
Neuroprotective Effects of Cholinesterase Inhibitors: Current Scenario in Therapies for Alzheimer’s Disease and Future Perspectives
Abstract
Alzheimer’s disease (AD) is a slowly progressive neurodegenerative disease conceptualized as a continuous process, ranging from mild cognitive impairment (MCI), to the mild, moderate, and severe clinical stages of AD dementia. AD is considered a complex multifactorial disease. Currently, the use of cholinesterase inhibitors (ChEI), such as tacrine, donepezil, rivastigmine, and galantamine, has been the main treatment for AD patients. Interestingly, there is evidence that ChEI also promotes neuroprotective effects, bringing some benefits to AD patients. The mechanisms by which the ChEI act have been investigated in AD. ChEI can modulate the PI3K/AKT pathway, which is an important signaling cascade that is capable of causing a significant functional impact on neurons by activating cell survival pathways to promote neuroprotective effects. However, there is still a huge challenge in the field of neuroprotection, but in the context of unravelling the details of the PI3K/AKT pathway, a new scenario has emerged for the development of more efficient drugs that act on multiple protein targets. Thus, the mechanisms by which ChEI can promote neuroprotective effects and prospects for the development of new drug candidates for the treatment of AD are discussed in this review.
INTRODUCTION
Alzheimer’s disease (AD) is an irreversible neurodegenerative disease. AD is conceptualized as a continuous process, spanning from normal cognition to mild cognitive impairment (MCI), followed by progression from the mild, moderate, and severe clinical stages of AD dementia [1–3]. AD is the most common cause of dementia worldwide. It is estimated that over 139 million people worldwide will develop dementia by 2050, and among them, about 50–60% will develop AD [4]. AD is characterized by neuritic plaques and neurofibrillary tangles as a result of the accumulation of extracellular aggregated amyloid-β (Aβ) and intracellular aggregation of hyperphosphorylated tau protein, respectively [5, 6].
The loss of cholinergic neurons in the brain leads to impairment of cholinergic transmission, being the main cause of a cognitive decline in patients with AD [6]. Due to the essential role of acetylcholine (ACh) in cognitive function, a cholinergic hypothesis was raised for the pathogenesis of AD, based on the progressive loss of limbic and neocortical cholinergic innervation and reduced ACh synthesis [7]. Since ACh is involved in several physiological processes (such as memory, attention, learning, sensory information, and other critical functions), the degeneration of cholinergic neurons in the brain leads to reduced ACh levels, affecting cholinergic transmission, generating cognitive deficits [1, 8]. Therefore, the acetylcholinesterase enzyme (AChE), which hydrolyzes the neurotransmitter acetylcholine, has become an important therapeutic target for AD [6]. Different AChE inhibitors (AChEIs) were developed based on the cholinergic hypothesis [8], and three drugs are currently available for the treatment of AD: donepezil, galantamine, and rivastigmine. However, the effectiveness of these drugs is limited since they reduce AD symptoms, but they are unable to stop disease progression [6, 9]. There are several literature reports showing that some AChEIs demonstrate neuroprotective effects that were unrelated to enzyme inhibition [1, 10, 11]. Thus, new therapeutic strategies are focusing not only on the amplification of cholinergic activity but also on modulating non-cholinergic functions; these strategies have been emerging to develop new disease-modifying agents for AD treatment [12].
This review addresses the mechanisms by which cholinesterase (ChE) inhibitors promote neuroprotective effects, which are capable of bringing benefits to patients with AD. In addition, we also show new perspectives in the development of potential drug candidates endowed with neuroprotective capacity, which have been designed for AD therapy.
CHOLINESTERASE ENZYMES
ChE is a ubiquitous class of serine hydrolases that cleaves choline esters. There are two forms of ChE (encoded by two distinct genes): AChE, which hydrolyzes the neurotransmitter acetylcholine, and butyrylcholinesterase (BuChE) [13, 14]. Although both enzymatic forms exhibit similar catalytic activities, they differ in ionic or hydrophobic interactions [14].
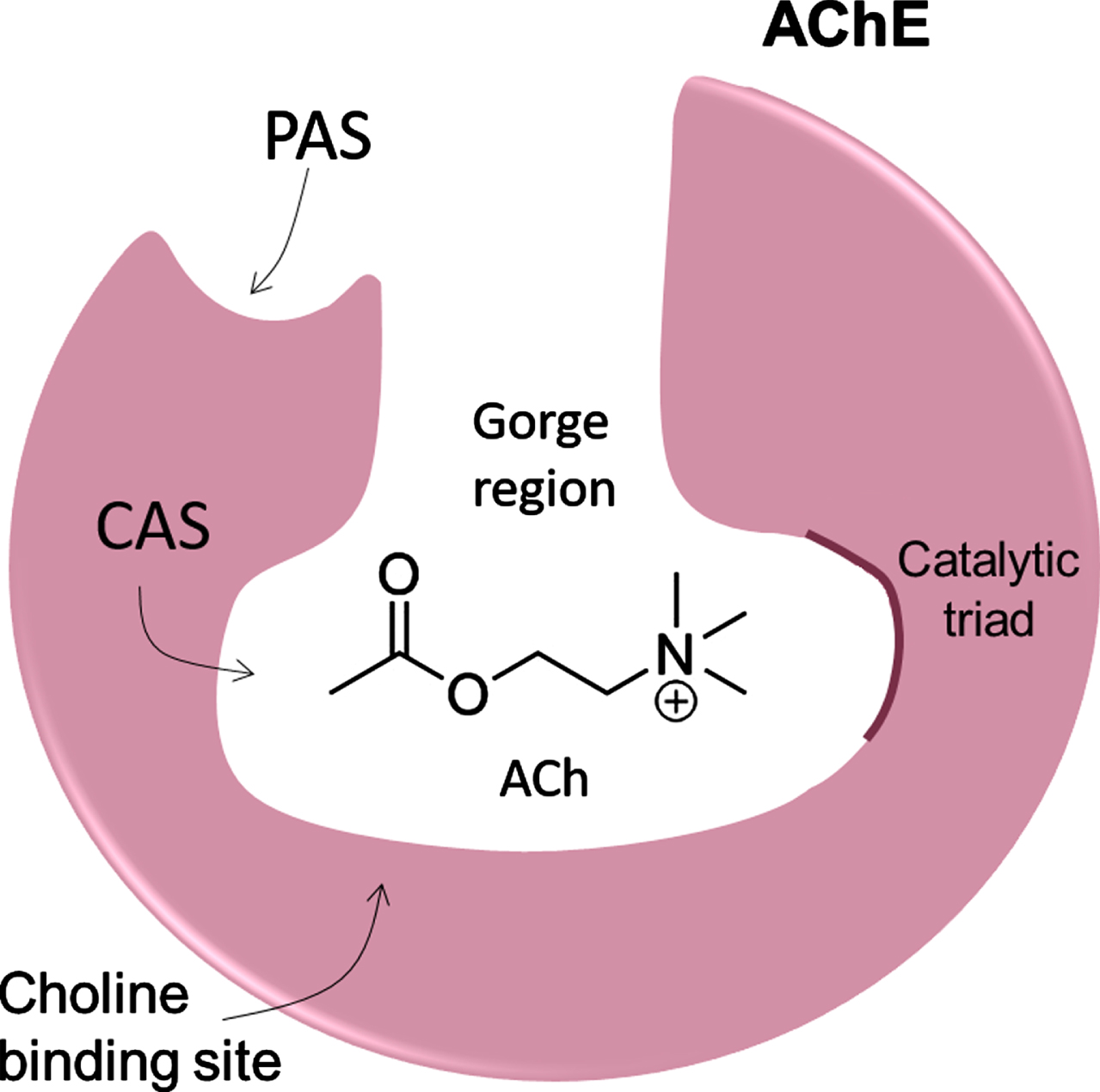
AChE is primarily located at neuromuscular junctions and cholinergic synapses in the central nervous system (CNS), where it catalyzes the hydrolysis of ACh into choline and acetate, making it responsible for terminating ACh-mediated synaptic transmission with high catalytic efficiency [9, 13]. AChE is structurally composed of an active catalytic site (CAS), the peripheral anionic site (PAS), and the gorge region (Fig. 1) [9]. The CAS site is located at the bottom of the gorge region, which is about 5 Å wide and 20 Å deep, lined with up to 14 conserved aromatic residues, whose rings constitute ∼70% of the gorge surface [15, 16]. CAS is the local where the hydrolysis reaction occurs and contains a catalytic triad (H440-E327-S200) and the critical aromatic residues, W84 and F330. The PAS is located near the entrance to the gorge, being composed of Y70, Y121, and W279 aromatic residues that are essential components of this site. PAS is a low-affinity binding site essential to forwarding ACh and to controlling substrate specificity at the gorge [9, 15, 16].
Fig. 1
Schematic structure of AChE showing the gorge region, active catalytic site (CAS), and the peripheral anionic site (PAS).
BuChE, also known as pseudocholinesterase or nonspecific cholinesterase, can hydrolyze larger substrates such as succinylcholine and acetylcholine, and, in particular, butyrylcholine [17, 18]. Similarly to AChE, BuChE is structurally formed by a 20 Å gorge lined with six aromatic amino acid residues, a CAS site, and a PAS site [19]; the catalytic triad is located at the bottom of the gorge, composed of S226, H466, and E353 [17]. Unlike AChE, in BuChE, the choline-binding site was replaced by Ala (A328), and the peripheral site was reduced to only two residues (D70 and Y332). These changes are likely responsible for the activation of the substrate characteristic of BuChE. In the acyl pocket of AChE, the Phe residues were replaced by less bulky aliphatic residues (L286 and V288); in BuChE, it increases the volume of the pouch, allowing the accommodation of larger substrates [18].
AChE is mainly from the neuronal origin, whereas BuChE is primarily originated from glial cells [19]. Under normal conditions, ACh is predominantly decomposed by AChE, while BuChE plays a supportive and functional role [19, 20]. Thus, the development of novel ChE inhibitors (ChEI) has been a promising approach for AD therapy [9]. However, it has been reported that BuChE levels increase as the AChE level decreases in AD patients at late-stage [21]. Furthermore, there is evidence that both AChE and BuChE have secondary non-cholinergic functions, which include the deposition of Aβ peptides in the form of senile plaques and the accumulation of neurofibrillary tangles in the brain of patients with AD [21]. Therefore, the development of new selective AChE and BuChE inhibitors is of crucial importance for new therapeutic modalities in AD [19, 22].
INTERACTIONS OF ChE WITH Aβ PEPTIDES
Aβ peptides are the main constituent of senile plaques and an important neurotoxic agent in AD [23]. AChE forms stable complexes with Aβ peptides during the enzyme assembly into filaments, consequently accelerating this process and stimulating the fibril elongation [24, 25]. This may be one of the reasons by which AChE is consistently found in deposits of amyloid plaques [26–28]. The interaction between AChE and Aβ peptides occurs due to the involvement of a small hydrophobic peptide that contains a conserved tryptophan (W279) located at the PAS site of AChE, contributing to the formation of a highly toxic complex [23, 28, 29]. In vitro studies showed that the action of AChEIs directed towards the PAS site are capable of inhibiting the AChE effect on the assembly of Aβ filaments, whereas inhibitors of the AChE CAS site did not show the same effect [25]. BuChE is also associated with amyloid plaques and it co-localizes with the Aβ peptide [28]. In contrast to AChE, reports show that BuChE inversely associates with Aβ peptides and delays the beginning of their assembly, decreasing the rate of in vitro Aβ fibril formation [30]. The PAS site of BuChE lacks three out of four aromatic residues that are found in the PAS site of AChE, thus exhibiting an inverse biochemical property [30].
Several studies indicate that AChE-Aβ complexes promote major neurotoxic effects when tested in vitro or in vivo, compared to treatment with Aβ peptides. Experiments performed in PC12 neuronal cell line demonstrated that treatments with AChE-Aβ complexes showed greater cytotoxicity than fibrillar Aβ complexes [24, 31, 32]. Interestingly, Reyes et al. (2004) found an increase in neurite network dystrophy, increased neuronal apoptosis, and a sustained increase in intracellular Ca2 + in hippocampal neurons from rats treated with AChE-Aβ complexes. There is evidence that AChE strongly stimulate mouse Aβ aggregation in vitro, resulting in Aβ-AChE complexes that are more toxic than Aβ fibrils [33], and altogether, these studies may suggest that AChE could play an important role in neurodegeneration [31]. Accordingly, AChEIs that selectively act at the PAS site of the enzyme also prevent the formation of AChE-AB complexes, as well as the acceleration of Aβ peptide assembly into filaments. Thus, AChEI seems to be an attractive and promising target for the development of new potential candidates for anti-AD therapeutic drugs [33, 34].
TRADITIONAL CHOLINESTERASE INHIBITORS
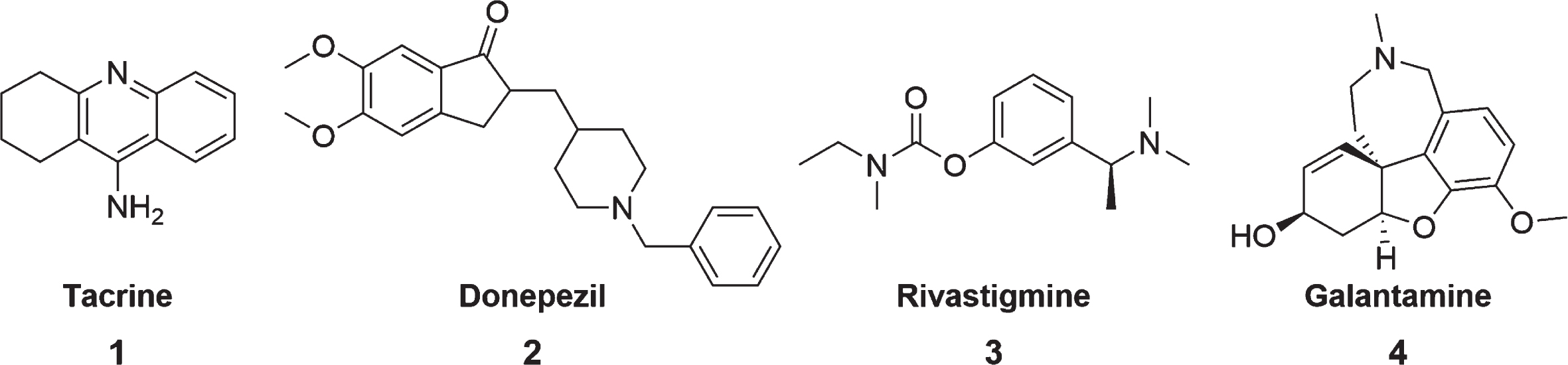
The various physiological alterations occurring in AD patients culminate in a reduction of ACh synthesis, as well as a decrease in cholinergic receptors, thus causing a reduction in cholinergic transmission [1]. Thus, ChEI was developed to block the catalytic action of the AChE, resulting in increased levels of ACh in the synaptic cleft and activation of cholinergic receptors [1, 5]. Thus, Tacrine 1, donepezil 2, rivastigmine 3, and galantamine 4 (Fig. 2) were the first drugs used in the clinic [34].
Fig. 2
Structures of the traditional ChEI.
Tacrine, an acridine derivative, was the first drug approved by the Food and Drug Administration (FDA) in 1993 to treat AD [35]. It is a potent reversible and non-competitive AChE/BuChE inhibitor [35, 36]. Tacrine inhibits AChE through its interaction with the CAS site [6]. Despite being an excellent cholinesterase inhibitor, soon after its registration, tacrine was withdrawn from the market due to adverse side effects, such as hepatotoxicity and low bioavailability [1, 35–37]. Nausea, vomiting, loss of appetite, and diarrhea were also common side effects caused by regular tacrine administration [6]. However, its chemical structure has been widely used to design new compounds that are capable of inhibiting ChE, with lower side effects and toxicity [35].
Donepezil is widely prescribed for mild, moderate, and severe AD and has been considered a first-line treatment against this disease since 1996 [38, 39]. It is a reversible, mixed inhibitor that exhibits competitive and non-competitive AChE activity [40, 41]. Donepezil is capable of simultaneously inhibiting both CAS and PAS sites of AChE [40]. It has a selective activity for AChE, but only a modest effect on BuChE, unlike other compounds such as rivastigmine and tacrine [41]. In addition to inhibiting AChE, donepezil also has activities at molecular and cellular levels in almost all stages involved in the pathogenesis of AD [6, 41]. However, adverse events can occur during its use, being significant at higher dosage (10 mg/d) compared to lower dosage forms (5 mg/d) [42]. Symptoms can range from appetite loss, vomiting, nausea, diarrhea, and rhinitis. However, gastrointestinal side effects are mainly observed [42].
Rivastigmine was approved for the treatment of patients with mild to moderate AD in 2000. It is a pseudo-irreversible carbamate-type of AChE and BuChE inhibitor, being selective for the CNS compared to peripheral tissues [8, 43, 44]. It is classified as a pseudo-irreversible inhibitor because it binds to AChE that cleaves the rivastigmine molecule. A covalent carbamoyl-AChE complex is formed, preventing the catalysis of acetylcholine and temporally inactivating the enzyme [5, 43]. Rivastigmine was initially administered orally, but due to gastrointestinal adverse events [44], the transdermal patch emerged, providing a continuous and well-controlled administration, reducing fluctuations in plasma concentration, with minor side effects [8, 44].
Galantamine is an alkaloid belonging to the Amaryllidaceae family, isolated from the Galanthus woronowii plant and indicated for treating mild to moderate AD [45, 46]. It is a reversible competitive and selective inhibitor of AChE interacting with the anionic subsite and the aromatic gorge, having only slight activity in the inhibition of BuChE [5, 44, 46]. Galantamine has CNS selectivity with little effect on peripheral tissues [45]. Furthermore, it is an allosteric modulator of nicotinic acetylcholine receptors (nAChRs) [45]. Although its clinical efficacy is equivalent to that of donepezil [47], galantamine seems to cause more side effects, especially gastrointestinal symptoms, compared to other AChE drugs [46].
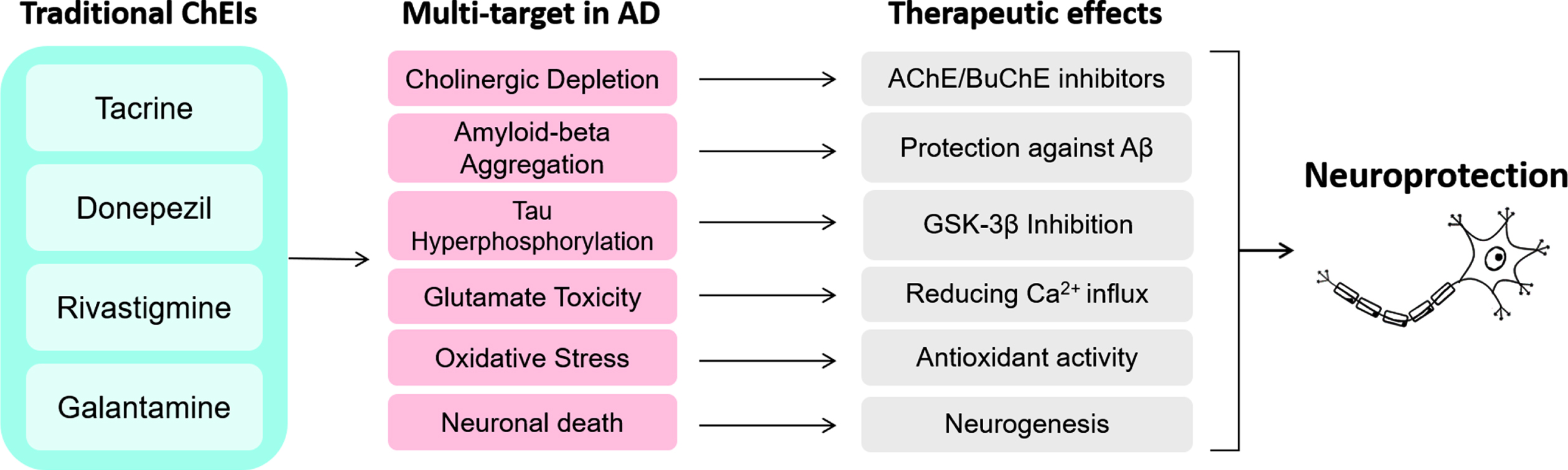
Fig. 3
Activities of traditional ChEI on multi-targets in AD may lead to various therapeutic effects, which altogether provide neuroprotection.
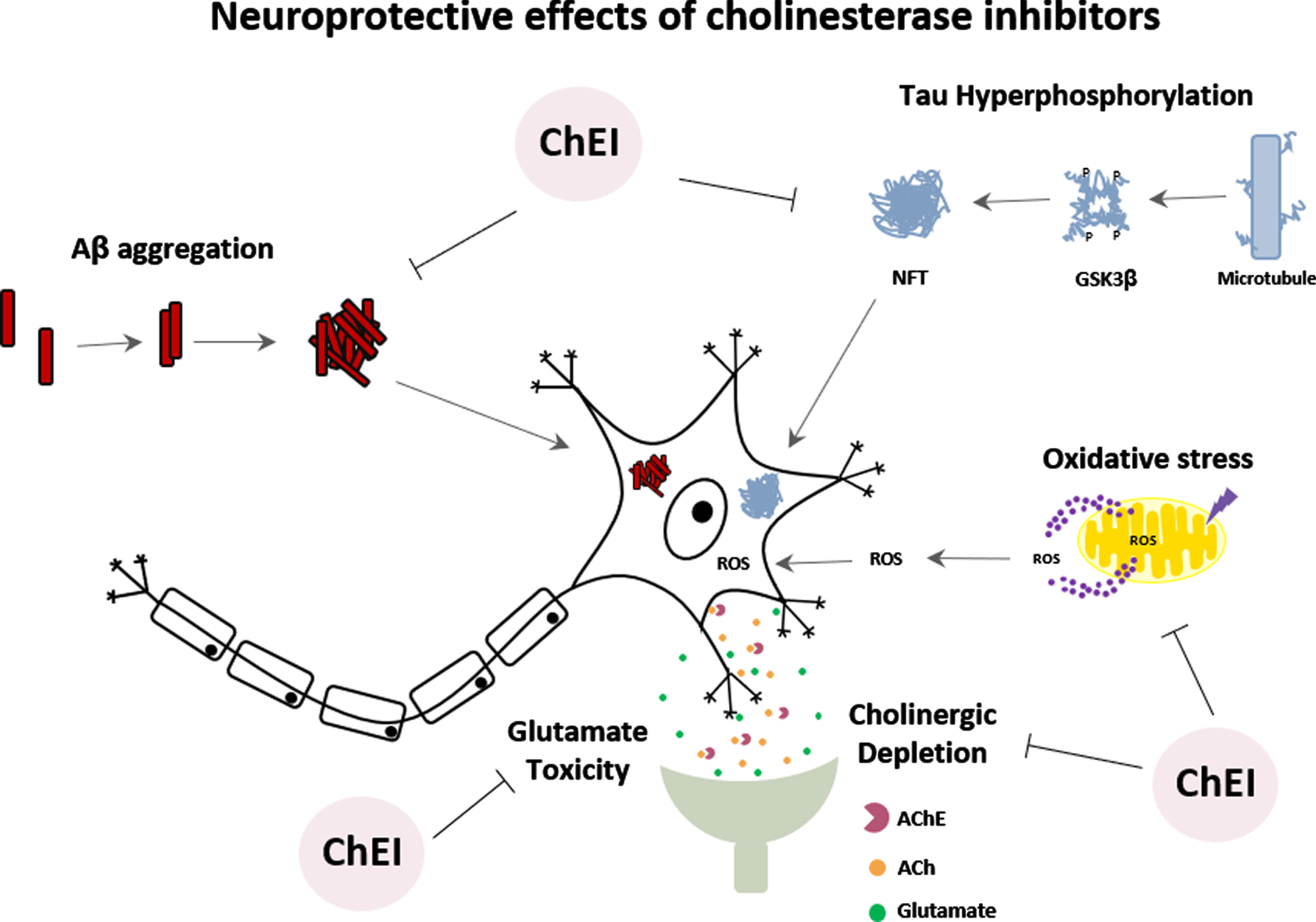
NEUROPROTECTIVE EFFECT OF TRADITIONAL ChE INHIBITORS
The mechanisms underlying the action of AChEIs drugs (donepezil, tacrine, rivastigmine, and galantamine) have been investigated not only for their effects on AChE inhibition but also for their ability to promote neuroprotection against cell damage. However, there still remains an immense challenge in the field of neuroprotection [11].
The Aβ cascade hypothesis, the most cited hypothesis in AD pathology, focuses on the abnormal processing of amyloid-β protein precursor (AβPP), leading to imbalance between the production and clearance of Aβ, generating an excess of Aβ aggregation and neurotoxicity [34, 48]. Several studies conducted in neuronal cultures treated with donepezil [49–51], tacrine [52, 53], rivastigmine [54], and galantamine [55–58] showed neuroprotective effects, such as increased cell viability, reduction of neuronal death and release of inflammatory mediators against Aβ toxicity. In animal models, donepezil was able to induce cognitive and behavioral improvements, as well as decrease the deposition of Aβ peptide and reduction of neuronal death [49, 59–62]. Other studies showed that donepezil, rivastigmine, and galantamine could promote neuroprotection and reduce Aβ deposition [63–66]. Furthermore, AD patients treated with donepezil revealed improvements in cognitive functions and decreased levels of Aβ in peripheral blood cells [67].
Another hypothesis is based on tau protein hyperphosphorylation leading to the formation of intracellular neurofibrillary tangles (NFTs) in the brain of AD patients [68]. Glycogen synthase kinase-3β (GSK-3β) is one of the central kinases responsible for tau hyperphosphorylation. Studies suggest that the neuroprotective effects of donepezil would also be occurring through the inhibition of GSK-3β activity [50, 69]. Interestingly, donepezil, rivastigmine, and galantamine showed a neuroprotective effect against the toxicity of okadaic acid, an inducer of tau protein hyperphosphorylation, reducing neuronal death [70, 71]. Treatment of AD patients with donepezil for six months caused an increase in phosphorylated GSK-3β (inactive enzyme), which would reflect a reduction in tau protein phosphorylation [72].
The cortical neurodegeneration of AD is also attributed to glutamate-induced neurotoxicity [10]. Several studies have shown that donepezil protects neurons against glutamate-induced toxicity by reducing Ca2 + influx, caspase-3 activation, and neuronal death [73–75]. There are reports showing that tacrine and galantamine can also reduce glutamate-induced cell death [10, 74, 76].
Under conditions of tissue injury, such as ischemia, the generation of oxidative stress can cause damage to all major cellular molecules [77]. On the other hand, under oxygen-glucose deprivation, assays performed in primary cultures of rat cortical neurons showed that donepezil caused protective effects against ischemic damage; however, galantamine, tacrine, and rivastigmine did not promote the same effect [75]. A protective effect of galantamine against oxidative damage has been observed in neuronal cultures [56, 78, 79]. Furthermore, in vitro pre-treatment with donepezil could protect endothelial cells against damage induced by hydrogen peroxide [77]. In the last decade, several reports in the literature have focused on the ability of ChEI to induce neuronal recovery by increasing neurogenesis and neuritogenesis. Several studies demonstrated that donepezil could promote neuronal differentiation and neurite growth in vitro and in vivo [80–84]. Similar effects were also found for tacrine, galantamine, and rivastigmine [85–90].
The importance of ChEIs is mainly centered on their mechanisms related to neuroprotection, and its capability of acting on different targets or processes in AD (Fig. 3). The ChEIs action occurs by modulating the cell survival pathway to promote neuroprotective effects. Most of these effects induced by ChEI are related to the stimulation of nAChRs, downregulation of NMDA receptors, and consequently, induction of the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) and mitogen-activated protein kinase (MAPK) signaling pathways [10, 73, 91]. PI3K/AKT pathway plays a significant functional impact on synaptic plasticity, neuronal polarity, neurotransmission, use-dependent translation, metabolic control and stress responses, including DNA repair in the brain [92, 93]. Thus, the PI3K/AKT pathway has been increasingly investigated regarding its role on the induction of neuroprotective effects.
PI3K/AKT PATHWAY MEDIATING THE MULTI-TARGET EFFECTS IN AD
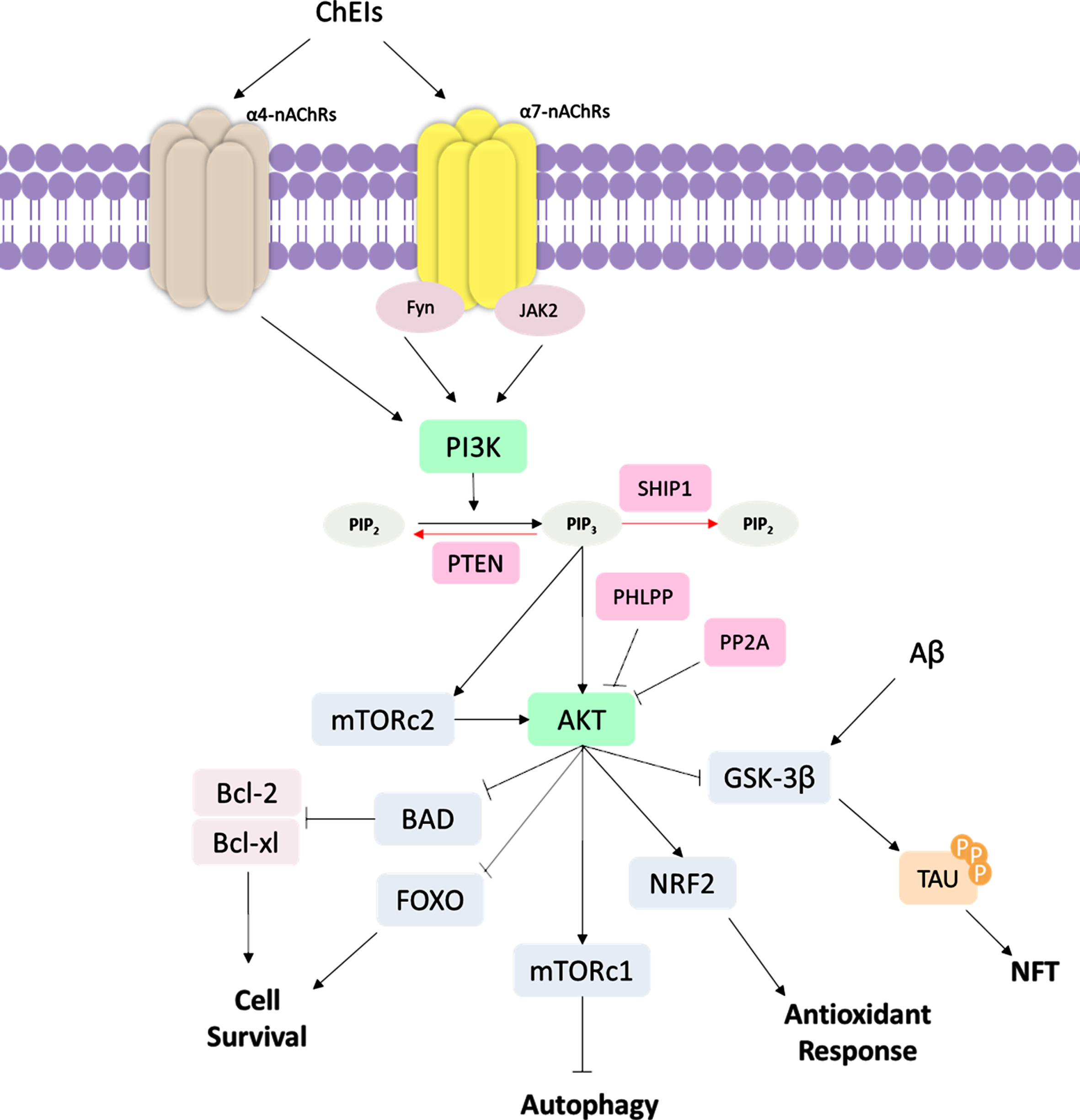
PI3K/AKT pathway provides a link between several pathological processes in AD, such as ageing “itself”, glucose metabolism, Aβ aggregation, tau hyperphosphorylation, synapse loss, oxidative stress, inflammation, and neuronal death [94]. The PI3K/AKT pathway can be induced by AChEI through signaling by nAChRs reported in in vitro studies [95–98]. To date, about seventeen subunits of nAChRs have been described [99]. In mammalian brain cells, the most commonly found nAChRs are the α4β2 heteromeric (α4β2-nAChR) and the α7 homomeric subunit–α7 nicotinic ACh receptor (α7-nAChR) [100], whose importance in AD treatment has been widely examined. For example, the stimulation of α7-nAChRs contributes to memory formation and consolidation [96] and cellular recovery after Aβ-induced neurotoxicity in mice [97]. On the other hand, the blockade of α7-nAChRs induces cognitive deficits in mice [98]. In addition to inhibiting AChE, there is evidence that ChEI interacts with α7-nAChRs, which in conjunction with tyrosine kinase Fyn and Janus-activated kinase 2 (JAK2), lead to the activation of PI3K signaling cascade and induction of neuroprotective effects [95, 101]. PI3K is a plasma membrane-associated kinase protein composed of a p85 regulatory subunit and a p110 catalytic subunit. Upon activation, PI3K catalyzes the conversion of phosphatidylinositol (3,4)-bisphosphate (PIP2) into phosphatidylinositol (3,4,5)-trisphosphate (PIP3), which further activates several downstream proteins, such as AKT [102]. Activated AKT modulates key biological processes that are essential for maintaining cellular metabolism and cell survival [103].
Notoriously, AKT plays an important role in the regulation of cell death through the activation of proteins, such as the B-cell lymphoma 2 (Bcl-2) protein family [104]. Bcl-2 protein family includes Bcl-2-associated death promoter (BAD), Bcl-2 Associated X-protein (BAX), Bcl-2, and B-cell lymphoma-extra large (Bcl-xL) proteins that localize within the mitochondrial membrane [105]. Whereas Bad and Bax are pro-apoptotic proteins that can trigger the release of cytochrome C or caspase, thus inducing apoptosis, Bcl-2 and Bcl-xL are anti-apoptotic proteins that contribute to cell survival [105]. Furthermore, AKT also acts on different processes, such as stress response, antioxidant defense and autophagy, through the inhibition of FOXO transcription factors [106, 107]. There are reports showing that Aβ oligomers alter the PI3K-AKT signaling [108, 109], thus suggesting this pathway as a potential therapeutic target for new drugs. Actually, studies in AD patients demonstrated a downregulation of PI3K and its downstream targets [110, 111].
Of particular interest in AD, AKT normally inhibits GSK-3β and activates the nuclear factor (erythroid-derived 2)-like 2 (NRF2) protein, which is the main transcription regulator of antioxidant genes [112]. As a consequence of GSK-3β activation, there is an increase in Aβ production, hyperphosphorylation of tau protein, and consequently, NFTs formation [113, 114]. In this context, GSK-3β seems to be an interesting, promising target for designing new drugs, under the hypothesis that its inhibition might impact several alterations in AD; some of them have been associated with reduced levels of Aβ, tau protein, and neuronal death [115, 116]. An interesting finding is that activated GSK-3β promotes NRF2 downregulation [117]. Since the role of the NRF2 gene is related to the antioxidant system, NRF2-deficient mice present increased levels of oxidative stress, phosphorylated tau, Aβ accumulation [118], and also significant cognitive deficits [119]. Interestingly, studies on mouse AD models showed that drugs targeting NRF2 are potential neuroprotectors and can ameliorate cognition in mice [120, 121].
Another key protein in the PI3K/AKT pathway is the mammalian target of rapamycin (mTOR), which is phosphorylated and activated by AKT. mTOR is the catalytic subunit of two multi-protein complexes: mTOR complex 1 (mTORc1) and 2 (mTORc2) [122]. mTORc1 plays a crucial role in preventing the initiation of autophagy, which is an essentially biological process responsible for the clearance of misfolded and aggregated proteins, such as Aβ and tau, two proteins considered as hallmarks of AD. Enhanced mTORc1 activity is associated with reduced Aβ clearance, accumulation of Aβ aggregates [123, 124], tau protein and increased NFT formation [125, 126] due to dysfunctional autophagy. mTORc1 can also upregulate AβPP processing, enhancing Aβ formation [127]. On the contrary, mTOR inhibitors have ameliorated cognitive function [124], learning capacity and memory [123], thus suggesting that mTOR might be a potential target for AD treatment. mTORc2 can phosphorylate AKT and induce AKT signaling. Additionally, mTOR plays a role in the regulation of synaptic plasticity and neurotransmission [128]. It has been demonstrated that the coordination between mTORc1 and mTORc2 is necessary for the correct functioning of AKT signaling [122].
To counterbalance the PI3K-AKT pathway, other proteins, such as phosphatase and tensin homolog (PTEN) protein and Src homology domain-containing inositol 5′-phosphatase 1 (SHIP1) are required as negative regulators of AKT signaling; these proteins induce the dephosphorylation of PIP3 into PIP2 [129]. In addition, PH domain and leucine-rich repeat protein phosphatase (PHLPP) and protein phosphatase 2A (PP2A) are also downregulators of AKT by dephosphorylating AKT [129]. Inhibition of PTEN in AD mouse models is associated with several consequences, such as reduced expression of Bax protein, decreased apoptosis, lower levels of DNA fragmentation, reduced endoplasmic reticulum stress [130], and rescue of normal synaptic function and cognition [131]. Noteworthy, reduced activation of PTEN and increased AKT phosphorylation might be involved in neuritogenesis and neurodifferentiation [84]. Accordingly, targeting PTEN to induce AKT activation might be a potential approach to be investigated in AD therapy (Fig. 4).
Fig. 4
Schematic representation of PI3K/AKT signaling pathway ChEI binding leads to the stimulation of α4 and α7 nicotinic acetylcholine receptors (nAChRs); subsequently, occurs the activation of tyrosine kinase Fyn and Janus-activated kinase 2 (JAK2), leading to the activation of phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K). PI3K converts phosphatidylinositol (3,4)-bisphosphate (PIP2) into phosphatidylinositol (3,4,5)-trisphosphate (PIP3), which activates protein kinase B (AKT). mTOR Complex 2 (mTORC2) also activate AKT signaling. PI3K/AKT pathway regulates several cellular functions, such as inhibition of Glycogen synthase kinase-3β (GSK-3β) that affects tau hyperphosphorylation, inhibition of Forkhead box O (FOXO) and Bcl-2-associated death promoter (BAD) proteins, which are cell survival regulators, such as B-cell lymphoma 2 (Bcl-2) and Bcl-2, and B-cell lymphoma-extra large (Bcl-xL). AKT also activates the mTOR Complex 1 (mTORc1) autophagy regulator and NRF2, which promotes antioxidant response. In AD, the Aβ also acts by inducing GSK-3β activity, increased NFT formation. The PI3K/AKT signaling pathway can be regulated in several ways. The Phosphatase and tensin homolog (PTEN) protein and Src homology domain-containing inositol 5′-phosphatase 1 (SHIP1) induce the dephosphorylation of PIP3 into PIP2, being negative regulators of PI3K/AKT signaling. PH domain and leucine-rich repeat protein phosphatase (PHLPP) and protein phosphatase 2A (PP2A) are also downregulators of AKT protein.
Over the years, the accumulation of literature information pointed out the increasing importance of the PI3K/AKT pathway in the pathogenesis of AD. Altogether, several key proteins acting on this pathway can be selected as potential targets for the development of multi-target-directed ligands (MTDLs), thus affecting simultaneously different stages of the signaling cascade [132]. Thus, several compounds, that act in the PI3K/AKT signaling pathway, have been developed for the treatment of AD. DL0410, a novel dual cholinesterase inhibitor, is a MTDL small molecule [132]. Studies have shown that DL0410 improves cognitive deficits, and also mitochondrial function; besides, the activation of the AKT/GSK-3β and MAPK/ERK signaling pathway promotes the reduction of Aβ that can result in the induction of synaptic transmission [132–134].
Treatment with EVP-6124 (encenicline), an α7-nAChR partial agonist, in patients with mild to moderate AD at Phase I (NCT00766363) and II (NCT01073228) led to improvements in cognitive and clinical functioning, being safe and well-tolerated [135]. However, Phase III studies (NCT01969136) have been suspended due to clinical hold. Another potent and selective α7-nAChR antagonist, ABT-126, showed cognitive efficacy in animal models [138]. ABT-126 was investigated in Phase I (NCT00867399) and Phase II (NCT00948909) studies, demonstrating to be well-tolerated and safe, but a significant cognitive improvement was not observed in patients with mild to moderate AD [136].
Phase I (NCT00948259) and II (NCT01350362) studies with NP031112, a GSK3 protein inhibitor, carried out in patients with mild and moderate AD demonstrated that the treatment was well tolerated, but without any clinical benefit [137, 138]. Another selective inhibitor of GSK3, AZD1080, tested in animal studies significantly reduced tau phosphorylation, reversing cognitive deficits [139].
Hence, in the context of unravelling the details of the PI3K/AKT signaling pathway regarding its role linked to AD pathology and neuroprotective effects caused by certain therapeutic drugs, data in the literature on the outcome of clinical applications are still scarce; but in spite of this, a new scenario emerges towards the development of more efficient drugs, which act on multiple protein targets. However, the consequences of inhibiting multiple targets must be investigated with caution, since the effects on more than one target may likely extend to different cellular processes, and these must be balanced against the desirable effects in terms of targeting the inherent changes to AD pathology.
NEXT-GENERATION ChE INHIBITORS
There is a growing interest in the development of potential MTDL drugs for the treatment of neurodegenerative diseases, such as AD. This comes from the perception that for this disease, the pathophysiology may result from an impairment of a complex intracellular network, mainly involving important signaling cascade, such as the PI3K/AKT pathway, as aforementioned. In this context, relying on a single target or process will probably not achieve the desired result, mainly due to the existence of cross-signaling mechanisms, as well as a positive and negative control of molecular pathways. Thus, interventions towards more than one step of this intricate molecular signaling network have great potential to overcome the pathological changes that occur in AD, although there are many limitations and the need to control biological responses and undesirable effects.
Although all initial AChEI drugs have been used as scaffolds to propose novel therapeutic compounds, a significant amount of these potential drugs relied on tacrine 1 to guide the research on anti-AD drug discovery. It is still of interest nowadays, even though this compound was discontinued in 2013 due to its hepatotoxicity [6, 140]. Some novel tacrine-like hybrids were recently reported [6, 140, 149–152, 141–148]. Compounds with the highest ChE inhibition are represented in Fig. 5, with their IC50 displayed in Table 1. It is worth mentioning that modifications in the tetrahydroaminacrine system have been proposed not only to increase the effectiveness of ChE inhibition, but also aiming to reduce hepatotoxic effects [6, 45, 140]. Besides, many structures are endowed not only to achieve ChE inhibition, but also to obtain antioxidant properties, metal chelation, blockage of Aβ42 self-aggregation, inhibition of BACE1, HDAC, or NMDA receptors, aiming to prevent neurodegeneration [141–143, 147, 149, 151, 153]. Although there is a great interest in developing rivastigmine and galantamine analogues [45, 140], significant efforts regarding the anti-cholinergic approach rely mostly on tacrine and donepezil modifications. Thus, here we focused on these kinds of structures.
Fig. 5
Chemical structure of tacrine and most potent tacrine-based hybrids as ChE inhibitors described in the last years. The color pink highlights the 1,2,3,4-tetrahydroakridin-9-amine rings as derivatives in each structure.
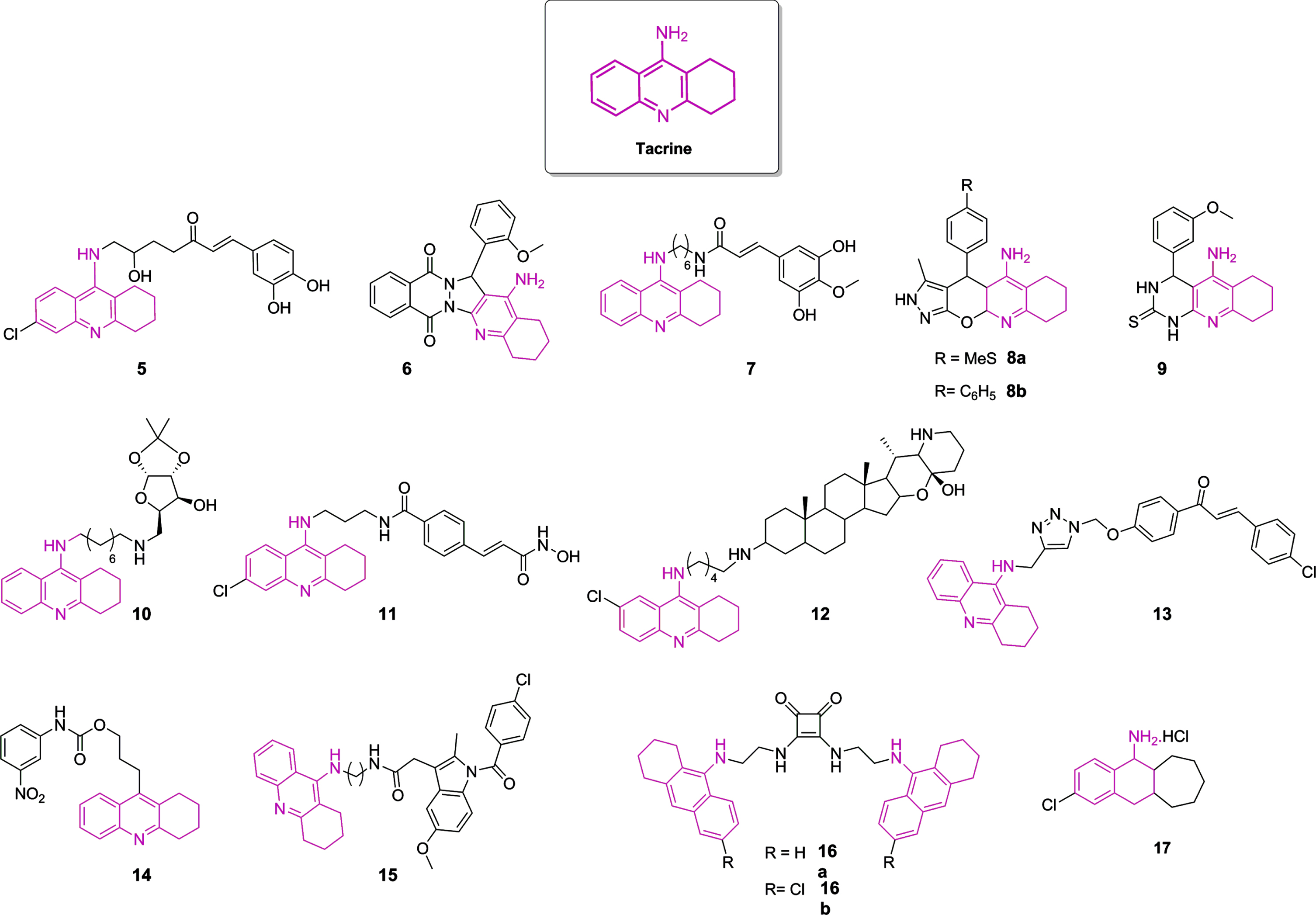
Table 1
Inhibitory activities (IC50 nM) of AChE and BuChE and selectivity index of novel tacrine-like compounds
| Compounds | IC50 (nM) | Ratio of BuChE/AChE IC50 | Multi-targeted ligand | Ref | |
| AChE | BuChE | ||||
| Tacrine | 174.00 | 32.00 | 5.40 | — | [151] |
| 5 | 150.00 | 36.00 | 0.24 | BACE1 inhibition, Neuroprotection against glutamate toxicity | [145] |
| 6 | 49.00 | 93280.00 | 1903.67 | Neuroprotection and Aβ aggregation inhibitory activities | [146] |
| 7 | 3.90 | 24.30 | 6.23 | Inhibition of Aβ 42 self-aggregation and fibril formation, Antioxidant activity, Neuroprotection | [147] |
| 8a | 580.00 | >66050 | >113.88 | — | [148] |
| 8b | 440.00 | >61020 | >138.68 | — | |
| 9 | 3050.00 | 3190.00 | 1.05 | Moderate-to-potent calcium channel blockers | [149] |
| 10 | 2.20 | 4.93 | 2.24 | — | [150] |
| 11 | 0.12 | 361.52 | 3012.67 | HDAC inhibition, Cu2+ chelating properties | [151] |
| 12 | 26.00 | 9.00 | 0.35 | — | [152] |
| 13 | 259.00 | – | – | Antioxidant activity | [141] |
| 14 | 22.15 | 6.96 | 0.31 | — | [142] |
| 15 | 10.00 | 57.00 | 5.70 | Antioxidant activity | [143] |
| 16a | 13.00 | 21.00 | 1.62 | — | [144] |
| 16b | 2.00 | 110.00 | 55.00 | — | |
| 17 | 33.40 | 62.00 | 1.86 | NMDA receptors inhibition | [153] |
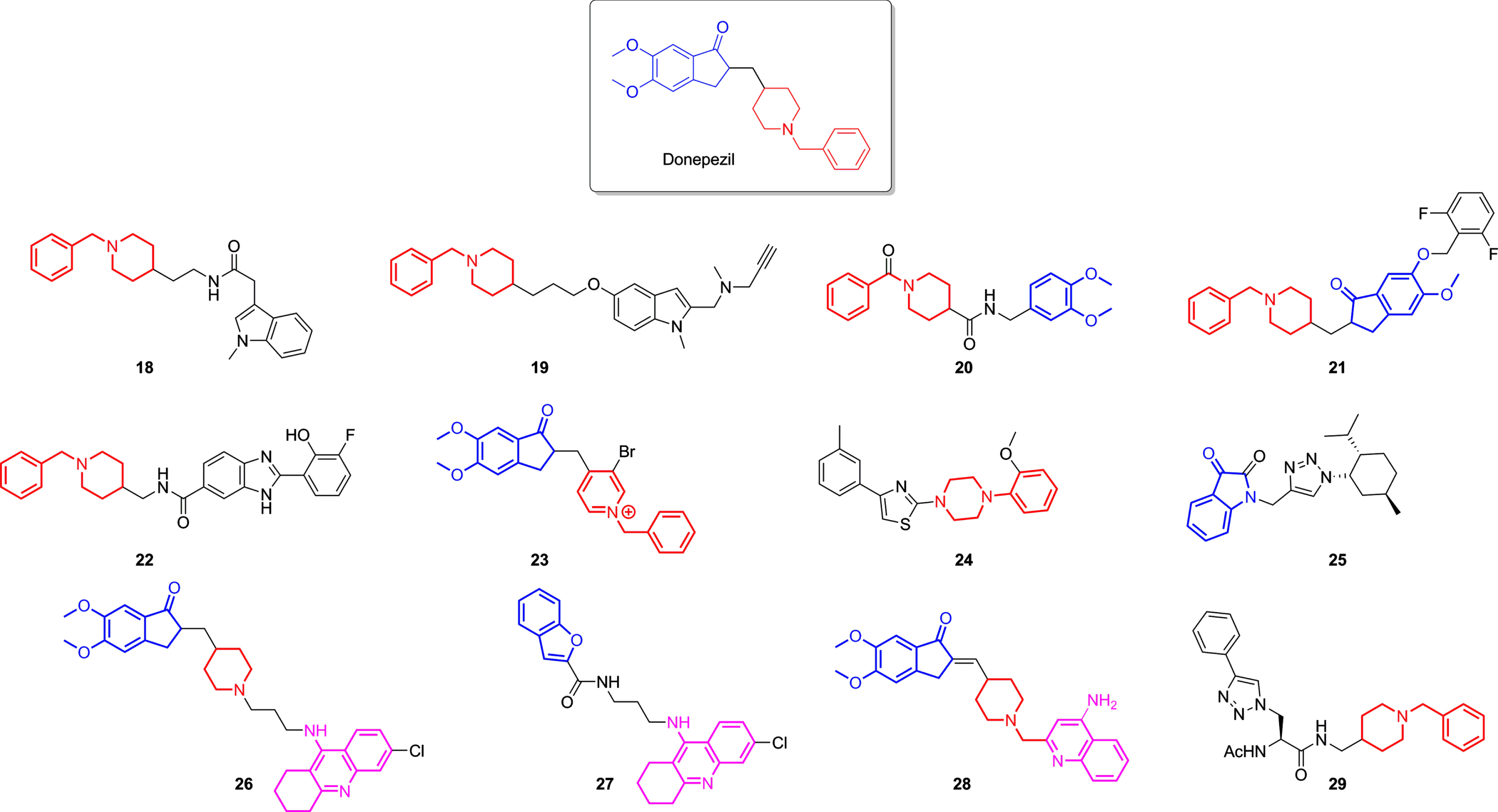
Donepezil-based derivatives were also designed and synthesized as AChEI. Their structure modification mainly relied upon replacing dimethoxy indanone fragment, responsible for the interaction with the PAS site, while preserving the benzylpiperidine moiety, which fulfils the CAS site [140, 154]. The hybrid molecular strategy was also employed, leading not only to novel selective ChEIs but also MTDLs, which possess other non-cholinergic effects, such as inhibiting MAO 19, BACE-1 20 and 21 [156, 157], chelating metal and diminishing ROS production 27 [158], or even inhibiting Aβ aggregation 22, 26, 27, 28, and 29 [12, 158–161]. Nevertheless, the hybridization of donepezil with tacrine was also attempted, originating potent compounds (Table 2) that conserved indanone, piperidine, and tetrahydroaminacrine moieties 26 and to structures that interchanged one of these moieties to an isostere, for example, 27, which replaced the indanone group to a benzofuran, as demonstrated in Fig. 6.
Table 2
Inhibitory activities (IC50 nM) of AChE and BuChE of donepezil-like compounds
| Compounds | IC50 (nM) | Multi-targeted ligand | Ref | |
| AChE | BuChE | |||
| Donepezil | 30.00 | 4660.00 | Aβ aggregation inhibition; BACE1 inhibition; MAO B inhibition; | [142, 155, 157] |
| 18 | 1420.00 | 0.25 | — | [162] |
| 19 | 350.00 | 460.00 | MAO inhibition | [155] |
| 20 | 4.10 | – | BACE-1 inhibition, metal chelating | [156] |
| 21 | 16.00 | – | BACE-1 inhibition | [157] |
| 22 | 1670.00 | – | Aβ aggregation inhibition, metal chelating | [159] |
| 23 | 0.36 | – | Antioxidant activity | [163] |
| 24 | 1147.00 | – | — | [164] |
| 25 | – | 510.00 | weak Aβ aggregation inhibition | [165] |
| 26 | 0.27 | – | Aβ antiaggregating activity | [160] |
| 27 | 120.00 | – | Aβ aggregation inhibition, metal chelating | [158] |
| 28 | 14.00 | 3690.00 | Neuroprotection against Aβ 42 | [12] |
| 29 | >10,000 | 0.17 | — | [161] |
Fig. 6
Donepezil and some of the most potent ChE inhibitors derived from donepezil lately developed. Highlighted in blue are the indanone fragments, in red benzylpiperidines moieties, and in pink the 1,2,3,4-tetrahydroakridin-9-amine rings in each structure.
As mentioned above, despite ChE inhibition is still considered an important strategy for anti-AD therapy, interest in novel MTDLs designed for AD treatment is a crucial aspect in the search for effective outcomes. Despite the neuroprotective effects reported for ChEIs, the effectiveness of drugs traditionally used in the clinic is modest, and there is an immense challenge in the search for more effective drugs capable of slowing the progression of AD, in addition to controlling the symptoms of the disease (Fig. 7). Thus, the search for understanding the molecular mechanisms of action of these compounds remains, highlighting the relevance of designing new drugs capable of reaching multiple targets in AD.
Fig. 7
Multi-target effects of cholinesterase inhibitors on different pathways involved in the development of Alzheimer’s disease.
CONCLUSIONS
AD is considered a complex multifactorial disease. The use of cholinesterase inhibitors (donepezil, rivastigmine, galantamine) is currently the main treatment modality. Several in vitro and in vivo studies have shown that traditional ChEI is provided with cholinergic activities as well as activity against various AD molecular targets, but their effects in reducing AD progression are still limited. In the field of therapeutic strategies for AD patients, it is extremely important to understand the signaling pathways and cellular mechanisms that are relevant for inducing neuroprotection. In this context, a substantial progress has been observed and, for the short-term future, the expected amount of information may provide the essential basis for the development of new highly effective multi-target-directed ligand drugs designed to improve cognitive functions and reduce AD neurodegeneration.
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Breijyeh Z , Karaman R ((2020) ) Comprehensive review on Alzheimer’s disease: Causes and treatment. Molecules 25: , 5789. |
[2] | Davis M , O’Connell T , Johnson S , Cline S , Merikle E , Martenyi F , Simpson K ((2018) ) Estimating Alzheimer’s disease progression rates from normal cognition through mild cognitive impairment and stages of dementia. Curr Alzheimer Res 15: , 777–788. |
[3] | Sperling RA , Aisen PS , Beckett LA , Bennett DA , Craft S , Fagan AM , Iwatsubo T , Jack CR , Kaye J , Montine TJ , Park DC , Reiman EM , Rowe CC , Siemers E , Stern Y , Yaffe K , Carrillo MC , Thies B , Morrison-Bogorad M , Wagster MV , Phelps CH ((2011) ) Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 7: , 280–292. |
[4] | Alzheimer’s Disease International (ADI). Dementia statistics. https://www.alzint.org/about/dementia-facts-figures/dementia-statistics/ |
[5] | Moss DE ((2020) ) Improving anti-neurodegenerative benefits of acetylcholinesterase inhibitors in Alzheimer’s disease: Are irreversible inhibitors the future? Int J Mol Sci 21: , 3438. |
[6] | Sharma K ((2019) ) Cholinesterase inhibitors as Alzheimer’s therapeutics. Mol Med Rep 20: , 1479–1487. |
[7] | Hampel H , Mesulam MM , Cuello AC , Farlow MR , Giacobini E , Grossberg GT , Khachaturian AS , Vergallo A , Cavedo E , Snyder PJ , Khachaturian ZS ((2018) ) The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 141: , 1917–1933. |
[8] | Kandiah N , Pai M-C , Senanarong V , Looi I , Ampil E , Park KW , Karanam AK , Christopher S ((2017) ) Rivastigmine: The advantages of dual inhibition of acetylcholinesterase and butyrylcholinesterase and its role in subcortical vascular dementia and Parkinson’s disease dementia. Clin Interv Aging 12: , 697–707. |
[9] | Eckroat TJ , Manross DL , Cowan SC ((2020) ) Merged tacrine-based, multitarget-directed acetylcholinesterase inhibitors 2015–present: Synthesis and biological activity. Int J Mol Sci 21: , 5965. |
[10] | Akaike A , Takada-Takatori Y , Kume T , Izumi Y ((2010) ) Mechanisms of neuroprotective effects of nicotine and acetylcholinesterase inhibitors: Role of α4 and α7 receptors in neuroprotection. J Mol Neurosci 40: , 211–216. |
[11] | Benfante R , Di Lascio S , Cardani S , Fornasari D ((2019) ) Acetylcholinesterase inhibitors targeting the cholinergic anti-inflammatory pathway: A new therapeutic perspective in aging-related disorders. Aging Clin Exp Res 33: , 823–834. |
[12] | Chierrito TPCC , Pedersoli-Mantoani S , Roca C , Requena C , Sebastian-Perez V , Castillo WO , Moreira NCSS , Pérez C , Sakamoto-Hojo ET , Takahashi CS , Jiménez-Barbero J , Cañada FJ , Campillo NE , Martinez A , Carvalho I , Mantoani SP , Roca C , Requena C , Sebastian V , Castillo WO , Moreira NCSS , Pérez C , Sakamoto-Hojo ET , Takahashi CS , Jiménez-Barbero J , Cañada FJ , Campillo NE , Martinez A , Carvalho I ((2017) ) From dual binding site acetylcholinesterase inhibitors to allosteric modulators: A new avenue for disease-modifying drugs in Alzheimer’s disease. Eur J Med Chem 139: , 773–791. |
[13] | Lionetto MG , Caricato R , Calisi A , Giordano ME , Schettino T ((2013) ) Acetylcholinesterase as a biomarker in environmental and occupational medicine: New insights and future perspectives. Biomed Res Int 2013: , 321213. |
[14] | Talesa VN ((2001) ) Acetylcholinesterase in Alzheimer’s disease. Mech Ageing Dev 122: , 1961–1969. |
[15] | Silman I , Sussman JL ((2008) ) Acetylcholinesterase: How is structure related to function? Chem Biol Interact 175: , 3–10. |
[16] | Xu Y , Cheng S , Sussman JL , Silman I , Jiang H ((2017) ) Computational studies on acetylcholinesterases. Molecules 22: , 1324. |
[17] | Darvesh S , Hopkins DA , Geula C ((2003) ) Neurobiology of butyrylcholinesterase. Nat Rev Neurosci 4: , 131–138. |
[18] | Johnson G , Moore SW ((2012) ) Why has butyrylcholinesterase been retained? Structural and functional diversification in a duplicated gene. Neurochem Int 61: , 783–797. |
[19] | Li Q , Yang H , Chen Y , Sun H ((2017) ) Recent progress in the identification of selective butyrylcholinesterase inhibitors for Alzheimer’s disease. Eur J Med Chem 132: , 294–309. |
[20] | Lane RM , Potkin SG , Enz A Targeting acetylcholinesterase and butyrylcholinesterase in dementia. Int J Neuropsychopharmacol 9: , 101–124. |
[21] | Anand P , Singh B ((2013) ) A review on cholinesterase inhibitors for Alzheimer’s disease. Arch Pharm Res 36: , 375–399. |
[22] | Ciro A , Park J , Burkhard G , Yan N , Geula C ((2012) ) Biochemical differentiation of cholinesterases from normal and Alzheimer’s disease cortex. Curr Alzheimer Res 9: , 138–143. |
[23] | Dinamarca MC , Sagal JP , Quintanilla RA , Godoy JA , Arrázola MS , Inestrosa NC ((2010) ) Amyloid-β-Acetylcholinesterase complexes potentiate neurodegenerative changes induced by the Aβ peptide. Implications for the pathogenesis of Alzheimer’s disease. Mol Neurodegener 5: , 4. |
[24] | Alvarez A , Alarcón R , Opazo C , Campos EO , Muñoz FJ , Calderón FH , Dajas F , Gentry MK , Doctor BP , De Mello FG , Inestrosa NC ((1998) ) Stable complexes involving acetylcholinesterase and amyloid-β peptide change the biochemical properties of the enzyme and increase the neurotoxicity of Alzheimer’s fibrils. J Neurosci 18: , 3213–3223. |
[25] | Carvajal FJ , Inestrosa NC ((2011) ) Interactions of AChE with Aβ aggregates in Alzheimer’s brain: Therapeutic relevance of IDN 5706. Front Mol Neurosci 4: , 19. |
[26] | Alvarez A , Opazo C , Alarcón R , Garrido J , Inestrosa NC ((1997) ) Acetylcholinesterase promotes the aggregation of amyloid-β-peptide fragments by forming a complex with the growing fibrils. J Mol Biol 272: , 348–361. |
[27] | Morán MA , Mufson EJ , Gómez-Ramos P ((1993) ) Colocalization of cholinesterases with β amyloid protein in aged and Alzheimer’s brains. Acta Neuropathol 85: , 362–369. |
[28] | Inestrosa NC , Dinamarca MC , Alvarez A ((2008) ) Amyloid-cholinesterase interactions. FEBS J 275: , 625–632. |
[29] | Price D , Dorandish S , Williams A , Iwaniec B , Stephens A , Marshall K , Guthrie J , Heyl D , Evans HG ((2020) ) Humanin blocks the aggregation of amyloid-β induced by acetylcholinesterase, an effect abolished in the presence of IGFBP-3. Biochemistry 59: , 1981. |
[30] | Diamant S , Podoly E , Friedler A , Ligumsky H , Livnah O , Soreq H ((2006) ) Butyrylcholinesterase attenuates amyloid fibril formation in vitro. Proc Natl Acad Sci U S A 103: , 8628–8633. |
[31] | Muñoz FJ , Inestrosa NC ((1999) ) Neurotoxicity of acetylcholinesterase amyloid β-peptide aggregates is dependent on the type of Aβ peptide and the AChE concentration present in the complexes. FEBS Lett 450: , 205–209. |
[32] | Bonnefont AB , Muñoz FJ , Inestrosa NC ((1998) ) Estrogen protects neuronal cells from the cytotoxicity induced by acetylcholinesterase-amyloid complexes. FEBS Lett 441: , 220–224. |
[33] | Reyes AE , Chacón MA , Dinamarca MC , Cerpa W , Morgan C , Inestrosa NC ((2004) ) Acetylcholinesterase-Aβ complexes are more toxic than Aβ fibrils in rat hippocampus: Effect on rat β-amyloid aggregation, laminin expression, reactive astrocytosis, and neuronal cell loss. Am J Pathol 164: , 2163–2174. |
[34] | Briggs R , Kennelly SP , O’Neill D ((2016) ) Drug treatments in Alzheimer’s disease. Clin Med (Lond) 16: , 247–253. |
[35] | Przybyłowska M , Kowalski S , Dzierzbicka K , Inkielewicz-Stepniak I ((2019) ) Therapeutic potential of multifunctional tacrine analogues. Curr Neuropharmacol 17: , 472–490. |
[36] | Horak M , Holubova K , Nepovimova E , Krusek J , Kaniakova M , Korabecny J , Vyklicky L , Kuca K , Stuchlik A , Ricny J , Vales K , Soukup O ((2017) ) The pharmacology of tacrine at N-methyl-d-aspartate receptors. Prog Neuropsychopharmacol Biol Psychiatry 75: , 54–62. |
[37] | Watkins PB , Zimmerman HJ , Knapp MJ , Gracon SI , Lewis KW ((1994) ) Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA 271: , 992–998. |
[38] | Cacabelos R ((2007) ) Donepezil in Alzheimer’s disease: From conventional trials to pharmacogenetics. Neuropsychiatr Dis Treat 3: , 303–333. |
[39] | Doody RS , Corey-Bloom J , Zhang R , Li H , Ieni J , Schindler R ((2012) ) Safety and tolerability of donepezil at doses up to 20mg/day. Drugs Aging 25: , 163–174. |
[40] | Silva MA , Kiametis AS , Treptow W ((2020) ) Donepezil inhibits acetylcholinesterase via multiple binding modes at room temperature. J Chem Inf Model 60: , 3463–3471. |
[41] | II JTB , Dell’Acqua S , Thach DQ , Sessler JL ((2018) ) Classics in chemical neuroscience: Donepezil. ACS Chem Neurosci 10: , 155–167. |
[42] | Adlimoghaddam A , Neuendorff M , Roy B , Albensi BC ((2018) ) A review of clinical treatment considerations of donepezil in severe Alzheimer’s disease. CNS Neurosci Ther 24: , 876–888. |
[43] | Onor ML , Trevisiol M , Aguglia E ((2007) ) Rivastigmine in the treatment of Alzheimer’s disease: An update. Clin Interv Aging 2: , 17–32. |
[44] | Szeto JYY , Lewis SJG ((2016) ) Current treatment options for Alzheimer’s disease and Parkinson’s disease dementia. Curr Neuropharmacol 14: , 326–338. |
[45] | Marucci G , Buccioni M , Ben DD , Lambertucci C , Volpini R , Amenta F ((2021) ) Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 190: , 108352. |
[46] | Čolović MB , Krstić DZ , Lazarević-Pašti TD , Bondžić AM , Vasić VM ((2013) ) Acetylcholinesterase inhibitors: Pharmacology and toxicology. Curr Neuropharmacol 11: , 315–335. |
[47] | Seltzer B ((2010) ) Galantamine-ER for the treatment of mild-to-moderate Alzheimer’s disease. Clin Interv Aging 5: , 1–6. |
[48] | Selkoe DJ , Hardy J ((2016) ) The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8: , 595–608. |
[49] | Kim HG , Moon M , Choi JG , Park G , Kim AJ , Hur J , Lee KT , Oh MS ((2014) ) Donepezil inhibits the amyloid-beta oligomer-induced microglial activation in vitro and in vivo.. Neurotoxicology 40: , 23–32. |
[50] | Noh M-Y , Koh S-H , Kim Y , Kim HY , Cho GW , Kim SH ((2009) ) Neuroprotective effects of donepezil through inhibition of GSK-3 activity in amyloid-β-induced neuronal cell death. J Neurochem 108: , 1116–1125. |
[51] | Takada-Takatori Y , Nakagawa S , Kimata R , Nao Y , Mizukawa Y , Urushidani T , Izumi Y , Akaike A , Tsuchida K , Kume T ((2019) ) Donepezil modulates amyloid precursor protein endocytosis and reduction by up-regulation of SNX33 expression in primary cortical neurons. Sci Rep 9: , 1922. |
[52] | Xiao XQ , Wang R , Tang XC ((2000) ) Huperzine A and tacrine attenuate-amyloid peptide-induced oxidative injury. J Neurosci Res 61: , 564–596. |
[53] | Svensson A-L , Nordberg A ((1998) ) Tacrine and donepezil attenuate the neurotoxic effect of Aβ(25-35) in rat PC12 cells. Neuroreport 9: , 1519–1522. |
[54] | Bailey JA , Ray B , Greig NH , Lahiri DK ((2011) ) Rivastigmine lowers Aβ and increases sAPPα levels, which parallel elevated synaptic markers and metabolic activity in degenerating primary rat neurons. PLoS One 6: , 21954. |
[55] | Takata K , Kitamura Y , Saeki M , Terada M , Kagitani S , Kitamura R , Fujikawa Y , Maelicke A , Tomimoto H , Taniguchi T , Shimohama S ((2010) ) Galantamine-induced amyloid-β clearance mediated via stimulation of microglial nicotinic acetylcholine receptors. J Biol Chem 285: , 40180. |
[56] | Melo JB , Sousa C , Garção P , Oliveira CR , Agostinho P ((2009) ) Galantamine protects against oxidative stress induced by amyloid-beta peptide in cortical neurons. Eur J Neurosci 29: , 455–464. |
[57] | Jiang S , Zhao Y , Zhang T , Lan J , Yang J , Yuan L , Zhang Q , Pan K , Zhang K ((2018) ) Galantamine inhibits β-amyloid-induced cytostatic autophagy in PC12 cells through decreasing ROS production. Cell Prolif 51: , e12427. |
[58] | Castillo WO , Aristizabal-Pachon AF , de Lima Montaldi AP , Sakamoto-Hojo ET , Takahashi CS ((2016) ) Galanthamine decreases genotoxicity and cell death induced by β-amyloid peptide in SH-SY5Y cell line. Neurotoxicology 57: , 291–297. |
[59] | Zheng H , Niu S , Zhao H , Li S , Jiao J ((2018) ) Donepezil improves the cognitive impairment in a tree shrew model of Alzheimer’s disease induced by amyloid-β1-40 via activating the BDNF/TrkB signal pathway. Metab Brain Dis 33: , 1961–1974. |
[60] | Easton A , Sankaranarayanan S , Tanghe A , Terwel D , Lin AX , Hoque N , Bourin C , Gu H , Ahlijanian M , Bristow L ((2013) ) Effects of sub-chronic donepezil on brain Abeta and cognition in a mouse model of Alzheimer’s disease. Psychopharmacology (Berl) 230: , 279–289. |
[61] | Dong H , Yuede CM , Coughlan CA , Murphy KM , Csernansky JG ((2009) ) Effects of donepezil on amyloid-β and synapse density in the Tg2576 mouse model of Alzheimer’s disease. Brain Res 1303: , 169–178. |
[62] | Tsunekawa H , Noda Y , Mouri A , Yoneda F , Nabeshima T ((2008) ) Synergistic effects of selegiline and donepezil on cognitive impairment induced by amyloid beta (25-35). Behav Brain Res 190: , 224–232. |
[63] | Geerts H ((2005) ) Indicators of neuroprotection with galantamine. Brain Res Bull 64: , 519–524. |
[64] | Wu Z , Zhao L , Chen X , Cheng X , Zhang Y ((2015) ) Galantamine attenuates amyloid-β deposition and astrocyte activation in APP/PS1 transgenic mice. Exp Gerontol 72: , 244–250. |
[65] | Saito T , Hisahara S , Iwahara N , Emoto MC , Yokokawa K , Suzuki H , Manabe T , Matsumura A , Suzuki S , Matsushita T , Kawamata J , Sato-Akaba H , Fujii HG , Shimohama S ((2019) ) Early administration of galantamine from preplaque phase suppresses oxidative stress and improves cognitive behavior in APPswe/PS1dE9 mouse model of Alzheimer’s disease. Free Radic Biol Med 145: , 20–32. |
[66] | Furukawa-Hibi Y , Alkam T , Nitta A , Matsuyama A , Mizoguchi H , Suzuki K , Moussaoui S , Yu Q-S , Greig NH , Nagai T , Yamada K ((2011) ) Butyrylcholinesterase inhibitors ameliorate cognitive dysfunction induced by amyloid-β peptide in mice. Behav Brain Res 225: , 222–229. |
[67] | Ma Y , Ji J , Li G , Yang S , Pan S ((2018) ) Effects of donepezil on cognitive functions and the expression level of β-amyloid in peripheral blood of patients with Alzheimer’s disease. Exp Ther Med 15: , 1875–1878. |
[68] | Zhang P , Xu S , Zhu Z , Xu J ((2019) ) Multi-target design strategies for the improved treatment of Alzheimer’s disease. Eur J Med Chem 176: , 228–247. |
[69] | Noh M-Y , Koh SH , Kim S-M , Maurice T , Ku S-K , Kim SH ((2013) ) Neuroprotective effects of donepezil against Aβ42-induced neuronal toxicity are mediated through not only enhancing PP2A activity but also regulating GSK-3β and nAChRs activity. J Neurochem 127: , 562–574. |
[70] | Buendia I , Parada E , Navarro E , León R , Negredo P , Egea J , López MG ((2015) ) Subthreshold concentrations of melatonin and galantamine improves pathological AD-hallmarks in hippocampal organotypic cultures. Mol Neurobiol 53: , 3338–3348. |
[71] | Arias E , Gallego-Sandín S , Villarroya M , García AG , López MG ((2005) ) Unequal neuroprotection afforded by the acetylcholinesterase inhibitors galantamine, donepezil, and rivastigmine in SH-SY5Y neuroblastoma cells: Role of nicotinic receptors. J Pharmacol Exp Ther 315: , 1346–1353. |
[72] | Talib LL , Hototian SR , Joaquim HPG , Forlenza OV , Gattaz WF ((2015) ) Increased iPLA2 activity and levels of phosphorylated GSK3B in platelets are associated with donepezil treatment in Alzheimer’s disease patients. Eur Arch Psychiatry Clin Neurosci 65: , 701–706. |
[73] | Shen H , Kihara T , Hongo H , Wu X , Kem WR , Shimohama S , Akaike A , Niidome T , Sugimoto H ((2010) ) Neuroprotection by donepezil against glutamate excitotoxicity involves stimulation of alpha7 nicotinic receptors and internalization of NMDA receptors. Br J Pharmacol 161: , 127–139. |
[74] | Takada Y , Yonezawa A , Kume T , Katsuki H , Kaneko S , Sugimoto H , Akaike A ((2003) ) Nicotinic acetylcholine receptor-mediated neuroprotection by donepezil against glutamate neurotoxicity in rat cortical neurons. J Pharmacol Exp Ther 306: , 772–777. |
[75] | Akasofu S , Kosasa T , Kimura M , Kubota A ((2003) ) Protective effect of donepezil in a primary culture of rat cortical neurons exposed to oxygen–glucose deprivation. Eur J Pharmacol 472: , 57–63. |
[76] | Kihara T , Sawada H , Nakamizo T , Kanki R , Yamashita H , Maelicke A , Shimohama S ((2004) ) Galantamine modulates nicotinic receptor and blocks Aβ-enhanced glutamate toxicity. Biochem Biophys Res Commun 325: , 976–982. |
[77] | Huang Z , Guo W , Zhang L , Song S , Hao C , Duan J ((2012) ) Donepezil protects endothelial cells against hydrogen peroxide-induced cell injury. CNS Neurosci Ther 18: , 185–187. |
[78] | Triana-Vidal LE , Carvajal-Varona SM ((2013) ) Protective effect of galantamine against oxidative damage using human lymphocytes: A novel in vitro model. Arch Med Res 44: , 85–92. |
[79] | Romero A , Egea J , García AG , López MG ((2010) ) Synergistic neuroprotective effect of combined low concentrations of galantamine and melatonin against oxidative stress in SH-SY5Y neuroblastoma cells. J Pineal Res 49: , 141–148. |
[80] | Imamura O , Arai M , Dateki M , Oishi K , Takishima K ((2020) ) Donepezil-induced oligodendrocyte differentiation is mediated through estrogen receptors. J Neurochem 155: , 494–507. |
[81] | Klionsky DJ , Abdelmohsen K , Abe A , Abedin MJ , Abeliovich H , Arozena AA , Adachi H , Adams CM , Adams PD , Adeli K , et al. ((2016) ) Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12: , 1–222. |
[82] | Sortino MA , Frasca G , Chisari M , Platania P , Chiechio S , Vancheri C , Copani A , Canonico PL ((2004) ) Novel neuronal targets for the acetylcholinesterase inhibitor donepezil. Neuropharmacology 47: , 1198–1204. |
[83] | Kotani S , Yamauchi T , Teramoto T , Ogura H ((2008) ) Donepezil, an acetylcholinesterase inhibitor, enhances adult hippocampal neurogenesis. Chem Biol Interact 175: , 227–230. |
[84] | Moreira dos NC S , Lima de JEB F , Chierrito TPC , Carvalho I , Sakamoto-Hojo ET ((2020) ) Novel hybrid acetylcholinesterase inhibitors induce differentiation and neuritogenesis in neuronal cells in vitro through activation of the AKT pathway. J Alzheimers Dis 78: , 353–370. |
[85] | Terada K , Migita K , Matsushima Y , Sugimoto Y , Kamei C , Matsumoto T , Mori M , Matsunaga K , Takata J , Karube Y ((2018) ) Cholinesterase inhibitor rivastigmine enhances nerve growth factor-induced neurite outgrowth in PC12 cells via sigma-1 and sigma-2 receptors. PLoS One 13: , e0209250. |
[86] | Islam MR , Moriguchi S , Tagashira H , Fukunaga K ((2014) ) Rivastigmine improves hippocampal neurogenesis and depression-like behaviors via 5-HT1A receptor stimulation in olfactory bulbectomized mice. Neuroscience 272: , 116–130. |
[87] | Taupin P ((2010) ) Adult neurogenesis and neural stem cells as a model for the discovery and development of novel drugs. Expert Opin Drug Discov 5: , 921–925. |
[88] | Jin K , Zhu Y , Sun Y , Mao XO , Xie L , Greenberg DA ((2002) ) Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc Natl Acad Sci U S A 99: , 11946–11950. |
[89] | Kita Y , Ago Y , Takano E , Fukada A , Takuma K , Matsuda T ((2012) ) Galantamine increases hippocampal insulin-like growth factor 2 expression via α7 nicotinic acetylcholine receptors in mice. Psychopharmacology (Berl) 225: , 543–551. |
[90] | Bailey JA , Lahiri DK ((2010) ) A novel effect of rivastigmine on presynaptic proteins and neuronal viability in a neurodegeneration model of fetal rat primary cortical cultures and its implication in Alzheimer’s disease. J Neurochem 112: , 843. |
[91] | Takada-Takatori Y , Kume T , Ohgi Y , Izumi Y , Niidome T , Fujii T , Sugimoto H , Akaike A ((2008) ) Mechanism of neuroprotection by donepezil pretreatment in rat cortical neurons chronically treated with donepezil. J Neurosci Res 86: , 3575–3583. |
[92] | O’Neill C ((2013) ) PI3-kinase/Akt/mTOR signaling: Impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp Gerontol 48: , 647–653. |
[93] | Fakhri S , Iranpanah A , Gravandi MM , Moradi SZ , Ranjbari M , Majnooni MB , Echeverría J , Qi Y , Wang M , Liao P , Farzaei MH , Xiao J ((2021) ) Natural products attenuate PI3K/Akt/mTOR signaling pathway: A promising strategy in regulating neurodegeneration. Phytomedicine 91: , 153664. |
[94] | Kumar M , Bansal N ((2021) ) Implications of phosphoinositide 3-kinase-Akt (PI3K-Akt) pathway in the pathogenesis of Alzheimer’s disease. Mol Neurobiol 59: , 354–385. |
[95] | Takada-Takatori Y , Kume T , Sugimoto M , Katsuki H , Sugimoto H , Akaike A ((2006) ) Acetylcholinesterase inhibitors used in treatment of Alzheimer’s disease prevent glutamate neurotoxicity via nicotinic acetylcholine receptors and phosphatidylinositol 3-kinase cascade. Neuropharmacology 51: , 474–486. |
[96] | Viel TA , Caetano AL , Albuquerque MS , Araujo MS , Buck HS ((2012) ) Chronic infusion of amyloid-beta peptide and sustained attention altered alpha7 nicotinic receptor density in the rat brain. Curr Alzheimer Res 9: , 1210–1220. |
[97] | Telles-Longui M , Mourelle D , Schöwe NM , Cipolli GC , Malerba HN , Buck HS , Viel TA ((2019) ) α7 nicotinic ACh receptors are necessary for memory recovery and neuroprotection promoted by attention training in amyloid-β-infused mice. Br J Pharmacol 176: , 3193–3205. |
[98] | Andriambeloson E , Huyard B , Poiraud E , Wagner S ((2014) ) Methyllycaconitine- and scopolamine-induced cognitive dysfunction: Differential reversal effect by cognition-enhancing drugs. Pharmacol Res Perspect 2: , e00048. |
[99] | Ho TNT , Abraham N , Lewis RJ ((2020) ) Structure-function of neuronal nicotinic acetylcholine receptor inhibitors derived from natural toxins. Front Neurosci 14: , 1209. |
[100] | Dani JA ((2015) ) Neuronal nicotinic acetylcholine receptor structure and function and response to nicotine. Int Rev Neurobiol 124: , 3–19. |
[101] | Kume T , Takada-Takatori Y ((2018) ) Nicotinic acetylcholine receptor signaling: Roles in neuroprotection. In Nicotinic Acetylcholine Receptor Signaling in Neuroprotection, Springer Singapore, pp. 59–71. |
[102] | Gabbouj S , Ryhänen S , Marttinen M , Wittrahm R , Takalo M , Kemppainen S , Martiskainen H , Tanila H , Haapasalo A , Hiltunen M , Natunen T ((2019) ) Altered insulin signaling in Alzheimer’s disease brain –special emphasis on PI3K-Akt pathway. Front Neurosci 13: , 629. |
[103] | Long HZ , Cheng Y , Zhou ZW , Luo HY , Wen DD , Gao LC ((2021) ) PI3K/AKT signal pathway: A target of natural products in the prevention and treatment of Alzheimer’s disease and Parkinson’s disease. Front Pharmacol 12: , 648636. |
[104] | Asnaghi L , Calastretti A , Bevilacqua A , D’Agnano I , Gatti G , Canti G , Delia D , Capaccioli S , Nicolin A ((2004) ) Bcl-2 phosphorylation and apoptosis activated by damaged microtubules require mTOR and are regulated by Akt. Oncogene 23: , 5781–5791. |
[105] | Liu R , Chen Y , Liu G , Li C , Song Y , Cao Z , Li W , Hu J , Lu C , Liu Y ((2020) ) PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers. Cell Death Dis 11: , 797. |
[106] | Zhang X , Tang N , Hadden TJ , Rishi AK ((2011) ) Akt, FoxO and regulation of apoptosis. Biochim Biophys Acta 1813: , 1978–1986. |
[107] | White MF ((2014) ) IRS2 integrates insulin/IGF1 signalling with metabolism, neurodegeneration and longevity. Diabetes Obes Metab 16: (Suppl 1), 4–15. |
[108] | Chen TJ , Wang DC , Chen SS ((2009) ) Amyloid-β interrupts the PI3K-Akt-mTOR signaling pathway that could be involved in brain-derived neurotrophic factor-induced Arc expression in rat cortical neurons. J Neurosci Res 87: , 2297–2307. |
[109] | Magrané J , Rosen KM , Smith RC , Walsh K , Gouras GK , Querfurth HW ((2005) ) Intraneuronal β-amyloid expression downregulates the Akt survival pathway and blunts the stress response. J Neurosci 25: , 10960–10969. |
[110] | Liu Y , Liu F , Grundke-Iqbal I , Iqbal K , Gong CX ((2011) ) Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J Pathol 225: , 54–62. |
[111] | Steen E , Terry BM , Rivera J. E , Cannon JL , Neely TR , Tavares R , Xu XJ , Wands JR , de la Monte SM ((2005) ) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease –is this type 3 diabetes? J Alzheimers Dis 7: , 63–80. |
[112] | He F , Ru X , Wen T ((2020) ) NRF2, a transcription factor for stress response and beyond. Int J Mol Sci 21: , 4777. |
[113] | Noble W , Hanger DP , Miller CCJ , Lovestone S ((2013) ) The importance of tau phosphorylation for neurodegenerative diseases. Front Neurol 4: , 83. |
[114] | Sims-Robinson C , Kim B , Rosko A , Feldman EL ((2010) ) How does diabetes accelerate Alzheimer disease pathology? Nat Rev Neurol 6: , 551–559. |
[115] | Griebel G , Stemmelin J , Lopez-Grancha M , Boulay D , Boquet G , Slowinski F , Pichat P , Beeské S , Tanaka S , Mori A , Fujimura M , Eguchi J ((2019) ) The selective GSK3 inhibitor, SAR502250, displays neuroprotective activity and attenuates behavioral impairments in models of neuropsychiatric symptoms of Alzheimer’s disease in rodents. Sci Rep 9: , 18045. |
[116] | Llorens-Martín M , Jurado J , Hernández F , Ávila J ((2014) ) GSK-3β, a pivotal kinase in Alzheimer disease. Front Mol Neurosci 7: , 46. |
[117] | Rada P , Rojo AI , Chowdhry S , McMahon M , Hayes JD , Cuadrado A ((2011) ) SCF/beta-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol 31: , 1121–1133. |
[118] | Rojo AI , Pajares M , Rada P , Nuñez A , Nevado-Holgado AJ , Killik R , Van Leuven F , Ribe E , Lovestone S , Yamamoto M , Cuadrado A ((2017) ) NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and tau pathology. Redox Biol 13: , 444–451. |
[119] | Branca C , Ferreira E , Nguyen TV , Doyle K , Caccamo A , Oddo S ((2017) ) Genetic reduction of Nrf2 exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. Hum Mol Genet 26: , 4823–4835. |
[120] | Bahn G , Park JS , Yun UJ , Lee YJ , Choi Y , Park JS , Baek SH , Choi BY , Cho YS , Kim HK , Han J , Sul JH , Baik SH , Lim J , Wakabayashi N , Bae SH , Han JW , Arumugam TV , Mattson MP , Jo DG ((2019) ) NRF2/ARE pathway negatively regulates BACE1 expression and ameliorates cognitive deficits in mouse Alzheimer’s models. Proc Natl Acad Sci U S A 116: , 12516–12523. |
[121] | Kim HV , Kim HY , Ehrlich HY , Choi SY , Kim DJ , Kim YS ((2013) ) Amelioration of Alzheimer’s disease by neuroprotective effect of sulforaphane in animal model. Amyloid 20: , 7–12. |
[122] | Liu GY , Sabatini DM ((2020) ) mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol 21: , 183–203. |
[123] | Li L , Zhang S , Zhang X , Li T , Tang Y , Liu H , Yang W , Le W ((2013) ) Autophagy enhancer carbamazepine alleviates memory deficits and cerebral amyloid-beta pathology in a mouse model of Alzheimer’s disease. Curr Alzheimer Res 10: , 433–441. |
[124] | Spilman P , Podlutskaya N , Hart MJ , Debnath J , Gorostiza O , Bredesen D , Richardson A , Strong R , Galvan V ((2010) ) Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-β levels in a mouse model of Alzheimer’s disease. PLoS One 5: , e9979. |
[125] | Silva MC , Nandi GA , Tentarelli S , Gurrell IK , Jamier T , Lucente D , Dickerson BC , Brown DG , Brandon NJ , Haggarty SJ ((2020) ) Prolonged tau clearance and stress vulnerability rescue by pharmacological activation of autophagy in tauopathy neurons. Nat Commun 11: , 3258. |
[126] | Hamano T , Gendron TF , Causevic E , Yen SH , Lin WL , Isidoro C , Deture M , Ko LW ((2008) ) Autophagic-lysosomal perturbation enhances tau aggregation in transfectants with induced wild-type tau expression. Eur J Neurosci 27: , 1119–1130. |
[127] | Son SM , Song H , Byun J , Park KS , Jang HC , Park YJ , Mook-Jung I ((2012) ) Altered APP processing in insulin-resistant conditions is mediated by autophagosome accumulation via the inhibition of mammalian target of rapamycin pathway. Diabetes 61: , 3126–3138. |
[128] | Ma T , Hoeffer CA , Capetillo-Zarate E , Yu F , Wong H , Lin MT , Tampellini D , Klann E , Blitzer RD , Gouras GK ((2010) ) Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer’s disease. PLoS One 5: , e12845. |
[129] | Hers I , Vincent EE , Tavaré JM ((2011) ) Akt signalling in health and disease. Cell Signal 23: , 1515–1527. |
[130] | Cui W , Wang S , Wang Z , Wang Z , Sun C , Zhang Y ((2017) ) Inhibition of PTEN attenuates endoplasmic reticulum stress and apoptosis via activation of PI3K/AKT pathway in Alzheimer’s disease. Neurochem Res 42: , 3052–3060. |
[131] | Knafo S , Sánchez-Puelles C , Palomer E , Delgado I , Draffin JE , Mingo J , Wahle T , Kaleka K , Mou L , Pereda-Perez I , Klosi E , Faber EB , Chapman HM , Lozano-Montes L , Ortega-Molina A , Ordóñez-Gutiérrez L , Wandosell F , Viña J , Dotti CG , Hall RA , Pulido R , Gerges NZ , Chan AM , Spaller MR , Serrano M , Venero C , Esteban JA ((2016) ) PTEN recruitment controls synaptic and cognitive function in Alzheimer’s models. Nat Neurosci 19: , 443–453. |
[132] | Zhou D , Zhou W , Song JK , Feng ZY , Yang RY , Wu S , Wang L , Liu AL , Du GH ((2016) ) DL0410, a novel dual cholinesterase inhibitor, protects mouse brains against Aβ-induced neuronal damage via the Akt/JNK signaling pathway. Acta Pharmacol Sin 37: , 1401. |
[133] | Zhou W , Lian WW , Yan R , Jia H , Xu LJ , Wang L , Liu AL , Du GH ((2019) ) DL0410 ameliorates cognitive deficits in APP/PS1 transgenic mice by promoting synaptic transmission and reducing neuronal loss. Acta Pharmacol Sin 41: , 599–611. |
[134] | Pang X , Zhao Y , Song J , Kang D , Wu S , Wang L , Liu A , Du G ((2019) ) Pharmacokinetics, excretion and metabolites analysis of DL0410, a dual-acting cholinesterase inhibitor and histamine-3 receptor antagonist. Mol Med Rep 20: , 1103–1112. |
[135] | Deardorff WJ , Shobassy A , Grossberg GT ((2015) ) Safety and clinical effects of EVP-6124 in subjects with Alzheimer’s disease currently or previously receiving an acetylcholinesterase inhibitor medication. Expert Rev Neurother 15: , 7–17. |
[136] | Gault LM , Ritchie CW , Robieson WZ , Pritchett Y , Othman AA , Lenz RA ((2015) ) A phase 2 randomized, controlled trial of the α7 agonist ABT-126 in mild-to-moderate Alzheimer’s dementia. Alzheimers Dement (N Y) 1: , 81. |
[137] | Lovestone S , Boada M , Dubois B , Hüll M , Rinne JO , Huppertz HJ , Calero M , Andrés MV , Gómez-Carrillo B , León T , Del Ser T ((2015) ) A phase II trial of Tideglusib in Alzheimer’s disease. J Alzheimers Dis 45: , 75–88. |
[138] | Del Ser T , Steinwachs KC , Gertz HJ , Andrés MV , Gómez-Carrillo B , Medina M , Vericat JA , Redondo P , Fleet D , León T ((2013) ) Treatment of Alzheimer’s disease with the GSK-3 inhibitor Tideglusib: A pilot study. J Alzheimers Dis 33: , 205–215. |
[139] | Georgievska B , Sandin J , Doherty J , Mörtberg A , Neelissen J , Andersson A , Gruber S , Nilsson Y , Schött P , Arvidsson PI , Hellberg S , Osswald G , Berg S , Fälting J , Bhat RV ((2013) ) AZD1080, a novel GSK3 inhibitor, rescues synaptic plasticity deficits in rodent brain and exhibits peripheral target engagement in humans. J Neurochem 125: , 446–456. |
[140] | Mishra P , Kumar A , Panda G ((2019) ) Anti-cholinesterase hybrids as multi-target-directed ligands against Alzheimer’s disease (1998-2018). Bioorg Med Chem 27: , 895–930. |
[141] | Digiacomo M , Chen Z , Wang S , Lapucci A , Macchia M , Yang X , Chu J , Han Y , Pi R , Rapposelli S ((2015) ) Synthesis and pharmacological evaluation of multifunctional tacrine derivatives against several disease pathways of AD. Bioorg Med Chem Lett 25: , 807–810. |
[142] | Jalili-Baleh L , Nadri H , Moradi A , Bukhari SNA , Shakibaie M , Jafari M , Golshani M , Homayouni Moghadam F , Firoozpour L , Asadipour A , Emami S , Khoobi M , Foroumadi A ((2017) ) New racemic annulated pyrazolo[1,2- b]phthalazines as tacrine-like AChE inhibitors with potential use in Alzheimer’s disease. Eur J Med Chem 139: , 280–289. |
[143] | Li G , Hong G , Li X , Zhang Y , Xu Z , Mao L , Feng X , Liu T ((2018) ) Synthesis and activity towards Alzheimer’s disease in vitro: Tacrine, phenolic acid and ligustrazine hybrids. Eur J Med Chem 148: , 238–254. |
[144] | Derabli C , Boualia I , Abdelwahab AB , Boulcina R , Bensouici C , Kirsch G , Debache A ((2018) ) A cascade synthesis, in vitro cholinesterases inhibitory activity and docking studies of novel Tacrine-pyranopyrazole derivatives. Bioorg Med Chem Lett 28: , 2481–2484. |
[145] | Chioua M , Buzzi E , Moraleda I , Iriepa I , Maj M , Wnorowski A , Giovannini C , Tramarin A , Portali F , Ismaili L , López-Alvarado P , Bolognesi ML , Jóźwiak K , Menéndez JC , Marco-Contelles J , Bartolini M ((2018) ) Tacripyrimidines, the first tacrine-dihydropyrimidine hybrids, as multi-target-directed ligands for Alzheimer’s disease. Eur J Med Chem 155: , 839–846. |
[146] | Lopes JPB , Silva L , da Costa Franarin G , Antonio Ceschi M , Seibert Lüdtke D , Ferreira Dantas R , de Salles CMC , Paes Silva F Jr , Roberto Senger M , Alvim Guedes I , Emmanuel Dardenne L ((2018) ) Design, synthesis, cholinesterase inhibition and molecular modelling study of novel tacrine hybrids with carbohydrate derivatives. Bioorganic Med Chem 26: , 5566–5577. |
[147] | Xu A , He F , Zhang X , Li X , Ran Y , Wei C , James Chou C , Zhang R , Wu J ((2020) ) Tacrine-hydroxamate derivatives as multitarget-directed ligands for the treatment of Alzheimer’s disease: Design, synthesis, and biological evaluation. Bioorg Chem 98: , 103721. |
[148] | Borioni JL , Cavallaro V , Murray AP , Peñéñory AB , Puiatti M , García ME ((2021) ) Design, synthesis and evaluation of cholinesterase hybrid inhibitors using a natural steroidal alkaloid as precursor. Bioorg Chem 111: , 104893. |
[149] | Rani A , Singh A , Kaur J , Singh G , Bhatti R , Gumede N , Kisten P , Singh P , Sumanjit , Kumar V ((2021) ) 1H-1,2,3-triazole grafted tacrine-chalcone conjugates as potential cholinesterase inhibitors with the evaluation of their behavioral tests and oxidative stress in mice brain cells. Bioorg Chem 114: , 105053. |
[150] | Ozten O , Zengin Kurt B , Sonmez F , Dogan B , Durdagi S ((2021) ) Synthesis, molecular docking and molecular dynamics studies of novel tacrine-carbamate derivatives as potent cholinesterase inhibitors. Bioorg Chem 115: , 105225–105225. |
[151] | Zawada K , Czarnecka K , Girek M , Kręcisz P , Trejtnar F , Mandíková J , Jończyk J , Bajda M , Staśkiewicz M , Wójtowicz P , Dziubek K , Skibiński R , Szymański P ((2021) ) New hybrids of tacrine and indomethacin as multifunctional acetylcholinesterase inhibitors. Chem Pap 75: , 249–264. |
[152] | Svobodova B , Mezeiova E , Hepnarova V , Hrabinova M , Muckova L , Kobrlova T , Jun D , Soukup O , Jimeno ML , Marco-Contelles J , Korabecny J ((2019) ) Exploring structure-activity relationship in tacrine-squaramide derivatives as potent cholinesterase inhibitors. Biomolecules 9: , 379. |
[153] | Gorecki L , Misiachna A , Damborsky J , Dolezal R , Korabecny J , Cejkova L , Hakenova K , Chvojkova M , Karasova JZ , Prchal L , Novak M , Kolcheva M , Kortus S , Vales K , Horak M , Soukup O ((2021) ) Structure-activity relationships of dually-acting acetylcholinesterase inhibitors derived from tacrine on N-methyl-d-Aspartate receptors. Eur J Med Chem 219: , 113434. |
[154] | Mohsin ul N A , Ahmad M ((2020) ) Donepezil: A review of the recent structural modifications and their impact on anti-Alzheimer activity. Brazilian J Pharm Sci 56: , 1–16. |
[155] | Unzeta M , Esteban G , Bolea I , Fogel WA , Ramsay RR , Youdim MBH , Tipton KF , Marco-Contelles J ((2016) ) Multi-target directed donepezil-like ligands for Alzheimer’s disease. Front Neurosci 10: , 205. |
[156] | Gabr MT , Abdel-Raziq MS ((2018) ) Design and synthesis of donepezil analogues as dual AChE and BACE-1 inhibitors. Bioorg Chem 80: , 245–252. |
[157] | Green KD , Fosso MY , Garneau-Tsodikova S ((2018) ) Multifunctional donepezil analogues as cholinesterase and BACE1 inhibitors. Molecules 23: , 3252. |
[158] | Fancellu G , Chand K , Tomás D , Orlandini E , Piemontese L , Silva DF , Cardoso SM , Chaves S , Santos MA ((2020) ) Novel tacrine-benzofuran hybrids as potential multi-target drug candidates for the treatment of Alzheimer’s disease. J Enzyme Inhib Med Chem 35: , 211–226. |
[159] | Chaves S , Resta S , Rinaldo F , Costa M , Josselin R , Gwizdala K , Piemontese L , Capriati V , Pereira-Santos AR , Cardoso SM , Santos MA ((2020) ) Design, synthesis, and in vitro evaluation of hydroxybenzimidazole-donepezil analogues as multitarget-directed ligands for the treatment of Alzheimer’s disease. Molecules 25: , 985. |
[160] | Camps P , Formosa X , Galdeano C , Gómez T , Muñoz-Torrero D , Scarpellini M , Viayna E , Badia A , Clos MV , Camins A , Pallàs M , Bartolini M , Mancini F , Andrisano V , Estelrich J , Lizondo M , Bidon-Chanal A , Luque FJ ((2008) ) Novel donepezil-based inhibitors of acetyl- and butyrylcholinesterase and acetylcholinesterase-induced β-amyloid aggregation. J Med Chem 51: , 3588–3598. |
[161] | de Andrade P , Mantoani SP , Gonçalves Nunes PS , Magadán CR , Pérez C , Xavier DJ , Hojo ETS , Campillo NE , Martínez A , Carvalho I ((2019) ) Highly potent and selective aryl-1,2,3-triazolyl benzylpiperidine inhibitors toward butyrylcholinesterase in Alzheimer’s disease. Bioorganic Med Chem 27: , 931–943. |


