the reverse direction to LRP1, P-glycoprotein, and PICALM), is found at significantly higher levels in cerebral endothelial cells in humans with AD, as compared to those without AD [21].
5. Apolipoprotein E4 (APOE4), a well-established inherited AD risk factor [22], is associated with slower efflux of ApoE-Aβ complexes, as compared with other APOE isoforms, together with impaired degradation and increased deposition and aggregation of Aβ itself. This is thought to be a primary reason why Aβ is more prone to forming amyloid plaques in individuals possessing APOE4, as compared to those with APOE2 and APOE3 [18, 23].
Any viable model of AD needs to take into account the importance of AβPP, Aβ, and other proteolytic products of AβPP, and the core evidence behind the amyloid cascade hypothesis. However, the amyloid cascade hypothesis does not provide a comprehensive explanation of AD.
Problems with the amyloid cascade hypothesis
Aβ is certainly a primary cause of neurodegeneration in AD in many cases. However, there are several aspects of AD which are not well-accounted by the amyloid cascade hypothesis. These are discussed in the Supplementary Material, and include:
1. Poor correlation between amyloid plaque distribution and disease progression.
2. Evidence that late-onset forms of AD (LOAD) do not always display elevated production of Aβ.
3. Questions about how so many diverse lifestyle risk factors for AD (hypertension [24], obesity [25–27], diabetes [28], smoking [29], stress [30], sleep deprivation [31], and head injury [32]) can all promote AD by increasing Aβ levels.
4. Lack of a convincing explanation why aging is the main risk factor for AD [33], specifically why the normal functioning of a small number of proteins should be so vulnerable to the aging process, and result in a disease that affects such a large number of elderly people, while leaving earlier age groups largely unaffected.
Viewing AD as a mechanical/structural disease
Most common diseases of aging involve some form of structural/mechanical damage, rather than changes in metabolic pathways, and the gradual accumulation of such damage explains why they only tend to emerge in old age. For example: hemorrhagic stroke and cardiovascular disease [34, 35], osteoporosis [36], cataracts and macular degeneration [37, 38], hearing loss [39], osteoarthritis [40], and cancer (given that it typically involves progressive damage to DNA) [41].
The next sections outline how viewing AD also as a structural damage-related disease could unlock an explanation for AD which is compatible with the amyloid hypothesis as well as with all the above-mentioned AD-associated risk factors.
Evidence of blood-brain barrier (BBB) disruption in AD
One extensively researched structural aspect of AD is the BBB, and substantial evidence exists to demonstrate an association of AD with BBB damage/disruption [42–47].
This includes postmortem and imaging studies of AD-affected brains [16], showing evidence of infiltration into the brain of plasma-associated cells and proteins that should not be there, as well as cerebral microbleeds, basement membrane thinning,and other direct evidence of neurovascular dysfunction. Similarly, many biofluid studies, looking at biomarkers of BBB disruption in live human subjects, have shown a good correlation between such disruption and the degree of AD progression. These are discussed in more detail in the Supplementary Material.
Some further support (although not conclusive proof) for BBB disruption in AD comes from the fact that AD brains commonly stain for Evans Blue, which is normally substantially excluded by the BBB [44, 48–50]. Also, in the reverse direction, S100 calcium-binding protein B, normally only found only in the central nervous system (CNS) [51], has been shown to be present in blood plasma (i.e., outside of the brain) in AD cases [52].
Further evidence, from a different perspective, of an association of BBB disruption with AD, relates to chronic traumatic encephalopathy (CTE). This progressive degenerative condition commonly affects athletes (especially those who have participated in contact sports, such as boxing, soccer, rugby and American football) and others with a history of brain trauma, especially concussion [53]. CTE typically shows many similarities with AD [53]. These include severe memory deficits, large-scale neuronal loss, extensive NFTs, and, frequently in advanced cases, diffuse amyloid plaques [53]. Crucially, CTE is characterized by BBB disruption, including disruption of tight junctions (TJs) and of BBB basement membrane integrity [53–57].
Finally, the many risk factors for LOAD mentioned previously, including aging, APOE4, hypertension, diabetes, lowered serum levels of sex hormones, diabetes, obesity, smoking, chronic sleep deprivation, stress, and head injury, are also all associated with vascular damage, including damage to the BBB [26, 27, 58–68].
The role of Aβ in BBB disruption
From the above evidence, AD is strongly linked with disruption of the BBB. So, what causes this disruption, and how does it associate with Aβ accumulation?
There is, in fact, substantial experimental evidence of Aβ itself directly damaging the BBB in humans, rodents, and other animals [46, 69–72]. Damage includes alterations in TJ protein distribution and expression in brain endothelial cells [46, 71–76], increases in matrix metalloproteinase expression [74, 77, 78], oxidative stress [76, 79], and possibly apoptosis [80–82] and dysregulation of calcium homoeostasis [46, 80]. Finally, there is further, less direct, evidence that Aβ can damage the BBB, for example, in cases of cerebral amyloid angiopathy (CAA) [74, 76, 77, 81, 83]. This is a disease of small and medium blood vessels of the brain (or wider CNS) and meninges, caused (in the majority of cases) by Aβ deposits in the blood vessel walls, and characterized by micro-hemorrhages [84]. CAA shares many pathogenetic features with AD (especially in terms of the amyloidogenic pathway), with both diseases frequently co-occurring, such that around 80–90% of AD cases show some degree of CAA [83–87]. But critically, it does not require plaque formation.
Aβ can be found on either side of the BBB. This means that it can damage the BBB from both sides. This, in turn, may help to explain evidence suggesting that externally sourced Aβ may be a major constituent of amyloid plaques found within AD brains, having passed through a compromised BBB [88], as mentioned in the Supplementary Material.
A recent theory, based on studies in mice and humans [88, 89], has proposed that Aβ-related BBB damage results from external assault by excessive numbers of Aβ-containing lipoproteins, which might be expected from a diet high in saturated fats and cholesterol. Among other evidence, this is based on the fact that Aβ is a regulatory protein involved in lipoprotein formation, primarily of triacyl glyceride (TAG)-enriched lipoproteins originating from absorptive enterocytes [90]. Building on this, since these lipoproteins will also contain high levels of Aβ, it is possible that they may contribute to cerebral amyloid plaque formation, as well as to BBB damage [88].
The above evidence suggests that Aβ might have a dual role in AD progression, firstly, by damaging the BBB from either side, and secondly, by causing neurodegeneration in the brain, in the form of amyloid plaques or as individual molecules or oligomers, including triggering NFTs, as described in the amyloid hypothesis.
But, as explained above, BBB disruption may result from any one or more of the AD-associated risk factors, allowing Aβ into the brain to cause neurodegeneration. This includes Aβ, alone or with others of these risk factors, but does not require it.
The lipid factor identified by Alois Alzheimer
A damaged BBB will clearly not just enable Aβ to enter the brain. Many other molecules and larger structures will also be allowed through. This could explain an additional feature of AD, which was identified by Alois Alzheimer in his original research.
In describing the first diagnosed case of AD, Alzheimer makes frequent reference to “adipose saccules”, “lipoid granules”, and similar lipid-based aggregations within both glial and neuronal cells, as well as to amyloid plaques, NFTs, and general brain degeneration [1, 91]. Lipids are a class of biomolecules that do not dissolve (or not easily) in water. They include fatty acids, glycerolipids, glycerophospholipids, and sphingolipids, as well as cholesterol and other sterols. (See the Lipid Maps database for a comprehensive list [92]). Descriptions of these lipid aggregations appear almost as frequently in Alzheimer’s writings as do the references to what have become known as amyloid plaques and NFTs.
Likewise, colleagues of Alzheimer, describing this and other AD cases, refer to “abundant deposits of fatty substances and pigments”, and “major accumulation of lipoid products in the ganglion cells, glia and vasculature” [91].
It is clear from Alzheimer’s writings that he regarded “this degree of lipid degeneration”, as he described it, to be as much a hallmark of the disease we now call AD as the associated plaques and NFTs [91].
Evidence of a role for lipids in AD
A growing body of research supports this association between AD and lipids:
1. As mentioned above, one of the biggest risk factors for AD, after aging, is having the E4 isoform of apolipoprotein E (i.e., APOE4) [22], which is a major component in lipoprotein transport of lipids within the body. Similarly, as mentioned earlier, one of the other biggest AD risk factors, Aβ, has been identified as a regulatory component of chylomicrons, a class of lipoprotein which serves to transport TAGs and other lipids from the intestine to the liver [93, 94]. Thus, two of the most important risk factors for AD (after aging) both have important roles in lipid transport.
2. Genes for another apolipoprotein, apolipoprotein J (also known as clusterin), and for the ABCA7 (ATP-binding cassette sub-family A member 7) protein, involved in lipid homoeostasis, have also been identified as susceptibility loci for AD [95–97].
3. Elevated serum levels of cholesterol, triglycerides, and apolipoprotein B (ApoB) have all been associated with higher risk of getting AD [98–100]. So, taken together, lipids, lipid transport, and homoeostasis all play an important role in AD progression.
4. There is also substantial experimental data pointing to the direct involvement of cholesterol in Aβ formation [101, 102], not least the high levels of cholesterol and other lipids found within amyloid plaques [103, 104]. Also, NFTs occur extensively in Niemann Pick type C (NPC), another disease associated with dementia, which is caused by internal cholesterol accumulation within cells [105]. Substantially elevated levels of cholesterol are found in NFT-bearing neurons in both NPC and AD [106].
5. Finally, brains of those who have died from, or with, AD contain unexpectedly high levels of albumin [107–109] (which transports free fatty acids (FFAs) within the bloodstream) and ApoB (another major component of lipoprotein-mediated lipid transport). ApoB is found, along with Aβ, within amyloid plaques, and also in NFTs [110–112].
Substantial levels of these lipid transport proteins, albumin and ApoB, should not be found within the brain compartment in normal circumstances, as they are both generally, but not conclusively, believed to only be expressed outside of it, and should be excluded from entering it by the BBB [113–119].
This suggests that the “lipid degeneration” that Alois Alzheimer referred to may have its origins outside the brain, and results from some kind of external lipid incursion through damaged portions of the BBB.
The unexpected role of cholesterol and FFAs
So, which of these invading lipids are most likely to cause AD? For several reasons, cholesterol and FFAs are the most likely candidates. This is partly because of their large numbers as a proportion of readily available plasma lipids [120], but mainly because their presence in excess numbers within the brain could explain a number of brain-associated anomalies seen in AD, including plaques, NFTs, lipid accumulation, neuroinflammation, and bioenergetic changes.
At first sight this second assertion makes little sense, because cholesterol and FFAs are essential to the survival of the brain, and pose no danger, in normal quantities, to it. So how does damage to the BBB turn such lipids into a threat? To explain this, it is helpful to understand the important role of BBB in respect of lipids.
The BBB: a barrier between two different lipid systems
It would appear from its unique architecture that the BBB’s main purpose is to exclude certain cells and molecules from the brain compartment (meaning the brain parenchyma and interstitial fluids) and the wider CNS. This architecture is described in detail in the Supplementary Material.
Because of its architecture, the BBB is known to substantially prevent lipids passing through it, certainly those that remain bound to, or within, their normal transport partners [121–124].
In addition, there are different lipid transport systems either side of the BBB, as will be explained in much more detail below, with major differences in the types of lipoprotein transporters employed. These assemblies transport cholesterol, fatty acids (FAs), and other lipid classes, all of them water-insoluble, within blood plasma and other extracellular watery fluids.
In the blood plasma compartment, such lipoproteins tend to be relatively large and contain large quantities of lipids [125], and there is also abundant non-lipoprotein transport of FFAs, mostly bound to albumin [114]. Within the brain compartment, separated from its external blood supply by the BBB, lipoproteins tend to be relatively small and lipid-poor [117], and there is little evidence of non-lipoprotein FFA transport [126].
The main reason for these differences in lipid transport on each side of the BBB lies in differences in availability of key transport proteins—principally, apolipoproteins, which determine the size and lipid content of lipoproteins, and serum albumin, the main non-lipoprotein FFA transporter. It is this difference which underlies the lipid invasion model, as explained in the next section.
The lipid invasion model
The differing lipid transport systems explain the apparent paradox, referred to above, whereby external lipids, especially FFAs and cholesterol-rich lipoproteins, which are so necessary for the normal functioning and survival of brain cells, may start to pose a threat to their normal functioning and survival, if they break through the BBB. These external lipids are (for the most part) transported in a different way to what is normally seen within the brain compartment [114, 117, 118, 122, 127], and their flow is often much less tightly controlled [118, 126, 128], so brain cells exposed to them become vulnerable. Neurons and inflammatory microglial cells, especially, become overloaded and overstimulated. This is explained in more detail in later sections.
Over time, this lipid invasion will result in various forms of lipid-associated brain pathology. In the case of invading FFAs this will take three main forms: 1) changes in brain bioenergetics, oxidative stress, lipid peroxidation, and mitochondrial damage resulting from excess FFA accumulation within neurons; 2) neuroinflammation; 3) anterograde amnesia (AA) resulting from tonic inhibition and disruption of neurogenesis. These are all characteristics that have been associated with AD [129–131].
Other characteristics of AD, such as changes in endosomal-lysosomal pathway disruption, cerebral amyloidosis and NFT formation can also be explained by lipid influx in the form of external lipoproteins. With the exception of high-density lipoprotein (HDL), these plasma lipoproteins are typically much larger than CNS lipoproteins, and therefore much richer in cholesterol, which has also been linked with AD [102, 132, 133], particularly in connection with amyloidosis and NFTs.
The lipid invasion model argues that failure of the BBB allows lipids in the plasma compartment to enter the brain compartment and cause various forms of damage, including AD, because these lipids are transported, stored, and regulated in very different ways to resident lipids.
How the lipid invasion model fits in with other lipid- and BBB-based explanations of AD
Numerous articles have shown an association between BBB dysfunction and AD, many of them suggesting some form of causal link [43, 134, 135]. Likewise, many articles have shown an association between lipids and AD [91, 136, 137]. A few of these have suggested that some form of lipid dysfunction may have a role in AD progression [136, 137]. But it appears that only one group of researchers (from Curtin University of Technology, Perth, Australia, subsequently referred to as “the Curtin explanation”), have sought to explain AD in a way that can be said to link these two aspects together [138]. This has already been referred to earlier, and links dietary fats (specifically saturated FAs and cholesterol) with BBB dysfunction and amyloid plaque formation, providing a compelling explanation for the presence of ApoB within these plaques [93, 94, 111, 138].
However, this explanation is largely limited to plaque formation and does not include a more general role for external lipids in AD progression, in terms of cerebral amyloidosis, NFT formation, AA, and the other AD-associated pathologies mentioned earlier. Therefore, by explaining the origins of AD in terms both of BBB disruption and subsequent external lipid invasion, and in showing how all aspects of disease progression are primarily driven by such lipid incursions, the lipid invasion model provides a new approach to understanding AD.
Similarities between AD and alcohol-associated brain damage
An additional reason for identifying BBB damage and subsequent lipid influx as the driver of AD is the similarity between the overall structural pattern of neurodegeneration seen in AD and that seen in alcohol-related brain damage (ARBD), resulting from chronic exposure of the brain to ethanol.
Ethanol passes relatively easily through the BBB. Once in the brain compartment, as will be explained in later sections, it will have some of the same overall effects as overexposure to FFAs, namely neuroinflammation, tonic inhibition, and neurodegeneration. However, unlike AD, ARBD does not involve exposure of the brain to external FAs and, even more importantly, cholesterol-rich lipoproteins. This explains why the amyloid plaques, NFTs, and endosomal-lysosomal abnormalities seen in AD are not seen in ARBD [139], while some other features of AD, including neuroinflammation and (somewhat limited) neurodegeneration and inhibition of neurogenesis, are seen in ARBD [140–144].
There is also extensive evidence that the detrimental effects observed in the brain from chronic alcohol exposure are the result not only of neurodegeneration resulting from neuroinflammation, but also (judged from what is seen in rodent models) of reduced levels of neurogenesis [140, 141, 143, 145], similar to what is seen in AD.
These similarities and differences yield important insights into the etiology of AD, which reinforce the model, particularly increases in neuroinflammation, neurogenetic inhibition, and AA, all mediated by FFAs. A more detailed discussion of this evidence appears in the Supplementary Material.
Following the above outline of the lipid invasion model, the next section provides a more detailed explanation and justification.
TECHNICAL EVIDENCE AND MORE DETAILED EXPLANATION OF THE LIPID INVASION MODEL
This section first looks at differences in lipid metabolism between the brain and plasma compartments of the body and the importance of this to the model. Then the many ways in which the BBB becomes damaged will be explained. Finally, the consequences for the brain compartment of the lipid incursions that result from such BBB disruption will be examined. This includes changes to brain energetics, neuroinflammation, neurodegeneration, AA, endosomal-lysosomal disorder, lipid accumulations, amyloid plaques, and NFTs.
Differences between lipid metabolism on either side of the BBB
There are many differences in lipid metabolism either side of the BBB. This is most apparent in the case of FAs and cholesterol. (Here, FAs denote all fatty acids, including esterified FAs such as TAGs, as well as FFAs, which are not esterified.) Because these differences in metabolism and transport are so critical to the lipid invasion model, accounting for many of the pathological hallmarks observed in AD, they are explained in detail below.
Fatty acid metabolism
For efficient transport within plasma, the vast majority of FAs, being highly hydrophobic, must travel within lipoproteins, or must be bound to the protein serum albumin, to improve solubility [114, 127].
Immediately after eating, dietary FAs are bound to glycerol as TAGs and then transported within the class of lipoproteins known as chylomicrons. These chylomicrons constitute a major proportion of the plasma transport pool [127, 146]. At the same time, high blood glucose levels associated with satiety lead to hepatic neogenesis of FFAs and glycerol, with the resulting TAGs being transported in the blood within very low-density lipoproteins (VLDLs) [127, 146]. During subsequent plasma transport most of the TAGs within chylomicrons and VLDLs are taken up by tissues, principally adipocytes and muscle cells [147, 148].
The chylomicrons and VLDLs are relatively large (typically within a range of 100–1000 nm and 30–80 nm, respectively [127, 146]) and lipid-rich by virtue of their association with ApoB isoforms. Such lipoprotein-mediated FA transport appears to allow only very restricted access to the postnatal brain compartment across the BBB. This is partly because ApoB is (according to current orthodoxy) synthesized only in the liver and in enterocytes, but not in the CNS [115, 127]. But more importantly, it is because of the architecture of the BBB, mentioned earlier and detailed in the Supplementary Material [117, 149–151]. This substantially excludes larger lipoproteins, with only much smaller, less lipid-rich, HDL appearing to cross the BBB in any quantity [118].
During the fasting state, adipocytes release stored FFAs directly back into the bloodstream, with the majority being subsequently bound to serum albumin [114, 127]. Because serum albumin is created almost exclusively in the liver [114, 152, 153] and cannot pass readily through the BBB [113, 154, 155], it has until recently been assumed that albumin-bound FFAs must also be largely excluded from the brain, in the same way as external lipoprotein-associated FAs. More recent findings suggest that more FFAs are taken up by the brain compartment through the BBB than originally envisaged [156–159].
At first sight, this would appear to invalidate the claim by the lipid invasion model that invading FFAs from the plasma compartment, entering the brain through a disrupted BBB, cause many of the pathologies seen in AD. Why are such pathologies not seen even when the BBB is not disrupted? This question is addressed in the next section.
FAs and the brain. Despite this evidence of external plasma-originating FFAs entering the brain compartment, it is clear that, almost uniquely among organs, the brain in humans and other animals does not rely on FAs (certainly in albumin-bound FFA form) as a primary energy source [156, 160]. This is despite the fact that the brain has a high energy requirement, and other organs with high energy needs, such as the heart and kidney, preferentially oxidize FAs [156, 161], reflecting the wide availability of FFAs in the bloodstream, and the fact that they provide about twice the energy content of glucose and similar sugars [162]. Instead, during the fasting state when glucose availability is low, the liver will typically transform such plasma FFAs into much smaller ketone bodies, which, having been transported through the BBB, are used in preference to FFAs as an energy source by the brain [163–165].
The most likely reason why the brain does not use whole FFAs extensively for its energy needs is that ATP generation from FFAs requires substantially more oxygen than from alternative energy substrates such as glucose, lactate, butyrate, and ketone bodies [156]. As a consequence, such FFA usage would increase the risk of brain hypoxia, as well as generating higher levels of oxidative stress, thus proving toxic to neurons [128, 156, 162, 166]. Another possible reason is that the rate of ATP generation from FFAs is slower than from the other mentioned energy substrates, meaning that FFAs may not be able to yield ATP fast enough (or flexibly enough) for rapidly firing neurons, especially under conditions of sustained activity [156].
What happens to those FFAs that make it across the BBB from the plasma compartment into the brain compartment? As explained in detail in the Supplementary Material, the evidence strongly suggests that the vast majority will be taken up by astrocytes. Typically, these will be sequestrated within lipid droplets inside the host astrocyte, with most being converted into ketone bodies or the much shorter FFA butyrate, and used as fuel, either by the host astrocyte or by local neurons and other neighboring brain cells [128]. Others of these FFAs will be transported onwards in esterified form (mainly as TAGs or phospholipids) within lipoproteins, for use by other neighboring cells. Certainly, given that albumin transport is no longer available to these extravasated FFAs, and given the absence of any obvious alternatives to albumin in the CNS [156, 167–169], some form of lipoprotein-mediated transport seems the most likely alternative.
However, there are important differences between lipoprotein transport in the CNS, and lipoprotein transport in the plasma compartment. These differences have critical implications if the BBB becomes disrupted and are explained in more detail in the next section.
Lipoprotein transport of lipids inside and outside the brain/CNS. In contrast to what is seen in plasma, the principal apolipoproteins expressed in the CNS (including Apo E, D, and J [117, 170]) associate into lipoprotein particles that are relatively small (typically less than 20 nm) and contain modest amounts of lipids [127, 168, 171]. Such CNS lipoprotein particles tend to resemble HDL [117, 146, 168, 171] much more than the larger ApoB-associated lipoproteins that predominate outside the CNS.
Furthermore, astrocytes are known to be a principal source of many of these CNS-originating apolipoproteins, particularly Apo E and J [117, 171, 172], and lipoproteins have been isolated from the conditioned medium of astrocytic cultures [170]. They can then be assembled into HDL-like (or even smaller) lipoproteins within the astrocyte body, and secreted into the interstitial fluid of the brain compartment, for onward transport and uptake by neurons and glial cells [173].
From this description, it would appear that FA transport and metabolism in the CNS is very different from that seen in the rest of the body. In particular, there appears to be little, if any, non-lipoprotein FA transport in the CNS and, on average, CNS lipoproteins are much smaller than their plasma equivalents. In many respects, FA transport seems more tightly controlled in the brain compartment than outside it [118, 126, 128]. Certainly, it is hard to see how such differences could be maintained without a BBB that is substantially intact.
Major differences either side of the BBB are also seen in cholesterol transport and regulation, as outlined in the next section. Later sections will explain the implications such differences in lipid organization will have, as predicted by the lipid invasion model, when the BBB becomes disrupted, and how this all relates to AD.
Cholesterol metabolism either side of the BBB
Numerous studies in humans and other higher animals have shown that, except in very early fetal development, almost all cholesterol in the CNS is of local origin, relying on endogenous de novo biosynthesis rather than external, lipoprotein-mediated provision [117, 122, 150, 151]. This appears to be true for a wide range of animals, including birds and mammals, with much of cholesterol production for neuronal consumption being delegated to local astrocytes [117, 122, 174].
Moreover, cholesterol turnover in the mature CNS is very low, typically only around 5% of the turnover seen in the rest of the body [122, 150, 151]. A major reason for this is that a large proportion of such cholesterol remains locked up within the insulating myelin sheath that permanently encases the axons of many neurons, particularly within the white matter of the brain [124]. Much of this myelination takes place early in organismal development [175]. Any remaining freely circulating cholesterol within the brain is kept at low levels by various mechanisms, including removal across the BBB in the form of 24S-hydroxycholesterol [122, 176].
In the rest of the body (and thus on the other side of the BBB), a large proportion of cholesterol is either of dietary origin or else the result of neogenesis in the liver [127, 146]. From there much of it is transported in the same large, lipid-rich, ApoB-containing lipoproteins (i.e., chylomicrons, VLDLs, and low-density lipoproteins (LDLs)) that also transport dietary and liver-derived FAs [115, 127, 146], albeit chylomicrons typically contain relatively little cholesterol. For this reason, as well as the properties of the BBB itself (as explained in the Supplementary Material), much cholesterol of non-CNS origin is unable to cross the BBB [117, 150, 151, 177].
By contrast, within the brain compartment and wider CNS, cholesterol is transported within the same HDL-like lipoproteins. As stated, such lipoproteins tend to be small, compared to many of their plasma counterparts, typically containing only modest amounts of cholesterol and other lipids [127].
Overall differences in lipid transport either side of the BBB
Overall, it is clear that, from birth onwards, in humans and other animals [178], the BBB separates two compartments, the brain/CNS and external systemic/plasma compartments, with very different lipid systems [122, 179]. Compared to the rest of the body, the mature CNS compartment is distinguished by a much lower circulation of lipids, with apparently restricted external lipid supplementation and a set of lipoproteins that are noticeably smaller and less lipid-rich. Much of this difference can be accounted for by the BBB (which substantially prevents lipoproteins crossing from one compartment into another), and by the fact that ApoB and albumin (or their functional equivalents) are not (according to current orthodoxy) produced in the brain compartment.
Given that this distinction appears to have first emerged comparatively early in vertebrate evolution [180, 181], it seems plausible that serious disruption to the BBB will have lipid-related consequences. This can be inferred from the fact that the mature brain compartment has evolved for so long to function in an environment low in circulating lipids compared with the rest of the body. And, given the relative volumes of the two compartments, it seems likely the brain compartment will be the most vulnerable to lipid incursion if no longer substantially isolated from the rest of the body by the BBB.
Above the evidence for BBB disruption in AD (from postmortem, biofluid and imaging studies, and CTE) was provided. However, this still leaves the question of how the disruption occurs, and how this relates to the many identified AD risk factors. This is explained in the next section.
The causes of BBB disruption in the lipid invasion model
Understanding how the BBB becomes disrupted in AD is central to the lipid invasion model. It largely explains why AD has all the risk factors it has, and why the disease is so much more common in old age.
As stated earlier, almost all the major identified risk factors for AD (aging, Aβ, APOE4, head injury, hypertension, diabetes, obesity, smoking, lowered sex hormones, sleep deprivation, and stress) have been shown to also be causally associated with the structural and functional disruption of the BBB. However, each one of the mentioned risk factors causes the BBB disruption via different mechanisms.
These mechanisms are detailed in the Supplementary Material, and include reduced junctional strength between BBB endothelial cells (especially TJs) [27, 45, 65, 76, 82, 182–190], inflammation and oxidative stress [191–194], lowered pericyte numbers and activity levels [135, 195–201], impaired astrocyte activity [189, 202, 203], and chronic dysregulated neogenesis [82, 185, 189, 204]. In addition, alterations in expression levels and activity of matrix metallopeptidases (MMPs) and basement membrane (BM) proteins drive changes in BM thickness, among other BBB-associated alterations [59, 74, 78, 184, 186, 189, 205–207]. In many cases more than one such mechanism may be involved, some of which may interact. For instance, APOE4-associated BBB breakdown has been shown to be driven by activation of a pro-inflammatory pathway in pericytes, acting on MMP9, which cleave TJ, as well as BM proteins [182, 195, 208]. Similarly, some AD risk factors such as smoking and sleep deprivation have been shown to damage the BBB by causing inflammation, which in turn causes reductions in expression of TJ proteins or redistribution of such proteins around endothelial cells [191, 192, 194, 209].
However, this is an area of intensive research, and many details remain to be determined.
BBB disruption as a biomarker for AD
Having outlined the various mechanisms by which the BBB may be disrupted by most risk factors for AD, it is important to clarify further why such BBB disruption is relevant to AD. This is because, as well as being directly associated with FAD and LOAD (as inferred from postmortem, biofluid and imaging studies, and from CTE, as outlined earlier), BBB breakdown has been shown to be an early biomarker of human cognitive decline, irrespective of Aβ reactivity and APOE status [210]. And there is plenty of reason to believe this is not a coincidence. Recent studies suggest that, perhaps because of zonal differences in BBB composition and other properties (as shown in mice and postmortem human brains [211, 212]), the aging brain is especially prone to BBB damage in the hippocampal and wider medial temporal lobe regions. These regions are, of course, known to be particularly associated with memory formation and cognition. Such zonal differences of the BBB, may reflect differences in the properties of pericytes within the BBB [63, 210]. Whatever the reason, it suggests that these brain regions will be subject to greater levels of lipid invasion than other, less affected regions. Further research is needed into such zonal differences to establish a more complete, and more quantitative picture.
Aging, AD, and BBB disruption
Collectively, these facts provide a simple reason, according to the model, why LOAD incidence is closely correlated with age. Not only is the wear and tear of aging itself directly associated with loss of BBB integrity, but so are most of the risk factors—including diabetes/obesity, hypertension, smoking, stress, sleep deprivation, and declining sex hormones—whose cumulative effects on the BBB may reasonably be expected to build up with age for those affected.
Similarly, the above account suggests a plausible explanation for why inherited forms of AD (i.e., FAD) occur earlier but much less commonly than non-inherited forms (i.e., LOAD). This is because FAD is the product of excess Aβ production, resulting from rare genetic mutations, but commencing relatively early in life [213]. By contrast, LOAD is the product of many factors, the majority of whose effects can be expected to result in equivalent cumulative levels of damage only later in life, with a mean age of onset around 20 years later than that of inherited forms [213].
Wider implications of the different mechanisms of BBB disruption in FAD and LOAD
In both FAD and LOAD, the resulting AD-associated neuropathology is explained by the model in terms of the resulting invasion of external lipids through the disrupted BBB. Except that in FAD, earlier Aβ-driven BBB disruption means that this invasion occurs much sooner than in LOAD, and, according to the model, it is this earlier external lipid invasion, and not so much the direct actions of Aβ on the brain, that primarily explain the earlier AD onset and progression typically seen in FAD. As mentioned above, it is these invading lipids that primarily drive the changes to brain energetics, neuroinflammation, neurodegeneration, AA, amyloid plaques, and NFTs seen in AD, rather than raised Aβ levels.
The role of APOE4 in BBB disruption and in AD
According to the model, APOE4 can be expected to have a potential role in both FAD and LOAD. In the case of FAD, it is known to reduce Aβ clearance from the brain [18, 23], to impair Aβ degradation [214], and to increase Aβ deposition and aggregation [215]. Such increased deposition is evident not just within amyloid plaques but also within cerebral blood vessels [214, 216], where postmortem studies in humans and animals show that it is associated with increased prevalence of CAA, BBB dysfunction, and other consequences leading to impaired blood vessel integrity [214, 216–220]. Therefore, in FAD, APOE4 contributes to BBB disruption both indirectly, by promoting Aβ-mediated disruption, and directly, via other mechanisms associated with APOE4-mediated BBB disruption (as described in the Supplementary Material).
In the case of LOAD, APOE4 may be viewed as just one of a number of AD risk factors that contribute to BBB dysfunction, as outlined earlier in this section. But it could also be that the same mechanisms by which APOE4 is believed to aggravate Aβ-mediated BBB damage in FAD (as outlined in the previous paragraph), also act on physiologically normal levels of Aβ to cause similar BBB damage, albeit at a lower rate than seen in FAD. Taken together with the BBB-disrupting mechanisms that APOE4 is associated with (as detailed in the Supplementary Material), this suggests that APOE4 may be much more damaging to the BBB than other LOAD-associated risk factors. Certainly, according to the model, this would seem to provide an obvious reason, why homozygous carriers of APOE4 have a two- to five-increased risk of developing LOAD, compared to non-APOE4 carriers, and with a substantially earlier age of onset [22].
Curtin explanation of BBB disruption in AD
As mentioned previously, there is possibly a further means by which high levels of Aβ may contribute to BBB damage, but in a different way to that described above; one that is relevant to both FAD and LOAD, and yet wholly consistent with the lipid invasion model. This is when Aβ-mediated disruption of the BBB originates from outside the brain, the Aβ being transported within triglyceride-rich lipoproteins, as explained in detail in the Supplementary Material. However, such disruption is not a requirement of the model.
Whatever the cause of BBB disruption (including Aβ-mediated damage), the model argues that the consequences for the brain, resulting from external lipid invasion, will be the same, thus explaining why so many disparate risk factors (aging, Aβ, APOE4, hypertension, diabetes, lowered sex hormone levels, obesity, smoking, stress, sleep deprivation, and head injury) are associated with the same disease, i.e., AD.
The next three main sections explain how, according to the model, this external lipid invasion of the brain results in the collection of neuropathologies that characterize AD.
AD-relevant consequences of FFA influx to the brain
This section and the next principally focus on the consequences of external FFA influx in terms of changes to brain energetics, increased neuroinflammation, neurodegeneration, AA, and inhibition of neurogenesis. Then the effects of exposure to excessive levels of cholesterol within certain externally-derived lipoproteins is explained.
FFA-mediated changes to brain bioenergetics and oxidative stress
External fatty acid ingress into the brain will substantially alter local brain bioenergetics, which are normally characterized by glucose/lactate-dominated energy metabolism [221, 222]. In its place, according to the model, such bioenergetics will be pushed in the direction of ketone metabolism, a shift that has been widely reported in AD [223, 224]. However, this differs from other accounts of glucose hypometabolism in AD. These tend to focus on age-related faulty mitochondrial bioenergetics, accumulation of Aβ within mitochondria coupled with oxidative stress, and other such factors [224], all driving a shift from glucose-driven bioenergetics towards a ketogenic/β-oxidation pathway. According to the lipid invasion model it is, in many ways, the other way round.
The underlying mechanism behind this bioenergetic shift, and its implications, is explained in more detail in the Supplementary Material. Put simply, it is argued that invading external FFAs will mainly be taken up by astrocytes, most being converted into ketone bodies and used to meet neuronal energy needs rather than those of the host astrocyte. The resulting increase in ketone availability will tend to inhibit glycolysis and drive a local bioenergetic shift towards ketone metabolism.
Nevertheless, some of the invading FFAs in interstitial fluids will be taken up by neurons, which have very limited capacities for β-oxidation or lipid sequestration [128, 156, 225, 226], leaving neuronal mitochondria directly exposed to these FFAs. Such FFA exposure has been shown to have various negative consequences for affected mitochondria, including interfering with electron transport, collapsing the electrochemical proton gradient and uncoupling oxidative phosphorylation [156, 227, 228]. As well as leading to a reduction in the rate of ATP production and in the calcium ion retention capacity of these mitochondria [156, 228], these FFA-associated deficits will also lead to increased reactive oxygen species production, lipid peroxidation, and oxidative stress, opening of the mitochondrial permeability transition pore and release of pro-apoptotic factors [156, 227–229]. Together with glucose hypometabolism, all these above-mentioned changes are seen in AD [166, 230–233].
Finally, recent evidence of non-mitochondrial oxidative phosphorylation within the myelin sheath, involving subunits of electron transport chain complexes and of F1F0-ATP synthase [234], suggests that this sheath may also be highly vulnerable to oxidative stress, as a result of exposure of these electron transport chain elements to intact FFAs [235], i.e., in much the same way as described for neuronal mitochondria. White matter loss is, of course, widely associated with AD, its occurrence coinciding with, and often preceding, the appearance of other AD markers [236]. It may be that this exposure to invading FFAs provides a primary explanation for such progressive local demyelination in AD.
Having shown how, according to the model, external FFA invasion disrupts normal brain bioenergetics in a similar ketogenetic direction as seen in AD, the next section shows how external FFAs can also trigger the neuroinflammation (defined as inflammation of the brain and other nervous tissue) seen in the disease.
FFA-mediated neuroinflammation
In order to appreciate how FFAs might cause neuroinflammation in AD (with accompanying damage to the brain), as predicted by the lipid invasion model, it is useful to understand firstly how such neuroinflammation is driven by exposure to excess ethanol in ARDB.
Extensive research (principally in rodents, but echoed by findings in plasma and in postmortem brains of human alcoholics) has established that neuroinflammation is an important cause of ethanol-induced neurodegeneration [145, 237–239], and that microglia are central agents of such inflammation [145, 237, 240, 241]. This central role is perhaps unsurprising, given that the “immune-privileged” status conferred on the brain compartment by the BBB leaves microglia as the primary immune cell [242, 243], a role not seen as a rule in macrophages in the rest of the body. Their ability to perform this role seems to depend in large part on being abnormally sensitive to a wide range of ligands [243–245], and this, in turn, helps to explain why chronic ethanol, largely unobstructed by the BBB, causes such extensive inflammatory damage to the brain compartment over time [246]. Additionally, the mechanism through which this occurs suggests that FFAs, provided they could pass through the BBB in quantity, would have similar inflammatory effects, since both are known to powerfully activate the same critical receptor.
Ethanol activation of microglia [246] is accompanied by upregulation of the transcription factor NF-κB [247, 248] and other macromolecules known to be involved in inflammation and in the immune response. The evidence (at least in mice) suggests that toll-like receptors, particularly toll-like receptor 4 (TLR4, a receptor that binds bacterial lipopolysaccharide), are central to such activation and the subsequent neuroinflammation [248, 249].
If TLR4 is central to ethanol-induced neuroinflammation, then there seems every reason to think that FFAs entering the brain compartment would have similar neuroinflammatory effects. Saturated (but not, apparently, unsaturated) FAs are known to activate TLR4 in macrophages, leading, in turn, to activation of NF-κB and the other pro-inflammatory molecules just mentioned [250, 251]. And TLR4 activation in adipocytes by saturated FAs (and perhaps by some unsaturated FAs) is an essential step in lipid-induced type 2 diabetes mellitus [250, 252], which is now thought to be substantially inflammatory in nature [252–254]. In support of this, knockdown or ablation of TLR4 has been shown to inhibit both FFA-induced and ethanol-induced inflammation [248, 250–252], as well as protecting against FA-induced diabetes.
Given how responsive microglia are to pathological stimuli [255–258], one could reasonably expect activation by both ethanol and FFAs to result in far more vigorous inflammatory activity than seen in other parts of the body. And, while the relative affinities of ethanol and FFAs for TLR4 have yet to be determined, the fact that saturated fatty acyl groups are known (at least from animal studies) to be crucial to TLR4 recognition of lipopolysaccharide (TLR4’s principal pathogenic ligand) [259] suggests that FFAs should have a substantially higher affinity than ethanol for TLR4. Thus, the relatively low levels of FFAs seen in human plasma (generally agreed to fall within an average range of 0.3–0.6 mM [260, 261]) should be sufficient to generate a steady level of neuroinflammation, following major BBB damage, especially if they are accompanied by pathogen-associated lipopolysaccharide, as seen in ethanol-induced liver injury [262]. So it may be FFAs, rather than TLR4 stimulation by amyloid [263], that are the primary driver of microglial-based neuroinflammation in LOAD.
Whatever the case, oxidative stress and neuroinflammation are not the only drivers of neurodegeneration, either in ARBD or AD. Another important factor is inhibition of neurogenesis, which can account for the very specific areas of atrophy seen in both diseases and the failure to recover, particularly in the case of AD. Again, the ethanol-induced mechanisms thought to underlie such inhibition in ARBD will most likely also be triggered by exposure to FFAs—another key prediction of the lipid invasion model of AD. The sections below explain this in much greater detail.
FFA-mediated neurodegeneration and AA
In the previous section, it was shown how ethanol and FFAs induce neuroinflammation by similar mechanisms. The same can also be said of neurodegeneration, with many of the insights gained about ethanol-induced neurodegeneration also being applicable to AD.
Much of ethanol-mediated neurodegeneration has been linked (at least in rats) to inhibition of neurogenesis [142, 239], with many studies suggesting that neurogenetic deficits are almost as important a factor as neuroinflammation in such degeneration [239]. Here too, TLR4, and other ethanol-sensitive toll-like receptors, are likely to have an important inhibitory role [264, 265], diminishing proliferation of adult neuronal progenitor cells and restricting neuronal differentiation. Such inhibition would obviously be most apparent in the main adult neurogenic niches, i.e., the subgranular and subventricular zones (SGZ and SVZ), which provide new neurons and glial cells to (respectively) the hippocampus and the olfactory bulb [266]. This could explain deficiencies in learning and olfaction common to both AD and ARBD [267–274], given that such receptors should also be sensitive to FFAs, as explained in the previous section.
In addition, current evidence indicates that the overall level of neurodegeneration in both cases is determined almost as much by the relentlessness of the ethanol assault as by the concentrations involved [140, 142, 239]. Thus, one can reasonably infer that constant exposure of the brain compartment to plasma levels of FFAs is likely to overwhelm the brain’s capacity to recover, especially in the elderly. Such a conclusion is further supported by evidence that inhibition of neurogenesis, by both ethanol and FFAs, does not need to rely on the TLR4 receptor alone, and may, in fact, depend more on GABAergic effects. The relevance of this to the lipid invasion model is explained in the next section.
GABAergic effects
Recent research (in postmortem rodent and human brains) has indicated a possible role for the inhibitory neurotransmitter gamma-aminobutyric acid (GABA) in the development of AD [275–277], with a number of possible mechanisms being suggested. One such mechanism, GABA-induced tonic inhibition within the hippocampus, provides an obvious explanation of why AD is characteristically associated with AA. However, the proposed source of this excess GABA within hippocampal-resident reactive astrocytes, does not have much support in the literature, either for AD or ARBD.
The lipid invasion model provides an alternative mechanism, extending beyond tonic inhibition. This mechanism accounts for the coexistence of AA in AD and ARBD, as well as other similarities, including the similar patterns of neurodegeneration within the two major neurogenic niches, i.e., the SGZ and SVZ, as outlined in the previous section. Underlying this common mechanism is the proven affinity of ethanol, and very likely affinity of FFAs, for GABAA receptors (GABAARs), as well as the recently-discovered role of high-affinity extrasynaptic GABAARs in both tonic inhibition and anesthesia-associated amnesia. This mechanism is explained more fully in the next section.
FFAs as anesthetics
From the 1950s onward, Samson and Dahl, and other groups, showed that injection of FFAs induced light general anesthesia in a range of mammals [278–281]. Anesthetic potency increases with FFA chain length, and thus hydrophobicity, up to an undetermined cut-off, in line with the well-established Meyer-Overton correlation [278, 279, 282, 283]. Such potencies fall within the low millimolar range (expressed both as moles per liter and moles per kilogram of body weight) and show similar potencies to structurally comparable 1-alcohols (including ethanol) [284], as well as to alkanes [285] and aldehydes [286]. All of which suggests that plasma levels of FFAs may be only an order of magnitude or so less than general anesthetic potency levels.
Given the general correlation between hydrophobicity and anesthetic potency first described (independently) by Meyer and Overton [287], it would perhaps be surprising if FFAs did not show similar anesthetic potencies to structurally very similar fatty alcohols/alkanols [287–290]. Nor, given the established anesthetic properties of various steroids [291, 292], should it be a surprise that other lipids might display similar properties.
Anesthesia, AA, and extrasynaptic GABAARs
The immediate significance of lipids’ anesthetic properties to dementia, in terms of the model, lies in the fact that the vast majority of anesthetic agents are known to cause AA, at concentrations well below those needed for clinical anesthesia [287, 293, 294]. Such low-level anesthesia-induced AA is now known to involve extrasynaptic GABAARs [293, 294], whose subunit composition (including either α5 or δ subunits) gives them sufficient sensitivity to respond to low levels of ambient GABA [295]. It is the resulting low-level inhibitory currents, collectively termed “tonic inhibition”, which are associated with AA [296–298]. (By contrast lower-affinity synaptic GABAARs, with different subunit compositions, respond only to the higher concentrations of GABA released within their associated synapses, with the resulting phasic inhibition causing the other, later, anesthetic effects [287, 294, 299], including analgesia, immobility, and unconsciousness.) In support of this, pharmacological and genetic knockdown of extrasynaptic α5- and δ-containing GABAARs in mice has been shown to improve performance on learning and memory tasks [300–302], possibly by lowering the threshold for long-term potentiation [303–305].
The reason for this is that GABAARs have associated ion channels, which become permeable to chloride (and, to a lesser extent, HCO3) ions, in response to GABA ligation [306–308]. Upon such activation, chloride ions flow through these GABAAR channels in a direction determined by their electrochemical gradient. Since mature neurons maintain an excess of chloride ions externally, the normal response to GABA binding is therefore for these negative ions to flow in through the GABAAR channels, increasing the negative membrane potential and thereby hyperpolarizing (i.e. inhibiting) the affected neuron [307, 309]. Tonic inhibition is the extrasynaptic form of this [310, 311]. The majority of anesthetic agents (including those that are only weakly anesthetic, such as ethanol) are known to enhance this GABA binding, acting primarily as positive allosteric modulators [312, 313]. Accordingly, they tend to inhibit normal activity in mature neurons of the CNS [312, 314, 315], resulting in the various anesthetic effects, among the earliest of which is AA—the form of amnesia primarily associated with AD, at least in its early stages.
Extrasynaptic GABAARs and neurogenesis
However, recent research has shown that the same high-affinity extrasynaptic GABAARs that mediate tonic inhibition in mature neurons [295, 316] also play a significant role in neurogenesis and neuronal plasticity [317, 318]. In support of this, pharmacological and genetic suppression of tonic GABA inhibition in animals, including by downregulation of extrasynaptic GABAAR activity, is associated with marked improvements in functional recovery after stroke [302, 319]. This is in agreement with findings in animal studies that suggest that increased GABA tonic inhibitory currents, in the days after stroke, hinder recovery [302, 320].
Since extrasynaptic GABAARs containing the δ-subunit are known to be especially sensitive to positive modulation by ethanol [321, 322], this may explain the alcohol-mediated neurodegeneration seen in ARBD. As explained earlier, disruption of neurogenesis appears to be critical to the neurodegenerative effects of ethanol upon the brain. Specifically, studies (primarily in rodents and humans) suggest that chronic exposure of the brain to ethanol is characterized from comparatively early on by erosion of the hippocampal region [141, 239], loss of interneurons (the primary product of neurogenesis [323]), AA [267, 268], and olfactory deficits [269, 270].
An obvious explanation for these findings is inhibition of neurogenesis in the SGZ and SVZ, given that the former supplies neurons to other hippocampal regions [266, 324], while the latter is known to replenish the olfactory bulb interneurons via the rostral migratory stream [266, 325]. Much evidence suggests that FFAs have, on average, similar, if not higher, anesthetic potency levels to ethanol [278, 288, 290, 326–328]. This implies a similar affinity for GABAARs, so it may well be that chronic exposure of the brain to excess FFAs over many years will have similar results. This would provide an explanation of why AD and ARBD share these hallmark effects on the brain.
The possible mechanisms for this are discussed in the Supplementary Material, with much still to be determined.
Implications of GABAAR-mediated neurogenic inhibition for FFAs
Whatever the precise mechanism may be, assuming that ethanol inhibition of neurogenesis in the SVZ and SGZ is mediated by GABAARs, then FFAs are likely to have a similar inhibitory effect. This is because, as with ethanol, a number of studies (in animals) point towards GABAARs as the principal target and mediator of FFA’s limited anesthetic properties. This would seem the most obvious reason why n-alkanes, n-alcohols, and n-aldehydes, which are structurally very similar to FFAs, share anesthetic properties in common (as alluded to earlier). As with FFAs, the anesthetic potency of these macromolecules increases with chain length, but only up to a certain “cut off” length [284, 285, 290, 329–331]. This, together with direct evidence that the n-alcohols act on GABAARs [330, 332], as does the endogenous FFA anesthetic, oleamide [333–335], suggests a common binding site. More direct evidence for this comes from the observed antagonizing effects of long-chain FFAs on GABAAR-mediated anesthesia by (much stronger) volatile anesthetics [336, 337], along with other evidence of direct interactions between FFAs and GABAARs [338–340].
Taken together, this evidence suggests that FFAs, entering the brain through a damaged BBB (and therefore greatly in excess of the brain’s normal circulating levels), will, if maintained over the long-term, tend to seriously disrupt neurogenesis by acting on GABAARs. Given the presence of major sites of neurogenesis in the SGZ and SVZ, this will principally manifest itself in AA and olfactory deficits. As stated earlier, and explained in more detail in the Supplementary Material, AA and olfactory deficits are both seen in AD, but also in ARBD—the latter driven by excess exposure to ethanol, which is known to act on GABAARs. This, then, credibly accounts for the similarities between AD and ARBD mentioned earlier.
The last two main sections have focused on external FFAs, and how they are predicted by the lipid invasion model to cause various forms of neuropathology (changes in brain bioenergetics, neuroinflammation, AA, and inhibition of neurogenesis) seen in AD. The next section now looks at the role of external cholesterol in the model, and how it accounts for cerebral amyloidogenesis, endosomal-lysosomal pathology, and NFT formation, which are three other major characteristics of AD.
AD-specific consequences of brain exposure to external cholesterol
If the above account explains many of the similarities seen between AD and ARBD, it does not explain why, unlike ARBD, AD is characterized by profuse plaques and tangles. The lipid invasion model of AD explains this by the fact that the BBB has to be disrupted for external FFAs to substantially enter the brain, unlike in ARBD, where ethanol can pass through the BBB relatively unhindered [341]. Consequently, in AD the brain is also exposed to other molecules from which it is normally protected, including lipoproteins, which are much larger and more lipid-laden than those normally found within the CNS compartment.
There are good reasons to think that such lipoproteins may account for the amyloid plaques and NFTs that characterize AD, as a result of excess exposure of the brain to cholesterol, as explained in the next section. Not least of these reasons is the presence of ApoB within amyloid plaques and NFTs, as mentioned earlier. This is because ApoB-containing lipoproteins, which are normally excluded from the brain compartment by the BBB, typically contain higher levels of cholesterol than are normally seen in this compartment.
The role of excess cholesterol in amyloidogenesis
Substantial evidence (including from postmortem human AD brain tissue) suggests that cholesterol may have a role in causing amyloid plaques, as seen in AD. Cholesterol does this by increasing production of amyloidogenic Aβ from AβPP at the expense of the production of alternative non-amyloidogenic fragments that normally occurs [101, 102, 342]. Increasing levels of cholesterol stimulates an amyloidogenic pathway (which involves β- and γ-secretases, two proteases involved in AβPP proteolysis), while at the same time inhibiting a non-amyloidogenic pathway involving α-secretase in place of β-secretase [102, 133, 343] (Fig. 1). By contrast, cholesterol depletion inhibits the amyloidogenic pathway and enhances non-amyloidogenic processing, resulting in lower levels of Aβ [343, 344], as shown by experiments in human cell lines.
Fig. 1
Comparison of amyloidogenic and non-amyloidogenic processing of AβPP.
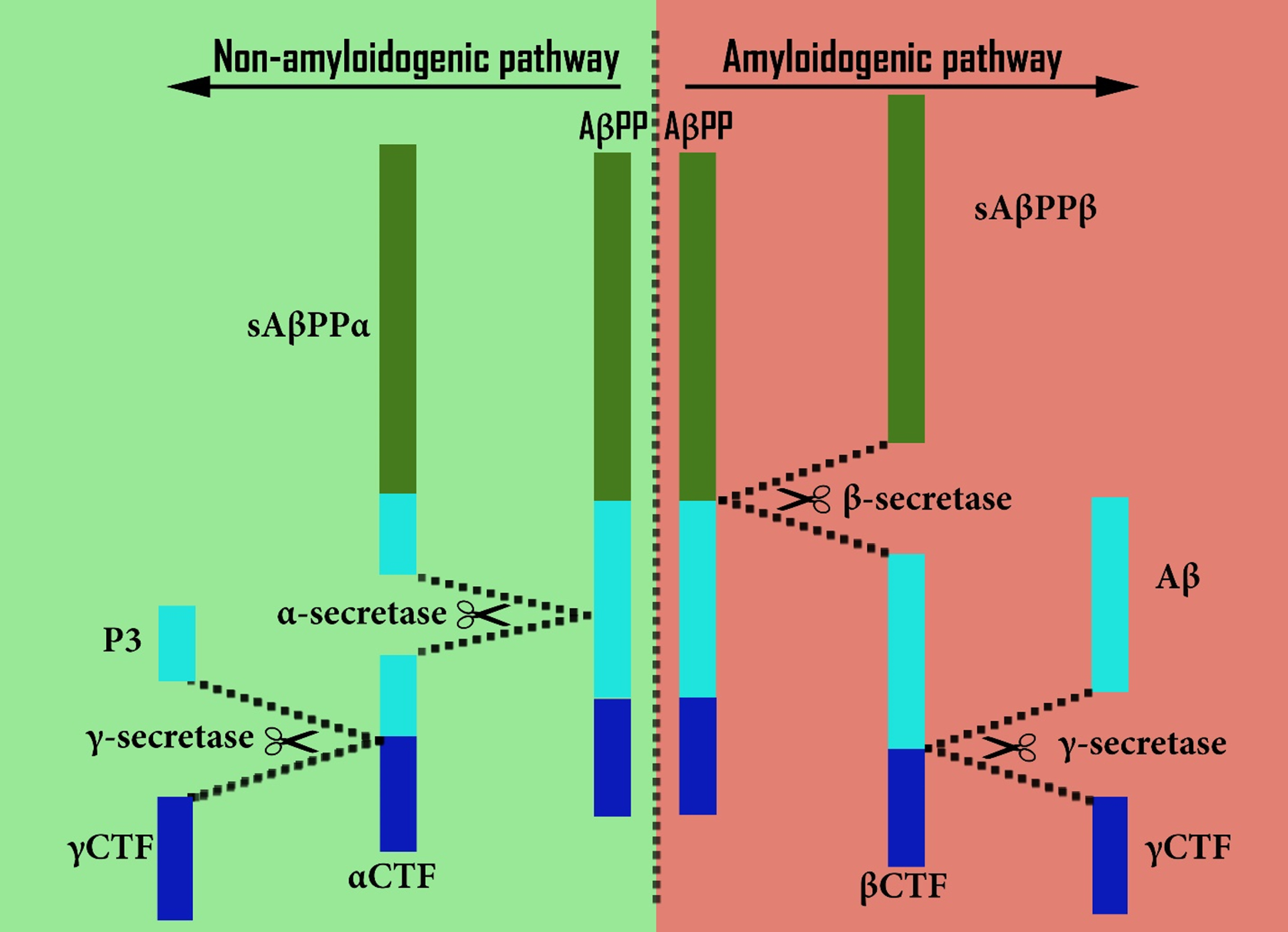
An explanation for this appears to be that amyloidogenic processing occurs within cholesterol-rich lipid rafts [345–348] (especially in early endosomes [346, 349], Figs. 1 and 2c–g), while non-amyloidogenic processing occurs in the main phospholipid-rich region of the neuronal plasma membrane [102, 350] (Figs. 1 and 2a, 2b). This suggests that an important part of cholesterol’s influence on amyloidogenic processing may be a consequence of its essential role as a major constituent of these lipid rafts, a conclusion that is well-evidenced in the literature [345, 346, 351]. Whatever the underlying mechanism, raised cholesterol levels would plausibly explain the increased amyloidogenesis seen in AD.
Fig. 2
Endosomal-lysosomal pathway and amyloidogenesis (not to scale). Non-amyloidogenic pathway: (a) In phospholipid-rich/cholesterol-poor plasma membrane (PM) regions, AβPP is first preferentially cleaved by α-secretase, then (b) within vesicles by γ-secretase, resulting in the P3 and γCTF peptides. Amyloidogenic pathway: (c) In cholesterol-rich PM regions, AβPP is instead first preferentially cleaved by β-secretase within vesicles, resulting in sAβPPβ and βCTF peptides. (d) Within early/late endosomes βCTF is further cleaved by γ-secretase, (e) resulting in Aβ, as well as γCTF. Normally, such downstream AβPP cleavage products then follow much the same route as cholesterol via late endosomes, (f) lysosomes, (g) the endoplasmic reticulum and/or Golgi apparatus, returning back to the PM. However, excess βCTF levels (resulting from excessive AβPP levels or β-secretase activity, or from downregulated γ-secretase activity) may lead to endosomal-lysosomal pathology (h), characterized by numerous excessively large early endosomes. In the same way, excessive cholesterol uptake may result in a similar pathology, possibly by overstimulating β-secretase activity. (i) This pathway stalling at the early endosome stage appears to result from βCTF-mediated excessive Rab5 activation, involving APPL1 stabilization. (j) Finally, defective NPC1- or NPC2-mediated cholesterol transport may lead to a similar “lipid traffic jam”, this time principally characterized by enlarged late endosomes.
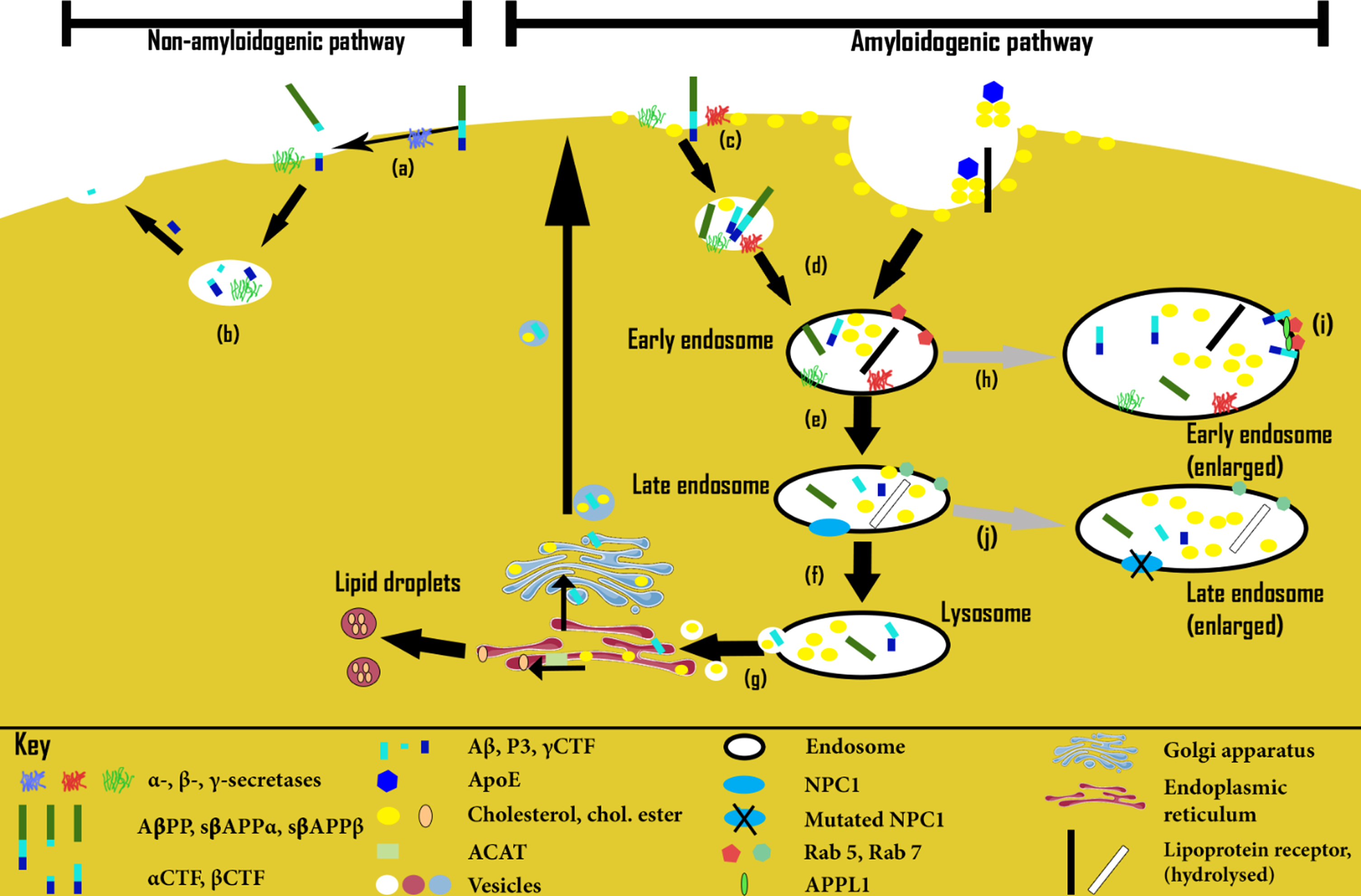
Certainly, some studies indicate that brain cholesterol levels may be raised in human AD brains, compared to non-demented brains [102, 352–354], although not all studies concur [355]. That cholesterol may be directly associated with amyloid plaque formation is supported by brain imaging studies, which show Aβ collocated with cholesterol within amyloid deposits in brain samples from AD-affected humans and other species [102, 104, 356].
The role of excess cholesterol in endosomal-lysosomal pathway abnormality
Indirect evidence of raised brain cholesterol levels as a causal factor in AD comes from studies of human AD brains. Such brains show abnormalities in the endosomal-lysosomal system compared to normal brains, together with NFTs [357, 358]. Such endosomal pathway overactivity and compartmental enlargement appears to be an early marker in AD, especially in pyramidal neurons, populations of which are known to be vulnerable in AD [346, 359–361].
Interestingly, a very similar pathology is also seen in mouse and other models of DS [349, 357, 362, 363]. However, at least in the case of one mouse model, such pathology was seen to emerge only following lipoprotein-mediated cholesterol treatment [349], suggesting that cholesterol is a crucial causal factor. Thus, cholesterol appears to be critical to the endosomal-lysosomal disorder characteristic of AD, suggesting that differences in the way it is supplied to neurons (and possibly other types of brain cell) could be a causal factor.
Further support for this conclusion comes from a number of studies in NPC (mentioned earlier). This is a neurological disorder characterized by faulty cholesterol transport and the presence of NFTs [105], in which endosomal-lysosomal pathology is also observed [364]. Such studies, while often contradictory in their results, collectively point to various failings in cholesterol uptake, transport, and recycling, and in abnormal endosomal-lysosomal pathway behavior. These failings include excessive uptake of exogenous LDL-derived cholesterol [365], excessive synthesis of endogenous cholesterol [365], enlarged early endosomes [366, 367], accumulation of unesterified cholesterol in late endosomes and lysosomes [367, 368], defective post-lysosomal cholesterol transport [369], and redistribution of lysosomal hydrolases to early endosomes [366].
Such reports commonly claim that other aspects of cholesterol internalization (and endosomal-lysosomal pathway behavior) appear to be normal, particularly in the case of initial cholesterol uptake and early endosome behavior [367].
However, a very similar phenotype was observed in a Chinese hamster ovary (CHO) cell mutant, which had a normal copy of NPC1 (the late endosome/lysosome-residing protein most commonly associated with NPC [367]) and of the HE/NPC2 protein (also associated with NPC, although less commonly), yet which still exhibited NPC-like pathology [364]. In this mutant, late sterol trafficking was reported to be normal despite obvious cholesterol accumulation in late endosomes/lysosomes [364]. Instead, cholesterol build-up occurred as a result of much-increased LDL binding, probably leading to cholesterol uptake being in excess of the normal capacity of the cell to dispose of it. Evidence in support of such excess cholesterol uptake includes the finding that LDL starvation of this mutant resulted in the disappearance of the cholesterol-laden aberrant late endosome compartment (characteristic also of NPC) that had previously been observed, only for this compartment to reappear with the restoration of LDL feeding. This provides further evidence that excess cholesterol uptake may lie behind the endosomal-lysosomal abnormalities seen in AD.
Another study, using a human fibroblast model, appears to provide further evidence for this conclusion. It found endosomal-lysosomal pathology in a number of inherited sphingolipid-storage disorders [370]. In almost all cases such pathology showed strong similarities with that seen in NPC, with a marked reduction in the accumulation of both cholesterol and a representative sphingolipid within the Golgi complex. Instead, increased accumulation was seen within many punctate cytoplasmic structures that also appeared to be associated with the NPC1 protein [370].
The authors of this latter study conclude that the observed pathology most likely results from a build-up of cholesterol (which is known to associate with high affinity to sphingolipids [371, 372]) within endosomes and lysosomes. This is because the reported pathology was seen to disappear following cholesterol depletion, being replaced with normal endosomal-lysosomal behavior [370]. However the same pathology could also be induced in normal cells by application of excess external cholesterol in the form of LDL [370]. This is similar to what was described for the CHO mutant mentioned above [364]. It is also in line with another study linking raised levels of plasma membrane cholesterol with correspondingly enlarged early endosomes in hippocampal neurons [373].
As stated earlier, LDL is not normally seen in the brain compartment (since it requires ApoB) and tends to be both larger in size and more cholesterol-rich than the HDL-like lipoproteins typically seen there [127, 170]. This suggests that externally-sourced cholesterol, supplied in excess of normal brain compartment levels—or in lipoproteins much more enriched in cholesterol than those normally seen in the brain compartment—may be a causal factor of AD-related endosomal abnormalities and of amyloidosis, at least in LOAD. Certainly, this would seem the most obvious explanation for why ApoB should be found within both amyloid plaques and NFTs [110, 111], as mentioned above, together with very high neuronal levels of cholesterol associated with NFTs in both NPC and AD [106], and within amyloid plaques [103, 104]. It may also help explain several lines of evidence that point towards close proximity of amyloid plaques and brain capillaries [374], especially damaged ones [375], in postmortem human brains with AD.
In further support of this hypothesis, inhibition of CYP46A1 (a protein indirectly responsible for cholesterol clearance from the brain through the BBB [376, 377]) in mouse hippocampal neurons has been shown to lead to accumulation of neuronal cholesterol. This, in turn, is associated with a distinctive AD-like pathology, including marked changes in endosomes (increasing both in size and number), Aβ peptide production, tau phosphorylation, endoplasmic reticulum stress and apoptosis, and eventually hippocampal atrophy and cognitive impairment [378, 379].
The role of the β-secretase-induced C-terminal fragment (βCTF)
Certainly, this interpretation, that such AD pathology results from excessive cholesterol uptake from larger lipoproteins of plasma origin, fits in well with the evidence that excessive cholesterol stimulates amyloidogenic processing of AβPP within early endosomes, given that cellular LDL-cholesterol uptake is known to be dependent on the endosomal-lysosomal pathway, by way of receptors possibly bound within lipid rafts [127, 346, 380, 381]. Furthermore, AβPP seems to be central to endosomal-lysosomal pathology, as the latter can be induced in AβPP-overexpressing human fibroblasts, or by AβPP’s C-terminal fragment βCTF. This is the fragment that remains after β–secretase cleavage of AβPP [346, 363], but prior to γ–secretase cleavage (Figs. 1 and 2), both cleavage events known to take place in early endosomes (Fig. 2d, e) [349, 357].
βCTF levels appear to be crucial to endosomal-lysosomal pathology, seemingly by stabilizing and amplifying Rab5 signaling within early endosomes, so that the latter become abnormally enlarged [346, 382–387]. This mechanism is explained in more detail in the Supplementary Material.
Thus, taken collectively, the evidence appears to explain the endosomal-lysosomal pathology seen in DS dementia and in many forms of AD, by two different mechanisms, explained as follows:
In the case of DS dementia, and some forms of FAD resulting from AβPP or secretase mutations, the pathology is likely to be primarily the result of βCTF overproduction. (In DS this results from overexpression of AβPP.) In the case of LOAD, over-supply of cholesterol originating from outside the brain, results in preferential upregulation of β-secretase [43]. This results in excess βCTF levels in both cases, leading to early endosomal enlargement and subsequent endosomal-lysosomal pathway abnormalities (Fig. 2h). Amyloidosis inevitably follows in all but a few cases, with NFTs presumably resulting from this amyloidosis, or from a failure of cholesterol transport, by a similar mechanism to that seen in NPC.
DISCUSSION
So far, evidence to support a new explanation of AD progression in which breakdown of the BBB results in an invasion into the brain compartment of external plasma-associated lipids has been presented. According to the model, it is this lipid invasion, acting in different ways on brain cells, rather than the BBB damage itself, that causes the pathologies characteristic of the disease.
Structural/mechanical failure as the initial cause of the disease
Interestingly, the explanation of AD proposed by the lipid invasion model has parallels in some other areas of medicine, where the integrity of physiological/structural barriers has been compromised. Nearly 500 years ago, Paracelsus famously stated that ‘All things are poison and nothing is without poison; only the dose makes a thing not a poison.’ This is usually understood in an absolute sense, but there is often an additional compartmental dimension to what constitutes a “poisonous” dose, which is why water in our lungs causes drowning, despite the rest of our bodies being substantially made up of this molecule, and why “good” bacteria leaking through our gut wall can lead to fatal infection. It is also why so-called “bad cholesterol”, i.e., LDL cholesterol, causes atherosclerosis—not because the cholesterol within LDL particles is in any way abnormal, but because damage to the arterial wall allows this cholesterol to enter the “wrong” compartment in excessive quantities.
The lipid invasion model argues that something similar happens in AD, certainly in the case of LOAD. Despite the brain being heavily composed of fatty acids and cholesterol, no different to those found elsewhere in the body, their entry into the brain compartment from the external plasma compartment puts them into another “wrong” compartment, one in which large, lipid-rich, ApoB-containing lipoproteins, and FFAs transported outside of lipoproteins, do not exist. As a result, many of these invading lipids can be said to act like a slow poison to the brain, causing a collection of pathologies that we identify as AD.
Lipids as drivers of all AD pathologies
The lipid invasion model argues that lipids are the drivers of all AD pathologies. Specifically, FFAs cause bioenergetic changes, oxidative stress, and other forms of lipotoxicity, neuroinflammation, neurodegeneration, inhibition of neurogenesis, and AA. Invading ApoB-containing lipoproteins cause endosomal-lysosomal disorder, amyloidosis, and NFT formation (as explained earlier). Figure 3 shows a general summary of the model.
Fig. 3
Summary diagram of lipid invasion model, showing basic differences between normal and AD-affected lipid transport. Diagram does not show bioenergetic changes or GABAergic aspects (tonic inhibition and inhibition of neurogenesis). Figure not to scale.
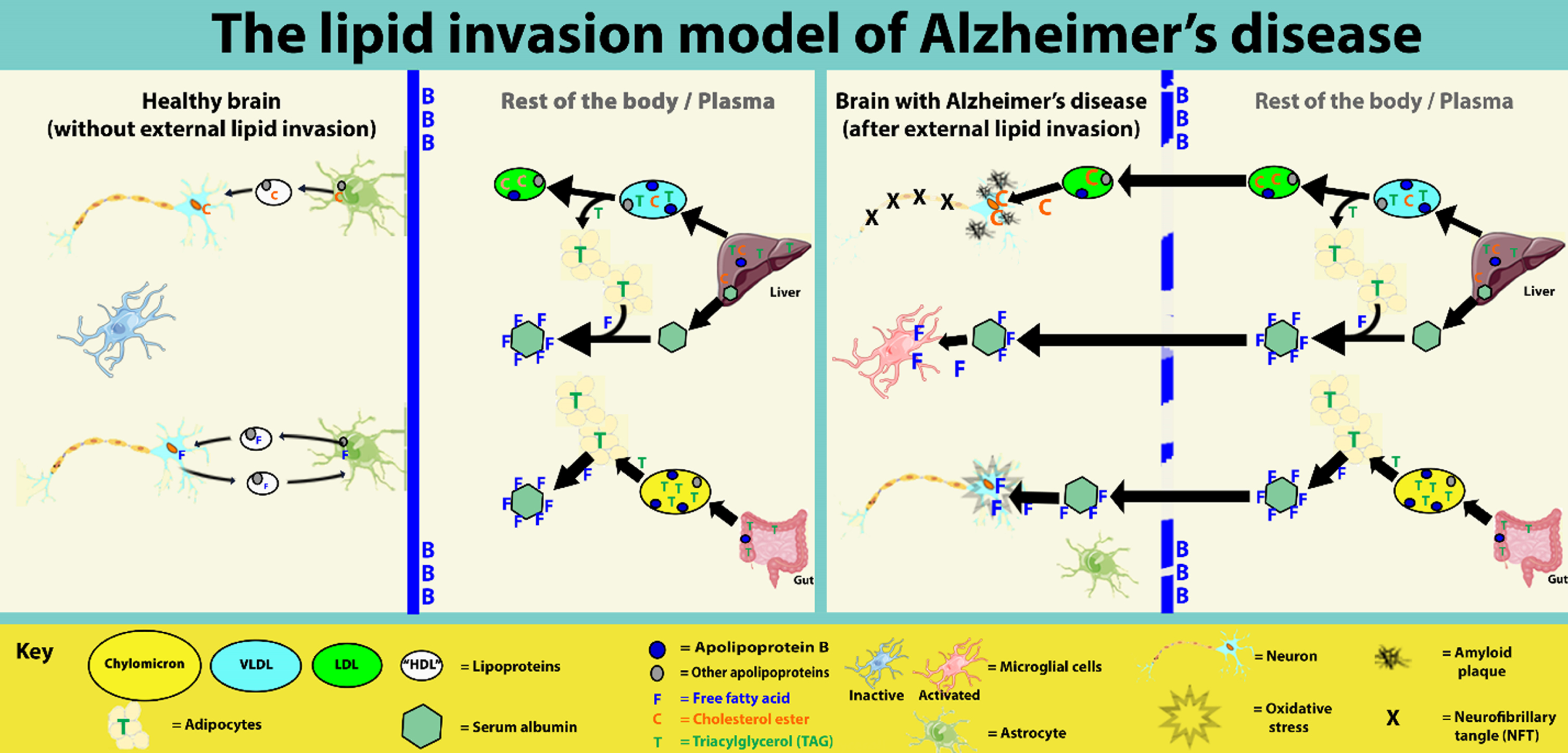
Of course, individually, many of these pathologies can be explained very adequately in other ways, as they are in the amyloid hypothesis. For instance, as described above, endosomal-lysosomal pathology may result from βCTF overproduction, which may, in turn, be the result of AβPP overexpression, secretase mutations, or other amyloid-associated anomalies. Similarly, APOE4 has been shown to be associated with lipid accumulation in various cells, including astrocytes and other glial cells [173, 388, 389]. It may be that such accumulation is a standard cerebral inflammatory response that protects neurons from oxidative stress and lipotoxicity [128, 390].
However, the lipid invasion model does not totally exclude previous explanations of AD. Rather, it builds on both the cholinergic and amyloid hypotheses, and on other lipid- and BBB-based explanations. As the previous main section sets out to show, the value of the model comes from the fact that it can account for all AD-associated pathologies taken collectively, and that it also explains some other puzzling phenomena associated with AD. In particular, it explains the presence of ApoB within plaques and NFTs, the close proximity of plaques with damaged capillaries, and the presence of non-cerebral proteins within the brain, as well as providing a comprehensive explanation for why such disparate risk factors as Aβ, aging, APOE4, hypertension, brain trauma, type II diabetes, smoking, and long-term sleep deprivation are all associated with increased risk of developing AD. As is made clear in earlier, this is because all these AD risk factors are also known to contribute to BBB disruption. Finally, as outlined previously, and explained in detail in the Supplementary Material, the lipid invasion model also provides a very coherent explanation for the similarities and differences seen between AD and ARBD.
Further aspects of the model
Several issues arising out of the model are discussed in detail in the Supplementary Material. These include discussions of the roles of Aβ and APOE4 in the model, followed by an account of how the model explains apparent gender differences in AD risk, the exponential increase in age-specific AD incidence, and the observed inverse correlation between AD and cancer. Finally, there is discussion of how FFA stimulation of extrasynaptic GABAARs may also help explain AD-associated disruption of the patient’s biological clock, in a similar way to which it explains AA.
Ultimately, for a disease as complex as AD, it is highly unlikely that all aspects of the disease can be explained by a single hypothesis. However, the lipid invasion model provides a more complete explanation than is currently available.
Further work required
Clearly further research into how the BBB is disrupted in AD is needed, including much more detailed studies into how each of the risk factors causes such damage, how relatively damaging each is to the BBB, and which areas of the BBB are most vulnerable to such damage. Such detailed information will be a minimum prerequisite for a quantitative empirical validation of the overall model. Similarly, much more work is needed to confirm that invading external lipids cause the AD-associated brain pathologies in all the ways proposed by the model. As alluded earlier, a recent paper has questioned the current orthodoxy that ApoB is not expressed in the brain compartment, based on magneto-fluorescent assays and RNA sequencing [119]. However, no evidence of LDL or other ApoB-containing lipoproteins within the CNS was reported in this paper. This, together with the fact that ApoB has not been detected previously in the CNS, suggests that its normal expression levels are likely to be much lower than in plasma. Certainly, further work is required to confirm this ApoB presence, and, if confirmed, to determine where and how much ApoB is expressed, and what the implications are for the model. Interestingly, in the same paper, CSF ApoB levels were reported to correlate well with early tau dysregulation.
Potential extensions of the model
The lipid invasion model may have wider implications for several other neurodegenerative diseases, including Parkinson’s disease (PD). A number of studies (including human postmortem and in vitro) have shown that raised levels of cholesterol under highly oxidative conditions can lead to the accumulation of α-synuclein, which is the principal component of Lewy bodies (signature macromolecules found in both PD and Lewy body dementia) [391, 392]. It is known that dopamine metabolism is strongly linked to oxidative stress [393], providing a compelling reason why PD is seen to be so strongly associated with the dopaminergic substantia nigra pars compacta region of the brain.
One of these studies has shown that α-synuclein aggregation can result from cholesterol accumulation within lysosomes [392]. Such accumulation can be induced in vitro by treatment of dopamine-producing neuroblastoma cells with MPP+ (a molecule known to produce PD-like pathology) or with U18666A, a molecule which prevents NPC1-mediated cholesterol exit from lysosomes and late endosomes. As described above, such faults in the cholesterol transport protein NPC1 commonly cause the neurodegenerative disease NPC, which is associated with the same endosomal-lysosomal abnormalities and NFTs seen in AD [394]. So, it could be that PD results from a similar lipid overload predicted to occur in AD by the model.
Similarly, amyotrophic lateral sclerosis (ALS, also known as Lou Gehrig’s or motor neuron disease) has been shown to be strongly-linked to lipid anomalies [395], including lipid droplet abnormalities and a bioenergetic switch from glucose to lipid usage [396], similar to what is seen in AD, and predicted by the model. Thus, in the small number of cases where concussion-related brain damage, rather than genetic mutations or other factors, are believed to have caused ALS [397, 398], it may be that lipid invasion, by exposing the motor cortex region [399] and other motor-related regions [398] of the brain to abnormally delivered lipids, provides an explanatory link.
Collectively, this evidence suggests that lipid invasion may account for a proportion of non-genetic PD and ALS cases. These PD cases would typically result from direct trauma (or associated shear forces) damaging the BBB in the vicinity of the substantia nigra, explaining why boxers are particularly prone to this disease [400, 401], whereas the ALS cases would result from similar BBB damage in the motor cortex and other motor-related regions of the brain, explaining why players of certain other contact sports (most obviously soccer players) appear to be over-represented among ALS cases [402–404].
More generally, not least because of the particular vulnerability of the hippocampal BBB region to damage, lipid invasion will manifest as AD. Nevertheless, one would expect a large degree of overlap, especially between AD and the other two diseases, as clearly seems to be the case [53]. Further investigation into this, and to how the model might extend to other neurodegenerative diseases, is needed.
CONCLUSION
The lipid invasion model offers a new hypothesis for AD, providing a comprehensive explanation of the observed neuropathologies associated with the disease, including the lipid irregularities first described by Alois Alzheimer himself. In explaining the disease as a result of lipid invasion, following damage to the BBB, it also plausibly explains the wide range of risk factors now identified with AD, most of which are associated with damage to, or greater vulnerability of, the BBB.
By pinpointing structural damage to the BBB as the critical initial step for disease progression, the model places AD more comfortably alongside other common diseases of aging, including stroke, atherosclerosis, and arthritis, in which structural failure is also critical. In doing so, it provides a clear explanation why the disease is so common in old age yet so comparatively rare in younger age groups.
A clear conclusion to be drawn from the model is that the primary focus in prevention and early treatment of AD should be on protecting, and if possible, repairing the BBB. Diagnosing BBB disruption at the earliest stage is critical to fighting AD, certainly in the case of LOAD, since relying on other signs, in the form of mild cognitive impairment, plaques, or NFTs, is quite likely, in most cases, to result in treatment starting too late to prevent further serious cognitive decline.
It is also clear from the model that, at later stages of the disease, as cognitive impairments appear, additional treatments will be needed to address external lipid invasion.
As well as new predictions, including a suggested role for invading plasma FFAs, acting on extrasynaptic GABAARs, in AA, the model also offers new insights into other AD-associated pathologies. These insights into neurotoxicity, neuroinflammation, endosomal-lysosomal disruption, amyloidogenic AβPP processing, and NFTs create potential new pathways to additional beneficial treatments.
As a result, the lipid invasion model provides a new explanation for the disease and offers new perspectives for further research into its diagnosis, prevention and treatment, as well as possibly providing new insights into other neurodegenerative diseases such as PD and ALS/motor neuron disease.
ACKNOWLEDGMENTS
The author would like to thank the following who provided helpful advice and support during the writing of this article: Dr. Francesco Tamagnini; Cherry Mill, MA (Oxon); Dr. Richard D. Jurd; Dr. Preethi Premkumar; Professor Peter Jeavons; Dr. Michael Kirk-Smith; Robin Thomas, FIChemE; Thomas Elliott, MA (Oxon); Carmel Connell, BA (Hons); and Mrs. Eveline Rudge.
I would also like to thank the reviewers for their very helpful critiques and suggestions in the preparation of this paper.
FUNDING
The author has no funding to report.
CONFLICT OF INTEREST
The author has no conflict of interest to report.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/ADR-210299.
REFERENCES
[1] | Alzheimer A, Stelzmann RA , Schnitzlein HN , Murtagh FR ((1995) ) An English translation of Alzheimer’s 1907 paper, “Über eine eigenartige Erkankung der Hirnrinde.”, Clin Anat 8: , 429–431. |
[2] | DeTure MA , Dickson DW ((2019) ) The neuropathological diagnosis of Alzheimer’s disease, Mol Neurodegener 14: , 32. |
[3] | Suzuki K , Parker CC , Pentchev PG , Katz D , Ghetti B , D’Agostino AN , Carstea ED ((1995) ) Neurofibrillary tangles in Niemann-Pick disease type C, Acta Neuropathol 89: , 227–238. |
[4] | Malek-Ahmadi M , Chen K , Perez SE , Mufson EJ ((2019) ) Cerebral amyloid angiopathy and neuritic plaque pathology correlate with cognitive decline in elderly non-demented individuals, J Alzheimers Dis 67: , 411–422. |
[5] | Götz J , Ittner LM ((2008) ) Animal models of Alzheimer’s disease and frontotemporal dementia, Nat Rev Neurosci 9: , 532–544. |
[6] | OECD ((2013) ) Dementia prevalence. In OECD, Health at a Glance 2013: OECD Indicators. OECD Publishing. |
[7] | Hardy J ((2006) ) A hundred years of Alzheimer’s disease research, Neuron 52: , 3–13. |
[8] | Castellani RJ , Perry G ((2012) ) Pathogenesis and disease-modifying therapy in Alzheimer’s disease: The flat line of progress, Arch Med Res 43: , 694–698. |
[9] | National Institute on Aging, How Is Alzheimer’s Disease Treated? https://www.nia.nih.gov/health/how-alzheimers-disease-treated |
[10] | Wallis C ((2019) ) It’s time to shift tactics on Alzheimer’s disease, Sci Am 22: , https://www.scientificamerican.com/article/its-time-to-shift-tactics-on-alzheimers-disease/. |
[11] | Contestabile A ((2011) ) The history of the cholinergic hypothesis, Behav Brain Res 221: , 334–340. |
[12] | Frölich L ((2002) ) The cholinergic pathology in Alzheimer’s disease–discrepancies between clinical experience and pathophysiological findings, J Neural Transm (Vienna) 109: , 1003–1013. |
[13] | Pimplikar SW ((2009) ) Reassessing the amyloid cascade hypothesis of Alzheimer’s disease, Int J Biochem Cell Biol 41: , 1261–1268. |
[14] | Wu L , Rosa-Neto P , Hsiung GY , Sadovnick AD , Masellis M , Black SE , Jia J , Gauthier S ((2012) ) Early-onset familial Alzheimer’s disease (EOFAD), Can J Neurol Sci 39: , 436–445. |
[15] | Nieuwenhuis-Mark RE ((2009) ) Diagnosing Alzheimer’s dementia in Down syndrome: Problems and possible solutions, Res Dev Disabil 30: , 827–838. |
[16] | Sweeney MD , Sagare AP , Zlokovic BV ((2018) ) Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders, Nat Rev Neurol 14: , 133–150. |
[17] | Lam FC , Liu R , Lu P , Shapiro AB , Renoir JM , Sharom FJ , Reiner PB ((2001) ) β-Amyloid efflux mediated by p-glycoprotein, J Neurochem 76: , 1121–1128. |
[18] | Ma Q , Zhao Z , Sagare AP , Wu Y , Wang M , Owens NC , Verghese PB , Herz J , Holtzman DM , Zlokovic BV ((2018) ) Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-β42 by LRP1-dependent apolipoprotein E isoform-specific mechanism, Mol Neurodegener 13: , 57. |
[19] | Wei W , Bodles-Brakhop AM , Barger SW ((2016) ) A role for P-glycoprotein in clearance of Alzheimer amyloid β-peptide from the brain, Curr Alzheimer Res 13: , 615–620. |
[20] | Zhao Z , Sagare AP , Ma Q , Halliday MR , Kong P , Kisler K , Winkler EA , Ramanathan A , Kanekiyo T , Bu G , Owens NC , Rege SV , Si G , Ahuja A , Zhu D , Miller CA , Schneider JA , Maeda M , Maeda T , Sugawara T , Ichida JK , Zlokovic BV ((2015) ) Central role for PICALM in amyloid–β blood–brain barrier transcytosis and clearance, Nat Neurosci 18: , 978–987. |
[21] | Miller MC , Tavares R , Johanson CE , Hovanesian V , Donahue JE , Gonzalez L , Silverberg GD , Stopa EG ((2008) ) Hippocampal RAGE immunoreactivity in early and advanced Alzheimer’s disease, Brain Res 1230: , 273–280. |
[22] | Liu CC , Liu CC , KanekiyoT , Xu H , Bu G ((2013) ) Apolipoprotein E and Alzheimer disease: Risk, mechanisms, and therapy, Nat Rev Neurol 9: , 106–118. |
[23] | Deane R , Sagare A , Hamm K , Parisi M , Lane S , Finn MB , Holtzman DM , Zlokovic BV ((2008) ) apoE isoform–specific disruption of amyloid β peptide clearance from mouse brain, J Clin Invest 118: , 4002–4013. |
[24] | Kivipelto M , Laakso MP , Tuomilehto J , Nissinen A , Soininen H ((2002) ) Hypertension and hypercholesterolaemia as risk factors for Alzheimer’s disease: Potential for pharmacological intervention, CNS Drugs 16: , 435–444. |
[25] | Whitmer RA , Gunderson EP , Barrett-Connor E , Quesenberry CP , Yaffe K ((2005) ) Obesity in middle age and future risk of dementia: A 27 year longitudinal population based study, BMJ 330: , 1360. |
[26] | Kivipelto M , Ngandu T , Fratiglioni L , Viitanen M , Kåreholt I , Winblad B , Helkala EL , Tuomilehto J , Soininen H , Nissinen A ((2005) ) Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease, Arch Neurol 62: , 1556–1560. |
[27] | Rhea EM , Salameh TS , Logsdon AF , Hanson AJ , Erickson MA , Banks WA ((2017) ) Blood-brain barriers in obesity, AAPS J 19: , 921–930. |
[28] | Jayaraj RL , Azimullah S , Beiram R ((2020) ) Diabetes as a risk factor for Alzheimer’s disease in the Middle East and its shared pathological mediators, Saudi J Biol Sci 27: , 736–750. |
[29] | Durazzo TC , Mattsson N , Weiner MW ; Alzheimer’s Disease Neuroimaging Initiative ((2014) ) Smoking and increased Alzheimer’s disease risk: A review of potential mechanisms, Alzheimers Dement 10: , S122–S145. |
[30] | Justice NJ ((2018) ) The relationship between stress and Alzheimer’s disease, Neurobiol Stress 8: , 127–133. |
[31] | Macedo AC , Balouch S , Tabet N ((2017) ) Is sleep disruption a risk factor for Alzheimer’s disease? J Alzheimer Dis 58: , 993–1002. |
[32] | Gottlieb S ((2000) ) Head injury doubles the risk of Alzheimer’s disease, BMJ 321: , 1100. |
[33] | Guerreiro R , Bras J ((2015) ) The age factor in Alzheimer’s disease, Genome Med 7: , 106. |
[34] | Unnithan AKA , Mehta P ((2021) ) Hemorrhagic stroke. In StatPearls, StatPearls Publishing, Treasure Island (FL). |
[35] | Cohn JN ((1995) ) Structural changes in cardiovascular disease, Am J Cardiol 76: , 34E–37E. |
[36] | Osterhoff G , Morgan EF , Shefelbine SJ , Karim L , McNamara LM , Augat P ((2016) ) Bone mechanical properties and changes with osteoporosis, Injury 47: (Suppl 2), S11–S20. |
[37] | Michael R , Bron AJ ((2011) ) The ageing lens and cataract: A model of normal and pathological ageing, Philos Trans R Soc Lond B Biol Sci 366: , 1278–1292. |
[38] | Bonilha VL ((2008) ) Age and disease-related structural changes in the retinal pigment epithelium, Clin Ophthalmol 2: , 413–424. |
[39] | Paplou V , Schubert NMA , Pyott SJ ((2021) ) Age-related changes in the cochlea and vestibule: Shared patterns and processes, Front Neurosci 15: , 680856. |
[40] | Cox LG , van Rietbergen B , van Donkelaar CC , Ito K ((2011) ) Bone structural changes in osteoarthritis as a result of mechanoregulated bone adaptation: A modeling approach, Osteoarthritis Cartilage 19: , 676–682. |
[41] | Li Y , Roberts ND , Wala JA , Shapira O , Schumacher SE , Kumar K , Khurana E , Waszak S , Korbel JO , Haber JE , Imielinski M ; PCAWG Structural Variation Working Group, Weischenfeldt J , Beroukhim R , Campbell PJ ; PCAWG Consortium ((2020) ) Patterns of somatic structural variation in human cancer genomes, Nature 578: , 112–121. |
[42] | Iadecola C , Gorelick PB ((2003) ) Converging pathogenic mechanisms in vascular and neurodegenerative dementia, Stroke 34: , 335–337. |
[43] | Ujiie M , Dickstein DL , Carlow DA , Jefferies WA ((2003) ) Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model, Microcirculation 10: , 463–470. |
[44] | Dickstein DL , Biron KE , Ujiie M , Pfeifer CG , Jeffries AR , Jefferies WA ((2006) ) Abeta peptide immunization restores blood-brain barrier integrity in Alzheimer disease, FASEB J 20: , 426–433. |
[45] | Popescu BO , Toescu EC , Popescu LM , Bajenaru O , Muresanu DF , Schultzberg M , Bogdanovic N ((2009) ) Blood-brain barrier alterations in ageing and dementia, J Neurol Sci 283: , 99–106. |
[46] | Kook SY , Hong HS , Moon M , Ha CM , Chang S , Mook-Jung I ((2012) ) Aβ1–42-RAGE interaction disrupts tight junctions of the blood–brain barrier via Ca2+-Calcineurin signaling, J Neurosci 32: , 8845–8854. |
[47] | Montagne A , Zhao Z , Zlokovic BV ((2017) ) Alzheimer’s disease: A matter of blood–brain barrier dysfunction? J Exp Med 214: , 3151–3169. |
[48] | Paul J , Strickland S , Melchor JP ((2007) ) Fibrin deposition accelerates neurovascular damage and neuroinflammation in mouse models of Alzheimer’s disease, J Exp Med 204: , 1999–2008. |
[49] | Cortes-Canteli M , Strickland S ((2009) ) Fibrinogen, a possible key player in Alzheimer’s disease, J Thromb Haemost 7: (Suppl 1), 146–150. |
[50] | Saunders NR , Dziegielewska KM , Møllgård K , Habgood MD ((2015) ) Markers for blood-brain barrier integrity: How appropriate is Evans blue in the twenty-first century and what are the alternatives? Front Neurosci 9: , 385. |
[51] | Marchi N , Cavaglia M , Fazio V , Bhudia S , Hallene K , Janigro D ((2004) ) Peripheral markers of blood-brain barrier damage, Clin Chim Acta 342: , 1–12. |
[52] | Takechi R , Galloway S , Pallebage-Gamarallage MM , Wellington CL , Johnsen RD , Dhaliwal SS , Mamo JC ((2010) ) Differential effects of dietary fatty acids on the cerebral distribution of plasma-derived apo B lipoproteins with amyloid-β, Br J Nutr 103: , 652–662. |
[53] | Stein TD , Alvarez VE , McKee AC ((2014) ) Chronic traumatic encephalopathy: A spectrum of neuropathological changes following repetitive brain trauma in athletes and military personnel, Alzheimers Res Ther 6: , 4. |
[54] | Chodobski A , Zink BJ , Szmydynger-Chodobska J ((2011) ) Blood–brain barrier pathophysiology in traumatic brain injury, Transl Stroke Res 2: , 492–516. |
[55] | Doherty CP , O’Keefe E , Wallace E , Loftus T , Keaney J , Kealy J , Humphries MM , Molloy MG , Meaney JF , Farrell M , Campbell M ((2016) ) Blood–brain barrier dysfunction as a hallmark pathology in chronic traumatic encephalopathy, J Neuropathol Exp Neurol 75: , 656–662. |
[56] | Johnson VE , Weber MT , Xiao R , Cullen DK , Meaney DF , Stewart W , Smith DH ((2018) ) Mechanical disruption of the blood–brain barrier following experimental concussion, Acta Neuropathol 135: , 711–726. |
[57] | Farrell M , Aherne S , O’Riordan S , O’Keeffe E , Greene C , Campbell M ((2019) ) Blood-brain barrier dysfunction in a boxer with chronic traumatic encephalopathy and schizophrenia, Clin Neuropathol 38: , 51–58. |
[58] | Salloway S , Gur T , Berzin T , Tavares R , Zipser B , Correia S , Hovanesian V , Fallon J , Kuo-Leblanc V , Glass D , Hulette C , Rosenberg C , Vitek M , Stopa E ((2002) ) Effect of APOE genotype on microvascular basement membrane in Alzheimer’s disease, J Neurol Sci 203-204: , 183–187. |
[59] | Mazzone P , Tierney W , Hossain M , Puvenna V , Janigro D , Cucullo L ((2010) ) Pathophysiological impact of cigarette smoke exposure on the cerebrovascular system with a focus on the blood-brain barrier: Expanding the awareness of smoking toxicity in an underappreciated area, Int J Environ Res Public Health 7: , 4111–4126. |
[60] | Prasad S , Sajja RK , Naik P , Cucullo L ((2014) ) Diabetes mellitus and blood-brain barrier dysfunction: An overview, J Pharmacovigil 2: , 125. |
[61] | Alluri H , Wiggins-Dohlvik K , Davis ML , Huang JH , Tharakan B ((2015) ) Blood-brain barrier dysfunction following traumatic brain injury, Metab Brain Dis 30: , 1093–1104. |
[62] | Girouard H ((2016) ) Hypertension and the Brain as an End-Organ Target, Springer. |
[63] | Montagne A , Nation DA , Sagare AP , Barisano G , Sweeney MD , Chakhoyan A , Pachicano M , Joe E , Nelson AR , D’Orazio LM , Buennagel DP , Harrington MG , Benzinger TLS , Fagan AM , Ringman JM , Schneider LS , Morris JC , Reiman EM , Caselli RJ , Chui HC , Tcw J , Chen Y , Pa J , Conti PS , Law M , Toga AW , Zlokovic BV ((2020) ) APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline, Nature 581: , 71–76. |
[64] | Hurtado-Alvarado G , Velázquez-Moctezuma J , Gómez-González B ((2017) ) Chronic sleep restriction disruptsinterendothelial junctions in the hippocampus and increasesblood–brain barrier permeability, J Microsc 268: , 28–38. |
[65] | Dudek KA , Dion-Albert L , Lebel M , LeClair K , Labrecque S , Tuck E , Perez CF , Golden SA , Tamminga C , Turecki G , Mechawar N , Russo SJ , Menard C ((2020) ) Molecular adaptations of the blood–brain barrier promote stress resilience vs. depression, Proc Natl Acad Sci U S A 117: , 3326–3336. |
[66] | Welcome MO , Mastorakis NE ((2020) ) Stress-induced blood brain barrier disruption: Molecular mechanisms and signaling pathways, Pharmacol Res 157: , 104769. |
[67] | Sohrabji F ((2018) ) Guarding the blood–brain barrier: A role for estrogen in the etiology of neurodegenerative disease, Gene Expr 13: , 311–319. |
[68] | Ahmadpour D , Grange-Messent V ((2021) ) Involvement of testosterone signaling in the integrity of the neurovascular unit in the male: Review of evidence, contradictions, and hypothesis, Neuroendocrinology 111: , 403–420. |
[69] | Jancsó G , Domoki F , Sántha P , Varga J , Fischer J , Orosz K , Penke B , Becskei A , Dux M , Tóth L ((1998) ) Beta-amyloid (1-42)peptide impairs blood-brain barrier function after intracarotidinfusion in rats, Neurosci Lett 253: , 139–141. |
[70] | Farkas IG , Czigner A , Farkas E , Dobó E , Soós K , Penke B , Endrész V , Mihály A ((2003) ) Beta-amyloid peptide-inducedblood-brain barrier disruption facilitates T-cell entry into the ratbrain, Acta Histochem 105: , 115–125. |
[71] | Tai LM , Holloway KA , Male DK , Loughlin AJ , Romero IA ((2010) ) Amyloid-β-induced occludin down-regulation and increased permeability in human brain endothelial cells is mediated by MAPK activation, J Cell Mol Med 14: , 1101–1112. |
[72] | Gosselet F , Saint-Pol J , Candela P , Fenart L ((2013) ) Amyloid-β peptides, Alzheimer’s disease and the blood-brain barrier, Curr Alzheimer Res 10: , 1015–1033. |
[73] | Ohtsuki S , Sato S , Yamaguchi H , Kamoi M , Asashima T , Terasaki T ((2007) ) Exogenous expression of claudin-5 induces barrier properties in cultured rat brain capillary endothelial cells, J Cell Physiol 210: , 81–86. |
[74] | Hartz AM , Bauer B , Soldner EL , Wolf A , Boy S , Backhaus R , Mihaljevic I , Bogdahn U , Klünemann HH , Schuierer G , Schlachetzki F ((2012) ) Amyloid-β contributes to blood–brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy, Stroke 43: , 514–523. |
[75] | Marco S , Skaper SD ((2006) ) Amyloid β-peptide1–42 alters tight junction protein distribution and expression in brain microvessel endothelial cells, Neurosci Lett 401: , 219–224. |
[76] | Carrano A , Hoozemans JJ , van der Vies SM , Rozemuller AJ , van Horssen J , de Vries HE ((2011) ) Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy, Antioxid Redox Signal 15: , 1167–1178. |
[77] | Lee JM , Yin K , Hsin I , Chen S , Fryer JD , Holtzman DM , Hsu CY , Xu J ((2005) ) Matrix metalloproteinase-9 in cerebral-amyloid-angiopathy-related hemorrhage, J Neurol Sci 229-230: , 249–254. |
[78] | Brkic M , Balusu S , Wonterghem EV , Gorlé N , Benilova I , Kremer A , Hove IV , Moons L , Strooper BD , Kanazir S , Libert C , Vandenbroucke RE ((2015) ) Amyloid β oligomers disrupt blood–CSF barrierintegrity by activating matrix metalloproteinases, J Neurosci 35: , 12766–12778. |
[79] | Thomas T , McLendon C , Sutton ET , Thomas G ((1997) ) Cerebrovascular endothelial dysfunction mediated by beta-amyloid, Neuroreport 8: , 1387–1391. |
[80] | Blanc EM , Toborek M , Mark RJ , Hennig B , Mattson MP ((1997) ) Amyloid beta-peptide induces cell monolayer albumin permeability, impairs glucose transport, and induces apoptosis in vascular endothelial cells, J Neurochem 68: , 1870–1881. |
[81] | Fossati S , Ghiso J , Rostagno A ((2012) ) Insights into caspase-mediated apoptotic pathways induced by amyloid-β in cerebral microvascular endothelial cells, Neurodegener Dis 10: , 324–328. |
[82] | Biron KE , Dickstein DL , Gopaul R , Jefferies WA ((2011) ) Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer’s disease, PLoS One 6: , e23789. |
[83] | Magaki S , Tang Z , Tung S , Williams CK , Lo D , Yong WH , Khanlou N , Vinters HV ((2018) ) The effects of cerebral amyloid angiopathy on integrity of the blood-brain barrier, Neurobiol Aging 70: , 70–77. |
[84] | Viswanathan A , Greenberg SM ((2011) ) Cerebral amyloid angiopathy in the elderly, Ann Neurol 70: , 871–880. |
[85] | Ghiso J , Tomidokoro Y , Revesz T , Frangione B , Rostagno A ((2010) ) Cerebral amyloid angiopathy and Alzheimer’s disease, Hirosaki Igaku 61: , S111–S124. |
[86] | Bergeron C , Ranalli PJ , Miceli PN ((1987) ) Amyloid angiopathy in Alzheimer’s disease, Can J Neurol Sci 14: , 564–569. |
[87] | Thal DR , Griffin WS , de Vos RA , Ghebremedhin E ((2008) ) Cerebral amyloid angiopathy and its relationship to Alzheimer’s disease, Acta Neuropathol 115: , 599–609. |
[88] | Pallebage-Gamarallage MM , Takechi R , Lam V , Galloway S , Dhaliwal S , Mamo JC ((2010) ) Post-prandial lipid metabolism, lipid-modulating agents and cerebrovascular integrity: Implications for dementia risk, Atheroscler Suppl 11: , 49–54. |
[89] | Mamo JC , Jian L , James AP , Flicker L , Esselmann H , Wiltfang J ((2008) ) Plasma lipoprotein beta-amyloid in subjects with Alzheimer’s disease or mild cognitive impairment, Ann Clin Biochem 45: , 395–403. |
[90] | Galloway S , Takechi R , Pallebage-Gamarallage MM , Dhaliwal SS , Mamo JC ((2009) ) Amyloid-β colocalizes with apolipoprotein B in absorptive cells of the small intestine, Lipids Health Dis 8: , 46. |
[91] | Foley P ((2010) ) Lipids in Alzheimer’s disease: A century-old story, Biochim Biophys Acta 1801: , 750–753. |
[92] | LIPID MAPS® Lipidomics Gateway. |
[93] | Galloway S , Jian L , Johnsen R , Chew S , Mamo JCL ((2007) ) [beta]-Amyloid or its precursor protein is found in epithelial cells of the small intestine and is stimulated by high-fat feeding, J Nutr Biochem 18: , 279–284. |
[94] | Takechi R , Galloway S , Pallebage-Gamarallage MM , Mamo JC ((2008) ) Chylomicron amyloid-beta in the aetiology of Alzheimer’s disease, Atheroscler Suppl 9: , 19–25. |
[95] | Gosselet F ((2011) ) The mysterious link between cholesterol andAlzheimer’s disease: Is the blood-brain barrier a suspect? JAlzheimers Dis Parkinsonism 1: , 103e. |
[96] | Lambert JC , Heath S , Even G , Campion D , Sleegers K , Hiltunen M , Combarros O , Zelenika D , Bullido MJ , Tavernier B , Letenneur L , Bettens K , Berr C , Pasquier F , Fiévet N , Barberger-Gateau P , Engelborghs S , De Deyn P , Mateo I , Franck A , Helisalmi S , Porcellini E , Hanon O ; European Alzheimer’s Disease Initiative Investigators, de Pancorbo MM , Lendon C , Dufouil C , Jaillard C , Leveillard T , Alvarez V , Bosco P , Mancuso M , Panza F , Nacmias B , Bossù P , Piccardi P , Annoni G , Seripa D , Galimberti D , Hannequin D , Licastro F , Soininen H , Ritchie K , Blanché H , Dartigues JF , Tzourio C , Gut I , Van Broeckhoven C , Alpérovitch A , Lathrop M , Amouyel P ((2009) ) Genome-wide association study identifies variants at CLU andCR1 associated with Alzheimer’s disease, Nat Genet 41: , 1094–1099. |
[97] | Hollingworth P , Harold D , Sims R , Gerrish A , Lambert JC , Carrasquillo MM , Abraham R , Hamshere ML , Pahwa JS , Moskvina V , et al. ((2011) ) Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease, Nat Genet 43: , 429–435. |
[98] | Bereczki E , Bernát G , Csont T , Ferdinandy P , Scheich H , Sántha M ((2008) ) Overexpression of human Apolipoprotein B-100induces severe neurodegeneration in transgenic mice, J ProteomeRes 7: , 2246–2252. |
[99] | Caramelli P , Nitrini R , Maranhão R , Lourenço ACG , Damasceno MC , Vinagre C , Caramelli B ((1999) ) Increased apolipoprotein B serum concentration in Alzheimer’s disease, Acta Neurol Scand 100: , 61–63. |
[100] | Solomon A , Kivipelto M , Wolozin B , Zhou J , Whitmer RA ((2009) ) Midlife serum cholesterol and increased risk of Alzheimer’s and vascular dementia three decades later, Dement Geriatr Cogn Disord 28: , 75–80. |
[101] | Nicholson AM , Ferreira A ((2010) ) Cholesterol and neuronal susceptibility to beta-amyloid toxicity, Cogn Sci (Hauppauge) 5: , 35–56. |
[102] | Xiong H , Callaghan D , Jones A , Walker DG , Lue LF , Beach TG , Sue LI , Woulfe J , Xu H , Stanimirovic DB , Zhang W ((2008) ) Cholesterol retention in Alzheimer’s brain is responsible for high β- and γ-secretase activities and Aβ production, Neurobiol Dis 29: , 422–437. |
[103] | Kiskis J , Fink H , Nyberg L , Thyr J , Li JY , Enejder A ((2015) ) Plaque-associated lipids in Alzheimer’s diseased brain tissue visualized by nonlinear microscopy, Sci Rep 5: , 13489. |
[104] | Mori T , Paris D , Town T , Rojiani AM , Sparks DL , Delledonne A , Crawford F , Abdullah LI , Humphrey JA , Dickson DW , Mullan MJ ((2001) ) Cholesterol accumulates in senile plaques of Alzheimer disease patients and in transgenic APPsw mice, J Neuropathol Exp Neurol 60: , 778–785. |
[105] | Saito Y , Suzuki K , Nanba E , Yamamoto T , Ohno K , Murayama S ((2002) ) Niemann-Pick type C disease: Accelerated neurofibrillary tangle formation and amyloid beta deposition associated with apolipoprotein E epsilon 4 homozygosity, Ann Neurol 52: , 351–355. |
[106] | Distl R , Treiber-Held S , Albert F , Meske V , Harzer K , Ohm TG ((2003) ) Cholesterol storage and tau pathology in Niemann-Pick type C disease in the brain, J Pathol 200: , 104–111. |
[107] | Bowman GL , Quinn JF ((2008) ) Alzheimer’s disease and the blood-brain barrier: Past, present and future, Aging Health 4: , 47–55. |
[108] | Skillbäck T , Delsing L , Synnergren J , Mattsson N , Janelidze S , Nägga K , Kilander L , Hicks R , Wimo A , Winblad B , Hansson O , Blennow K , Eriksdotter M , Zetterberg H ((2017) ) CSF/serum albumin ratio in dementias: A cross-sectional study on 1861 patients, Neurobiol Aging 59: , 1–9. |
[109] | Sweeney MD , Sagare AP , Zlokovic BV ((2015) ) Cerebrospinal fluid biomarkers of neurovascular dysfunction in mild dementia and Alzheimer’s disease, J Cereb Blood Flow Metab 35: , 1055–1068. |
[110] | Namba Y , Tsuchiya H , Ikeda K ((1992) ) Apolipoprotein B immunoreactivity in senile plaque and vascular amyloids and neurofibrillary tangles in the brains of patients with Alzheimer’s disease, Neurosci Lett 134: , 264–266. |
[111] | Takechi R , Galloway S , Pallebage-Gamarallage M , Wellington C , Johnsen R , Mamo JC ((2009) ) Three-dimensional colocalization analysis of plasma-derived apolipoprotein B with amyloid plaques in APP/PS1 transgenic mice, Histochem Cell Biol 131: , 661–666. |
[112] | Lam V , Takechi R , Pallebage-Gamarallage MM , Galloway S , Mamo JC ((2011) ) Colocalisation of plasma derived apo B lipoproteins with cerebral proteoglycans in a transgenic-amyloid model of Alzheimer’s disease, Neurosci Lett 492: , 160–164. |
[113] | Banks WA ((2006) ) The dam breaks: Disruption of the blood-brain barrier in diabetes mellitus, Am J Physiol Heart Circ Physiol 291: , H2595–H2596. |
[114] | van der Vusse GJ ((2009) ) Albumin as fatty acid transporter, Drug Metab Pharmacokinet 24: , 300–307. |
[115] | Young SG ((1990) ) Recent progress in understanding apolipoprotein B, Circulation 82: , 1574–1594. |
[116] | Takechi R , Galloway S , Pallebage-Gamarallage MM , Lam V , Dhaliwal SS , Mamo JC ((2013) ) Probucol prevents blood–brain barrier dysfunction in wild-type mice induced by saturated fat or cholesterol feeding, Clin Exp Pharmacol Physiol 40: , 45–52. |
[117] | Elliott DA , Weickert CS , Garner B ((2010) ) Apolipoproteins in the brain: Implications for neurological and psychiatric disorders. Clin Lipidol 51: , 555–573. |
[118] | Wang H , Eckel RH ((2014) ) What are lipoproteins doing in the brain? Trends Endocrinol Metab 25: , 8–14. |
[119] | Picard C , Nilsson N , Labonté A , Auld D , Rosa-Neto P ; Alzheimer’s Disease Neuroimaging Initiative, Ashton NJ , Zetterberg H , Blennow K , Breitner JCB , Villeneuve S , Poirier J ; PREVENT-AD research group ((2021) ) Apolipoprotein B is a novel marker for early tau pathology inAlzheimer’s disease, Alzheimers Dement, doi: 10.1002/alz.12442 |
[120] | Quehenberger O , Dennis EA ((2011) ) The human plasma lipidome, N Engl J Med 365: , 1812–1823. |
[121] | Jeske DJ , Dietschy JM ((1980) ) Regulation of rates of cholesterol synthesis in vivo in the liver and carcass of the rat measured using [3H]water, J Lipid Res 21: , 364–376. |
[122] | Dietschy JM , Turley SD ((2004) ) Cholesterol metabolism in the central nervous system during early development and in the mature animal, J Lipid Res 45: , 1375–1397. |
[123] | Hamilton J , Brunaldi K ((2007) ) A model for fatty acid transport into the brain, J Mol Neurosci 33: , 12–17. |
[124] | Zhang J , Liu Q ((2015) ) Cholesterol metabolism and homeostasis in the brain, Protein Cell 6: , 254–264. |
[125] | Feingold KR , Grunfeld C ((2000) ) Introduction to lipids and lipoproteins. In Endotext, FeingoldKR, AnawaltB, BoyceA, ChrousosG, DunganK, GrossmanA, HershmanJM, KaltsasG, KochC, KoppP, KorbonitsM, McLachlanR, MorleyJE, NewM, PerreaultL, PurnellJ, RebarR, SingerF, TrenceDL, VinikA, WilsonDP, eds. MDText.com, Inc., South Dartmouth (MA). |
[126] | Bruce KD , Zsombok A , Eckel RH ((2017) ) Lipid processing in the brain: A key regulator of systemic metabolism. Front Endocrinol (Lausanne) 8: , 60. |
[127] | Vance DE , Vance JE ((2008) ), Biochemistry of lipids, lipoproteins, and membranes, Elsevier. |
[128] | Ioannou MS , Jackson J , Sheu SH , Chang CL , Weigel AV , Liu H , Pasolli HA , Xu CS , Pang S , Matthies D , Hess HF , Lippincott-Schwartz J , Liu Z ((2019) ) Neuron-astrocyte metabolic coupling protects against activity-induced fatty acid toxicity, Cell 177: , 1522–1535.e14. |
[129] | Markesbery WR ((1997) ) Oxidative stress hypothesis in Alzheimer’s disease, Free Radic Biol Med 23: , 134–147. |
[130] | Hensley K ((2010) ) Neuroinflammation in Alzheimer’s disease: Mechanisms, pathologic consequences, and potential for therapeutic manipulation, J Alzheimers Dis 21: , 1–14. |
[131] | Moreno-Jiménez EP , Flor-García M , Terreros-Roncal J , Rábano A , Cafini F , Pallas-Bazarra N , Ávila J , Llorens-Martín M ((2019) ) Adult hippocampal neurogenesis isabundant in neurologically healthy subjects and drops sharply inpatients with Alzheimer’s disease, Nat Med 25: , 554–560. |
[132] | Simons M , Keller P , Dichgans J , Schulz JB ((2001) ) Cholesterol and Alzheimer’s disease: Is there a link? Neurology 57: , 1089–1093. |
[133] | Wolozin B ((2004) ) Cholesterol and the biology of Alzheimer’s disease, Neuron 41: , 7–10. |
[134] | Zenaro E , Piacentino G , Constantin G ((2017) ) The blood-brain barrier in Alzheimer’s disease, Neurobiol Dis 107: , 41–56. |
[135] | Montagne A , Barnes SR , Sweeney MD , Halliday MR , Sagare AP , Zhao Z , Toga AW , Jacobs RE , Liu CY , Amezcua L , Harrington MG , Chui HC , Law M , Zlokovic BV ((2015) ) Blood-brain barrier breakdown in the aging human hippocampus, Neuron 85: , 296–302. |
[136] | Di Paolo G , Kim TW ((2011) ) Linking lipids to Alzheimer’s disease: Cholesterol and beyond, Nat Rev Neurosci 12: , 284–296. |
[137] | Kao YC , Ho PC , Tu YK , Jou IM , Tsai KJ ((2020) ) Lipids and Alzheimer’s disease, Int J Mol Sci 21: , 1505. |
[138] | Takechi R , Galloway S , Pallebage-Gamarallage MM , Lam V , Mamo JC ((2010) ) Dietary fats, cerebrovasculature integrity and Alzheimer’s disease risk, Prog Lipid Res 49: , 159–170. |
[139] | Wiegmann C , Mick I , Brandl EJ , Heinz A , Gutwinski S ((2020) ) Alcohol and dementia – what is the link? A systematic review, Neuropsychiatr Dis Treat 16: , 87–99. |
[140] | Nixon K ((2006) ) Alcohol and adult neurogenesis: Roles in neurodegeneration and recovery in chronic alcoholism, Hippocampus 16: , 287–295. |
[141] | Morris SA , Eaves DW , Smith AR , Nixon K ((2010) ) Alcohol inhibition of neurogenesis: A mechanism of hippocampal neurodegeneration in an adolescent alcohol abuse model, Hippocampus 20: , 596–607. |
[142] | Nixon K , Crews FT ((2002) ) Binge ethanol exposure decreases neurogenesis in adult rat hippocampus, J Neurochem 83: , 1087–1093. |
[143] | Fadda F , Rossetti ZL ((1998) ) Chronic ethanol consumption: From neuroadaptation to neurodegeneration, Prog Neurobiol 56: , 385–431. |
[144] | Crews FT , Lawrimore CJ , Walter TJ , Coleman LG Jr ((2017) ) The role of neuroimmune signaling in alcoholism, Neuropharmacology 122: , 56–73. |
[145] | Crews FT ((2008) ) Alcohol-related neurodegeneration and recovery, Alcohol Res Health 31: , 377–388. |
[146] | Rang HP ((2012) ) 23. Atherosclerosis and lipoprotein metabolism. In Rang & Dale’s pharmacology, Churchill Livingstone, Edinburgh. |
[147] | Brindley DN ((1991) ) Chapter 6 Metabolism of triacylglycerols. In New Comprehensive Biochemistry, VanceDE, VanceJE, eds. Elsevier, pp. 171–203. |
[148] | Ahmadian M , E Duncan R , Jaworski K , Sarkadi-Nagy E , Sul HS ((2007) ) Triacylglycerol metabolism in adipose tissue, Future Lipidol 2: , 229–237. |
[149] | Beffert U , Danik M , Krzywkowski P , Ramassamy C , Berrada F , Poirier J ((1998) ) The neurobiology of apolipoproteins and their receptors in the CNS and Alzheimer’s disease, Brain Res Brain Res Rev 27: , 119–142. |
[150] | Björkhem I , Meaney S ((2004) ) Brain cholesterol: Long secret life behind a barrier, Arterioscler Thromb Vasc Biol 24: , 806–815. |
[151] | Orth M , Bellosta S ((2012) ) Cholesterol: Its regulation and role in central nervous system disorders, Cholesterol 2012: , 292598. |
[152] | Ballmer PE ((2001) ) Causes and mechanisms of hypoalbuminaemia, Clin Nutr 20: , 271–273. |
[153] | Schiff ER , Maddrey WC , Sorrell MF ((2011) ) Chapter 2: Laboratory Tests. In Schiff’s Diseases of the Liver, John Wiley & Sons. |
[154] | Nag S ((2003) ) Pathophysiology of blood-brain barrier breakdown. In The blood-brain barrier: Biology and research protocols, Humana Press, pp. 97. |
[155] | Banks WA ((2008) ) Developing drugs that can cross the blood-brain barrier: Applications to Alzheimer’s disease, BMC Neurosci 9: , S2. |
[156] | Schönfeld P , Reiser G ((2013) ) Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain, J Cereb Blood Flow Metab 33: , 1493–1499. |
[157] | Karmi A , Iozzo P , Viljanen A , Hirvonen J , Fielding BA , Virtanen K , Oikonen V , Kemppainen J , Viljanen T , Guiducci L , Haaparanta-Solin M , Någren K , Solin O , Nuutila P ((2010) ) Increased brain fatty acid uptake in metabolic syndrome, Diabetes 59: , 2171–2177. |
[158] | Panov A , Orynbayeva Z , Vavilin V , Lyakhovich V ((2014) ) Fatty acids in energy metabolism of the central nervous system, Biomed Res Int 2014: , 472459. |
[159] | Murphy EJ ((2017) ) The blood–brain barrier and protein-mediated fatty acid uptake: Role of the blood–brain barrier as a metabolic barrier, J Neurochem 141: , 324–329. |
[160] | Jha MK , Morrison BM ((2018) ) Glia-neuron energy metabolism in health and diseases: New insights into the role of nervous system metabolic transporters, Exp Neurol 309: , 23–31. |
[161] | Johnson RC , Young SK , Cotter R , Lin L , Rowe WB ((1990) ) Medium-chain-triglyceride lipid emulsion: Metabolism and tissue distribution, Am J Clin Nutr 52: , 502–508. |
[162] | Speijer D , Manjeri GR , Szklarczyk R ((2014) ) How to deal with oxygen radicals stemming from mitochondrial fatty acid oxidation, Philos Trans R Soc Lond B Biol Sci 369: , 20130446. |
[163] | Sokoloff L ((1973) ) Metabolism of ketone bodies by the brain, Annu Rev Med 24: , 271–280. |
[164] | Owen OE ((2005) ) Ketone bodies as a fuel for the brain during starvation, Biochem Mol Biol Educ 33: , 246–251. |
[165] | Yang H , Shan W , Zhu F , Wu J , Wang Q ((2019) ) Ketone bodies in neurological diseases: Focus on neuroprotection and underlying mechanisms, Front Neurol 10: , 585. |
[166] | Sultana R , Perluigi M , Butterfield DA ((2013) ) Lipid peroxidation triggers neurodegeneration: A redox proteomics view into the Alzheimer disease brain, Free Radic Biol Med 62: , 157–169. |
[167] | Olsson Y , Klatzo I , Sourander P , Steinwall O ((1968) ) Blood-brain barrier to albumin in embryonic new born and adult rats, Acta Neuropathol 10: , 117–122. |
[168] | Roheim PS , Carey M , Forte T , Vega GL ((1979) ) Apolipoproteins in human cerebrospinal fluid, Proc Natl Acad Sci U S A 76: , 4646–4649. |
[169] | Cipolla MJ ((2009) ) Barriers of the CNS. In The Cerebral Circulation, Morgan & Claypool Life Sciences. |
[170] | Danik M , Champagne D , Petit-Turcotte C , Beffert U , Poirier J ((1999) ) Brain lipoprotein metabolism and its relation to neurodegenerative disease, Crit Rev Neurobiol 13: , 357–407. |
[171] | Ladu MJ , Reardon C , Eldik LV , Fagan AM , Bu G , Holtzman D , Getz GS ((2000) ) Lipoproteins in the central nervous system, Ann N Y Acad Sci 903: , 167–175. |
[172] | Mahley RW , Weisgraber KH , Huang Y ((2006) ) Apolipoprotein E4: A causative factor and therapeutic target in neuropathology, including Alzheimer’s disease, Proc Natl Acad Sci U S A 103: , 5644–5651. |
[173] | Farmer BC , Kluemper J , Johnson LA ((2019) ) Apolipoprotein E4 alters astrocyte fatty acid metabolism and lipid droplet formation, Cells 8: , 182. |
[174] | Pfrieger FW ((2003) ) Outsourcing in the brain: Do neurons deend on cholesterol delivery by astrocytes? Bioessays 25: , 72–78. |
[175] | Deoni SCL , Dean DC , O’Muircheartaigh J , Dirks H , Jerskey BA ((2012) ) Investigating white matter development in infancy and early childhood using myelin water faction and relaxation time mapping, Neuroimage 63: , 1038–1053. |
[176] | Lütjohann D , von Bergmann K ((2003) ) 24S-hydroxycholesterol: A marker of brain cholesterol metabolism, Pharmacopsychiatry 36: (Suppl 2), S102–S106. |
[177] | Kay AD , Day SP , Nicoll JA , Packard CJ , Caslake MJ ((2003) ) Remodelling of cerebrospinal fluid lipoproteins after subarachnoid hemorrhage, Atherosclerosis 170: , 141–146. |
[178] | Saunders N , Habgood M , Dziegielewska , Saunders N ((1999) ) Barrier mechanisms in the brain, I. Adult brain, Clin Exp Pharmacol Physiol 26: , 11–19. |
[179] | Pardridge WM , Mietus LJ ((1980) ) Palmitate and cholesterol transport through the blood-brain barrier, J Neurochem 34: , 463–466. |
[180] | Abbott NJ ((2005) ) Dynamics of CNS barriers: Evolution, differentiation, and modulation, Cell Mol Neurobiol 25: , 5–23. |
[181] | Bundgaard M , Abbott NJ ((2008) ) All vertebrates started out with a glial blood-brain barrier 4-500 million years ago, Glia 56: , 699–708. |
[182] | Bell RD , Winkler EA , Singh I , Sagare AP , Deane R , WU Z , Holtzman DM , Betsholtz C , Armulik A , Sallstrom J , Berk BC , Zlokovic BV ((2012) ) Apolipoprotein E controls cerebrovascular integrity via cyclophilin A, Nature 485: , 512–516. |
[183] | Salameh TS , Mortell WG , Logsdon AF , Butterfield DA , Banks WA ((2019) ) Disruption of the hippocampal and hypothalamic blood–brain barrier in a diet-induced obese model of type II diabetes: Prevention and treatment by the mitochondrial carbonic anhydrase inhibitor, topiramate, Fluids Barriers CNS 16: , 1. |
[184] | Pelisch N , Hosomi N , Ueno M , Nakano D , Hitomi H , Mogi M , Shimada K , Kobori H , Horiuchi M , Sakamoto H , Matsumoto M , Kohno M , Nishiyama A ((2011) ) Blockade of AT1 receptors protects the blood–brain barrier and improves cognition in Dahl salt-sensitive hypertensive rats, Am J Hypertens 24: , 362–368. |
[185] | Naik P , Cucullo L ((2015) ) Pathobiology of tobacco smoking and neurovascular disorders: Untied strings and alternative products, Fluids Barriers CNS 12: , 25. |
[186] | Hurtado-Alvarado G , Domínguez-Salazar E , Pavon L , Velázquez-Moctezuma J , Gómez-González B ((2016) ) Blood-brain barrier disruption induced by chronic sleep loss:Low-grade inflammation may be the link, J Immunol Res 2016: , e4576012. |
[187] | Abrahamson EE , Ikonomovic MD ((2020) ) Brain injury-induced dysfunction of the blood brain barrier as a risk for dementia, Exp Neurol 328: , 113257. |
[188] | Weber CM , Clyne AM ((2021) ) Sex differences in the blood–brain barrier and neurodegenerative diseases, APL Bioeng 5: , 011509. |
[189] | Venkat P , Chopp M , Chen J ((2017) ) Blood–brain barrier disruption, vascular impairment, and ischemia/reperfusion damage in diabetic stroke, J Am Heart Assoc 6: , e005819. |
[190] | Yamamoto M , Guo DH , Hernandez CM , Stranahan AM ((2019) ) Endothelial Adora2a activation promotes blood–brain barrier breakdown and cognitive impairment in mice with diet-induced insulin resistance, J Neurosci 39: , 4179–4192. |
[191] | Hossain M , Sathe T , Fazio V , Mazzone P , Weksler B , Janigro D , Rapp E , Cucullo L ((2009) ) Tobacco smoke: A critical etiological factor for vascular impairment at the blood–brain barrier, Brain Res 1287: , 192–205. |
[192] | Zielinski MR , Kim Y , Karpova SA , McCarley RW , Strecker RE , Gerashchenko D ((2014) ) Chronic sleep restriction elevates brain Interleukin-1 beta and tumor necrosis factor-alpha and attenuates brain-derived neurotrophic factor expression, Neurosci Lett 580: , 27–31. |
[193] | Pan W , Zadina JE , Harlan RE , Weber JT , Banks WA , Kastin AJ ((1997) ) Tumor necrosis Factor-α: A neuromodulator in the CNS, Neurosci Biobehav Rev 21: , 603–613. |
[194] | Wang Y , Jin S , Sonobe Y , Cheng Y , Horiuchi H , Parajuli B , Kawanokuchi J , Mizuno T , Takeuchi H , Suzumura A ((2014) ) Interleukin-1β induces blood–brain barrier disruption by downregulating sonic hedgehog in astrocytes, PLoS One 9: , e110024. |
[195] | Main BS , Villapol S , Sloley SS , Barton DJ , Parsadanian M , Agbaegbu C , Stefos K , McCann MS , Washington PM , Rodriguez OC , Burns MP ((2018) ) Apolipoprotein E4 impairs spontaneous blood brain barrier repair following traumatic brain injury, Mol Neurodegener 13: , 17. |
[196] | Jo DH , Kim JH , Heo JI , Kim JH , Cho CH ((2013) ) Interaction between pericytes and endothelial cells leads to formation of tight junction in hyaloid vessels, Mol Cells 36: , 465–471. |
[197] | Yang Y , Rosenberg GA ((2011) ) Blood–brain barrier breakdown in acute and chronic cerebrovascular disease, Stroke 42: , 3323–3328. |
[198] | Tagami M , Nara Y , Kubota A , Fujino H , Yamori Y ((1990) ) Ultrastructural changes in cerebral pericytes and astrocytes of stroke-prone spontaneously hypertensive rats, Stroke 21: , 1064–1071. |
[199] | Hurtado-Alvarado G , Cabañas-Morales AM , Gómez-Gónzalez B ((2014) ) Pericytes: Brain-immune interface modulators, Front Integr Neurosci 7: , 80. |
[200] | Patrick P , Price TO , Diogo AL , Sheibani N , Banks WA , Shah GN ((2015) ) Topiramate protects pericytes from glucotoxicity: Role for mitochondrial CA VA in cerebromicrovascular disease in diabetes, J Endocrinol Diabetes 2: , https://www.symbiosisonlinepublishing.com/endocrinology-diabetes/endocrinology-diabetes23.php. |
[201] | Winkler EA , Sagare AP , Zlokovic BV ((2014) ) The pericyte: A forgotten cell type with important implications for Alzheimer’s disease? Brain Pathol 24: , 371–386. |
[202] | Daneman R , Prat A ((2015) ) The blood–brain barrier, Cold Spring Harb Perspect Biol 7: , a020412. |
[203] | Argaw AT , Zhang Y , Snyder BJ , Zhao ML , Kopp N , Lee SC , Raine CS , Brosnan CF , John GR ((2006) ) IL-1β regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program, J Immunol 177: , 5574–5584. |
[204] | Viggars AP , Wharton SB , Simpson JE , Matthews FE , Brayne C , Savva GM , Garwood C , Drew D , Shaw PJ , Ince PG ((2011) ) Alterations in the blood brain barrier in ageing cerebral cortex in relationship to Alzheimer-type pathology: A study in the MRC-CFAS population neuropathology cohort, Neurosci Lett 505: , 25–30. |
[205] | Nair SA , Jagadeeshan S , Indu R , Sudhakaran PR , Pillai MR ((2012) ) How intact is the basement membrane? Role of MMPs. In Biochemical Roles of Eukaryotic Cell Surface Macromolecules, SudhakaranPR, SuroliaA, eds. Springer, New York, NY, pp. 215–232. |
[206] | Dokken B ((2008) ) The pathophysiology of cardiovascular disease and diabetes: Beyond blood pressure and lipids, Diabetes Spectr 21: , 160–165. |
[207] | Thomsen MS , Routhe LJ , Moos T ((2017) ) The vascular basement membrane in the healthy and pathological brain, J Cereb Blood Flow Metab 37: , 3300–3317. |
[208] | Yang Y , Estrada EY , Thompson JF , Liu W , Rosenberg GA ((2007) ) Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat, J Cereb Blood Flow Metab 27: , 697–709. |
[209] | Pan W , Xiang S , Tu H , Kastin AJ ((2006) ) Cytokines interact with the blood-brain barrier. In Blood-Brain Barriers, John Wiley & Sons, Ltd, pp. 247–264. |
[210] | Nation DA , Sweeney MD , Montagne A , Sagare AP , D’Orazio LM , Pachicano M , Sepehrband F , Nelson AR , Buennagel DP , Harrington MG , Benzinger TLS , Fagan AM , Ringman JM , Schneider LS , Morris JC , Chui HC , Law M , Toga AW , Zlokovic BV ((2019) ) Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction, Nat Med 25: , 270–276. |
[211] | Vanlandewijck M , He L , Mäe MA , Andrae J , Ando K , Del Gaudio F , Nahar K , Lebouvier T , Laviña B , Gouveia L , Sun Y , Raschperger E , Räsänen M , Zarb Y , Mochizuki N , Keller A , Lendahl U , Betsholtz C ((2018) ) A molecular atlas of cell types and zonation in the brain vasculature, Nature 554: , 475–480. |
[212] | Zhao L , Li Z , Vong JSL , Chen X , Lai HM , Yan LYC , Huang J , Sy SKH , Tian X , Huang Y , Chan HYE , So HC , Ng WL , Tang Y , Lin WJ , Mok VCT , Ko H ((2020) ) Pharmacologically reversible zonation-dependent endothelial cell transcriptomic changes with neurodegenerative disease associations in the aged brain, Nat Commun 11: , 4413. |
[213] | Koedam EL , Lauffer V , van der Vlies AE , van der Flier WM , Scheltens P , Pijnenburg YA ((2010) ) Early-versus late-onset Alzheimer’s disease: More than age alone, J Alzheimers Dis 19: , 1401–1408. |
[214] | Safieh M , Korczyn AD , Michaelson DM ((2019) ) ApoE4: An emerging therapeutic target for Alzheimer’s disease, BMC Med 17: , 64. |
[215] | Dorey E , Chang N , Liu QY , Yang Z , Zhang W ((2014) ) Apolipoprotein E, amyloid-beta, and neuroinflammation in Alzheimer’s disease, Neurosci Bull 30: , 317–330. |
[216] | Premkumar DR , Cohen DL , Hedera P , Friedland RP , Kalaria RN ((1996) ) Apolipoprotein E-epsilon4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with Alzheimer’s disease, Am J Pathol 148: , 2083–2095. |
[217] | Olichney JM , Hansen LA , Galasko D , Saitoh T , Hofstetter CR , Katzman R , Thal LJ ((1996) ) The apolipoprotein E epsilon 4 allele is associated with increased neuritic plaques and cerebral amyloid angiopathy in Alzheimer’s disease and Lewy body variant, Neurology 47: , 190–196. |
[218] | Alonzo NC , Hyman BT , Rebeck GW , Greenberg SM ((1998) ) Progression of cerebral amyloid angiopathy: Accumulation of amyloid-beta40 in affected vessels, J Neuropathol Exp Neurol 57: , 353–359. |
[219] | Fryer JD , Taylor JW , DeMattos RB , Bales KR , Paul SM , Parsadanian M , Holtzman DM ((2003) ) Apolipoprotein E markedly facilitates age-dependent cerebral amyloid angiopathy and spontaneous hemorrhage in amyloid precursor protein transgenic mice, J Neurosci 23: , 7889–7896. |
[220] | Rannikmäe K , Kalaria RN , Greenberg SM , Chui HC , Schmitt FA , Samarasekera N , Al-Shahi Salman R , Sudlow CL ((2014) ) APOE associations with severe CAA-associated vasculopathic changes – collaborative meta-analysis, J Neurol Neurosurg Psychiatry 85: , 300–305. |
[221] | Dienel GA , Hertz L ((2001) ) Glucose and lactate metabolism during brain activation, J Neurosci Res 66: , 824–838. |
[222] | Demetrius LA , Magistretti PJ , Pellerin L ((2015) ) Alzheimer’s disease: The amyloid hypothesis and the Inverse Warburg effect, Front Physiol 5: , 522. |
[223] | Ding F , Yao J , Rettberg JR , Chen S , Brinton RD ((2013) ) Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer’s mouse brain: Implication for bioenergetic intervention. PLoS One 8: , e79977. |
[224] | Yao J , Rettberg JR , Klosinski LP , Cadenas E , Brinton RD ((2011) ) Shift in brain metabolism in late onset Alzheimer’s disease: Implications for biomarkers and therapeutic interventions, Mol Aspects Med 32: , 247–257. |
[225] | Bird MI , Munday LA , Saggerson ED , Clark JB ((1985) ) Carnitine acyltransferase activities in rat brain mitochondria. Bimodal distribution, kinetic constants, regulation by malonyl-CoA and developmental pattern, Biochem J 226: , 323–330. |
[226] | Yang SY , He XY , Schulz H ((1987) ) Fatty acid oxidation in rat brain is limited by the low activity of 3-ketoacyl-coenzyme A thiolase, J Biol Chem 262: , 13027–13032. |
[227] | Di Paola M , Lorusso M ((2006) ) Interaction of free fatty acids with mitochondria: Coupling, uncoupling and permeability transition, Biochim Biophys Acta 1757: , 1330–1337. |
[228] | Wojtczak L , Schönfeld P ((1993) ) Effect of fatty acids on energy coupling processes in mitochondria, Biochim Biophys Acta 1183: , 41–57. |
[229] | Schönfeld P , Wojtczak L ((2008) ) Fatty acids as modulators of the cellular production of reactive oxygen species, Free Radic Biol Med 45: , 231–241. |
[230] | Hoyer S , Nitsch R , Oesterreich K ((1991) ) Predominant abnormality in cerebral glucose utilization in late-onset dementia of the Alzheimer type: A cross-sectional comparison against advanced late-onset and incipient early-onset cases, J Neural Transm Park Dis Dement Sect 3: , 1–14. |
[231] | Costantini LC , Barr LJ , Vogel JL , Henderson ST ((2008) ) Hypometabolism as a therapeutic target in Alzheimer’s disease, BMC Neurosci 9: (Suppl 2), S16. |
[232] | Bojarski L , Herms J , Kuznicki J ((2008) ) Calcium dysregulation in Alzheimer’s disease, Neurochem Int 52: , 621–633. |
[233] | Kim J , Yang Y , Song SS , Na JH , Oh KJ , Jeong C , Yu YG , Shin YK ((2014) ) Beta-amyloid oligomers activate apoptotic BAK pore for cytochrome c release, Biophys J 107: , 1601–1608. |
[234] | Bartolucci M , Ravera S , Garbarino G , Ramoino P , Ferrando S , Calzia D , Candiani S , Morelli A , Panfoli I ((2015) ) Functional expression of electron transport chain and FoF1-ATP synthase in optic nerve myelin sheath, Neurochem Res 40: , 2230–2241. |
[235] | Ravera S , Bartolucci M , Cuccarolo P , Litamè E , Illarcio M , Calzia D , Degan P , Morelli A , Panfoli I ((2015) ) Oxidative stress in myelin sheath: The other face of the extramitochondrial oxidative phosphorylation ability, Free Radic Res 49: , 1156–1164. |
[236] | Papuć E , Rejdak K ((2018) ) The role of myelin damage inAlzheimer’s disease pathology, Arch Med Sci 16: , 345–351. |
[237] | Syapin PJ , Hickey WF ((2006) ) Alcohol brain damage and neuroinflammation: Is there a connection? Alcohol Clin Exp Res 29: , 1080–1089. |
[238] | Blanco AM , Guerri C ((2007) ) Ethanol intake enhances inflammatory mediators in brain: Role of glial cells and TLR4/IL-1RI recetors. Front Biosci 12: , 2616–2630. |
[239] | Crews FT , Nixon K ((2009) ) Mechanisms of neurodegeneration and regeneration in alcoholism, Alcohol Alcohol 44: , 115–127. |
[240] | Zhao YN , Wang F , Fan YX , Ping GF , Yang JY , Wu CF ((2013) ) Activated microglia are implicated in cognitive deficits, neuronal death, and successful recovery following intermittent ethanol exposure, Behav Brain Res 236: , 270–282. |
[241] | Walter TJ , Crews FT ((2017) ) Microglial depletion alters the brain neuroimmune response to acute binge ethanol withdrawal, J Neuroinflammation 14: , 86. |
[242] | Kaur G , Han SJ , Yang I , Crane C ((2010) ) Microglia and central nervous system immunity, Neurosurg Clin N Am 21: , 43–51. |
[243] | Yang I , Han SJ , Kaur G , Crane C , Parsa AT ((2010) ) The role of microglia in central nervous system immunity and glioma immunology, J Clin Neurosci 17: , 6–10. |
[244] | Gehrmann J , Matsumoto Y , Kreutzberg GW ((1995) ) Microglia: Intrinsic immuneffector cell of the brain, Brain Res Brain Res Rev 20: , 269–287. |
[245] | Dissing-Olesen L , Ladeby R , Nielsen HH , Toft-Hansen H , Dalmau I , Finsen B ((2007) ) Axonal lesion-induced microglial proliferation and microglial cluster formation in the mouse, Neurosci 149: , 112–122. |
[246] | Crews FT , Vetreno RP ((2014) ) Neuroimmune basis of alcoholic brain damage, Int Rev Neurobiol 118: , 315–357. |
[247] | Zou J , Crews F ((2010) ) Induction of innate immune gene expression cascades in brain slice cultures by ethanol: Key role of NF-κB and proinflammatory cytokines, Alcohol Clin Exp Res 34: , 777–789. |
[248] | Alfonso-Loeches S , Pascual-Lucas M , Blanco AM , Sanchez-Vera I , Guerri C ((2010) ) Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage, J Neurosci 30: , 8285–8295. |
[249] | Fernandez-Lizarbe S , Montesinos J , Guerri C ((2013) ) Ethanol induces TLR4/TLR2 association, triggering an inflammatory response in microglial cells, J Neurochem 126: , 261–273. |
[250] | Chait A , Kim F ((2010) ) Saturated fatty acids and inflammation: Who pays the toll? Arterioscler Thromb Vasc Biol 30: , 692–693. |
[251] | Wang Z , Liu D , Wang F , Liu S , Zhao S , Ling EA , Hao A ((2012) ) Saturated fatty acids activate microglia via Toll-like receptor 4/NF-κB signalling, Br J Nutr 107: , 229–241. |
[252] | Shi H , Kokoeva MV , Inouye K , Tzameli I , Yin H , Flier JS ((2006) ) TLR4 links innate immunity and fatty acid-induced insulin resistance, J Clin Invest 116: , 3015–3025. |
[253] | Wellen KE , Hotamisligil GS ((2005) ) Inflammation, stress, and diabetes, J Clin Invest 115: , 1111–1119. |
[254] | Donath MY , Shoelson SE ((2011) ) Type 2 diabetes as an inflammatory disease, Nat Rev Immunol 11: , 98–107. |
[255] | Kreutzberg GW ((1996) ) Microglia: A sensor for pathological events in the CNS, Trends Neurosci 19: , 312–318. |
[256] | Rock RB , Gekker G , Hu S , Sheng WS , Cheeran M , Lokensgard JR , Peterson PK ((2004) ) Role of microglia in central nervous system infections, Clin Microbiol Rev 17: , 942–964. |
[257] | Rangaraju S , Gearing M , Jin LW , Levey A ((2015) ) Potassium channel Kv1.3 is highly expressed by microglia in human Alzheimer’s disease, J Alzheimers Dis 44: , 797–808. |
[258] | Lenz KM , Nelson LH ((2018) ) Microglia and beyond: Innate immune cells as regulators of brain development and behavioral function, Front Immunol 9: , 698. |
[259] | Hwang D ((2001) ) Modulation of the expression of cyclooxygenase-2 by fatty acids mediated through toll-like receptor 4-derived signaling pathways, FASEB J 15: , 2556–2564. |
[260] | Belfort R , Mandarino L , Kashyap S , Wirfel K , Pratipanawatr T , Berria R , DeFronzo RA , Cusi K ((2005) ) Dose-response effect of elevated plasma free fatty acid on insulin signaling, Diabetes 54: , 1640–1648. |
[261] | Huber AH , Kleinfeld AM ((2017) ) Unbound free fatty acid profiles in human plasma and the unexpected absence of unbound palmitoleate, J Lipid Res 58: , 578–585. |
[262] | Nagy LE ((2003) ) Recent insights into the role of the innate immune system in the development of alcoholic liver disease, Exp Biol Med (Maywood) 228: , 882–890. |
[263] | Walter S , Letiembre M , Liu Y , Heine H , Penke B , Hao W , Bode B , Manietta N , Walter J , Schulz-Schuffer W , Fassbender K ((2007) ) Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease, Cell Physiol Biochem 20: , 947–956. |
[264] | Barak B , Feldman N , Okun E ((2014) ) Toll-like receptors as developmental tools that regulate neurogenesis during development: An update, Front Neurosci 8: , 272. |
[265] | Crews FT , Walter TJ , Coleman LG , Vetreno RP ((2017) ) Toll-like receptor signaling and stages of addiction, Psychopharmacology (Berl) 234: , 1483–1498. |
[266] | Ming GL , Song H ((2011) ) Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron 70: , 687–702. |
[267] | White AM , Signer ML , Kraus CL , Swartzwelder HS ((2004) ) Experiential aspects of alcohol-induced blackouts among college students, Am J Drug Alcohol Abuse 30: , 205–224. |
[268] | Sanday L , Patti CL , Zanin KA , Fernandes-Santos L , Oliveira LC , Kameda SR , Tufik S , Frussa-Filho R ((2013) ) Ethanol-induced memory impairment in a discriminative avoidance task is state-dependent, Alcohol Clin Exp Res 37: (Suppl 1), E30–E39. |
[269] | Ditraglia GM , Press DS , Butters N , Jernigan TL , Cermak LS , Velin RA , Shear PK , Irwin M , Schuckit M ((1991) ) Assessment of olfactory deficits in detoxified alcoholics, Alcohol 8: , 109–115. |
[270] | Collins MA , Corso TD , Neafsey EJ ((1996) ) Neuronal degeneration in rat cerebrocortical and olfactory regions during subchronic “binge” intoxication with ethanol: Possible explanation for olfactory deficits in alcoholics, Alcohol Clin Exp Res 20: , 284–292. |
[271] | Mesholam RI , Moberg PJ , Mahr RN , Doty RL ((1998) ) Olfaction in neurodegenerative disease: A meta-analysis of olfactory functioning in Alzheimer’s and Parkinson’s diseases, Arch Neurol 55: , 84–90. |
[272] | Murphy C , Gilmore MM , Seery CS , Salmon DP , Lasker BR ((1990) ) Olfactory thresholds are associated with degree of dementia in Alzheimer’s disease, Neurobiol Aging 11: , 465–469. |
[273] | Hodges J ((1998) ) The amnestic prodrome of Alzheimer’s disease, Brain 121: , 1601–1602. |
[274] | Weintraub S , Wicklund AH , Salmon DP ((2012) ) The neuropsychological profile of Alzheimer disease, Cold Spring Harb Perspect Med 2: , a006171. |
[275] | Rissman RA , Mobley WC ((2011) ) Implication for treatment: GABAA receptors in aging, Down syndrome and Alzheimer’s disease, J Neurochem 117: , 613–622. |
[276] | Wu Z , Guo Z , Gearing M , Chen G ((2014) ) Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s disease model, Nat Commun 5: , 4159. |
[277] | Jo S , Yarishkin O , Hwang YJ , Chun YE , Park M , Woo DH , Bae JY , Kim T , Lee J , Chun H , Park HJ , Lee DY , Hong J , Kim HY , Oh SJ , Park SJ , Lee H , Yoon BE , Kim Y , Jeong Y , Shim I , Bae YC , Cho J , Kowall NW , Ryu H , Hwang E , Kim D , Lee CJ ((2014) ) GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease, Nat Med 20: , 886–896. |
[278] | Dahl DR , Dahl N , Samson FE Jr ((1956) ) A study on the narcotic action of the short chain fatty acids, J Clin Invest 35: , 1291–1298. |
[279] | White RP , Samson FE ((1956) ) Effects of fatty acid anions on the electroencephalogram of unanesthetized rabbits, Am J Physiol 186: , 271–274. |
[280] | Matsuzaki M , Takagi H ((1967) ) Sleep induced by sodium butyrate in the cat, Brain Res 4: , 206–222. |
[281] | McCandless DW ((1985) ) Octanoic acid-induced coma and reticular formation energy metabolism, Brain Res 335: , 131–137. |
[282] | Dahl DR ((1968) ) Short chain fatty acid inhibition of rat brain Na-K adenosine triphosphatase, J Neurochem 15: , 815–820. |
[283] | Perlman BJ , Goldstein DB ((1984) ) Membrane-disordering potency and anticonvulsant action of valproic acid and other short-chain fatty acids, Mol Pharmacol 26: , 83–89. |
[284] | Alifimoff JK , Firestone LL , Miller KW ((1989) ) Anaesthetic potencies of primary alkanols: Implications for the molecular dimensions of the anaesthetic site, Br J Pharmacol 96: , 9–16. |
[285] | Hau KM , Connell DW , Richardson BJ ((2002) ) A study of the biological partitioning behavior of n-Alkanes and n-Alkanols in causing anesthetic effects, Regul Toxicol Pharmacol 35: , 273–279. |
[286] | Deneer JW , Seinen W , Hermens JLM ((1988) ) The acute toxicity of aldehydes to the guppy, Aquatic Toxicol 12: , 185–192. |
[287] | Evers AS , Crowder CM ((2009) ) Mechanisms of anesthesia and consciousness. In Clinical Anesthesia, Lippincott Williams & Wilkins, pp. 95–114. |
[288] | Ueda I , Suzuki A ((1998) ) Is there a specific receptor for anesthetics? Contrary effects of alcohols and fatty acids on phase transition and bioluminescence of firefly luciferase, Biophys J 75: , 1052–1057. |
[289] | Matsuki H , Suzuki A , Kamaya H , Ueda I ((1999) ) Specific and non-specific binding of long-chain fatty acids to firefly luciferase: Cutoff at octanoate, Biochim Biophys Acta 1426: , 143–150. |
[290] | Frangopol PT , Mihailescu D ((2001) ) Interactions of some local anesthetics and alcohols with membranes, Colloids Surf B Biointerfaces 22: , 3–22. |
[291] | Kappas A , Palmer RH ((1963) ) Selected aspects of steroid pharmacology, Pharmacol Rev 15: , 123–167. |
[292] | Belelli D , Lambert JJ ((2005) ) Neurosteroids: Endogenous regulators of the GABAA receptor, Nat Rev Neurosci 6: , 565–575. |
[293] | Orser BA ((2007) ) Lifting the fog around anesthesia, Sci Am 296: , 54–61. |
[294] | Bonin RP , Orser BA ((2008) ) GABAA receptor subtypes underlying general anesthesia, Pharmacol Biochem Behav 90: , 105–112. |
[295] | Brickley SG , Mody I ((2012) ) Extrasynaptic GABAA receptors: Their function in the CNS and implications for disease, Neuron 73: , 23–34. |
[296] | Cheng VY , Martin LJ , Elliott EM , Kim JH , Mount HT , Taverna FA , Roder JC , MacDonald JF , Bhambri A , Collinson N , Wafford KA , Orser BA ((2006) ) α5GABAA receptors mediate the amnestic but not sedative-hypnotic effects of the general anesthetic etomidate, J Neurosci 26: , 3713–3720. |
[297] | Nutt DJ , Besson M , Wilson SJ , Dawson GR , Lingford-Hughes AR ((2007) ) Blockade of alcohol’s amnestic activity in humans by an [alpha]5 subtype benzodiazepine receptor inverse agonist, Neuropharmacology 53: , 810–820. |
[298] | Sikka PK , Beaman ST , Street JA ((2015) ) Basic Clinical Anesthesia, Springer. |
[299] | Farrant M , Nusser Z ((2005) ) Variations on an inhibitory theme: Phasic and tonic activation of GABA(A) receptors, Nat Rev Neurosci 6: , 215–229. |
[300] | Collinson N , Kuenzi FM , Jarolimek W , Maubach KA , Cothliff R , Sur C , Smith A , Otu FM , Howell O , Atack JR , McKernan RM , Seabrook GR , Dawson GR , Whiting PJ , Rosahl TW ((2002) ) Enhanced learning and memory and altered GABAergic synaptic transmission in mice lacking the alpha 5 subunit of the GABAA receptor, J Neurosci 22: , 5572–5580. |
[301] | Shen H , Sabaliauskas N , Sherpa A , Fenton AA , Stelzer A , Aoki C , Smith SS ((2010) ) A critical role for alpha4betadelta GABAA receptors in shaping learning deficits at puberty in mice, Science 327: , 1515–1518. |
[302] | Clarkson AN , Huang BS , Macisaac SE , Mody I , Carmichael ST ((2010) ) Reducing excessive GABA-mediated tonic inhibition promotes functional recovery after stroke, Nature 468: , 305–309. |
[303] | Liu Y , Namba T , Liu J , Suzuki R , Shioda S , Seki T ((2010) ) Glial fibrillary acidic protein-expressing neural progenitors give rise to immature neurons via early intermediate progenitors expressing both glial fibrillary acidic protein and neuronal markers in the adult hippocampus, Neuroscience 166: , 241–251. |
[304] | Martin LJ , Zurek AA , MacDonald JF , Roder JC , Jackson MF , Orser BA ((2010) ) α5GABAA receptor activity sets the threshold for long-term potentiation and constrains hippocampus-dependent memory, J Neurosci 30: , 5269–5282. |
[305] | Whissell PD , Eng D , Lecker I , Martin LJ , Wang DS , Orser BA ((2013) ) Acutely increasing δGABAA receptor activity impairs memory and inhibits synaptic plasticity in the hippocampus, Front Neural Circuits 7: , 146. |
[306] | Grover LM , Lambert NA , Schwartzkroin PA , Teyler TJ ((1993) ) Role of HCO3- ions in depolarizing GABAA receptor-mediated responses in pyramidal cells of rat hippocampus, J Neurophysiol 69: , 1541–1555. |
[307] | Li K , Xu E ((2008) ) The role and the mechanism of γ-aminobutyric acid during central nervous system development, Neurosci Bull 24: , 195–200. |
[308] | Sigel E , Steinmann ME ((2012) ) Structure, function, and modulation of GABAA receptors, J Biol Chem 287: , 40224–40231. |
[309] | Kaila K ((1994) ) Ionic basis of GABAA receptor channel function in the nervous system, Prog Neurobiol 42: , 489–537. |
[310] | Petrini EM , Marchionni I , Zacchi P , Sieghart W , Cherubini E ((2004) ) Clustering of extrasynaptic GABAA receptors modulates tonic inhibition in cultured hippocampal neurons, J Biol Chem 279: , 45833–45843. |
[311] | Jia F , Pignataro L , Schofield CM , Yue M , Harrison NL , Goldstein PA ((2005) ) An extrasynaptic GABAA receptor mediates tonic inhibition in thalamic VB neurons, J Neurophysiol 94: , 4491–4501. |
[312] | Orser BA , McAdam LC , Roder S , MacDonald JF ((1998) ) General anaesthetics and their effects on GABAA receptor desensitization, Toxicol Lett 100-101: , 217–224. |
[313] | Krasowski MD ((2003) ) Contradicting a unitary theory of general anesthetic action: A history of three compounds from 1901 to 2001, Bull Anesth Hist 21: , 1, 4-8, 21 passim. |
[314] | Krasowski MD , Harrison NL ((1999) ) General anaesthetic actions on ligand-gated ion channels, Cell Mol Life Sci 55: , 1278–1303. |
[315] | MacIver MB ((2014) ) Anesthetic agent-specific effects on synaptic inhibition, Anesth Analg 119: , 558–569. |
[316] | Yeung JYT , Canning KJ , Zhu G , Pennefather P , MacDonald JF , Orser BA ((2003) ) Tonically activated GABAA receptors in hippocampal neurons are high-affinity, low-conductance sensors for extracellular GABA, Mol Pharmacol 63: , 2–8. |
[317] | Liu X , Wang Q , Haydar TF , Bordey A ((2005) ) Nonsynaptic GABA signaling in postnatal subventricular zone controls GFAP-expressing progenitor proliferation, Nat Neurosci 8: , 1179–1187. |
[318] | Bordey A ((2007) ) Enigmatic GABAergic networks in adult neurogenic zones, Brain Res Rev 53: , 124–134. |
[319] | Paik NJ , Yang E ((2014) ) Role of GABA plasticity in stroke recovery, Neural Regen Res 9: , 2026–2028. |
[320] | Clarkson AN ((2012) ) Perisynaptic GABA receptors the overzealous protector, Adv Pharmacol Sci 2012: , 708428. |
[321] | Wei W , Faria LC , Mody I ((2004) ) Low ethanol concentrations selectively augment the tonic inhibition mediated by Δ subunit-containing GABAA receptors in hippocampal neurons, J Neurosci 24: , 8379–8382. |
[322] | Meera P , Olsen RW , Otis TS , Wallner M ((2010) ) Alcohol- and alcohol antagonist-sensitive human GABAA receptors: Tracking δ subunit incorporation into functional receptors, Mol Pharmacol 78: , 918–924. |
[323] | Mandyam CD ((2013) ) Neurogenesis and addictive disorders. In Biological Research on Addiction: Comprehensive Addictive Behaviors and Disorders, Academic Press, pp. 760. |
[324] | Eriksson PS , Perfilieva E , Björk-Eriksson T , Alborn AM , Nordborg C , Peterson DA , Gage FH ((1998) ) Neurogenesis in the adult human hippocampus, Nat Med 4: , 1313–1317. |
[325] | Lim DA , Alvarez-Buylla A ((2016) ) The adult Ventricular–Subventricular Zone (V-SVZ) and Olfactory Bulb (OB) neurogenesis, Cold Spring Harb Perspect Biol 8: , a018820. |
[326] | Walker CO , McCandless DW , McGarry JD , Schenker S ((1970) ) Cerebral energy metabolism in short-chain fatty acid-induced coma, J Lab Clin Med 76: , 569–583. |
[327] | Pringle MJ , Brown KB , Miller KW ((1981) ) Can the lipid theories of anesthesia account for the cutoff in anesthetic potency in homologous series of alcohols? Mol Pharmacol 19: , 49–55. |
[328] | Wong SM , Fong E , Tauck DL , Kendig JJ ((1997) ) Ethanol as a general anesthetic: Actions in spinal cord, Eur J Pharmacol 329: , 121–127. |
[329] | Chiou JS , Ma SM , Kamaya H , Ueda I ((1990) ) Anesthesia cutoff phenomenon: Interfacial hydrogen bonding, Science 248: , 583–585. |
[330] | Wick MJ , Mihic SJ , Ueno S , Mascia MP , Trudell JR , Brozowski SJ , Ye Q , Harrison NL , Harris RA ((1998) ) Mutations of γ-aminobutyric acid and glycine receptors change alcohol cutoff: Evidence for an alcohol receptor? Proc Natl Acad Sci U S A 95: , 6504–6509. |
[331] | Lugli AK , Yost CS , Kindler CH ((2009) ) Anaesthetic mechanisms: Update on the challenge of unravelling the mystery of anaesthesia, Eur J Anaesthesiol 26: , 807–820. |
[332] | Davies M ((2003) ) The role of GABAA receptors in mediating the effects of alcohol in the central nervous system, J Psychiatry Neurosci 28: , 263–274. |
[333] | Lees G , Edwards MD , Hassoni AA , Ganellin CR , Galanakis D ((1998) ) Modulation of GABA(A) receptors and inhibitory synaptic currents by the endogenous CNS sleep regulator cis-9,10-octadecenoamide (cOA), Br J Pharmacol 124: , 873–882. |
[334] | Laws D , Verdon B , Coyne L , Lees G ((2001) ) Fatty acid amides are putative endogenous ligands for anaesthetic recognition sites in mammalian CNS, Br J Anaesth 87: , 380–384. |
[335] | Coyne L , Lees G , Nicholson RA , Zheng J , Neufield KD ((2002) ) The sleep hormone oleamide modulates inhibitory ionotropic receptors in mammalian CNS in vitro, Br J Pharmacol 135: , 1977–1987. |
[336] | Hanada R , Tatara T , Iwao Y ((2004) ) Antagonizing potencies of saturated and unsaturated long-chain free fatty acids to isoflurane in goldfish, J Anesth 18: , 89–93. |
[337] | Yamakura T ((2004) ) Volatile anesthetic antagonism by long-chain free fatty acids, J Anesth 18: , 71–72. |
[338] | Koenig JA , Martin IL ((1992) ) Effect of free fatty acids on GABAA receptor ligand binding, Biochem Pharmacol 44: , 11–15. |
[339] | Witt M , Nielsen M ((1994) ) Characterization of the influence of unsaturated free fatty acids on brain GABA/benzodiazepine receptor binding in vitro, J Neurochem 62: , 1432–1439. |
[340] | Zhang L , Xiong W ((2009) ) Modulation of the Cys-loop ligand-gated ion channels by fatty acid and cannabinoids. In Vitamins & Hormones, Academic Press, pp. 315–335. |
[341] | Laterra J , Keep R , Betz LA , Goldstein GW ((1999) ) Blood-brain barrier. Basic Neurochemistry: Molecular, Cellular and Medical Aspects, 6th edition, SiegelGJ, AgranoffBW, AlbersRW, FisherSK, UhlerMD, eds. Lippincott-Raven, Philadelphia. |
[342] | Bodovitz S , Klein WL ((1996) ) Cholesterol modulates -secretase cleavage of amyloid precursor protein, J Biol Chem 271: , 4436–4440. |
[343] | Kojro E , Gimpl G , Lammich S , Marz W , Fahrenholz F ((2001) ) Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha-secretase ADAM 10, Proc Natl Acad Sci U S A 98: , 5815–5820. |
[344] | Simons M , Keller P , De Strooper B , Beyreuther K , Dotti CG , Simons K ((1998) ) Cholesterol depletion inhibits the generation of β-amyloid in hippocampal neurons, Proc Natl Acad Sci U S A 95: , 6460–6464. |
[345] | Ehehalt R , Keller P , Haass C , Thiele C , Simons K ((2003) ) Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts, J Cell Biol 160: , 113–123. |
[346] | Nixon RA ((2017) ) Amyloid precursor protein and endosomal–lysosomal dysfunction in Alzheimer’s disease: Inseparable partners in a multifactorial disease, FASEB J 31: , 2729–2743. |
[347] | Habchi J , Chia S , Galvagnion C , Michaels TCT , Bellaiche MMJ , Ruggeri FS , Sanguanini M , Idini I , Kumita JR , Sparr E , Linse S , Dobson CM , Knowles TPJ , Vendruscolo M ((2018) ) Cholesterol catalyses Aβ42 aggregation through a heterogeneous nucleation pathway in the presence of lipid membranes, Nat Chem 10: , 673–683. |
[348] | Rushworth JV , Hooper NM ((2010) ) Lipid rafts: Linking Alzheimer’s amyloid-β production, aggregation, and toxicity at neuronal membranes, Int J Alzheimers Dis 2011: , 603052. |
[349] | Arriagada C , Astorga C , Atwater I , Rojas E , Mears D , Caviedes R , Caviedes P ((2007) ) Endosomal abnormalities related to amyloid precursor protein in cholesterol treated cerebral cortex neuronal cells derived from trisomy 16 mice, an animal model of Down syndrome, Neurosci Lett 423: , 172–177. |
[350] | Grimm MOW , Haupenthal VJ , Rothhaar TL , Zimmer VC , Grösgen S , Hundsdörfer B , Lehmann J , Grimm HS , Hartmann T ((2013) ) Effect of different phospholipids on α-secretase activity in the non-amyloidogenic pathway of Alzheimer’s disease, Int J Mol Sci 14: , 5879–5898. |
[351] | Vetrivel KS , Thinakaran G ((2010) ) Membrane rafts in Alzheimer’s disease beta-amyloid production, Biochim Biophys Acta 1801: , 860–867. |
[352] | Kivipelto M , Helkala E-L , Laakso MP , Hänninen T , Hallikainen M , Alhainen K , Soininen H , Tuomilehto J , Nissinen A ((2001) ) Midlife vascular risk factors and Alzheimer’s disease in later life: Longitudinal, population based study, BMJ 322: , 1447–1451. |
[353] | Jin P , Pan Y , Pan Z , Xu J , Lin M , Sun Z , Chen M , Xu M ((2018) ) Alzheimer-like brain metabolic and structural features in cholesterol-fed rabbit detected by magnetic resonance imaging, Lipids Health Dis 17: , 61. |
[354] | Wingo TS , Cutler DJ , Wingo AP , Le N-A , Rabinovici GD , Miller BL , Lah JJ , Levey AI ((2019) ) Association of early-onset Alzheimer disease with elevated low-density lipoprotein cholesterol levels and rare genetic coding variants of APOB, JAMA Neurol 76: , 809–817. |
[355] | Ledesma MD , Dotti CG ((2005) ) The conflicting role of brain cholesterol in Alzheimer’s disease: Lessons from the brain plasminogen system, Biochem Soc Symp 72: , 129–138. |
[356] | Burns MP , Noble WJ , Olm V , Gaynor K , Casey E , LaFrancois J , Wang L , Duff K ((2003) ) Co-localization of cholesterol, apolipoprotein E and fibrillar Aβ in amyloid plaques, Mol Brain Res 110: , 119–125. |
[357] | Cataldo AM , Peterhoff CM , Troncoso JC , Gomez-Isla T , Hyman BT , Nixon RA ((2000) ) Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: Differential effects of APOE genotype and presenilin mutations, Am J Pathol 157: , 277–286. |
[358] | Xu W , Fang F , Ding J , Wu C ((2018) ) Dysregulation of Rab5-mediated endocytic pathways in Alzheimer’s disease, Traffic 19: , 253–262. |
[359] | Cataldo A , Hamilton D , Barnett J , Paskevich P , Nixon R ((1996) ) Properties of the endosomal-lysosomal system in the human central nervous system: Disturbances mark most neurons in populations at risk to degenerate in Alzheimer’s disease, J Neurosci 16: , 186–199. |
[360] | Morrison JH , Hof PR ((2002) ) Selective vulnerability of corticocortical and hippocampal circuits in aging and Alzheimer’s disease, Prog Brain Res 136: , 467–486. |
[361] | Fu H , Hardy J , Duff KE ((2018) ) Selective vulnerability in neurodegenerative diseases, Nat Neurosci 21: , 1350–1358. |
[362] | Cataldo AM , Mathews PM , Boiteau AB , Hassinger LC , Peterhoff CM , Jiang Y , Mullaney K , Neve RL , Gruenberg J , Nixon RA ((2008) ) Down syndrome fibroblast model of Alzheimer-related endosome pathology: Accelerated endocytosis promotes late endocytic defects, Am J Pathol 173: , 370–384. |
[363] | Jiang Y , Mullaney KA , Peterhoff CM , Che S , Schmidt SD , Boyer-Boiteau A , Ginsberg SD , Cataldo AM , Mathews PM , Nixon RA ((2010) ) Alzheimer’s-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition, Proc Natl Acad Sci U S A 107: , 1630–1635. |
[364] | Frolov A , Srivastava K , Daphna-Iken D , Traub LM , Schaffer JE , Ory DS ((2001) ) Cholesterol overload promotes morphogenesis of a Niemann-Pick C (NPC)-like compartment independent of inhibition of NPC1 or HE1/NPC2 function, J Biol Chem 276: , 46414–46421. |
[365] | Liscum L , Faust JR ((1987) ) Low density lipoprotein (LDL)-mediated suppression of cholesterol synthesis and LDL uptake is defective in Niemann-Pick type C fibroblasts, J Biol Chem 262: , 17002–17008. |
[366] | Jin L-W , Shie F-S , Maezawa I , Vincent I , Bird T ((2004) ) Intracellular accumulation of amyloidogenic fragments of amyloid-beta precursor protein in neurons with Niemann-Pick type C defects is associated with endosomal abnormalities, Am J Pathol 164: , 975–985. |
[367] | Nixon RA ((2004) ) Niemann-Pick Type C disease and Alzheimer’s disease, Am J Pathol 164: , 757–761. |
[368] | Sobo K , Le Blanc I , Luyet P-P , Fivaz M , Ferguson C , Parton RG , Gruenberg J , van der Goot FG ((2007) ) Late endosomal cholesterol accumulation leads to impaired intra-endosomal trafficking, PLoS One 2: , e851. |
[369] | Roff CF , Goldin E , Comly ME , Cooney A , Brown A , Vanier MT , Miller SP , Brady RO , Pentchev PG ((1991) ) Type C Niemann-Pick disease: Use of hydrophobic amines to study defective cholesterol transport, Dev Neurosci 13: , 315–319. |
[370] | Puri V , Watanabe R , Dominguez M , Sun X , Wheatley CL , Marks DL , Pagano RE ((1999) ) Cholesterol modulates membrane traffic along the endocytic pathway in sphingolipid-storage diseases, Nat Cell Biol 1: , 386–388. |
[371] | Brown RE ((1998) ) Sphingolipid organization in biomembranes: What physical studies of model membranes reveal, J Cell Sci 111 (Pt 1): , 1–9. |
[372] | Lönnfors M , Doux JPF , Killian JA , Nyholm TKM , Slotte JP ((2011) ) Sterols have higher affinity for sphingomyelin than for phosphatidylcholine bilayers even at equal acyl-chain order, Biophys J 100: , 2633–2641. |
[373] | Cossec J-C , Marquer C , Panchal M , Lazar AN , Duyckaerts C , Potier M-C ((2010) ) Cholesterol changes in Alzheimer’s disease: Methods of analysis and impact on the formation of enlarged endosomes, Biochim Biophys Acta 1801: , 839–845. |
[374] | Kawai M , Kalaria RN , Harik SI , Perry G ((1990) ) The relationship of amyloid plaques to cerebral capillaries in Alzheimer’s disease, Am J Pathol 137: , 1435–1446. |
[375] | Perlmutter LS , Chui Helena Chang ((1990) ) Microangiopathy, the vascular basement membrane and Alzheimer’s disease: A review, Brain Res Bull 24: , 677–686. |
[376] | Lütjohann D , Breuer O , Ahlborg G , Nennesmo I , Sidén A , Diczfalusy U , Björkhem I ((1996) ) Cholesterol homeostasis in human brain: Evidence for an age-deendent flux of 24S-hydroxycholesterolfrom the brain into the circulation. Proc Natl Acad Sci U S A 93: , 9799–9804. |
[377] | Lund EG , Guileyardo JM , Russell DW ((1999) ) cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain, Proc Natl Acad Sci U S A 96: , 7238–7243. |
[378] | Djelti F , Braudeau J , Hudry E , Dhenain M , Varin J , Bièche I , Marquer C , Chali F , Ayciriex S , Auzeil N , Alves S , Langui D , Potier M-C , Laprevote O , Vidaud M , Duyckaerts C , Miles R , Aubourg P , Cartier N ((2015) ) CYP46A1 inhibition, brain cholesterol accumulation and neurodegeneration pave the way for Alzheimer’s disease, Brain 138: , 2383–2398. |
[379] | Ayciriex S , Djelti F , Alves S , Regazzetti A , Gaudin M , Varin J , Langui D , Bièche I , Hudry E , Dargère D , Aubourg P , Auzeil N , Laprévote O , Cartier N ((2017) ) Neuronal cholesterol accumulationinduced by Cyp46a1 down-regulation in mouse hippocampus disruptsbrain lipid homeostasis, Front Mol Neurosci 10: , 211. |
[380] | Sun S , Zu X , Tuo Q , Chen L , Lei X , Li K , Tang C , Liao D ((2010) ) Caveolae and caveolin-1 mediate endocytosis and transcytosis of oxidized low density lipoprotein in endothelial cells, Acta Pharmacol Sin 31: , 1336–1342. |
[381] | Pompey S , Zhao Z , Luby-Phelps K , Michaely P ((2013) ) Quantitative fluorescence imaging reveals point of release for lipoproteins during LDLR-dependent uptake, J Lipid Res 54: , 744–753. |
[382] | Miaczynska M , Christoforidis S , Giner A , Shevchenko A , Uttenweiler-Joseph S , Habermann B , Wilm M , Parton RG , Zerial M ((2004) ) APPL proteins link Rab5 to nuclear signal transduction via an endosomal compartment, Cell 116: , 445–456. |
[383] | Zhu G , Chen J , Liu J , Brunzelle JS , Huang B , Wakeham N , Terzyan S , Li X , Rao Z , Li G , Zhang XC ((2007) ) Structure of the APPL1 BAR-PH domain and characterization of its interaction with Rab5, EMBO J 26: , 3484–3493. |
[384] | Gorvel JP , Chavrier P , Zerial M , Gruenberg J ((1991) ) rab5 controls early endosome fusion in vitro, Cell 64: , 915–925. |
[385] | Grbovic OM , Mathews PM , Jiang Y , Schmidt SD , Dinakar R , Summers-Terio NB , Ceresa BP , Nixon RA , Cataldo AM ((2003) ) Rab5-stimulated up-regulation of the endocytic pathway increases intracellular β-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Aβ production, J Biol Chem 278: , 31261–31268. |
[386] | Mishra A , Eathiraj S , Corvera S , Lambright DG ((2010) ) Structural basis for Rab GTPase recognition and endosome tethering by the C2H2 zinc finger of Early Endosomal Autoantigen 1 (EEA1), Proc Natl Acad Sci U S A 107: , 10866–10871. |
[387] | Kim S , Sato Y , Mohan PS , Peterhoff C , Pensalfini A , Rigoglioso A , Jiang Y , Nixon RA ((2016) ) Evidence that the rab5 effector APPL1 mediates APP-βCTF-induced dysfunction of endosomes in Down syndrome and Alzheimer’s disease, Mol Psychiatry 21: , 707–716. |
[388] | Farmer BC , Walsh AE , Kluemper JC , Johnson LA ((2020) ) Lipid droplets in neurodegenerative disorders, Front Neurosci 14: , 742. |
[389] | Lin Y-T , Seo J , Gao F , Feldman HM , Wen H-L , Penney J , Cam HP , Gjoneska E , Raja WK , Cheng J , Rueda R , Kritskiy O , Abdurrob F , Peng Z , Milo B , Yu CJ , Elmsaouri S , Dey D , Ko T , Yankner BA , Tsai L-H ((2018) ) APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types, Neuron 98: , 1141–1154.e7. |
[390] | Derk J , Hernandez KB , Rodriguez M , He M , Koh H , Abedini A , Li H , Fenyö D , Schmidt AM ((2018) ) Diaphanous 1 (DIAPH1) is highly expressed in the aged human medial temporal cortex and upregulated in myeloid cells during Alzheimer’s disease, J Alzheimers Dis 64: , 995–1007. |
[391] | Bosco DA , Fowler DM , Zhang Q , Nieva J , Powers ET , Wentworth P , Lerner RA , Kelly JW ((2006) ) Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate α-synuclein fibrilization, Nat Chem Biol 2: , 249–253. |
[392] | Eriksson I , Nath S , Bornefall P , Giraldo AMV , Öllinger K ((2017) ) Impact of high cholesterol in a Parkinson’s disease model: Prevention of lysosomal leakage versus stimulation of α-synuclein aggregation, Eur J Cell Biol 96: , 99–109. |
[393] | Meiser J , Weindl D , Hiller K ((2013) ) Complexity of dopamine metabolism, Cell Commun Signal 11: , 34. |
[394] | Lu F , Liang Q , Abi-Mosleh L , Das A , De Brabander JK , Goldstein JL , Brown MS ((2015) ) Identification of NPC1 as the target of U18666A, an inhibitor of lysosomal cholesterol export and Ebola infection, eLife 4: , e12177. |
[395] | Schmitt F , Hussain G , Dupuis L , Loeffler J-P , Henriques A ((2014) ) A plural role for lipids in motor neuron diseases: Energy, signaling and structure, Front Cell Neurosci 8: , 25. |
[396] | Pennetta G , Welte MA ((2018) ) Emerging links between lipid droplets and motor neuron diseases, Dev Cell 45: , 427–432. |
[397] | Wright DK , Liu S , van der Poel C , McDonald SJ , Brady RD , Taylor L , Yang L , Gardner AJ , Ordidge R , O’Brien TJ , Johnston LA , Shultz SR ((2017) ) Traumatic brain injury results in cellular, structural and functional changes resembling motor neuron disease, Cereb Cortex 27: , 4503–4515. |
[398] | McKee AC , Gavett BE , Stern RA , Nowinski CJ , Cantu RC , Kowall NW , Perl DP , Hedley-Whyte ET , Price B , Sullivan C , Morin P , Lee H-S , Kubilus CA , Daneshvar DH , Wulff M , Budson AE ((2010) ) TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy, J Neuropathol Exp Neurol 69: , 918–929. |
[399] | Rosenbohm A , Kassubek J , Weydt P , Marroquin N , Volk AE , Kubisch C , Huppertz H-J , Weber M , Andersen PM , Weishaupt JH , Ludolph AC , ALS Schwaben Register Group ((2014) ) Can lesions to the motor cortex induce amyotrophic lateral sclerosis? J Neurol 261: , 283–290. |
[400] | Wright P , Zvartau-Hind M ((2012) ) 27 - Organic psychiatry and epilepsy. In Core Psychiatry (Third Edition), WrightP, SternJ, PhelanM, eds. W.B. Saunders, Oxford, pp. 391–420. |
[401] | Lolekha P , Phanthumchinda K , Bhidayasiri R ((2010) ) Prevalence and risk factors of Parkinson’s disease in retired Thai traditional boxers, Mov Disord 25: , 1895–1901. |
[402] | Chiò A , Benzi G , Dossena M , Mutani R , Mora G ((2005) ) Severely increased risk of amyotrophic lateral sclerosis among Italian professional football players, Brain 128: , 472–476. |
[403] | Mackay DF , Russell ER , Stewart K , MacLean JA , Pell JP , Stewart W ((2019) ) Neurodegenerative disease mortality among former professional soccer players, N Engl J Med 381: , 1801–1808. |
[404] | Blecher R , Elliott MA , Yilmaz E , Dettori JR , Oskouian RJ , Patel A , Clarke A , Hutton M , McGuire R , Dunn R , DeVine J , Twaddle B , Chapman JR ((2019) ) Contact sports as a risk factor for amyotrophic lateral sclerosis: A systematic review, Global Spine J 9: , 104–118. |

